 Giacomo Canesin
Giacomo Canesin Seyed M. Hejazi
Seyed M. Hejazi Kenneth D. Swanson
Kenneth D. Swanson Barbara Wegiel
Barbara Wegiel- 1Department of Surgery, Cancer Research Institute and Transplant Institute, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA, United States
- 2Brain Tumor Center and Neuro-Oncology Unit, Beth Israel Deaconess Medical Center, Boston, MA, United States
Heme is one of the most abundant molecules in the body acting as the functional core of hemoglobin/myoglobin involved in the O2/CO2 carrying in the blood and tissues, redox enzymes and cytochromes in mitochondria. However, free heme is toxic and therefore its removal is a significant priority for the host. Heme is a well-established danger-associated molecular pattern (DAMP), which binds to toll-like receptor 4 (TLR4) to induce immune responses. Heme-derived metabolites including the bile pigments, biliverdin (BV) and bilirubin (BR), were first identified as toxic drivers of neonatal jaundice in 1800 but have only recently been appreciated as endogenous drivers of multiple signaling pathways involved in protection from oxidative stress and regulators of immune responses. The tissue concentration of heme, BV and BR is tightly controlled. Heme oxygenase-1 (HO-1, encoded by HMOX1) produces BV by heme degradation, while biliverdin reductase-A (BLVR-A) generates BR by the subsequent conversion of BV. BLVR-A is a fascinating protein that possesses a classical protein kinase domain, which is activated in response to BV binding to its enzymatic site and initiates the downstream mitogen-activated protein kinases (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways. This links BLVR-A activity to cell growth and survival pathways. BLVR-A also contains a bZip DNA binding domain and a nuclear export sequence (NES) and acts as a transcription factor to regulate the expression of immune modulatory genes. Here we will discuss the role of heme-related immune response and the potential for targeting the heme system for therapies directed toward hepatitis and cancer.
Introduction
Many human diseases are associated with immune dysfunctions affecting the host ability to control inflammation. Under normal physiological conditions, the activation of pro-inflammatory processes is resolved by the controlled response to reinstate tissue homeostasis. For this reason, cells and organisms have evolved to integrate protective molecular mechanisms that specifically counter inflammatory effector functions in order to threshold the response and re-establish homeostasis upon inflammatory resolution (1, 2). The details of heme metabolism have only been appreciated in the last 30 years while bile pigments were known a century earlier. Heme is a tetrapyrrolic porphyrin ring that coordinates an Fe2+ atom and is catabolized by heme oxygenases (HO-1, encoded by HMOX1 or HO-2, encoded by HMOX2) into biliverdin (BV), iron, and carbon monoxide (CO) (3–5). The subsequent conversion of BV to bilirubin (BR) is catalyzed by biliverdin reductases (BLVR), for which two isoforms exist in humans, BLVR-A and BLVR-B (Figure 1). These enzymatic reactions can be best visualized by the colorful stages occurring during bruising: (i) the initial dark purple is due to heme release from damaged red blood cells (RBC), (ii) while the green color corresponds to BV, (iii) and finally the yellow color is BR which is also responsible for the yellow pigmentation evident during jaundice.
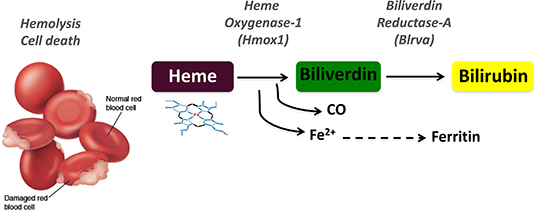
Figure 1. Schematic representation of the heme degradation pathway. Damaged red blood cells release free heme, which is converted to biliverdin by heme oxygenase-1 (HO-1, HMOX1) in a reaction that generates carbon monoxide (CO) and iron (Fe2+). Biliverdin is in turn converted to bilirubin by biliverdin reductase A (BLVR-A).
BV and BR possess strong antioxidant, anti-inflammatory and protective properties (6) and have recently been shown to participate as regulators of inflammatory reactions in sepsis and organ injury. BR exerts general immunosuppressive effects with possibly important clinical consequences, as evidenced by multiple experimental as well as clinical studies (7–11). In humans, BR is the most prominent bile pigment and is very effective at counteracting cellular oxidative stress, protecting cells against lipid oxidation, attenuating oxidative damage to proteins, and acting as a scavenger of nitric oxide (NO) and reactive oxygen species (ROS) (12–14). Consistent with this, mildly elevated plasma BR levels have been negatively correlated with the risk for atherosclerosis, stroke (15, 16), cancer (17, 18), and inflammatory diseases including ulcerative colitis or Crohn's disease (19–23). This strongly suggests an immunomodulatory role for BR. Under physiologic conditions, for disposal from the body, BR is conjugated to glucuronic acid in the liver by bilirubin UDP glucuronosyl transferase (UGT1A1) and then secreted within the bile into the intestinal lumen (24). In the colon, BR is de-conjugated by microbial-produced enzymes and can be partially reabsorbed into the circulation, but it has also been suggested to contribute locally to gut immune homeostasis by modulating T cell activation (25, 26). Furthermore, recent work showed that BR acts as a scavenger of superoxide within neural synapses, protecting against excitotoxicity and neuronal death during neurostimulation with ligands such as with NMDA (27).
The physiological turnover of heme derived from RBC generates both BV and BR. In adults, HO-1-mediated heme cleavage occurs predominantly on the alpha-meso carbon to generate BV. Subsequently, BLVR-A, the predominant BLVR isoform in adults, converts BV to the BR-IXα isoform of BR (28, 29). Conversely, BLVR-B is the dominant isoform found in the fetus and produces the BR-IXβ isoform (30, 31). The highest levels of BR-IXβ are found in fetal bile, indicating that heme catabolism in utero differs from that in adults (32). Thus, unconjugated BR-IXβ is the first bilirubin pigment to appear in bile during fetal development, being observed as early as at 14 weeks gestation (33). At 16 weeks gestation, small amounts of unconjugated BR-IXα are also detected in human fetal bile, indicating the maturation of liver-uptake and biliary-secretion mechanisms (33). Furthermore, BR-IXβ accounts for 60–95% of the unconjugated bilirubin in the first sample of excreted meconium, but its amount decreases rapidly during the first 5 days in full-term newborns while declining more slowly in preterm neonates (34, 35). This may be related to the fact that BR-IXβ cannot easily cross the placenta and it needs to be excreted into bile without previous conjugation to glucuronic acid (36).
Heme is the primary inducer of HMOX1 gene expression. Since the main function of HO-1 is to degrade heme, this results in a negative feedback mechanism for maintaining cellular homeostasis under stress conditions, HMOX1 expression will be driven in cells and tissues where excess heme is present until the excess heme is cleared (37). HO-1 expression is also induced by other stressors, including UV radiation, hormones, endotoxins, and cytokines. HO-1 exerts anti-inflammatory, anti-apoptotic and anti-proliferative actions in various cell types, including endothelial cells and macrophages (38, 39). This provides a basis for how the heme catabolic pathway may be necessary for preventing tissue injuries in several disease states, from endotoxic shock to ischemia/reperfusion injury, vascular injury, and hepatitis (40–46). Similarly, BLVR are also critical enzymes in the heme catabolic pathway by removing BV. Although BV is a non-toxic molecule, mammalians evolved to remove it within minutes as shown using exogenous administration of BV. Among the reasons why BV is removed is the need for the strong antioxidant BR and/or the necessity to act as a ligand for BLVR-A triggering signaling through PI3K-Akt (47). Functional ligands, as BV, have a short half-life to prevent chronic signaling. Importantly, BLVR-A has been found on the cell surface (BLVRsurf) where it initiates signaling cascades within the cytoplasm upon extracellular BV-binding (47). BV initiates the activation of tyrosine kinase domain of BLVR-A. Interestingly, BLVR-A possesses dual specificity protein kinase activity (48–50) that plays important roles not only in response to BV (47) but also in the insulin/insulin-like growth factor 1 (IGF1)-signaling pathways, with effects on insulin action, glucose uptake, signal transduction and gene expression (48, 51). Additionally, BLVR-A kinase activity is responsible for the production of IL-10 via PI3K/Akt activation upon binding of BV to BLVR-A in the membrane (29, 47). Through its kinase activity domain, BLVR-A inhibits total glycogen synthase kinase 3β (GSK3β) activity downstream of Akt activation, which supports a role for it in many cellular functions including the modulation of immune response or inflammation regulated by nuclear factor (NF)-κB (NF-κB) (52–54). A recent study by Sharma and colleagues showed that loss of BLVR-A impairs a neuroprotective Akt-mediated inhibition of GSK-3β in response to oxidative stress, thus contributing to early stage Alzheimer's disease (55). However, BLVR kinase activity is dispensable for BLVR-dependent PKC activation. In this settings, BLVR acts as a scaffold to stabilize the active conformation of the PKC (56). This scaffolding role of BLVR may promote the assembly of elaborate signal transduction complexes that facilitate the phosphorylation and subsequent activation of MAPK Erk1/2, either via MEK1/2 or via PKC (56–58).
BLVR-A possesses additional activities and distinct signaling capabilities, which makes it a highly pleiotropic and multifaceted protein (47, 48, 59, 60) (Figure 2). BLVR-A has a direct transcriptional control activity due to a bZip DNA binding domain in its C-terminal domain (61) (Figure 2). Thus, both HO-1 and BLVR act as oxidative stress and inflammatory response enzymes, but also key signaling molecules and are considered to play important roles in response to and protection against cellular stress (62–67). Therefore, understanding the biology of bile pigments and the mechanism of action of BLVR is central to a full comprehension of tissue homeostasis and many immune-associated pathologies.
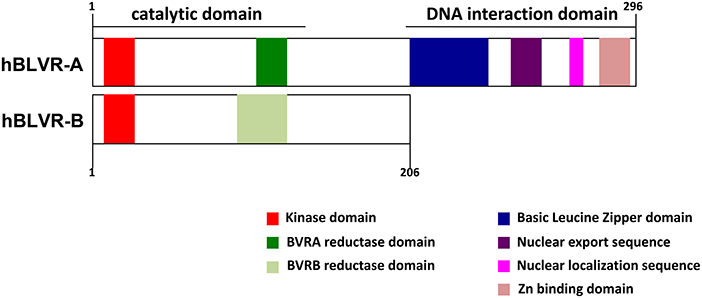
Figure 2. Domain structures of human BLVR-A and BLVR-B. A schematic comparison of the structure of human BLVR-A and BLVR-B shows a similar kinase and catalytic domain for both enzymes. The two isoforms differ in the C-terminus, where BLVR-A contains a bZip DNA binding domain, a nuclear localization sequence and a nuclear export sequence that are not present within BLVR-B.
HO-1 And Blvr Enzymes: Expression And Modes Of Action
HO-1 is a ubiquitous and evolutionary conserved protein that in mammalian cells localizes mainly to the smooth endoplasmic reticulum (ER) and cytosol, where it primarily degrades heme (68). However, recent evidence suggests an association of HO-1 with other intracellular membranes, including the inner mitochondrial membrane and plasma membrane caveolae (69). HO-1 expression is detected at basal levels in the liver and spleen, which are major sites of iron recycling from hemoglobin (Hb) of senescent erythrocytes phagocyted by macrophages (70). For this reason, liver and spleen tissue macrophages exhibit constitutively high levels of activity and gene expression of HMOX1 under physiological conditions (71–73). However, HMOX1 can be induced at the transcriptional level in various other tissues by multiple stimuli, including ROS, heavy metals and its own substrate, heme (74).
