
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 26 February 2020
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00254
This article is part of the Research TopicPerinatal ImmunobiologyView all 34 articles
 Valeria Garcia-Flores1,2
Valeria Garcia-Flores1,2 Roberto Romero1,3,4,5,6,7
Roberto Romero1,3,4,5,6,7 Amy-Eunice Furcron1,2
Amy-Eunice Furcron1,2 Dustyn Levenson1,2Jose Galaz1,2Chengrui Zou1,2Sonia S. Hassan1,2,8,9Chaur-Dong Hsu1,2,9David Olson10
Dustyn Levenson1,2Jose Galaz1,2Chengrui Zou1,2Sonia S. Hassan1,2,8,9Chaur-Dong Hsu1,2,9David Olson10 Gerlinde A. S. Metz11
Gerlinde A. S. Metz11 Nardhy Gomez-Lopez1,2,12*
Nardhy Gomez-Lopez1,2,12*Maternal stress is a well-established risk factor for preterm birth and has been associated with adverse neonatal outcomes in the first and subsequent generations, including increased susceptibility to disease and lasting immunological changes. However, a causal link between prenatal maternal stress and preterm birth, as well as compromised neonatal immunity, has yet to be established. To fill this gap in knowledge, we used a murine model of prenatal maternal stress across three generations and high-dimensional flow cytometry to evaluate neonatal adaptive immunity. We report that recurrent prenatal maternal stress induced preterm birth in the first and second filial generations and negatively impacted early neonatal growth. Strikingly, prenatal maternal stress induced a systematic reduction in T cells and B cells, the former including regulatory CD4+ T cells as well as IL-4- and IL-17A-producing T cells, in the second generation. Yet, neonatal adaptive immunity gained resilience against prenatal maternal stress by the third generation. We also show that the rate of prenatal maternal stress-induced preterm birth can be reduced upon cessation of stress, though neonatal growth impairments persisted. These findings provide evidence that prenatal maternal stress causes preterm birth and affects neonatal immunity across generations, adverse effects that can be ameliorated upon cessation.
Stress can best be understood as the inability to adapt to environmental demands, namely acute and chronic stressors, and is known to cause adverse health outcomes (1). These demands range from traumas to daily nuisances, and the degree of experienced stress varies based on genetic, regulatory, and social factors (1). The underlying physiology of stress is well-understood by way of the hypothalamic-pituitary-adrenal (HPA) axis, ultimately leading to the secretion of glucocorticoids into the blood stream (2). Pregnant women are particularly vulnerable to stress given homeostatic adaptations during this period (3). Indeed, maternal stress is a well-established risk factor for preterm birth (4), the leading cause of perinatal morbidity and mortality worldwide (5, 6). Prenatal maternal stress has also been associated with physiological, neurological, and psychological consequences in the offspring (7–14).
The intra-uterine period is a window of vulnerability in the development of the fetal immune system (15). Hence, prenatal maternal stress is associated with increased susceptibility to disease and lasting immunological changes in the offspring (16, 17). Previous reports have shown that prenatal maternal stress contributes to an increased risk of immune-related disorders such as asthma (18) and allergies (19, 20) in children. Several potential and non-exclusive mechanisms whereby prenatal maternal stress induces adverse neonatal outcomes have been suggested, including epigenetic alterations (21) and dysregulation of the maternal/fetal HPA axis (22). Previous descriptive studies have also suggested that maternal stress impacts neonatal immunity (23). However, the mechanisms underlying the effects of prenatal maternal stress on neonatal adaptive immunity are poorly understood.
The deleterious consequences of prenatal maternal stress not only affect the first generation of newborns, but also may be transmitted across subsequent generations (24, 25). In fact, recent animal studies have shown that gestational stress across generations has downstream effects on the endocrine and metabolic pathways (26, 27). Importantly, intergenerational maternal stress gradually shortens the length of gestation (26) and affects physiological and molecular processes in both the mother and offspring (28).
In the current study, we used mice to evaluate the adverse effects of prenatal maternal stress on the timing of delivery across three generations. In addition, we performed deep immunophenotyping of the neonatal adaptive immune system to determine the lasting adverse effects of prenatal maternal stress across generations. Lastly, we evaluated whether the cessation of stress reverts the adverse pregnancy and neonatal outcomes induced by prenatal maternal stress.
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA), bred in the animal care facility at the C.S. Mott Center for Human Growth and Development, Wayne State University, Detroit, MI, and housed under a circadian cycle (light/dark = 12:12 h). Several generations were inbred in our animal facility prior to initiating the study. Dams between 8 and 12 weeks of age were mated with males of proven fertility, also 8 to 12 weeks of age; the dams were checked between 8:00 and 9:00 a.m. daily for the appearance of a vaginal plug, indicating 0.5 days post coitum (dpc), at which point female mice were removed from the mating cages and housed separately. Pregnancy was confirmed by a weight gain of ≥2 g at 12.5 dpc. All mouse experiments were approved by the Institutional Animal Care and Use Committee at Wayne State University (Protocol No. A-09-08-12 and A-07-03-15). The authors adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Three generations of mice were bred (Figure 1A) and divided into several groups: (1) Non-stressed controls; (2) Female mice stressed during gestation (F0-S); (3) Pregnant daughters stressed during gestation (F1-SS); and (4) pregnant granddaughters stressed during gestation (F2-SSS). Females from each litter were used to breed subsequent generations, and each generation was composed of subjects from several different mothers and fathers. Three different cohorts of mice were utilized in this study; the first two to obtain observational data in two different years (2013 and 2014) and the third to determine maternal corticosterone levels and perform immunophenotyping of the neonates (2014–2015). Each group, including the control group, refers to an amalgamation of these cohorts. It is worth mentioning that we did not include one control group for every generation because we used in-bred mice under consistent conditions and followed the laws that mandate replacement alternatives, reduction alternatives, and refinement alternatives (The Three R's) in scientific research (29).
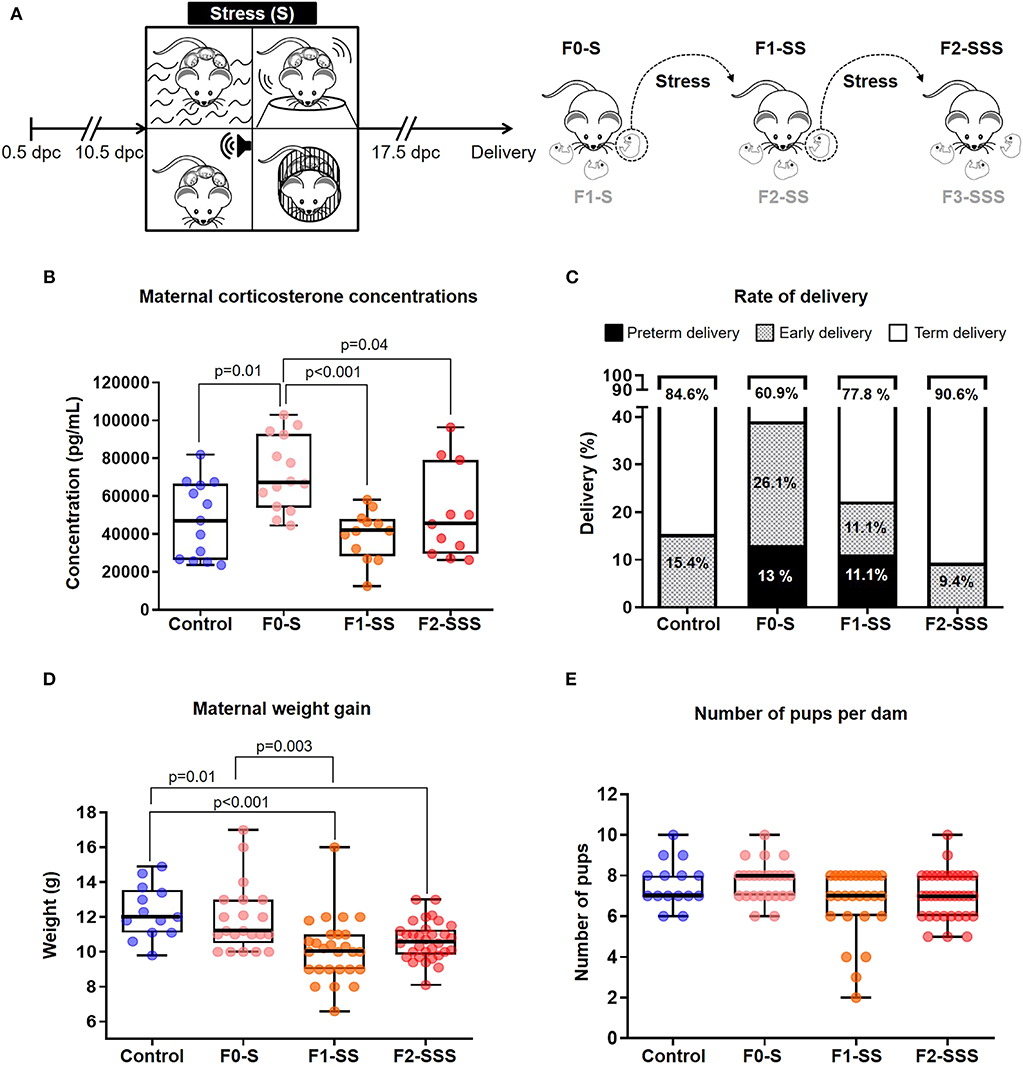
Figure 1. Maternal outcomes of prenatal maternal stress across generations. (A) Experimental design of alternating, unpredictable stress procedure. (B) Maternal corticosterone levels across generations 1 week after delivery (control n = 13; F0-S n = 14; F1-SS n = 12; F2-SSS n = 11). (C) Rates of delivery in each generation, specified as preterm, early, and term delivery (control n = 13; F0-S n = 23; F1-SS n = 27; F2-SSS n = 32). (D) Maternal weight gain across generations (control n = 13; F0-S n = 21; F1-SS n = 27; F2-SSS n = 32). (E) Number of pups per dam across generations (control n = 15; F0-S n = 25; F1-SS n = 27; F2-SSS n = 36). For box plots, mid-lines indicate medians, boxes indicate interquartile ranges, and whiskers indicate min-max range.
