 Stefano Regis
Stefano Regis Alessandra Dondero
Alessandra Dondero Fabio Caliendo
Fabio Caliendo Cristina Bottino
Cristina Bottino Roberta Castriconi
Roberta Castriconi- 1Laboratory of Clinical and Experimental Immunology, IRCCS Istituto Giannina Gaslini, Genoa, Italy
- 2Department of Experimental Medicine, University of Genoa, Genoa, Italy
- 3Department of Biological Engineering, Synthetic Biology Center, Massachusetts Institute of Technology, Cambridge, MA, United States
- 4Centre of Excellence for Biomedical Research, CEBR, University of Genoa, Genoa, Italy
TGF-β is a potent immunosuppressive cytokine that severely affects the function of NK cells. Tumor cells can take advantage of this ability, enriching their surrounding microenvironment with TGF-β. TGF-β can alter the expression of effector molecules and of activating and chemokine receptors, influence metabolism, induce the NK cell conversion toward the less cytolytic ILC1s. These and other changes possibly occur by the induction of complex gene expression programs, involving epigenetic mechanisms. While most of these programs are at present unexplored, the role of certain transcription factors, microRNAs and chromatin changes determined by TGF-β in NK cells start to be elucidated in human and/or mouse NK cells. The deep understanding of these mechanisms will be useful to design therapies contributing to restore the full NK function.
Introduction
NK cells are innate lymphocytes able to recognize and kill virus-infected and tumor cells. They are equipped with a set of inhibitory and activating receptors, whose integrated signaling selectively directs their activity (1). While inhibitory receptors bind molecules such as HLA class I or immune checkpoint ligands, activating receptors interact with stress molecules induced or upregulated by tumor transformation or viral infection. Several cytokines, like IL-2, IL-12, IL-15, and IL-18, influence NK cell activity. Stimulated NK cells can, in turn, produce other cytokines, including IFN-γ and TNF-α (1). Traditionally, two populations of NK cells are described, CD56bright, prevailing in secondary lymphoid organs, producing large amounts of cytokines, and CD56dim, abundant in peripheral blood, characterized by high cytotoxic potential. However, recent views consider the spectrum of NK cell diversity quite wider (2–4). Moreover, NK cells represent the cytolytic members of a large, innate lymphoid cell family (ILC) that includes ILC1, poor cytolytic cells sharing with NK cells the ability to produce IFN-γ, ILC2, secreting IL-5 and IL-13, and ILC3, producing IL-7 and IL-22 (5, 6).
The activity of NK cells can be strongly affected by TGF-β, an immunomodulatory cytokine with a prominent role in both innate and adaptive immune responses (7, 8). In particular, TGF-β is a negative regulator of IFN-γ production (9) and decreases the surface level of the activating receptors NKG2D and NKp30, reducing the cytotoxic ability of NK cells (10) and impairing their antitumor function. Accordingly, mice lacking the expression of the TGF-β receptor 2 (TGFBR2) in their NK cells show improved suppression of metastases compared to control mice (11). TGF-β, also alters the surface expression of the chemokine receptors CXCR3, CXCR4, CX3CR1, with a possible impact on NK cell migration and recruitment (12). Surprisingly, some of the modulatory effects of TGF-β are potentiated by IL-18, via p38/MAPK pathway (13). The negative regulatory role of TGF-β also extends to different metabolic pathways including glycolysis and mitochondrial processes (14, 15).
TGF-β acts on target cells via specific receptors composed of TGFBR1 and TGFBR2 subunits, which can transduce the signal phosphorylating SMAD2 and SMAD3 proteins. These molecules bind to SMAD4 forming heterotrimeric transcriptional complexes that accumulate in the nucleus, where they activate or repress the transcription of sets of target genes. Other SMAD-independent non-canonical pathways proceed via PI3K and MAPK (16).
TGF-β signaling has been associated with epigenetic alterations (16), i.e., reversible and heritable changes not associated with DNA sequence mutation. These include DNA methylation, post-translational modification of chromatin histones as well as changes in the expression of transcription factors and noncoding RNAs (17).
Several epigenetic modifications contributing to the deep influence of TGF-β on the NK cell function, including chromatin remodeling, induction or repression of transcription factors, and post-transcriptional regulation of gene expression by miRNAs, have been described, and are here reviewed.