In silico analysis of the expression of HMOX1, HMOX2, BLVR-A, and BLVR-B across the ImmGen expression data (http://rstats.immgen.org/MyGeneSet) demonstrated that while BLVR-B is generally expressed at highest levels during fetal development, both isoforms of BLVR and HMOX1 are expressed by immune cells, primarily within the myeloid compartment in the adult (Figure 3). HMOX2 showed less distinct pattern of expression (Figure 3). We and others have reported high levels of BLVR-A expression within the reticulo-endothelial system and phagocytic mononuclear populations in the spleen and liver (47, 60, 75). Our previous findings demonstrated that BLVR-A expression is also responsive to both LPS and BV treatment in macrophages (29, 76). Additionally, analysis of data obtained from www.immuneprofiling.org in whole blood cells showed that BLVRA, BLVR-A is increased in human blood cells following stimulation with various cytokines, DAMPs and pathogen-associated molecular patterns (PAMPs) (Figure 4). The highest induction of BLVR-A was observed after treatment with E. coli, Flagellin (TLR5 ligand), PolyI:C (TLR7 ligand), R837, influenza virus, IFNα2b or IFNβ. These data suggest a role for BLVR-A in infection with bacteria or viruses. Interestingly, BV seems to interfere with the replication of hepatitis C virus (HCV) by inducing the expression of interferon alpha2 and alpha17, thus triggering an antiviral interferon response (77). A similar antiviral effect has been demonstrated for BR against human herpes simplex virus type 1 (HSV-1) and the enterovirus EV71, indicating that bile pigments and BLVR enzymes can have an important antiviral effect and might improve antiviral therapy (78).
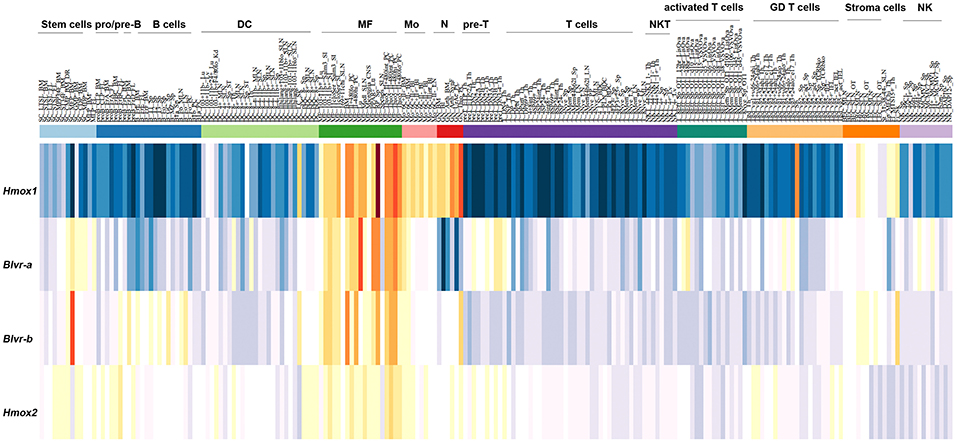
Figure 3. Expression pattern of HMOX1, HMOX2, BLVR-A, and BLVR-B mRNA in immune cells as reported in the ImmGen expression data software (http://rstats.immgen.org/MyGeneSet).
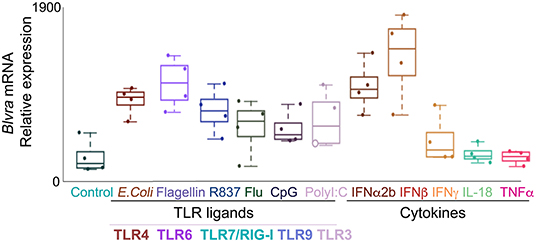
Figure 4. Expression of BLVR-A in human leukocytes. qPCR analysis of BLVR-A in peripheral blood mononuclear cells after stimulation with toll-like receptor ligands and cytokines. Results represent the mean ± s.d.
The heme degradation pathway operates in the cytoplasm, where both HO-1 and BLVR are located as either free proteins or associated with endoplasmic reticulum (ER) membranes (79). However, the conversion of BV to BR has been shown to occur in various cellular compartments in response to cellular stress (29, 47, 80). BLVR-A is also found in the nucleus (57, 61), mitochondria (81) and several reports show that its translocation between different cellular compartments is controlled by post-translational modifications such as nitrosylation or phosphorylation (47, 60, 82).
BLVR-A possesses a bZip DNA binding sequence, a nuclear export sequence (NES) and a nuclear localization sequence (NLS) that together enable its bidirectional nuclear transport (Figure 2) (57, 61, 83). BLVR-A is a member of the “leucine zipper” family of transcription factors driving activator protein 1 (AP-1)-regulated genes, such as HMOX1 (3, 28, 82, 84). BLVR-A can also regulate the expression of genes involved in cell growth, differentiation and survival, which suggests that perturbation of its function may play a role in various disorders and pathologies (48, 84–86).
Since BLVR-B lacks these functions, it is unlikely to possess significant signaling properties or transcription factor activity, representing a major functional difference between the two isoforms (Figure 2) (87, 88). Using a combination of proteomic and transcriptomic approaches, BLVR-B was recently discovered as a novel marker of intraplaque hemorrhage and carotid atherosclerosis, indicating a possible role for its involvement in cardiovascular physiology and disease (89). Interestingly, BLVR-B expression was detected in CD163+CD68+ myeloid cells at both the mRNA and protein levels, but not in lymphocytes, endothelial or smooth muscle cells, which suggests that it somehow controls inflammation in this pathology (89). Thus, both BLVR isoforms play unique roles in the host homeostasis and as such their abnormal expression may lead to pathologies such as autoimmune disease, excessive immune response or cancer.
HO-1 And Blvr In Inflammatory Responses
HO-1 was initially recognized primarily for its role in heme catabolism and iron recycling, but it has also been described as an enzyme with important anti-inflammatory, anti-oxidant and cytoprotective properties (90). Increased HO-1 expression in tissue is commonly associated with increased inflammation or oxidative stress, as seen in models of acute lung injury or ischemia-reperfusion (91). These anti-inflammatory effects are largely exerted by heme by-products like CO, which exhibits a broad range of immunomodulatory properties in in vivo models of endotoxemia, microbial sepsis and organ injury, among others (92, 93). Similarly, HO-1 modulation or application of low concentrations of CO (250 ppm) to cultured macrophages challenged with LPS were shown to reduce the expression of pro-inflammatory cytokines (TNF-α, IL-1β) and simultaneously stimulate the production of the anti-inflammatory cytokine IL-10 through the stimulation of p38 MAPK activity (69, 92, 94). Additionally, previous work demonstrated that macrophage-generated CO can increase ATP release by bacteria, driving the activation of the Nacht, LRR, and PYD domains-containing protein 3 (NALP3) inflammasome in macrophages, thus promoting their bactericidal effector functions (95). Nevertheless, HO-1 up-regulation in macrophages may not always be beneficial: while HO-1 derived CO promotes bacterial clearance by increasing macrophage activity and phagocytosis (96, 97), HO-1 induction has been linked to the intracellular survival of specific pathogens due to reduced inflammatory cytokine production or increased iron availability (98–100). For example, a dual role of heme-derived metabolic signals has been shown in malaria: HO-1 induction amplifies malaria-infection associated liver damage (101), but it also promotes disease tolerance during “the blood stage of the disease” (102). Furthermore, HO-1 induction is known to polarize macrophages into an anti-inflammatory M2 phenotype, which could also be detrimental in infections with pathogens that favor an M2 environment (103). Thus, this duality underlines the complexity of heme catabolism and the role of HO-1 as a critical mediator of innate immune response, indicating that the therapeutic potential of HO-1 may depend on both its expression, its enzymatic activity and the stage of the inflammatory response or disease (104–106).
The role of BLVR-A in inflammation has been primarily described in myeloid cells including macrophages, where this enzyme is expressed as a cell surface protein (47). Both macrophage BLVR-A expression and phosphorylation are increased upon LPS-treatment which leads to anti-inflammatory responses by stimulating PI3K-Akt-driven IL-10 production (47). In the presence of endotoxic stress and induction of NO, macrophage expressed-BLVR-A can also become S-nitrosylated by eNOS (endothelial nitric oxide synthase)-derived NO. This modification leads to its translocation to the nucleus, where it binds directly to the TLR4 gene promoter and represses its expression (60). These mechanisms have been shown to be important in promoting hepatic diseases such as non-alcoholic fatty liver disease (NAFLD) or non-alcoholic steatohepatitis (NASH), where LPS- and TLR4-associated inflammation plays a significant role (107). NAFLD is characterized by excessive accumulation of fatty acids in the form of microdeposits and its pathophysiology is not fully understood, but insulin resistance and oxidative stress are thought to factor significantly in its progression toward NASH (108). Recent work suggests the regulation of hepatic metabolism by the BLVR-A-GSKβ-PPARα axis where BLVR-A inhibits the phosphorylation of GSK3β resulting in the decreased activation of its substrate, PPARα (109). In this study, mice lacking BLVR-A in hepatocytes showed increased GSK3β activity and PPARα levels, with higher levels of plasma glucose and insulin and reduced glycogen storage (109). Interestingly, BR binds directly to PPARα and elevated total serum BR levels have been reported to negatively correlate with onset of the disease in NAFLD and NASH patients (110–113). These data indicate a major role of BLVR-A in hepatic lipid metabolism and associated inflammation, also supporting an additional role for BR as a protective factor against the progression and development of chronic liver disease. Of note, therapies directed at increasing the activity of BLVR-A or at regulating BR metabolism may prove useful for the treatment of NAFLD (107, 114). A role for BLVR-A in inflammation has also been described in other diseases, such as in germinal matrix hemorrhage (GMH), a neurologic event with high morbidity and mortality in pre-term infants (115). In this model Zhang and colleagues revealed that by suppressing TLR4 expression, BLVR-A induces the phosphorylation of eNOS in the spleen, modulating the inflammatory response and decreasing neutrophil infiltration into the brain after GMH (115). More recently, a BV/BLVR-A regulatory mechanism that controls TLR4 activation by direct/indirect interaction has been also identified in human leukocytes, suggesting that a fully functional signaling of BLVR could counter undesired TLR4 signaling and related inflammation (116).
BLVR-A has been shown to play a critical role in M2 macrophage polarization both in vitro and in response to renal ischemia-reperfusion injury in vivo (117). In addition, we have recently shown that BV reduces pro-inflammatory cytokine release and inhibits LPS-mediated C5aR expression in macrophages through the mTOR signaling pathway, further supporting a role for BV as an endogenous anti-inflammatory modulator (75). Moreover, using a mouse mutant for conditional BLVR-A deletion in macrophages, we identified a specific set of genes whose expression is altered in the absence of BLVR-A. Among these, the expression of C-X-C motif chemokine 10 (CXCL10 also known as IP-10) and chemokine C-C motif ligand 5 (CCL5, also known as RANTES) was elevated in BLVR-A deficient macrophages in response to LPS and this was associated with an increase in C5aR expression and chemotaxis toward C5a (76). Altogether, these studies point to a pivotal role of BLVR-A in controlling the inflammatory response in endotoxemia and beyond, with macrophages and T cells likely participating most in such a mechanism. Interestingly, BR is implicated in the control of T cell function by increasing number of T regulatory cells (26).