Prenatal stress was applied daily from 10.5 to 17.5 dpc without interruption. The four stress procedures were adapted from well-established rodent stress models and included swimming (30, 31), restraint (30, 31), shaking (32), and white noise (33). Two of the four procedures were applied daily in an alternating, unpredictable sequence from 8:00 a.m. to 4:00 p.m. Stress treatments were performed in a designated room other than the housing facility. For the swimming procedure, a small tub was filled with room temperature water (~22°C). Mice were individually placed in the tub and made to swim for 5 min (with a rest of 10 s). The water was deep enough to prevent the feet or tail from contacting the bottom of the tub (Link to Supplementary Video 1). For the restraint procedure, mice were placed inside of a Plexiglas restrainer (2.5 cm inner diameter) in a standing position without compression of the body for a period of 20 min. The container had perforated ends to allow ventilation (Link to Supplementary Video 2). For the shaking procedure, mice were placed inside of a transparent, plastic 1-liter beaker and held on a low-speed vortex (speed 9) for a period of 5 min (Link to Supplementary Video 3). For the white noise procedure, mice were placed in a clean cage free of bedding. The cage was placed in a fume hood with a high-speed fan faced away from the cage generating white noise for 20 min (Link to Supplementary Video 4). Following the stress procedure on 16.5 dpc, dams were placed under recording via video camera (Sony Corporation, Tokyo, Japan) until delivery to evaluate maternal and neonatal outcomes. The number of feces produced during each procedure was recorded as a readout of stress response (Supplementary Figure 1).
Gestational length was utilized as a pregnancy parameter, defined as the day mice were plugged (0.5 dpc) until the time the first pup was delivered. Based on the gestational length, the rate of delivery was categorized as (1) preterm delivery occurring 17–18.5 dpc; (2) early delivery occurring 18.5–19 dpc; and (3) term delivery occurring after 19 dpc. The rate of each delivery group was represented by the number of females delivering within that timeframe among the total number of mice within that generation. Maternal weight gain was also measured, defined as the total weight gained from the day mice were plugged until 16.5 dpc. The number of pups per dam was also recorded. Additionally, the occurrence of neonatal mortality at birth was recorded, defined as the number of pups born dead among the total litter size. After delivery, the mother and her pups were kept under observation and offspring weights were recorded 1, 2, and 3 weeks after birth.
One week after delivery, dams (stressed or non-stressed controls) were euthanized to collect peripheral blood by intra-cardiac puncture between 9:00 and 10:00 a.m. The peripheral blood was centrifuged at 1,300 × g for 10 min at 4°C after collection, and serum was separated to be stored at −20°C until analysis. Total corticosterone concentrations were measured using a multiple species Cortisol Immunoassay Kit (Cat #IRAAKT2546, Innovative Research, Inc., Novi, MI, USA), according to the manufacturer's instructions. The colorimetric reaction was read using a programmable spectrophotometer (SpectraMax M5 Multi-Mode Microplate Reader, Molecular Devices, Sunnyvale, CA, USA). The assay sensitivity was 17.3 pg/mL, according to the manufacturer.
Neonates born to stressed or non-stressed control dams were euthanized 1 week after birth and spleens were collected. The spleens were dissociated using glass slides and 1X phosphate-buffered saline (PBS), filtered using FACS buffer (BSA 0.1%, Sodium Azide 0.05%, and 1X PBS), and centrifuged at 1,300 × g for 10 min at 4°C to obtain a cell suspension and to perform immunophenotyping. The splenic cell suspensions were incubated with CD16/CD32 (FcγIII/II Receptor; BD Biosciences, San Jose, CA, USA) for 10 min, followed by extracellular and/or intracellular staining for immunophenotyping (Supplementary Table 1). For the staining of T cell and B cell populations, either the FoxP3 Staining Buffer Kit (Cat # 00-5523-00, eBiosciences, San Diego, CA, USA) or the Cytofix/Cytoperm Fixation/Permeabilization Solution (Cat# 554714, BD Biosciences) was used prior to intra-nuclear or intra-cellular staining, respectively. For the staining of CD71+ erythroid cells, the 1X FACS Lysing Solution (BD Biosciences) was used. Upon completion of the staining procedures, cell pellets were washed with 1X PBS and re-suspended in 0.5 mL FACS buffer. Samples were acquired using the BD LSRFortessa® Flow Cytometer (BD Biosciences) and analyzed with BD FACSDiva® Software Version 7.0 (BD Biosciences). The analysis and figures were performed using FlowJo software version 10 (FlowJo, LLC, Ashland, OR, USA). The absolute number of cells was determined using CountBright absolute counting beads (Molecular Probes, Eugene, OR, USA).
Immunophenotyping of T cell populations included the identification of naïve (CD3+CD44–CD62L+), memory (CD3+CD44+CD62L+), and effector (CD3+CD44+CD62L−) T cells [CD4+ and CD8+ naïve T cells (CD4+CD44−CD62L+; CD8+CD44−CD62L+, respectively), CD4+ and CD8+ memory T cells (CD4+CD44+CD62L+; CD8+CD44+CD62L+), and CD4+ and CD8+ effector T cells (CD4+CD44+CD62L−; CD8+CD44+CD62L−)]. We also identified the following T cell subsets: conventional T cells (CD3+), CD4+ T cells (CD3+CD4+), CD8+ T cells (CD3+CD8+), regulatory CD4+ T cells (CD3+CD4+CD25+FoxP3+), CD8+FoxP3+ cells (CD3+CD8+CD25+FoxP3+), Th1 cells (CD3+CD4+IFNγ+), Th2 cells (CD3+CD4+IL-4+), Th17 cells (CD3+CD4+IL-17A+), CD8+IL-17A+ T cells (CD3+CD8+IL-17A+), CD8+IFNγ+ cells (CD3+CD8+IFNγ+), and CD8+IL-4+ cells (CD3+CD8+IL-4+). B cell subsets were identified as: total B cells (B220+), B1-like cells (B220+CD5+), and B2-like cells (B220+CD23+). Lastly, CD71+ erythroid cells (CD3-CD71+TER119+) were also identified.
In a third cohort, first-generation daughters born to prenatally stressed mothers were not subjected to the prenatal stress model during their pregnancies (F1-SNS). This cessation of intergenerational stress was done in comparison to mice from the same litter that continued to undergo the prenatal stress model. Maternal and neonatal outcomes were recorded via video camera and the weights of the offspring were recorded at weeks 1, 2, and 3.
Data were analyzed using SPSS Statistics Software Version 19.0, 2010 (IBM, Armonk, NY, USA). A Shapiro–Wilk test was performed to determine whether the data was normally distributed. For Figures 1–6, multiple comparisons were performed using ANOVA or Kruskal-Wallis tests with corresponding post-hoc tests. For Figure 7, the two groups were compared using a Student's t-test. An adjusted p ≤ 0.05 was considered statistically significant.
First, we created a model of multiple, unpredictable stressors that included swimming, shaking, white noise, and restraint procedures (Figure 1A). We validated the efficacy and lack of desensitization to our alternating model by fecal pellet quantification (Supplementary Figure 1), a well-established indicator of murine stress (34–36). Continuously marked fecal production was observed throughout the procedure between generations, suggesting an incessant stress response. To further corroborate our stress model, we quantified maternal corticosterone concentrations in serum, a known hormonal biomarker of stress (37). It was found that the first generation of dams (F0-S) had the highest corticosterone concentration compared to the second (F1-SS) and third (F2-SSS) generations (Figure 1B), suggesting that the first generation suffers from acute stress while the subsequent generations suffer from chronic stress.
