 A. Leoni Swart
A. Leoni Swart Hubert Hilbi
Hubert Hilbi- Faculty of Medicine, Institute of Medical Microbiology, University of Zürich, Zurich, Switzerland
Legionella pneumophila is the causative agent of a severe pneumonia called Legionnaires' disease. The environmental bacterium replicates in free-living amoebae as well as in lung macrophages in a distinct compartment, the Legionella-containing vacuole (LCV). The LCV communicates with a number of cellular vesicle trafficking pathways and is formed by a plethora of secreted bacterial effector proteins, which target host cell proteins and lipids. Phosphoinositide (PI) lipids are pivotal determinants of organelle identity, membrane dynamics and vesicle trafficking. Accordingly, eukaryotic cells tightly regulate the production, turnover, interconversion, and localization of PI lipids. L. pneumophila modulates the PI pattern in infected cells for its own benefit by (i) recruiting PI-decorated vesicles, (ii) producing effectors acting as PI interactors, phosphatases, kinases or phospholipases, and (iii) subverting host PI metabolizing enzymes. The PI conversion from PtdIns(3)P to PtdIns(4)P represents a decisive step during LCV maturation. In this review, we summarize recent progress on elucidating the strategies, by which L. pneumophila subverts host PI lipids to promote LCV formation and intracellular replication.
Legionella Pneumophila—An Amoebae-Resistant Environmental Bacterium
Legionella spp. are obligate aerobic, Gram-negative bacteria, which are ubiquitously found in technical and natural water systems, where they colonize different niches (1, 2). The facultative intracellular bacteria replicate in planktonic form as well as in biofilms (3–5), and they infect environmental predators such as nematodes (6–9) and protozoa (10–12). Complex, ecologically relevant interactions take place in the aquatic niches inhabited by Legionella spp.; e.g., nematode larvae rupture Legionella-infected amoebae and thus are exposed to a highly virulent form of the bacterial pathogen (9).
Upon inhalation of contaminated water droplets, Legionella bacteria reach the lung, where they replicate in and destroy alveolar macrophages, thus causing a potentially fatal pneumonia termed Legionnaires' disease (2). The clinically most relevant and best studied species is Legionella pneumophila; yet, Legionella longbeachae is prevalent in some parts of the world, too (13). The spread of Legionella spp. predominantly occurs through environmental sources; however, a probable person-to-person transmission of L. pneumophila, resulting in the death of the two people involved, was recently reported (14).
Legionella pneumophila replicates intracellularly in amoebae and macrophages by exploiting evolutionarily conserved pathways (15, 16). The pathogen forms a unique, degradation-resistant compartment, the Legionella-containing vacuole (LCV), wherein which bacterial replication takes place. The LCV does neither acidify nor fuse with lysosomes, but communicates with several vesicle trafficking pathways including the endosomal, secretory, and retrograde routes (17–21). At later steps of pathogen vacuole maturation, the LCV tightly and continuously associates with the endoplasmic reticulum (ER). Small GTPases of the Arf (22, 23), Rab (24, 25), Ran (26), and Rap (27) families regulate LCV formation and intracellular replication of L. pneumophila. Moreover, large GTPases implicated in eukaryotic membrane fusion and fission play a role in L. pneumophila infection. Atlastin3 (Atl3/Sey1), an ER tubule-resident large GTPase that catalyzes homotypic ER fusions, promotes ER remodeling around LCVs, pathogen vacuole expansion and intracellular bacterial replication (28). Dynamin1-like GTPase (Dnm1l), a mitochondrial large GTPase, mediates L. pneumophila-induced mitochondrial fragmentation and inhibition of host cell respiration (29).
LCV formation requires the Icm/Dot (intracellular multiplication/defective organelle trafficking) type IVB secretion system (T4SS), which is conserved among Legionella spp., and in the case of L. pneumophila translocates more than 300 different “effector” proteins into host cells (30, 31). In eukaryotic cells, the effector proteins subvert essential process such as signal transduction, cytoskeleton dynamics and membrane trafficking (17, 32–37). Distinct effector proteins have been shown to target the small GTPases Arf1 (22), Rab1 (38–41) or Ran (26, 42), the retromer coat complex (43–46), the vacuolar H+-ATPase (47), the autophagy machinery (48–50), or phosphoinositide (PI) lipids (35, 51, 52). Here, we focus on how L. pneumophila subverts host PI lipids to promote LCV formation and intracellular replication.
Phosphoinositide lipids—Regulators of Organelle Identity and Membrane Dynamics
Phosphoinositides are minor constituents of eukaryotic membranes (<10% of all phospholipids), but this low abundance class of lipids exert pivotal functions for cellular organelle identity, membrane dynamics and vesicle trafficking (53–56). Accordingly, the production, turnover, interconversion, and subcellular localization of PI lipids are tightly regulated by eukaryotic cells. The core compound of PI lipids is phosphatidylinositol (PtdIns), comprising a diacylglycerol (DAG) moiety and a D-myo-inositol 1-phosphate head group facing the cytoplasmic side of membranes (Figure 1). PtdIns can be reversibly phosphorylated at the positions 3, 4, and/or 5 of the inositol ring, giving rise to seven different mono- or poly-phosphorylated derivatives (53–56). These reactions are catalyzed by organelle-specific PI metabolizing enzymes (PI kinases and PI phosphatases), the activity of which controls compartmentalization and vesicle trafficking within the cell (57, 58).
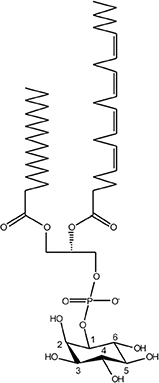
Figure 1. Chemical structure of phosphoinositide lipids. The core moiety of phosphoinositide (PI) lipids is phosphatidylinositol (PtdIns), comprising diacylglycerol (DAG), and D-myo-inositol 1-phosphate. The inositol head group is reversibly phosphorylated by organelle-specific PI kinases and PI phosphatases at the positions 3, 4, and/or 5, giving rise to seven different mono- or poly-phosphorylated derivatives.
PI lipids, jointly with small GTPases in their active GTP-bound form, recruit peripheral membrane proteins harboring distinct PI-binding motifs, such as the PH, PX, FYVE, ENTH/ANTH, or FERM domains (59). Hence, lipid-protein co-incidence detection, along with specific adaptor proteins, determines organelle identity and vesicle trafficking routes in eukaryotic cells (54, 60). PI-metabolizing enzymes are usually recruited to the cytoplasmic side of cellular membranes by small GTPases; e.g., the endosomal small GTPase Rab5 recruits and activates the class III phosphatidylinositol 3-kinase (PI3K) to produce PtdIns(3)P from PtdIns (61). The small GTPases themselves are localized and activated by specific guanine nucleotide exchange factors (GEFs), which concomitantly displace the guanine nucleotide dissociation inhibitor (GDI) protein from the small GTPase, thus allowing the membrane association of the GTPase. To switch off the signal, the inactivation of small GTPases is catalyzed by specific GTPase activating proteins (GAPs) (61).
