 Amol Suryawanshi
Amol Suryawanshi Mohamed S. Hussein2
Mohamed S. Hussein2 Santhakumar Manicassamy
Santhakumar Manicassamy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 February 2020
Sec. Cancer Immunity and Immunotherapy
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.00122
This article is part of the Research Topic Dendritic Cell-based Immunotherapy in Solid and Haematologic Tumors View all 10 articles
Dendritic cells (DCs) control the strength and quality of antigen-specific adaptive immune responses. This is critical for launching a robust immunity against invading pathogens while maintaining a state of tolerance to self-antigens. However, this also represents a fundamental barrier to anti-tumor immune responses and cancer immunotherapy. DCs in the tumor microenvironment (TME) play a key role in this process. The factors in the TME and signaling networks that program DCs to a regulatory state are not fully understood. Recent advances point to novel mechanisms by which the canonical Wnt signaling cascade in DCs regulates immune suppression, and the same pathway in tumors is associated with the evasion of anti-tumor immunity. Here, we review these recent advances in the context of the pleiotropic effects of the Wnts in shaping anti-tumor immune responses by modulating DC functions. In addition, we will discuss how Wnt/β-catenin pathway in DCs can be targeted for successful cancer immunotherapy.
Dendritic cells (DCs) control the strength and quality of the adaptive immune response (1, 2). This is critical for launching robust immunity against invading pathogens while maintaining a state of tolerance to self-antigen (3, 4). This dichotomy assumes a particular significance in tumor immune surveillance, as tumors actively suppress immune response through multiple mechanisms by creating tolerance to their own antigen (2, 4). This also represents a fundamental barrier to successful cancer immune therapy (2, 4). Accumulating evidence suggest that DCs play a fundamental role in driving immune suppression against tumor-associated antigens (TAAs) (5–7). We now know that there are multiple subpopulations of DCs that differentially regulate anti-tumor immune responses, and that these subsets display tremendous functional plasticity in response to instructive signals from the tumor microenvironment (TME) and tumor vaccines (5–7). Although there has been much progress in understanding DCs-driven immune suppression, signaling networks and transcription factors within DCs that regulate these responses are not fully understood. Thus, understanding the molecular mechanisms by which DCs fine tune the anti-tumor immunity will be useful in the rational design of therapies against various tumors. Emerging studies suggest a fundamental role for the Wnt signaling cascade in shaping the functions of immune cells in the TME, particularly DCs, in this process. In addition, recent studies have shown that tumor cell-intrinsic Wnt signaling plays a key role in the evasion of anti-tumor immunity in several human cancers. Here, we review these studies, highlight unanswered questions, and offer a conceptual framework for understanding the Wnt-signaling-mediated control of anti-tumor immunity.
The TME is a distinctive niche that contains not only malignant cells but also cells of the immune system, the tumor vasculature, lymphatics, fibroblasts, perivascular stromal cells, and extracellular matrix components (8). Wnts are secreted lipid-modified cysteine-rich glycoproteins, and the TME contains high levels of the Wnt family of ligands (Wnts) (9). In the TME, malignant cells, stromal cells, DCs, and macrophages secrete Wnts (10). In humans, there are at least 19 different Wnt proteins all within 350–400 amino acids in length and 10 different cognate Frizzled (Fzd) receptors (9). The composition of Wnt proteins varies depending on the types of tumors. For example, Wnt3a and Wnt5a are expressed at higher levels in melanoma, whereas Wnt1 is highly expressed in lung adenocarcinoma. Wnts can exert autocrine effects on tumor cells and paracrine effects on immune cells. Although there has been much progress in understanding the role of the Wnt pathway in tumor development, progression, and metastasis, the role of this pathway in regulating anti-tumor immunity is only beginning to become better defined in the past few years.
Wnt ligands bind to Fzd receptors and activate multiple signaling pathways that includes the canonical and non-canonical pathways (9). Co-receptors Low-density lipoprotein receptor-related protein 5 (LRP5) and LRP6 are critical for canonical Wnt signaling and β-catenin, a multifunctional protein, is a central component of this pathway (9). In the absence of Wnt-signaling, β-catenin is sequestered by adenomatus polyposis coli (APC)/Axin complex, leading to its phosphorylation by glycogen synthase kinase 3β (GSK-3β), which targets it for degradation via the ubiquitin–proteosome pathway (Figure 1). Wnt-signaling inactivates APC/Axin complex, resulting in the accumulation and translocation of unphosphorylated free β-catenin to the nucleus. In the nucleus, β-catenin interacts with T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors and regulates transcription of several target genes (Figure 1) (9). In addition, Wnts can activate a number of non-canonical pathways, such as the planar cell polarity pathway and the Wnt-Ca++ pathways that activates several transcription factors, such as NFAT, AP1, JUN, CREB by β-catenin-independent mechanisms (Figure 1) (9). Recent reports show that tumor-intrinsic Wnt/β-catenin signaling facilitates immune evasion, whereas immune-cell intrinsic Wnt/β-catenin signaling drives immune suppression. However, the role of non-canonical Wnt signaling in regulating anti-tumor immune responses remains largely unexplored.
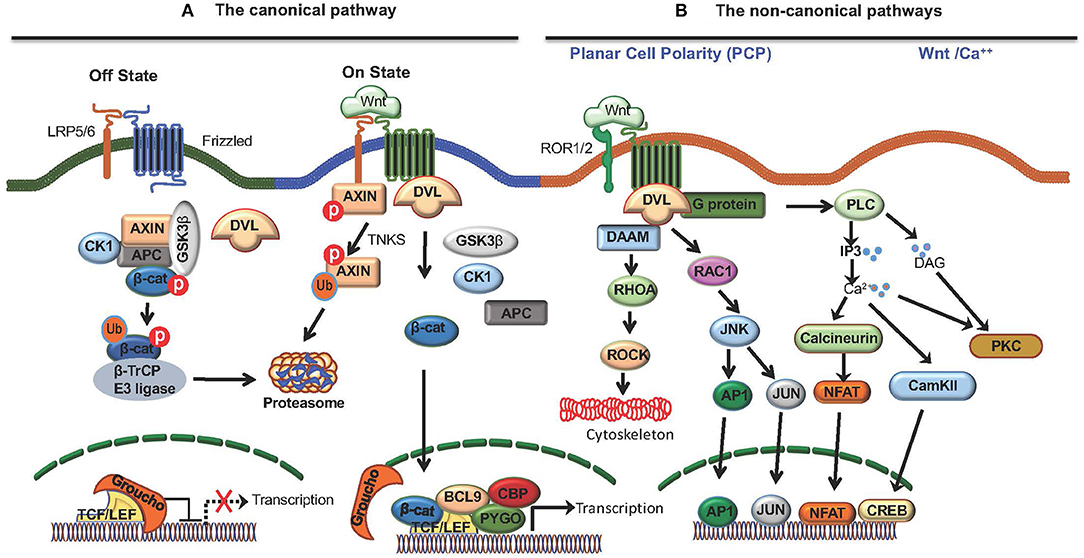
Figure 1. The Wnt signaling pathways. Wnt ligands bind to Frizzled (Fzd) receptors and activate the canonical Wnt pathway that is dependent on co-receptors LRP5/6 and β-catenin, and the non-canonical Wnt pathway that is independent of β-catenin. (A) The canonical Wnt pathway. In the absence of Wnt signaling (off state), free β-catenin in the cytoplasm is sequestered by adenomatus polyposis coli (APC)/Axin/CK1α/GSK3β complex, leading to its phosphorylation by glycogen synthase kinase 3β (GSK-3β) which targets it for degradation via the ubiquitin–proteosome pathway. Wnts binding Fzd receptors and LRP5/6 co-receptors (on state) recruits DVL resulting in disassembly of (APC)/Axin/CK1α/GSK3β complex, and the accumulation and translocation of unphosphorylated β-catenin to the nucleus. β-catenin translocation to nucleus results in the displacement of the co-repressor Groucho on the TCF/LEF transcription factor and the recruitment of co-activators, such as BCL9, CBP, and PYGO resulting in the transcription of target genes. Moreover, cytoplasmic TNKS ubiquitinates Axin, targeting it for proteasomal degradation and causing disassembly of the β-catenin destruction complex. (B) Wnts also activate non-canonical pathways, such as the planar cell polarity pathway and the Wnt-Ca++ pathway. The planar cell polarity pathway regulates cytoskeletal organization through Rhoa and Rock whereas the Wnt-Ca++ pathway activates several transcription factors, such as NFAT, AP1, JUN, CREB by β-catenin-independent mechanisms.