Next, we investigated whether prenatal stress could induce preterm birth or early delivery. This research question was based on strong associations between stress and preterm delivery (38–44). We found that stress prompted preterm delivery and early delivery in the first and second generations (Figure 1C). Specifically, the rate of preterm birth in the first generation (F0-S) and second generation (F1-SS) was 13% (3/23) and 11.1% (3/27), respectively. Although some of the controls experienced early delivery, this rate was elevated in the first stressed generation (F0-S 26.1% 6/23 vs. controls 15.4% 2/13). The second (F1-SS) and third (F2-SSS) generations also displayed early delivery (F1-SS 11.1% 3/27 & F2-SSS 9.4% 3/32), but these rates were similar to controls (15.4% 2/13). These data show that prenatal maternal stress can induce preterm birth in the first and second generations; however, such an effect was not observed in the third generation.
Given that stress can cause a reduction in litter size and resorption of implanted embryos (33), we investigated whether prenatal stress impacts maternal weight gain across generations. Consistently, it was found that total weight gain successively decreased throughout the generations (Figure 1D), a likely consequence of the decreased numbers of pups per dam (Figure 1E, non-significant). The data provide supporting evidence showing that prenatal maternal stress affects overall reproductive health.
Next, we investigated the impact of prenatal maternal stress on the immediate and long-term health of the offspring (Figure 2A). We first measured the rate of mortality at birth and found no significant differences amongst the stressed groups compared to controls (Figure 2B). However, we continued to monitor the growth of the offspring and found that neonatal weight was significantly decreased in each generation within the first week of life (Figure 2C). A similar non-significant trend was consistently observed at weeks 2 and 3 (Figure 2C). Indeed, pups born to stressed dams seemed smaller compared to controls (data not shown). This data shows that while prenatal maternal stress does not induce neonatal mortality, it does impact neonatal growth in early life.
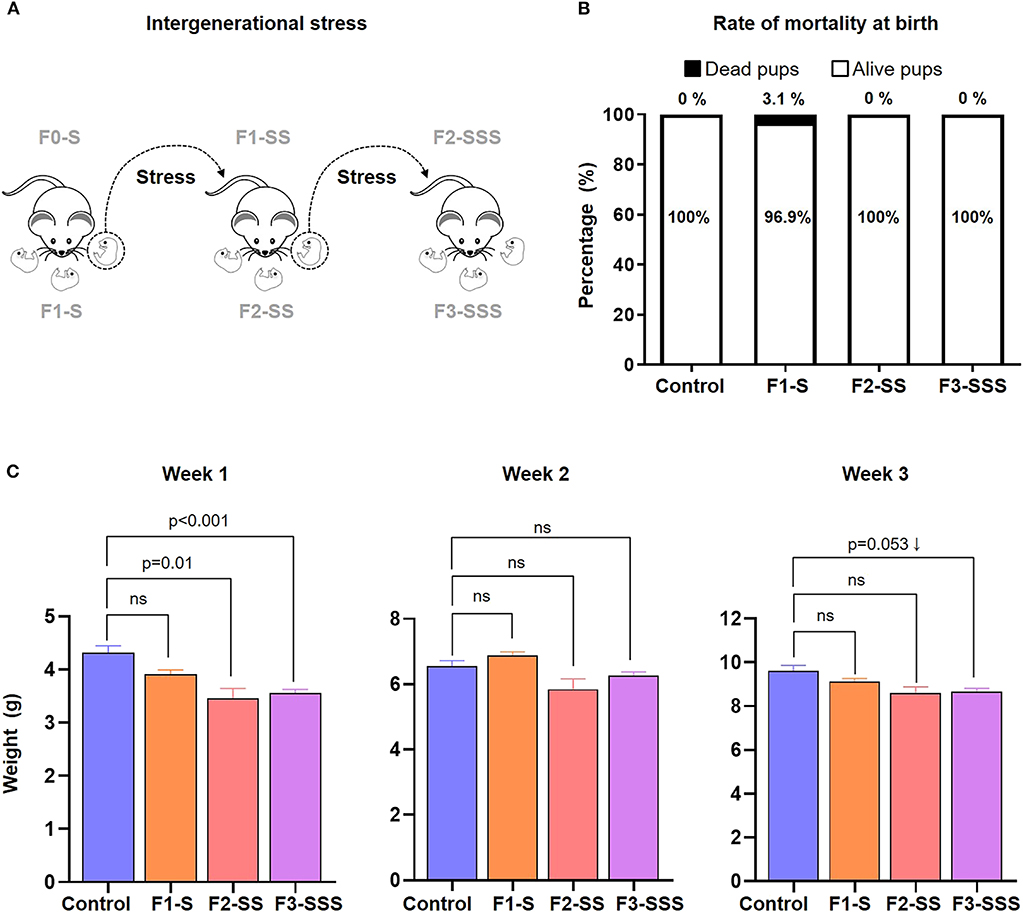
Figure 2. Neonatal outcomes of prenatal maternal stress across generations. (A) Experimental design of neonates born to stressed dams across generations. (B) Rate of neonatal mortality at birth in each generation (control n = 10; F1-S n = 19; F2-SS n = 19; F3-SSS n = 30). (C) Growth trajectory of neonates in the first 3 weeks after birth (control n = 5 litters; F1-S n = 8 litters; F2-SS n = 2 litters; F3-SSS n = 21 litters). Data are presented as mean ± standard error of the mean.
Thus far, our findings show that stress compromises early offspring growth development in the second (F1-SS) and third (F2-SSS) generations of stressed dams. Therefore, we further explored any potential immunological detriments in F2-SS and F3-SSS neonates, born, respectively, to F1-SS and F2-SSS stressed dams. To measure the effect of cumulative prenatal maternal stress on immunocompetence, we first quantified neonatal T cell populations using flow cytometry (Figure 3A). In general, T cells can be divided into naïve, effector, and memory populations; these states are acquired through early recognition of self- or non-self-antigens (e.g., early-life microbiota) (45–48). We found that naïve T cells and naïve CD8+ T cells were significantly reduced in the F2-SS generation, while a downward trend was seen in naïve CD4+ T cells (Figures 3B–D). This observed reduction was overcome in the F3-SSS generation (Figures 3B,D). The memory T cell population was not changed in either the F2-SS or F3-SSS generation compared to controls (Figures 3E–G). There were non-significant alterations in the total effector T cells (Figures 3H–J). These results show that prenatal maternal stress reduces the pool of neonatal naïve T cells in the second generation, but not in the third generation.
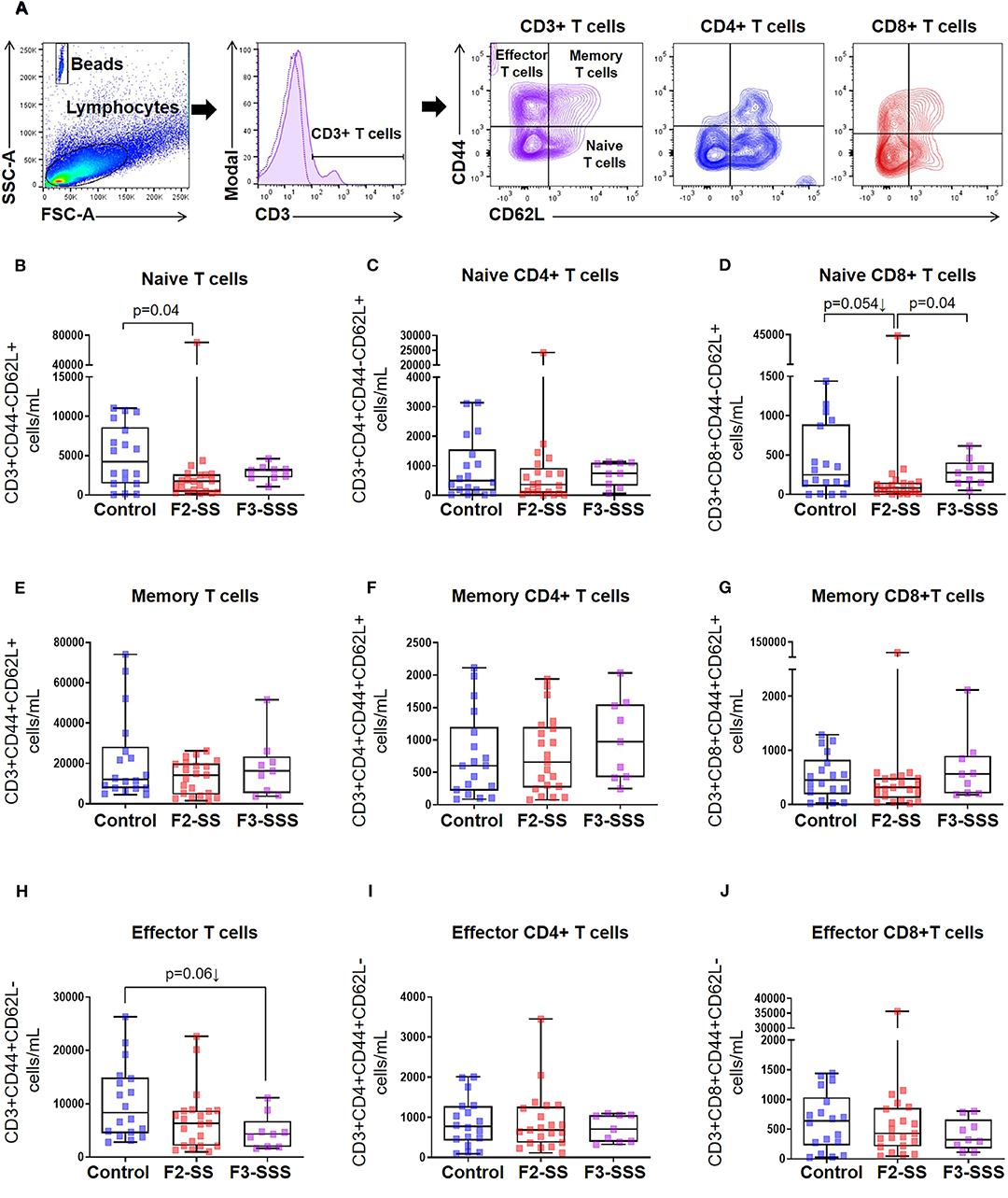
Figure 3. Immunophenotyping of naïve, memory, and effector T cells in prenatally stressed neonates. (A) Gating strategy used to identify T cell subsets. (B–D) Number of naïve T cells, (E–G) memory T cells, and (H–J) effector T cells (control n = 19; F2-SS n = 21; F3-SSS n = 9). Mid-lines indicate medians, boxes indicate interquartile ranges, and whiskers indicate min–max range.