HO-1 And Blvr In Cancer
HO-1 is normally expressed in the spleen and liver, but it can also be induced in many other organs/tissues by a variety of stimuli, including heme, ROS levels or hypoxia (118). In cancer, HO-1-overexpression has been reported in leukemia and in several solid tumors, including glioblastoma, melanoma, and hepatocellular carcinoma (119–121). Interestingly, both renal clear cell carcinoma and sarcoma patients with high HMOX1 expression exhibited better survival rates than those with low HMOX1 expression, while the opposite is true for thymoma patients, indicating the complexity of HO-1 biology in cancer [Figure 5, data based on kmplot.com; (122)] In many tumors HO-1 is suggested to act as a survival molecule, promoting cancer cell growth, metastasis, angiogenesis and resistance to chemotherapy (123, 124). However, HO-1 expression was found to be decreased in patients with early-stage non-small cell lung cancer (125) and its induction increased cell death and inhibited the migratory ability of hepatocellular carcinoma (HCC) cells (126, 127), suggesting tumor type specific effects. Specifically, HO-1 seems to hinder HCC progression via downregulation of miR-30d/miR-107 expression, a mechanism that also involves PI3K/AKT and MAPK/ERK pathways (128). HO-1 activity may help to mitigate DNA damage, gene mutation and carcinogenesis resulting from excessive ROS levels; we have previously showed that HO-1/CO facilitate DNA damage repair via ATM-γH2AX mechanism in normal cells (129). However, HO-1 was also shown to promote chemotherapy-induced cell death in cancer cells (130). There is also evidence that HO-1 activity can result in iron accumulation, which several epidemiological and experimental studies associate with increased cancer incidence and risk, tumor initiation, growth and metastasis (131, 132). Since iron is highly reactive and it continuously exchanges between its different oxidized forms, excess iron induces free radical formation, lipid peroxidation, DNA and protein damages, with important consequences in carcinogenesis (133). Emerging evidence has also revealed that HO-1-induced iron levels can impact ferroptosis, a form of oxidative cell death that plays a critical role in the pathogenesis of diseases involving iron overload, such as cancer (134). Recent data indicated a crucial role of tumor-associated macrophages (TAMs)-derived iron within the tumor microenvironment in disease progression, implying that HO-1 expressed in these cells plays profound roles in modulating tumor microenvironment and promoting metastasis (135). Of note, we showed that HO-1 in TAMs is critical for regulating epithelial-mesenchymal transition (EMT) and metastatic outgrowth in prostate cancer (136).
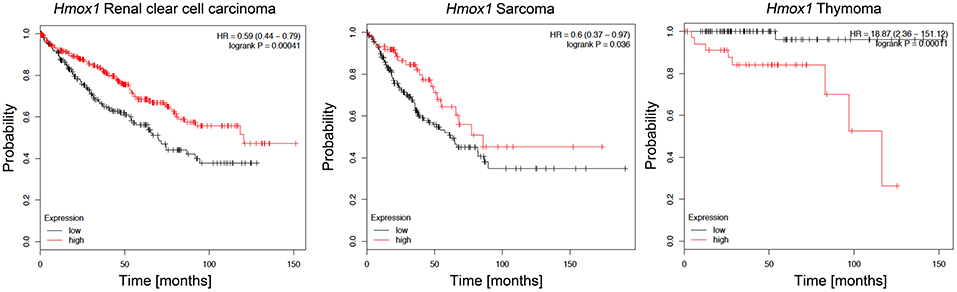
Figure 5. Kaplan-Meier survival plots for patients stratified by HMOX1 expression. Kaplan-Meier plots were obtained by using the Kaplan-Meyer plotter online database tool (122). Patients were stratified for high and low expression of HMOX1 in renal clear cell carcinoma, sarcoma, and thymoma patients.
Although HO-1 is normally resident on the endoplasmic reticulum, it has been detected in the nuclei of prostate, lung, and oral cancer tumor cells, where it is correlated with tumor progression (137–139). However, the pathophysiological relevance of this finding and any specific mechanisms involved are not yet fully understood. It is well-known that HO-1 undergoes proteolytic cleavage with subsequent nuclear translocation under stress conditions in vitro, and this promotes tumor growth and invasion independently of its enzymatic activity (138, 140). Nevertheless, HO-1 lacks a DNA binding domain and whether it can interact with transcriptional control factors or chromatin proteins to impact the expression of specific genes related to cancer progression is still a matter of debate and deserves further investigation (141).
Importantly, HO-1 can also impact cancer progression by participating in both innate and adaptive immune responses in the tumor microenvironment. By suppressing the expression of proinflammatory cytokines (i.e., TNF-α) and promoting the expression of immunosuppressive cytokines (i.e., IL-10), HO-1 significantly modulates the immune regulatory functions of myeloid cells that control TME maintenance, for example by promoting inflammation-associated angiogenesis through up-regulation of VEGF expression in macrophages (141, 142). A more recent study showed that IL-6-driven expression of HO-1 in TAMs facilitated transendothelial migration and metastatic spread of breast cancer cells, suggesting that HO-1+ macrophages can significantly influence TAMs phenotype and cancer progression (143).
Since various BLVR functions are related to signaling and gene expression that regulates cell growth, differentiation and survival, BLVR-A and BLVR-B may be directly involved in the development and progression of cancer (83). Supporting this, high levels of BLVR-A expression have been reported in malignancies including skin, breast, lung, and liver cancers (51, 144, 145), while BLVR-B is highly expressed in esophageal carcinoma, leukemia, and hepatocellular carcinoma (146–150). Importantly, BLVR-B was found to be most highly expressed at the tumor invasive margin in endometrial carcinoma, suggesting a role for this protein in cancer invasion (151). Indeed, BLVR-A has also been reported to affect the cell morphology and processes involved in EMT, a common characteristic of metastatic cancers (49, 51, 152). Additionally, a few studies have identified both BLVR-A and BLVR-B levels as potential biomarkers for the diagnosis, prognosis or treatment of prostate, pancreas, and vaginal carcinomas (153–155). Our analyses based on available gene expression data (kmplot.com) suggests that similar to HO-1 (above), there may be a role for BLVR in patient survival dependent on cancer type. High BLVR-A expression confers survival benefits in patients with cervical squamous cell carcinoma or endometrial carcinoma, while low expression levels are more beneficial for patients with liver hepatocellular carcinoma (Figure 6). High levels of BLVR-B expression in cervical squamous cell carcinoma and sarcoma patients is also correlated with better survival than those with lower BLVR-B expression. However, the opposite appears true for lung adenocarcinoma patients (Figure 6). Elevation of BLVR-A expression in tumor cells has been linked to cancer-associated hypoxia (83). Gibbs et al. showed that BLVR-A expression is driven by HIF1-α induction (156) and a similar mechanisms for regulation of BLVR-A levels in hypoxic conditions was seen in pulmonary arterial smooth muscle cell (PASMC) and human glioblastoma cells (157, 158). The role of BLVR in cancer is closely related to oxidative stress: the knockdown of BLVR-A resulted in a significant increase in intracellular ROS levels in glioblastoma cells and resulted in increased HMOX-1 expression (50, 158, 159). In addition, nuclear BLVR-A acts as a transcription factor and binds directly to ARE/AP1 and ATF2/CRE DNA sequences or in complex with Erk1/2/Elk or Nrf2/ARE, affecting multiple signaling pathways involved in cancer progression (160). Finally, BLVR-A overexpression enhanced drug resistance and protection against chemotherapeutics (cisplatin and doxorubicin) (50, 158). Therefore, due to their regulatory effects on signaling and transcription, BLVR and its related bile pigments may impact both the development and progression of cancer.
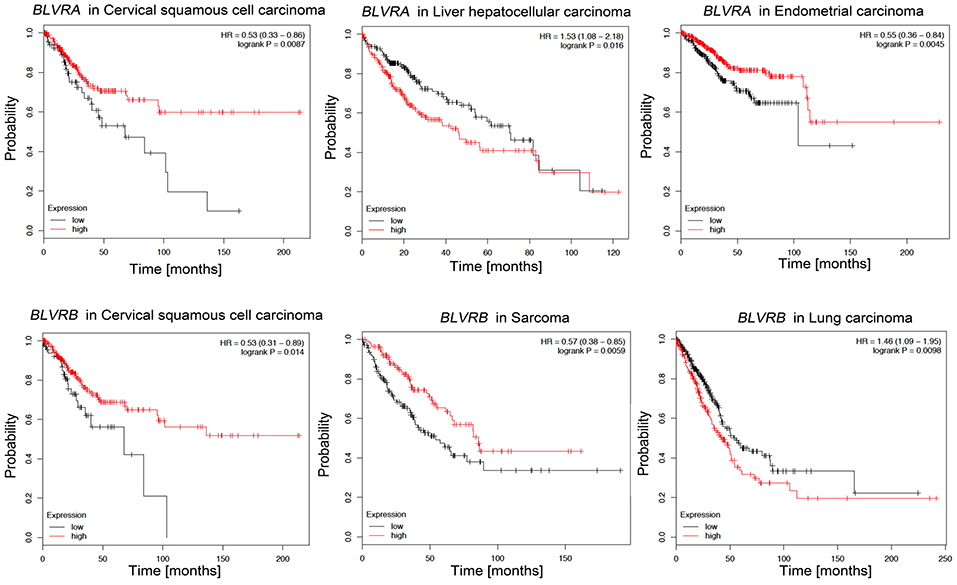
Figure 6. Kaplan-Meier survival plots for patients stratified by BLVR-A and BLVR-B expression. Kaplan-Meier plots were obtained by using the Kaplan-Meyer plotter online database tool (122). Patients were stratified for high and low expression of BLVR-A in cervical squamous cell carcinoma, endometrial carcinoma and liver hepatocellular carcinoma; and for high and low expression of BLVR-B in cervical squamous cell carcinoma, sarcoma, and lung adenocarcinoma.
While direct evidence for a role of BLVR in the TME has not been shown, there is indirect evidence for its influence on immunosenescence, polarization of macrophages and function of T cells in ways that are known to contribute to carcinogenesis. Several reports have demonstrated that elevated BR is toxic (161–163) and may damage erythrocytes and neurons and can induce cell death (164–167). BR can activate astrocytes and neurons to release soluble factors through MAPKs and NF-κB that ultimately reduce the production of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 by microglia, which is important for the regulation of immunology within the central nervous system (168, 169). Furthermore, our recent data suggest that BV stimulates removal of cellular debris by macrophages and is required for the HMOX1-mediated protection against immunosenescence in myeloid cells (170). BR also has immuno-modulatory and anti-inflammatory properties through the induction of necrosis and apoptosis in mature immune cells (9, 171–173). In T helper type 17 (Th17) cells, BR exerts its immunomodulatory properties through AhR-dependent upregulation of CD39, with beneficial effects for patients with inflammatory bowel disease (IBD) (26). Moreover, mildly elevated BR levels modulate intracellular signaling pathways involved in immunosuppression and protect against diseases associated with increased oxidative stress (86, 160, 174–176). For example, exogenous BR supplementation suppressed DAMP release and altered cytokine profiles in a model of pancreatic islet transplantation, showing overall cytoprotective and antioxidant effects and suggesting that this method could be used to improve outcomes after allograft transplantation (8). In the same model, BR treatment of donors is known to lower the levels of iNOS and IL-1β and to increase islet quality, resulting in lower inflammatory response in the recipients, which also improves transplantation outcomes (177, 178). This suggests that targeting BR metabolism and/or BLVR enzymatic activity could have important consequences in the tissue microenvironment and could be a potential therapeutic approach for metabolic, cardiovascular, oncogenic and neurological disorders as well.