DCs also play a fundamental role in maintaining the balance between immunity and tolerance (1, 3). DCs can be classified into distinct subsets, based on their phenotype, microenvironmental localizations, and functions. A detailed discussion of DC subsets and their influence on adaptive immunity is outside the scope of the present review, and the reader is referred elsewhere (5, 6). Briefly, in mice, multiple subsets of DCs exist in both lymphoid (CD8α+ DCs; pDCs; CD11b+ DCs) and non-lymphoid tissues (CD103+ DCs; CD11b+ DCs; pDCs). The TME contains all these major DC subsets. CD8α+/CD103+ are the migratory DCs that excel in transporting tumor-associated antigens (TAAs) to the lymph nodes in a CCR7-dependent manner (7, 11–13). They also display enhanced ability to prime and cross-present tumor antigens to CD8+ T cells. In general, CD11b+ DCs are more effective at driving CD4+ helper T cell responses; however, the role of these DCs in tumor immunity remain largely unexplored (14). The human counterparts of CD8α+/CD103+ and CD11b+ DCs have been identified, and they are CD141+ and CD1c+ DCs (14). Moreover, these DC subsets produce chemokines that are important for recruitment of CTLs to tumors (5, 7). Tumors also contain pDCs that are capable of producing high levels of type I interferons (IFN-I) in response to viral infection (5, 7, 15). Emerging studies have shown pDCs can drive effective anti-tumor specific immune responses where certain TLR ligands are used as tumor vaccine adjuvants (16). However, their role in anti-tumor immunity is still unclear and debatable.
Robust anti-tumor immune response is dependent on several factors, such as the degree of maturation and activation of DCs, their ability to capture tumor cells and tumor-associated (TAAs), them trafficking to tumors and tumor draining lymph nodes (TDLNs), and type of factors they produce in the TME. However, DCs within the TME are often perceived as tolerogenic or immunosuppressive. In general, it is believed that instructive signals within the TME program DCs to a tolerogenic or immunosuppressive state rather than to an inflammatory state. A key issue is the nature of the instructive signals and molecules within the TME that promote regulatory DCs. Emerging studies have shown that canonical Wnt signaling plays a key role in shaping anti-tumor immune responses by modulating DC functions (Table 1; Figure 2). Here, we will discuss how Wnts shape key functions of DC in the TME that influence robust T cell- mediated cancer immunity.

Table 1. Evidence for involvement of the Wnt/β-catenin pathway in regulating immune suppression and immune cell exclusion.
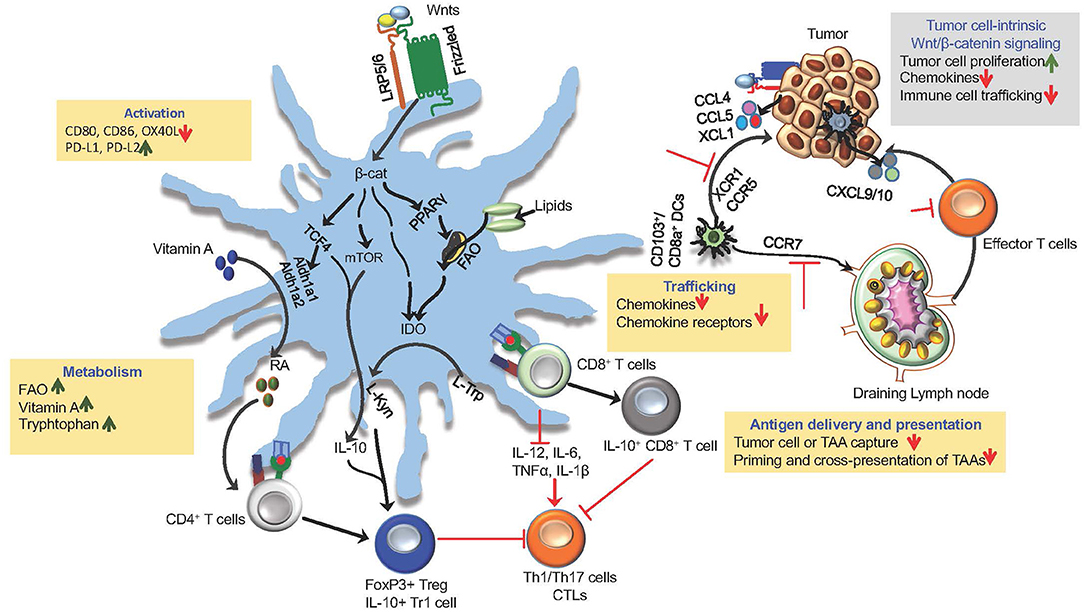
Figure 2. Wnt/β-catenin signaling in DCs and tumors in driving immune suppression and immune evasion. The canonical Wnt signaling shape anti-tumor immune responses by modulating DC functions, such as activation, trafficking, capturing and cross-presenting TAAs and expression of immune regulatory factors (RA, IL-10, IDO, TGF-β). Activation of β-catenin and its down-stream mediators mTOR and TCF4 in tumor DCs through Wnt-LRP5/6 signaling leads to the induction of anti-inflammatory factors RA, IDO, and IL-10 that are critical for promoting Treg response to tumor antigens. Wnts regulate trafficking of DCs to tumor and tumor-draining lymph nodes (TDLNs) by regulating the expression of chemokine receptors CCR5, XCR1, and CCR7. Within the TME, DC-intrinsic Wnt signaling limits the recruitment of CTLs by suppressing the expression of chemokines CXCL9/10. Wnt signaling leads to metabolic alterations in DCs that programs them to a regulatory state. Wnt-β-catenin-PPARγ-mediated signaling shifts DC metabolism from glycolysis to FAO by upregulating the expression of the carnitine palmitoyltransferase-1A (CPT1A) fatty acid transporter. In addition, Wnt-β-catenin-TCF signaling in tumor DCs promote vitamin A metabolism through the induction of enzymes involved in RA synthesis and tryptophan metabolism through the induction of IDO. The canonical Wnt signaling pathway in tumor cells promotes tumor growth and immune evasion via chemokines (CCL4, XCL1, CCL5) that are critical for the recruitment and accumulation of DCs within the TME.