Transcription Factors and Chromatin Remodeling
TGF-β affects NK cell function inducing relevant transcription programs, which can involve events of chromatin remodeling, through the action of various transcription factors; the landscape of these changes is presumably wide and mostly unexplored. Studies have been performed on the SMAD3-dependent regulation of T-bet and IFN-γ expression, on NK cell metabolism and conversion of NK cells to ILC1s (Figure 1).
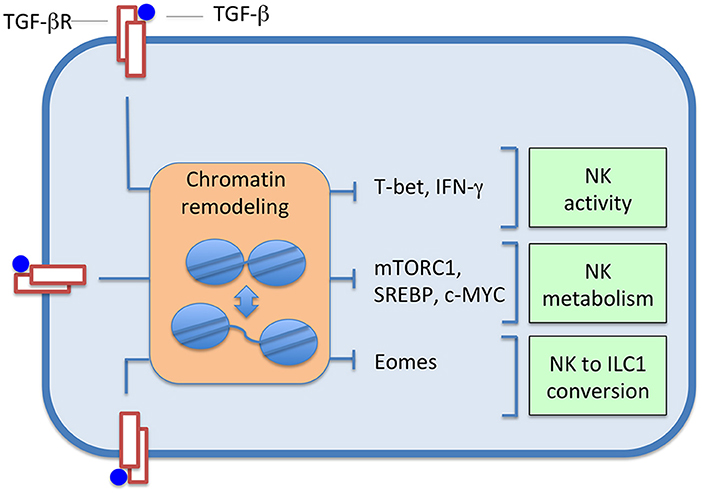
Figure 1. TGF-β induces changes in the expression of several transcription factors, and, consequently, in sets of controlled genes. Effects, often accompanied by chromatin alterations, range from inhibition of NK cell activity, to depression of cell metabolism, up to the induced conversion of NK cells to ILC1s.
T-bet-IFN-γ Pathway
Pro-inflammatory cytokines positively regulate the production of IFN-γ and the expression of the transcription factor T-bet, which, as the other T-box protein Eomes, has a relevant role in development and function of NK cells (18). Acting as an immunosuppressive cytokine, TGF-β antagonizes the effects of pro-inflammatory cytokines, down-regulating T-bet and IFN-γ via SMAD3 (9, 19). In line with this evidence, SMAD3-deficient mouse NK cells produced more IFN-γ (9), and TGFBR2-deficient mouse NK cells stimulated with IL-15 showed increased T-bet expression (11). Interestingly, chromatin remodeling has been associated with the T-bet-dependent regulation of IFN-γ gene expression in CD4 T cells polarizing toward TH1 cells (20). Moreover, it has been shown that T-bet, besides its ability to interact with the IFN-γ promoter and with several enhancers, can recruit histone-modifying complexes inducing epigenetic modifications at the IFN-γ locus (20, 21). In response to activating receptor engagement, CD56dim human NK cells showed increased expression of T-bet and IFN-γ, compared to CD56bright NK cells. High IFN-γ production was accompanied by a reduced CpG methylation pattern and histone modifications of the IFN-γ gene promoter (22). Moreover, epigenetic remodeling of the IFNG conserved non-coding sequence (CNS) 1, located upstream of the human IFN-γ promoter, has been documented in NKG2Chi “adaptive” NK cells, which expand in CMV seropositive individuals (23).
Interestingly, “TGF-β-imprinted” NK cells, produced by prolonged IL-2 and TGF-β stimulation of activated NK cells, contrarily to acutely TGF-β-treated NK cells, exhibited a pro-inflammatory phenotype, with abundant production of IFN-γ and TNF-α persisting after removal of TGF-β (24). The TGF-β pathway was altered in these cells, as indicated by the down-regulation of SMAD3, whose locus showed reduced chromatin accessibility, and of T-bet.
Of note, TGF-β can inhibit IFN-γ expression both by a T-bet dependent and a T-bet independent manner (9). In the latter context, Tang et al. (25) reported that SMAD3 suppresses transcription of IFN-γ via E4BP4, which is an important transcription factor for the NK cell lineage commitment (26).
From a more general perspective, the intimate link between T-bet and chromatin modifications is underlined by a recent study exploring chromatin and gene expression changes associated with viral infection in mouse CD8 T cells. The authors reported that T-box and Runx genes have been shown to act as critical mediators of chromatin remodeling during CD8 T cell activation (27).
