 Yosuke Kunishita1
Yosuke Kunishita1 Ryusuke Yoshimi1*
Ryusuke Yoshimi1* Reikou Kamiyama1Daiga Kishimoto1Koji Yoshida1Eijin Hashimoto1Takaaki Komiya1Natsuki Sakurai1Yumiko Sugiyama2
Reikou Kamiyama1Daiga Kishimoto1Koji Yoshida1Eijin Hashimoto1Takaaki Komiya1Natsuki Sakurai1Yumiko Sugiyama2 Yohei Kirino1
Yohei Kirino1 Keiko Ozato3Hideaki Nakajima1
Keiko Ozato3Hideaki Nakajima1- 1Department of Stem Cell and Immune Regulation, Yokohama City University Graduate School of Medicine, Yokohama, Japan
- 2Center for Rheumatic Diseases, Yokohama City University Medical Center, Yokohama, Japan
- 3Program in Genomics of Differentiation, National Institute of Child Health and Human Development, National Institute of Health, Bethesda, MD, United States
TRIM21 is one of the autoantigens that reacts with an anti-SS-A antibody (Ab) present in patients with systemic lupus erythematosus (SLE) and Sjögren's syndrome. TRIM21 is thought to play a role in B-cell proliferation and apoptosis, among other activities. Here we examined a pathological role of TRIM21 in SLE. Trim21-deficient MRL/lpr mice were generated by backcrossing Trim21-deficient C57BL/6 mice to MRL/lpr mice. The levels of serum anti-dsDNA Ab and urine protein at 28 weeks of age were significantly higher in Trim21-deficient MRL/lpr mice as compared to wild-type MRL/lpr mice (p = 0.029 and 0.003, respectively). Resting B cells from Trim21-deficient mice showed significantly higher abilities to differentiate into plasmablasts and to produce Ab as compared with control mice. Due to the reduction of TRIM21-mediated ubiquitylation, IRF5 protein expression was increased in Trim21-deficient MRL/lpr mice (p = 0.021), which correlated with increased plasmablast generation and immunoglobulin production. B cells from SLE patients with anti-TRIM21 Ab seropositivity also showed a significantly higher ability to differentiate into plasmablasts as compared with those without anti-TRIM21 Ab or healthy controls. These results suggest that TRIM21 dysfunction contributes to SLE pathogenesis by promoting B-cell differentiation, for which anti-TRIM21 Ab may be partly responsible.
Introduction
Systemic lupus erythematosus (SLE) is a chronic, systemic autoimmune disease of unknown cause. The etiology of SLE is thought to be multifactorial and to include contributions from both environmental factors, such as hormones (1), ultraviolet light (2), and infection (3), and genetic susceptibility which has been investigated by genome-wide association studies (4). As the disease is characterized by B-cell hyperactivity with the presence of increased immunoglobulin and a variety of autoantibodies in patients' sera, modulating B-cell function has been considered as a promising therapeutic approach (5, 6).
Tripartite motif containing 21 (TRIM21), a ~52-kDa protein also known as Ro52/SS-A, is one of major autoantigens recognized by anti-SS-A autoantibody present in sera of autoimmune diseases such as SLE and Sjögren's syndrome (7–9). TRIM21 belongs to the TRIM family, in which proteins are commonly comprised of a RING-finger domain, one or two B-box domains and a coiled-coil domain (10, 11).
Previous studies have revealed that TRIM21 is expressed ubiquitously but predominantly in hematopoietic cells and induced by type I and type II interferons (IFNs) (9, 11, 12). TRIM21 ubiquitylates IFN regulatory factors (IRFs) via its E3 ubiquitin ligase activity on the RING-finger domain and regulates the production of type I IFN and IL-12p40 (13–18). It has been reported that the TRIM21 overexpression in a B-cell line leads to reduced growth and increased apoptosis after activation (19, 20).
Two groups have disrupted the murine Trim21 gene to analyze the function of TRIM21. We showed that NF-κB-dependent proinflammatory cytokine production is increased in Trim21-deficient C57BL/6 mice (17). Another group reported that IL-23-dependent Th17 differentiation and production of proinflammatory cytokines and type I IFN are enhanced and thereby contact dermatitis is evoked in Trim21-deficient C57BL/6 mice (16). These results suggest that TRIM21 has physiological roles for suppressing the excess inflammatory response and keeping homeostasis.
It has been shown that TRIM21 is expressed at higher levels in peripheral blood mononuclear cells (PBMCs) in SLE patients as compared to healthy controls (21). Although the role of TRIM21 in SLE pathogenesis is still unknown, previous studies strongly suggest that TRIM21 can act as a suppressor for autoimmune and inflammatory response and its dysfunction can cause the pathological state of SLE.
In this study, we generated and analyzed MRL/lpr lupus-prone mice in which Trim21 gene is deleted to investigate the function of TRIM21 in autoimmune pathogenesis. The mice showed worsening SLE pathology with significantly increased autoantibody production and urine protein relative to wild-type MRL/lpr mice. We found the aberrant B-cell differentiation is accompanied by increased expression of transcription factors, IRF5 and BLIMP-1, which are crucial for B-cell differentiation and antibody (Ab) production (22–24). These factors have also been identified by SLE genome-wide association studies (25, 26). Similar to the results from mouse gene disruption studies, B cells from SLE patients with seropositivity of anti-TRIM21 Ab also indicated significantly higher ability to differentiate into plasmablasts and to produce Ab as compared with controls. Together, our results point that TRIM21 dysfunction promotes aberrant B-cell differentiation and Ab production in some SLE patients, which may be associated with anti-TRIM21 Ab.
Materials and Methods
Mice
Trim21−/−C57BL/6 mice have been described previously (17). C57BL/6JJmsSlc, MRL/MpJJmsSlc+/+ (MRL/+) and MRL/MpJJmsSlclpr/lpr (MRL/lpr) mice were purchased from Japan SLC (Hamamatsu, Japan). Trim21−/−C57BL/6 mice were backcrossed to MRL/lpr mice for more than 10 generations to produce Trim21−/−MRL/lpr mice. All mice were maintained under specific pathogen-free conditions within the animal facility at Yokohama City University, and female mice were used in all experiments. A comprehensive mouse genotyping examination was performed by ICLAS monitoring Center (Kawasaki, Japan). All experiments of skin-draining lymph nodes (sdLNs) were performed using bilateral axillary and inguinal lymph nodes. All animal experiment protocols were approved by the animal protocol ethics committee of Yokohama City University.
Patients
Seventeen patients with SLE (16 women and one man), who fulfilled the revised 1997 American College of Rheumatology criteria for SLE (27), and five healthy controls (4 women and one man) were enrolled in the study. The study was conducted in accordance with the Declaration of Helsinki, and informed consent was obtained from all patients and healthy controls before study enrollment. The study design was approved by the ethics committee of Yokohama City University Hospital (B100701027).
B Cell Preparation and Culture
Mice CD43− resting B cells were isolated from the spleen of 8-week-old mice using Mouse B cell Isolation Kit (Miltenyi Biotec, Bergisch-Gladbach, Germany), according to the manufacturer's protocols. Isolated resting B cells were cultured in RPMI-1640 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (MP Biomedicals, Santa Ana, CA, USA), 1 mM sodium pyruvate (Wako, Osaka, Japan), 10 mM HEPES (Gibco, Waltham, MA, USA), 100 μg/ml streptomycin, 100 U/ml penicillin (Gibco), and 1 mM 2-mercaptoethanol (Gibco). For some experiments, resting B cells were stimulated with 1 μg/ml anti-mouse IgM (Jackson ImmunoResearch, West Grove, PA, USA), 1 μg/ml anti-mouse CD40 (BioLegend, San Diego, CA, USA), 100 μg/ml poly(I:C) (Tocris Bioscience, Minneapolis, MN, USA), and/or 50 μg/ml imiquimod (AdooQ BioScience, Irvine, CA, USA). After incubation for 24 or 72 h, the cells were immediately used for flow cytometric analyses, and the supernatants were stocked at −80°C until use. The cell viability was assessed 24 h after stimulation using CellTiter-Blue Cell Viability Assay (Promega, Madison, WI, USA), according to the manufacturer's protocols.
Human PBMCs were separated by density gradient centrifugation using Lympholyte-H (Cedarlane, Burlington, Canada). Human CD43− resting B cells were isolated from PBMCs using MojoSort Human B Cell (CD43−) Isolation Kit (BioLegend), according to the manufacturer's protocols. Isolated resting B cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, 10 mM HEPES, 100 μg/ml streptomycin, 100 U/ml penicillin, and 1 mM 2-mercaptoethanol. For some experiments, resting B cells were stimulated with 1 μg/ml anti-human IgM Ab (BioLegend), 1 μg/ml recombinant human CD40L (BioLegend), 100 μg/ml poly(I:C), and/or 50 μg/ml imiquimod (R837; InvivoGen, San Diego, CA, USA). After incubation for 24 h, the cells were immediately used for flow cytometric analyses, and the supernatants were stocked at – 80°C until use.
T Cell Preparation
Mice CD3+ T cells were isolated from the spleen and sdLN of 8-week-old mice using Mouse CD3ε MicroBead Kit (Miltenyi Biotec), according to the manufacturer's protocols. Gene expression of T-cell transcription factor in the isolated CD3+ T cells were evaluated using quantitative real-time RT-PCR (qPCR).
qPCR
Total RNA was extracted with RNeasy Mini Kit (Qiagen, Hilden, Germany), and complementary DNA (cDNA) was prepared with SuperScript II Reverse Transcriptase (Invitrogen, Waltham, MA, USA). Fast SYBR Green Master Mix (Applied Biosystems, Waltham, MA, USA) was used for amplification of sample cDNA, and the data were analyzed by the StepOnePlus Real-Time PCR Systems (Applied Biosystems), according to the manufacturer's instructions. Transcript levels were standardized by the expression of hypoxanthine phosphoribosyltransferase (Hprt) mRNA. Primer sequences are listed in Table 1.

Table 1. Mouse primer list used in qPCR.
Flow Cytometric Analysis
Spleen and sdLNs were collected from mice and passed through 40-μm nylon cell strainer (Falcon, Corning, NY, USA) to obtain a single cell. Red blood cells were removed by hemolysis using RBC Lysis Buffer (BioLegend), according to the manufacturer's protocols. The Fc of harvested cells and cultured B cells were blocked with anti-mouse CD16/CD32 Ab (BD Biosciences) for 10 min at 4°C. Then the cells were stained with Abs to surface markers for 20 min at 4°C. For intracellular staining, cultured B cells were treated with Fixation Buffer (BioLegend) and Intracellular Staining Permeabilization Wash Buffer (BioLegend), according to the manufacturer's protocols. After permeabilization, cells were stained with Abs to intracellular antigen for 20 min at 4°C.
For human PBMCs, CD43− resting B cells, and cultured B cells, the FcR were blocked with anti-human FcR Ab (Human TruStain FcX; BioLegend) for 10 min at 4°C. The cell staining was performed according to the method of standardizing immunophenotyping for the human immunology project (36).
Zombie Aqua dye (BioLegend) or Zombie Green dye (BioLegend) were used for detecting dead cells in samples, according to the manufacturer's protocols. All gates were set using fluorescence minus one control. Stained cells were analyzed on BD FACSCanto II instrument (BD Biosciences, Franklin Lakes, NJ, USA), and data were analyzed using FlowJo software version 10 (FlowJo, Ashland, OR, USA).
Multiple Soluble Analyte Immunoassay
The level of cytokines and immunoglobulins in cell supernatants were measured by bead-based immunoassay. The concentration of mice cytokines, immunoglobulins, and human immunoglobulins were measured using LEGENDplex Mouse Inflammation Panel, Mouse IFN-α Capture Bead, Mouse Anti-Virus Response Panel Standard, Mouse Anti-Virus Response Panel Detection Antibodies, Mouse Immunoglobulin Isotyping Panel and Human Immunoglobulin Isotyping Panel (BioLegend), according to the manufacturer's protocols.