The different PIs preferentially localize to distinct subcellular compartments and pathways [(53, 54, 62); Figure 2]. Accordingly, PtdIns(4)P and in particular PtdIns(4,5)P2 are enriched at the plasma membrane, where PtdIns(3,4,5)P3 and PtdIns(3,4)P2 transiently accumulate upon signal transduction events and during phagocytosis. PtdIns(3)P is the “signpost” PI lipid of the endocytic pathway, and is enriched on phagosomes and early endosomes, as well as on autophagosomes and multivesicular bodies, which like late endosomes and lysosomes are also decorated with PtdIns(3,5)P2. PtdIns(4)P is the hallmark PI lipid of the secretory pathway and predominantly localizes to the Golgi apparatus and secretory vesicles (53, 54, 56, 62). This PI lipid is formed from PtdIns on the ER and together with PtdIns(3)P also regulates phagosome-lysosome fusion (63).
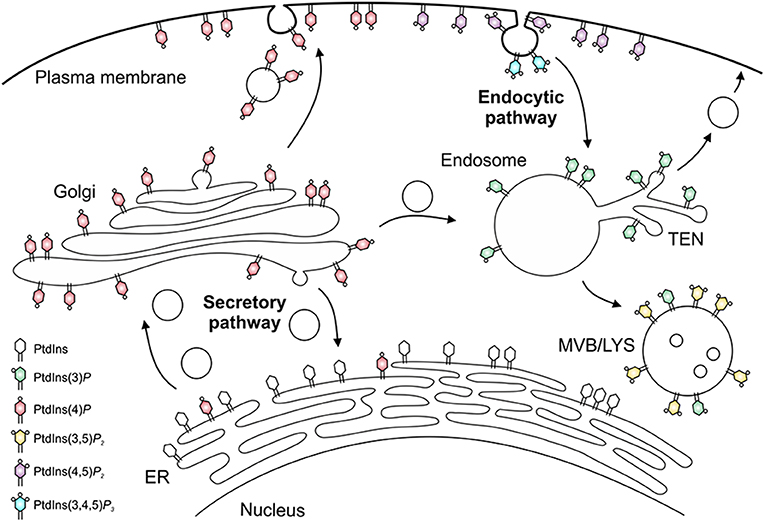
Figure 2. Subcellular distribution of phosphoinositides. The subcellular distribution of phosphoinositide lipids is primarily arranged around the cellular dichotomy of the secretory and endocytic vesicle trafficking pathways. In the secretory pathway, PtdIns(4)P is synthesized in the endoplasmic reticulum (ER) and Golgi apparatus, localizes to secretory vesicles, and finally accumulates at the plasma membrane, where it is converted to PtdIns(4,5)P2 and, transiently, to PtdIns(3,4,5)P3. In the endocytic pathway, PtdIns(3)P decorates early endosomes and the tubular endosomal network (TEN), and is converted to PtdIns(3,5)P2 on multivesicular bodies (MVB), late endosomes and lysosomes (LYS).
On certain compartments and along some vesicle trafficking pathways, distinct PIs are functionally coupled, i.e., the product of a given PI-metabolizing enzyme is the substrate of a subsequent modification. This occurs, e.g., in the endocytic pathway, where PtdIns(3)P is phosphorylated to yield PtdIns(3,5)P2, as well as in the secretory pathway, where PtdIns(4)P serves as the precursor of PtdIns(4,5)P2 at the plasma membrane. In turn, PtdIns(4,5)P2 is phosphorylated by class I PI3K to transiently yield PtdIns(3,4,5)P3 during phagocytosis.
Eukaryotic PI Kinases Implicated in Uptake and Endocytosis of L. Pneumophila
PtdIns(3,4,5)P3 and PtdIns(3)P are produced by class I or class III PI3Ks and are major regulators of phagocytosis or the endocytic pathway, respectively. Using the haploid social soil amoeba Dictyostelium discoideum, genetic and pharmacological disruption of class I PI3Ks indicated that these kinases are largely dispensable for uptake of wild-type L. pneumophila, but required for uptake of an icm/dot mutant strain (51, 64). Moreover, using D. discoideum producing a fluorescent probe for PtdIns(3,4,5)P3, live-cell microscopy revealed that this PI lipid accumulated at bacterial entry sites and was cleared within approximately 40 s after uptake, regardless of whether the amoebae were infected with wild-type or icm/dot mutant L. pneumophila. In parallel, plasma membrane PtdIns(4,5)P2 disappeared from the uptake sites (65).
Similar to amoebae, the uptake of L. pneumophila wild-type, but not the icm/dot mutant strain by replication-permissive human U937 macrophage-like cells was not affected by the class I PI3K inhibitor wortmannin (66, 67). In contrast, wortmannin or LY294002 inhibited the uptake of wild-type as well as icm/dot mutant L. pneumophila by non-permissive murine J774A.1 macrophages (64, 66, 67). The Icm/Dot T4SS controls the uptake of L. pneumophila by phagocytes (68, 69); however, no effectors implicated in the process have been identified. These results suggest that during uptake of L. pneumophila class I PI3Ks are activated and the pathogen evades/inhibits downstream processes in an Icm/Dot-dependent manner to form the replication-permissive compartment.
Dictyostelium discoideum mutant strains were also used to examine the role of endosomal PI kinases, PI phosphatases and phospholipases for intracellular growth of L. pneumophila. Wild-type L. pneumophila replicated more efficiently in D. discoideum lacking two or five class I PI3Ks (51, 64) or in amoebae lacking PIKfyve (70), a PI 5-kinase, which is recruited through its FYVE domain to early endosomes, where it phosphorylates PtdIns(3)P to yield PtdIns(3,5)P2. While it is not clear how lower levels of PtdIns(3,4,5)P3 promote the intracellular replication of L. pneumophila, the reduction of PtdIns(3,5)P2 impairs the bactericidal endocytic pathway, which restricts bacterial killing and thus benefits the pathogen (70). The disruption of D. discoideum PTEN (phosphatase and tensin homolog), a PI phosphatase antagonizing PI3Ks, reduces the uptake of L. pneumophila but does not affect intracellular growth (64). Finally, the inhibition of D. discoideum PLC (Phospholipase C), a hydrolase cleaving PI(4,5)P2 to yield DAG and inositol 1,4,5-phosphate (IP3), also abolishes the uptake of L. pneumophila, but again has no effect on bacterial replication (64).