DC maturation and activation is an important step in inducing robust immune response against tumors. Immature or tolerogenic DCs facilitate tolerance toward tumors whereas immunogenic/inflammatory DCs facilitate robust anti-tumor responses (1, 49). Marked differences were observed in maturation and activation of status between tolerogenic/regulatory DCs and immunogenic/inflammatory DCs (1, 3). DCs in the TME have been shown to exhibit immature state or tolerogenic state (7, 49). Immature DC or regulatory DCs express markedly lower levels of costimulatory molecules (CD80, CD86, and CD40) whereas the immunogenic or inflammatory DCs express markedly higher levels costimulatory molecules (1, 3). DCs recognize a diverse array of microbial structures through multiple receptors collectively known as pattern recognition receptors (PRRs) (50). In addition, DCs can recognize damage-associated molecular patterns (DAMPs) and other endogenous ligands that are released from dying tumor cells through PPRs (51–53). PRRs include Toll-like receptors (TLRs), C-type lectin like receptors (CLRs), RIG-I like receptors (RLRs), and Nod-like receptors (NLRs) (50). TLR ligands have gained great interest in cancer immunotherapy in the recent years for their potential use as vaccine adjuvants (16, 54). PRR-mediated signaling controls DC functions, such as antigen uptake, antigen presentation, activation, and cytokine production that are critical for anti-tumor immunity (50). In general, PRR engagement potently activates DCs by upregulating the surface expression of maturation markers, such as MHCII, CD80, CD83, and CD86 (50). Even though, PRR ligands are there in the TME (51, 52), tumor-associated DCs display markedly decreased expression of co-stimulatory molecules (49). Emerging studies have shown an important role for Wnts in regulating maturation and activation of tumor-associated DCs. Earlier ex vivo studies on human and murine DCs have shown that exposure to Wnt1, Wnt3a, and Wnt5a that activates β-catenin can program DCs to a regulatory state with decreased expression of co-stimulatory molecules (17, 20, 21). Similar observations were made with murine and human DCs upon blocking GSK3β activation (a negative regulator of β-catenin) or activating β-catenin in DCs (55, 56). Such Wnt-conditioned regulatory DCs failed to upregulate co-stimulatory molecules even in response to TLR ligands (10, 57). Further, mechanistic studies have shown that the canonical Wnt signaling can negatively regulate the inflammatory pathways, such as the NF-kB and MAPK pathways, which are critical for DC activation (58). Accordingly, tumor DCs lacking LRP5/6 or β-catenin isolated from knockout mouse models displayed increased activation with upregulated expression of co-stimulatory molecule and decreased expression of co-inhibitory molecules (PD-L1, PD-L2) (21, 22). Furthermore, studies using small molecule inhibitors of canonical Wnt signaling in tumor bearing mice showed an augmented DCs activation with an increased expression of co-stimulatory molecules and decreased expression of co-inhibitory molecules (21, 22, 56). Collectively, these studies show that Wnt/β-catenin signaling interferes with DC maturation and activation in the TME.
The migration of DCs is essential for tumor immune surveillance (5, 6). This involves, DCs migrating to tumor tissues, capturing and endocytosing dead tumor cells or cellular debris, and transporting TAAs to TDLNs where they prime and activate tumor-specific T cells (7, 11–13). This is dependent on the expression of specific chemokine receptors on DCs and its cognate chemokine ligand expression within the TME and TDLNs. The migration of DCs to TDLNs requires CCR7 expression whereas the recruitment of DCs to the TME is dependent on chemokines, such as CCL4, CCL5, and XCL1 (5). However, only a small fraction of DCs end up migrating to tumor tissue and subsequently to TDLNs. This is due to factors in the TME that control the expression of chemokine receptors and chemokines. Recent studies have shown that Wnts in the TME regulate DC trafficking by regulating chemokine receptors and chemokines via two different mechanisms. First, DC-intrinsic Wnt-signaling regulates its migration to tumors and TDLNs. Evidence supporting this come from studies showing that conditional deletion of either LRP5/6 or β-catenin in DCs in mice lead to marked increase in the number of DCs in TDLNs and TME. Furthermore, similar observations were made upon treating tumor-bearing mice with pharmacological inhibitors of the Wnt-β-catenin pathway (21, 22). Tumor DCs also produce chemokines that are critical for accumulation of T cells within the TME (5). A recent study on lung adenocarcinoma (LUND) has shown that tumor DC-intrinsic Wnt signaling plays a key role in blocking T cell infiltration into the tumors and driving cross-tolerance to tumor antigens (17). Mechanistically, Wnt1-mediated β-catenin signaling in tumor DCs resulted in transcriptional silencing of CC/CXC chemokines that are critical for recruiting effector T cells to the TME (17). In line with these observations, other studies have shown that the pharmacological blocking of the canonical Wnt signaling in DCs result in increased accumulation of effector T cells within the TME (21, 22). Second, tumor-intrinsic Wnt-signaling regulates the evasion of anti-tumor immunity by regulating the expression of chemokines that are critical for recruitment and accumulation of DCs in the TME (10). In this context, it was shown that an active Wnt-β-catenin signaling in melanoma cells suppresses production of CCL4 (C-C motif chemokine ligand 4) and consequently reduces migration and accumulation of CD103+ DCs in the TME (46). CD103+ DCs play an important role in recruitment of CTLs in TME through their production of CXCL9 (C-X-C motif chemokine ligand 9) and CXCL10 (46, 47). Collectively, these studies suggest that Wnt/β-catenin signaling regulates trafficking of DCs and T cells to the tumor tissue by regulating the expression of chemokine receptors and chemokines.
Effective cross-priming of tumor-specific CD8+ T cells by DCs involves efficient capture and cross-presentation of tumor-associated antigens (2, 4, 5). However, DCs within the TME are less efficient in cross-priming CD8+ T cells (2, 4, 5). In addition to regulating DC trafficking, recent studies with fluorescently labeled proteins have shown that in the absence of the canonical Wnt signaling, DCs are more efficient in capturing TAAs and actively transporting them to the TDLNs (21, 22). These findings are further supported by observations that LRP5/6- or β-catenin- deficient tumor DCs are more potent in capturing TAAs and are robust in priming and cross-presenting TAAs to CD8+ T cells (23–25, 59). Similar observations were made upon pharmacologically blocking canonical Wnt signaling in tumor DCs (17, 23–25, 27, 48, 59). In contrast, Wnt-conditioned DCs or tumor DCs expressing a constitutively active form of β-catenin is less potent in capturing and cross-presenting TAAs (17, 21, 22, 27, 48). Collectively, these studies show that the activation of canonical Wnt signaling pathway in tumor DCs suppresses efficient capture of tumor-associated antigens and cross-priming of CD8+ T cells.
Aberrant Wnt signaling leads to metabolic alterations in cancer cells that are critical for their survival and proliferation (60). Interestingly, Wnt signaling also plays an important role in the metabolic reprogramming of DCs in the TME (60, 61). Cellular metabolic pathways play a critical role in modulating the functions of DCs (61–63). Emerging evidence show that potential metabolic differences exist among DC subsets and also between tolerogenic and immunogenic DCs (60, 62). Immature or tolerogenic DCs show a catabolic metabolism that is manifested by increased oxidative phosphorylation, fatty acid oxidation (FAO), and glutaminolysis (61, 63, 64). In contrast, immunogenic or inflammatory DCs display an anabolic metabolism that is marked by increased glycolysis (61, 63, 64). Accumulating evidence show that tolerogenic response to tumors is also related to the metabolic dysfunction and metabolic reprograming of immune cells within the TME (26, 61, 63, 64). A recent study has shown that Wnt-mediated signaling shifts DC metabolism from glycolysis to FAO by upregulating the expression of the carnitine palmitoyltransferase-1A (CPT1A), an enzyme important for the transport of fatty acids into mitochondria (26). Furthermore, this study also revealed that this metabolic shift is dependent on the activation of β-catenin and PPAR-γ in DCs (26). In addition, other studies have shown that the canonical Wnt signaling in tumor DCs promote the metabolism of vitamin A and tryptophan through the induction of enzymes involved in retinoic acid (RA) synthesis and indoleamine 2,3-dioxygenase-1 (IDO) (22, 25). Collectively, these studies have conclusively demonstrated that Wnt-mediated metabolic shift is critical for programming DCs to the regulatory state in the TME.