Measurement of Proteinuria and Serum Autoantibodies
Urine was collected from individual mice every 2 weeks from 8 weeks of age for over 16 h in metabolic cages (Shinano Manufacturing Co., Tokyo, Japan). The concentration of urine albumin and creatinine were measured using DC Protein Assay Reagent (Bio-Rad Laboratories, Hercules, CA, USA) and Creatinine Parameter Assay Kit (R&D Systems, Minneapolis, MN, USA), respectively, and the urine albumin-creatinine ratio was quantified.
Mouse serum anti-dsDNA Ab was measured using an enzyme-linked immunosorbent assay (ELISA) (Shibayagi, Shibukawa, Japan). Human serum anti-TRIM21 Ab was measured using an ELISA kit (Dr. Fooke Laboratorien, Neuss, Germany), according to the manufacturer's protocols.
Western Blot Analysis
Mouse CD43− resting B cells, which were stimulated with 1 μg/ml anti-mouse IgM Ab and 100 μg/ml poly(I:C) for 72 h, were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate and 1% Nonidet P-40) in the presence of a protease inhibitor cocktail (Sigma-Aldrich). After 30 min incubation, the cells were sonicated by ultrasonication (Emerson Electric, St. Louis, MO, USA). Supernatants were collected after centrifugation at 15,000 g for 30 min and mixed with SDS sample buffer with 2-mercaptoethanol (Sigma-Aldrich). After 5 min boiling, the proteins were separated by NuPAGE 4–12% Bis-Tris gel electrophoresis (Invitrogen) and transferred onto polyvinylidene fluoride (PVDF) membrane (Merck Millipore). Anti-IRF4 (1:1,000 dilution, BioLegend), anti-IRF5 (1:1,000 dilution, Abcam, Cambridge, MA, USA), anti-IRF8 (1:1,000 dilution, Abcam), anti-ubiquitin (1:2,000 dilution, BioLegend), and anti-β-Actin antibodies (1:4,000 dilution, BioLegend) were used to detect the specific proteins. Chemiluminescence was developed using ECL Select Western Blotting Detection Reagent (GE Healthcare Life Sciences, Pittsburgh, PA, USA) and exposed to the LAS-3000 Mini Imaging System (FUJIFILM, Tokyo, Japan).
Ubiquitylation Assay
Mice CD43− resting B cells were stimulated with 1 μg/ml anti-mouse IgM Ab and 100 μg/ml poly(I:C) for 72 h. Cultures were treated with 25 μM MG-132 (Merck Millipore, Darmstadt, Germany) for 2 h before harvest. Extracts were immunoprecipitated with an anti-IRF5 Ab (Santa Cruz Biotechnology, Dallas, TX, USA) bound to protein G agarose beads (Cell Signaling Technology, Danvers, MA, USA). Immunoprecipitants were analyzed by Western blot using anti-ubiquitin Ab.
Abs
Anti-CD3 (clone 17A2, catalog # 100227), anti-CD138 (clone 281-2, catalog # 142523), anti-CD317 (clone 927, catalog # 127023), anti-I-Ab (clone M5/114.15.2, catalog # 107631), anti-IgD (clone 11-26c.2a, catalog # 405725), anti-CD4 (clone RM4-5, catalog # 100539), anti-CD5 (clone 53-7.3, catalog # 100623), anti-CD8a (clone 53-6.7, catalog # 100733), anti-CD93 (clone AA4.1, catalog # 136511), anti-IgM (clone RMM-1, catalog # 406511), anti-B220 (clone RA3-6B2, catalog # 103235), anti-F4/80 (clone BM8, catalog # 123127), anti-CD1d (clone 1B1, catalog # 123521), anti-CD11b (clone M1/70, catalog # 101211), anti-CD11c (clone N418, catalog # 117309), anti-CD19 (clone 6D5, catalog # 115512), anti-Siglec H (clone 551, catalog # 129611), anti-Blimp-1 (clone 5E7, catalog # 150003) and anti-Bcl6 (clone 7D1, catalog # 358507) Abs were purchased from BioLegend and used for flow cytometric analyses of murine cells.
Anti-CD24 (clone ML5, catalog # 311105), anti-CD19 (clone HIB19, catalog # 302229), anti-CD27 (clone M-T271, catalog # 356411), anti-CD38 (clone HB-7, catalog # 356605), anti-CD20 (clone 2H7, catalog # 302357), anti-CD3 (clone UCHT1, catalog # 300433), and anti-IgD (clone IA6-2, catalog # 348220) Abs were purchased from BioLegend and used for flow cytometric analyses of human cells.
Statistics
Statistical analysis was performed using Prism software version 6.0 (GraphPad Software, La Jolla, CA, USA) and SPSS Statistics software version 22 (IBM, Armonk, NY, USA). Unpaired two-tailed Student's t-test was used for parametric analysis of continuous variables. Chi-square test or Fisher's exact was used for analysis of categorical variables. A log-rank test was used for evaluating the survival curves statistically. A p < 0.05 was considered significant in all analyses.
Results
TRIM21 Deficiency Promotes Exacerbation of Lupus-Like Symptoms in MRL/lpr Mice
To verify the pathological role of TRIM21 in the autoimmune response, we first examined whether the expression of TRIM21 is influenced by an autoimmune condition. MRL/lpr mice, which are model mice spontaneously developing lupus-like symptoms, including anti-TRIM21 Ab production (37, 38), expressed the transcript of Trim21 ubiquitously as shown in the previous study using C57BL/6 mice (17). However, the Trim21 transcript tended to be expressed more preferably in lymphoid tissues such as thymus and spleen in MRL/lpr mice as compared to their control, MRL/+ mice (Figure 1A).
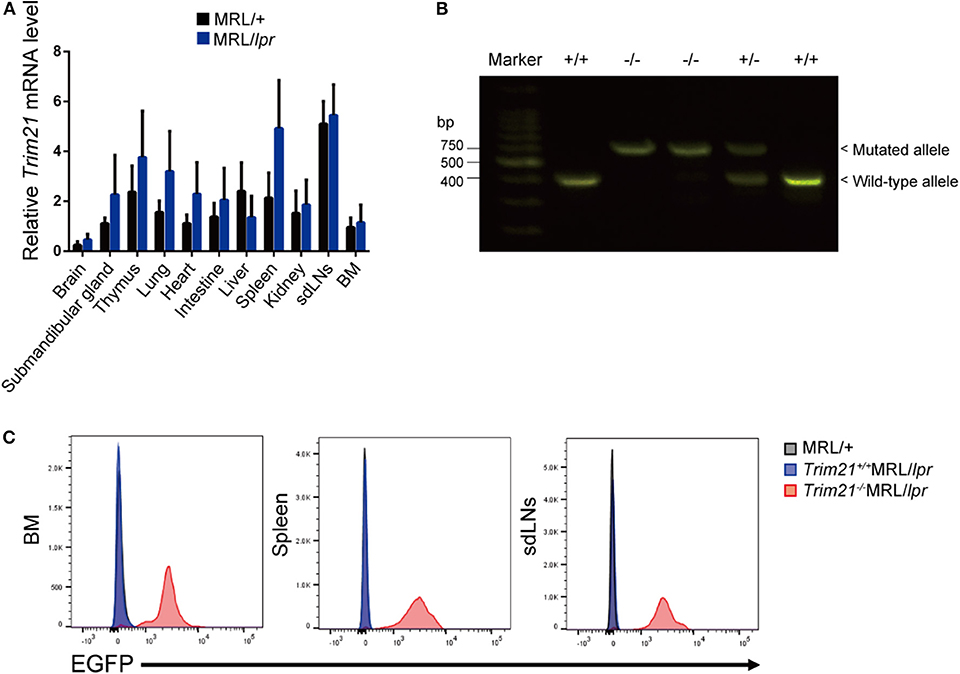
Figure 1. Trim21 expression in tissues from MRL/lpr mice and generation of Trim21−/−MRL/lpr mice. (A) Trim21 mRNA expression in tissues from MRL/+ and MRL/lpr mice at 8 weeks of age. (n = 4 in each group). (B) The genotype of Trim21 is determined by PCR of genomic DNA from the tail. Arrows indicate the position of bands to detect the wild-type and mutated alleles. (C) Flow cytometric analyses reveal the EGFP expression by the mutated alleles in cells from BM, spleen and sdLNs of the 8-week-old Trim21−/−MRL/lpr mice. Data are representatives of at least three independent experiments. Values are shown as means ± SEM.
The genetic background of Trim21+/−MRL/lpr mice was confirmed by genome scanning based on the microsatellite short tandem repeat analysis (Figure 2). The mice were genotyped by PCR analysis (Figure 1B), and Trim21-deleted allele was confirmed by the expression of an enhanced GFP (EGFP) reporter gene replacing Trim21 exons in cells from the immune tissues using flow cytometric analysis (Figure 1C).
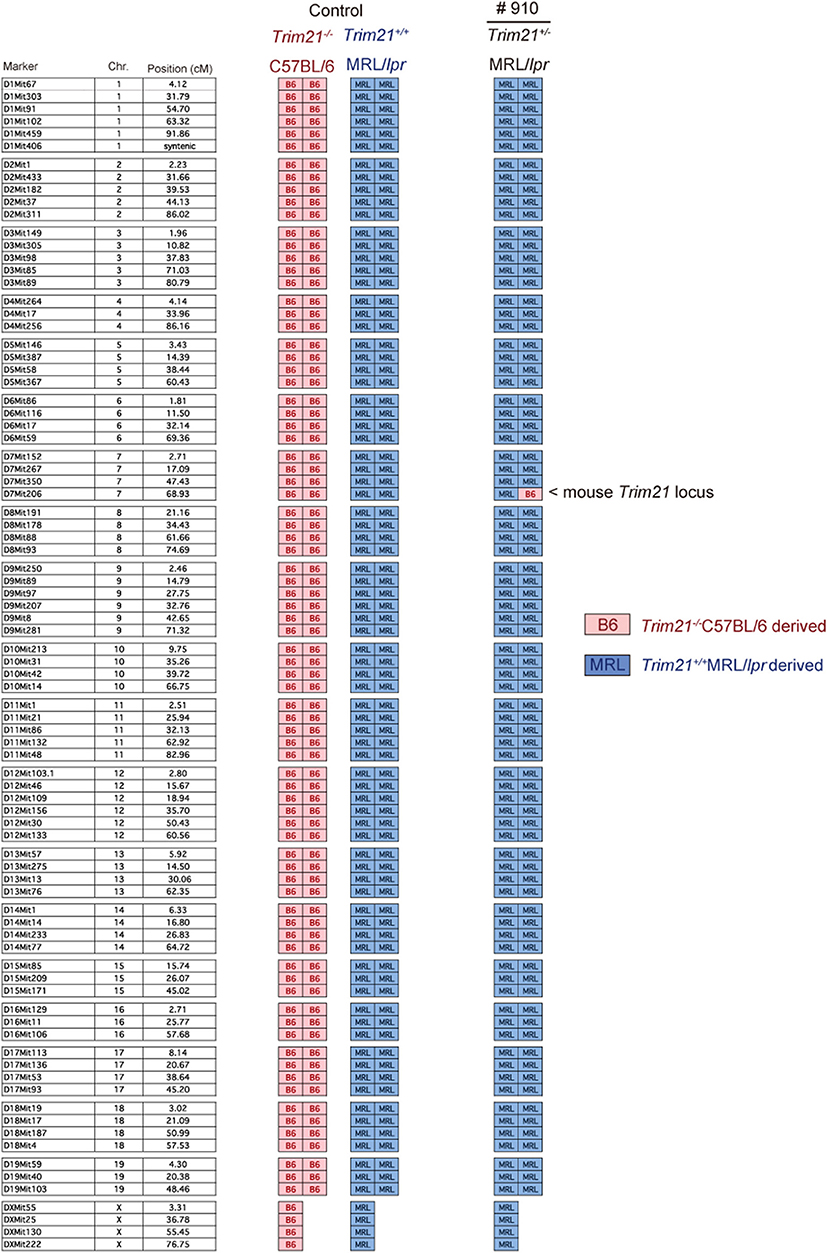
Figure 2. Genome scanning of backcrossed Trim21+/−MRL/lpr mice. A comprehensive examination of the genotype of Trim21+/−MRL/lpr mouse (Mouse # 910) generated by back-crossing for more than ten generations. Chr., chromosome.