Phosphoinositide Conversion on the Legionella-Containing Vacuole
PtdIns(3)P accumulates on LCVs within 1 min after uptake, regardless of whether the vacuole contains wild-type or icm/dot mutant L. pneumophila (71). However, while phagosomes containing icm/dot mutant bacteria remain decorated with PtdIns(3)P, more than 80% of wild-type LCVs gradually lose this PI within 2 h. Concomitantly, major membrane rearrangements take place with PtdIns(3)P-positive membranes being segregated from the LCV and compacted at the cell center. PtdIns(4)P, on the other hand, transiently localizes to early phagosomes harboring wild-type or icm/dot mutant L. pneumophila, but is cleared within minutes after uptake. During the following 2 h, PtdIns(4)P steadily accumulates only on wild-type LCVs, which for at least 8 h maintain a discrete PtdIns(4)P identity spatially separated from the calnexin-positive ER. PtdIns(4)P decorates the LCV for a prolonged time (18 h p. i. and beyond) up to when the bacteria exit from the pathogen vacuole and the infected cell (71). Taken together, within 2 h post-infection, the LCV undergoes a PI conversion, replacing the endosomal PtdIns(3)P with the secretory PtdIns(4)P (Figure 3). Importantly, the LCV PI conversion occurs prior to and independently from ER recruitment, and the two compartments appear to remain separate throughout the intracellular life of L. pneumophila.
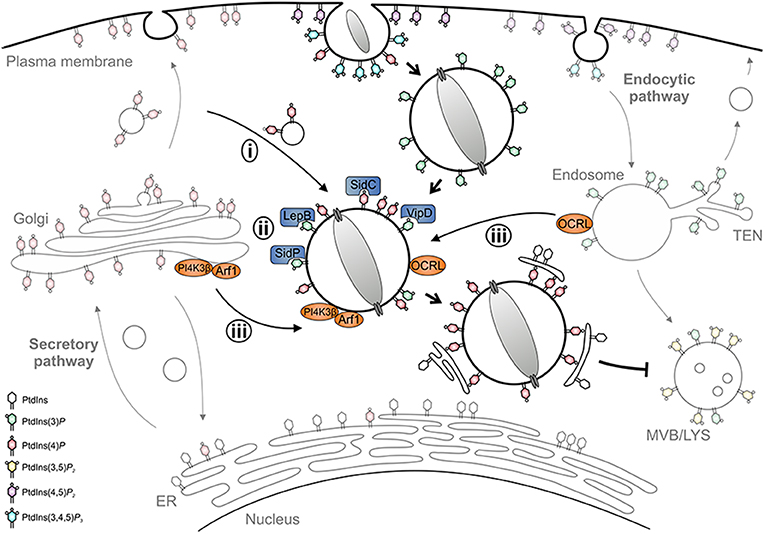
Figure 3. LCV formation and phosphoinositide conversion. The Legionella-containing vacuole (LCV) is a replication-permissive compartment disconnected from the bactericidal endocytic pathway and tightly associated with the ER. LCV formation is governed by a PI conversion from endosomal PtdIns(3)P to secretory PtdIns(4)P. L. pneumophila subverts the LCV PI pattern (i) by recruiting and selectively retaining PI-decorated vesicles, (ii) by producing effectors acting as PI interactors (SidC), kinases (LepB), phosphatases (SidP), or phospholipases (VipD), and (iii) by subverting host PI kinases (PI4KIIIβ) and phosphatases (OCRL).
Mechanistically, the PI conversion on the LCV possibly proceeds along several, mutually non-exclusive pathways: (i) the LCV might communicate and selectively retain PI-decorated vesicles, (ii) L. pneumophila might produce (Icm/Dot-secreted) effectors acting directly as PI interactors, phosphatases or kinases, and/or (iii) the pathogen might subvert host PI metabolizing enzymes (Figure 3). Indeed, using D. discoideum producing fluorescent PtdIns(3)P and PtdIns(4)P probes in tandem, we recently showed by high-resolution real-time confocal laser scanning microscopy that nascent LCVs continuously capture and accumulate PtdIns(4)P-positive vesicles derived from the trans-Golgi network (72). The sustained association of the PtdIns(4)P-positive vesicles, but not the LCV-vesicle interactions per se, require a functional T4SS. Thus, L. pneumophila exploits the cellular dynamics of vesicle-bound PtdIns(4)P for LCV formation. At different stages of infection L. pneumophila effectors might modulate the host PI pattern in different ways (73).
As outlined below in detail, L. pneumophila Icm/Dot-translocated effector proteins subvert PI lipids (i) by directly binding PIs (SidC, SidM, RidL, LtpM), (ii) by acting as bacterial PI phosphatases (SidF, SidP), PI kinases (LepB, LegA5), or phospholipases (VipD, PlcC, LpdA), or (iii) by recruiting eukaryotic PI phosphatases or kinases (RalF, SidM). Currently, no effector has been described, which directly modulates the activity of host PI-metabolizing enzyme. In general, L. pneumophila effectors determining the LCV PI pattern might act either in cis (on the LCV membrane) or in trans (in a distance from the LCV). In fact, a number of these effectors have been shown to act in cis, in agreement with their exceptional affinity for specific PI receptors (40, 74–76).
Phosphoinositide Anchors for L. Pneumophila Effectors
Legionella pneumophila Icm/Dot substrates translocated to the cytoplasmic face of the LCV can bind to the pathogen vacuole as peripheral membrane protein [e.g., RalF; (77, 78)], as intrinsic membrane protein [e.g., MavN; (79, 80)], through host cell prenylation of a C-terminal CAAX motif [e.g., LegG1, AnkB, LpdA; (81–83)], or through PI lipids [e.g., SidC, SidM, RidL, LtpM; (44, 84, 85); Figure 4]. PI lipids bind a plethora of eukaryotic proteins through distinct domains (59), none of which was identified in L. pneumophila effector proteins. However, L. pneumophila produces a battery of effector proteins, which bind through novel domains to PtdIns(4)P (SidC, SdcA, SidM, Lpg1101, Lpg2603, AnkX, LidA) and/or PtdIns(3)P (LepB, RidL, SetA, LtpD, LtpM, RavD, RavZ, AnkX, LidA) (Table 1).