NK Cell Metabolism: mTORC1, SREBP, and c-MYC
TGF-β can sensibly reduce NK cell metabolism affecting glycolysis and oxidative phosphorylation, which are considerably increased in cytokine-stimulated NK cells and support effector functions (11). The serine/threonine kinase mTOR is part of the mTORC1 complex, a master regulator of various metabolic processes in different cell types. mTORC1 activity is important for activation-induced metabolic and functional responses in NK cells (14). TGF-β was found to inhibit mTOR-dependent activities in NK stimulated by IL-15 (11). Moreover, mouse TGFBR2-defective NK cells stimulated with IL-15 showed increased mTOR-dependent activities. mTORC1 also contributed to the regulation of the SREBP (28) and c-MYC (29) transcription factors, which are key controllers of glycolysis and oxidative phosphorylation in cytokine-stimulated NK cells (14).
In humans, it has been reported that TGF-β, acting by the canonical pathway, significantly decreases the rate of IL-2-induced mitochondrial metabolism, and inhibits the glycolysis in IL-2 stimulated human NK cells independently by mTORC1 (14, 30).
NK-ILC1 Cell Conversion
The potency of TGF-β action is further highlighted by its ability to induce the conversion of NK cells to ILC1s (31, 32). Although NK cells and ILC1s are both IFN-γ producers and functionally dependent on T-bet (5), they are developmentally distinct. ILC1s are not found in blood and lymphoid organs, having instead tissue-resident features (33) and being weakly cytolytic (5). Moreover, while mature NK cells express Eomes, ILC1s do not.
Gao et al. (31) reported that, in the tumor microenvironment, TGF-β can induce the conversion of mouse NK cells to an NK-ILC1 intermediate cell type (intILC1s) and, finally, to ILC1s; the switch is accompanied by the down-regulation of the transcription factor Eomes. Compared to NK cells, intILC1s and ILC1s are unable to control tumor growth and metastasis. Therefore, TGF-β, inducing the conversion, inhibits cancer immunosurveillance by a new mechanism of immune evasion. Cortez et al. (32) generated SMAD4-deficient NK cells, which showed features of ILC1s, had impaired effector functions and were unable to control tumor metastasis, thus indicating that SMAD4 is a negative regulator of the NK-ILC1s conversion. In particular, SMAD4 was shown to hamper the conversion of NK cells to ILC1s inhibiting a non-canonical TGFBR1-mediated pathway.
As stated by the RNAseq-based transcriptome data obtained by NKs, intILC1s, and ILC1s cell populations (31), the NK to ILC1 conversion induced by TGF-β involves sets of genes, presumably governed by still undefined, complex, transcriptional and epigenetic programs. Given the reduction of Eomes expression during cell conversion and its potential role as chromatin remodeler, we can speculate that it may have a relevant role in this event of cell plasticity. In a different context, similar considerations can be done for T-bet, which is reduced by TGF-β in human liver resident NK cells, this reduction being fundamental for the maintenance of the Eomeshi T-betlo phenotype of this subtype of NK cells (4).
Notably, together with IL-23, TGF-β guides the conversion ILC1 toward ILC3, which can occur in mucosal tissues (34). TGF-β increases the expression of the transcription factors T-bet and Aiolos. Interestingly, Aiolos, like the other IKZF members, can suppress target gene expression altering chromatin accessibility via histones deacetylation (35). By this mechanism, it suppresses the ILC3 specific genes IL-22 and RORγt, promoting the ILC3-ILC1 conversion (34).
miRNAs
Besides chromatin remodeling and transcription factor manipulation, TGF-β can affect the NK cell function acting on the miRNA machinery, exploiting its capacity to influence gene expression at the post-transcriptional level. Distinct miRNA-containing pathways deregulating the expression of receptors or transcription factors have been described (Figure 2) (36).