After observing an alteration in the naive T cells of neonates born to stressed dams, we further investigated the impact of prenatal maternal stress on T cell subsets in F2-SS and F3-SSS neonates, born, respectively, to F1-SS and F2-SSS stressed dams (Figure 4A). We first broadly looked at conventional T cells and found a reduction in T cells (Figure 4B), CD4+ helper T cells (Figure 4C), and CD8+ cytotoxic T cells (Figure 4D) in the F2-SS neonates compared to controls. These reductions were all overcome by the F3-SSS generation. Since regulatory T cells play a critical role in neonatal development (49–51), we investigated whether prenatal maternal stress alters such a T cell subset. Interestingly, we found that regulatory CD4+ T cells tended to decrease in F2-SS neonates compared to controls, but this effect was restored in the F3-SSS generation (Figure 4E). Given that CD8+ T cells also express the transcriptional factor FoxP3 and seem to have regulatory properties (52–54), we also investigated whether such cells were altered in neonates born to stressed dams. No significant differences were seen in CD8+ cells expressing FoxP3 (Figure 4F). Furthermore, we quantified the Th1, Th2, and Th17 cell subsets of these neonates. In the Th1 cell subset, no differences were observed between the F2-SS and F3-SSS groups compared to controls (Figure 4G). However, the Th2 and Th17 cell types were significantly reduced in the F2-SS generation, but these reductions were phenotypically overcome in the F3-SSS generation (Figures 4H,I). The Th2 and Th17 findings mirrored our data in CD8+ T cells expressing IL-4 or IL-17A (Figures 4K,L). However, no differences were seen in the cytotoxic T cells expressing IFNγ (Figure 4J). In summary, prenatal maternal stress causes a systematic reduction of CD4+ T cells and, to a lesser extent, CD8+ T cells, expressing the cytokines IL-4 and IL-17A in the second generation, but such effects are restored in the third generation.

Figure 4. Immunophenotyping of T cell subsets in prenatally stressed neonates. (A) Gating strategy used to identify T cell subsets. (B) Number of conventional T cells, (C) CD4+ T cells, (D) CD8+ T cells, (E) regulatory CD4+ T cells or CD4+ Tregs, (F) CD8+FoxP3+ T cells, (G) Th1 cells, (H) Th2 cells, (I) Th17 cells, (J) CD8+IFNγ+ T cells, (K) CD8+IL-4+ T cells, (L) CD8+IL-17A+ T cells (control n = 19; F2-SS n = 21; F3-SSS n = 9). Mid-lines indicate medians, boxes indicate interquartile ranges, and whiskers indicate min–max range.
In addition to T cells, neonatal immunocompetence requires the function of B cells, and given their immaturity, B cells are quite distinct from those present in adults (55). B cells are generally divided into two predominant subsets: B2 cells that circulate through the blood and secondary lymphoid tissues and respond to antigens (56), and B1 cells that produce IgM and IgA for protection against pathogens (57–59). Using flow cytometry, we investigated the total number of B cells as well as B1-like and B2-like cell subsets in neonates born to prenatally stressed dams or controls (Figure 5A). Total B cells and the evaluated B cell subsets were decreased in F2-SS neonates compared to controls (Figures 5B–D). Yet, there was a re-establishment of total and B cell subsets in the F3-SSS generation to values comparable to controls (Figures 5B–D). These results indicate that prenatal maternal stress causes a systematic reduction in the B cell repertoire, which is restored in the third generation.
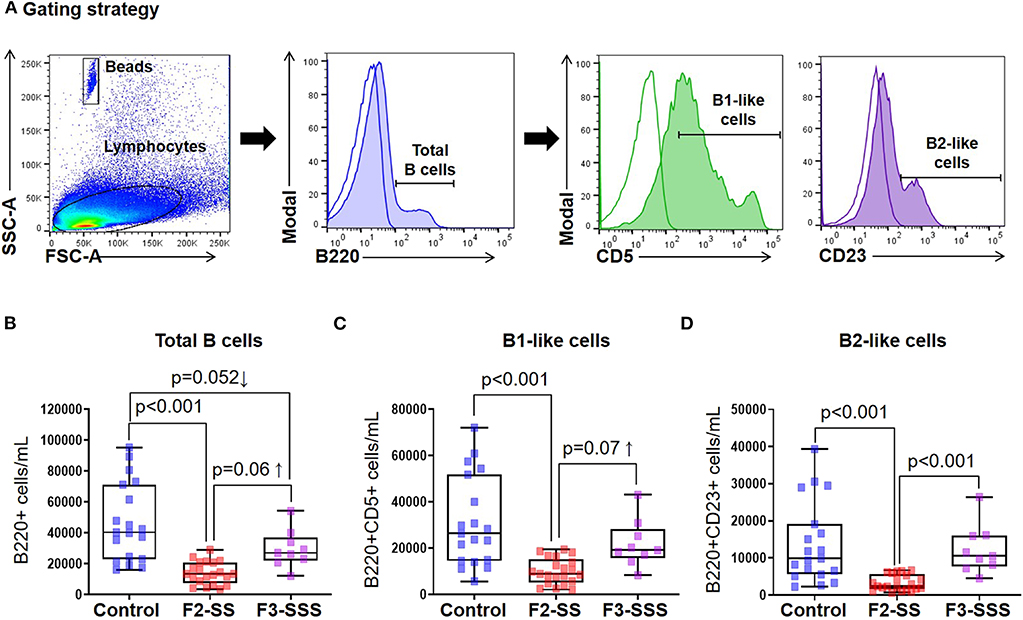
Figure 5. Immunophenotyping of B cells in prenatally stressed neonates. (A) Gating strategy used to identify B cells. (B) Total number of B cells, (C) B1-like cells, and (D) B2-like cells (control n = 19; F2-SS n = 21; F3-SSS n = 9). Mid-lines indicate medians, boxes indicate interquartile ranges, and whiskers indicate min–max range.
Growing evidence shows that neonatal immunity depends on the critical immunosuppressive function of CD71+ nucleated erythroid cells (60–65). Indeed, we have shown that cord blood CD71+ erythroid cells play a central role in the modulation of inflammatory responses of neonates, which may be defective in those delivered prematurely (66, 67). Therefore, we investigated whether prenatal maternal stress alters the number of CD71+ erythroid cells in neonates (Figure 6A). To our surprise, neonatal CD71+ erythroid cells were unchanged in the F2-SS and F3-SSS generations (Figure 6B). The data show that prenatal maternal stress does not affect the neonatal CD71+ erythroid cells in number, yet further studies are required to investigate whether maternal stress alters the functionality of such cells.
