 Cai Zhang
Cai Zhang Yuan Hu
Yuan Hu Chongdeng Shi
Chongdeng Shi- Institute of Immunopharmaceutical Sciences, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Shandong University, Jinan, China
Natural killer (NK) cells are important innate cytotoxic lymphocytes with a rapid and efficient capacity to recognize and kill tumor cells. In recent years, adoptive transfer of autologous- or allogeneic-activated NK cells has become a promising cellular therapy for cancer. However, the therapeutic efficiency is encouraging in hematopoietic malignancies, but disappointing in solid tumors, for which the use of NK-cell-based therapies presents considerable challenges. It is difficult for NK cells to traffic to, and infiltrate into, tumor sites. NK cell function, phenotype, activation, and persistence are impaired by the tumor microenvironment, even leading to NK cell dysfunction or exhaustion. Many strategies focusing on improving NK cells' durable persistence, activation, and cytolytic activity, including activation with cytokines or analogs, have been attempted. Modifying them with chimeric antigen receptors further increases the targeting specificity of NK cells. Checkpoint blockades can relieve the exhausted state of NK cells. In this review, we discuss how the cytolytic and effector functions of NK cells are affected by the tumor microenvironment and summarize the various immunotherapeutic strategies based on NK cells. In particular, we discuss recent advances in overcoming the suppressive effect of the tumor microenvironment with the aim of enhancing the clinical outcome in solid tumors treated with NK-cell-based immunotherapy.
Introduction
The important innate cytotoxic lymphocytes, natural killer (NK) cells, show rapid, and efficient cytolytic activity to recognize and kill both virally infected and transformed cells. They exert effector functions relying on germline-encoded receptors for their activation, without the need for prior exposure to the antigen. NK cells possess an extensive repertoire of receptors (activating and inhibitory) that recognize altered protein expression on target cells, thereby controlling the cytolytic function. Uniquely, these receptors can distinguish normal from transformed cells via “missing-self” or “induced self” recognition models (1, 2). The balance between activating and inhibitory receptors tightly governs the inhibition or activation of NK cells, and inhibitory signals from inhibitory receptors usually predominate over activation signals for the maintenance of homeostasis (3, 4). NK-cell-activating receptors recognize stress-induced molecules on target cells to prime NK cell activation. The downregulation of inhibitory ligands and the expression of ligands that activate receptors on tumor cells trigger the activation of NK cells to kill these abnormal cells. Both human and mouse NK cells express a CD16-activating receptor, which mediates antibody-dependent cellular cytotoxicity (ADCC) upon binding to the Fc portion of antibodies. To stimulate responses to target cells, NK-cell-activating receptors, such as NK group 2D (NKG2D), CD226, and the natural cytotoxicity receptors NKp30, NKp44, and NKp46, are considered the most relevant receptors. Once activated, NK cells lyse tumor cells through the release of the cytotoxic molecules perforin and granzyme, upregulating the expression of the Fas ligand and tumor necrosis factor-related apoptosis inducing ligand, and the secretion of cytokines such as interferon gamma (IFN-γ) and TNF alpha (TNF-α).
In addition to their direct cytotoxic potential against target cells, NK cells also exhibit immunoregulatory functions. NK cells contribute to the homeostasis of the immune system and initiate or promote the activation and effector functions of other innate and adaptive immune cells via secretion of cytokines and chemokines, or through direct cell–cell contact (5, 6). NK cells can shape adaptive immune responses through cross-talk with other immune cells such as T cells, B cells, and dendritic cells (DCs). NK cells promote Th1 polarization by producing IFN-γ. They also indirectly enhance the immune responses of adaptive T cells by promoting DC maturation (6, 7). Emerging evidence suggests that NK cells enhance CD8+ T cells function and/or ameliorate their exhaustion (8–10). NK cell function is proven to be critical for the antitumor or antiviral effects of CD8+ T cells, even with checkpoint blockade therapy (11).
Recently, it was found that NK cells have features of adaptive immune cells, particularly the memory-like responses (12–18). A long-lived murine NK cell population had been reported to be able to self-renew and survive in a lymphopenic environment for more than 6 months (19). To date, three types of long-lived memory NK cells have been identified: hepatic-liver-resident NK cells, cytomegalovirus (CMV)-specific NK cells, and cytokine-induced memory-like NK cells (20–22). They usually express high levels of NKG2C and have the capacity for long-term in vivo proliferation and persistence. Upon restimulation with antigens or cytokines, memory-like NK cells undergo clonal-like expansion followed by longevity, self-renewal, and recall responses (13, 23, 24). Recently, the transcription factor interferon regulatory factor 8 has been found to orchestrate the adaptive NK cell response against CMV infection (25). A recent study showed that naive NK cells could be induced to functionally convert into tumor-induced memory-like NK cells by priming using acute myeloid leukemia or pediatric acute B-cell leukemia specimens (14). These tumor-induced memory-like NK cells exhibit certain similarities to cytokine-induced memory-like NK cells and CMV-specific NK cells; however, more importantly, they show significant differences, such as higher tumor-specific cytotoxicity and increased synthesis of perforins, but not IFN-γ secretion. These NK cell adaptive features are promising for the future use of immunotherapy to treat cancers and infective diseases.
NK cells' critical role in immunosurveillance and their powerful antitumor efficacy have prompted their use in many clinical trials to control tumor growth via their effector capacity. However, although the results have been encouraging in hematological malignancies, there has been less success for solid tumors. Indeed, solid tumors present considerable challenges to the application of NK-cell-based therapies. For example, it is difficult for NK cells to traffic and infiltrate into the tumor sites. NK cell function, activation, and phenotype are impaired by the tumor microenvironment, even rendering NK cells dysfunctional or exhausted. Thus, strategies to improve the cytolytic activity, durable persistence, and activation of NK cells have been developed. In the present review, we discuss how the cytolytic and effector functions of NK cells are affected by the tumor microenvironment. We also summarize the various immunotherapeutic strategies based on NK cells, especially the recent attempts to improve NK-cell-based immunotherapy clinical outcomes against solid tumors by overcoming the suppressive effect of the tumor microenvironment.
Effect of the Tumor Microenvironment on NK Cells' Cytolytic Function
NK-cell-based immunotherapies, particularly the adoptive transfer of autologous or allogeneic NK cells, or gene-modified NK cells, have been used widely in clinical trials and have shown great promise for different hematological malignancies (26, 27). However, for patients with solid tumors, the outcomes of adoptive NK cell infusions have been disappointing. There are considerable challenges for NK cell therapy to treat patients with solid tumors. One of the major challenges is the difficulty of NK cells to traffic to the tumor location and infiltrate into the tumor. This poor ability of NK cells to infiltrate into solid tumors limits the clinical outcome of adoptive NK cell infusion. Enhanced infiltration of NK cells into tumor lesions has been associated with good prognosis for patients with diverse types of solid cancer (28, 29).
Another major challenge comes from the tumor microenvironment, which impairs the phenotype, activation, persistence, and function of NK cells. Accumulating data have shown that tumor-infiltrating NK cells exhibit poor cytotoxic capacity, accompanied by downregulation of activating receptors and upregulation of inhibitory receptors, compared with NK cells in non-tumor tissues (4, 30, 31). The tumor microenvironment is a complex network comprising regulatory T cells (Tregs), tumor-associated macrophages (TAMs), regulatory γδT cells, myeloid-derived suppressor cells (MDSCs), soluble factors, the extracellular matrix, and suppressive molecules expressed on tumor cells (32–35). NK cell proliferation and antitumor activity are suppressed by tumor cell secretion of various immunosuppressive factors, including prostaglandin E2, indoleamine 2,3-dioxygenase (IDO), interleukin 10 (IL-10), transforming growth factor-β (TGF-β), and vascular endothelial growth factor. The growth of many types of solid tumors promotes the expansion of immunosuppressive cells, including Tregs, MDSCs, and TAMs. Tumor cells also secrete chemokines, such as C–X–C motif chemokine ligand 8 or C–C motif chemokine ligand 2, to promote Tregs, TAM, or MDSCs accumulation at the tumor sites. By producing TGF-β and IL-10, or via direct cell-to-cell interaction, these immunosuppressive cells inhibit intratumoral NK cell cytotoxicity (36–38). Tregs directly inhibit NK cell cytolytic functions via the production of TGF-β and also reduce the expression of activating receptors NKG2D and natural cytotoxicity triggering receptor 3 (NKp30) through membrane bound TGF-β (39, 40). MDSCs suppress NK cell cytotoxicity and cytokine secretion via membrane-bound TGF-β on MDSCs in a cell-contact-dependent manner or dependent on NKp30 on NK cells (39, 41). Several proinflammatory cytokines have also been reported to contribute to MDSC-mediated NK cell inhibition, resulting in reduced NK cytotoxicity, decreased expression of activating receptor NKGD or NKR, and limited release of IFN-γ (42). These soluble factors include TGF-β, IDO, nitric oxide (NO), and adenosine (38, 43–45). Adenosine is an important immunosuppressive molecule that is released at high levels by MDSCs in response to hypoxia and inflammation of the tumor microenvironment. In tumors sites, high levels of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) and 5′-nucleotidase ecto (CD73) are expressed by MDSCs, which results in markedly increased levels of adenosine secretion. NK cell antitumor activities are inhibited by adenosine via limiting IFN-γ/TNF-α release, inhibiting Fas ligand and perforin-mediated cytotoxic activity and blocking granzyme exocytosis (38, 46). In patients with hepatocellular carcinoma, a substantial proportion of CD11b−CD27−NK subsets with an inactive and immature phenotype was reported to accumulate in tumor tissues and render tumor-infiltrating NK cells less tumoricidal, which was also associated with tumor progression (47). High levels of inhibitory molecules, including programmed death ligand 1 (PD-L1) or PD-L2, are expressed on tumor cells, antigen-presenting cells, immunosuppressive cells, and stromal cells in the tumor microenvironment, thus preventing the activation of NK cells through binding with their respective inhibitory receptors on NK cells, which results in exhaustion and dysfunction of NK cells (48, 49). Among the stromal cells, cancer-associated fibroblasts (CAFs) are the major cells that affect the antitumor capacity of NK cells. CAFs secrete IDO or prostaglandin E2 to decrease NK cell expression of NKG2D, and secrete TGF-β to reduce the expression of NKG2D, NKp30, and NKp44 (50, 51). CAFs also inhibit the killing activity of NK cells by downregulating the level of the poliovirus receptor (poliovirus receptor/CD155) (52).