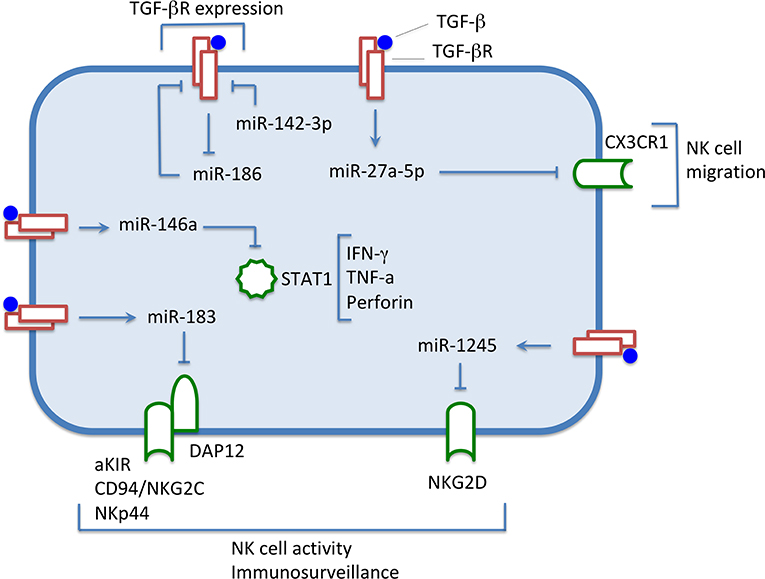
Figure 2. Under the influence of TGF-β, levels of expression of some miRNAs are increased, down-regulating the expression of surface molecules and transcription factors involved in NK cell activity. Conversely, miRNA-186 level decreases, thus preventing the down-regulation of the TGF-β receptor expression, and maintaining the NK cells responsiveness to the immunomodulatory cytokine. miR-142-3p down-regulates TGFBR1, thus modulating the TGF-β signaling.
miR-1245
NKG2D is a C-type lectin-like activating receptor expressed on NK and CD8 T cells, which promotes the elimination of transformed and pathogen-infected cells. In humans, it recognizes stress-inducible ligands belonging to the MIC (MICA and MICB) and ULBP (ULBP1-6) families (37).
The surface expression of NKG2D is significantly down-regulated in NK cells by TGF-β1 or TGF-β2 (10). Espinoza et al. (38) reported that TGF-β1 causes an increase of miR-1245 in human NK cells. Overexpression of miR-1245 induced a down-regulation of NKG2D at the cell surface without affecting the other activating receptors NKp30, NKp44, and NKp46. Moreover, NK cells overexpressing miR-1245 had lower cytotoxicity against target cells expressing NKG2D ligands. MiR-1245 was shown to directly target and down-regulate the NKG2D mRNA. Interestingly, a polymorphism in the NKG2D 3′-UTR region occurs in the region complementary to the miR-1245 seed region (39). NK cells carrying this polymorphism exhibit a reduced TGF-β-mediated modulation of NKG2D activity, which can be due to less efficient targeting of the miRNA. Importantly, the NKG2D variants seem to influence immunosurveillance capability and risk of cancer development (40).
miR-183
DAP12 (also known as KARAP or TYROBP) is a 12 kDa transmembrane protein containing an immune tyrosine-based activation motif (ITAM) in its cytoplasmic domain (41). DAP12 is a signaling molecule associating with different activating receptors, including the activating KIRs, CD94/NKG2C, and NKp44, the latter expressed by NK cells upon activation and by a subset of ILC3 (42).
Donatelli et al. (43) reported that TGF-β1 down-regulates the DAP12 level in human NK cells. In particular, it up-regulated miR-183, which was shown to directly target DAP12 mRNA. The NK92 cell line overexpressing miR-183 had reduced DAP12, decreased NKp44 surface levels, and was less efficient in killing the Raji Burkitt lymphoma cell line.
Interestingly, NK cells infiltrating lung cancers were shown to have a diminished DAP12 expression (43). Recently, a mechanism of immune evasion has been described in human lung cancer cells (44). TGF-β appears to act simultaneously on NK cells and tumor cells via miR-183, with the result to inhibit the NK cells' anti-tumor activity by down-regulating DAP12, in NK cells, and NKG2D ligands (MICA and MICB), in cancer cells.
miR-27a-5p
CX3CR1 is a chemokine receptor expressed by different immune cell types, including NK cells, which binds CX3CL1 (also known as fractalkine). CX3CR1 drives, with other chemokine receptors, NK cells localization at a steady state in peripheral tissues, and promotes their migration under inflammatory conditions (45). Moreover, CX3CR1, CXCR4, and S1P5 regulate NK cells homing and egress from the bone marrow (46, 47).