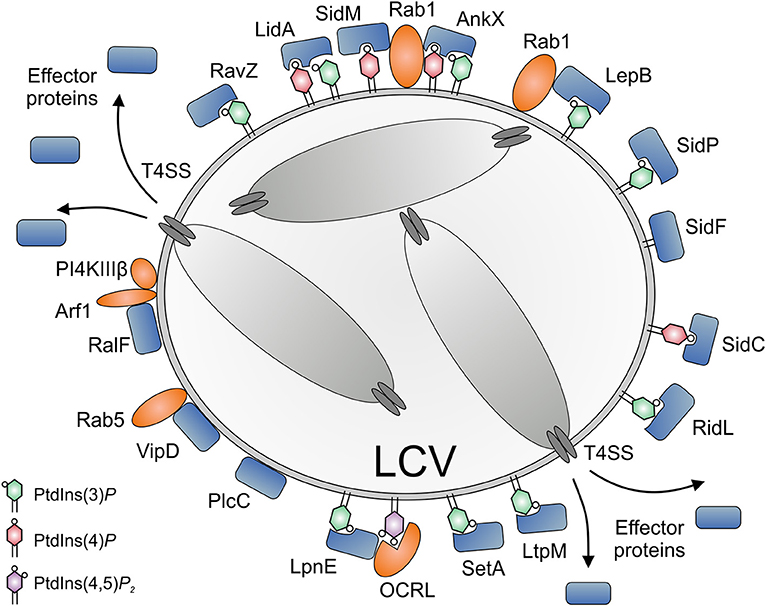
Figure 4. Subversion of host PI lipids by L. pneumophila effector proteins. L. pneumophila effector proteins translocated by the Icm/Dot T4SS subvert PI lipids on the Legionella-containing vacuole (LCV) (i) by directly binding PIs (SidC, SidM, AnkX, LidA, RidL, SetA, LtpM), (ii) by acting as bacterial PI phosphatases (SidF, SidP), PI kinases (LepB, LegA5), or phospholipases (VipD, PlcC, LpdA), or (iii) by recruiting eukaryotic PI phosphatases or kinases (RalF, SidM). PtdIns(4)P is bound by SidC (ubiquitin ligase) and SidM (Rab1 GEF/AMPylase). LidA and the Rab1 phosphocholinase AnkX bind PtdIns(3)P as well as PtdIns(4)P. PtdIns(3)P is bound by RidL (retromer inhibitor) and RavZ (Atg8/LC3 protease), as well as by SetA and LtpM (glycosyltransferases) and LepB (Rab1 GAP, PI 4-kinase). SidF and SidP are PI 3-phosphatases. VipD and PlcC function as a Rab5-activated phospholipase A1 or a Zn2+ metallophospholipase C, respectively. LpnE is secreted by an unknown mechanism and binds PtdIns(3)P as well as the host PI 5-phosphatase OCRL. The GEF RalF activates the small GTPase Arf1, which in turn recruits the host PI 4-kinase IIIβ (PI4KIIIβ). OCRL and PI4KIIIβ produce PtdIns(4)P from PtdIns(4,5)P2 or PtdIns, respectively.
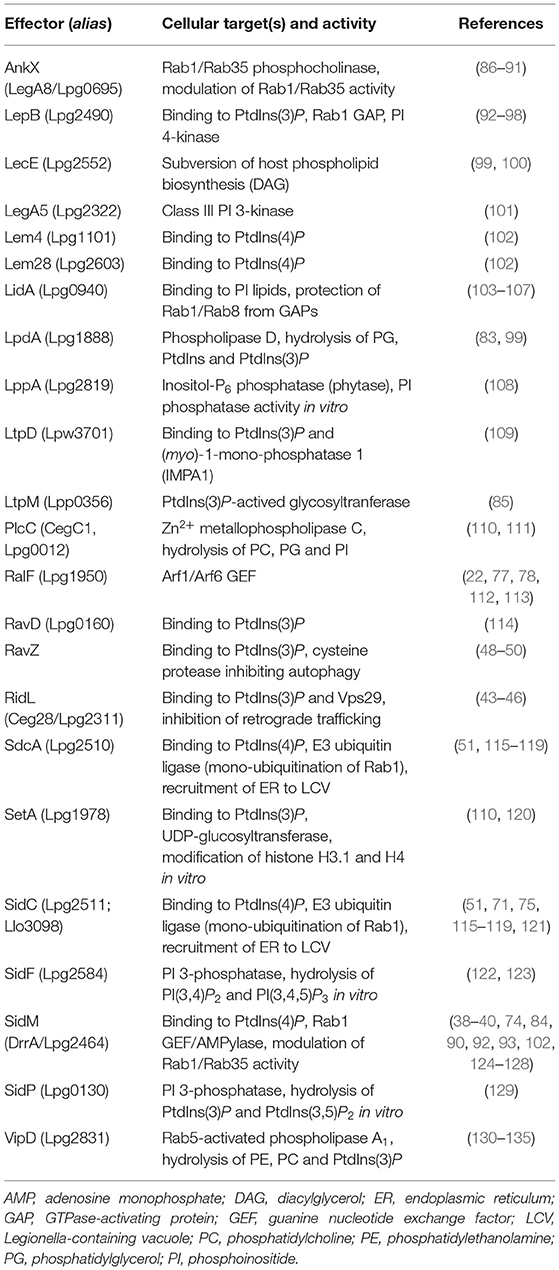
Table 1. L. pneumophila T4SS-translocated effectors targeting host PI lipids.
The L. pneumophila Icm/Dot substrate SidC and its paralogue SdcA localize to the LCV membrane (115) and almost exclusively bind to PtdIns(4)P [(51); Figure 4 and Table 1]. The 105 kDa effector proteins harbor a unique 20 kDa C-terminal domain termed P4C [PtdIns(4)P-binding domain of SidC], which does not show similarity to any eukaryotic PI-binding motif and was used as a PtdIns(4)P probe in eukaryotic cells (116, 136). SidC and the P4C domain are conserved in Legionella longbeachae, where the 111 kDa effector represents the major PtdIns(4)P binding protein (75). The SidC orthologs of L. pneumophila and L. longbeachae bind PtdIns(4)P with a low dissociation constant (Kd) of ca. 240 or 70 nM, respectively. The crystal structure of SidC revealed a unique PtdIns(4)P-binding domain essential for targeting the effector to the pathogen vacuole (137).