NK-Cell-Based Tumor Immunotherapy
Cytokine-Activated NK Cells in Tumor Immunotherapy
NK cells' unique capability to distinguish normal from transformed cells, and their rapid and efficient killing capacity, have led to them becoming an attractive option for tumor immunotherapy and have shown great promise recently. The initial attempts comprised adoptive transfer of peripheral blood-derived autologous or allogeneic NK cells or those stimulated and expanded with cytokines, before injection into patients. In most cases, autologous or allogeneic NK cells were stimulated to activate and proliferate using cytokines (IL-2 or IL-15), in the presence of feeder cells genetically engineered to express costimulatory molecules or cytokines (IL-21 and IL-15) in the culture. The NK cells were then transferred into the patient, followed by the administration of IL-2 to maintain the expansion and function of the infused NK cells (20, 26). Encouraging results were achieved using adoptive transfer of haploidentical NK cells or allogeneic NK cells from killer cell immunoglobulin like receptor (KIR) mismatched donors, leading to graft vs. host disease (GVHD) in patients with hematological cancers. However, these approaches are undesirable to treat patients with solid tumors. The safety of adoptive transfer of autologous or allogeneic NK cells has been proved; however, efficacy is limited by their poor proliferation and persistence in vivo and the increase in Tregs related to IL-2 administration (53). Administration of exogenous IL-15 has been shown to improve survival, in vivo persistence, and therapeutic efficacy without inducing the survival and expansion of Tregs (54, 55). A human NKG2D–IL-15 fusion protein could efficiently bind to major histocompatibility complex class I polypeptide-related sequence+ tumor cells and stimulated NK cell activity by transpresentation of IL-15, thus exhibiting higher control efficiency of the growth of xenografted human gastric cancer via the NK cell recruitment and activation (55). In addition, membrane-bound IL-15 expressed on NK cells (mbIL-15 NK) or IL15 gene-modified NK cells have been investigated and displayed the ability to survive and proliferate without exogenous cytokines, as well as superior cytotoxicity against both hematological neoplasms and solid tumors (56–59). Moreover, an IL15-IL15Rα-Sushi-Fc fusion protein (ALT-803), termed a superagonist, in which the IL-15Ra sushi domain is complexed with IL-15, potently enhances NK cell survival and cytotoxic activity compared with that induced by native IL-15. In preclinical studies, ALT-803 enhanced memory CD8+ T cell subpopulations and specific NK expansion, promoted the secretion of IFN-γ, and improved NK cell function in multiple animal models, including B cell lymphoma, glioblastoma, colon cancer, and ovarian cancer (60–62). Phase I or II clinical trial evaluating its safety and efficacy in patients with both hematological neoplasms (e.g., relapsed or refractory multiple myeloma, acute myelogenous leukemia, acute lymphoblastic leukemia, and myelodysplastic syndromes) and solid tumors (NCT01885897, NCT01946789, NCT02099539, NCT03054909) (63, 64) or in combination with NK cell adoptive therapy (NCT02465957, NCT02890758), or with nivolumab (NCT02523469) (65) are currently ongoing. New expansion methods are being exploited to obtain large numbers of NK cells with enhanced cytotoxic activity (66). Notably, pre-activation of human peripheral blood-derived NK cells (PB-NK) using cytokine combinations, such as IL-18, IL-15, and IL-13, were observed to induce human NK cell differentiation into memory-like NK cells (67–69). These human memory-like NK cells expressed NKG2C, NKG2A, and killer cell lectin like receptor D1 (CD94); however, they had reduced expression of inhibitory KIRs. The pre-activated NK cells express the high-affinity IL-2 receptor and thus are more sensitive to low concentrations of IL-2, such that in IL-2 culture, they proliferate more rapidly and NK cell recovery is higher. The peculiar features of cytokine-induced memory-like NK cells are antigen unspecific with increased proliferative capacity, long-term survival, and in vivo persistence, enhanced production of IFN-γ and higher cytotoxicity during ex vivo restimulation. Thus, cytokine-induced memory-like NK cells are becoming an attractive tool for antitumor immunotherapy. Recently, several phase I and II clinical trials of cytokine-induced memory-like NK cells have been performed in patients with relapsed or refractory acute myeloid leukemia (AML) after allogeneic hematopoietic cell transplant (NCT03068819, NCT02782546) or myelodysplastic syndrome (NCT01898793) sponsored by Washington University School of Medicine and have shown robust antitumor capacity (67). In addition to PB-NK, human embryonic stem cells, induced pluripotent stem cells (iPSCs), and bone marrow or umbilical-cord blood are being studied as alternative sources of therapeutic NK cells and are showing great promise (70).
CAR-Modified NK Cells in Tumor Immunotherapy
Recently, the development of chimeric antigen receptor (CAR)-T cells represents a breakthrough in cancer immunotherapy, particularly to treat hematological malignancies. Transfusion of CAR-T cells targeting CD19 to treat relapsed B-cell acute lymphoblastic leukemia ALL (B-ALL) and certain types of relapsed non-Hodgkin's lymphoma obtain as high as 70–90% clinical complete response rates. Thus, CD19-CAR-T cells have been approved by the Food and Drug Administration (FDA) to treat refractory ALL and diffuse large B-cell lymphoma. Currently, multiple CAR-T cells targeting different surface tumor antigens are undergoing clinical development to treat other hematological malignancies and several solid tumors (71, 72). However, there are still a number of obstacles that limit their clinical application. The main problems with CAR-T cell therapy are cytokine release syndrome, neurotoxicity, and on-target/off-tumor effects. To prevent GVHD, CAR-T cells are usually prepared from autologous peripheral blood, which is costly, time consuming, and personalized. NK cells modified with CAR (CAR-NK) are potentially safer than CAR-T cells in that infusion of CAR-NK cells seldom results in cytokine release syndrome because the NK cells secrete mainly restricted levels of granulocyte-macrophage colony-stimulating factor and IFN-γ, while the pro-inflammatory cytokines IL-1 and IL-6 are seldom secreted. The low likelihood of triggering GVHD upon allogeneic infusion means that CAR-NK cells may be prepared as an “off-the-shelf” product and are not restricted to autologous cells. Moreover, natural recognition receptors that recognize stress-induced ligands independent of CARs are retained on CAR-NK cells, including CD226, NKp30, NKp46, NKG2D, and NKp44. NK cells express FcγRIII (CD16) that can mediate ADCC. Therefore, CAR-NK cells are able to induce tumor cell lysis in CAR-dependent and CAR-independent manners, not only further promoting their killing activity but also decreasing the possibility of loss of CAR-targeting antigen-related relapse (73, 74). Intriguingly, activating signaling mediated by CAR is able to overcome the uneducated NK cell hyporesponsiveness, thereby mediating target cell killing. Indeed, CAR-mediated activation is able to overcome the dominant inhibition associated with NKG2A-mediated signaling, thereby enhancing NK cells' antitumor responses (75, 76).
To date, CAR-NK cells are being studied in ongoing clinical trials to evaluate their safety and efficacy for both hematological malignancies and solid tumors, which have shown promising results (Table 1). The main targets for hematological malignancy include CD19, CD22, CD33, and CD7 for relapsed and refractory leukemia and lymphoma (NCT01974479, NCT02742727, NCT02892695, NCT02944162, NCT03056339, NCT03579927, NCT03690310, NCT03692767, and NCT03824951) and B-cell maturation antigen for multiple myeloma (NCT03940833). The targets of current ongoing CAR-NK cells for solid cancer in clinical trials include mucin 1, human epidermal growth factor receptor 2, NKG2D ligands, roundabout guidance receptor 1 (ROBO1), and Mesothelin (NCT02839954, NCT03383978, NCT03415100, NCT03692637, NCT03692663, NCT03931720, NCT03940820, NCT03941457).