It has been shown that TGF-β1 released by neuroblastoma tumor cells down-regulated the surface expression of CX3CR1 in human NK cells (12). Afterward, TGF-β1 was found to up-regulate in NK cells the miR-23a-27a-24-2 cluster, encoding, among others, miR-27a-5p, that has been demonstrated to target CX3CR1 (48). In agreement with this finding (12), NK cells cultured in the presence of neuroblastoma cells exhibited a significant inverse correlation between miR-27a-5p and CX3CR1 mRNA expression (48). Notably, an unusual CX3CR1low/neg phenotype has been described in circulating CD56dim NK cells and in NKp46+ bone marrow resident NK cells from high-risk NB patients (12).
miR-186
Neviani et al. (49) reported that miR-186 down-regulates TGFBR1 and TGFBR2 preventing the TGF-β-dependent inhibition of human NK cells. MiR-186 was found to be down-regulated in TGF-β-treated NK cells as well as in neuroblastoma cells. Importantly, by receiving miR-186, carried by NK exosomes, neuroblastoma cells down-regulated the oncogenic proteins MYCN and AURKA. Therefore, delivery of miR-186 to neuroblastoma and NK cells may hamper both tumorigenic potential and inhibition of NK cells.
miR-142
MiR-142 is a critical regulator of ILC biology. It is induced by IL-15 and mediates modulation of TGFBR1. miR-142−/− knockout mice showed increased TGF-β signaling compared to normal controls, as indicated by the augmented expression of TGFBR1 and functionally related genes in NK/ILC1 cells from spleen and bone marrow (50). Interestingly, miR-142−/− mice presented an altered number and functionality of NK cells, with increased susceptibility to cytomegalovirus infection. On the contrary, an increase of ILC1-like cells was observed, possibly related to the increased TGF-β signaling and NK-ILC1 conversion.
miR-146a
Xu et al. (51) reported that human miR-146a, which is up-regulated by TGF-β in NK cells, targets STAT1, a transcription factor involved in interferons signal transduction, which contributes to NK cell cytotoxicity (52). miR-146 overexpression lowered the production of effector molecules as IFN-γ, TNF-α, and perforin. Moreover, the expression level of miR-146a negatively correlated with NK cell-mediated cytotoxicity (51).
Concluding Remarks
TGF-β can exert its immunosuppressive action influencing NK cells at different levels. Acting by transcription factors, TGF-β can hamper NK cell function inhibiting the T-bet-IFN-γ pathway and negatively influences cell metabolism, thus hampering cell activation. Using miRNAs, TGF-β down-regulates activating receptors, receptor-associated signaling molecules, and chemokine receptors, thus inhibiting NK cell cytotoxicity and recruitment in tissues. Acting by still undefined combined transcriptional and epigenetic programs, TGF-β converts NK cells to ILC1, which are less cytolytic against tumor cells. Moreover, we can speculate that long non-coding RNA (lncRNAs), which are known to be involved in TGF-β-driven fibrosis, epithelial-mesenchymal transition and cancer progression (53, 54), may represent additional mediators of still undefined TGF-β-dependent immune-modulatory functions on NK cells.
Studying how TGF-β alters the NK/ILC biology through epigenetic mechanisms could be an important area of investigation to understand its functions in the tumor microenvironment and in other diseases containing TGF-β rich milieus, such as fibrosis. In this context, it will be also important the development of molecular approaches aimed to control the TGF-β signaling. Several strategies have been recently elaborated for the therapeutic inhibition of the TGF-β pathway, particularly to improve the antitumor immune responses. Combination therapy with an inhibitor of TGFβ type I receptor (TGFβR1) kinase, galunisertib, anti-GD2 antibody and ex vivo activated NK cells, has been used by Tran et al. (55) in a mouse model of neuroblastoma with positive results. Genetically modified SMAD3-silenced human NK-92 cells inhibited cancer progression in two xenograft mouse models with human hepatoma and melanoma (56). NK cells genetically modified to express a truncated TGFBR2 receptor fused to the DAP12 activation domain exhibited higher cytotoxic activity against neuroblastoma (57). A deeper knowledge of the mechanisms by which TGF-β acts on NK cells will be useful to improve therapeutic strategies aimed to efficiently restore NK cell activity or to increase the NK cell pool for an effective antitumor response.
Author Contributions
All authors contributed to the writing of the manuscript.