Targeting Blvr In Cancer And Immune Diseases
Since BLVR plays a major role in cellular signaling and gene expression by promoting oxidative and immune homeostasis it is therefore considered a potential therapeutic target for a variety of diseases (66). Recent studies employed BLVR-based peptides to inhibit BLVR-A kinase activity or its interaction with growth-promoting kinases (83). Two human BLVR-A-derived peptides (FGFPAFSG and KKRILHCLGL) have been shown to inhibit the activity of Erk1/2 by blocking the formation of the complexes that include BLVR, MEK1 and Erk1/2 or BLVR, PKCδ and Erk1/2 (57, 58, 179, 180). Regulating the activity of BLVR and its ability to form such complexes may block the activation of Erk1/2 upstream, offering a novel approach to slow the growth of tumor cells with hyper-activated Erk1/2 or to treat any other inflammatory disease related to its activation (83, 181). Another BLVR-derived peptide (SFHFKSGSL) was described as a potent PKCδ inhibitor, with the ability to induce apoptosis by disrupting the cell membrane integrity (182). This further supports a potential therapeutic application of BLVR-based peptides in tumors showing excessive PKCδ activation, such as non-small cell lung cancer or breast cancer (58, 182–184). Two BLVR-derived peptides have been demonstrated to bind to the insulin receptor kinase (IRK) and alter its secondary structure, with important effects on glucose and insulin metabolism (185). Specifically, the KYCCSRK peptide was able to stimulate glucose uptake and to increase insulin function, while the KEDQYMKMTV peptide inhibited IRK activity and glucose uptake (185). BLVR is a substrate for IRK and it shares regulatory motifs and sequences with proteins that function in insulin and insulin-like growth factor-1 (IGF-1) pathways (48, 58, 82, 186). BLVR-A protein levels and activation are significantly reduced in peripheral blood mononuclear cells from obese patients and are associated with impaired insulin signaling, obesity, metabolic syndrome, NASH and visceral adipose tissue inflammation, indicating that BLVR-A modulation could be a therapeutic approach to obesity prevention and care (187). These peptides have been delivered successfully both in vitro and in vivo, supporting their potential as a novel therapeutic approach to control abnormal glucose metabolism and insulin resistance (185, 188–190). A major limitation of a peptide-based therapy is the short half-life of peptides in the circulation, which can result in a lower delivery compared to antibody-drug conjugates (191, 192). Hence, more effort needs to be made on improving the pharmacokinetics as well as the therapeutic efficacy of peptides or their derivatives in vivo. Alternative administration routes such as respiratory, local (intra-tumor injection) and site-specific delivery should also be considered (193). The use of enhancers or nano/micro particulate carriers also represents a promising approach for peptide-targeted delivery (194, 195). Interestingly, a recent report showed that bilirubin-based nanoparticles may represent a valid carrier option for many small peptides whose therapeutic efficacy is limited by their short circulation half-life (196) and this method could therefore be used for a more efficient delivery of BLVR-based peptides in vivo. Furthermore, BR-based nanoparticles have been tested in several diseases and conditions for their efficacy in tumor targeting and drug release (197–202). A second limitation of a peptide-based therapy is that BLVR kinase activities cannot be completely separated from its reductase activity and the use of BLVR-based peptides can affect both pathways simultaneously. Thus, the possible side effects that will likely compromise BLVR reductase activity in healthy cells need to be addressed. These effects might be more evident in macrophages or vascular endothelial cells, where BLVR has a significant reductase activity but also mediates homeostatic signaling regulating immune responses (47, 203). Such an issue highlights the importance of using peptide delivery systems capable of targeting specific cellular compartments to reduce the potential for systemic side effects.
In addition to peptides, BLVR-A modulation can also be achieved by using small molecules. In a recent study, several FDA-approved compounds were screened and tested for their effects on BLVR-A activity in the context of hyperbilirubinemia (204). After the initial characterization of safety profile and oral absorption, only two compounds were selected for further studies. However, one of them failed to reduce bilirubin levels, while the other proved to be hepatotoxic in rats (204). More BLVR-A inhibitors could be evaluated for their structure-activity relation to produce even more potent inhibitors. Moreover, the availability of the BLVR-A conditional and total knockout mice will further help dissect the role of this pathway in various diseases. The manipulation of BLVR can also be an attractive strategy to overcome treatment-related multidrug resistance, a serious concern in patients treated with chemotherapy, in whom the tumor cells develop resistance to multiple classes of chemotherapeutic agents (205). A few studies showed that overexpression of BLVR-A can enhance multidrug resistance, while its inhibition can overcome multidrug resistance and re-establish drug sensitivity (50, 158, 206).
Concluding Remarks
In conclusion, heme catabolism and its resulting products exert significant control of both homeostatic and pathophysiologic processes. BLVR-A and BLVR-B are multifunctional proteins with activities ranging from enzymatic activity to signaling kinases and regulators of transcription. In conjunction with HO-1 and heme metabolites, BLVR provides a cytoprotective and immunomodulatory mechanism for the cell and the host. Therefore, it is pivotal to understand the molecular mechanisms regulating heme metabolism for the maintenance of homeostasis and the impact of these proteins as drivers in various immune pathologies. Similarly, increased awareness of the role heme metabolites play and their modes of regulation in response to various stressors will likely prove to be extremely useful in the treatment of several pathologies.
Author Contributions
GC and SH wrote a first draft. KS and BW edited the text. GC and BW prepared the figures.
Funding
Our studies were supported by funding from: NIDDK R01 DK104714-01 to BW.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. (2012) 335:936–41. doi: 10.1126/science.1214935
3. Kim SY, Kang HT, Choi HR, Park SC. Biliverdin reductase A in the prevention of cellular senescence against oxidative stress. Exp Mol Med. (2011) 43:15–23. doi: 10.3858/emm.2011.43.1.002
4. Schluchter WM, Glazer AN. Characterization of cyanobacterial biliverdin reductase. Conversion of biliverdin to bilirubin is important for normal phycobiliprotein biosynthesis. J Biol Chem. (1997) 272:13562–9. doi: 10.1074/jbc.272.21.13562
5. Wegiel B, Nemeth Z, Correa-Costa M, Bulmer AC, Otterbein LE. Heme oxygenase-1: a metabolic nike. Antioxid Redox Signal. (2014) 20:1709–22. doi: 10.1089/ars.2013.5667
6. Stocker R, McDonagh AF, Glazer AN, Ames BN. Antioxidant activities of bile pigments: biliverdin and bilirubin. Methods Enzymol. (1990) 186:301–9. doi: 10.1016/0076-6879(90)86123-D
7. Jangi S, Otterbein L, Robson S. The molecular basis for the immunomodulatory activities of unconjugated bilirubin. Int J Biochem Cell Biol. (2013) 45:2843–51. doi: 10.1016/j.biocel.2013.09.014
8. Adin CA, Vangundy ZC, Papenfuss TL, Xu F, Ghanem M, Lakey J, et al. Physiologic doses of bilirubin contribute to tolerance of islet transplants by suppressing the innate immune response. Cell Transplant. (2017) 26:11–21. doi: 10.3727/096368916X692096
9. Liu Y, Li P, Lu J, Xiong W, Oger J, Tetzlaff W, et al. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J Immunol. (2008) 181:1887–97. doi: 10.4049/jimmunol.181.3.1887
10. Wu J, Ma J, Fan S-T, Schlitt HJ, Tsui T-Y. Bilirubin derived from heme degradation suppresses MHC class II expression in endothelial cells. Biochem Biophys Res Commun. (2005) 338:890–6. doi: 10.1016/j.bbrc.2005.10.021
11. Sundararaghavan VL, Binepal S, Stec DE, Sindhwani P, Hinds TD. Bilirubin, a new therapeutic for kidney transplant? Transplant Rev. (2018) 32:234–40. doi: 10.1016/j.trre.2018.06.003
12. Stocker R, Ames BN. Potential role of conjugated bilirubin and copper in the metabolism of lipid peroxides in bile. Proc Natl Acad Sci USA. (1987) 84:8130–4. doi: 10.1073/pnas.84.22.8130
13. Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci USA. (1987) 84:5918–22. doi: 10.1073/pnas.84.16.5918
14. Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. (2004) 6:841–9. doi: 10.1089/1523086041797999
15. Novotný L, Vítek L. Inverse relationship between serum bilirubin and atherosclerosis in men: a meta-analysis of published studies. Exp Biol Med. (2003) 228:568–71. doi: 10.1177/15353702-0322805-29
16. Chen W, Maghzal GJ, Ayer A, Suarna C, Dunn LL, Stocker R. Absence of the biliverdin reductase-a gene is associated with increased endogenous oxidative stress. Free Radic Biol Med. (2018) 115:156–65. doi: 10.1016/j.freeradbiomed.2017.11.020
17. Jiang D, Shi J, Yuan M, Duan X, Li L, Li Q. Levels of serum bilirubin in small cell lung cancer and non-small cell lung cancer patients. Cell Mol Biol. (2018) 64:71–6. doi: 10.14715/cmb/2018.64.6.13
18. Liu X, Meng QH, Ye Y, Hildebrandt MAT, Gu J, Wu X. Prognostic significance of pretreatment serum levels of albumin, LDH and total bilirubin in patients with non-metastatic breast cancer. Carcinogenesis. (2015) 36:243–8. doi: 10.1093/carcin/bgu247
19. Schieffer KM, Bruffy SM, Rauscher R, Koltun WA, Yochum GS, Gallagher CJ. Reduced total serum bilirubin levels are associated with ulcerative colitis. PLoS ONE. (2017) 12:e0179267. doi: 10.1371/journal.pone.0179267
20. Leníček M, Duricová D, Hradsky O, Dušátková P, Jirásková A, Lukáš M, et al. The relationship between serum bilirubin and Crohn's disease. Inflamm Bowel Dis. (2014) 20:481–7. doi: 10.1097/01.MIB.0000440817.84251.98
21. de Vries HS, Te Morsche RHM, Jenniskens K, Peters WHM, de Jong DJ. A functional polymorphism in UGT1A1 related to hyperbilirubinemia is associated with a decreased risk for Crohn's disease. J Crohns Colitis. (2012) 6:597–602. doi: 10.1016/j.crohns.2011.11.010
22. Su Q, Li X, Mo W, Yang Z. Low serum bilirubin, albumin, and uric acid levels in patients with Crohn's disease. Medicine. (2019) 98:e15664. doi: 10.1097/MD.0000000000015664
23. Juping D, Yuan Y, Shiyong C, Jun L, Xiuxiu Z, Haijian Y, et al. Serum bilirubin and the risk of rheumatoid arthritis. J Clin Lab Anal. (2017) 31:e22118. doi: 10.1002/jcla.22118
24. Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. (2014) 146:1625–38. doi: 10.1053/j.gastro.2014.03.047
25. Hamoud A-R, Weaver L, Stec DE, Hinds TD. Bilirubin in the liver–gut signaling axis. Trends Endocrinol Metab. (2018) 29:140–50. doi: 10.1016/j.tem.2018.01.002
26. Longhi MS, Vuerich M, Kalbasi A, Kenison JE, Yeste A, Csizmadia E, et al. Bilirubin suppresses Th17 immunity in colitis by upregulating CD39. JCI Insight. (2017) 2:e92791. doi: 10.1172/jci.insight.92791
27. Vasavda C, Kothari R, Malla AP, Tokhunts R, Lin A, Ji M, et al. Bilirubin links heme metabolism to neuroprotection by scavenging superoxide. Cell Chem Biol. (2019). doi: 10.1016/j.chembiol.2019.07.006
28. Miralem T, Hu Z, Torno MD, Lelli KM, Maines MD. Small interference RNA-mediated gene silencing of human biliverdin reductase, but not that of heme oxygenase-1, attenuates arsenite-mediated induction of the oxygenase and increases apoptosis in 293A kidney cells. J Biol Chem. (2005) 280:17084–92. doi: 10.1074/jbc.M413121200
29. Wegiel B, Otterbein LE. Go green: the anti-inflammatory effects of biliverdin reductase. Front Pharmacol. (2012) 3:47. doi: 10.3389/fphar.2012.00047
30. Pereira PJB, Macedo-Ribeiro S, Párraga A, Pérez-Luque R, Cunningham O, Darcy K, et al. Structure of human biliverdin IXbeta reductase, an early fetal bilirubin IXbeta producing enzyme. Nat Struct Biol. (2001) 8:215–20. doi: 10.1038/84948
31. Cunningham O, Gore MG, Mantle TJ. Initial-rate kinetics of the flavin reductase reaction catalysed by human biliverdin-IXbeta reductase (BVR-B). Biochem J. (2000) 345(Pt 2):393–9. doi: 10.1042/bj3450393
33. Aziz S, Kotal P, Leroy P, Servaes R, Eggermont E, Fevery J, et al. Bilirubin-IXα and-IXβ pigments, coproporphyrins and bile acids in meconium and stools from full-term and preterm neonates during the first month of life. Acta Paediatr. (2001) 90:81–7. doi: 10.1080/080352501750064923
34. Aziz S, Anjum S, Rehman AU, Ata-ur-Rehman S, Akram DS, Naqvi SAA, et al. Bilirubin pigments in the first meconium of newborn infants. J Pak Med Assoc. (2005) 55:188–92.