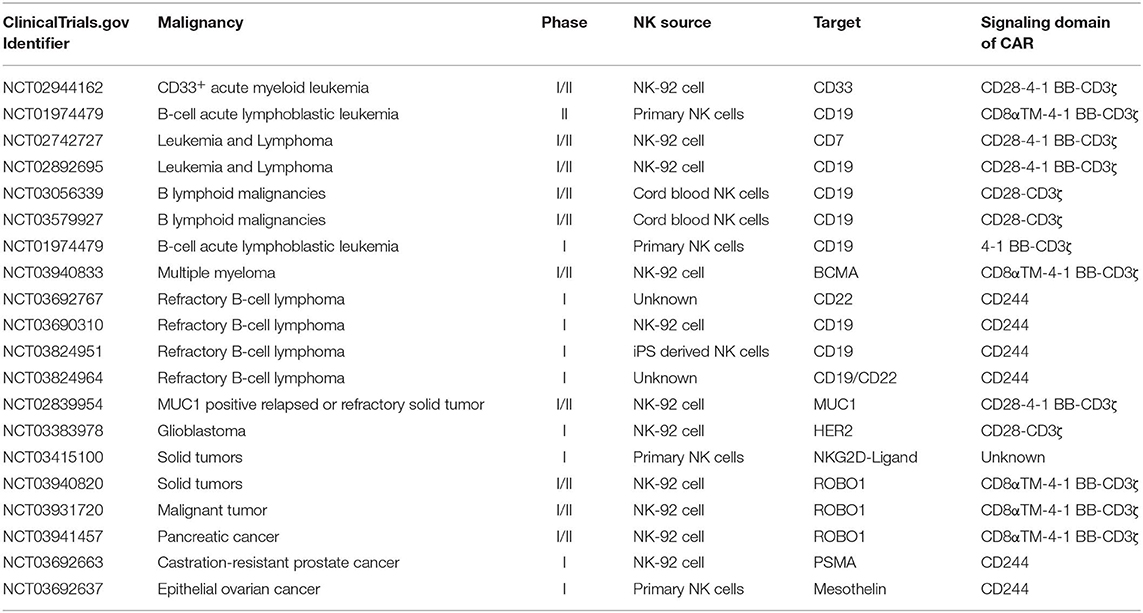
Table 1. Current clinical trials of CAR-NK cells.
The main challenges to the clinical applications of CAR-NK cells include their low transfection efficiency, low in vivo persistence, difficulty to infiltrate into the solid tumor sites, and suppression from the tumor microenvironment. Although the viral transduction technology for NK cells has been improved recently, the transfection efficiency for primary NK cells remains unsatisfactory (77, 78). Non-viral vectors, an alternative gene transfer systems, particularly the Sleeping Beauty (SB) transposon system, could mediate stable transgene expression (79). CAR transgenes have been successfully transfected into T cells via electroporation using the SB transposon system (80, 81). Several phase I clinical trials have been sponsored by MD Anderson Cancer for the treatment of patients with advanced non-Hodgkin's lymphoma, ALL, and chronic lymphocytic leukemia by infusion of SB-modified CD19-specific CAR-T cells following hematopoietic stem cell transplantation (HSCT) (NCT00968760, NCT01362452, NCT01497184, NCT01653717, and NCT02529813). The results identified 84% CAR expression as well as the feasibility and safety of the SB system (81). An UltraCAR-T co-expressing CAR, mbIL15, and a kill switch introduced by SB transposon system using electroporation was approved by FDA for investigational new drug (IND) application to initiate a phase I study for the treatment of patients with advanced platinum-resistant ovarian cancer. The piggyBac transposon system has also been utilized recently to introduce the CAR gene into T or NK cells. Nanoparticle delivery technology has been harnessed to improve the transduction efficiency and augment the antitumor efficacy of immunotherapy (82). Using a T-cell-targeted nanoparticle to deliver the CAR gene, Smith et al. report one potential novel and interesting technology to establish in situ programming of CD19-CAR-T cells to treat leukemia (83). This approach contains several features: anti-CD3e F(ab′)2 fragments targeting T cells were coupled on the surface of nanoparticles; efficient nuclear delivery was achieved by biodegradable polymer functionalized with nuclear localization signals and microtubule-associated sequences; and stable chromosomal integration was facilitated by the piggyBac transposon system. The authors demonstrated that the nanoparticle-reprogrammed CAR-T cells have similar antitumor therapeutic efficacy to conventional lentiviral-based CAR-T therapy in a B-cell ALL mouse model. Moreover, the particle synthesis is relatively simple, the product is stable, and can be lyophilized for extended storage, with low cost (84, 85). The piggyBac transposon system was also used to introduce NKG2D. CAR-presenting vectors were transfected into NK92 cells to generate NKG2D CAR-NK cells, and efficacy was evaluated in a human lung cancer xenograft model. These non-viral engineered NKG2D CAR-NK cells in combination with CD73 blockade showed significant synergistic therapeutic efficacy (86). The SB transposon system to generate CAR-T cells was found to be safe in recent clinical trials (NCT00968760, NCT01362452, NCT01497184, NCT01653717, and NCT02529813). Therefore, it might be feasible to use the SB or piggyBac transposon system combined with nanoparticle delivery technology to establish CAR-NK cells for clinical therapy, although the potential risks and benefits need to be carefully evaluated.
The CAR constructs in most CAR-NK are similar to those in CAR-T cells, except for the unique signaling, and activation features of NK cells. Thus, designing optimized CAR structures suitable for NK cells has been pursued. The NK-cell-activating receptor NKG2D recognizes multiple ligands, such as UL-16-binding proteins, major histocompatibility complex class I chain-related A, and MICB, which are present in increased abundance on the surface of viral-infected cells and many tumor cells. The first NK-cell-based CAR were NKG2D-expressing CAR-NK cells containing a NKG2D–DNAX activation protein 10 (DAP10)–CD3ζ construct, which have shown encouraging results in vitro and in a mouse model of osteosarcoma (87). This NKG2D CAR can recognize ~90% of human tumor types that express NKG2D ligands. Notably, several studies have demonstrated that NKG2D ligands are expressed on immunosuppressive cells, such as MDSCs and Tregs; therefore, NKG2D-expressing CAR-NK cells not only directly kill NKG2D+ tumor cells but also could eradicate immunosuppressive cells in the tumor environment (88). Other NK-activating receptors (NKp30 and DNAM-1)-based CAR could be designed using a similar strategy, having advantage that one CAR could target several of tumor types that express the corresponding ligands. Prostate stem cell antigen (PSCA)-DAP12 CAR, targeting the prostate stem cell antigen, was expressed in primary NK cells and the YTS-NK cell line and improved the specific cytotoxicity compared with PSCA-CD3ζ-based CAR NK cells (89). Interestingly, no additional costimulatory signaling molecules for in vitro cytotoxicity and activation were required by PSCA-DAP12 CAR. A panel of NK-cell-specific CAR constructs comprising various combinations of NK-cell-specific activating domains were screened for NK cell activation recently (90). The results showed that for optimal antigen-specific NK cell signaling and NK-cell-mediated cytotoxicity, the intracellular 2B4 domain, the CD3ζ signaling domain, and the transmembrane NKG2D are necessary. Based on these CAR constructs, mesothelin-targeted CAR-NK cells were constructed using human-induced pluripotent stem cells, and the antitumor activity was examined in vitro and using a murine ovarian cancer xenograft model. The NK-specific CAR-NK cells exhibited a markedly augmented cytolytic capacity, a significantly lower tumor burden, and prolonged survival compared with a traditional T-cell CAR construct expressing CAR-NK cells (90). Notably, the in vivo expansion and survival of the NK cells was improved as a result of the NK-cell-specific CAR-mediated signaling.
To increase the specificity, efficacy, and safety of CAR-NK cells, as well as to overcome suppression by the tumor microenvironment, the strategies and experience from CAR-T studies could be applied to CAR-NK research. The targets used for CAR-NK research in preclinical studies include CD19, CD20, CD138, CD5, CD2 subset 1 (CS1), NKG2D ligand, glucosylceramidase beta (GD2), HER-2, epidermal growth factor receptor (EGFR), EGFRvIII, epithelial cell adhesion molecule 1, glypican 3, and guanine nucleotide-binding protein alpha-7 (91). For example, IL15 was introduced into CAR design to increase CAR-NK cells' antitumor capacity and persistence (92). CAR-NK cells can be engineered with chemokine receptors or chemokines to enhance NK cell homing and infiltrating to tumor sites.
The sources of NK cells include PB-NK cells, umbilical cord blood NK cells, and the NK cell line NK-92. However, the use of PB-NK cells is limited because primary NK cells are generally hard to genetically modify; therefore, the transduction efficacy is very low, even with viral vectors, and in the adoptive transfer setting, the allogeneic NK cells can survive for only a few weeks, limiting their antitumor efficacy (93). Recently, stem-cell-derived NK cells, such as those induced from human embryonic stem cells or iPSCs, have showed promise as candidates to develop off-the-shelf adoptive NK cell therapy. They have a similar phenotype and function to primary NK cells with an efficient capacity to kill both hematological malignancies and solid tumor cells. More importantly, they are homogenous, reproducible, and amenable to genetic modification, and can be easily expanded on the clinical scale (70, 94–96).