Funding
This work from our laboratories has been supported by Associazione Italiana per la Ricerca sul Cancro (A.I.R.C.), Ministero dell'Istruzione, dell'Università e della Ricerca (M.I.U.R), and 100008-2018-SS-DIP_001 project (Department of Experimental Medicine, School of Medical and Pharmaceutical Science, University of Genova).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Björkström NK, Ljunggren H-G, Michaëlsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. (2016) 16:310–20. doi: 10.1038/nri.2016.34
2. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
3. Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. (2018) 49:971–86.e5. doi: 10.1016/j.immuni.2018.09.009
4. Harmon C, Jameson G, Almuaili D, Houlihan DD, Hoti E, Geoghegan J, et al. Liver-derived TGF-β maintains the EomeshiTbetlo phenotype of liver resident natural killer cells. Front Immunol. (2019) 10:1502. doi: 10.3389/fimmu.2019.01502
5. Spits H, Bernink JH, Lanier L. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol. (2016) 17:758–64. doi: 10.1038/ni.3482
6. Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol. (2013) 13:145–9. doi: 10.1038/nri3365
7. Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
8. David CJ, Massagué J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat Rev Mol Cell Biol. (2018) 19:419–35. doi: 10.1038/s41580-018-0007-0
9. Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, et al. Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity. (2006) 24:575–90. doi: 10.1016/j.immuni.2006.03.016
10. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci USA. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
11. Viel S, Marçais A, Guimaraes FS-F, Loftus R, Rabilloud J, Grau M, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. (2016) 9:ra19. doi: 10.1126/scisignal.aad1884
12. Castriconi R, Dondero A, Bellora F, Moretta L, Castellano A, Locatelli F, et al. Neuroblastoma-derived TGF-β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol. (2013) 190:5321–8. doi: 10.4049/jimmunol.1202693
13. Casu B, Dondero A, Regis S, Caliendo F, Petretto A, Bartolucci M, et al. Novel immunoregulatory functions of IL-18, an accomplice of TGF-β1. Cancers. (2019) 11:E75. doi: 10.3390/cancers11010075
14. O'Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. (2019) 19:282–90. doi: 10.1038/s41577-019-0139-2
15. Viel S, Besson L, Marotel M, Walzer T, Marçais A. Regulation of mTOR, metabolic fitness, and effector functions by cytokines in natural killer cells. Cancers. (2017) 9:E132. doi: 10.3390/cancers9100132
16. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. (2012) 13:616–30. doi: 10.1038/nrm3434
17. Bugide S, Janostiak R, Wajapeyee N. Epigenetic mechanisms dictating eradication of cancer by natural killer cells. Trends Cancer. (2018) 4:553–66. doi: 10.1016/j.trecan.2018.06.004
18. Zhang J, Marotel M, Fauteux-Daniel S, Mathieu A-L, Viel S, Marçais A, et al. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur J Immunol. (2018) 48:738–50. doi: 10.1002/eji.201747299
19. Trotta R, Col JD, Yu J, Ciarlariello D, Thomas B, Zhang X, et al. TGF-β utilizes SMAD3 to inhibit CD16-mediated IFN-γ production and antibody-dependent cellular cytotoxicity in human NK cells. J Immunol. (2008) 181:3784–92. doi: 10.4049/jimmunol.181.6.3784
20. Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. (2009) 9:91–105. doi: 10.1038/nri2487
21. Miller SA, Weinmann AS. Molecular mechanisms by which T-bet regulates T-helper cell commitment. Immunol Rev. (2010) 238:233–46. doi: 10.1111/j.1600-065X.2010.00952.x
22. Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, et al. NK cells gain higher IFN-γ competence during terminal differentiation. Eur J Immunol. (2014) 44:2074–84. doi: 10.1002/eji.201344072