As previously reported (39), Faslpr mutation shortened life-span (Figure 3A). Both Trim21+/+MRL/lpr and Trim21−/−MRL/lpr mice started to die at around 24 weeks of age and ~90% dead by 60 weeks of age. There was no significant difference in survival between them. Although splenomegaly and LN swelling were observed in both Trim21+/+MRL/lpr and Trim21−/−MRL/lpr mice, there was no significant difference in the weights of spleens and sdLNs between the two genotype mice (Figure 3B). On the other hand, the titer of serum anti-dsDNA Ab and daily urine protein were significantly increased over time in Trim21−/−MRL/lpr mice as compared with Trim21+/+MRL/lpr mice (Figures 3C,D). In particular, two severe cases of urine protein appeared in Trim21−/−MRL/lpr mice at 28 weeks while no such severe cases were seen in Trim21+/+MRL/lpr mice (Figure 3D).
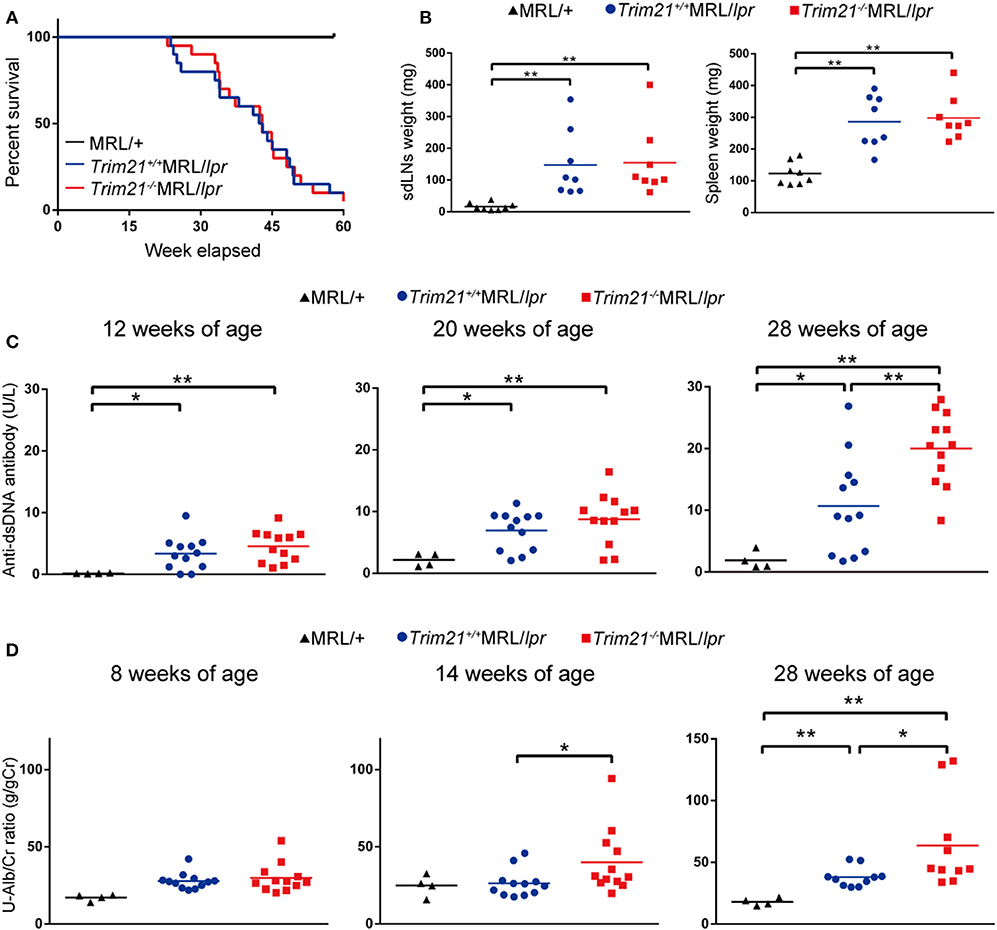
Figure 3. TRIM21 deficiency promotes exacerbation of lupus-like symptoms in MRL/lpr mice. (A) Survival rates of mice (MRL/+; n = 4, Trim21+/+MRL/lpr; n = 20 and Trim21−/−MRL/lpr; n = 20). (B) Weights of sdLNs and spleens at 10 weeks of age (n = 8 in each group). (C) Concentrations of serum anti-dsDNA Ab were measured by ELISA at 12, 20, and 28 weeks (MRL/+; n = 4, Trim21+/+MRL/lpr; n = 12 and Trim21−/−MRL/lpr; n = 12). (D) Proteinuria (albumin-creatinine ratio) was measured at 8, 14, and 28 weeks (MRL/+; n = 4, Trim21+/+MRL/lpr; n = 12 and Trim21−/−MRL/lpr; n = 12). Statistically significant data (*p < 0.05 and **p < 0.01) by Student's t-test, respectively.
Taken together, these results indicate that TRIM21 deficiency promotes exacerbation of systemic autoimmunity in MRL/lpr mice.
TRIM21 Deficiency Increases B Cells in the Spleen of Lupus Model Mice
It has been reported that autoantibody production and urine protein due to glomerulonephritis in MRL/lpr mice depend on B cell-intrinsic MyD88 signal (40). To examine whether the autoimmune pathogenesis in Trim21−/−MRL/lpr mice is associated with B-cell abnormalities, we investigated the populations of leukocyte subsets in spleens and sdLNs by flow cytometric analysis. The number of B220+B19+ B cells was significantly higher in spleens from Trim21−/−MRL/lpr mice as compared to those from Trim21+/+MRL/lpr mice (p = 0.008; Figure 4A). The difference is mainly reflected by the increased number of IgM+IgD+CD19+ mature B cells in Trim21−/−MRL/lpr mice (Figure 4A). Although there was no significant difference in the number of B220+B19+ B cells in sdLNs between Trim21+/+MRL/lpr mice and Trim21−/−MRL/lpr mice, the number of CD19−CD138+ plasma cell was significantly higher in Trim21−/−MRL/lpr mice as compared to Trim21+/+MRL/lpr mice (p = 0.009; Figure 4B). There were no significant differences in the activation markers, CD80 and CD86, in B cells from spleens and sdLNs between Trim21+/+ and Trim21−/−MRL/lpr mice (Figures 4A,B).
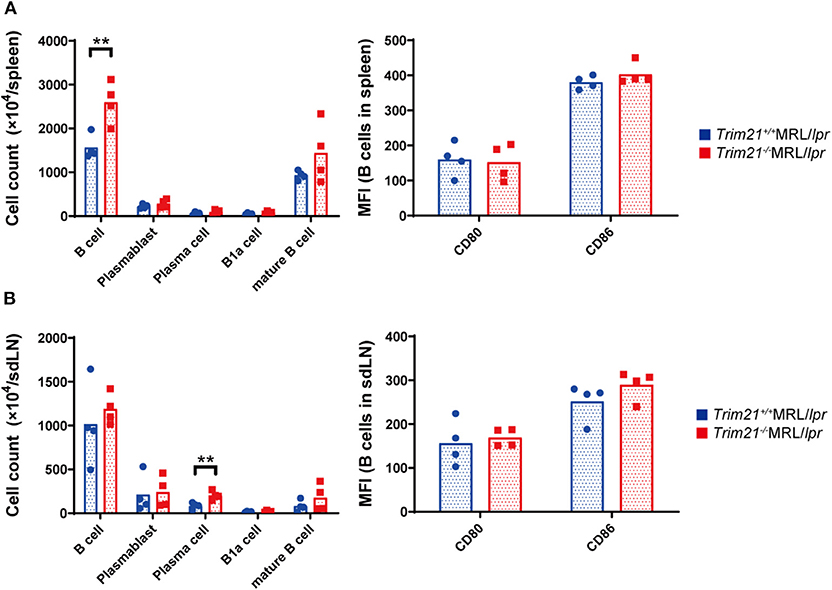
Figure 4. The comparison of B cell subset populations between Trim21+/+ and Trim21−/−MRL/lpr mice. Flow cytometric analyses and qPCR analyses were performed for spleen or sdLN cells at 10 weeks of age (n = 4 in each group). (A,B) B-cell subsets in spleens and sdLNs were classified by surface markers as follows: B cells; B220+CD19+, plasmablasts; CD19+CD138+, plasma cells; CD19−CD138+, B-1a cells; IgM+CD5+CD1d− and mature B cells; IgM+IgD+CD19+. Mean fluorescence intensities (MFIs) of CD80 and CD86 were measured in B220+CD19+ B cells. Statistically significant data (**p < 0.01) by Student's t-test.
It is known that various immune cells are activated due to an increase in abnormal autoreactive T cells in MRL/lpr mice (41). There was no significant difference in the proportion of CD4+ T cells and CD8+ T cells, and gene expressions of transcription factors related to Th1 (Tbet), Th2 (Gata3), Th17 (Rorc), Treg (Foxp3), and Tfh (Bcl6) subsets in CD3+ T cells from spleens and sdLNs between Trim21+/+MRL/lpr and Trim21−/−MRL/lpr mice (Figures 5A,B).