Notably, mesothelin-targeted CAR-NK cells prepared from human iPSCs showed significantly inhibited tumor growth, prolonged survival, and markedly augmented cytotoxicity in a xenograft model of ovarian cancer (90). Thus, iPSC-derived CAR-NK cells could be a promising resource to generate standardized, off-the-shelf, and clinical-scale adoptive immunotherapeutic products. Moreover, human iPSC-NK cells could be genetically modified to further improve their in vivo persistence, overcome the immunosuppressive tumor microenvironment, and enhance their antitumor capacity. iPSC-NK cells could also be harnessed in combination with other therapies (such as checkpoint blockade). Clinical trials treating cancer with human iPSC-NK cells are currently under IND application. The safety concerns for iPSC-NK cells include possible immune responses against allogenic iPSCs and their potential for malignant transformation. HLA-E-expressing or HLA knockout together with overexpression of CD47 hypoimmunogenic iPSCs have been generated recently to evade immune rejection by host cells (97, 98).
Antibody-Based NK Cell Therapy
ADCC of NK Cells Mediated by Tumor-Specific Antibodies
One of the main killing mechanism for antitumor effects of NK cells is ADCC, which involves the binding of the Fc portion of an antibody (Ab) to CD16 on NK cells. Most tumor-targeting Ab drugs exert their antitumor effect by promoting NK-cell ADCC. Studies have tried to increase the affinity of Abs to Fc receptors on NK cells to enhance ADCC by modifying Abs via mutagenesis or glycosylation (99). Expression high-affinity CD16 FcγRIII in NK cells is also a strategy used to enhance ADCC-mediated antitumor effects. To enhance NK-mediated ADCC using Cetuximab, Trastuzumab, and Pertuzumab, IL-2 and the high-affinity CD16 FcγRIIIa (158V) allele were engineered into NK-92 cells (named haNK) (100). Phase I and II clinical trials of haNK, alone, or in combination with anti-PD-L1 Ab (avelumab), a cancer vaccine, or super-IL-15, are ongoing for the treatment of triple negative breast cancer, squamous cell carcinoma, Merkel cell carcinoma, pancreatic cancer, and other types of cancers (NCT03027128, NCT03387085, NCT03387111, NCT03853317, and NCT03586869). CAR-modified haNK (also named target-activated NK-92) cells targeting CD19 and PD-L1 are currently under investigation and have recently been approved for IND by the FDA.
Potentiation of NK Cell Activity Using Bi- or Trispecific Killer Engagers
Molecules comprising single-chain variable fragments against activating receptors on NK cells and tumor-associated antigen, termed bi- or trispecific killer cell engagers (BiKEs or TriKEs), have been developed to create an immune connection between NK and tumor cells, thus promoting NK-mediated killing of tumor cells. Several NK-based BiKEs or TriKEs are currently in preclinical and clinical development. CD16-directed BiKEs CD16 × 19 and CD16 × 33 and TriKE CD16 × 19 × 22 were shown to specifically stimulate NK cell activation via CD16, which triggers NK cell cytolytic activity and cytokine secretion to fight lymphoma and leukemia (101, 102). A combination comprising an ADAM metallopeptidase domain 17 inhibitor further enhanced the function of NK cells by preventing CD16 shedding from the membrane of NK cells (102). Multiple other CD16-directed BiKEs, such as CD16 × Erb-B2 receptor tyrosine kinase 2, CD16 × human epidermal growth factor receptor 2/neu, CD16 × EGFR, CD16 × carcinoembryonic antigen, and CD16 × EpCAM, are under investigation (103). A bi-specific Ab-recognizing CS1-NKG2D, which comprises an anti-NKG2D scFv and an anti-CS1 scFv, showed enhanced cytotoxicity and cytokine production from NK cells and significantly prolonged their survival in a multiple myeloma xenograft NOD-SCIDIL2γc−/− (NSG) mouse model (104). A novel CD30/CD16A tetravalent bispecific Ab (AFM13), which has a longer half-life compared with that of other bi-/trispecific Abs, is currently under evaluation in a phase II clinical trial to treat patients with relapsed/refractory Hodgkin's lymphomas (NCT03192202) (105, 106).
To augment the in vivo expansion and survival of NK cells, a TriKE was constructed having a modified IL-15 cross-linker (107, 108). CD16 × IL-15 × CD33 displayed markedly enhanced NK cytotoxicity against AML and better cell persistence in vivo compared with those of BiKEs. Importantly, it is expected to target both malignant cells and CD33+ MDSCs, thus exerting more efficient antitumor effect (107). Currently, CD16 × IL-15 × CD33 TriKE is being evaluated in phase I and II clinical trials in patients with advanced systemic mastocytosis, refractory/relapsed acute myeloid leukemia, or CD33-expressing high-risk myelodysplastic syndromes (NCT03214666). CD16 × IL-15 × Epcam TriKE, CD16 × IL-15 × CD133 TriKEs, and CD16 × IL-15 × CD133 × Epcam TetraKE are under investigation to treat colorectal cancer and have shown significantly enhanced cytokine secretion, NK cell proliferation, and lytic degranulation (108–110). More recently, trifunctional NK cell engagers targeting two activating receptors, NKp46 and CD16, on NK cells and a tumor antigen (CD19, CD20, or EGFR) on cancer cells were generated by Prof. Vivier and his colleagues (111). By cotargeting NKp46 and CD16, which led to full NK cell activation, the trifunctional NK cell engagers showed more efficient promotion of NK cell activation and cytotoxicity in vitro and more effective control of tumor growth in mouse models of solid and invasive tumors compared with those of current clinically available therapeutic antibodies. Remarkably, TriKEs represent a cost-effective and versatile platform for the incorporation of novel targeting molecules and could potentially boost NK cell function. For example, introducing Abs against checkpoint receptors or TGF-β to construct TriKEs might enhance NK cell efficacy by reversing the immunosuppression from the tumor microenvironment (TME) (112).
Releasing the Inhibition on NK Cells by Targeting the Immune Checkpoints
Recently, monoclonal antibodies (mAbs) targeting immune checkpoint ligands or receptors have been applied to markedly enhance the tumor-suppressed T-cell immune response, leading to the improved control of several types of cancer. Similarly, a variety of immune checkpoints receptors are expressed in NK cells, including KIRs, NKG2A, T-cell immunoglobulin- and mucin-domain-containing molecule 3, programmed cell death 1 (PD-1), lymphocyte activation gene-3, and T-cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domains (TIGIT). These inhibitory receptors are often induced or upregulated on tumor-infiltrating NK cells and affect the antitumor function of NK cells upon interaction with their respective ligands, even leading to dysfunction or exhaustion of NK cells (113). Functionally exhausted NK cells show less proliferation, reduced cytolytic activity, and downregulated cytokine secretion, thus losing the ability to attack tumor cells. Therapeutic blockade of the immune checkpoint receptors or ligands with mAbs can restore the antitumor function of NK cells, which have shown great promise for NK-cell-based tumor immunotherapy (114).
The first attempt was blockade of HLA-KIR interactions to ameliorate NK cell inhibition and enhance NK cell activity using the anti-KIR antibody, IPH2101, to treat leukemia, lymphoma, and multiple myeloma, which demonstrated efficacy and safety (115). The second generation, fully human immunoglobulin G4 anti-KIR antibody, IPH2102, was well-tolerated in patients with hematological malignancies and solid tumors (116). Anti-KIR antibodies combined with other immunotherapies, for example, anti-PD-1 and anticytotoxic T-lymphocyte-associated protein 4 Abs, to treat solid tumors are currently ongoing (NCT03203876, NCT03347123).
In various cancers, such as cervical cancer, breast cancer, hepatocellular carcinoma, and lung cancer, high NKG2A expression was identified inn tumor-infiltrating NK and T cells, which contributes to exhaustion of these cells and predicts a poor prognosis (49). Therapeutic blockade of NKG2A could improve NK cell dysfunction in both hematological malignancies and solid tumors (117, 118). In particular, a humanized anti-NKG2A Ab, Monalizumab (IPH2201), was reported to augment NK cell activity against certain types of tumor cell (B-cell lymphoma, solid tumors, and T-cell lymphoma) and rescue the effector and activation functions CD8+ T and NK cells, especially in combination with PD-L1 blockade (118). A phase I and II clinical trial of IPH2201 and Cetuximab, with or without anti-PD-L1 Abs, in patients with human papillomavirus (+) and (–) recurrent or metastatic squamous cell carcinoma of the head and neck is ongoing (NCT02643550). Its safety was evaluated as well-tolerated for the combination therapy. The interim results of the phase II trial for treatment efficacy showed that combination therapy comprising IPH2201 and Cetuximab resulted in 31% partial response, 54% stable disease, and 11% progressive disease (118). Therefore, anti-NKG2A mAbs are proposed as promising checkpoint inhibitors that could enhance antitumor immunity via unleashing the potential of both T and NK cells (113, 119).