LCVs harboring an L. pneumophila ΔsidC-sdcA mutant strain recruit the ER slower and to a smaller extent; yet, the formation of the spatially separated PtdIns(4)P-positive limiting LCV membrane is not affected (28, 51, 65, 116). The interaction with the ER is catalyzed by a 70 kDa N-terminal fragment of SidC (116). The crystal structure of the N-terminal fragment revealed a novel fold (117, 121), comprising a catalytic Cys-His-Asp triad, which is essential for SidC to promote the polyubiquitination of protein substrates on the LCV (118). Indeed, SidC and SdcA act as E3 ubiquitin ligases, which show a broad and non-overlapping specificity for ubiquitin-conjugating E2 enzymes (118, 119). Hence, the L. pneumophila effector SidC links and subverts two different eukaryotic pathways, phosphoinositide and ubiquitination signaling.
In L. pneumophila-infected phagocytes, SidC decorates the LCV selectively, uniformly and in copious amounts (51, 116). We exploited this feature to isolate LCVs from homogenates of infected host cells by establishing a two-step procedure comprising immuno-affinity enrichment with an anti-SidC antibody, followed by Histodenz density gradient centrifugation (138, 139). Using this protocol, intact LCVs were isolated from D. discoideum amoeba (28, 140), murine RAW 264.7 macrophage-like cells (24, 27) and bone marrow-derived primary macrophages (141). The isolated LCVs were utilized for biochemical fusion experiments (28) and proteomics analysis (24, 27, 140, 141), which identified small GTPases and their effectors (Rab family, Rap1, Ran, RanBP1), large GTPases, components of the endosomal and late secretory trafficking pathways, as well as protein or lipid kinases and phosphatases. LCV localization of some of these proteins was confirmed by fluorescence microscopy using D. discoideum strains producing the corresponding GFP-fusion proteins (24, 26–28, 140, 142).
The Icm/Dot substrate SidM (alias DrrA) localizes to the LCV membrane early during L. pneumophila infection (92) and is the major PtdIns(4)P-binding protein, as it was exclusively identified as such in a non-biased pulldown approach [(84); Figure 4 and Table 1]. In lysates of L. pneumophila ΔsidM, no other PI-binding protein (not even SidC) was identified. The 73 kDa effector protein harbors the 12 kDa C-terminal domain P4M [PtdIns(4)P-binding domain of SidM], which does not show similarity to any eukaryotic PI-binding motif or the P4C domain of SidC, but is shared with two other effectors, Lpg1101 (alias Lem4) and Lpg2603 (alias Lem28) [(102); Table 1]. The P4M domain has been ectopically produced and used as a PtdIns(4)P probe in eukaryotic cells (143) and Drosophila photoreceptor cells (144). The crystal structure of SidM and biochemical analysis revealed a unique PtdIns(4)P-binding domain and a very high binding affinity (Kd = 4–18 nM) (40, 74).
SidM, i.e., its central domain, exerts GEF activity toward Rab1-GDI complexes, thus leading to GTP loading and Rab1 activation on LCV membranes (38, 39, 92, 124–127). Moreover, the N-terminal domain of SidM catalyzes the covalent attachment of AMP to Rab1, a reaction termed AMPylation (128), which renders Rab1(GTP) inaccessible to GAPs and causes the constitutive activation of the small GTPase on LCVs (93). The AMPylation reaction is reversible, and the L. pneumophila effector protein SidD can remove the AMP residue from Rab1 by a deAMPylation reaction (145–147). The removal of the covalent modification allows the GAP LepB to inactivate Rab1 (92, 94). Through activation of Rab1, SidM catalyzes the non-canonical pairing of plasma membrane t-SNARE syntaxin proteins (present on the LCV membrane) with the ER-localized v-SNARE protein Sec22b (148, 149). Thus, the SidM-catalyzed activation of Rab1 seems to promote the tethering and fusion of the LCV with ER-derived vesicles, which has been described many years ago (150, 151). In summary, the L. pneumophila effector SidM links and subverts two different eukaryotic pathways, phosphoinositide and small GTPase signaling.
The Icm/Dot substrate LidA supports SidM-dependent recruitment of Rab1 to LCVs (39) and preferentially binds to PtdIns(3)P or with lower affinity to PtdIns(4)P [(84, 103); Figure 4 and Table 1]. The 83 kDa effector targets Rab1 and several other host Rab GTPases (152, 153) and binds with high affinity to the GDP- and GTP-bound as well as the AMPylated form of Rab1, thus stabilizing the active conformation of the GTPase and preventing inactivation by GAPs (39, 104, 105).
The Icm/Dot substrate AnkX localizes to LCVs and binds with apparently similar affinity to PtdIns(3)P and PtdIns(4)P [(154); Figure 4 and Table 1]. AnkX covalently attaches a phosphocholine moiety to GDP-bound Rab1 and Rab35 in a process termed phosphocholination, which stabilizes inactive Rab1 at the LCV membrane (86, 87, 155). The CDP-choline-dependent activity of AnkX is reversed by the Icm/Dot-secreted effector Lem3, which dephosphocholinates Rab1 (88, 155).
The Icm/Dot substrate RidL specifically binds PtdIns(3)P and localizes to the LCV, juxtaposed to where the polar Icm/Dot T4SS connects to the pathogen vacuole membrane [(44); Figure 4 and Table 1]. RidL binds the Vps29 subunit of the retromer coat complex, inhibits retrograde trafficking and thereby promotes intracellular bacterial replication (19, 20). Structural studies revealed that a hydrophobic β-hairpin in the N-terminal domain of RidL interacts with Vps29, thus displacing the Rab7 GAP TBC1D5 [a regulator of retrograde trafficking; (43, 45, 46)].
The Icm/Dot substrate RavZ targets autophagosomes and binds PtdIns(3)P on high-curvature membranes trough a C-terminal domain [(49); Figure 4 and Table 1]. RavZ inhibits autophagy by deconjugating Atg8/LC3 from phosphatidylethanolamine (PE) (48). In contrast to the eukaryotic deconjugating factor Atg4, the cysteine protease RavZ irreversible decouples Atg8 from PE by hydrolyzing the amide bond between the C-terminal glycine and an adjacent aromatic amino acid in Atg8.
The Icm/Dot substrates SetA (110, 120) and LtpM (85) localize to LCVs and endosomes through C-terminal PtdIns(3)P-binding domains (Figure 4 and Table 1). The N-terminal domains of these effectors show similarities with glycosyl transferases, and indeed, the purified enzymes were found to exhibit glycohydrolase and glycosyltransferase activity in vitro, using UDP-glucose as a sugar donor. Intriguingly, PtdIns(3)P activates the glycosyltransferase activity of LtpM (85).