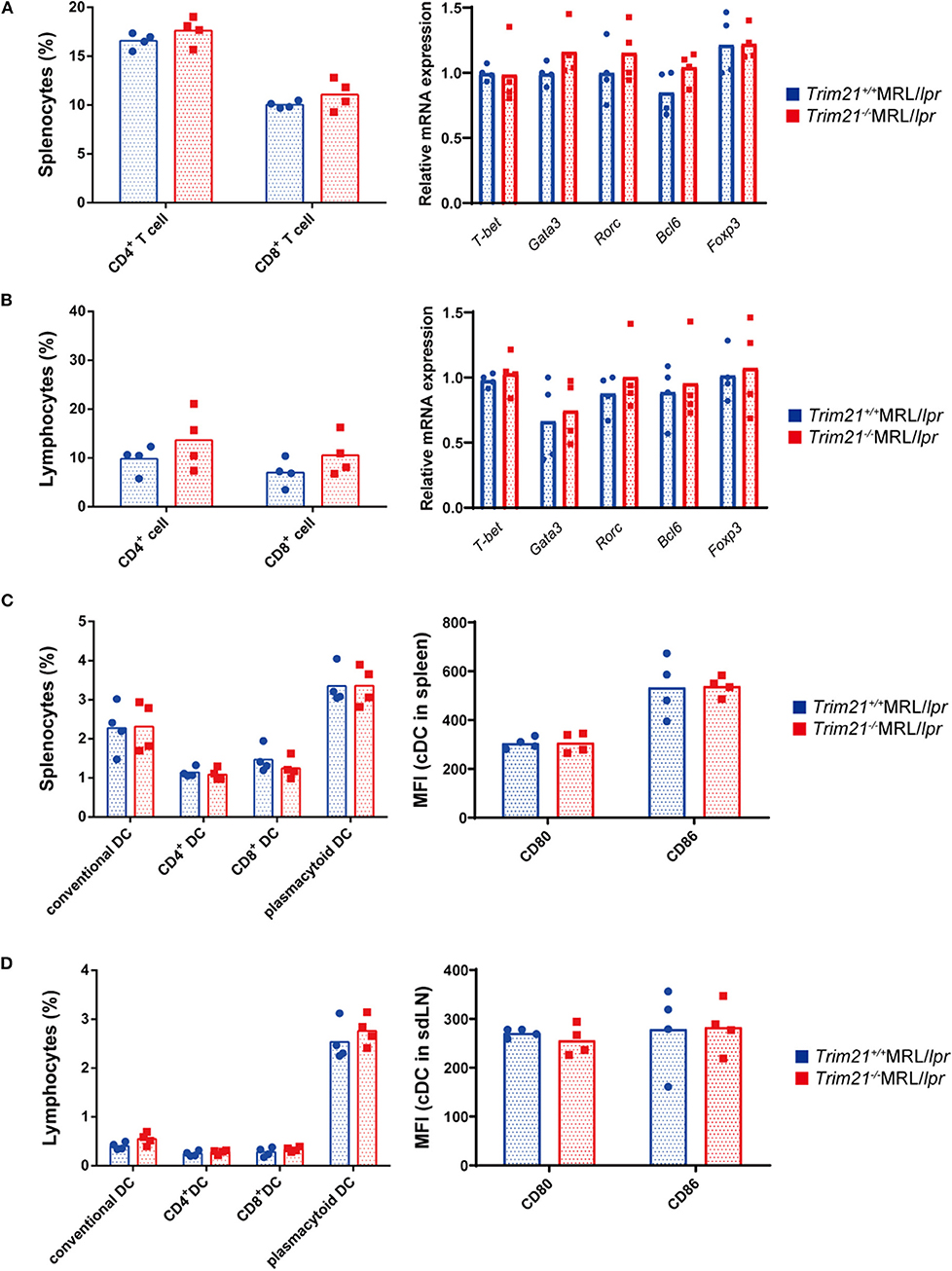
Figure 5. The comparison of T cell and DC subset populations between Trim21+/+ MRL/lpr mice and Trim21−/−MRL/lpr mice. Flow cytometric analyses and qPCR analyses were performed for sdLN cells at 10 weeks of age (n = 4 in each group). (A,B) T-cell subsets in spleen and sdLNs were classified by surface markers as follows: CD4+ T cell; CD3+CD4+ and CD8+ T cell; CD3+CD8+. mRNA expression levels of various T cell transcription factors in CD3+ T cells from spleens and sdLNs were measured by qPCR. (C,D) DC subsets in spleen and sdLNs were classified by surface markers as follows: cDC, I-Ab+B220−CD11c+; CD4+ DC, CD3−CD4+CD11c+; CD8+ DC, CD3−CD8+CD11c+; and pDC, CD317+B220+SiglecH+. MFIs of CD80 and CD86 were measured in cDCs.
Dendritic cells (DCs) are also known to be important for autoimmune pathology like SLE. There were no significant differences in the proportions of conventional DCs (cDCs), CD4+ DCs, CD8+ DCs, and plasmacytoid DCs (pDCs), and the expression levels of activation markers, CD80 and CD86, in cDCs from spleens and sdLNs between Trim21+/+MRL/lpr and Trim21−/−MRL/lpr mice (Figures 5C,D).
These results suggested that B cells, rather than DCs and T cells, are essential in the autoimmune pathology of Trim21−/−MRL/lpr mice.
TRIM21 Deficiency Promotes Aberrant B-Cell Differentiation and Ab Production in MRL/lpr Mice
Although we found that mature B and plasma cells were increased in lymphoid tissues of Trim21−/−MRL/lpr mice, it is unknown whether they are due to an intrinsic abnormality of B cells or some abnormal state which can affect B-cell differentiation indirectly. To verify B cell-intrinsic function in Trim21−/−MRL/lpr mice, we isolated resting state B cells, which are CD43 negative, from spleens and stimulated them by BCR signal (anti-IgM Ab), CD40L signal (anti-CD40 Ab), and/or TLR3/7 ligands. Surprisingly, resting B cells from Trim21−/−MRL/lpr mice showed differentiation to plasmablasts in a significantly higher rate at 24 or 72 h after stimulation as compared to those from Trim21+/+MRL/lpr mice (Supplemental Figures 1A,B and Figures 6A,B). Not only in the absence but also in the presence of CD40L signal, known to be related to T cell-dependent B-cell activation, the B-cell differentiation into plasmablasts was enhanced by TRIM21 deficiency. The results suggest that TRIM21 has roles in B-cell differentiation both in T cell-dependent and T cell-independent manners [Figure 6B; (42, 43)].
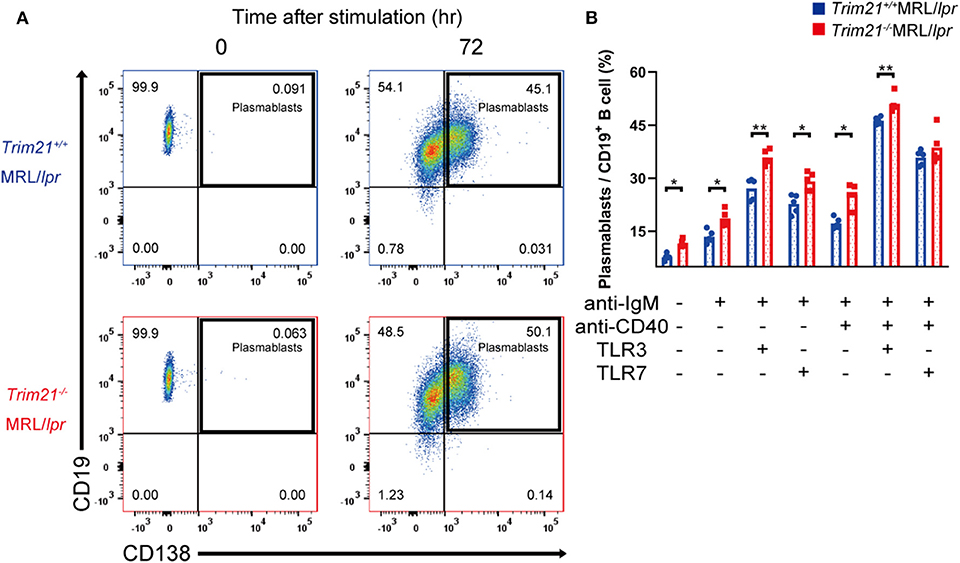
Figure 6. TRIM21 deficiency promotes aberrant B cell differentiation in MRL/lpr mice. (A) Flow cytometric analysis shows an increased rate of CD19+CD138+ plasmablasts at 72 h after stimulation with anti-IgM Ab and TLR3 ligand in Trim21−/−MRL/lpr mice. (B) Percentage of plasmablasts at 72 h after stimulation with anti-IgM Ab, anti-CD40 Ab, and/or TLR3/7 ligands (n = 4 in each group). Statistically significant data (*p < 0.05 and **p < 0.01) by Student's t-test. See also Supplemental Figure 1.
There could be the possibility that the rate of plasmablasts was influenced by cell life-span because some previous reports showed that TRIM21 acts on apoptosis-related molecules such as BCL-2 and c-FLIP (19, 20). However, there was no significant difference in the survival of B cells at 24 h after stimulation between Trim21+/+MRL/lpr and Trim21−/− MRL/lpr mice (Supplemental Figure 1C).
Next, we investigated the effect of TRIM21 deletion in Ab production by stimulating the resting B cells with anti-IgM Ab, anti-CD40 Ab, and/or TLR3/7 ligands. The concentrations of IgG1, IgG2a, and IgA in the cell supernatants, in which the stimulated resting B cells were cultured for 24 or 72 h, were significantly higher in Trim21−/−MRL/lpr mice as compared to Trim21+/+ MRL/lpr mice in multiple stimulation conditions (Supplemental Figure 2 and Figure 7).
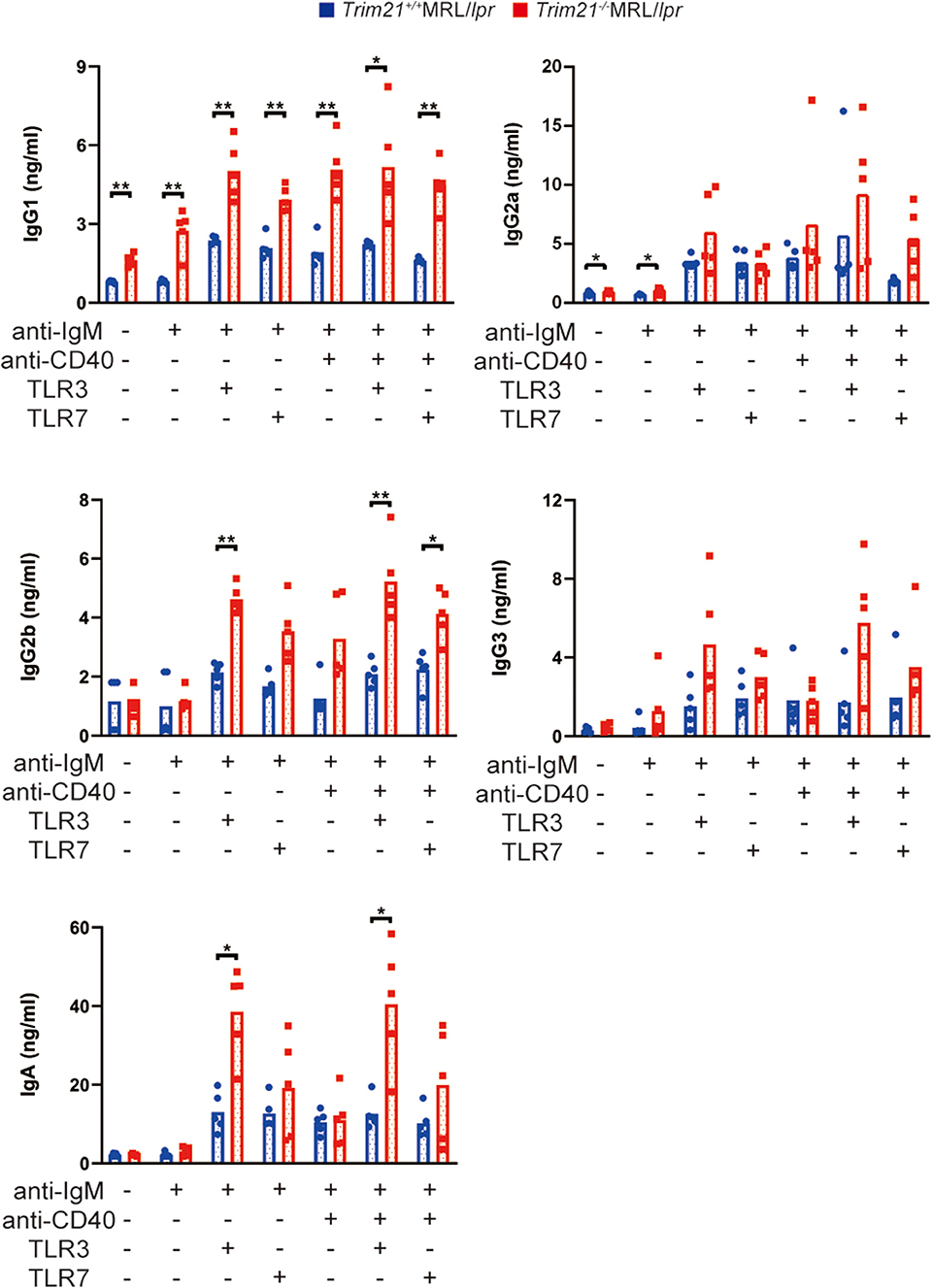
Figure 7. TRIM21 deficiency promotes aberrant Ab production in MRL/lpr mice. Concentrations of IgG1, IgG2a, IgG2b, IgG3, IgA, and IgM in culture supernatants of resting B cell stimulated with anti-IgM Ab, anti-CD40 Ab, and/or TLR3/7 ligands for 72 h were measured by multiple soluble analyte immunoassays (n = 4 in each group). Statistically significant data (*p < 0.05 and **p < 0.01) by Student's t-test, respectively. See also Supplemental Figure 2.