23. Luetke-Eversloh M, Hammer Q, Durek P, Nordström K, Gasparoni G, Pink M, et al. Human cytomegalovirus drives epigenetic imprinting of the IFNG locus in NKG2Chi natural killer cells. PLoS Pathog. (2014) 10:e1004441. doi: 10.1371/journal.ppat.1004441
24. Foltz JA, Moseman JE, Thakkar A, Chakravarti N, Lee DA. TGFβ imprinting during activation promotes natural killer cell cytokine hypersecretion. Cancers. (2018) 10:E423. doi: 10.3390/cancers10110423
25. Tang PM-K, Zhou S, Meng X-M, Wang Q-M, Li C-J, Lian G-Y, et al. Smad3 promotes cancer progression by inhibiting E4BP4-mediated NK cell development. Nat Commun. (2017) 8:14677. doi: 10.1038/ncomms14677
26. Male V, Nisoli I, Kostrzewski T, Allan DSJ, Carlyle JR, Lord GM, et al. The transcription factor E4bp4/Nfil3 controls commitment to the NK lineage and directly regulates Eomes and Id2 expression. J Exp Med. (2014) 211:635–42. doi: 10.1084/jem.20132398
27. van der Veeken J, Zhong Y, Sharma R, Mazutis L, Dao P, Pe'er D, et al. Natural genetic variation reveals key features of epigenetic and transcriptional memory in virus-specific CD8 T cells. Immunity. (2019) 50:1202–17.e7. doi: 10.1016/j.immuni.2019.03.031
28. Assmann N, O'Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. (2017) 18:1197–206. doi: 10.1038/ni.3838
29. Loftus RM, Assmann N, Kedia-Mehta N, O'Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. (2018) 9:2341. doi: 10.1038/s41467-018-04719-2
30. Zaiatz-Bittencourt V, Finlay DK, Gardiner CM. Canonical TGF-β signaling pathway represses human NK cell metabolism. J Immunol. (2018) 200:3934–41. doi: 10.4049/jimmunol.1701461
31. Gao Y, Souza-Fonseca-Guimaraes F, Bald T, Ng SS, Young A, Ngiow SF, et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat Immunol. (2017) 18:1004–15. doi: 10.1038/ni.3800
32. Cortez VS, Ulland TK, Cervantes-Barragan L, Bando JK, Robinette ML, Wang Q, et al. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat Immunol. (2017) 18:995–1003. doi: 10.1038/ni.3809
33. Fuchs A. ILC1s in tissue inflammation and infection. Front Immunol. (2016) 7:104. doi: 10.3389/fimmu.2016.00104
34. Cella M, Gamini R, Sécca C, Collins PL, Zhao S, Peng V, et al. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat Immunol. (2019) 20:980–91. doi: 10.1038/s41590-019-0425-y
35. Koipally J, Renold A, Kim J, Georgopoulos K. Repression by Ikaros and Aiolos is mediated through histone deacetylase complexes. EMBO J. (1999) 18:3090–100. doi: 10.1093/emboj/18.11.3090
36. Regis S, Caliendo F, Dondero A, Bellora F, Casu B, Bottino C, et al. Main NK cell receptors and their ligands: regulation by microRNAs. Allergy. (2018) 2:98–112. doi: 10.3934/Allergy.2018.2.98
37. Zingoni A, Molfetta R, Fionda C, Soriani A, Paolini R, Cippitelli M, et al. NKG2D and its ligands: “one for all, all for one.” Front Immunol. (2018) 9:476. doi: 10.3389/fimmu.2018.00476
38. Espinoza JL, Takami A, Yoshioka K, Nakata K, Sato T, Kasahara Y, et al. Human microRNA-1245 down-regulates the NKG2D receptor in natural killer cells and impairs NKG2D-mediated functions. Haematologica. (2012) 97:1295–303. doi: 10.3324/haematol.2011.058529
39. Espinoza JL, Nguyen VH, Ichimura H, Pham TTT, Nguyen CH, Pham TV, et al. A functional polymorphism in the NKG2D gene modulates NK-cell cytotoxicity and is associated with susceptibility to Human Papilloma Virus-related cancers. Sci Rep. (2016) 6:39231. doi: 10.1038/srep39231
40. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res. (2006) 66:563–70. doi: 10.1158/0008-5472.CAN-05-2776
41. Tomasello E, Vivier E. KARAP/DAP12/TYROBP: three names and a multiplicity of biological functions. Eur J Immunol. (2005) 35:1670–7. doi: 10.1002/eji.200425932