The Icm/Dot substrates LtpD (109) and RavD (114) also localize to the LCV through C-terminal PtdIns(3)P-binding domains (Table 1). LtpD might bind to the inositol monophosphatase IMPA1, which has indeed been detected on isolated LCVs (140). LpnE is a 41 kDa L. pneumophila virulence factor that binds to PtdIns(3)P and the eukaryotic PI 5-phosphatase OCRL (see below) [(156); Figure 4 and Table 1]. The Sel1 repeat-containing LpnE is secreted independently of the Icm/Dot T4SS or the Lsp T2SS and promotes uptake of L. pneumophila by phagocytes and intracellular replication (157, 158). Finally, a recent bioinformatics-based screen identified three novel PtdIns(3)P-binding domains, which are present in at least 14 known Icm/Dot substrates, including LepB and RavZ (95).
L. Pneumophila Phosphoinositide Phosphatases, Kinases, and Phospholipases
Legionella pneumophila produces Icm/Dot-translocated effector proteins, which directly modify PI lipids by acting as PI phosphatases, PI kinases or phospholipases (Figure 4). The Icm/Dot substrate SidF localizes to the LCV at early time points of infection (2 h) [(122, 123); Figure 4 and Table 1]. The crystal structure of the N-terminal catalytic domain in complex with its substrate PtdIns(3,4)P2 revealed a positively charged groove in the catalytic center, similar to other PI phosphatases harboring the “CX5R” motif (123). The 102 kDa effector SidF harbors two predicted C-terminal transmembrane motifs, which anchor the protein to the LCV membrane. SidF specifically hydrolyses in vitro PtdIns(3,4)P2 and PtdIns(3,4,5)P3 typically occurring on early phagosomes, and it likely contributes to the production of PtdIns(4)P on LCVs, since vacuoles harboring L. pneumophila ΔsidF accumulate lower amounts of the PtdIns(4)P-binding effector SidC. Yet, the ΔsidF mutant strain is not impaired for intracellular growth.
The Icm/Dot substrate SidP acts as a PI 3-phosphatase in vitro and converts PtdIns(3,5)P2 to PtdIns(5)P as well as PtdIns(3)P to PtdIns (Figure 4 and Table 1). However, its PI-phosphatase activity was not assessed in L. pneumophila-infected cells, and a ΔsidP mutant strain is not impaired for intracellular growth (129). The crystal structure of SidP from L. longbeachae revealed three distinct domains: a large N-terminal catalytic domain, an appendage domain inserted into the catalytic domain, and a C-terminal α-helical domain. Based largely on biochemical studies, SidF and SidP were postulated to produce PtdIns(4)P and hydrolyze PtdIns(3)P on LCVs, thus contributing to the PI conversion on the pathogen vacuole.
The Icm/Dot substrate LepB is a Rab1 GAP (see above), but also shows PI 4-kinase activity specific for PtdIns(3)P [(96); Figure 4 and Table 1]. The effector might contribute to the production of PtdIns(4)P on LCVs, since pathogen vacuoles harboring L. pneumophila ΔlepB accumulate lower amounts of the PtdIns(4)P-binding effector SidC. LepB was proposed to convert PtdIns(3)P on LCVs into PtdIns(3,4)P, which could be hydrolyzed by SidF to yield PtdIns(4)P (96). Interestingly, the Icm/Dot substrate LegA5 (159), a membrane-associated effector toxic for yeast (110, 160), was recently found to be a wortmannin-insensitive, class III-like PI 3-kinase [(101); Table 1]. In fact, LegA5 might be a PI 3-kinase producing PtdIns(3)P on LCVs as a substrate for the PI 4-kinase LepB.
The Icm/Dot substrate LppA is another example of a CX5R motif PI phosphatase hydrolyzing in vitro PtdIns(3,4)P2, PtdIns(4,5)P2, and PtdIns(3,4,5)P3 to yield PtdIns(4)P [(108); Table 1]. While LppA appeared like an ideal candidate to produce PtdIns(4)P on LCVs, live-cell microscopy using GFP-P4C as a PtdIns(4)P probe indicated that LppA does not affect the LCV PI pattern. Instead, LppA is a T4SS-translocated hexakisphosphate inositol phosphatase (phytase), which degrades the micronutrient chelator phytate (indeed produced by amoebae), and thereby promotes the intracellular growth of L. pneumophila. Given that the L. pneumophila genome encodes more than 400 proteins with the CX5R (PI) phosphatase signature (123), other (PI) phosphatases are likely produced by the pathogen.
The Icm/Dot substrates VipD, PlcC, and LpdA are lipases, which possess broad range activity against phospholipids including mono-phosphorylated PIs (Figure 4 and Table 1). VipD was identified as an Icm/Dot substrate that impairs membrane trafficking in yeast (130, 131). The effector hydrolyzes PE as well as phosphatidylcholine (PC) (132) and, intriguingly, binds Rab5 as well as Rab22 and acts as a Rab5-activated phospholipase A1 (133–135). Accordingly, VipD removes PtdIns(3)P from endosomal membranes and thus might promote the evasion of the endocytic pathway by LCVs (133, 134). Analogously, the Icm/Dot substrate PlcC (alias CegC1) is a metallophospholipase C, which hydrolyzes a broad spectrum of lipids including PC, phosphatidylglycerol (PG), and PtdIns (111). The effector can degrade PtdIns(3)P and likely destabilizes target membranes. Finally, the Icm/Dot substrate LpdA is a phospholipase D that binds to membranes through C-terminal prenylation and hydrolyzes PG, PtdIns and PtdIns(3)P as well as PtdIns(4)P yielding phosphatidic acid (PA) (83). While LpdA does not seem to affect the cellular PI pattern, the phospholipase triggers Golgi fragmentation.
Subversion of Host Phosphoinositide Kinases and Phosphatases by L. Pneumophila
In addition to directly modulating PI lipids, L. pneumophila effectors also subvert the host cell PI pattern indirectly by targeting eukaryotic PI phosphatases and kinases (Figure 4). The PtdIns(3)P-binding virulence factor LpnE binds mammalian OCRL (Oculocerebrorenal syndrome of Lowe) and its Dictyostelium homolog Dd5P4 (D. discoideum 5-phosphatase 4) via their N-terminal domains (156). The interaction of LpnE with OCRL was recently confirmed by size exclusion chromatography and supported by the crystal structure of the bacterial protein (161). OCRL and Dd5P4 are PI 5-phosphatases, which hydrolyse PtdIns(4,5)P2 and PtdIns(3,4,5)P3 to yield PtdIns(4)P and PtdIns(3,4)P2, respectively (162, 163). Dd5P4 is likely catalytically active on LCVs and increases the PtdIns(4)P available for binding by effectors such as SidC or SidM (156). Consequently, LpnE might increase the concentration of PtdIns(4)P on LCVs by recruiting OCRL/Dd5P4, and thereby promote PI conversion. L. pneumophila grows more efficiently in D. discoideum lacking Dd5P4, and thus, the pleiotropic PI 5-phosphatase restricts intracellular bacterial growth. Mechanistic details of this process are not known, but Dd5P4 modulates the recruitment of calnexin, Rab1 and retromer components to LCVs, which might account for growth restriction (156, 164).