Based on the above results, it is conceivable that TRIM21 deficiency leads to an intrinsic abnormality of B cells that induce aberrant B-cell differentiation and Ab production in MRL/lpr mice.
TRIM21 Deficiency Induces Overexpression of Blimp-1 and Suppression of Bcl6 via Overexpression of IRF5 Due to the Decreased Ubiquitylation
IRF5 is the transcription factor known as one of the most essential factors for B-cell differentiation (23, 24). IRF5 promotes the differentiation of the late B cell and regulates transcription factors necessary for differentiation into activated B cells, plasmablasts and plasma cells (24). In the differentiation into antibody-producing cells (APCs) such as plasmablasts and plasma cells, IRF5 regulates the expression of IRF4, consequently promotes the expression of BLIMP-1, which drives mature B cells to differentiate into APCs, and thereby suppresses the expression of BCL-6, which is known as an inhibitor of APCs development (44). Furthermore, it has been reported that TRIM21 ubiquitylates IRF5 protein and promotes its degradation from the overexpression experiments using human embryonic kidney (HEK) 293 cell line (45).
Although the E3 ubiquitin ligase activity of TRIM21 for IRF5 is not known in primary B cells, we hypothesized that TRIM21 regulates plasma cell development via IRF5 ubiquitylation. In the ubiquitylation assay, there was no significant difference between Trim21+/+ and Trim21−/− MRL/lpr mice in the total amount of ubiquitylated proteins in resting B cells at 72 h after stimulation with anti-IgM Ab and TLR3 ligand (Figure 8A). On the other hand, ubiquitylated IRF5 was remarkably decreased in Trim21−/−MRL/lpr mice as compared to Trim21+/+MRL/lpr mice (Figure 8A). Instead, the unubiquitylated IRF5 protein was significantly increased in Trim21−/−MRL/lpr mice as compared to Trim21+/+MRL/lpr mice (p = 0.021; Figures 8A,B). Furthermore, there was no significant difference in Irf5 mRNA expression in resting B cells after stimulation with anti-IgM and TLR3 ligand for 72 h (Figure 8C), also suggesting that TRIM21 negatively regulates IRF5 protein expression via post-translational modifications.
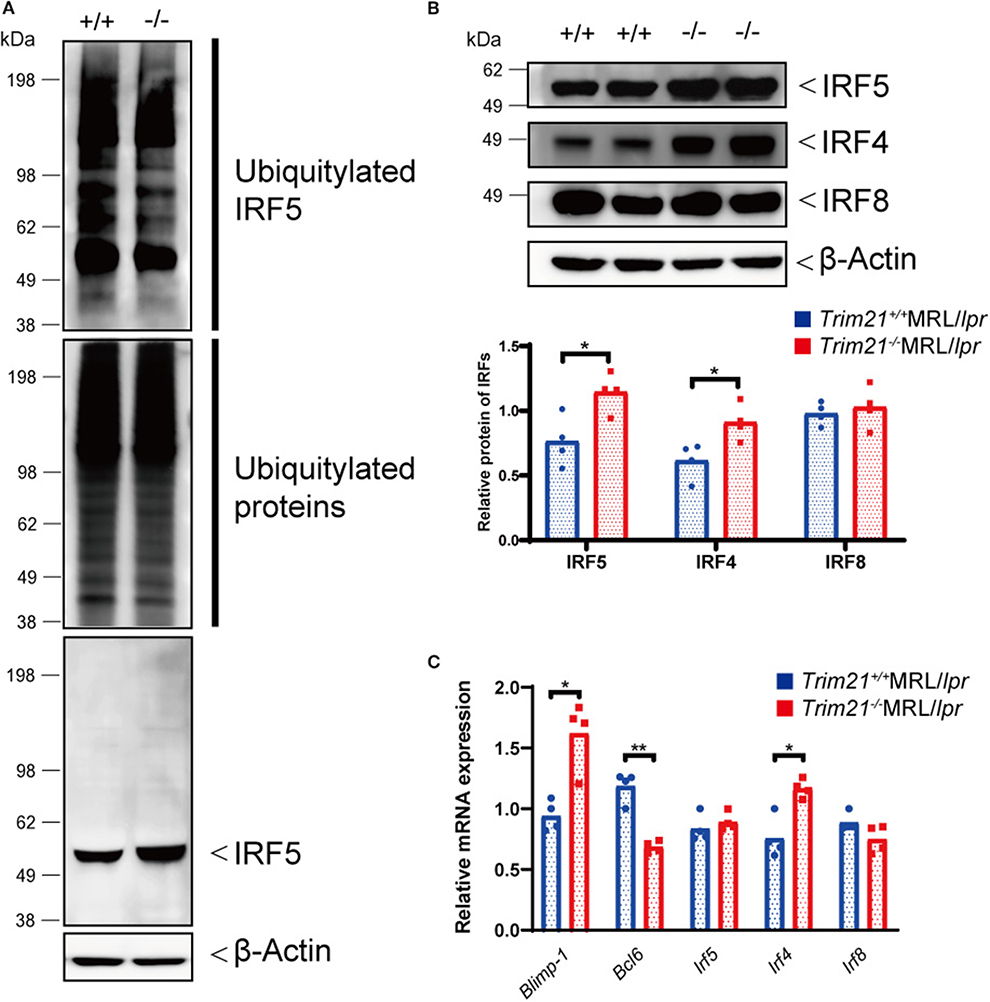
Figure 8. TRIM21 deficiency causes overexpression of IRF5 due to decreased ubiquitylation. (A) Extracts from resting B cells from Trim21+/+ and Trim21−/−MRL/lpr mice, which were stimulated with anti-IgM Ab and TLR3 ligand for 72 h, were immunoprecipitated with anti-IRF5 Ab and blotted with anti-ubiquitin Ab. Ubiquitylated IRF5 protein in the total cell lysate is decreased in the total extracts from Trim21−/−MRL/lpr mice as compared with Trim21+/+MRL/lpr mice (top panel). The amount of total ubiquitylated proteins is not different in two genotypes (the second panel). The amount of unubiquitylated IRF5 protein was higher in cells from Trim21−/−MRL/lpr mice as compared with Trim21+/+MRL/lpr mice (the third panel). β-Actin was used as a loading control (the lowest panel). (B) Western blot analysis shows that the expression level of IRF5, IRF4, and IRF8 protein in resting B cells at 72 h after stimulation with anti-IgM Ab and TLR3 ligand is higher in Trim21−/−MRL/lpr mice as compared to Trim21+/+MRL/lpr mice (n = 4 in each group). (C) mRNA expression levels of Blimp-1, Bcl6, Irf5, Irf4, and Irf8 in resting B cells at 72 h after stimulation with anti-IgM Ab and TLR3 ligand (n = 4 in each group) in qPCR. Statistically significant (*p < 0.05 and **p < 0.01) data by Student's t-test, respectively.
We further investigated the change in factors important for B-cell differentiation downstream region of IRF5 by Trim21 deletion. IRF4 and IRF8 are also known to be essential transcription regulators in B-cell differentiation (44, 46, 47). Irf4 has already been reported as one of the target genes of IRF5 (24). Both mRNA and protein of IRF4 showed significantly higher expression levels in B cells from Trim21−/−MRL/lpr mice at 72 h after stimulation as compared to those from Trim21+/+MRL/lpr mice (Figures 8B,C). On the other hand, there were no significant differences in the mRNA and protein expression levels of IRF8 between the two genotype mice (Figures 8B,C). IRF4 has already been shown to suppress BCL6 expression and promote BLIMP1 expression (48, 49). The mRNA expression of Blimp1 was significantly increased, and the expression of Bcl6 mRNA was significantly decreased in B cells from Trim21−/−MRL/lpr mice as compared to those from Trim21+/+MRL/lpr mice (Figure 8C). The flow cytometric analysis using intracellular staining showed significantly higher expression of BLIMP-1, and significantly lower expression of BCL-6 at the protein level in B cells from Trim21−/−MRL/lpr mice as compared to Trim21+/+MRL/lpr mice (p = 0.014 and 0.049, respectively; Figures 9A,B).
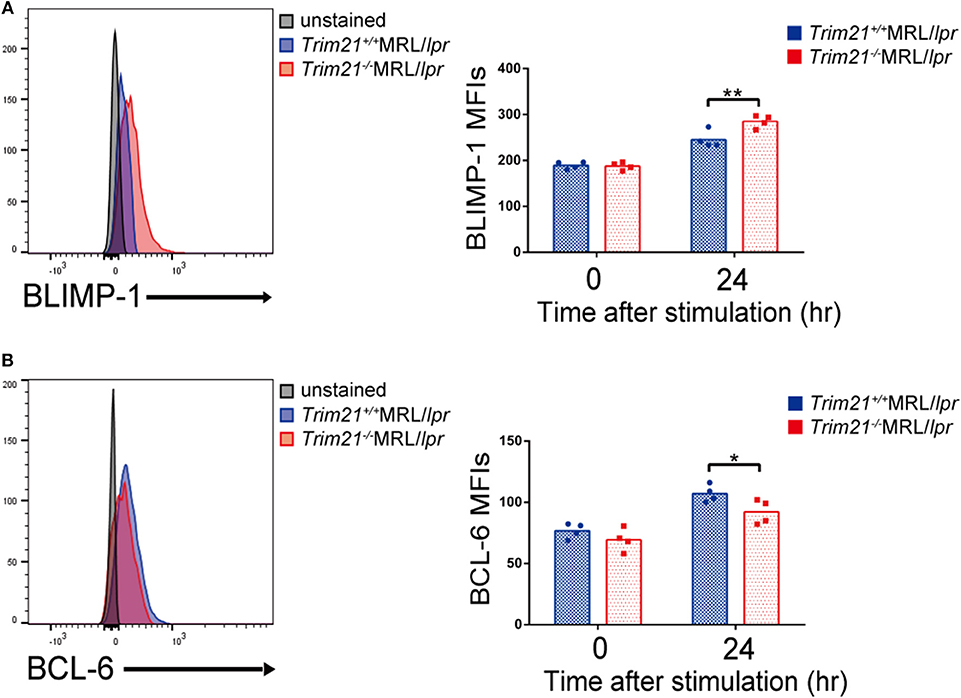
Figure 9. TRIM21 deficiency promotes BLIMP-1 overexpression and suppresses BCL-6 expression. (A,B) Flow cytometric analyses show that the intracellular expression level of BLIMP-1 is increased (A) and that of BCL-6 is decreased (B) by TRIM21 deficiency in resting B cells at 24 h after stimulation with anti-IgM Ab and TLR3 ligand (n = 4 in each group). Values are shown as means ± SEM. Statistically significance data (*p < 0.05 and **p < 0.01) by Student's t-test, respectively.
Together with the previous papers, these results suggest that TRIM21 negatively regulates the expression of IRF5 protein, decreases IRF4 expression, and thereby induces both BLIMP-1 suppression and BCL-6 expression, resulting in suppression of aberrant B-cell differentiation and Ab overproduction during lupus-like pathological condition.
Aberrant B-Cell Differentiation and Ab Production in SLE Patients With Anti-TRIM21 Ab
Our previous study showed that TRIM21 mRNA and protein in PBMCs of patients with SLE were significantly higher than those of healthy controls and that the expression level of TRIM21 mRNA was well-correlated with disease activities of SLE (21). It is conceivable that the high expression of IFN signature in SLE may be responsible for increased expression of TRIM21 as its suppression mechanism. Furthermore, our previous study showed that a negative correlation is observed between the expression of IFN signature mRNA and TRIM21 mRNA in healthy controls and SLE patients without anti-TRIM21 Ab who are considered to have normal function of TRIM21, but the correlation disappeared in SLE patients with anti-TRIM21 Ab (21). In this context, it was suggested that the function of TRIM21 is impaired by anti-TRIM21 Ab in patients with SLE. Therefore, we examined the difference of function of B cells between SLE patients with and without anti-TRIM21 Ab.