35. Aziz S, Leroy P, Servaes R, Eggermont E, Fevery J. Bilirubin-IXβ is a marker of meconium, like zinc coproporphyrin. J Pediatr Gastroenterol Nutr. (2001) 32:287–92. doi: 10.1097/00005176-200103000-00010
36. Macias RI, Marin JJ, Serrano MA. Excretion of biliary compounds during intrauterine life. World J Gastroenterol. (2009) 15:817. doi: 10.3748/wjg.15.817
37. Bilban M, Haschemi A, Wegiel B, Chin BY, Wagner O, Otterbein LE. Heme oxygenase and carbon monoxide initiate homeostatic signaling. J Mol Med. (2008) 86:267–79. doi: 10.1007/s00109-007-0276-0
38. Soares MP, Usheva A, Brouard S, Berberat PO, Gunther L, Tobiasch E, et al. Modulation of endothelial cell apoptosis by heme oxygenase-1-derived carbon monoxide. Antioxid Redox Signal. (2002) 4:321–9. doi: 10.1089/152308602753666370
39. Soares MP, Seldon MP, Gregoire IP, Vassilevskaia T, Berberat PO, Yu J, et al. Heme oxygenase-1 modulates the expression of adhesion molecules associated with endothelial cell activation. J Immunol. (2004) 172:3553–63. doi: 10.4049/jimmunol.172.6.3553
40. Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. (2003) 24:449–55. doi: 10.1016/S1471-4906(03)00181-9
41. Tsuchihashi S, Fondevila C, Kupiec-Weglinski JW. Heme oxygenase system in ischemia and reperfusion injury. Ann Transplant. (2004) 9:84–7. doi: 10.1097/01.mot.0000125488.13679.cd
42. Rensing H, Jaeschke H, Bauer I, Pätau C, Datene V, Pannen BHJ, et al. Differential activation pattern of redox-sensitive transcription factors and stress-inducible dilator systems heme oxygenase-1 and inducible nitric oxide synthase in hemorrhagic and endotoxic shock. Crit Care Med. (2001) 29:1962–71. doi: 10.1097/00003246-200110000-00019
43. Otterbein L, Sylvester SL, Choi AM. Hemoglobin provides protection against lethal endotoxemia in rats: the role of heme oxygenase-1. Am J Respir Cell Mol Biol. (1995) 13:595–601. doi: 10.1165/ajrcmb.13.5.7576696
44. Neto JS, Nakao A, Kimizuka K, Romanosky AJ, Stolz DB, Uchiyama T, et al. Protection of transplant-induced renal ischemia-reperfusion injury with carbon monoxide. Am J Physiol Physiol. (2004) 287:F979–89. doi: 10.1152/ajprenal.00158.2004
45. Ishikawa K. Heme oxygenase-1 against vascular insufficiency: roles of atherosclerotic disorders. Curr Pharm Des. (2003) 9:2489–97. doi: 10.2174/1381612033453767
46. Bauer I, Vollmar B, Jaeschke H, Rensing H, Kraemer T, Larsen R, et al. Transcriptional activation of heme oxygenase-1 and its functional significance in acetaminophen-induced hepatitis and hepatocellular injury in the rat. J Hepatol. (2000) 33:395–406. doi: 10.1016/S0168-8278(00)80275-5
47. Wegiel B, Baty CJ, Gallo D, Csizmadia E, Scott JR, Akhavan A, et al. Cell surface biliverdin reductase mediates biliverdin-induced anti-inflammatory effects via phosphatidylinositol 3-kinase and Akt. J Biol Chem. (2009) 284:21369–78. doi: 10.1074/jbc.M109.027433
48. Lerner-Marmarosh N, Shen J, Torno MD, Kravets A, Hu Z, Maines MD. Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc Natl Acad Sci USA. (2005) 102:7109–14. doi: 10.1073/pnas.0502173102
49. Zhang M, Song S, Yi Z, Zhao X, Fu L, Wang L, et al. Human biliverdin reductase promotes EMT through the ERK1/2 signal pathway in breast cancer. Eur J Pharmacol. (2016) 788:45–53. doi: 10.1016/j.ejphar.2016.06.019
50. Florczyk U, Golda S, Zieba A, Cisowski J, Jozkowicz A, Dulak J. Overexpression of biliverdin reductase enhances resistance to chemotherapeutics. Cancer Lett. (2011) 300:40–7. doi: 10.1016/j.canlet.2010.09.003
51. Liu F, Song S, Yi Z, Zhang M, Li J, Yang F, et al. HGF induces EMT in non-small-cell lung cancer through the hBVR pathway. Eur J Pharmacol. (2017) 811:180–90. doi: 10.1016/j.ejphar.2017.05.040
52. Miralem T, Lerner-Marmarosh N, Gibbs PEM, Jenkins JL, Heimiller C, Maines MD. Interaction of human biliverdin reductase with Akt/protein kinase B and phosphatidylinositol-dependent kinase 1 regulates glycogen synthase kinase 3 activity: a novel mechanism of Akt activation. FASEB J. (2016) 30:2926–44. doi: 10.1096/fj.201600330RR
53. Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. (2014) 55:28–38. doi: 10.1016/j.jbior.2014.04.001
54. Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFκB and the essentialness of NFκB for the oncogenicity of PI3K and Akt. Int J cancer. (2009) 125:2863–70. doi: 10.1002/ijc.24748
55. Sharma N, Tramutola A, Lanzillotta C, Arena A, Blarzino C, Cassano T, et al. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer's disease. Neurobiol Dis. (2019) 125:176–89. doi: 10.1016/j.nbd.2019.02.003
56. Maines DM. Potential application of biliverdin reductase and its fragments to modulate insulin/IGF-1/MAPK/PI3-K signaling pathways in therapeutic settings. Curr Drug Targets. (2010) 11:1586–94. doi: 10.2174/1389450111009011586
57. Lerner-Marmarosh N, Miralem T, Gibbs PEM, Maines MD. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc Natl Acad Sci USA. (2008) 105:6870–5. doi: 10.1073/pnas.0800750105
58. Gibbs PEM, Miralem T, Lerner-Marmarosh N, Tudor C, Maines MD. Formation of ternary complex of human biliverdin reductase-protein kinase Cδ-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-κB, and inducible nitric-oxidase synthase (iNOS). J Biol Chem. (2012) 287:1066–79. doi: 10.1074/jbc.M111.279612
59. Maines MD. Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid Redox Signal. (2007) 9:2187–96. doi: 10.1089/ars.2007.1805
60. Wegiel B, Gallo D, Csizmadia E, Roger T, Kaczmarek E, Harris C, et al. Biliverdin inhibits Toll-like receptor-4 (TLR4) expression through nitric oxide-dependent nuclear translocation of biliverdin reductase. Proc Natl Acad Sci USA. (2011) 108:18849–54. doi: 10.1073/pnas.1108571108
61. Ahmad Z, Salim M, Maines MD. Human biliverdin reductase is a leucine zipper-like DNA-binding protein and functions in transcriptional activation of heme oxygenase-1 by oxidative stress. J Biol Chem. (2002) 277:9226–32. doi: 10.1074/jbc.M108239200
62. Nakao A, Otterbein LE, Overhaus M, Sarady JK, Tsung A, Kimizuka K, et al. Biliverdin protects the functional integrity of a transplanted syngeneic small bowel. Gastroenterology. (2004) 127:595–606. doi: 10.1053/j.gastro.2004.05.059
63. Fondevila C, Katori M, Lassman C, Carmody I, Busuttil RW, Bach FH, et al. Biliverdin protects rat livers from ischemia/reperfusion injury. Transplant Proc. (2003) 35:1798–9. doi: 10.1016/S0041-1345(03)00720-6
64. Fondevila C, Shen X-D, Tsuchiyashi S, Yamashita K, Csizmadia E, Lassman C, et al. Biliverdin therapy protects rat livers from ischemia and reperfusion injury. Hepatology. (2004) 40:1333–41. doi: 10.1002/hep.20480
65. Baranano DE, Rao M, Ferris CD, Snyder SH. Biliverdin reductase: a major physiologic cytoprotectant. Proc Natl Acad Sci USA. (2002) 99:16093–8. doi: 10.1073/pnas.252626999
66. Florczyk UM, Jozkowicz A, Dulak J. Biliverdin reductase: new features of an old enzyme and its potential therapeutic significance. Pharmacol Rep. (2008) 60:38–48.