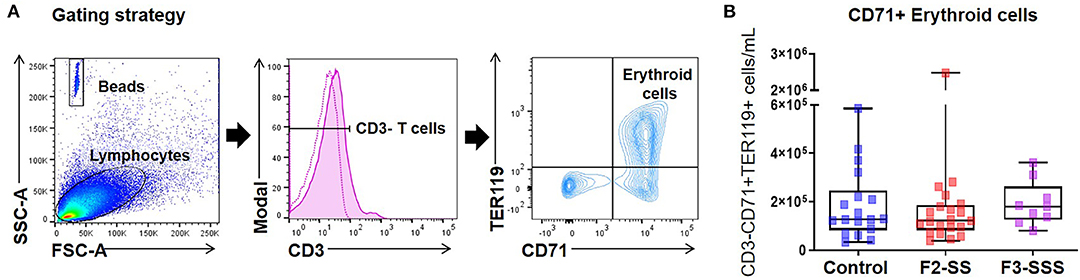
Figure 6. Immunophenotyping of CD71+ erythroid cells in prenatally stressed neonates. (A) Gating strategy used to identify CD71+ erythroid cells. (B) Number of CD71+ erythroid cells (control n = 18; F2-SS n = 21; F3-SSS n = 9). Mid-lines indicate medians, boxes indicate interquartile ranges, and whiskers indicate min–max range.
Lastly, we investigated the inheritability of stress-induced alterations and evaluated whether these consequences could be overcome by the cessation of stress. We separated the litter delivered by an F0-S mother into two groups: one group that continued with intergenerational stressors (F1-SS) and another group in which stress was ceased (F1-SNS) (Figure 7A). We then compared the rate of preterm birth between the stressed F1-SS group and cessation F1-SNS group. Importantly, the cessation of stress resulted in all dams delivering at term or having an early delivery, which is comparable to controls. Specifically, the rate of preterm birth in F1-SS dams was 11.1% (3/27) whereas no animals delivered preterm in the control and cessation groups (Figure 7B). The rate of early delivery in the cessation group, 17.6% (3/17), was similar to that of controls, 15.4% (2/13) (Figure 7B). We further examined the impact of the cessation of stress on neonatal growth and did not find any significant differences between the F2-SS and F2-SNS neonates, indicating that the weight reduction observed in neonates born to stressed dams (Figure 2C) was not recovered with cessation of stress (Figure 7C). The data show that prenatal maternal stress-induced preterm birth can be fully attenuated by the interruption of stressful stimuli in the second generation, yet neonatal growth may still be impacted.
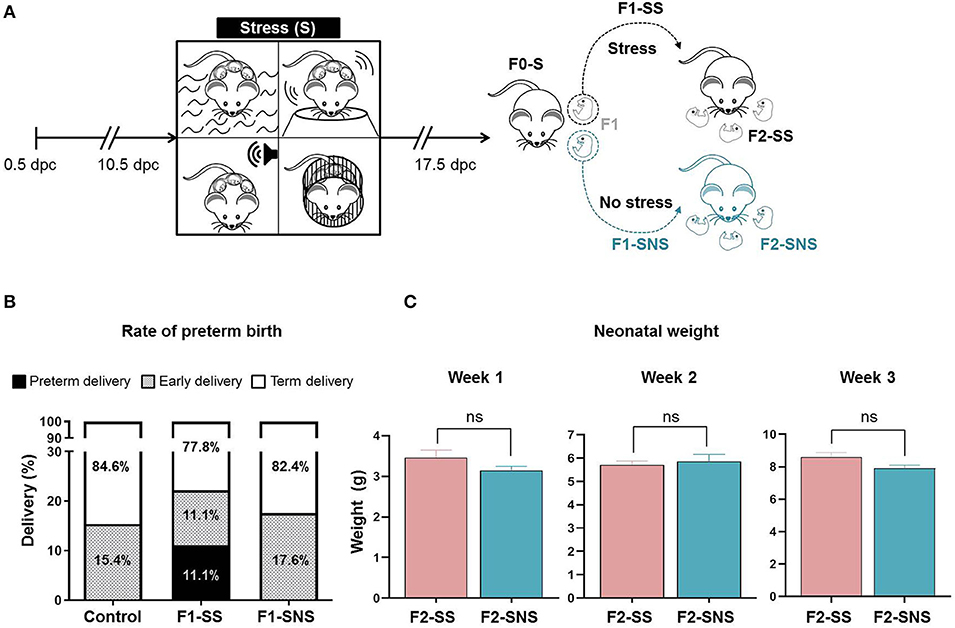
Figure 7. Experimental outcome of stress cessation cohort. (A) Experimental design of stress procedure. Animals of the parental generation (F0-S) were stressed and their pregnant daughters were either stressed (F1-SS) or not stressed (F1-SNS). (B) Rates of delivery in the control, F1-SS, and F1-SNS groups (control n = 13; F1-SS n = 27; F1-SNS n = 17). (C) Growth trajectory of F2-SS and F2-SNS neonates in the first 3 weeks after birth (F2-SS n = 2 litters; F2-SNS n = 10 litters). Data are presented as mean ± standard error of the mean.
The current study provides evidence that prenatal maternal stress: (1) induces preterm birth across the directly exposed first and second generations in mice, (2) impairs neonatal growth at weeks one, (3) reduces the number of neonatal conventional CD4+ and CD8+ T cells in the second and third generations, (4) alters the neonatal T-cell subsets (Th2 and Th17) in the second generation, but not in the third generation, (5) causes a systematic reduction in the number of neonatal B cells in the second generation, but not in the third generation, and (6) does not alter the number of neonatal CD71+ erythroid cells across generations. Importantly, we report that the cessation of prenatal maternal stress in the second generation can attenuate preterm birth; yet, this intervention does not restore neonatal growth impairment. These findings provide insights into the causal relationship between prenatal maternal stress and neonatal immunity, which is further discussed below.
Pregnancy itself can be classified as a stressor given that endocrine and immune adaptations, largely mediated by the placenta, must occur in order for the mother to support the developing fetus (15, 68). Under normal circumstances, the systemic levels of adrenocorticotropic hormone (ACTH) and corticotropin-releasing hormone (CRH) progressively rise throughout early gestation, followed by an exponential increase prior to parturition (69–71). External stressors can further activate the placental maternal pituitary-adrenal axis in a manner consistent with the classic endocrine response, leading to the production of glucocorticoids (72, 73). Therefore, external stressors are perceived to prematurely activate such a pathway, leading to preterm labor and birth (70–72, 74). Consistent with this hypothesis, in the current study, we report that systemic corticosterone levels were increased in dams stressed during their first pregnancy. However, corticosterone levels remained at their basal state during the second and third generations. This finding is consistent with the attenuation of cortisol increase that has been observed in both repeat exposures to stress, as well as in the offspring of stressed parents (75–77). The mechanism whereby this attenuation occurs involves epigenetic programming processes, namely, increased DNA methylation of the gene coding for the glucocorticoid receptor NR3C1 (78). Epigenetics modifications have also been held responsible for the effects of prenatal maternal stress on gestational length (26); however, whether these pathways include the regulation of glucocorticoid synthesis is unknown.
In the current study, we also report that stress during gestation modestly reduced the litter size in the directly exposed second and third generations, suggesting that fecundity is impacted by maternal stress (79, 80). Yet, all neonates born to stressed dams were viable, suggesting that prenatal maternal stress afflicts reproductive health but does not cause neonatal death. It is worth mentioning that the reduced litter size was not associated with a rise in corticosterone levels. Thus, we suggest that cumulative stress skews the HPA axis in a manner that is not reflected in systemic corticosterone levels. Importantly, we observed that prenatal maternal stress delayed the growth of neonates at week 1. These findings are in line with prior studies showing impaired offspring growth upon maternal stress (26, 81). These data could be explained by the substantial compilation of evidence suggesting that exposure to prenatal stress impacts health and disease susceptibility in the offspring (7–9, 13, 15–17, 82, 83).
Exposure to chronic stress impacts the adaptive immune system (84); thus, we investigated whether prenatal maternal stress alters neonatal adaptive immunity. In this study, we observed a consistent reduction in the pool of naïve and conventional T cells, as well as B cells, in neonates born to F2-SS dams. These findings are in line with previous reports showing that stress induces a decrease in the number of lymphocytes (i.e., lymphopenia) in subsets such as CD4+, CD8+, and B220+ cells (85, 86). A possible mechanism whereby prenatal maternal stress induces neonatal lymphopenia involves the triggering of a fetal systemic inflammatory response that, in turn, may cause involution of the lymphatic organs (87, 88).
Prenatal maternal stress also induced a neonatal immunosuppression-like syndrome characterized by reduced numbers of IL-4- and IL-17-expressing T cells, as well as regulatory CD4+ T cells, in the second generation. All of these T cell subsets have regulatory and anti-inflammatory functions (89–93); therefore, it is likely that prenatal maternal stress alters the T cell repertoire in utero. Yet, further research is required to investigate the mechanisms whereby prenatal maternal stress induces immunosuppression in neonates born to mothers and grandmothers who underwent stress during pregnancy.
Notably, we found that the neonatal immunosuppressive-like syndrome induced by prenatal maternal stress was overcome in the third generation. Indeed, neonates born to third generation stressed dams had greater numbers of Th2 cells, IL-4+ CD8+ T cells, and tended to have higher numbers of regulatory CD4+ T cells. To our knowledge, this is the first demonstration to show that chronic exposure to multigenerational stress boosts the neonatal immune system. We ought to propose that this response is observed as a compensatory mechanism against prenatal maternal stress.