The types of cytokines and other factors secreted by DCs program the differentiation of newly primed CD4+ and CD8+ T cells into effector T cell or regulatory T cells (1, 3). The TME contains higher levels of immune regulatory factors, such as IL-10, RA, and TGF-β that actively suppress differentiation and expansion of tumor-specific effector T cells (10, 57). Given the enormous burden of endogenous PRR ligands, DCs associated with the tumors express higher levels of immune regulatory factors that drive Treg response (10, 57). Recent studies have highlighted an important role for the Wnt signaling in tumor-associated DCs in regulating the expression of immune regulatory factors (21–26). Unlike DCs stimulated with microbial products, Wnt-conditioned DCs do not release immunostimulatory cytokines; instead they express IDO, IL-10, RA synthesizing enzymes and TGF-β1 (21–26). In contrast, tumor DCs that are deficient in LRP5/6 or β-catenin expressed markedly higher levels of IL-12, IL-6, IL-23, and TNF-α, and lower levels of immune regulatory factors (21, 22). These observations where further corroborated by studies showing that pharmacological blocking of the Wnt/β-catenin pathway in tumor DCs decreased the expression of RA synthesizing enzymes, IDO, IL-10, and TGF-β1 while markedly increased the expression of inflammatory cytokines (21–26).
The TME conditions DCs to acquire tolerogenic or immunosuppressive properties by activating the immune regulatory pathways (1, 5, 49). Although there has been much progress in understanding the role of DCs in inducing regulatory responses to tumor antigens, we understand very little about the regulatory signaling networks that program tumor DCs to become tolerogenic or immunosuppressive. In this context, there are emerging insights into the roles of the Wnt signaling network in DCs orchestrating tolerogenic responses to tumors.
First, a key mechanism by which Wnts in the TME drives immune suppression is through induction of vitamin A-metabolizing enzymes in DC via the β-catenin/TCF pathway (22). TME contains high levels of RA, and APCs are major producers of RA (22, 65). It is well-established that RA, an active metabolite of vitamin A, regulates a broad array of immune responses (66, 67). RA synthesis is a tightly regulated process that includes several key enzymes. Within cells, RA is produced from vitamin A (retinol) via a two-step enzymatic pathway where retinol is first oxidized to retinaldehyde (retinal) by alcohol dehydrogenases (ADH-1, -4, -5), which is next converted to RA by retinal dehydrogenases (Aldh1a1 and Aldh1a2) (50). In contrast to the gut DCs, DCs in the periphery do not express Aldh1a1 and Aldh1a2, but do constitutively express different isoforms of ADH, and hence, they lack the ability to convert vitamin A to RA (68, 69). However, within the TME, DCs express enzymes Aldh1a1 and Aldh1a2 and acquire the ability to metabolize Vitamin A to RA (22). RA produced by tumor DCs acts directly on CD4+ T cell cells, inducing Treg cell response while suppressing T effector cell response. Pharmacological blocking of RA synthesis or RA signaling affected the ability of tumor DCs to induce Treg (22). In line with these observations, DCs isolated form LRP5/6- or β-catenin- conditional knockout mice bearing B16F10, LLC, or EL4 tumors expressed markedly lower levels of vitamin A-metabolizing enzymes, and these DCs were less potent in inducing Tregs in response to tumor antigens (21, 22). Collectively these studies have shown that tumors, through DCs, exploit the LRP5/6-β-catenin-RA pathway as a mechanism of immune suppression by inducing regulatory T-cell responses.
A second key mechanism by which Wnt-β-catenin signaling in DCs promotes immune suppression is through the induction of IDO (25, 26). IDO is an immunoregulatory enzyme that catalyzes the degradation of the essential amino acid tryptophan into kynurenines (59). Previous work has identified that tumor DCs express IDO and depletion of tryptophan dampens T-cell proliferation and the generation of kynurenine drives Treg differentiation (59). However, the signaling pathways in DCs that control IDO expression and its activity remained unknown. In this context, recent studies have shown an important role for the Wnt5a-β-catenin-PPARγ signaling pathway in regulating IDO expression and activity (25, 26). Furthermore, abrogation or blocking this pathway in a transgenic murine melanoma model markedly reduced IDO expression and activity in DCs with augmented anti-tumor immune responses (25, 26). These data indicate that Wnt5a-conditioned DCs promote the differentiation of Tregs in an IDO-dependent manner and that this process serves to suppress melanoma immune surveillance. Since Wnt5a activates the non-canonical Wnt pathway in DCs (18, 19), further mechanistic studies are necessary to understand how Wnt5a activates β-catenin in DCs within the TME. Also, additional studies are warranted to understand whether β-catenin directly regulates IDO expression in DCs through TCF4 or PPARγ.
Finally, the Wnt-β-catenin pathway in DCs can drive T cell tolerance to tumors through the induction of IL-10. IL-10 is a key immunosuppressive cytokine that regulates a broad array of immune responses (70). Recent studies have shown that Wnt-β-catenin-dependent activation of mTOR and TCF4 in DCs regulates IL-10 expression (21, 23, 24). IL-10 produced by DCs exert autocrine effects to suppress cross-priming of tumor-specific CD8+ T cells (23). Furthermore, tumor DCs that are deficient in LRP5/6 or β-catenin express markedly lower levels of IL-10 and are more potent in cross-priming CD8+ T cells (21, 23, 24). In line with these observations, selective blocking of LRP5/6, β-catenin, and mTOR activation resulted in markedly reduced IL-10 production by DCs with augmented anti-tumor immune response in mice (21, 23, 24). However, further studies are warranted to understand mechanistically how β-catenin regulates mTOR activation in tumor DCs to induce IL-10. Collectively, these studies illustrate a critical role for the canonical Wnt signaling network in programming tumor DCs to induce regulatory response through induction of immune regulatory factors.