67. Sedlak TW, Saleh M, Higginson DS, Paul BD, Juluri KR, Snyder SH. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc Natl Acad Sci USA. (2009) 106:5171–6. doi: 10.1073/pnas.0813132106
68. Wilks A. Heme oxygenase: evolution, structure, and mechanism. Antioxid Redox Signal. (2002) 4:603–14. doi: 10.1089/15230860260220102
69. Ryter SW, Choi AMK. Targeting heme oxygenase-1 and carbon monoxide for therapeutic modulation of inflammation. Transl Res. (2016) 167:7–34. doi: 10.1016/j.trsl.2015.06.011
70. Maines MD, Trakshel GM, Kutty RK. Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. (1986) 261:411–9.
71. Bauer I, Wanner GA, Rensing H, Alte C, Miescher EA, Wolf B, et al. Expression pattern of heme oxygenase isoenzymes 1 and 2 in normal and stress-exposed rat liver. Hepatology. (1998) 27:829–38. doi: 10.1002/hep.510270327
72. Immenschuh S, Baumgart-Vogt E, Tan M, Iwahara S, Ramadori G, Fahimi HD. Differential cellular and subcellular localization of heme-binding protein 23/peroxiredoxin I and heme oxygenase-1 in rat liver. J Histochem Cytochem. (2003) 51:1621–31. doi: 10.1177/002215540305101206
73. Ryter SW, Alam J, Choi AMK. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. (2006) 86:583–650. doi: 10.1152/physrev.00011.2005
74. Otterbein LE, Foresti R, Motterlini R. Heme oxygenase-1 and carbon monoxide in the heart. Circ Res. (2016) 118:1940–59. doi: 10.1161/CIRCRESAHA.116.306588
75. Bisht K, Wegiel B, Tampe J, Neubauer O, Wagner K-H, Otterbein LE, et al. Biliverdin modulates the expression of C5aR in response to endotoxin in part via mTOR signaling. Biochem Biophys Res Commun. (2014) 449:94–9. doi: 10.1016/j.bbrc.2014.04.150
76. Bisht K, Canesin G, Cheytan T, Li M, Nemeth Z, Csizmadia E, et al. Deletion of biliverdin reductase A in myeloid cells promotes chemokine expression and chemotaxis in part via a complement C5a—C5aR1 pathway. J Immunol. (2019) 202:2982–90. doi: 10.4049/jimmunol.1701443
77. Lehmann E, El-Tantawy WH, Ocker M, Bartenschlager R, Lohmann V, Hashemolhosseini S, et al. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology. (2010) 51:398–404. doi: 10.1002/hep.23339
78. Santangelo R, Mancuso C, Marchetti S, Di Stasio E, Pani G, Fadda G. Bilirubin: an endogenous molecule with antiviral activity in vitro. Front Pharmacol. (2012) 3:36. doi: 10.3389/fphar.2012.00036
79. Gottlieb Y, Truman M, Cohen LA, Leichtmann-Bardoogo Y, Meyron-Holtz EG. Endoplasmic reticulum anchored heme-oxygenase 1 faces the cytosol. Haematologica. (2012) 97:1489–93. doi: 10.3324/haematol.2012.063651
80. Kim HP, Wang X, Galbiati F, Ryter SW, Choi AMK. Caveolae compartmentalization of heme oxygenase-1 in endothelial cells. FASEB J. (2004) 18:1080–9. doi: 10.1096/fj.03-1391com
81. Muhsain SNF, Lang MA, Abu-Bakar A. Mitochondrial targeting of bilirubin regulatory enzymes: an adaptive response to oxidative stress. Toxicol Appl Pharmacol. (2015) 282:77–89. doi: 10.1016/j.taap.2014.11.010
82. Tudor C, Lerner-Marmarosh N, Engelborghs Y, Gibbs PEM, Maines MD. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO - 1 gene expression by haematin. Biochem J. (2008) 413:405–16. doi: 10.1042/BJ20080018
83. Gibbs PEM, Miralem T, Maines MD. Biliverdin reductase: a target for cancer therapy? Front Pharmacol. (2015) 6:119. doi: 10.3389/fphar.2015.00119
84. Kravets A, Hu Z, Miralem T, Torno MD, Maines MD. Biliverdin reductase, a novel regulator for induction of activating transcription factor-2 and heme oxygenase-1. J Biol Chem. (2004) 279:19916–23. doi: 10.1074/jbc.M314251200
85. Salim M, Brown-Kipphut BA, Maines MD. Human biliverdin reductase is autophosphorylated, and phosphorylation is required for bilirubin formation. J Biol Chem. (2001) 276:10929–34. doi: 10.1074/jbc.M010753200
86. Kapitulnik J, Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci. (2009) 30:129–37. doi: 10.1016/j.tips.2008.12.003
87. Chu W-T, Nesbitt NM, Gnatenko DV, Li Z, Zhang B, Seeliger MA, et al. Enzymatic activity and thermodynamic stability of biliverdin IXβ reductase are maintained by an active site serine. Chem A Eur J. (2017) 23:1891–900. doi: 10.1002/chem.201604517
88. O'Brien L, Hosick PA, John K, Stec DE, Hinds TD. Biliverdin reductase isozymes in metabolism. Trends Endocrinol Metab. (2015) 26:212–20. doi: 10.1016/j.tem.2015.02.001
89. Matic LP, Jesus Iglesias M, Vesterlund M, Lengquist M, Hong M-G, Saieed S, et al. Novel multiomics profiling of human carotid atherosclerotic plaques and plasma reveals biliverdin reductase B as a marker of intraplaque hemorrhage. JACC Basic Transl Sci. (2018) 3:464–80. doi: 10.1016/j.jacbts.2018.04.001
90. Deshane J, Wright M, Agarwal A. Heme oxygenase-1 expression in disease states. Acta Biochim Pol. (2005) 52:273–84. doi: 10.18388/abp.2005_3440
91. Ryter SW, Choi AMK. Heme oxygenase-1/carbon monoxide: novel therapeutic strategies in critical care medicine. Curr Drug Targets. (2010) 11:1485–94. doi: 10.2174/1389450111009011485
92. Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, et al. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. (2000) 6:422–8. doi: 10.1038/74680
93. Soares MP, Hamza I. Macrophages and iron metabolism. Immunity. (2016) 44:492–504. doi: 10.1016/j.immuni.2016.02.016
94. Kim HP, Wang X, Zhang J, Suh GY, Benjamin IJ, Ryter SW, et al. Heat shock protein-70 mediates the cytoprotective effect of carbon monoxide: involvement of p38 beta MAPK and heat shock factor-1. J Immunol. (2005) 175:2622–9. doi: 10.4049/jimmunol.175.4.2622
95. Wegiel B, Larsen R, Gallo D, Chin BY, Harris C, Mannam P, et al. Macrophages sense and kill bacteria through carbon monoxide-dependent inflammasome activation. J Clin Invest. (2014) 124:4926–40. doi: 10.1172/JCI72853
96. Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, et al. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology. (2013) 144:789–98. doi: 10.1053/j.gastro.2012.12.025
97. Chung SW, Liu X, Macias AA, Baron RM, Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest. (2008) 118:239–47. doi: 10.1172/JCI32730
98. Scharn CR, Collins AC, Nair VR, Stamm CE, Marciano DK, Graviss EA, et al. Heme oxygenase-1 regulates inflammation and mycobacterial survival in human macrophages during Mycobacterium tuberculosis Infection. J Immunol. (2016) 196:4641–9. doi: 10.4049/jimmunol.1500434
99. Abdalla MY, Ahmad IM, Switzer B, Britigan BE. Induction of heme oxygenase-1 contributes to survival of Mycobacterium abscessus in human macrophages-like THP-1 cells. Redox Biol. (2015) 4:328–39. doi: 10.1016/j.redox.2015.01.012
100. Kelley VA, Schorey JS. Mycobacterium's arrest of phagosome maturation in macrophages requires Rab5 activity and accessibility to iron. Mol Biol Cell. (2003) 14:3366–77. doi: 10.1091/mbc.e02-12-0780
101. Epiphanio S, Mikolajczak SA, Gonçalves LA, Pamplona A, Portugal S, Albuquerque S, et al. Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine plasmodium liver infection. Cell Host Microbe. (2008) 3:331–8. doi: 10.1016/j.chom.2008.04.003
102. Jeney V, Ramos S, Bergman M-L, Bechmann I, Tischer J, Ferreira A, et al. Control of disease tolerance to malaria by nitric oxide and carbon monoxide. Cell Rep. (2014) 8:126–36. doi: 10.1016/j.celrep.2014.05.054
103. Vijayan V, Wagener FADTG, Immenschuh S. The macrophage heme-heme oxygenase-1 system and its role in inflammation. Biochem Pharmacol. (2018) 153:159–67. doi: 10.1016/j.bcp.2018.02.010
104. Naito Y, Takagi T, Higashimura Y. Heme oxygenase-1 and anti-inflammatory M2 macrophages. Arch Biochem Biophys. (2014) 564:83–8. doi: 10.1016/j.abb.2014.09.005
105. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. (2016) 73:3221–47. doi: 10.1007/s00018-016-2223-0
106. Jais A, Einwallner E, Sharif O, Gossens K, Lu TT-H, Soyal SM, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell. (2014) 158:25–40. doi: 10.1016/j.cell.2014.04.043
107. Weaver L, Hamoud A, Stec DE, Hinds TD. Biliverdin reductase and bilirubin in hepatic disease. Am J Physiol Liver Physiol. (2018) 314:G668–76. doi: 10.1152/ajpgi.00026.2018
108. Chitturi S, Farrell GC, Hashimoto E, Saibara T, Lau GKK, Sollano JD, et al. Non-alcoholic fatty liver disease in the Asia-Pacific region: definitions and overview of proposed guidelines. J Gastroenterol Hepatol. (2007) 22:778–87. doi: 10.1111/j.1440-1746.2007.05001.x
109. Hinds TD, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, et al. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem. (2016) 291:25179–91. doi: 10.1074/jbc.M116.731703
110. Stec DE, John K, Trabbic CJ, Luniwal A, Hankins MW, Baum J, et al. Bilirubin binding to PPARα inhibits lipid accumulation. PLoS ONE. (2016) 11:e0153427. doi: 10.1371/journal.pone.0153427
111. Kwak M-S, Kim D, Chung GE, Kang SJ, Park MJ, Kim YJ, et al. Serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. (2012) 18:383. doi: 10.3350/cmh.2012.18.4.383
112. Jang BK. Elevated serum bilirubin levels are inversely associated with nonalcoholic fatty liver disease. Clin Mol Hepatol. (2012) 18:357. doi: 10.3350/cmh.2012.18.4.357
113. Puri K, Nobili V, Melville K, Corte CD, Sartorelli MR, Lopez R, et al. Serum bilirubin level is inversely associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr. (2013) 57:114–8. doi: 10.1097/MPG.0b013e318291fefe
114. Hinds TD, Adeosun SO, Alamodi AA, Stec DE. Does bilirubin prevent hepatic steatosis through activation of the PPARα nuclear receptor? Med Hypotheses. (2016) 95:54–7. doi: 10.1016/j.mehy.2016.08.013
115. Zhang Y, Ding Y, Lu T, Zhang Y, Xu N, McBride DW, et al. Biliverdin reductase-A attenuated GMH-induced inflammatory response in the spleen by inhibiting toll-like receptor-4 through eNOS/NO pathway. J Neuroinflammation. (2018) 15:118. doi: 10.1186/s12974-018-1155-z
116. Zhang Z, Amorosa LF, Petrova A, Coyle S, Macor M, Nair M, et al. TLR4 counteracts BVRA signaling in human leukocytes via differential regulation of AMPK, mTORC1 and mTORC2. Sci Rep. (2019) 9:7020. doi: 10.1038/s41598-019-43347-8