42. Hoorweg K, Peters CP, Cornelissen F, Aparicio-Domingo P, Papazian N, Kazemier G, et al. Functional differences between human NKp44(-) and NKp44(+) RORC(+) innate lymphoid cells. Front Immunol. (2012) 3:72. doi: 10.3389/fimmu.2012.00072
43. Donatelli SS, Zhou J-M, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. TGF-β-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci USA. (2014) 111:4203–8. doi: 10.1073/pnas.1319269111
44. Trinh TL, Kandell WM, Donatelli SS, Tu N, Tejera MM, Gilvary DL, et al. Immune evasion by TGFβ-induced miR-183 repression of MICA/B expression in human lung tumor cells. OncoImmunology. (2019) 8:e1557372. doi: 10.1080/2162402X.2018.1557372
45. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
46. Sciumè G, De Angelis G, Benigni G, Ponzetta A, Morrone S, Santoni A, et al. CX3CR1 expression defines 2 KLRG1+ mouse NK-cell subsets with distinct functional properties and positioning in the bone marrow. Blood. (2011) 117:4467–75. doi: 10.1182/blood-2010-07-297101
47. Ponzetta A, Sciumè G, Benigni G, Antonangeli F, Morrone S, Santoni A, et al. CX3CR1 regulates the maintenance of KLRG1+ NK cells into the bone marrow by promoting their entry into circulation. J Immunol. (2013) 191:5684–94. doi: 10.4049/jimmunol.1300090
48. Regis S, Caliendo F, Dondero A, Casu B, Romano F, Loiacono F, et al. TGF-β1 downregulates the expression of CX3CR1 by inducing miR-27a-5p in primary human NK cells. Front Immunol. (2017) 8:868. doi: 10.3389/fimmu.2017.00868
49. Neviani P, Wise PM, Murtadha M, Liu CW, Wu C-H, Jong AY, et al. Natural killer–derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. (2019) 79:1151–64. doi: 10.1158/0008-5472.CAN-18-0779
50. Berrien-Elliott MM, Sun Y, Neal C, Ireland A, Trissal MC, Sullivan RP, et al. MicroRNA-142 Is critical for the homeostasis and function of type 1 innate lymphoid cells. Immunity. (2019) 51:479–90.e6. doi: 10.1016/j.immuni.2019.06.016
51. Xu D, Han Q, Hou Z, Zhang C, Zhang J. miR-146a negatively regulates NK cell functions via STAT1 signaling. Cell Mol Immunol. (2017) 14:712–20. doi: 10.1038/cmi.2015.113
52. Paolini R, Bernardini G, Molfetta R, Santoni A. NK cells and interferons. Cytokine Growth Factor Rev. (2015) 26:113–20. doi: 10.1016/j.cytogfr.2014.11.003
53. Tang PM-K, Zhang Y-Y, Lan H-Y. LncRNAs in TGF-β-driven tissue fibrosis. Non Coding RNA. (2018) 4:E26. doi: 10.3390/ncrna4040026
54. Hao Y, Baker D, Ten Dijke P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci. (2019) 20:E2767. doi: 10.3390/ijms20112767
55. Tran HC, Wan Z, Sheard MA, Sun J, Jackson JR, Malvar J, et al. TGFβR1 blockade with Galunisertib (LY2157299) enhances anti-neuroblastoma activity of the anti-GD2 antibody dinutuximab (ch14.18) with natural killer cells. Clin Cancer Res. (2017) 23:804–13. doi: 10.1158/1078-0432.CCR-16-1743
56. Wang Q-M, Tang PM-K, Lian G-Y, Li C, Li J, Huang X-R, et al. Enhanced cancer immunotherapy with Smad3-silenced NK-92 cells. Cancer Immunol Res. (2018) 6:965–77. doi: 10.1158/2326-6066.CIR-17-0491
Keywords: NK cells, TGF-β, epigenetic, transcription factors, microRNAs
Citation: Regis S, Dondero A, Caliendo F, Bottino C and Castriconi R (2020) NK Cell Function Regulation by TGF-β-Induced Epigenetic Mechanisms. Front. Immunol. 11:311. doi: 10.3389/fimmu.2020.00311
Received: 13 September 2019; Accepted: 07 February 2020;
Published: 25 February 2020.
Edited by:
Laurent Brossay, Brown University, United StatesReviewed by:
Frank Momburg, German Cancer Research Center (DKFZ), GermanyMarina Cella, Washington University School of Medicine in St. Louis, United States
Copyright © 2020 Regis, Dondero, Caliendo, Bottino and Castriconi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cristina Bottino, Y3Jpc3RpbmEuYm90dGlub0B1bmlnZS5pdA==