Seventeen patients with SLE and 5 healthy controls were enrolled in the study. Eleven and 6 of the patients showed seronegative and seropositive for anti-TRIM21 Ab, respectively. There were no significant differences in the background parameters including female ratio, age, disease duration, SLE activity, treatment, and laboratory data (Table 2).
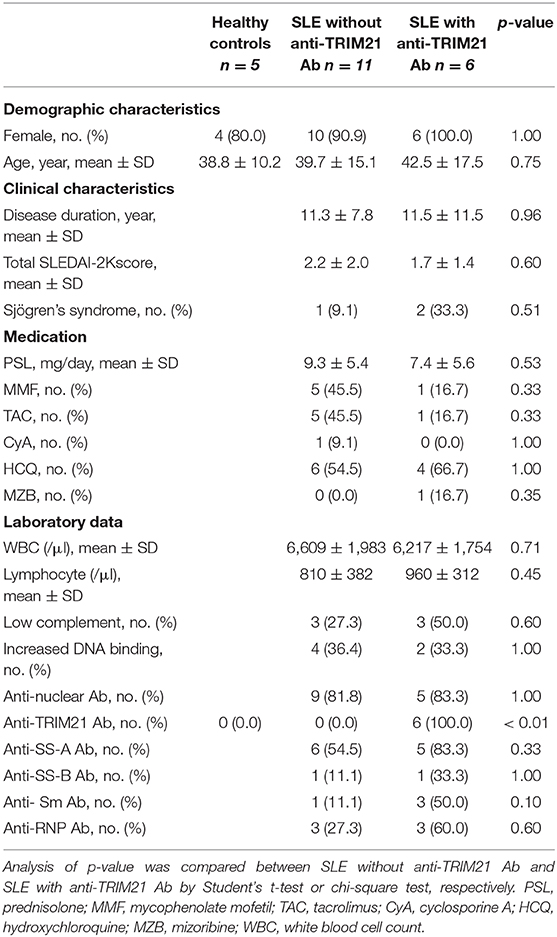
Table 2. Demographic and clinical characteristics in human subjects.
There was no significant difference in the proportion of CD19+ B cells in PBMCs among healthy controls, SLE patients without anti-TRIM21 Ab and SLE patients with anti-TRIM21 Ab (Figure 10A). The proportion of CD19+ B cells after magnetically activated cell sorting of CD43− resting B cell was more than 90% in all three groups (Figure 10A).
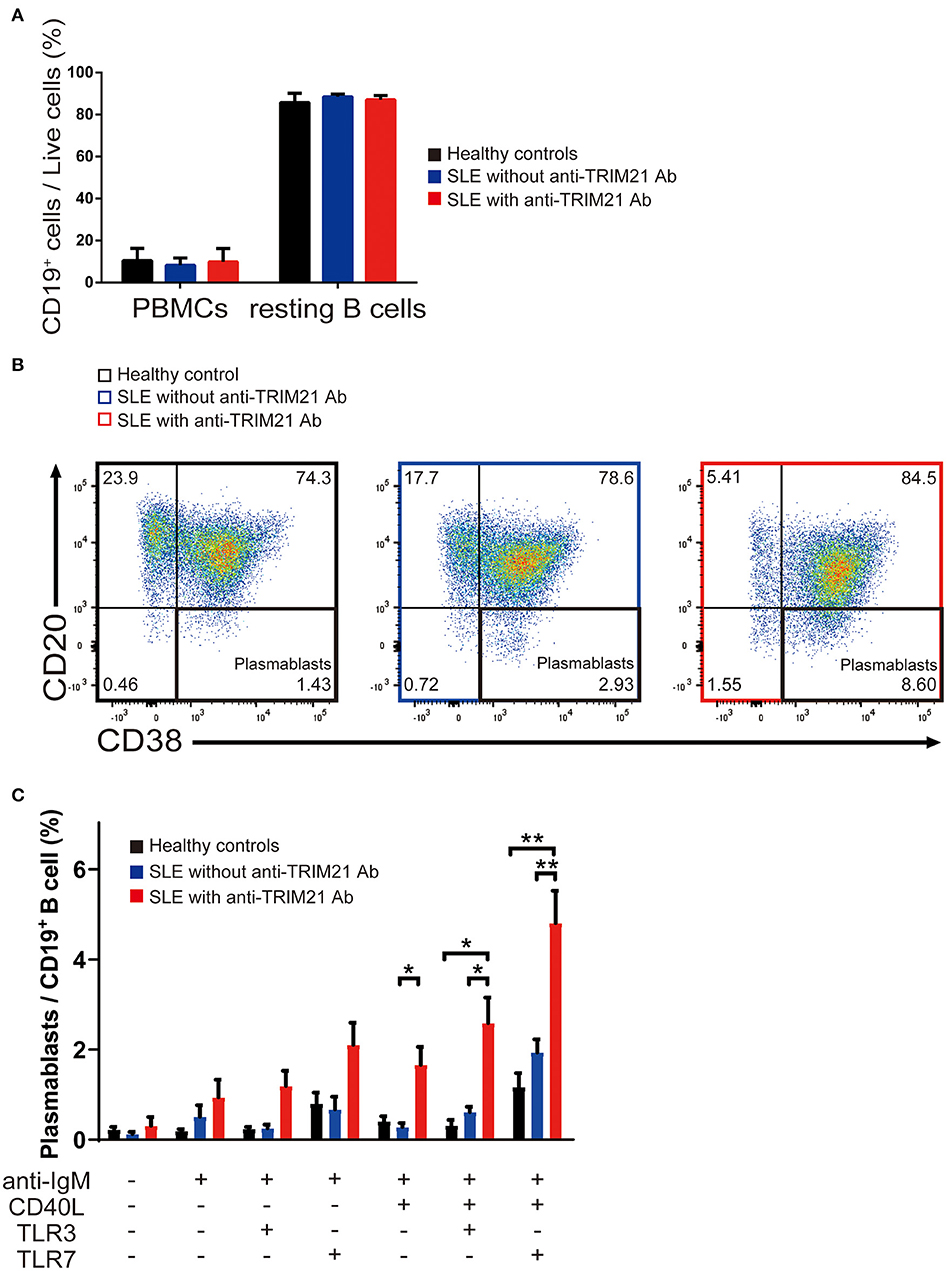
Figure 10. The seropositivity of anti-TRIM21 Ab is related to aberrant B cell differentiation in patients with SLE. (A) Percentages of CD19+ B cells in PBMCs and in resting B cells isolated by magnetic-activated cell sorting in SLE patients with anti-TRIM21 Ab, those without anti-TRIM21 Ab and healthy controls. (B) Flow cytometric analysis shows an increased rate of CD19+CD20−CD38+ plasmablasts at 24 h after stimulation with anti-IgM Ab, CD40L, and TLR7 ligand. The panels show the cells after gating with population positive to both CD19 and CD27. (C) Percentage of plasmablasts at 24 h after stimulation with anti-IgM Ab, CD40L, and/or TLR3/7 ligands in healthy controls (n = 5), SLE patients without anti-TRIM21 Ab seropositivity (n = 11) and those with anti-TRIM21 Ab seropositivity (n = 6). Values are shown as means ± SEM. Statistically significance data (*p < 0.05 and **p < 0.01) by Student's t-test, respectively.
CD43− resting B cell was stimulated by BCR signal (anti-IgM Ab), CD40L, and/or TLR3/7 ligands for 24 h and then tested for differentiation into CD19+CD20−CD38+ plasmablasts. As shown in Figures 10B,C, the percentage of plasmablasts was the highest in SLE patients with anti-TRIM21 Ab. Further, the percentage of plasmablasts was significantly higher in SLE patients without anti-TRIM21 Ab than that in healthy controls. Accordingly, the amounts of immunoglobulins produced in the culture was the highest in SLE patients with anti-TRIM21 Ab, followed by B-cell cultures from SLE patients without the Ab, and the lowest in healthy controls (Figure 11).
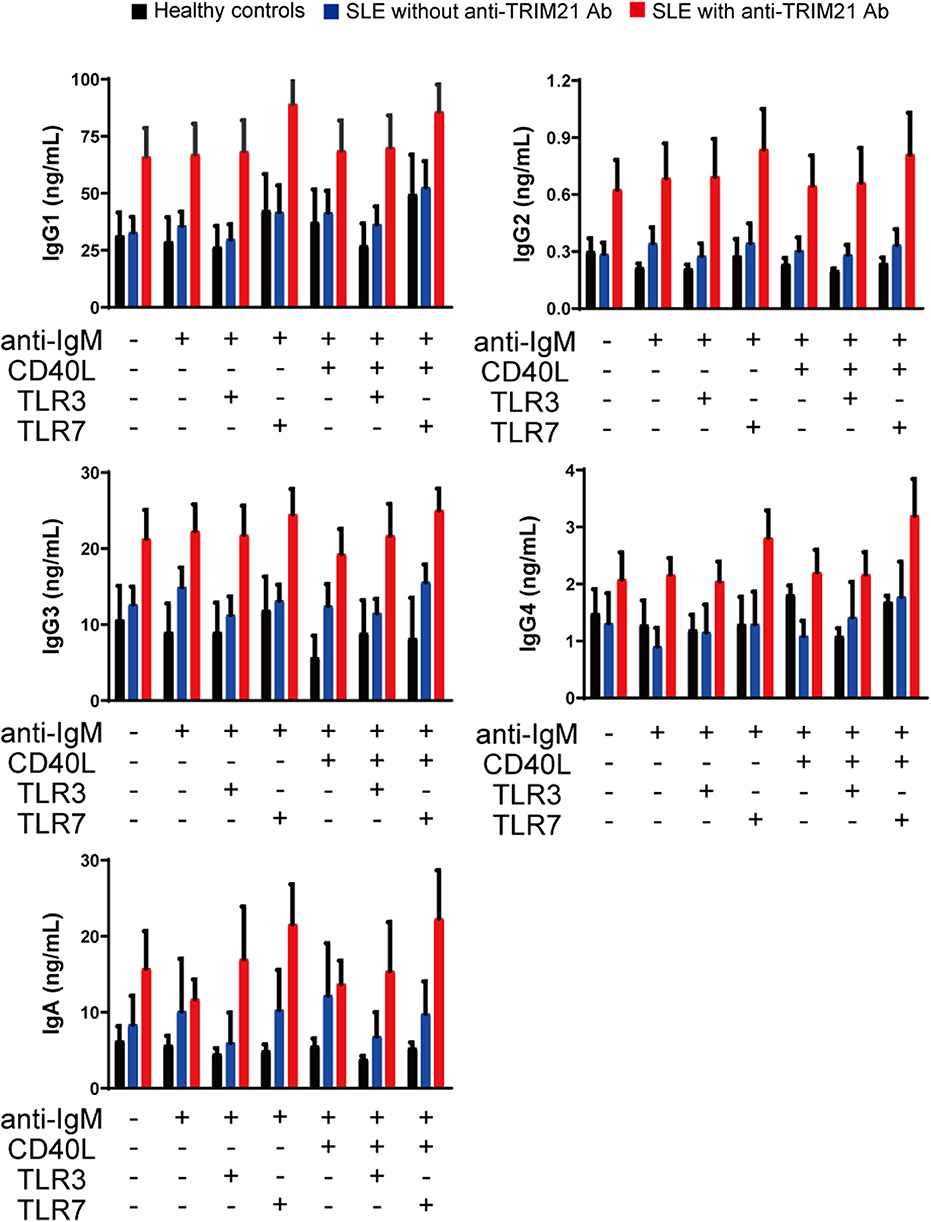
Figure 11. The immunoglobulin productivity in patients with SLE. Concentrations of IgG1, IgG2, IgG3, IgG4, IgA, and IgM in culture supernatants of human resting B cells stimulated with anti-IgM Ab, CD40L and/or TLR3/7 ligands were measured by multiple soluble analyte immunoassays (healthy controls; n = 5, SLE patients without anti-TRIM21 Ab; n = 11 and SLE patients with anti-TRIM21 Ab; n = 6). Values are shown as means ± SEM.