Tumor-infiltrating NK cells from patients with various cancers induce PD-1, which causes inducing functional failure of activated NK cells, which is associated with poor prognosis (120, 121). Blocking PD-1/PD-L1 signaling markedly increased the cytotoxicity and cytokine production of NK cells and significantly suppressed tumor growth in vivo (121). Several antibodies targeting PD-1 and PD-L1 have been used in clinical practice, and several others are under investigation to treat different solid and hematological malignancies (122). Various combination therapies of anti-PD-1/PD-L1 Abs with other checkpoint blockades, antiangiogenic bevacizumab, or chemotherapy have been explored recently in clinical trials. A current phase II clinical trial is assessing the effect of Pembrolizumab (an anti-PD-1 Ab) to induce changes to NK cell exhaustion and function in patients with unresectable melanoma at stage III or stage IV (NCT03241927).
Inhibitory receptors TIGIT and CD96 are regarded as new checkpoint receptors for NK cells and T cells. TIGIT competes with CD226 (DNAM-1) (an NK activating receptor) for the same set of ligands CD112 [Poliovirus receptor-related 2 (Herpesvirus entry mediator B)] and CD155 (poliovirus receptor), while CD96 shares binding of CD155 with CD226 and TIGIT but also binds to CD111 to directly inhibit NK cell function. Similar to the CD28/cytotoxic T-lymphocyte-associated protein 4 pathway, TIGIT and CD96, together with CD226, form a pathway in which CD226 functions as a costimulatory receptor, whereas TIGIT and CD96 act as coinhibitory receptors (123). The balance among the three receptors fine tunes the immune response against tumors. In patients with cancer, TIGIT and CD96 are upregulated in tumor-associated NK cells and promote NK cell functional exhaustion, accompanied by poor cytolytic potential and impaired cytokine production (124, 125). Blockade of TIGIT with Abs could effectively reverse NK cell exhaustion and enhance NK-cell-dependent antitumor immune responses in several tumor-bearing mouse models. Intriguingly, blockade of TIGIT could further promote tumor-specific T-cell immune responses and improve memory responses to tumor rechallenge (11). Blocking CD96–CD155 interaction could reverse NK- and T-cell exhaustion, and restore both NK- and T-cell-mediated antitumor immunity (125, 126). Recently, IL-1 receptor 8 long isoform (also known as single immunoglobulin IL-1R-related receptor) was identified as an NK cell checkpoint protein that regulates NK cell maturation and antitumor activity. Genetic blockade of IL-1 receptor 8 long isoform induces NK-cell-mediated resistance in hepatic carcinogenesis and liver or lung metastasis (127). Currently, the safety and efficacy of anti-TIGIT Ab, alone or in combination with anti-PD-1 or anti-PD-L1 Abs, are being evaluated in phase I and II clinical trials in patients with locally advanced or metastatic solid tumors, e.g., renal cell carcinoma, non-small cell lung cancer, breast cancer, squamous cell carcinoma of the head and neck, melanoma, and colorectal cancer (NCT03119428, NCT03563716, NCT03628677). Blocking Abs for other checkpoint proteins, including T-cell immunoglobulin- and mucin-domain-containing molecule 3 and lymphocyte activation gene-3, alone or in combination with other therapeutic approaches, are also currently in progress in patients in clinical trials or are under investigation for therapy of both hematological neoplasms and various solid tumors (e.g., NCT02060188, NCT02817633, NCT03307785, NCT03680508, NCT03250832, NCT03489369, NCT03625323, NCT03662659). Current clinical trials of checkpoint blockade antibodies to enhance NK cell antitumor efficacy are summarized in Table 2.
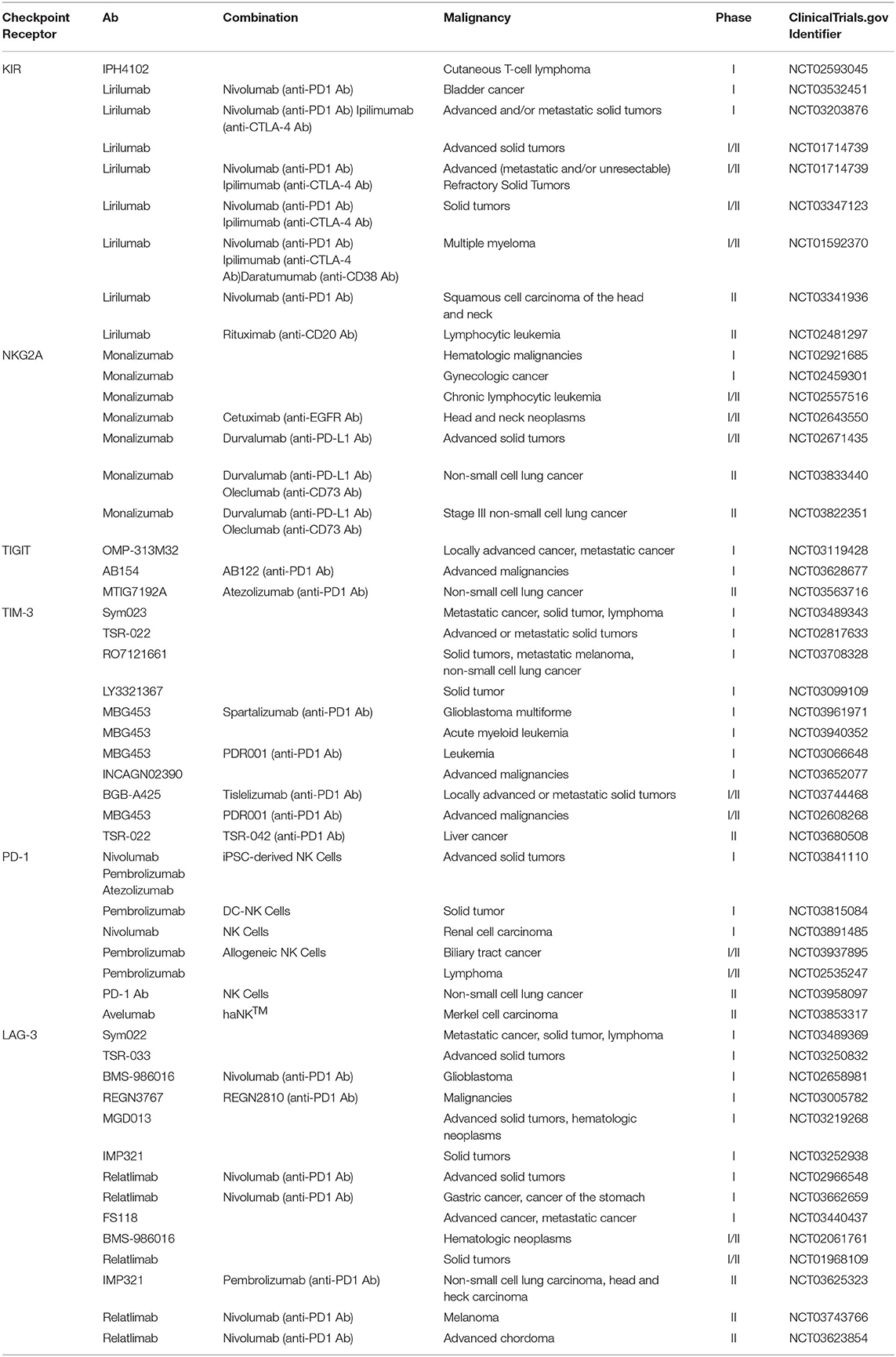
Table 2. Current clinical trials of checkpoint blockade molecules.
Other Strategies to Overcome the Suppression by the Tumor Microenvironment
The TME is a major obstacle for ensuring the optimum antitumor activity of NK cells, in which immunosuppressive cells and molecules limit NK cell activity. In addition, to overcome the suppression from inhibitory receptors or ligands by checkpoint blockade, neutralizing, or blocking suppressive cytokines secreted by tumor cells, immunosuppressive cells, or stromal cells in the TME is another key strategy. One of the most important immunosuppressive cytokine is TGF-β, which is secreted by tumor cells, Tregs, MDSCs, and other stromal cells in the TME to hamper the antitumor immune response. NK cell antitumor function is inhibited by TGF-β-mediated downregulation of the expression of NK-activating receptors NKp30 and NKG2D and also reduces the expression of NKG2D ligands on tumor cells, thus suppressing NK-mediated cytotoxic capacity and IFN-γ secretion (103). TGF-β also affects the development and differentiation of human NK cell subsets (128). Blockade of TGF-β signaling or neutralization of TGF-β could prevent NKG2D downregulation and restore the antitumor function of NK cells. Therapies that interrupt TGF-β signaling to enhance NK cell antitumor capacity are currently in clinical trials or are under investigation. The safety and efficacy of human TGF-β neutralizing mAb Fresolimumab (GC1008) and TGFβR1 inhibitor Galunisertib (LY2157299) have been evaluated in a phase I clinical trial to treat patients with advanced malignant melanoma, renal cell carcinoma, and other advanced solid tumors, and have obtained acceptable tolerability and safety results (NCT00356460, NCT01722825). Importantly, Galunisertib therapy could restore NKG2D and NKp30 expression on activated NK cells and enhanced NK cell cytotoxicity. Knockdown of TGFBR2 or SMAD3 in NK cells, engineering CB NK cells to express TGF-β dominant negative receptor II, or modifying NK cells using CAR containing TGF-β type II receptor extracellular and transmembrane domains, and the intracellular domain of NKG2D, are all under investigation and have shown great promise for the recovery of tumor-suppressed NK cell antitumor activity to treat patients with solid tumors (129–131).