The role of the Wnt signaling cascade in other immune cells in promoting tolerogenic response is poorly understood. Beyond the ability to modulate DC functions, Wnts in the TME can directly influence the development and effector functions of various immune cell types. Wnt-mediated immunological tolerance to tumors is often considered to occur by multiple processes. Emerging studies are beginning to provide insights into the mechanisms by which Wnt signaling cascade directly modulates immunological functions of other immune cells, such as macrophages, myeloid-derived suppressor cells (MDSCs), Tregs, CD4+ T cells, CD8+ T cells, and NK cells (71). There are several excellent studies and reviews that discuss extensively how the Wnt signaling pathway shape the functions of other immune cells (43, 44, 60, 71, 72) and will thus be discussed only briefly (Table 1). Tumor-infiltrating T cells express markedly higher levels of Wnt3a and β-catenin, and display dysfunctional and exhausted effector memory phenotype (28). Furthermore, Wnt-mediated activation of β-catenin/TCF1 pathway activation suppresses naïve T cell differentiation and terminal effector differentiation of CD8+ T cells (28, 32). Likewise, T cell-infiltrating human hepatocellular carcinoma and colorectal cancer are dysfunctional and show an exhausted effector memory phenotype (43, 44, 60). In line with these observations, Wnt3a neutralization in tumor-bearing mice controls tumor growth by augmenting the expansion of tumor-antigen-specific CD8+ T cells with enhanced effector functions (27). Furthermore, the induction of Wnt signaling is critical for maintaining stemness of memory CD8+ T cells and Treg-intrinsic β-catenin signaling is critical for Treg survival, migration and suppressive functions (29–31). IL-17A-producing CD4+ T (Th17) cells play an important role in the pathogenesis of colorectal cancer (33, 34). In murine colorectal cancer model, forced expression of β-catenin in CD4+ T cells caused increased IL-17A expression that favor tumor progression. Natural killer T (NKT) cells are specialized CD1d-restricted T cells that play a critical role in tumor immune surveillance (73). Cytokines produced by activated NKT cells regulate the functions of other immune cells in the TME. There is ample evidence that Wnts can modulate anti-tumor immune response through NKT cells by suppressing the expression IFNγ (42). In addition, the chemokines CCL5 and XCL1 produced by NK cells also contribute to the recruitment and accumulation of DCs, macrophages, and Tregs within the TME (74, 75). However, it is not known whether Wnt signaling in NK cells regulates the expression of these chemokines. Tumor-associated macrophages (TAMs) play an important role in tumor progression and immune suppression (76–78). Wnts regulate macrophage functions, such as adhesion, migration, and tissue recruitment (35). In addition, it is well-documented that Wnts produced by macrophages is critical for tissue development and repair (35). Emerging studies have shown that Wnts produced by macrophages contribute to tumor cell invasiveness and tumor growth (36, 37). Furthermore, active Wnt-β-catenin signaling in macrophages programs to M2-phenotype that drives cancer cell growth, migration, metastasis, and immunosuppression (37, 38). MDSCs are a heterogeneous mix of cells that expand during cancer and potently suppress T cell responses (79, 80). Interestingly, in the murine extraskeletal tumor model, the PLCγ2-β-catenin pathway plays an important role in tumor progression, suggesting an anti-tumorigenic role for this pathway in MDSCs (40, 41). Conversely, in EL4 tumor model, the MUC1-β-catenin pathway is critical for MDSC development, suggesting a key role in MDSC-mediated immune suppression instead of tumor progression (39). Collectively, these studies show that in addition to DCs, Wnts in the TME can modulate anti-tumor immunity by directly regulating the effector functions of other immune cells.
Accumulating evidence from studies involving human cancers suggest that enhanced Wnt signaling is associated with worst clinical outcomes. Hence, targeting the canonical Wnt signaling pathway is a promising approach to overcome immune evasion by tumors and to augment anti-tumor immunity by potently activating DCs. Pre-clinical studies have shown that the canonical Wnt signaling pathways can be targeted at four different levels to overcome tumor-mediated immune suppression and augment anti-tumor immunity. These strategies include (1) blocking ligand-receptor interaction, (2) blocking Fzd-LRP5/6 signaling (PORCN inhibitors), (3) promoting β-catenin degradation (tankyrase enzyme or TNKS inhibitors), and (4) blocking β-catenin-TCF interaction (β-catenin inhibitors) (Figure 3). Pharmacological inhibitors of the Wnt pathway exist and several of them are currently in clinical testing [extensively reviewed in (43–45)]. Here, we will briefly discuss pre-clinical studies related to effects of blocking the canonical Wnt pathway on anti-tumor immunity.
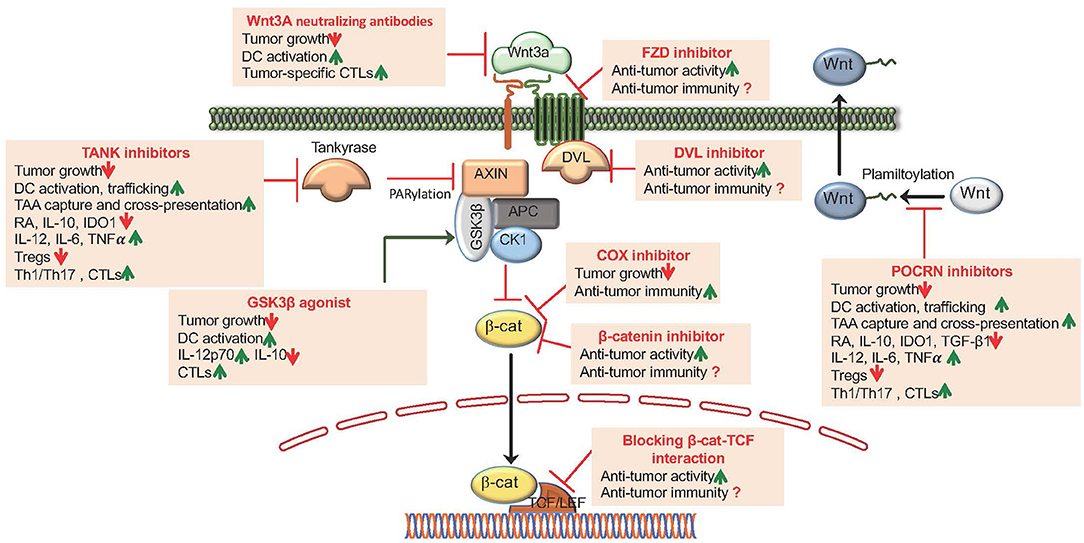
Figure 3. Pharmacological targeting of Wnt/β-catenin signaling augments anti-tumor activity and anti-tumor immune responses. The canonical Wnt signaling pathways can be targeted at four different levels: (1) Blocking Wnt ligand interaction with Fzd receptors by utilizing Wnt ligand antagonists that includes mAbs generated against the Wnts and Fzds and DKK1; (2) Blocking Fzd-LRP5/6 signaling by utilizing POCRN or DVL inhibitors; (3) Promoting β-catenin degradation by utilizing TNKS inhibitors, β-catenin-destruction complex activators that targets the Ser/Thr kinases CK1α or GSK3β and COX inhibitors and (4) Antagonizing β-catenin interaction with TCFs utilizing several small molecule modulators and synthetic inhibitors.
Emerging studies indicate a strong correlation between specific Wnt ligand expression based on the type of tumor (43–45). Thus, blocking specific Wnt ligand interaction with cognate Fzd receptors represents a potential strategy to restrain tumor cell proliferation while boosting the anti-tumor immunity (43–45). In this context, a previous study (81) using a murine model of prostate cancer has shown that administration of Wnt3a-neutralizing antibodies restrained tumor growth. However, the impact on anti-tumor immunity was not assessed in this study (81). In another critical study, it was shown that administration of Wnt3a-neutralizing antibodies in tumor bearing mice restrains tumor growth by potently activating DCs and boosting the expansion of tumor-specific CD8+ T cells with improved effector activities (27). Likewise, neutralizing or silencing Wnt1 in lung adenocarcinoma model augmented DC activation, resulting in increased recruitment and accumulation of CTLs in the TME (17). In addition, In addition, monoclonal antibodies (mAbs) that targets the different Fzd receptors are in preclinical stage and early clinical trails in humans (43–45). There are also several recently developed therapeutic agents targeting dickkopf family members (DKK1) that inhibit the binding of Wnts to co-receptors LRP5/6 (43–45). However, further studies are warranted to understand the immune consequence of DKK1 as therapeutic agents in tumor settings.