Importantly, in the mouse model, the cessation of stress in the second generation restores the timing of delivery. Our results in mice are partially in agreement with those reported in rats; cessation of prenatal maternal stress in rats does not fully restore timing of delivery (26). This discrepancy could be explained by variations in how different species respond to stress (94, 95). Yet, our finding in mice holds tangible and immediate public health relevance given the implication that daughters of stressed mothers may be able to manage the impacts of ancestral stress during their pregnancy. Additional research is warranted to investigate whether the cessation of stress impacts neonatal immunity given that neonatal growth impairment was not restored.
There are some limitations to the current study that we must acknowledge and address. First, using a murine model presents inevitable discrepancies given the fundamental mouse-human differences. However, studying stress in humans is very difficult due to a multitude of variables and uncontrollable co-factors, and can only be done from an epidemiological standpoint. Past studies have demonstrated that efficiently inducing stress in rodents is difficult due to their adaptability (96, 97), but we compensated for this condition by prolonging the period of stress and utilizing an unpredictable, multi-procedural stress schedule. Another limitation of our study is that we utilized a syngenic mating model, which does not allow us to evaluate the contribution of allo-antigenicity; yet, this syngenic model was chosen to maintain prenatal stress as our singular variable. It is worth mentioning that, in this study, we did not include non-stressed controls for every generation; therefore, our experimental design does not allow us to distinguish the transgenerational effects of stress in the second and third generations. An important strength of our study is that we performed two different cohorts of observational studies in which we observed very similar results; both cohorts included animals that delivered preterm. Yet, the execution of this study required a large investment of time and funds as well as the creation of a multidisciplinary team.
The data presented herein provides a causal link between prenatal maternal stress and preterm birth, as well as neonatal adaptive immunity, across generations. We report that the impact of ancestral prenatal maternal stress results in inheritable consequences, but these alterations can be mitigated by intervention, as well as progressive stress sensitization over time. These findings may hold clinical importance for individuals exposed to direct or ancestral chronic stress and their ability to overcome adverse pregnancy and neonatal outcomes.
The datasets generated for this study are available on request to the corresponding author.
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at Wayne State University.
VG-F and A-EF performed research and analyzed and interpreted data. RR analyzed and interpreted data and provided guidance in the experimental design. DL, JG, and CZ analyzed and interpreted data. C-DH, SH, DO, and GM provided intellectual input. NG-L designed research, analyzed and interpreted data, and provided supervision throughout the study. All authors participated in the writing of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by the Wayne State University Perinatal Initiative in Maternal, Perinatal and Child Health and the Perinatology Research Branch, Division of Obstetrics and Maternal-Fetal Medicine, Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, U.S. Department of Health and Human Services (NICHD/NIH/DHHS); and, in part, with federal funds from NICHD/NIH/DHHS under Contract No. HHSN275201300006C. RR has contributed to this work as part of his official duties as an employee of the United States Federal Government.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00254/full#supplementary-material
Supplementary Video 1. Pregnant mice exposed to swimming.
Supplementary Video 2. Pregnant mice exposed to restrain.
Supplementary Video 3. Pregnant mice exposed to shaking.
Supplementary Video 4. Pregnant mice exposed to white noise.
ACTH, adrenocorticotropic hormone; BSA, bovine serum albumin; CRH, corticotropic-releasing hormone; dpc, days post coitum; F0-S, pregnant mice stressed during gestation; F1-S, pregnant daughters that were stressed during gestation; F2-SS, pregnant granddaughters that were stressed during gestation; F1-SNS, prenatally stressed pregnant daughters who were not stressed during gestation; HPA, hypothalamic-pituitary-adrenal; IFN, interferon; Ig, immunoglobulin; IL, interleukin; PBS, phosphate-buffered saline.
1. Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. (1992) 267:1244–52. doi: 10.1001/jama.1992.03480090092034
2. Smith SM, Vale WW. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin Neurosci. (2006) 8:383–95.
3. Mulder EJ, Robles de Medina PG, Huizink AC, Van den Bergh BR, Buitelaar JK, Visser GH. Prenatal maternal stress: effects on pregnancy and the (unborn) child. Early Hum Dev. (2002) 70:3–14. doi: 10.1016/S0378-3782(02)00075-0
4. Austin MP, Leader L. Maternal stress and obstetric and infant outcomes: epidemiological findings and neuroendocrine mechanisms. Aust N Z J Obstet Gynaecol. (2000) 40:331–7. doi: 10.1111/j.1479-828X.2000.tb03344.x
5. Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller AB, Narwal R, et al. National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet. (2012) 379:2162–72. doi: 10.1016/S0140-6736(12)60820-4
6. Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, et al. Global, regional, and national causes of child mortality in 2000-13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. (2015) 385:430–40. doi: 10.1016/S0140-6736(14)61698-6
7. Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. Fetal nutrition and cardiovascular disease in adult life. Lancet. (1993) 341:938–41. doi: 10.1016/0140-6736(93)91224-A
8. Yehuda R, Schmeidler J, Giller EL Jr, Siever LJ, Binder-Brynes K. Relationship between posttraumatic stress disorder characteristics of Holocaust survivors and their adult offspring. Am J Psychiatry. (1998) 155:841–3. doi: 10.1176/ajp.155.9.1163
9. Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, et al. Coronary heart disease after prenatal exposure to the Dutch famine, 1944-45. Heart. (2000) 84:595–8. doi: 10.1136/heart.84.6.595
10. Entringer S, Wust S, Kumsta R, Layes IM, Nelson EL, Hellhammer DH, et al. Prenatal psychosocial stress exposure is associated with insulin resistance in young adults. Am J Obstet Gynecol. (2008) 199:498 e491–7. doi: 10.1016/j.ajog.2008.03.006
11. Entringer S, Buss C, Kumsta R, Hellhammer DH, Wadhwa PD, Wust S. Prenatal psychosocial stress exposure is associated with subsequent working memory performance in young women. Behav Neurosci. (2009) 123:886–93. doi: 10.1037/a0016265
12. Zucchi FC, Yao Y, Ward ID, Ilnytskyy Y, Olson DM, Benzies K, et al. Maternal stress induces epigenetic signatures of psychiatric and neurological diseases in the offspring. PLoS ONE. (2013) 8:e56967. doi: 10.1371/journal.pone.0056967
13. Devakumar D, Birch M, Osrin D, Sondorp E, Wells JC. The intergenerational effects of war on the health of children. BMC Med. (2014) 12:57. doi: 10.1186/1741-7015-12-57
14. McCreary JK, Truica LS, Friesen B, Yao Y, Olson DM, Kovalchuk I, et al. Altered brain morphology and functional connectivity reflect a vulnerable affective state after cumulative multigenerational stress in rats. Neuroscience. (2016) 330:79–89. doi: 10.1016/j.neuroscience.2016.05.046
15. Veru F, Laplante DP, Luheshi G, King S. Prenatal maternal stress exposure and immune function in the offspring. Stress. (2014) 17:133–48. doi: 10.3109/10253890.2013.876404
16. Wadhwa PD. Psychoneuroendocrine processes in human pregnancy influence fetal development and health. Psychoneuroendocrinology. (2005) 30:724–43. doi: 10.1016/j.psyneuen.2005.02.004
17. Solano ME, Jago C, Pincus MK, Arck PC. Highway to health; or How prenatal factors determine disease risks in the later life of the offspring. J Reprod Immunol. (2011) 90:3–8. doi: 10.1016/j.jri.2011.01.023
18. Cookson H, Granell R, Joinson C, Ben-Shlomo Y, Henderson AJ. Mothers' anxiety during pregnancy is associated with asthma in their children. J Allergy Clin Immunol. (2009) 123:847–53 e811. doi: 10.1016/j.jaci.2009.01.042
19. Peters JL, Cohen S, Staudenmayer J, Hosen J, Platts-Mills TA, Wright RJ. Prenatal negative life events increases cord blood IgE: interactions with dust mite allergen and maternal atopy. Allergy. (2012) 67:545–51. doi: 10.1111/j.1398-9995.2012.02791.x
20. Lim R, Fedulov AV, Kobzik L. Maternal stress during pregnancy increases neonatal allergy susceptibility: role of glucocorticoids. Am J Physiol Lung Cell Mol Physiol. (2014) 307:L141–8. doi: 10.1152/ajplung.00250.2013
21. Metz GA, Ng JW, Kovalchuk I, Olson DM. Ancestral experience as a game changer in stress vulnerability and disease outcomes. Bioessays. (2015) 37:602–11. doi: 10.1002/bies.201400217
22. Rakers F, Rupprecht S, Dreiling M, Bergmeier C, Witte OW, Schwab M. Transfer of maternal psychosocial stress to the fetus. Neurosci Biobehav Rev. (2017). doi: 10.1016/j.neubiorev.2017.02.019
23. Wright RJ, Visness CM, Calatroni A, Grayson MH, Gold DR, Sandel MT, et al. Prenatal maternal stress and cord blood innate and adaptive cytokine responses in an inner-city cohort. Am J Respir Crit Care Med. (2010) 182:25–33. doi: 10.1164/rccm.200904-0637OC
24. Buss C, Entringer S, Moog NK, Toepfer P, Fair DA, Simhan HN, et al. Intergenerational transmission of maternal childhood maltreatment exposure: implications for fetal brain development. J Am Acad Child Adolesc Psychiatry. (2017) 56:373–82. doi: 10.1016/j.jaac.2017.03.001
25. Ramo-Fernandez L, Boeck C, Koenig AM, Schury K, Binder EB, Gundel H, et al. The effects of childhood maltreatment on epigenetic regulation of stress-response associated genes: an intergenerational approach. Sci Rep. (2019) 9:983. doi: 10.1038/s41598-018-36689-2
26. Yao Y, Robinson AM, Zucchi FC, Robbins JC, Babenko O, Kovalchuk O, et al. Ancestral exposure to stress epigenetically programs preterm birth risk and adverse maternal and newborn outcomes. BMC Med. (2014) 12:121. doi: 10.1186/s12916-014-0121-6
27. Kiss D, Ambeskovic M, Montina T, Metz GA. Stress transgenerationally programs metabolic pathways linked to altered mental health. Cell Mol Life Sci. (2016) 73:4547–57. doi: 10.1007/s00018-016-2272-4
28. Verstraeten BSE, McCreary JK, Weyers S, Metz GAS, Olson DM. Prenatal two-hit stress affects maternal and offspring pregnancy outcomes and uterine gene expression in rats: match or mismatch? Biol Reprod. (2019) 100:195–207. doi: 10.1093/biolre/ioy166
29. Balls M, Goldberg AM, Fentem JH, Broadhead CL, Burch RL, Festing MF, et al. The three Rs: the way forward: the report and recommendations of ECVAM Workshop 11. Altern Lab Anim. (1995) 23:838–66.