Conclusion and Perspectives
Although there are still some challenges that limit the widespread use of NK-cell-based therapies, particular for solid tumors, advances in ex vivo expansion and activation technologies, genetic modification, and nanoparticle delivery technology will lead to novel therapeutic strategies to overcome the immune suppression from the TME of solid tumors, indicating that that NK cell therapy is achievable and promises to become a powerful method to treat cancers. In certain respects, NK cells have unique advantages over conventional T cells (73, 132). Notably, the use of NK cells could result in off-the-shelf allogeneic products to treat patients, thereby eliminating the necessity for personalized and patient-specific products in current CAR-T cell therapies. NK-cell-based therapy provides an alternative or complementary immunotherapy approach to T-cell therapy. It is worth exploring the utility of the selective expansion of suitable NK cell subsets and their delivery to the corresponding type of tumor. The longer lifespan of memory-like NK cells compared with that of PB-derived NK cells may make them ideal sources for NK cell therapy. Recently, CMV-induced adaptive memory NK cells were demonstrated as relatively resistant to Tregs and MDSCs' suppressive effects, which might have important clinical implications (133, 134). Remarkable benefits might be achieved by engineering the CAR protein into memory-like NK cells or a specific NK cell subset. In fact, CD19-specific CAR-engineered NKG2C+CD57+ adaptive NK cells showed an enhanced ability to kill CD19+ tumor cells compared with that of other NK subsets (75). Different combinations of multiple strategies could play an increasingly prominent role in clinical practice. For example, the use of checkpoint blockade to overcome the immunosuppressive effect within the TME could increase the endogenous NK cell antitumor ability and the function of adoptive transferred NK cells. Therefore, it is reasonable to combine checkpoint inhibition with adoptive transfer of allogeneic NK cells or CAR-transduced NK cells. Checkpoint receptor blockade in conjunction with BiKEs or TriKEs could increase antigen specificity and reverse immunosuppression, thus further enhancing the antitumor responses of NK cells.
Author Contributions
CZ and YH designed and conceived the review. YH and CS performed the literature search and analysis. CZ wrote the initial draft of the manuscript together with YH and CS. All authors revised and approved the final submitted version of the manuscript.
Funding
The National Natural Science Foundation of China (91842305, 81771686), the National Major Science and Technology Project for Control and Prevention of Major Infectious Diseases in China (2018ZX10301401), and the Shandong Provincial Key Research and Development Program (Major Scientific and Technological Innovation Project) (2019JZZY021013).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript.
References
1. Hilton HG, Parham P. Missing or altered self: human NK cell receptors that recognize HLA-C. Immunogenetics. (2017) 69:567–79. doi: 10.1007/s00251-017-1001-y
2. Komori HK, Meehan TF, Havran WL. Epithelial and mucosal gamma delta T cells. Curr Opin Immunol. (2006) 18:534–8. doi: 10.1016/j.coi.2006.06.001
3. Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. (2015) 15:243–54. doi: 10.1038/nri3799
4. Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. (2015) 12:292–302. doi: 10.1038/cmi.2014.91
5. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
6. Zhang C, Zhang J, Tian Z. The regulatory effect of natural killer cells: do “NK-reg cells” exist? Cell Mol Immunol. (2006) 3:241–54.
7. Moretta A, Marcenaro E, Parolini S, Ferlazzo G, Moretta L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. (2008) 15:226–33. doi: 10.1038/sj.cdd.4402170
8. Zheng M, Sun R, Wei H, Tian Z. NK cells help induce anti-hepatitis B virus CD8+ T cell immunity in mice. J Immunol. (2016) 196:4122–31. doi: 10.4049/jimmunol.1500846
9. Liu Y, Zheng J, Liu Y, Wen L, Huang L, Xiang Z, et al. Uncompromised NK cell activation is essential for virus-specific CTL activity during acute influenza virus infection. Cell Mol Immunol. (2018) 15:827–37. doi: 10.1038/cmi.2017.10
10. Tian Z, Gershwin ME, Zhang C. Regulatory NK cells in autoimmune disease. J Autoimmun. (2012) 39:206–15. doi: 10.1016/j.jaut.2012.05.006
11. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
12. O'Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity. (2015) 43:634–45. doi: 10.1016/j.immuni.2015.09.013
13. Rapp M, Wiedemann GM, Sun JC. Memory responses of innate lymphocytes and parallels with T cells. Semin Immunopathol. (2018) 40:343–55. doi: 10.1007/s00281-018-0686-9
14. Pal M, Schwab L, Yermakova A, Mace EM, Claus R, Krahl AC, et al. Tumor-priming converts NK cells to memory-like NK cells. Oncoimmunology. (2017) 6:e1317411. doi: 10.1080/2162402X.2017.1317411
15. Wu LS, Wang JY. Warm up, cool down, and tearing apart in NK cell memory. Cell Mol Immunol. (2018) 15:1095–7. doi: 10.1038/s41423-018-0188-7
16. O'Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. (2006) 7:507–16. doi: 10.1038/ni1332
17. Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol. (2010) 11:1127–35. doi: 10.1038/ni.1953
18. Nikzad R, Angelo LS, Aviles-Padilla K, Le DT, Singh VK, Bimler L, et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci Immunol. (2019) 4:eaat8116. doi: 10.1126/sciimmunol.aat8116
19. Sun JC, Beilke JN, Bezman NA, Lanier LL. Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J Exp Med. (2011) 208:357–68. doi: 10.1084/jem.20100479
20. Hu Y, Tian ZG, Zhang C. Natural killer cell-based immunotherapy for cancer: advances and prospects. Engineering. (2019) 5:106–14. doi: 10.1016/j.eng.2018.11.015
21. Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. (2013) 132:536–44. doi: 10.1016/j.jaci.2013.07.006
22. Beaulieu AM. Memory responses by natural killer cells. J Leukoc Biol. (2018) 104:1087–96. doi: 10.1002/JLB.1RI0917-366R
23. Geary CD, Sun JC. Memory responses of natural killer cells. Semin Immunol. (2017) 31:11–9. doi: 10.1016/j.smim.2017.08.012
24. Peng H, Tian Z. Natural killer cell memory: progress and implications. Front Immunol. (2017) 8:1143. doi: 10.3389/fimmu.2017.01143
25. Adams NM, Lau CM, Fan X, Rapp M, Geary CD, Weizman OE, et al. Transcription factor IRF8 orchestrates the adaptive natural killer cell response. Immunity. (2018) 48:1172–82.e6. doi: 10.1016/j.immuni.2018.04.018
26. Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. (2017) 31:37–54. doi: 10.1016/j.smim.2017.07.009
27. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. (2013) 10:230–52. doi: 10.1038/cmi.2013.10
28. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. (2013) 73:3499–510. doi: 10.1158/0008-5472.CAN-13-0371
29. Gras Navarro A, Bjorklund AT, Chekenya M. Therapeutic potential and challenges of natural killer cells in treatment of solid tumors. Front Immunol. (2015) 6:202. doi: 10.3389/fimmu.2015.00202
30. Sun C, Sun HY, Xiao WH, Zhang C, Tian ZG. Natural killer cell dysfunction in hepatocellular carcinoma and NK cell-based immunotherapy. Acta Pharmacol Sin. (2015) 36:1191–9. doi: 10.1038/aps.2015.41
31. Habif G, Crinier A, Andre P, Vivier E, Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
32. Zou W. Mechanistic insights into cancer immunity and immunotherapy. Cell Mol Immunol. (2018) 15:419–20. doi: 10.1038/s41423-018-0011-5
33. Yasinska IM, Goncalves Silva I, Sakhnevych S, Gibbs BF, Raap U, Fasler-Kan E, et al. Biochemical mechanisms implemented by human acute myeloid leukemia cells to suppress host immune surveillance. Cell Mol Immunol. (2018) 15:989–91. doi: 10.1038/s41423-018-0047-6
34. Nie X, Chen W, Zhu Y, Huang B, Yu W, Wu Z, et al. B7-DC (PD-L2) costimulation of CD4+ T-helper 1 response via RGMb. Cell Mol Immunol. (2018) 15:888–97. doi: 10.1038/cmi.2017.17
35. Chen H, He W. Human regulatory gammadeltaT cells and their functional plasticity in the tumor microenvironment. Cell Mol Immunol. (2018) 15:411–3. doi: 10.1038/cmi.2017.73
36. Trzonkowski P, Szmit E, Mysliwska J, Dobyszuk A, Mysliwski A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clin Immunol. (2004) 112:258–67. doi: 10.1016/S1521-6616(04)00119-6
37. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
38. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