The Icm/Dot substrates RalF and SidM possibly contribute indirectly to the modulation of the LCV PI pattern through the recruitment and activation of small host GTPases. RalF is an Arf1 GEF and activates the small GTPase on the LCV [(22, 112); Figure 4 and Table 1]. RalF harbors a C-terminal globular “capping” domain, which regulates GEF activity by auto-inhibition (77). Activated Arf1 recruits PI 4-kinase IIIβ (PI4KIIIβ) to the trans Golgi network (165), and hence, RalF might indirectly increase the PtdIns(4)P concentration on LCVs. Indeed, the depletion by RNA interference of PI4KIIIβ, but not PI4KIIIα or PI4KIIα decreases the amount of the PtdIns(4)P-binding effector SidC on LCVs, suggesting that in absence of PI4KIIIβ the level of PtdIns(4)P is reduced (84). Analogously, SidM recruits and activates Rab1 on LCVs (see above). Activated Rab1 (166) as well as Arf1 (167) recruit OCRL to endosomal membranes. Accordingly, SidM might not only bind to PtdIns(4)P, but also indirectly contribute to an increase of this PI on LCV membranes.
The Icm/Dot substrates LpdA and LecE localize to LCVs and might also indirectly modulate the LCV PI pattern by promoting DAG biosynthesis [(99); Table 1]. LpdA is a phospholipase D, which hydrolyzes PC to yield PA (see above). LecE enhances the activity of the eukaryotic PA phosphatase Pah1, which dephosphorylates PA yielding DAG. The second messenger DAG recruits protein kinase D (PKD) and its activator protein kinase C (PKC) to membranes. Activated PKD then interacts with PI4KIIIβ, thereby possibly also contributing to an increase in PtdIns(4)P on LCVs (99).
Conclusions and Outlook
Legionella pneumophila replicates intracellularly in phagocytes within an LCV, a complex compartment tightly associated with the ER. The nascent LCV undergoes a PI conversion from PtdIns(3)P to PtdIns(4)P, and thereby is rerouted from the bactericidal endocytic to the replication-permissive secretory pathway. To modulate the PI pattern in infected cells, L. pneumophila (i) recruits PI-decorated vesicles, (ii) produces effectors acting as PI interactors, phosphatases, kinases or phospholipases, or (iii) subverts host PI-metabolizing enzymes. To this end, at least 21 T4SS-translocated effector proteins have been shown to target the host PI metabolism (Table 1). Intriguingly, a number of these effectors harbor 2–3 different functional domains and link PI signaling to other pivotal cellular pathways, e.g., SidC (PI interactor, ubiquitin ligase), SidM (PI interactor, Rab1 GEF, Rab1 AMPylase), LepB (PI interactor, PI 4-kinase, Rab1 GAP), SetA and LtpM (PI interactor, glycosyltransferase), and VipD (Rab5 interactor, phospholipase). LCV formation and the contribution of PI lipids to this process are incompletely understood. Among the more than 300 T4SS-translocated effector proteins of L. pneumophila only about 50 have been thoroughly investigated. Future studies will focus on the structural, molecular and cellular characterization of novel effectors implicated in host cell PI pattern subversion, as well as on the spatiotemporal regulation of effector translocation and function.
Author Contributions
ALS and HH wrote the manuscript.
Funding
Research in the laboratory of HH was supported by the Swiss National Science Foundation (SNF; 31003A_153200, 31003A_175557), the University of Zürich, the Novartis Foundation for Medical-Biological Research, and the OPO foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
AMPylase, adenylyltransferase; DAG, diacylglycerol; Icm/Dot, intracellular multiplication/defective organelle trafficking; GAP, GTPase activating protein; GDI, guanine nucleotide dissociation inhibitor; GEF, guanine nucleotide exchange factor; LCV, Legionella-containing vacuole; OCRL, oculocerebrorenal syndrome of Lowe; PI, phosphoinositide; PI3/4/5K, PI 3-/4-/5-kinase; PtdIns, phosphatidylinositol; T4SS, type IV secretion system.
References
1. Hilbi H, Hoffmann C, Harrison CF. Legionella spp. outdoors: colonization, communication and persistence. Environ Microbiol Rep. (2011) 3:286–96. doi: 10.1111/j.1758-2229.2011.00247.x
2. Newton HJ, Ang DK, van Driel IR, Hartland EL. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin Microbiol Rev. (2010) 23:274–98. doi: 10.1128/CMR.00052-09
3. Abdel-Nour M, Duncan C, Low DE, Guyard C. Biofilms: the stronghold of Legionella pneumophila. Int J Mol Sci. (2013) 14:21660–75. doi: 10.3390/ijms141121660
4. Declerck P. Biofilms: the environmental playground of Legionella pneumophila. Environ Microbiol. (2010) 12:557–66. doi: 10.1111/j.1462-2920.2009.02025.x
5. Taylor M, Ross K, Bentham R. Legionella, protozoa, and biofilms: interactions within complex microbial systems. Microb Ecol. (2009) 58:538–47. doi: 10.1007/s00248-009-9514-z
6. Brassinga AK, Kinchen JM, Cupp ME, Day SR, Hoffman PS, Sifri CD. Caenorhabditis is a metazoan host for Legionella. Cell Microbiol. (2010) 12:343–61. doi: 10.1111/j.1462-5822.2009.01398.x
7. Komura T, Yasui C, Miyamoto H, Nishikawa Y. Caenorhabditis elegans as an alternative model host for Legionella pneumophila, and protective effects of Bifidobacterium infantis. Appl Environ Microbiol. (2010) 76:4105–8. doi: 10.1128/AEM.03021-09
8. Hilbi H, Weber SS, Ragaz C, Nyfeler Y, Urwyler S. Environmental predators as models for bacterial pathogenesis. Environ Microbiol. (2007) 9:563–75. doi: 10.1111/j.1462-2920.2007.01238.x
9. Rasch J, Kruger S, Fontvieille D, Unal CM, Michel R, Labrosse A, et al. Legionella-protozoa-nematode interactions in aquatic biofilms and influence of Mip on Caenorhabditis elegans colonization. Int J Med Microbiol. (2016) 306:443–51. doi: 10.1016/j.ijmm.2016.05.012