Using clinically relevant murine tumor models, studies have examined anti-tumor immune responses by blocking Fzd-LRP5/6 signaling. Porcupine (POCRN) is a membrane-bound-O-acetyltransferase enzyme that palmitoylates Wnts, which is critical for its interactions with co-receptors LRP5/6 and Fzd receptors (10). Treatment of mice with established B16-OVA tumors or EL4-OVA tumors with IWP-L6 or C59 delayed tumor growth, and this was due to marked increase in tumor-antigen-specific CD4+ and CD8+ effector T cells with reduced number of Tregs, IL-10+ Tr1, and IL-10+ CD8 T cells within the tumors (21). Furthermore, IWP-L6 or C59 treatment enhances the ability of DCs to capture and cross-present tumor antigens to CD8+ T cells (71). Similar effect was observed in murine models of melanoma, colorectal, and ovarian cancer using different POCRN inhibitors, RXC004 and WNT974 (82). In addition, POCRN inhibitor in combination with checkpoint inhibitors, such as anti-PD-1, or chemotherapy was also found to enhance anti-tumor immunity (82).
Third strategy to augment anti-tumor immune responses involves promoting β-catenin degradation using TNKS inhibitors (43–45). TNKS enzymes are members of PARP family that regulate the canonical Wnt signaling via PARylation of AXIN, a key component of β-catenin destruction complex. TNKS inhibitors promote β-catenin degradation by increasing the levels of AXIN (43–45). Recent studies have shown that treatment of melanoma or EG7 tumor-bearing mice with TNKS inhibitors XAV939 or JW55 markedly delayed tumor growth with augmented anti-tumor immunity (21, 22, 24). Likewise, recent ex vivo study on co-culture of LNCaP and PC-3 prostate cancer, cells with lymphocytes from prostrate cancer patients have shown that lymphocytes treated with XAV 939 are more potent in eliminating LNCaP and PC-3 prostate cancer cells (83). In addition, XAV 939 in combination with vaccines enhances anti-tumor immune responses by potently activating DCs in mice (83). Similarly, RNAi-mediated inhibition of β-catenin resulted in marked increase in anti-tumor immune response with reduced tumor burden in models of B16F10 melanoma, 4T1 mammary carcinoma, Neuro2A neuroblastoma, and renal adenocarcinoma (48). Another potential approach to promote β-catenin degradation is by activating GSK3β (55). In this context, a recent study using murine melanoma model has shown that intratumoral activation of GSK3β improved tumor immune surveillance that is associated with increased DC activation and CD8+ effector response (55).
Finally, another potential strategy to target the canonical Wnt pathway is blocking the interaction of β-catenin with its downstream transcription factors (43–45). Several small molecule modulators and synthetic inhibitors that antagonize β-catenin interaction with TCFs have been identified and tested on murine tumor models of myeloma, liver, colorectal, and breast cancer (43–45). These studies have shown that blocking the interaction of β-catenin and TCF interaction markedly reduced the tumor growth (43–45). However, the primary focuses of these studies are mostly to test the effect on cancer stem cells and tumor cells, but not on immune cells. In this context, a recent study has shown the peptide-mediated targeted blocking of β-catenin interaction with BCL9 co-factor and its downstream transcription factor shows robust anti-tumor efficacy across multiple murine tumor models (56, 84). Markedly, this treatment approach augmented intratumoral infiltration of cytotoxic T cells by reducing Tregs and increasing DCs and also sensitizing cancer cells to PD-1 inhibitors (84). Further preclinical studies are warranted to understand the effect of other pharmacological inhibitors that targets β-catenin and TCF interaction on anti-tumor immunity.
Thus, pharmacological blocking of the canonical Wnt pathway appears promising in preclinical models. However, the impacts of potential side effects are currently unclear, as this pathway plays an important role in several physiological processes and immune-mediated inflammatory diseases. Recent advances in the understanding of Wnt/β-catenin signaling in DCs and other immune cells present promising new therapeutic opportunities for targeted regulation of this pathway to overcome immune evasion by tumors and to augment anti-tumor immunity. In this context, targeted delivery of Wnt modulators specifically to DCs and/or tumor cells using nanoparticles and antibody drug conjugates represents a promising approach for the development of novel combinatorial anti-cancer immunotherapies. This will aid in overcoming the toxicity and potential side effects associated with Wnt inhibitors.
The central role of the Wnt pathway in regulating diverse biological processes has been appreciated for a long time. Even immunologists have recognized for decades that aberrant Wnt signaling occurs in several tumors. However, it is only recently that immunologists have begun to explore the cellular and molecular mechanisms by which the Wnt signaling pathway exerts its effects on the innate and adaptive immune systems. As evident from the discussion above, several recent observations have highlighted novel mechanisms by which the canonical Wnt signaling cascade in DCs regulates immune suppression, and the same pathway in tumors is associated with the evasion of anti-tumor immunity. In addition, preclinical studies have shown that targeting the canonical Wnt signaling pathway represents a promising approach to overcome immune evasion by tumors and promote anti-tumor immunity by potently activating DCs. However, several important questions remain unexplored (Table 2). For example, how Wnt signaling shape the innate immune functions of migratory DCs vs. non-migratory DCs in the TME? How are signals from dying cells (chemotherapy), DAMPs, and endogenous TLR ligands integrated with signals from Wnts, and what is the consequence on anti-tumor immunity? What are the effects of Wnt inhibitors plus TLR vaccine adjuvants on anti-tumor immune responses? What are the consequences of blocking the Wnt/β-catenin pathway using inhibitors as possible adjuvants plus ICI on anti-tumor immune responses? Furthermore, a major unanswered question is the extent to which the tumor-specific differences in Wnt-signaling impacts the evasion of anti-tumor immunity. Clearly, discovering answers to these questions is likely to unravel molecular mechanisms by which Wnts play a pervasive and central role in regulating anti-tumor immune response. This in turn is likely to be of great value in the design of immunotherapies against a whole range of human cancers.

Table 2. Some knowledge gaps in understanding how Wnts regulate anti-tumor immunity.
AS, MH, PP, and SM have performed bibliographic researches. AS, PP, and SM have drafted the manuscript.