30. Metz GA, Jadavji NM, Smith LK. Modulation of motor function by stress: a novel concept of the effects of stress and corticosterone on behavior. Eur J Neurosci. (2005) 22:1190–200. doi: 10.1111/j.1460-9568.2005.04285.x
31. Ward ID, Zucchi FC, Robbins JC, Falkenberg EA, Olson DM, Benzies K, et al. Transgenerational programming of maternal behaviour by prenatal stress. BMC Pregnancy Childbirth. (2013) 13(Suppl 1):S9. doi: 10.1186/1471-2393-13-S1-S9
32. Katz RJ. Animal model of depression: pharmacological sensitivity of a hedonic deficit. Pharmacol Biochem Behav. (1982) 16:965–8. doi: 10.1016/0091-3057(82)90053-3
33. Jafari Z, Faraji J, Mirza Agha B, Metz GAS, Kolb BE, Mohajerani MH. The adverse effects of auditory stress on mouse uterus receptivity and behaviour. Sci Rep. (2017) 7:4720. doi: 10.1038/s41598-017-04943-8
34. Lieblich I, Guttman R. Analysis of emotional defecation under severe and mild stress–evidence for genotype-situation interaction. Life Sci. (1968) 7:301–9. doi: 10.1016/0024-3205(68)90027-1
35. Bruell JH. Genetics and adaptive significance of emotional defecation in mice. Ann N Y Acad Sci. (1969) 159:825–30. doi: 10.1111/j.1749-6632.1969.tb12981.x
36. Larauche M, Gourcerol G, Million M, Adelson DW, Tache Y. Repeated psychological stress-induced alterations of visceral sensitivity and colonic motor functions in mice: influence of surgery and postoperative single housing on visceromotor responses. Stress. (2010) 13:343–54. doi: 10.3109/10253891003664166
37. Gong S, Miao YL, Jiao GZ, Sun MJ, Li H, Lin J, et al. Dynamics and correlation of serum cortisol and corticosterone under different physiological or stressful conditions in mice. PLoS ONE. (2015) 10:e0117503. doi: 10.1371/journal.pone.0117503
38. Lockwood CJ. Stress-associated preterm delivery: the role of corticotropin-releasing hormone. Am J Obstet Gynecol. (1999) 180(Pt 3):S264–6. doi: 10.1016/S0002-9378(99)70713-1
39. Challis JR, Smith SK. Fetal endocrine signals and preterm labor. Biol Neonate. (2001) 79:163–7. doi: 10.1159/000047085
40. Wadhwa PD, Culhane JF, Rauh V, Barve SS. Stress and preterm birth: neuroendocrine, immune/inflammatory, and vascular mechanisms. Matern Child Health J. (2001) 5:119–25. doi: 10.1023/A:1011353216619
41. Wadhwa PD, Culhane JF, Rauh V, Barve SS, Hogan V, Sandman CA, et al. Stress, infection and preterm birth: a biobehavioural perspective. Paediatr Perinat Epidemiol. (2001) 15(Suppl 2):17–29. doi: 10.1046/j.1365-3016.2001.00005.x
42. Hobel CJ. Stress and preterm birth. Clin Obstet Gynecol. (2004) 47:856–80. doi: 10.1097/01.grf.0000142512.38733.8c
43. Hobel CJ, Goldstein A, Barrett ES. Psychosocial stress and pregnancy outcome. Clin Obstet Gynecol. (2008) 51:333–48. doi: 10.1097/GRF.0b013e31816f2709
44. Wadhwa PD, Entringer S, Buss C, Lu MC. The contribution of maternal stress to preterm birth: issues and considerations. Clin Perinatol. (2011) 38:351–84. doi: 10.1016/j.clp.2011.06.007
45. Ahmed R, Gray D. Immunological memory and protective immunity: understanding their relation. Science. (1996) 272:54–60. doi: 10.1126/science.272.5258.54
46. Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. (1996) 272:60–6. doi: 10.1126/science.272.5258.60
47. Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. (1998) 281:96–9. doi: 10.1126/science.281.5373.96
48. Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Coughlin PE, McCrate S, et al. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci Immunol. (2017) 2:aao1314. doi: 10.1126/sciimmunol.aao1314
49. Adkins B, Leclerc C, Marshall-Clarke S. Neonatal adaptive immunity comes of age. Nat Rev Immunol. (2004) 4:553–64. doi: 10.1038/nri1394
50. Rueda CM, Wells CB, Gisslen T, Jobe AH, Kallapur SG, Chougnet CA. Effect of chorioamnionitis on regulatory T cells in moderate/late preterm neonates. Hum Immunol. (2015) 76:65–73. doi: 10.1016/j.humimm.2014.10.016
51. Pan H, Gazarian A, Dubernard JM, Belot A, Michallet MC, Michallet M. Transplant tolerance induction in newborn infants: mechanisms, advantages, and potential strategies. Front Immunol. (2016) 7:116. doi: 10.3389/fimmu.2016.00116
52. Cosmi L, Liotta F, Lazzeri E, Francalanci M, Angeli R, Mazzinghi B, et al. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood. (2003) 102:4107–14. doi: 10.1182/blood-2003-04-1320
53. Nakagawa T, Tsuruoka M, Ogura H, Okuyama Y, Arima Y, Hirano T, et al. IL-6 positively regulates Foxp3+CD8+ T cells in vivo. Int Immunol. (2010) 22:129–39. doi: 10.1093/intimm/dxp119
54. Gomez-Lopez N, Olson DM, Robertson SA. Interleukin-6 controls uterine Th9 cells and CD8(+) T regulatory cells to accelerate parturition in mice. Immunol Cell Biol. (2016) 94:79–89. doi: 10.1038/icb.2015.63
55. Basha S, Surendran N, Pichichero M. Immune responses in neonates. Expert Rev Clin Immunol. (2014) 10:1171–84. doi: 10.1586/1744666X.2014.942288
56. Kantor AB, Herzenberg LA. Origin of murine B cell lineages. Annu Rev Immunol. (1993) 11:501–38. doi: 10.1146/annurev.iy.11.040193.002441
57. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. (2011) 11:34–46. doi: 10.1038/nri2901
58. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. (2011) 208:67–80. doi: 10.1084/jem.20101499
59. Montecino-Rodriguez E, Dorshkind K. B-1 B cell development in the fetus and adult. Immunity. (2012) 36:13–21. doi: 10.1016/j.immuni.2011.11.017
60. Rincon MR, Oppenheimer K, Bonney EA. Selective accumulation of Th2-skewing immature erythroid cells in developing neonatal mouse spleen. Int J Biol Sci. (2012) 8:719–30. doi: 10.7150/ijbs.3764
61. Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, et al. Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature. (2013) 504:158–62. doi: 10.1038/nature12675
62. Elahi S. New insight into an old concept: role of immature erythroid cells in immune pathogenesis of neonatal infection. Front Immunol. (2014) 5:376. doi: 10.3389/fimmu.2014.00376
63. Dunsmore G, Bozorgmehr N, Delyea C, Koleva P, Namdar A, Elahi S. Erythroid suppressor cells compromise neonatal immune response against Bordetella pertussis. J Immunol. (2017) 199:2081–95. doi: 10.4049/jimmunol.1700742
64. Namdar A, Koleva P, Shahbaz S, Strom S, Gerdts V, Elahi S. CD71(+) erythroid suppressor cells impair adaptive immunity against Bordetella pertussis. Sci Rep. (2017) 7:7728. doi: 10.1038/s41598-017-07938-7
65. Shahbaz S, Bozorgmehr N, Koleva P, Namdar A, Jovel J, Fava RA, et al. CD71+VISTA+ erythroid cells promote the development and function of regulatory T cells through TGF-beta. PLoS Biol. (2018) 16:e2006649. doi: 10.1371/journal.pbio.2006649