117. Hu Z, Pei G, Wang P, Yang J, Zhu F, Guo Y, et al. Biliverdin reductase A (BVRA) mediates macrophage expression of interleukin-10 in injured kidney. Int J Mol Sci. (2015) 16:22621–35. doi: 10.3390/ijms160922621
118. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. (2010) 50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600
119. Doi K, Akaike T, Fujii S, Tanaka S, Ikebe N, Beppu T, et al. Induction of haem oxygenase-1 nitric oxide and ischaemia in experimental solid tumours and implications for tumour growth. Br J Cancer. (1999) 80:1945–54. doi: 10.1038/sj.bjc.6690624
120. Hjortsø MD, Andersen MH. The expression, function and targeting of haem oxygenase-1 in cancer. Curr Cancer Drug Targets. (2014) 14:337–47. doi: 10.2174/1568009614666140320111306
121. Salerno L, Romeo G, Modica MN, Amata E, Sorrenti V, Barbagallo I, et al. Heme oxygenase-1: a new druggable target in the management of chronic and acute myeloid leukemia. Eur J Med Chem. (2017) 142:163–78. doi: 10.1016/j.ejmech.2017.07.031
122. Nagy Á, Lánczky A, Menyhárt O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep. (2018) 8:11515. doi: 10.1038/s41598-018-29514-3
123. Li Volti G, Tibullo D, Vanella L, Giallongo C, Di Raimondo F, Forte S, et al. The heme oxygenase system in hematological malignancies. Antioxid Redox Signal. (2017) 27:363–77. doi: 10.1089/ars.2016.6735
124. Loboda A, Jozkowicz A, Dulak J. HO-1/CO system in tumor growth, angiogenesis and metabolism - targeting HO-1 as an anti-tumor therapy. Vascul Pharmacol. (2015) 74:11–22. doi: 10.1016/j.vph.2015.09.004
125. De Palma G, Mozzoni P, Acampa O, Internullo E, Carbognani P, Rusca M, et al. Expression levels of some antioxidant and epidermal growth factor receptor genes in patients with early-stage non-small cell lung cancer. J Nucleic Acids. (2010) 2010:1–6. doi: 10.4061/2010/147528
126. Lee SE, Yang H, Jeong S II, Jin Y-H, Park C-S, Park YS. Induction of heme oxygenase-1 inhibits cell death in crotonaldehyde-stimulated HepG2 cells via the PKC-δ-p38-Nrf2 pathway. PLoS ONE. (2012) 7:e41676. doi: 10.1371/journal.pone.0041676
127. Zou C, Zhang H, Li Q, Xiao H, Yu L, Ke S, et al. Heme oxygenase-1: a molecular brake on hepatocellular carcinoma cell migration. Carcinogenesis. (2011) 32:1840–8. doi: 10.1093/carcin/bgr225
128. Zou C, Zou C, Cheng W, Li Q, Han Z, Wang X, et al. Heme oxygenase-1 retards hepatocellular carcinoma progression through the microRNA pathway. Oncol Rep. (2016) 36:2715–22. doi: 10.3892/or.2016.5056
129. Otterbein LE, Hedblom A, Harris C, Csizmadia E, Gallo D, Wegiel B. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia-telangiectasia mutated (ATM) protein. Proc Natl Acad Sci USA. (2011) 108:14491–6. doi: 10.1073/pnas.1102295108
130. Wegiel B, Gallo D, Csizmadia E, Harris C, Belcher J, Vercellotti GM, et al. Carbon monoxide expedites metabolic exhaustion to inhibit tumor growth. Cancer Res. (2013) 73:7009–21. doi: 10.1158/0008-5472.CAN-13-1075
131. Torti SV, Manz DH, Paul BT, Blanchette-Farra N, Torti FM. Iron and cancer. Annu Rev Nutr. (2018) 38:97–125. doi: 10.1146/annurev-nutr-082117-051732
132. Wang B, Zhang J, Song F, Tian M, Shi B, Jiang H, et al. EGFR regulates iron homeostasis to promote cancer growth through redistribution of transferrin receptor 1. Cancer Lett. (2016) 381:331–40. doi: 10.1016/j.canlet.2016.08.006
133. Wang Y, Yu L, Ding J, Chen Y. Iron metabolism in cancer. Int J Mol Sci. (2018) 20:95. doi: 10.3390/ijms20010095
134. Chiang S-K, Chen S-E, Chang L-C. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci. (2018) 20:39. doi: 10.3390/ijms20010039
135. Jung M, Mertens C, Tomat E, Brüne B. Iron as a central player and promising target in cancer progression. Int J Mol Sci. (2019) 20:273. doi: 10.3390/ijms20020273
136. Nemeth Z, Li M, Csizmadia E, Döme B, Johansson M, Persson JL, et al. Heme oxygenase-1 in macrophages controls prostate cancer progression. Oncotarget. (2015) 6:33675–88. doi: 10.18632/oncotarget.5284
137. Sacca P, Meiss R, Casas G, Mazza O, Calvo JC, Navone N, et al. Nuclear translocation of haeme oxygenase-1 is associated to prostate cancer. Br J Cancer. (2007) 97:1683–9. doi: 10.1038/sj.bjc.6604081
138. Hsu F-F, Yeh C-T, Sun Y-J, Chiang M-T, Lan W-M, Li F-A, et al. Signal peptide peptidase-mediated nuclear localization of heme oxygenase-1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene. (2015) 34:2360–70. doi: 10.1038/onc.2014.166
139. Gandini NA, Fermento ME, Salomón DG, Blasco J, Patel V, Gutkind JS, et al. Nuclear localization of heme oxygenase-1 is associated with tumor progression of head and neck squamous cell carcinomas. Exp Mol Pathol. (2012) 93:237–45. doi: 10.1016/j.yexmp.2012.05.001
140. Lin Q, Weis S, Yang G, Weng Y-H, Helston R, Rish K, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem. (2007) 282:20621–33. doi: 10.1074/jbc.M607954200
141. Chau L-Y. Heme oxygenase-1: emerging target of cancer therapy. J Biomed Sci. (2015) 22:22. doi: 10.1186/s12929-015-0128-0
142. Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, et al. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. (2004) 103:761–6. doi: 10.1182/blood-2003-06-1974
143. Muliaditan T, Caron J, Okesola M, Opzoomer JW, Kosti P, Georgouli M, et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat Commun. (2018) 9:2951. doi: 10.1038/s41467-018-05346-7
144. Wei XX, Chan S, Kwek S, Lewis J, Dao V, Zhang L, et al. Systemic GM-CSF recruits effector t cells into the tumor microenvironment in localized prostate cancer. Cancer Immunol Res. (2016) 4:948–58. doi: 10.1158/2326-6066.CIR-16-0042
145. Arena V, Pennacchia I, Guerriero G, Mancuso C. The heme oxygenase/biliverdin reductase system in skin cancers. J Biol Regul Homeost Agents. (2018) 29:259–64.
146. Zhang J, Wang K, Zhang J, Liu SS, Dai L, Zhang J-Y. Using proteomic approach to identify tumor-associated proteins as biomarkers in human esophageal squamous cell carcinoma. J Proteome Res. (2011) 10:2863–72. doi: 10.1021/pr200141c
147. Silveira VS, Scrideli CA, Moreno DA, Yunes JA, Queiroz RGP, Toledo SC, et al. Gene expression pattern contributing to prognostic factors in childhood acute lymphoblastic leukemia. Leuk Lymphoma. (2013) 54:310–4. doi: 10.3109/10428194.2012.710330
148. Melle C, Ernst G, Scheibner O, Kaufmann R, Schimmel B, Bleul A, et al. Identification of specific protein markers in microdissected hepatocellular carcinoma. J Proteome Res. (2007) 6:306–15. doi: 10.1021/pr060439b
149. Huan L, Bao C, Chen D, Li Y, Lian J, Ding J, et al. MicroRNA-127-5p targets the biliverdin reductase B/nuclear factor-κB pathway to suppress cell growth in hepatocellular carcinoma cells. Cancer Sci. (2016) 107:258–66. doi: 10.1111/cas.12869
150. Kubícková KN, Subhanová I, Konícková R, Matousová L, Urbánek P, Parobková H, et al. Predictive role BLVRA mRNA expression in hepatocellular cancer. Ann Hepatol. (2016) 15:881–7. doi: 10.5604/16652681.1222104
151. Monge M, Doll A, Colas E, Gil-Moreno A, Castellvi J, Garcia A, et al. Subtractive proteomic approach to the endometrial carcinoma invasion front. J Proteome Res. (2009) 8:4676–84. doi: 10.1021/pr900390t
152. Zeng R, Yao Y, Han M, Zhao X, Liu X-C, Wei J, et al. Biliverdin reductase mediates hypoxia-induced EMT via PI3-kinase and Akt. J Am Soc Nephrol. (2008) 19:380–7. doi: 10.1681/ASN.2006111194
153. Pallua JD, Schaefer G, Seifarth C, Becker M, Meding S, Rauser S, et al. MALDI-MS tissue imaging identification of biliverdin reductase B overexpression in prostate cancer. J Proteomics. (2013) 91:500–14. doi: 10.1016/j.jprot.2013.08.003
154. Yu KH, Barry CG, Austin D, Busch CM, Sangar V, Rustgi AK, et al. Stable isotope dilution multidimensional liquid chromatography-tandem mass spectrometry for pancreatic cancer serum biomarker discovery. J Proteome Res. (2009) 8:1565–76. doi: 10.1021/pr800904z
155. Hellman K, Alaiya AA, Becker S, Lomnytska M, Schedvins K, Steinberg W, et al. Differential tissue-specific protein markers of vaginal carcinoma. Br J Cancer. (2009) 100:1303–14. doi: 10.1038/sj.bjc.6604975
156. Gibbs PEM, Miralem T, Maines MD. Characterization of the human biliverdin reductase gene structure and regulatory elements: promoter activity is enhanced by hypoxia and suppressed by TNF-α-activated NF-κB. FASEB J. (2010) 24:3239–54. doi: 10.1096/fj.09-144592
157. Song S, Wang S, Ma J, Yao L, Xing H, Zhang L, et al. Biliverdin reductase/bilirubin mediates the anti-apoptotic effect of hypoxia in pulmonary arterial smooth muscle cells through ERK1/2 pathway. Exp Cell Res. (2013) 319:1973–87. doi: 10.1016/j.yexcr.2013.05.015
158. Kim SS, Seong S, Lim SH, Kim SY. Biliverdin reductase plays a crucial role in hypoxia-induced chemoresistance in human glioblastoma. Biochem Biophys Res Commun. (2013) 440:658–63. doi: 10.1016/j.bbrc.2013.09.120
159. Atukeren P, Oner S, Baran O, Kemerdere R, Eren B, Cakatay U, et al. Oxidant and anti-oxidant status in common brain tumors: Correlation to TP53 and human biliverdin reductase. Clin Neurol Neurosurg. (2017) 158:72–6. doi: 10.1016/j.clineuro.2017.05.003
160. Gazzin S, Vitek L, Watchko J, Shapiro SM, Tiribelli C. A novel perspective on the biology of bilirubin in health and disease. Trends Mol Med. (2016) 22:758–68. doi: 10.1016/j.molmed.2016.07.004
161. Ostrow JD, Tiribelli C. New concepts in bilirubin neurotoxicity and the need for studies at clinically relevant bilirubin concentrations. J Hepatol. (2001) 34:467–70. doi: 10.1016/S0168-8278(00)00051-9
162. Brito MA, Lima S, Fernandes A, Falcão AS, Silva RFM, Butterfield DA, et al. Bilirubin injury to neurons: contribution of oxidative stress and rescue by glycoursodeoxycholic acid. Neurotoxicology. (2008) 29:259–69. doi: 10.1016/j.neuro.2007.11.002