AS was supported by funds from Auburn University Intramural Grants Program and Auburn University Research Initiative in Cancer (AURIC). We gratefully acknowledge the generous support of the National Institutes of Health (DK097271, DK123360) in our work.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Manicassamy S, Pulendran B. Dendritic cell control of tolerogenic responses. Immunol Rev. (2011) 241:206–27. doi: 10.1111/j.1600-065X.2011.01015.x
2. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. (2012) 12:265–77. doi: 10.1038/nrc3258
3. Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. (2010) 11:647–55. doi: 10.1038/ni.1894
4. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
5. Bottcher JP, Reis ESC. The role of type 1 conventional dendritic cells in cancer immunity. Trends Cancer. (2018) 4:784–92. doi: 10.1016/j.trecan.2018.09.001
6. Cancel JC, Crozat K, Dalod M, Mattiuz R. Are conventional type 1 dendritic cells critical for protective antitumor immunity and how? Front Immunol. (2019) 10:9. doi: 10.3389/fimmu.2019.00009
7. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
8. Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. (2012) 125:5591–6. doi: 10.1242/jcs.116392
9. Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. (2012) 149:1192–205. doi: 10.1016/j.cell.2012.05.012
10. Suryawanshi A, Tadagavadi RK, Swafford D, Manicassamy S. Modulation of inflammatory responses by Wnt/β-catenin signaling in dendritic cells: a novel immunotherapy target for autoimmunity and cancer. Front Immunol. (2016) 7:460. doi: 10.3389/fimmu.2016.00460
11. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing ccr7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
12. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
13. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. (2012) 21:402–17. doi: 10.1016/j.ccr.2012.01.008
14. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
15. Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. (2015) 15:471–85. doi: 10.1038/nri3865
16. Li JK, Balic JJ, Yu L, Jenkins B. TLR agonists as adjuvants for cancer vaccines. Adv Exp Med Biol. (2017) 1024:195–212. doi: 10.1007/978-981-10-5987-2_9
17. Kerdidani D, Chouvardas P, Arjo AR, Giopanou I, Ntaliarda G, Guo YA, et al. Wnt1 silences chemokine genes in dendritic cells and induces adaptive immune resistance in lung adenocarcinoma. Nat Commun. (2019) 10:1405. doi: 10.1038/s41467-019-09370-z
18. Valencia J, Hernandez-Lopez C, Martinez VG, Hidalgo L, Zapata AG, Vicente A, et al. Wnt5a skews dendritic cell differentiation to an unconventional phenotype with tolerogenic features. J Immunol. (2011) 187:4129–39. doi: 10.4049/jimmunol.1101243
19. Oderup C, LaJevic M, Butcher EC. Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J Immunol. (2013) 190:6126–34. doi: 10.4049/jimmunol.1203002
20. Bergenfelz C, Janols H, Wullt M, Jirstrom K, Bredberg A, Leandersson K. Wnt5a inhibits human monocyte-derived myeloid dendritic cell generation. Scand J Immunol. (2013) 78:194–204. doi: 10.1111/sji.12075
21. Hong Y, Manoharan I, Suryawanshi A, Shanmugam A, Swafford D, Ahmad S, et al. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology. (2016) 5:e1115941. doi: 10.1080/2162402X.2015.1115941
22. Hong Y, Manoharan I, Suryawanshi A, Majumdar T, Angus-Hill ML, Koni PA, et al. β-catenin promotes regulatory T-cell responses in tumors by inducing vitamin A metabolism in dendritic cells. Cancer Res. (2015) 75:656–65. doi: 10.1158/0008-5472.CAN-14-2377
23. Liang X, Fu C, Cui W, Ober-Blobaum JL, Zahner SP, Shrikant PA, et al. β-catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8(+) T cells. J Leukoc Biol. (2014) 95:179–90. doi: 10.1189/jlb.0613330
24. Fu C, Liang X, Cui W, Ober-Blobaum JL, Vazzana J, Shrikant PA, et al. β-catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc Natl Acad Sci USA. (2015) 112:2823–8. doi: 10.1073/pnas.1414167112
25. Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res. (2015) 3:1082–95. doi: 10.1158/2326-6066.CIR-14-0167
26. Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, et al. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. (2018) 48:147–60.e7. doi: 10.1016/j.immuni.2017.12.004
27. Pacella I, Cammarata I, Focaccetti C, Miacci S, Gulino A, Tripodo C, et al. Wnt3a neutralization enhances T-cell responses through indirect mechanisms and restrains tumor growth. Cancer Immunol Res. (2018) 6:953–64. doi: 10.1158/2326-6066.CIR-17-0713
28. Schinzari V, Timperi E, Pecora G, Palmucci F, Gallerano D, Grimaldi A, et al. Wnt3a/β-catenin signaling conditions differentiation of partially exhausted T-effector cells in human cancers. Cancer Immunol Res. (2018) 6:941–52. doi: 10.1158/2326-6066.CIR-17-0712
29. van Loosdregt J, Fleskens V, Tiemessen MM, Mokry M, van Boxtel R, Meerding J, et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. (2013) 39:298–310. doi: 10.1016/j.immuni.2013.07.019
30. Dai W, Liu F, Li C, Lu Y, Lu X, Du S, et al. Blockade of Wnt/β-catenin pathway aggravated silica-induced lung inflammation through Tregs regulation on Th immune responses. Mediators Inflamm. (2016) 2016:6235614. doi: 10.1155/2016/6235614
31. Sumida T, Lincoln MR, Ukeje CM, Rodriguez DM, Akazawa H, Noda T, et al. Activated β-catenin in Foxp3(+) regulatory T cells links inflammatory environments to autoimmunity. Nat Immunol. (2018) 19:1391–402. doi: 10.1038/s41590-018-0236-6
32. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. (2009) 15:808–13. doi: 10.1038/nm.1982
33. De Simone V, Pallone F, Monteleone G, Stolfi C. Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology. (2013) 2:e26617. doi: 10.4161/onci.26617
34. Kempski J, Brockmann L, Gagliani N, Huber S. TH17 cell and epithelial cell crosstalk during inflammatory bowel disease and carcinogenesis. Front Immunol. (2017) 8:1373. doi: 10.3389/fimmu.2017.01373
35. Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/β-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. (2018) 9:793. doi: 10.1038/s41419-018-0818-0
36. Linde N, Casanova-Acebes M, Sosa MS, Mortha A, Rahman A, Farias E, et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat Commun. (2018) 9:21. doi: 10.1038/s41467-017-02481-5
37. Pukrop T, Klemm F, Hagemann T, Gradl D, Schulz M, Siemes S, et al. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc Natl Acad Sci USA. (2006) 103:5454–9. doi: 10.1073/pnas.0509703103
38. Debebe A, Medina V, Chen CY, Mahajan IM, Jia C, Fu D, et al. Wnt/β-catenin activation and macrophage induction during liver cancer development following steatosis. Oncogene. (2017) 36:6020–29. doi: 10.1038/onc.2017.207
39. Poh TW, Bradley JM, Mukherjee P, Gendler SJ. Lack of Muc1-regulated β-catenin stability results in aberrant expansion of CD11b+Gr1+ myeloid-derived suppressor cells from the bone marrow. Cancer Res. (2009) 69:3554–62. doi: 10.1158/0008-5472.CAN-08-3806
40. Capietto AH, Kim S, Sanford DE, Linehan DC, Hikida M, Kumosaki T, et al. Down-regulation of PLCγ2-β-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J Exp Med. (2013) 210:2257–71. doi: 10.1084/jem.20130281
41. D'Amico L, Mahajan S, Capietto AH, Yang Z, Zamani A, Ricci B, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. (2016) 213:827–40. doi: 10.1084/jem.20150950
42. Kling JC, Jordan MA, Pitt LA, Meiners J, Thanh-Tran T, Tran LS, et al. Temporal regulation of natural killer T cell interferon gamma responses by β-catenin-dependent and -independent Wnt signaling. Front Immunol. (2018) 9:483. doi: 10.3389/fimmu.2018.00483
43. Haseeb M, Pirzada RH, Ain QU, Choi S. Wnt signaling in the regulation of immune cell and cancer therapeutics. Cells. (2019) 8:E1380. doi: 10.3390/cells8111380
44. Goldsberry WN, Londono A, Randall TD, Norian LA, Arend RC. A review of the role of Wnt in cancer immunomodulation. Cancers. (2019) 11:771. doi: 10.3390/cancers11060771
45. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. (2017) 36:1461–73. doi: 10.1038/onc.2016.304
46. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
47. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003
48. Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. RNAi-mediated β-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol Ther. (2018) 26:2567–79. doi: 10.1016/j.ymthe.2018.09.005
49. Dudek AM, Martin S, Garg AD, Agostinis P. Immature, semi-mature, and fully mature dendritic cells: toward a DC-cancer cells interface that augments anticancer immunity. Front Immunol. (2013) 4:438. doi: 10.3389/fimmu.2013.00438
50. Manicassamy S, Pulendran B. Modulation of adaptive immunity with Toll-like receptors. Semin Immunol. (2009) 21:185–93. doi: 10.1016/j.smim.2009.05.005
51. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. (2017) 17:97–111. doi: 10.1038/nri.2016.107
52. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol. (2015) 33:445–74. doi: 10.1146/annurev-immunol-032414-112043
53. Zelenay S, Reis e Sousa C. Adaptive immunity after cell death. Trends Immunol. (2013) 34:329–35. doi: 10.1016/j.it.2013.03.005
54. Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc Natl Acad Sci USA. (2014) 111:12294–9. doi: 10.1073/pnas.1400478111
55. Lopez Gonzalez M, Oosterhoff D, Lindenberg JJ, Milenova I, Lougheed SM, Martianez T, et al. Constitutively active GSK3β as a means to bolster dendritic cell functionality in the face of tumour-mediated immune suppression. Oncoimmunology. (2019) 8:e1631119. doi: 10.1080/2162402X.2019.1631119
56. Zizzari IG, Napoletano C, Botticelli A, Caponnetto S, Calabro F, Gelibter A, et al. TK inhibitor pazopanib primes DCs by downregulation of the β-catenin pathway. Cancer Immunol Res. (2018) 6:711–22. doi: 10.1158/2326-6066.CIR-17-0594
57. Swafford D, Manicassamy S. Wnt signaling in dendritic cells: its role in regulation of immunity and tolerance. Discov Med. (2015) 19:303–10.