66. Gomez-Lopez N, Romero R, Xu Y, Miller D, Unkel R, C MacKenzie T, et al. Umbilical cord CD71+ erythroid cells are reduced in neonates born to women in spontaneous preterm labor. Am J Reprod Immunol. (2016) 76:280–4. doi: 10.1111/aji.12556
67. Miller D, Romero R, Unkel R, Xu Y, Vadillo-Ortega F, Hassan SS, et al. CD71+ erythroid cells from neonates born to women with preterm labor regulate cytokine and cellular responses. J Leukoc Biol. (2018) 103:761–75. doi: 10.1002/JLB.5A0717-291RRR
68. Norwitz ER, Bonney EA, Snegovskikh VV, Williams MA, Phillippe M, Park JS, et al. Molecular regulation of parturition: the role of the decidual clock. Cold Spring Harb Perspect Med. (2015) 5:a023143. doi: 10.1101/cshperspect.a023143
69. Hobel CJ, Dunkel-Schetter C, Roesch SC, Castro LC, Arora CP. Maternal plasma corticotropin-releasing hormone associated with stress at 20 weeks' gestation in pregnancies ending in preterm delivery. Am J Obstet Gynecol. (1999) 180(Pt 3):S257–63. doi: 10.1016/S0002-9378(99)70712-X
70. Sfakianaki AK, Norwitz ER. Mechanisms of progesterone action in inhibiting prematurity. J Matern Fetal Neonatal Med. (2006) 19:763–72. doi: 10.1080/14767050600949829
72. Wadhwa PD, Dunkel-Schetter C, Chicz-DeMet A, Porto M, Sandman CA. Prenatal psychosocial factors and the neuroendocrine axis in human pregnancy. Psychosom Med. (1996) 58:432–46. doi: 10.1097/00006842-199609000-00006
73. Brunton PJ, Russell JA, Douglas AJ. Adaptive responses of the maternal hypothalamic-pituitary-adrenal axis during pregnancy and lactation. J Neuroendocrinol. (2008) 20:764–76. doi: 10.1111/j.1365-2826.2008.01735.x
74. Paarlberg KM, Vingerhoets AJ, Passchier J, Dekker GA, Heinen AG, van Geijn HP. Psychosocial predictors of low birthweight: a prospective study. Br J Obstet Gynaecol. (1999) 106:834–41. doi: 10.1111/j.1471-0528.1999.tb08406.x
75. Resnick HS, Yehuda R, Pitman RK, Foy DW. Effect of previous trauma on acute plasma cortisol level following rape. Am J Psychiatry. (1995) 152:1675–7. doi: 10.1176/ajp.152.11.1675
76. Yehuda R, LeDoux J. Response variation following trauma: a translational neuroscience approach to understanding PTSD. Neuron. (2007) 56:19–32. doi: 10.1016/j.neuron.2007.09.006
77. Radley JJ, Kabbaj M, Jacobson L, Heydendael W, Yehuda R, Herman JP. Stress risk factors and stress-related pathology: neuroplasticity, epigenetics and endophenotypes. Stress. (2011) 14:481–97. doi: 10.3109/10253890.2011.604751
78. Perroud N, Rutembesa E, Paoloni-Giacobino A, Mutabaruka J, Mutesa L, Stenz L, et al. The Tutsi genocide and transgenerational transmission of maternal stress: epigenetics and biology of the HPA axis. World J Biol Psychiatry. (2014) 15:334–45. doi: 10.3109/15622975.2013.866693
79. Prasad S, Tiwari M, Pandey AN, Shrivastav TG, Chaube SK. Impact of stress on oocyte quality and reproductive outcome. J Biomed Sci. (2016) 23:36. doi: 10.1186/s12929-016-0253-4
80. Downs CJ, Boan BV, Lohuis TD, Stewart KM. Investigating Relationships between reproduction, immune defenses, and cortisol in dall sheep. Front Immunol. (2018) 9:105. doi: 10.3389/fimmu.2018.00105
81. Burkus J, Kacmarova M, Kubandova J, Kokosova N, Fabianova K, Fabian D, et al. Stress exposure during the preimplantation period affects blastocyst lineages and offspring development. J Reprod Dev. (2015) 61:325–31. doi: 10.1262/jrd.2015-012
82. Entringer S, Buss C, Wadhwa PD. Prenatal stress, development, health and disease risk: a psychobiological perspective-2015 Curt Richter Award Paper. Psychoneuroendocrinology. (2015) 62:366–75. doi: 10.1016/j.psyneuen.2015.08.019
84. Dhabhar FS. Effects of stress on immune function: the good, the bad, and the beautiful. Immunol Res. (2014) 58:193–210. doi: 10.1007/s12026-014-8517-0
85. Fukui Y, Sudo N, Yu XN, Nukina H, Sogawa H, Kubo C. The restraint stress-induced reduction in lymphocyte cell number in lymphoid organs correlates with the suppression of in vivo antibody production. J Neuroimmunol. (1997) 79:211–7. doi: 10.1016/S0165-5728(97)00126-4
86. Abe S, Saito T, Sato T, Suzuki K. Stressors increase leptin receptor-expressing thymic epithelial cells in the infant/child thymus. Int J Legal Med. (2018) 132:1665–70. doi: 10.1007/s00414-018-1793-9
87. Di Naro E, Cromi A, Ghezzi F, Raio L, Uccella S, D'Addario V, et al. Fetal thymic involution: a sonographic marker of the fetal inflammatory response syndrome. Am J Obstet Gynecol. (2006) 194:153–9. doi: 10.1016/j.ajog.2005.05.036
88. Gotsch F, Romero R, Kusanovic JP, Mazaki-Tovi S, Pineles BL, Erez O, et al. The fetal inflammatory response syndrome. Clin Obstet Gynecol. (2007) 50:652–83. doi: 10.1097/GRF.0b013e31811ebef6
89. Muraille E, Leo O. Revisiting the Th1/Th2 paradigm. Scand J Immunol. (1998) 47:1–9. doi: 10.1111/j.1365-3083.1998-47-1.00383.x
90. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
91. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
92. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. (2005) 175:5–14.
93. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. (2005) 6:1133–41. doi: 10.1038/ni1261
94. McCreary JK, Erickson ZT, Hao Y, Ilnytskyy Y, Kovalchuk I, Metz GA. Environmental intervention as a therapy for adverse programming by ancestral stress. Sci Rep. (2016) 6:37814. doi: 10.1038/srep37814
95. Nagel C, Aurich C, Aurich J. Stress effects on the regulation of parturition in different domestic animal species. Anim Reprod Sci. (2019) 207:153–61. doi: 10.1016/j.anireprosci.2019.04.011
96. Pitman DL, Ottenweller JE, Natelson BH. Plasma corticosterone levels during repeated presentation of two intensities of restraint stress: chronic stress and habituation. Physiol Behav. (1988) 43:47–55. doi: 10.1016/0031-9384(88)90097-2
Keywords: preterm labor, neonates, offspring, birthweight, T cells
Citation: Garcia-Flores V, Romero R, Furcron A-E, Levenson D, Galaz J, Zou C, Hassan SS, Hsu C-D, Olson D, Metz GAS and Gomez-Lopez N (2020) Prenatal Maternal Stress Causes Preterm Birth and Affects Neonatal Adaptive Immunity in Mice. Front. Immunol. 11:254. doi: 10.3389/fimmu.2020.00254
Received: 26 November 2019; Accepted: 30 January 2020;
Published: 26 February 2020.
Edited by:
Sandra Maria Blois, Charité Medical University of Berlin, GermanyReviewed by:
Kristin Thiele, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2020 Garcia-Flores, Romero, Furcron, Levenson, Galaz, Zou, Hassan, Hsu, Olson, Metz and Gomez-Lopez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nardhy Gomez-Lopez, bmFyZGh5LmdvbWV6LWxvcGV6QHdheW5lLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.