These results indicate that B cells in patients with SLE, especially in those with anti-TRIM21 Ab, are defective in plasmablasts differentiation and Ab production. Anti-TRIM21 Ab may be related to the B-cell abnormalities via inhibition of TRIM21 function.
Discussion
TRIM21 has been shown to regulate the expression of various proinflammatory cytokine genes, type I IFN genes and IFN-inducible genes by modulating the signals from the innate immunoreceptor such as TLR via ubiquitylation of various IRF proteins (12, 45, 50). Previous gene disruption studies have not shown the pathological role of TRIM21 in an autoimmune state such as SLE (16, 17). Therefore, we made Trim21−/−MRL/lpr mouse widely used as a lupus mouse model and examined the phenotype change and the reactivity of immune cells to various TLR ligands (37). This is the first report that shows the pathological role of TRIM21 dysfunction in the lupus mouse model.
Flow cytometric analyses showed that B cells were significantly increased in the spleens and sdLNs by Trim21 gene disruption in lupus-prone mice. Interestingly, Brauner et al. reported that the prognosis was reduced when the Trim21 gene expression was decreased in patients with diffuse large B-cell lymphoma (51). The report suggests that the dysfunction of TRIM21 promotes abnormal activation and proliferation of B cells. Lazzari et al., showed by the overexpression experiments using cell lines that TRIM21 regulates the expression of IRF5 by ubiquitylation (45). IRF5 has been shown to be necessary for B-cell differentiation and Ab production of autoreactive B cells (23, 24, 52). The knockdown of IRF5 by siRNA transfection in human primary naïve B cells reduces plasmablast differentiation and Ab production (24). Our data are well-consistent with these previous studies because our ubiquitylation assay showed that TRIM21 deficiency leads to decreased ubiquitylation and increased expression of IRF5 in B cells. On the other hand, a recent study showed that the Irf5 risk alleles for SLE intrinsically affects the function of myeloid cells rather than that of B cells (53). Although the overexpression experiments are considered as somewhat artificial in general, not the risk haplotype but the levels of IRF5 expression may be important in B cell function.
We found the differences in B-cell differentiation and Ab production between the Trim21−/−MRL/lpr mice and the Trim21+/+MRL/lpr mice. B-cell activation can occur both in T cell-dependent and T cell-independent manners and DCs also play a role in B-cell differentiation via several functions including the production of B cell-activating factor belonging to the tumor necrosis factor family (BAFF) (54, 55). In our experiments, Trim21 deletion did not change the states of T cells and DCs. Moreover, we did not find any differences in the expression of CD80 and CD86, which are important for T cell-B cell interaction (56), in B cells between Trim21+/+ and Trim21−/−MRL/lpr mice. These results suggest that the enhanced B-cell proliferation and Ab production by Trim21 deletion is mainly caused by the abnormality of B cells themselves, and not by that of T cells or DCs.
Anti-SS-A Ab is observed in sera of patients with autoimmune diseases such as SLE and Sjögren's syndrome, and TRIM21 is one of the autoantigens that bind to anti-SS-A Ab (57, 58). We have shown that negative correlation between TRIM21 and type I IFNs expression, which is seen in PBMCs from healthy controls, disappeared in those from SLE patients, especially those with anti-TRIM21 Ab seropositivity (21). In this study, the differentiation from resting B cells to plasmablasts and Ab production were significantly increased in SLE patients with anti-TRIM21 Ab seropositivity as compared to those negative for anti-TRIM21 Ab. In combination with previously reported in vitro data that TRIM21 has a suppressive role in human B-cell proliferation (19, 20, 59), these results strongly suggest that the disruption of TRIM21 fu0nction can be caused partly by anti-TRIM21 Ab in patients with SLE. Consistent with this hypothesis, B-cell differentiation and Ab production were increased in the Trim21−/− MRL/lpr mice as compared with the Trim21+/+ MRL/lpr mice in our experiments. On the other hand, type I IFNs induce TRIM21 expression while TRIM21 itself suppresses the production of type I IFNs (14, 15, 17, 60). This suggests that the increased TRIM21 expression in SLE patients seems due to an increase in serum type I IFN concentration, and it can be considered as a negative feedback reaction to suppress type I IFN overproduction. Therefore, further increasing TRIM21 expression and suppressing the production of anti-TRIM21 Ab can be new treatment strategies for SLE.
How the extracellular anti-TRIM21 Ab can affect the function of intracellular TRIM21 proteins is an important point that has not been studied sufficiently yet. Further studies are needed to clarify this issue. It is possible that the extracellular anti-TRIM21 Ab directly invades the cell and binds to intracellular TRIM21, which may affect function. There were some reports that an anti-DNA Ab that recognizes DNA in the nucleus invades intracellularly from outside the cell via calreticulin or myosin-1 (61–63). Furthermore, it was recently reported that TRIM21 recruits incoming antibody-coated virus and targets it to the proteasome via its E3 ligase activity, resulting in degradation of virions in the cytosol of the infected cells (64). Another recent report shows that anti-TRIM21 autoantibodies inhibit the E3 ligase activity of TRIM21 by sterically blocking the E2/E3 interaction between TRIM21 and UBE2E1 (57). Thus, there is a possibility that serum anti-TRIM21 Ab enters the cytosolic compartment by some cue, interacts with TRIM21 protein, and interferes with its function.
In our experiment, Trim21−/−MRL/lpr mice showed significant increases in urinary protein and serum dsDNA Ab titers as compared to wild-type MRL/lpr mice. Therefore, it is certain that the Trim21 gene deletion has an impact on nephritis and autoantibody production in lupus pathogenesis. On the other hand, Trim21−/−MRL/lpr mice were not different from wild-type mice in the survival curve. The result suggests that various compensatory actions work against the changes associated with the Trim21 deletions to maintain life as a whole body. This may also be related to the fact that human SLE is a multifactorial disease.
During the preparation of this paper, Brauner et al. have reported that follicular B cells and Ab production were significantly increased by immunization of Trim21-deficient C57BL/6 mice with ovalbumin (65). Although the paper does not show an effect of TRIM21 deficiency on phenotype in disease model and the mechanism of B cell increase has not been investigated, their data are well-consistent with our results.
In conclusion, we showed that TRIM21 deficiency promotes aberrant B-cell differentiation and Ab production, leading to exacerbation of the lupus-like symptoms in lupus-prone mice. We also found that B cells from SLE patients with anti-TRIM21 Ab also have significantly higher abilities to differentiate into plasmablasts and to product Ab, suggesting that anti-TRIM21 Ab may be related to the TRIM21 dysfunction in human SLE pathogenesis. These findings suggest that TRIM21 and anti-TRIM21 Ab can be promising targets for SLE treatment.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by the ethics committee of Yokohama City University Hospital (B100701027). The patients/participants provided their written informed consent to participate in this study. This animal study was reviewed and approved by the Animal Protocol Ethics Committee of Yokohama City University.
Author Contributions
YKu performed experiments with assistance from RY, RK, DK, KY, EH, TK, and NS. YKu, RK, and DK backcrossed and generated the Trim21−/−MRL/lpr mice. YKu and RY conceived the project, designed the experiments, analyzed the results, prepared figures, and devised the manuscript. YS, YKi, KO, and HN coordinated and critically discussed research work. All the above-listed authors edited the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Nos. JP30849972 (YKu), JP26461468 (RY), and JP19K08914 (RY), the Japan Rheumatism Foundation (RY), Yokohama Foundation for Advancement of Medical Science (RY), the Takeda Science Foundation (RY), and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (RY). This study was partly supported by GSK Japan Research Grant 2019 (YKu) and the grant of Bristol-Myers K.K. (RY). The authors declare that this study received funding from GlaxoSmithKline K.K. and Bristol-Myers K.K. These funders were not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the colleagues in our laboratories for the helpful cooperation, suggestions, and meaningful discussions of this project. Notably, we thank Dr. Herbert C. Morse III for his help for generation of Trim21−/−C57BL/6 mice. We also thank Mr. Tom Kiper for his review of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2020.00098/full#supplementary-material
References
1. Cunningham M, Gilkeson G. Estrogen receptors in immunity and autoimmunity. Clin Rev Allergy Immunol. (2011) 40:66–73. doi: 10.1007/s12016-010-8203-5
2. Dorgham K, Amoura Z, Parizot C, Arnaud L, Frances C, Pionneau C, et al. Ultraviolet light converts propranolol, a nonselective beta-blocker and potential lupus-inducing drug, into a proinflammatory AhR ligand. Eur J Immunol. (2015) 45:3174–87. doi: 10.1002/eji.201445144
3. Qiu CC, Caricchio R, Gallucci S. Triggers of autoimmunity: the role of bacterial infections in the extracellular exposure of lupus nuclear autoantigens. Front Immunol. (2019) 10:2608. doi: 10.3389/fimmu.2019.02608
4. Cui Y, Sheng Y, Zhang X. Genetic susceptibility to SLE: recent progress from GWAS. J Autoimmun. (2013) 41:25–33. doi: 10.1016/j.jaut.2013.01.008
5. Liossis SN, Melissaropoulos K. Molecular abnormalities of the B cell in systemic lupus erythematosus are candidates for functional inhibition treatments. Expert Opin Pharmacother. (2014) 15:833–40. doi: 10.1517/14656566.2014.894976
6. Rawlings DJ, Metzler G, Wray-Dutra M, Jackson SW. Altered B cell signalling in autoimmunity. Nat Rev Immunol. (2017) 17:421–36. doi: 10.1038/nri.2017.24
7. Ben-Chetrit E, Chan EK, Sullivan KF, Tan EM. A 52-kD protein is a novel component of the SS-A/Ro antigenic particle. J Exp Med. (1988) 167:1560–71. doi: 10.1084/jem.167.5.1560
8. Frank MB, Itoh K, Fujisaku A, Pontarotti P, Mattei MG, Neas BR. The mapping of the human 52-kD Ro/SSA autoantigen gene to human chromosome 11, and its polymorphisms. Am J Hum Genet. (1993) 52:183–91.