58. Ma B, Hottiger MO. Crosstalk between Wnt/β-catenin and NF-κB signaling pathway during inflammation. Front Immunol. (2016) 7:378. doi: 10.3389/fimmu.2016.00378
59. Mellor AL, Munn DH. Creating immune privilege: active local suppression that benefits friends, but protects foes. Nat Rev Immunol. (2008) 8:74–80. doi: 10.1038/nri2233
60. El-Sahli S, Xie Y, Wang L, Liu S. Wnt signaling in cancer metabolism and immunity. Cancers. (2019) 11:904. doi: 10.3390/cancers11070904
61. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. (2010) 16:880–6. doi: 10.1038/nm.2172
62. Huang L, Xu H, Peng G. TLR-mediated metabolic reprogramming in the tumor microenvironment: potential novel strategies for cancer immunotherapy. Cell Mol Immunol. (2018) 15:428–37. doi: 10.1038/cmi.2018.4
63. Zitvogel L, Kroemer G. Targeting dendritic cell metabolism in cancer. Nat Med. (2010) 16:858–9. doi: 10.1038/nm0810-858
64. Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol. (2014) 192:2920–31. doi: 10.4049/jimmunol.1302801
65. Guo Y, Pino-Lagos K, Ahonen CA, Bennett KA, Wang J, Napoli JL, et al. A retinoic acid–rich tumor microenvironment provides clonal survival cues for tumor-specific CD8(+) T cells. Cancer Res. (2012) 72:5230–9. doi: 10.1158/0008-5472.CAN-12-1727
66. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The role of retinoic acid in tolerance and immunity. Immunity. (2011) 35:13–22. doi: 10.1016/j.immuni.2011.07.002
67. Manicassamy S, Pulendran B. Retinoic acid-dependent regulation of immune responses by dendritic cells and macrophages. Semin Immunol. (2009) 21:22–7. doi: 10.1016/j.smim.2008.07.007
68. Manicassamy S, Ravindran R, Deng J, Oluoch H, Denning TL, Kasturi SP, et al. Toll-like receptor 2-dependent induction of vitamin A-metabolizing enzymes in dendritic cells promotes T regulatory responses and inhibits autoimmunity. Nat Med. (2009) 15:401–9. doi: 10.1038/nm.1925
69. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of β-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. (2010) 329:849–53. doi: 10.1126/science.1188510
70. Ouyang W, O'Garra A. IL-10 family cytokines IL-10 and IL-22: from basic science to clinical translation. Immunity. (2019) 50:871–91. doi: 10.1016/j.immuni.2019.03.020
71. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol. (2019) 10:2293. doi: 10.3389/fimmu.2019.02293
72. Gattinoni L, Ji Y, Restifo NP. Wnt/β-catenin signaling in T-cell immunity and cancer immunotherapy. Clin Cancer Res. (2010) 16:4695–701. doi: 10.1158/1078-0432.CCR-10-0356
73. Nair S, Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. (2017) 8:1178. doi: 10.3389/fimmu.2017.01178
74. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37.e14. doi: 10.1016/j.cell.2018.01.004
75. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
76. Guerriero JL. Macrophages: the road less traveled, changing anticancer therapy. Trends Mol Med. (2018) 24:472–89. doi: 10.1016/j.molmed.2018.03.006
77. Cheung KJ, Ewald AJ. Illuminating breast cancer invasion: diverse roles for cell-cell interactions. Curr Opin Cell Biol. (2014) 30:99–111. doi: 10.1016/j.ceb.2014.07.003
78. Malsin ES, Kim S, Lam AP, Gottardi CJ. Macrophages as a source and recipient of Wnt signals. Front Immunol. (2019) 10:1813. doi: 10.3389/fimmu.2019.01813
79. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. (2009) 9:162–74. doi: 10.1038/nri2506
80. Anani W, Shurin MR. Targeting myeloid-derived suppressor cells in cancer. Adv Exp Med Biol. (2017) 1036:105–28. doi: 10.1007/978-3-319-67577-0_8
81. Li X, Placencio V, Iturregui JM, Uwamariya C, Sharif-Afshar AR, Koyama T, et al. Prostate tumor progression is mediated by a paracrine TGF-β/Wnt3a signaling axis. Oncogene. (2008) 27:7118–30. doi: 10.1038/onc.2008.293
82. Bhamra I, Armer R, Bingham M, Eagle C, Cook AE, Phillips C, et al. Porcupine inhibitor RXC004 enhances immune response in pre-clinical models of cancer. Cancer Res. (2018) 78 (Suppl. 13):3764.
83. Stakheev D, Taborska P, Strizova Z, Podrazil M, Bartunkova J, Smrz D. The WNT/β-catenin signaling inhibitor XAV939 enhances the elimination of LNCaP and PC-3 prostate cancer cells by prostate cancer patient lymphocytes in vitro. Sci Rep. (2019) 9:4761. doi: 10.1038/s41598-019-41182-5
Keywords: Wnt, dendritic cells, beta-catenin (β-catenin), tumor microenvironment (TME), immunotherapy, anti-tumor immunity, immunotherapy
Citation: Suryawanshi A, Hussein MS, Prasad PD and Manicassamy S (2020) Wnt Signaling Cascade in Dendritic Cells and Regulation of Anti-tumor Immunity. Front. Immunol. 11:122. doi: 10.3389/fimmu.2020.00122
Received: 10 June 2019; Accepted: 16 January 2020;
Published: 17 February 2020.
Edited by:
Riccardo Dolcetti, University of Queensland, AustraliaReviewed by:
Bozena Kaminska, Nencki Institute of Experimental Biology (PAS), PolandCopyright © 2020 Suryawanshi, Hussein, Prasad and Manicassamy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amol Suryawanshi, YW1vbC5zdXJ5YXdhbnNoaUBhdWJ1cm4uZWR1; Santhakumar Manicassamy, c21hbmljYXNzYW15QGF1Z3VzdGEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.