39. Hasmim M, Messai Y, Ziani L, Thiery J, Bouhris JH, Noman MZ, et al. Critical role of tumor microenvironment in shaping NK cell functions: implication of hypoxic stress. Front Immunol. (2015) 6:482. doi: 10.3389/fimmu.2015.00482
40. Castriconi R, Cantoni C, Della Chiesa M, Vitale M, Marcenaro E, Conte R, et al. Transforming growth factor beta 1 inhibits expression of NKp30 and NKG2D receptors: consequences for the NK-mediated killing of dendritic cells. Proc Natl Acad Sci U.S.A. (2003) 100:4120–5. doi: 10.1073/pnas.0730640100
41. Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. (2009) 50:799–807. doi: 10.1002/hep.23054
42. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: roles in tumor progression. Front Immunol. (2019) 10:771. doi: 10.3389/fimmu.2019.00771
43. Zhang J, Han X, Hu X, Jin F, Gao Z, Yin L, et al. IDO1 impairs NK cell cytotoxicity by decreasing NKG2D/NKG2DLs via promoting miR-18a. Mol Immunol. (2018) 103:144–55. doi: 10.1016/j.molimm.2018.09.011
44. Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric oxide production by myeloid-derived suppressor cells plays a role in impairing Fc receptor-mediated natural killer cell function. Clin Cancer Res. (2018) 24:1891–904. doi: 10.1158/1078-0432.CCR-17-0691
45. Zhai L, Ladomersky E, Lenzen A, Nguyen B, Patel R, Lauing KL, et al. IDO1 in cancer: a Gemini of immune checkpoints. Cell Mol Immunol. (2018) 15:447–57. doi: 10.1038/cmi.2017.143
46. Chiu DK, Tse AP, Xu IM, Di Cui J, Lai RK, Li LL, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. (2017) 8:517. doi: 10.1038/s41467-017-00530-7
47. Zhang QF, Yin WW, Xia Y, Yi YY, He QF, Wang X, et al. Liver-infiltrating CD11b(-)CD27(-) NK subsets account for NK-cell dysfunction in patients with hepatocellular carcinoma and are associated with tumor progression. Cell Mol Immunol. (2017) 14:819–29. doi: 10.1038/cmi.2016.28
49. Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunology. (2017) 6:e1264562. doi: 10.1080/2162402X.2016.1264562
50. Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U.S.A. (2009) 106:20847–52. doi: 10.1073/pnas.0906481106
51. Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. (2012) 318:154–61. doi: 10.1016/j.canlet.2011.12.020
52. Inoue T, Adachi K, Kawana K, Taguchi A, Nagamatsu T, Fujimoto A, et al. Cancer-associated fibroblast suppresses killing activity of natural killer cells through downregulation of poliovirus receptor (PVR/CD155), a ligand of activating NK receptor. Int J Oncol. (2016) 49:1297–304. doi: 10.3892/ijo.2016.3631
53. Fang F, Xiao W, Tian Z. Challenges of NK cell-based immunotherapy in the new era. Front Med. (2018) 12:440–50. doi: 10.1007/s11684-018-0653-9
54. Pillet AH, Theze J, Rose T. Interleukin (IL)-2 and IL-15 have different effects on human natural killer lymphocytes. Hum Immunol. (2011) 72:1013–7. doi: 10.1016/j.humimm.2011.07.311
55. Chen Y, Chen B, Yang T, Xiao W, Qian L, Ding Y, et al. Human fused NKG2D-IL-15 protein controls xenografted human gastric cancer through the recruitment and activation of NK cells. Cell Mol Immunol. (2017) 14:293–307. doi: 10.1038/cmi.2015.81
56. Jiang W, Zhang C, Tian Z, Zhang J. hIL-15-gene modified human natural killer cells (NKL-IL15) exhibit anti-human leukemia functions. J Cancer Res Clin Oncol. (2018) 144:1279–88. doi: 10.1007/s00432-018-2654-0
57. Jiang W, Zhang C, Tian Z, Zhang J. hIL-15 gene-modified human natural killer cells (NKL-IL15) augments the anti-human hepatocellular carcinoma effect in vivo. Immunobiology. (2014) 219:547–53. doi: 10.1016/j.imbio.2014.03.007
58. Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SM, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. (2014) 124:1081–8. doi: 10.1182/blood-2014-02-556837
59. Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol. (2017) 8:930. doi: 10.3389/fimmu.2017.00930
60. Rosario M, Liu B, Kong L, Collins LI, Schneider SE, Chen X, et al. The IL-15-based ALT-803 complex enhances FcgammaRIIIa-triggered NK cell responses and in vivo clearance of B cell lymphomas. Clin Cancer Res. (2016) 22:596–608. doi: 10.1158/1078-0432.CCR-15-1419
61. Mathios D, Park CK, Marcus WD, Alter S, Rhode PR, Jeng EK, et al. Therapeutic administration of IL-15 superagonist complex ALT-803 leads to long-term survival and durable antitumor immune response in a murine glioblastoma model. Int J Cancer. (2016) 138:187–94. doi: 10.1002/ijc.29686
62. Felices M, Chu S, Kodal B, Bendzick L, Ryan C, Lenvik AJ, et al. IL-15 super-agonist (ALT-803) enhances natural killer (NK) cell function against ovarian cancer. Gynecol Oncol. (2017) 145:453–61. doi: 10.1016/j.ygyno.2017.02.028
63. Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. (2018) 131:2515–27. doi: 10.1182/blood-2017-12-823757
64. Margolin K, Morishima C, Velcheti V, Miller JS, Lee SM, Silk AW, et al. Phase I trial of ALT-803, a novel recombinant IL15 complex, in patients with advanced solid tumors. Clin Cancer Res. (2018) 24:5552–61. doi: 10.1158/1078-0432.CCR-18-0945
65. Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. (2018) 19:694–704. doi: 10.1016/S1470-2045(18)30148-7
66. Sakamoto N, Ishikawa T, Kokura S, Okayama T, Oka K, Ideno M, et al. Phase I clinical trial of autologous NK cell therapy using novel expansion method in patients with advanced digestive cancer. J Transl Med. (2015) 13:277. doi: 10.1186/s12967-015-0632-8
67. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8:357ra123. doi: 10.1126/scitranslmed.aaf2341
68. Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood. (2012) 120:4751–60. doi: 10.1182/blood-2012-04-419283
69. Pahl JHW, Cerwenka A, Ni J. Memory-Like NK Cells: Remembering a previous activation by cytokines and NK cell receptors. Front Immunol. (2018) 9:2796. doi: 10.3389/fimmu.2018.02796
70. Wang K, Han Y, Cho WC, Zhu H. The rise of human stem cell-derived natural killer cells for cancer immunotherapy. Expert Opin Biol Ther. (2019) 19:141–8. doi: 10.1080/14712598.2019.1559293
71. Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. (2017) 27:38–58. doi: 10.1038/cr.2016.154
72. Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
73. Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. (2014) 3:e28147. doi: 10.4161/onci.28147
74. Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. (2018) 39:167–76. doi: 10.1038/aps.2017.125
75. Oei VYS, Siernicka M, Graczyk-Jarzynka A, Hoel HJ, Yang W, Palacios D, et al. Intrinsic functional potential of NK-cell subsets constrains retargeting driven by chimeric antigen receptors. Cancer Immunol Res. (2018) 6:467–80. doi: 10.1158/2326-6066.CIR-17-0207
76. Zhuang X, Long EO. CD28 homolog is a strong activator of natural killer cells for lysis of B7H7+ tumor cells. Cancer Immunol Res. (2019) 7:939–51. doi: 10.1158/2326-6066.CIR-18-0733
77. Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. (2015) 6:266. doi: 10.3389/fimmu.2015.00266
78. Kellner JN, Cruz CR, Bollard CM, Yvon ES. Gene modification of human natural killer cells using a retroviral vector. Methods Mol Biol. (2016) 1441:203–13. doi: 10.1007/978-1-4939-3684-7_17
79. Kebriaei P, Izsvak Z, Narayanavari SA, Singh H, Ivics Z. Gene therapy with the sleeping beauty transposon system. Trends Genet. (2017) 33:852–70. doi: 10.1016/j.tig.2017.08.008
80. Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. (2017) 31:186–94. doi: 10.1038/leu.2016.180
81. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest. (2016) 126:3363–76. doi: 10.1172/JCI86721
82. Sengupta M, Pal R, Nath A, Chakraborty B, Singh LM, Das B, et al. Anticancer efficacy of noble metal nanoparticles relies on reprogramming tumor-associated macrophages through redox pathways and pro-inflammatory cytokine cascades. Cell Mol Immunol. (2018) 15:1088–90. doi: 10.1038/s41423-018-0046-7
83. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotechnol. (2017) 12:813–20. doi: 10.1038/nnano.2017.57
84. Miller MA. Nanoparticles improve economic mileage for CARs. Sci Transl Med. (2017) 9:eaan2784. doi: 10.1126/scitranslmed.aan2784
85. Olweus J. Manufacture of CAR-T cells in the body. Nat Biotechnol. (2017) 35:520–1. doi: 10.1038/nbt.3898
86. Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. (2018) 6:136. doi: 10.1186/s40425-018-0441-8
87. Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. (2013) 73:1777–86. doi: 10.1158/0008-5472.CAN-12-3558
88. Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. (2019) 7:363–75. doi: 10.1158/2326-6066.CIR-18-0572
89. Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. (2015) 194:3201–12. doi: 10.4049/jimmunol.1400330
90. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92.e5. doi: 10.1016/j.stem.2018.06.002
91. Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. (2017) 25:1769–81. doi: 10.1016/j.ymthe.2017.06.012
92. Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. (2018) 32:520–31. doi: 10.1038/leu.2017.226
93. Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. (2018) 2018:4054815. doi: 10.1155/2018/4054815
94. Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. (2009) 113:6094–101. doi: 10.1182/blood-2008-06-165225
95. Knorr DA, Bock A, Brentjens RJ, Kaufman DS. Engineered human embryonic stem cell-derived lymphocytes to study in vivo trafficking and immunotherapy. Stem Cells Dev. (2013) 22:1861–9. doi: 10.1089/scd.2012.0608
96. Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. (2013) 2:274–83. doi: 10.5966/sctm.2012-0084
97. Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol. (2017) 35:765–72. doi: 10.1038/nbt.3860
98. Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol. (2019) 37:252–8. doi: 10.1038/s41587-019-0016-3
99. Koerner SP, Andre MC, Leibold JS, Kousis PC, Kubler A, Pal M, et al. An Fc-optimized CD133 antibody for induction of NK cell reactivity against myeloid leukemia. Leukemia. (2017) 31:459–69. doi: 10.1038/leu.2016.194
100. Jochems C, Hodge JW, Fantini M, Fujii R, Morillon YM 2nd, Greiner JW, et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget. (2016) 7:86359–73. doi: 10.18632/oncotarget.13411
101. Gleason MK, Verneris MR, Todhunter DA, Zhang B, McCullar V, Zhou SX, et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol Cancer Ther. (2012) 11:2674–84. doi: 10.1158/1535-7163.MCT-12-0692
102. Wiernik A, Foley B, Zhang B, Verneris MR, Warlick E, Gleason MK, et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin Cancer Res. (2013) 19:3844–55. doi: 10.1158/1078-0432.CCR-13-0505
103. Nayyar G, Chu Y, Cairo MS. Overcoming resistance to natural killer cell based immunotherapies for solid tumors. Front Oncol. (2019) 9:51. doi: 10.3389/fonc.2019.00051
104. Chan WK, Kang S, Youssef Y, Glankler EN, Barrett ER, Carter AM, et al. A CS1-NKG2D bispecific antibody collectively activates cytolytic immune cells against multiple myeloma. Cancer Immunol Res. (2018) 6:776–87. doi: 10.1158/2326-6066.CIR-17-0649
105. Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. (2015) 125:4024–31. doi: 10.1182/blood-2014-12-614636
106. Pahl JHW, Koch J, Gotz JJ, Arnold A, Reusch U, Gantke T, et al. CD16A activation of NK cells promotes NK cell proliferation and memory-like cytotoxicity against cancer cells. Cancer Immunol Res. (2018) 6:517–27. doi: 10.1158/2326-6066.CIR-17-0550
107. Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 trispecific killer engagers (TriKE) make natural killer cells specific to CD33+ targets while also inducing persistence, in vivo expansion, and enhanced function. Clin Cancer Res. (2016) 22:3440–50. doi: 10.1158/1078-0432.CCR-15-2710
108. Schmohl JU, Felices M, Taras E, Miller JS, Vallera DA. Enhanced ADCC and NK cell activation of an anticarcinoma bispecific antibody by genetic insertion of a modified IL-15 cross-linker. Mol Ther. (2016) 24:1312–22. doi: 10.1038/mt.2016.88
109. Schmohl JU, Felices M, Oh F, Lenvik AJ, Lebeau AM, Panyam J, et al. Engineering of anti-CD133 trispecific molecule capable of inducing NK expansion and driving antibody-dependent cell-mediated cytotoxicity. Cancer Res Treat. (2017) 49:1140–52. doi: 10.4143/crt.2016.491
110. Schmohl JU, Felices M, Todhunter D, Taras E, Miller JS, Vallera DA. Tetraspecific scFv construct provides NK cell mediated ADCC and self-sustaining stimuli via insertion of IL-15 as a cross-linker. Oncotarget. (2016) 7:73830–44. doi: 10.18632/oncotarget.12073
111. Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional natural killer cell engagers targeting NKp46 trigger protective tumor immunity. Cell. (2019) 177:1701–13 e16. doi: 10.1016/j.cell.2019.04.041
112. Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. (2017) 31:64–75. doi: 10.1016/j.smim.2017.07.011
113. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
114. Kim N, Kim HS. Targeting checkpoint receptors and molecules for therapeutic modulation of natural killer cells. Front Immunol. (2018) 9:2041. doi: 10.3389/fimmu.2018.02041
115. Benson DM Jr., Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, et al. A phase I trial of the anti-KIR antibody IPH2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin Cancer Res. (2015) 21:4055–61. doi: 10.1158/1078-0432.CCR-15-0304
116. Vey N, Karlin L, Sadot-Lebouvier S, Broussais F, Berton-Rigaud D, Rey J, et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget. (2018) 9:17675–88. doi: 10.18632/oncotarget.24832
117. McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ, et al. Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology. (2016) 5:e1226720. doi: 10.1080/2162402X.2016.1226720
118. Andre P, Denis C, Soulas C, Bourbon-Caillet C, Lopez J, Arnoux T, et al. Anti-NKG2A mAb is a checkpoint inhibitor that promotes anti-tumor immunity by unleashing both T and NK cells. Cell. (2018) 175:1731–43.e13. doi: 10.1016/j.cell.2018.10.014
119. Mingari MC, Pietra G, Moretta L. Immune checkpoint inhibitors: anti-NKG2A antibodies on board. Trends Immunol. (2019) 40:83–5. doi: 10.1016/j.it.2018.12.009
120. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vely F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
121. Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, et al. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. (2017) 36:6143–53. doi: 10.1038/onc.2017.209
122. Pesce S, Greppi M, Grossi F, Del Zotto G, Moretta L, Sivori S, et al. PD/1-PD-Ls checkpoint: insight on the potential role of NK cells. Front Immunol. (2019) 10:1242. doi: 10.3389/fimmu.2019.01242
123. Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. doi: 10.1111/imr.12518
124. Zhou XM, Li WQ, Wu YH, Han L, Cao XG, Yang XM, et al. Intrinsic expression of immune checkpoint molecule TIGIT could help tumor growth in vivo by suppressing the function of NK and CD8+ T cells. Front Immunol. (2018) 9:2821. doi: 10.3389/fimmu.2018.02821
125. Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology. (2019) 70:168–83. doi: 10.1002/hep.30347
126. Mittal D, Lepletier A, Madore J, Aguilera AR, Stannard K, Blake SJ, et al. CD96 is an immune checkpoint that regulates CD8+ T-cell antitumor function. Cancer Immunol Res. (2019) 7:559–71. doi: 10.1158/2326-6066.CIR-18-0637
127. Molgora M, Bonavita E, Ponzetta A, Riva F, Barbagallo M, Jaillon S, et al. IL-1R8 is a checkpoint in NK cells regulating anti-tumour and anti-viral activity. Nature. (2017) 551(7678):110–4. doi: 10.1038/nature24293
128. Allan DS, Rybalov B, Awong G, Zuniga-Pflucker JC, Kopcow HD, Carlyle JR, et al. TGF-beta affects development and differentiation of human natural killer cell subsets. Eur J Immunol. (2010) 40:2289–95. doi: 10.1002/eji.200939910
129. Wang QM, Tang PM, Lian GY, Li C, Li J, Huang XR, et al. Enhanced cancer Immunotherapy with Smad3-silenced NK-92 cells. Cancer Immunol Res. (2018) 6:965–77. doi: 10.1158/2326-6066.CIR-17-0491
130. Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-beta receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. (2017) 19:408–18. doi: 10.1016/j.jcyt.2016.12.005
131. Wang Z, Guo L, Song Y, Zhang Y, Lin D, Hu B, et al. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-betaR II and NKG2D. Cancer Immunol Immunother. (2017) 66:537–48. doi: 10.1007/s00262-017-1959-1
132. Stojanovic A, Cerwenka A. Checkpoint inhibition: NK cells enter the scene. Nat Immunol. (2018) 19:650–2. doi: 10.1038/s41590-018-0142-y
133. Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. (2016) 76:5696–706. doi: 10.1158/0008-5472.CAN-16-0839
Keywords: natural killer cells, tumor microenvironment, cytokine, CAR-NK, checkpoint blockade, tumor immunotherapy
Citation: Zhang C, Hu Y and Shi C (2020) Targeting Natural Killer Cells for Tumor Immunotherapy. Front. Immunol. 11:60. doi: 10.3389/fimmu.2020.00060
Received: 14 July 2019; Accepted: 10 January 2020;
Published: 19 February 2020.
Edited by:
Daniel Olive, Faculté de Médecine, Aix Marseille Université, FranceReviewed by:
Rui Sun, University of Science and Technology of China, ChinaWilliam K. Decker, Baylor College of Medicine, United States
Copyright © 2020 Zhang, Hu and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cai Zhang, Y2FpemhhbmdzZCYjeDAwMDQwO3NkdS5lZHUuY24=