9. Rhodes DA, Ihrke G, Reinicke AT, Malcherek G, Towey M, Isenberg DA, et al. The 52 000 MW Ro/SS-A autoantigen in Sjogren's syndrome/systemic lupus erythematosus (Ro52) is an interferon-gamma inducible tripartite motif protein associated with membrane proximal structures. Immunology. (2002) 106:246–56. doi: 10.1046/j.1365-2567.2002.01417.x
10. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J. (2001) 20:2140–51. doi: 10.1093/emboj/20.9.2140
11. Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. (2005) 3:799–808. doi: 10.1038/nrmicro1248
12. Ozato K, Shin DM, Chang TH, Morse HC III. TRIM family proteins and their emerging roles in innate immunity. Nat Rev Immunol. (2008) 8:849–60. doi: 10.1038/nri2413
13. Kong HJ, Anderson DE, Lee CH, Jang MK, Tamura T, Tailor P, et al. Cutting edge: autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF-8 and enhances cytokine expression in macrophages. J Immunol. (2007) 179:26–30. doi: 10.4049/jimmunol.179.1.26
14. Higgs R, Ni Gabhann J, Ben Larbi N, Breen EP, Fitzgerald KA, Jefferies CA. The E3 ubiquitin ligase Ro52 negatively regulates IFN-beta production post-pathogen recognition by polyubiquitin-mediated degradation of IRF3. J Immunol. (2008) 181:1780–6. doi: 10.4049/jimmunol.181.3.1780
15. Strandberg L, Ambrosi A, Espinosa A, Ottosson L, Eloranta ML, Zhou W, et al. Interferon-alpha induces up-regulation and nuclear translocation of the Ro52 autoantigen as detected by a panel of novel Ro52-specific monoclonal antibodies. J Clin Immunol. (2008) 28:220–31. doi: 10.1007/s10875-007-9157-0
16. Espinosa A, Dardalhon V, Brauner S, Ambrosi A, Higgs R, Quintana FJ, et al. Loss of the lupus autoantigen Ro52/Trim21 induces tissue inflammation and systemic autoimmunity by disregulating the IL-23-Th17 pathway. J Exp Med. (2009) 206:1661–71. doi: 10.1084/jem.20090585
17. Yoshimi R, Chang TH, Wang H, Atsumi T, Morse HC III, Ozato K. Gene disruption study reveals a nonredundant role for TRIM21/Ro52 in NF-kappaB-dependent cytokine expression in fibroblasts. J Immunol. (2009) 182:7527–38. doi: 10.4049/jimmunol.0804121
18. Manocha GD, Mishra R, Sharma N, Kumawat KL, Basu A, Singh SK. Regulatory role of TRIM21 in the type-I interferon pathway in Japanese encephalitis virus-infected human microglial cells. J Neuroinflammation. (2014) 11:24. doi: 10.1186/1742-2094-11-24
19. Jauharoh SN, Saegusa J, Sugimoto T, Ardianto B, Kasagi S, Sugiyama D, et al. SS-A/Ro52 promotes apoptosis by regulating Bcl-2 production. Biochem Biophys Res Commun. (2012) 417:582–7. doi: 10.1016/j.bbrc.2011.12.010
20. Zhang J, Fang L, Zhu X, Qiao Y, Yu M, Wang L, et al. Ro52/SSA sensitizes cells to death receptor-induced apoptosis by down-regulating c-FLIP(L). Cell Biol Int. (2012) 36:463–8. doi: 10.1042/CBI20110322
21. Kamiyama R, Yoshimi R, Takeno M, Iribe Y, Tsukahara T, Kishimoto D, et al. Dysfunction of TRIM21 in interferon signature of systemic lupus erythematosus. Mod Rheumatol. (2018) 28:993–1003. doi: 10.1080/14397595.2018.1436028
22. Savitsky D, Calame K. B-1 B lymphocytes require Blimp-1 for immunoglobulin secretion. J Exp Med. (2006) 203:2305–14. doi: 10.1084/jem.20060411
23. Lien C, Fang CM, Huso D, Livak F, Lu R, Pitha PM. Critical role of IRF-5 in regulation of B-cell differentiation. Proc Natl Acad Sci USA. (2010) 107:4664–8. doi: 10.1073/pnas.0911193107
24. De S, Zhang B, Shih T, Singh S, Winkler A, Donnelly R, et al. B cell-intrinsic role for IRF5 in TLR9/BCR-induced human B cell activation, proliferation, and plasmablast differentiation. Front Immunol. (2017) 8:1938. doi: 10.3389/fimmu.2017.01938
25. Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. (2009) 41:1228–33. doi: 10.1038/ng.468
26. Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. (2016) 48:323–30. doi: 10.1038/ng.3496
27. Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. (1997) 40:1725. doi: 10.1002/art.1780400928
28. Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, et al. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. (2007) 282:9358–63. doi: 10.1074/jbc.C600321200
29. Harada Y, Tanaka S, Motomura Y, Harada Y, Ohno S, Ohno S, et al. The 3' enhancer CNS2 is a critical regulator of interleukin-4-mediated humoral immunity in follicular helper T cells. Immunity. (2012) 36:188–200. doi: 10.1016/j.immuni.2012.02.002
30. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. (2008) 455:808–12. doi: 10.1038/nature07240
31. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
32. Duy C, Yu JJ, Nahar R, Swaminathan S, Kweon SM, Polo JM, et al. BCL6 is critical for the development of a diverse primary B cell repertoire. J Exp Med. (2010) 207:1209–21. doi: 10.1084/jem.20091299
33. Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell. (2010) 18:580–9. doi: 10.1016/j.ccr.2010.11.024
34. Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, et al. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. (2005) 102:15989–94. doi: 10.1073/pnas.0508327102
35. Ueda Y, Kayama H, Jeon SG, Kusu T, Isaka Y, Rakugi H, et al. Commensal microbiota induce LPS hyporesponsiveness in colonic macrophages via the production of IL-10. Int Immunol. (2010) 22:953–62. doi: 10.1093/intimm/dxq449
36. Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the human immunology project. Nat Rev Immunol. (2012) 12:191–200. doi: 10.1038/nri3158
37. Adachi M, Watanabe-Fukunaga R, Nagata S. Aberrant transcription caused by the insertion of an early transposable element in an intron of the Fas antigen gene of lpr mice. Proc Natl Acad Sci USA. (1993) 90:1756–60. doi: 10.1073/pnas.90.5.1756
38. Wahren M, Skarstein K, Blange I, Pettersson I, Jonsson R. MRL/lpr mice produce anti-Ro 52,000 MW antibodies: detection, analysis of specificity and site of production. Immunology. (1994) 83:9–15.
39. Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. (1978) 148:1198–215. doi: 10.1084/jem.148.5.1198
40. Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. (2013) 38:528–40. doi: 10.1016/j.immuni.2012.11.017
41. Hammond DM, Nagarkatti PS, Gote LR, Seth A, Hassuneh MR, Nagarkatti M. Double-negative T cells from MRL-lpr/lpr mice mediate cytolytic activity when triggered through adhesion molecules and constitutively express perforin gene. J Exp Med. (1993) 178:2225–30. doi: 10.1084/jem.178.6.2225
42. Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. (1998) 393:480–3. doi: 10.1038/31002
43. Jabara HH, Brodeur SR, Geha RS. Glucocorticoids upregulate CD40 ligand expression and induce CD40L-dependent immunoglobulin isotype switching. J Clin Invest. (2001) 107:371–8. doi: 10.1172/JCI10168
44. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
45. Lazzari E, Korczeniewska J, Ni Gabhann J, Smith S, Barnes BJ, Jefferies CA. TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PLoS ONE. (2014) 9:e103609. doi: 10.1371/journal.pone.0103609
46. Lu R, Medina KL, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. (2003) 17:1703–8. doi: 10.1101/gad.1104803
47. Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol. (2015) 15:172–84. doi: 10.1038/nri3814
48. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
49. Saito M, Gao J, Basso K, Kitagawa Y, Smith PM, Bhagat G, et al. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell. (2007) 12:280–92. doi: 10.1016/j.ccr.2007.08.011
50. Kawai T, Akira S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med. (2011) 3:513–27. doi: 10.1002/emmm.201100160
51. Brauner S, Zhou W, Backlin C, Green TM, Folkersen L, Ivanchenko M, et al. Reduced expression of TRIM21/Ro52 predicts poor prognosis in diffuse large B-cell lymphoma patients with and without rheumatic disease. J Intern Med. (2015) 278:323–32. doi: 10.1111/joim.12375
52. Savitsky DA, Yanai H, Tamura T, Taniguchi T, Honda K. Contribution of IRF5 in B cells to the development of murine SLE-like disease through its transcriptional control of the IgG2a locus. Proc Natl Acad Sci USA. (2010) 107:10154–9. doi: 10.1073/pnas.1005599107
53. Calise J, Marquez Renteria S, Gregersen PK, Diamond B. Lineage-specific functionality of an interferon regulatory factor 5 lupus risk haplotype: lack of B cell intrinsic effects. Front Immunol. (2018) 9:996. doi: 10.3389/fimmu.2018.00996
54. Grammer AC, Lipsky PE. B cell abnormalities in systemic lupus erythematosus. Arthritis Res Ther. (2003) 5(Suppl. 4):S22–27. doi: 10.1186/ar1009
55. Jego G, Pascual V, Palucka AK, Banchereau J. Dendritic cells control B cell growth and differentiation. Curr Dir Autoimmun. (2005) 8:124–39. doi: 10.1159/000082101
56. Chevrier S, Genton C, Malissen B, Malissen M, Acha-Orbea H. Dominant role of CD80-CD86 over CD40 and ICOSL in the massive polyclonal B cell activation mediated by LAT(Y136F) CD4(+) T cells. Front Immunol. (2012) 3:27. doi: 10.3389/fimmu.2012.00027
57. Espinosa A, Hennig J, Ambrosi A, Anandapadmanaban M, Abelius MS, Sheng Y, et al. Anti-Ro52 autoantibodies from patients with Sjogren's syndrome inhibit the Ro52 E3 ligase activity by blocking the E3/E2 interface. J Biol Chem. (2011) 286:36478–91. doi: 10.1074/jbc.M111.241786
58. Ghillani P, Andre C, Toly C, Rouquette AM, Bengoufa D, Nicaise P, et al. Clinical significance of anti-Ro52 (TRIM21) antibodies non-associated with anti-SSA 60kDa antibodies: results of a multicentric study. Autoimmun Rev. (2011) 10:509–13. doi: 10.1016/j.autrev.2011.03.004
59. Espinosa A, Zhou W, Ek M, Hedlund M, Brauner S, Popovic K, et al. The Sjogren's syndrome-associated autoantigen Ro52 is an E3 ligase that regulates proliferation and cell death. J Immunol. (2006) 176:6277–85. doi: 10.4049/jimmunol.176.10.6277
60. Higgs R, Lazzari E, Wynne C, Ni Gabhann J, Espinosa A, Wahren-Herlenius M, et al. Self protection from anti-viral responses–Ro52 promotes degradation of the transcription factor IRF7 downstream of the viral Toll-Like receptors. PLoS ONE. (2010) 5:e11776. doi: 10.1371/journal.pone.0011776
61. Zack DJ, Stempniak M, Wong AL, Taylor C, Weisbart RH. Mechanisms of cellular penetration and nuclear localization of an anti-double strand DNA autoantibody. J Immunol. (1996) 157:2082–8.
62. Yanase K, Smith RM, Puccetti A, Jarett L, Madaio MP. Receptor-mediated cellular entry of nuclear localizing anti-DNA antibodies via myosin 1. J Clin Invest. (1997) 100:25–31. doi: 10.1172/JCI119517
63. Seddiki N, Nato F, Lafaye P, Amoura Z, Piette JC, Mazie JC. Calreticulin, a potential cell surface receptor involved in cell penetration of anti-DNA antibodies. J Immunol. (2001) 166:6423–9. doi: 10.4049/jimmunol.166.10.6423
64. Mallery DL, McEwan WA, Bidgood SR, Towers GJ, Johnson CM, James LC. Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc Natl Acad Sci USA. (2010) 107:19985–90. doi: 10.1073/pnas.1014074107
Keywords: TRIM21, systemic lupus erythematosus, B cell, immunoglobulin, lupus model mouse
Citation: Kunishita Y, Yoshimi R, Kamiyama R, Kishimoto D, Yoshida K, Hashimoto E, Komiya T, Sakurai N, Sugiyama Y, Kirino Y, Ozato K and Nakajima H (2020) TRIM21 Dysfunction Enhances Aberrant B-Cell Differentiation in Autoimmune Pathogenesis. Front. Immunol. 11:98. doi: 10.3389/fimmu.2020.00098
Received: 06 September 2019; Accepted: 14 January 2020;
Published: 07 February 2020.
Edited by:
Stefania Gallucci, Temple University, United StatesReviewed by:
Betsy J. Barnes, Feinstein Institute for Medical Research, United StatesKevin M. Nickerson, University of Pittsburgh, United States
Copyright © 2020 Kunishita, Yoshimi, Kamiyama, Kishimoto, Yoshida, Hashimoto, Komiya, Sakurai, Sugiyama, Kirino, Ozato and Nakajima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryusuke Yoshimi, eW9zaGlyeXVAbWVkLnlva29oYW1hLWN1LmFjLmpw