163. Brito MA, Rosa AI, Falcão AS, Fernandes A, Silva RFM, Butterfield DA, et al. Unconjugated bilirubin differentially affects the redox status of neuronal and astroglial cells. Neurobiol Dis. (2008) 29:30–40. doi: 10.1016/j.nbd.2007.07.023
164. Alexandra Brito M, Silva RFM, Brites D. Bilirubin toxicity to human erythrocytes: a review. Clin Chim Acta. (2006) 374:46–56. doi: 10.1016/j.cca.2006.06.012
165. Seubert JM, Darmon AJ, El-Kadi AOS, D'Souza SJA, Bend JR. Apoptosis in murine hepatoma hepa 1c1c7 wild-type, C12, and C4 cells mediated by bilirubin. Mol Pharmacol. (2002) 62:257–64. doi: 10.1124/mol.62.2.257
166. Keshavan P, Schwemberger SJ, Smith DLH, Babcock GF, Zucker SD. Unconjugated bilirubin induces apoptosis in colon cancer cells by triggering mitochondrial depolarization. Int J cancer. (2004) 112:433–45. doi: 10.1002/ijc.20418
167. Rodrigues CMP, Solá S, Brites D. Bilirubin induces apoptosis via the mitochondrial pathway in developing rat brain neurons. Hepatology. (2002) 35:1186–95. doi: 10.1053/jhep.2002.32967
168. Silva SL, Osório C, Vaz AR, Barateiro A, Falcão AS, Silva RFM, et al. Dynamics of neuron-glia interplay upon exposure to unconjugated bilirubin. J Neurochem. (2011) 117:412–24. doi: 10.1111/j.1471-4159.2011.07200.x
169. Silva SL, Vaz AR, Barateiro A, Falcão AS, Fernandes A, Brito MA, Silva RFM, et al. Features of bilirubin-induced reactive microglia: from phagocytosis to inflammation. Neurobiol Dis. (2010) 40:663–75. doi: 10.1016/j.nbd.2010.08.010
170. Hedblom A, Hejazi SM, Canesin G, Choudhury R, Hanafy KA, Csizmadia E, et al. Heme detoxification by heme oxygenase-1 reinstates proliferative and immune balances upon genotoxic tissue injury. Cell Death Dis. (2019) 10:72. doi: 10.1038/s41419-019-1342-6
171. Haga Y, Tempero MA, Zetterman RK. Unconjugated bilirubin inhibits in vitro cytotoxic T lymphocyte activity of human lymphocytes. Biochim Biophys Acta. (1996) 1317:65–70. doi: 10.1016/0925-4439(96)00039-7
172. Síma P, Malá J, Miler I, Hodr R, Truxová E. The suppressive effect of continuous infusion of bilirubin on the immune response in mice. Folia Microbiol. (1980) 25:483–90. doi: 10.1007/BF02897214
173. Rola-Plezczynski M, Hensen SA, Vincent MM, Bellanti JA. Inhibitory effects of bilirubin on cellular immune responses in man. J Pediatr. (1975) 86:690–6. doi: 10.1016/S0022-3476(75)80352-0
174. Khan NM, Poduval TB. Immunomodulatory and immunotoxic effects of bilirubin: molecular mechanisms. J Leukoc Biol. (2011) 90:997–1015. doi: 10.1189/jlb.0211070
175. Tsai M-T, Tarng D-C. Beyond a measure of liver function—bilirubin acts as a potential cardiovascular protector in chronic kidney disease patients. Int J Mol Sci. (2018) 20:117. doi: 10.3390/ijms20010117
176. Lee H, Choi Y. Regenerative effects of heme oxygenase metabolites on neuroinflammatory diseases. Int J Mol Sci. (2018) 20:78. doi: 10.3390/ijms20010078
177. Montolio M, Biarnés M, Téllez N, Escoriza J, Soler J, Montanya E. Interleukin-1beta and inducible form of nitric oxide synthase expression in early syngeneic islet transplantation. J Endocrinol. (2007) 192:169–77. doi: 10.1677/joe.1.06968
178. Heitmeier MR, Arnush M, Scarim AL, Corbett JA. Pancreatic beta-cell damage mediated by beta-cell production of interleukin-1. A novel mechanism for virus-induced diabetes. J Biol Chem. (2001) 276:11151–8. doi: 10.1074/jbc.M009159200
179. Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. (1999) 13:163–75. doi: 10.1101/gad.13.2.163
180. Minden A, Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim Biophys Acta. (1997) 1333:F85–104. doi: 10.1016/S0304-419X(97)00018-8
181. Zhou L-F, Chen Q-Z, Yang C-T, Fu Z-D, Zhao S-T, Chen Y, et al. TRPC6 contributes to LPS-induced inflammation through ERK1/2 and p38 pathways in bronchial epithelial cells. Am J Physiol Physiol. (2018) 314:C278–88. doi: 10.1152/ajpcell.00117.2017
182. Miralem T, Lerner-Marmarosh N, Gibbs PEM, Tudor C, Hagen FK, Maines MD. The human biliverdin reductase-based peptide fragments and biliverdin regulate protein kinase Cδ activity. J Biol Chem. (2012) 287:24698–712. doi: 10.1074/jbc.M111.326504
183. Clark AS, West KA, Blumberg PM, Dennis PA. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCdelta promotes cellular survival and chemotherapeutic resistance. Cancer Res. (2003) 63:780–6.
184. McCracken MA, Miraglia LJ, McKay RA, Strobl JS. Protein kinase C delta is a prosurvival factor in human breast tumor cell lines. Mol Cancer Ther. (2003) 2:273–81.
185. Gibbs PEM, Lerner-Marmarosh N, Poulin A, Farah E, Maines MD. Human biliverdin reductase-based peptides activate and inhibit glucose uptake through direct interaction with the kinase domain of insulin receptor. FASEB J. (2014) 28:2478–91. doi: 10.1096/fj.13-247015
186. Maines MD, Miralem T, Lerner-Marmarosh N, Shen J, Gibbs PEM. Human biliverdin reductase, a previously unknown activator of protein kinase C βII. J Biol Chem. (2007) 282:8110–22. doi: 10.1074/jbc.M513427200
187. Cimini FA, Arena A, Barchetta I, Tramutola A, Ceccarelli V, Lanzillotta C, et al. Reduced biliverdin reductase-A levels are associated with early alterations of insulin signaling in obesity. Biochim Biophys Acta Mol Basis Dis. (2019) doi: 10.1016/j.bbadis.2019.02.021
188. Wu B, Liu X, Shen J. Old biliverdin reductase: Links to insulin resistance and may be a novel therapeutic target. Med Hypotheses. (2008) 71:73–6. doi: 10.1016/j.mehy.2008.02.007
189. Gibbs PEM, Miralem T, Lerner-Marmarosh N, Maines MD. Nanoparticle delivered human biliverdin reductase-based peptide increases glucose uptake by activating IRK/Akt/GSK3 axis: the peptide is effective in the cell and wild-type and diabetic Ob/Ob mice. J Diabetes Res. (2016) 2016:4712053. doi: 10.1155/2016/4712053
190. Lee SJ, Kang HK, Eum WS, Park J, Choi SY, Kwon HY. Tat-biliverdin reductase A protects INS-1 cells from human islet amyloid polypeptide-induced cytotoxicity by alleviating oxidative stress and ER stress. Cell Biol Int. (2017) 41:514–24. doi: 10.1002/cbin.10750
191. Kumar A, Mastren T, Wang B, Hsieh J-T, Hao G, Sun X. Design of a small-molecule drug conjugate for prostate cancer targeted theranostics. Bioconjug Chem. (2016) 27:1681–9. doi: 10.1021/acs.bioconjchem.6b00222
192. Chari RVJ, Miller ML, Widdison WC. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew Chemie Int Ed. (2014) 53:3796–827. doi: 10.1002/anie.201307628
193. Sinha V, Singh A, Kumar RV, Singh S, Kumria R, Bhinge J. Oral colon-specific drug delivery of protein and peptide drugs. Crit Rev Ther Drug Carrier Syst. (2007) 24:63–92. doi: 10.1615/CritRevTherDrugCarrierSyst.v24.i1.30
194. El-Andaloussi S, Holm T, Langel U. Cell-penetrating peptides: mechanisms and applications. Curr Pharm Des. (2005) 11:3597–611. doi: 10.2174/138161205774580796
195. Allémann E, Leroux J, Gurny R. Polymeric nano- and microparticles for the oral delivery of peptides and peptidomimetics. Adv Drug Deliv Rev. (1998) 34:171–89. doi: 10.1016/S0169-409X(98)00039-8
196. Lee S, Lee Y, Kim H, Lee DY, Jon S. Bilirubin nanoparticle-assisted delivery of a small molecule-drug conjugate for targeted cancer therapy. Biomacromolecules. (2018) 19:2270–7. doi: 10.1021/acs.biomac.8b00189
197. Kim JY, Lee DY, Kang S, Miao W, Kim H, Lee Y, Jon S. Bilirubin nanoparticle preconditioning protects against hepatic ischemia-reperfusion injury. Biomaterials. (2017) 133:1–10. doi: 10.1016/j.biomaterials.2017.04.011
198. Lee Y, Kim H, Kang S, Lee J, Park J, Jon S. Bilirubin nanoparticles as a nanomedicine for anti-inflammation therapy. Angew Chemie Int Ed. (2016) 55:7460–3. doi: 10.1002/anie.201602525
199. Kim DE, Lee Y, Kim M, Lee S, Jon S, Lee S-H. Bilirubin nanoparticles ameliorate allergic lung inflammation in a mouse model of asthma. Biomaterials. (2017) 140:37–44. doi: 10.1016/j.biomaterials.2017.06.014
200. Kim MJ, Lee Y, Jon S, Lee DY. PEGylated bilirubin nanoparticle as an anti-oxidative and anti-inflammatory demulcent in pancreatic islet xenotransplantation. Biomaterials. (2017) 133:242–52. doi: 10.1016/j.biomaterials.2017.04.029
201. Lee Y, Lee S, Jon S. Biotinylated bilirubin nanoparticles as a tumor microenvironment-responsive drug delivery system for targeted cancer therapy. Adv Sci. (2018) 5:1800017. doi: 10.1002/advs.201800017
202. Lee Y, Lee S, Lee DY, Yu B, Miao W, Jon S. Multistimuli-responsive bilirubin nanoparticles for anticancer therapy. Angew Chem Int Ed Engl. (2016) 55:10676–80. doi: 10.1002/anie.201604858
203. Jansen T, Hortmann M, Oelze M, Opitz B, Steven S, Schell R, et al. Conversion of biliverdin to bilirubin by biliverdin reductase contributes to endothelial cell protection by heme oxygenase-1-evidence for direct and indirect antioxidant actions of bilirubin. J Mol Cell Cardiol. (2010) 49:186–95. doi: 10.1016/j.yjmcc.2010.04.011
204. van Dijk R, Aronson SJ, de Waart DR, van de Graaf SF, Duijst S, Seppen J, et al. Biliverdinreductase inhibitors did not improve severe unconjugated hyperbilirubinemia in vivo. Sci Rep. (2017) 7:1646. doi: 10.1038/s41598-017-01602-w
205. Liscovitch M, Lavie Y. Cancer multidrug resistance: a review of recent drug discovery research. IDrugs. (2002) 5:349–55.
Keywords: heme, biliverdin reductases, inflammation, bilirubin, cancer, liver disease
Citation: Canesin G, Hejazi SM, Swanson KD and Wegiel B (2020) Heme-Derived Metabolic Signals Dictate Immune Responses. Front. Immunol. 11:66. doi: 10.3389/fimmu.2020.00066
Received: 01 November 2019; Accepted: 10 January 2020;
Published: 31 January 2020.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceReviewed by:
Stephan Immenschuh, Hannover Medical School, GermanyLibor Vitek, Charles University, Czechia
David Stec, University of Mississippi Medical Center, United States
Copyright © 2020 Canesin, Hejazi, Swanson and Wegiel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Barbara Wegiel, YndlZ2llbCYjeDAwMDQwO2JpZG1jLmhhcnZhcmQuZWR1