10. Fields BS. The molecular ecology of Legionellae. Trends Microbiol. (1996) 4:286–90. doi: 10.1016/0966-842X(96)10041-X
11. Hoffmann C, Harrison CF, Hilbi H. The natural alternative: protozoa as cellular models for Legionella infection. Cell Microbiol. (2014) 16:15–26. doi: 10.1111/cmi.12235
12. Swart AL, Harrison CF, Eichinger L, Steinert M, Hilbi H. Acanthamoeba and Dictyostelium as cellular models for Legionella infection. Front Cell Infect Microbiol. (2018) 8:61. doi: 10.3389/fcimb.2018.00061
13. Whiley H, Bentham R. Legionella longbeachae and legionellosis. Emerg Infect Dis. (2011) 17:579–83. doi: 10.3201/eid1704.100446
14. Correia AM, Ferreira JS, Borges V, Nunes A, Gomes B, Capucho R, et al. Probable person-to-person transmission of Legionnaires' disease. N Engl J Med. (2016) 374:497–8. doi: 10.1056/NEJMc1505356
15. Al-Quadan T, Price CT, Abu Kwaik Y. Exploitation of evolutionarily conserved amoeba and mammalian processes by Legionella. Trends Microbiol. (2012) 20:299–306. doi: 10.1016/j.tim.2012.03.005
16. Gomez-Valero L, Rusniok C, Cazalet C, Buchrieser C. Comparative and functional genomics of Legionella identified eukaryotic like proteins as key players in host-pathogen interactions. Front Microbiol. (2011) 2:208. doi: 10.3389/fmicb.2011.00208
17. Isberg RR, O'Connor TJ, Heidtman M. The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat Rev Microbiol. (2009) 7:13–24. doi: 10.1038/nrmicro1967
18. Asrat S, de Jesus DA, Hempstead AD, Ramabhadran V, Isberg RR. Bacterial pathogen manipulation of host membrane trafficking. Annu Rev Cell Dev Biol. (2014) 30:79–109. doi: 10.1146/annurev-cellbio-100913-013439
19. Personnic N, Bärlocher K, Finsel I, Hilbi H. Subversion of retrograde trafficking by translocated pathogen effectors. Trends Microbiol. (2016) 24:450–62. doi: 10.1016/j.tim.2016.02.003
20. Bärlocher K, Welin A, Hilbi H. Formation of the Legionella replicative compartment at the crossroads of retrograde trafficking. Front Cell Infect Microbiol. (2017) 7:482. doi: 10.3389/fcimb.2017.00482
21. Steiner B, Weber S, Hilbi H. Formation of the Legionella-containing vacuole: phosphoinositide conversion, GTPase modulation and ER dynamics. Int J Med Microbiol. (2018) 308:49–57. doi: 10.1016/j.ijmm.2017.08.004
22. Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. (2002) 295:679–82. doi: 10.1126/science.1067025
23. Goody RS, Itzen A. Modulation of small GTPases by Legionella. Curr Top Microbiol Immunol. (2013) 376:117–33. doi: 10.1007/82_2013_340
24. Hoffmann C, Finsel I, Otto A, Pfaffinger G, Rothmeier E, Hecker M, et al. Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell Microbiol. (2014) 16:1034–52. doi: 10.1111/cmi.12256
25. Sherwood RK, Roy CR. Autophagy evasion and endoplasmic reticulum subversion: the Yin and Yang of Legionella intracellular infection. Annu Rev Microbiol. (2016) 70:413–33. doi: 10.1146/annurev-micro-102215-095557
26. Rothmeier E, Pfaffinger G, Hoffmann C, Harrison CF, Grabmayr H, Repnik U, et al. Activation of Ran GTPase by a Legionella effector promotes microtubule polymerization, pathogen vacuole motility and infection. PLoS Pathog. (2013) 9:e1003598. doi: 10.1371/journal.ppat.1003598
27. Schmölders J, Manske C, Otto A, Hoffmann C, Steiner B, Welin A, et al. Comparative proteomics of purified pathogen vacuoles correlates intracellular replication of Legionella pneumophila with the small GTPase Ras-related protein 1 (Rap1). Mol Cell Proteomics. (2017) 16:622–41. doi: 10.1074/mcp.M116.063453
28. Steiner B, Swart AL, Welin A, Weber S, Personnic N, Kaech A, et al. ER remodeling by the large GTPase atlastin promotes vacuolar growth of Legionella pneumophila. EMBO Rep. (2017) 18:1817–36. doi: 10.15252/embr.201743903
29. Escoll P, Song OR, Viana F, Steiner B, Lagache T, Olivo-Marin JC, et al. Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host Microbe. (2017) 22:302–16.e7. doi: 10.1016/j.chom.2017.07.020
30. Burstein D, Amaro F, Zusman T, Lifshitz Z, Cohen O, Gilbert JA, et al. Genomic analysis of 38 Legionella species identifies large and diverse effector repertoires. Nat Genet. (2016) 48:167–75. doi: 10.1038/ng.3481
31. Gomez-Valero L, Rusniok C, Carson D, Mondino S, Perez-Cobas AE, Rolando M, et al. More than 18,000 effectors in the Legionella genus genome provide multiple, independent combinations for replication in human cells. Proc Natl Acad Sci USA. (2019) 116:2265–73. doi: 10.1073/pnas.1808016116
32. Hubber A, Roy CR. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol. (2010) 26:261–83. doi: 10.1146/annurev-cellbio-100109-104034
33. Hilbi H, Haas A. Secretive bacterial pathogens and the secretory pathway. Traffic. (2012) 13:1187–97. doi: 10.1111/j.1600-0854.2012.01344.x
34. Sherwood RK, Roy CR. A Rab-centric perspective of bacterial pathogen-occupied vacuoles. Cell Host Microbe. (2013) 14:256–68. doi: 10.1016/j.chom.2013.08.010
35. Haneburger I, Hilbi H. Phosphoinositide lipids and the Legionella pathogen vacuole. Curr Top Microbiol Immunol. (2013) 376:155–73. doi: 10.1007/82_2013_341
36. Finsel I, Hilbi H. Formation of a pathogen vacuole according to Legionella pneumophila: how to kill one bird with many stones. Cell Microbiol. (2015) 17:935–50. doi: 10.1111/cmi.12450
37. Qiu J, Luo ZQ. Legionella and Coxiella effectors: strength in diversity and activity. Nat Rev Microbiol. (2017) 15:591–605. doi: 10.1038/nrmicro.2017.67
38. Murata T, Delprato A, Ingmundson A, Toomre DK, Lambright DG, Roy CR. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol. (2006) 8:971–7. doi: 10.1038/ncb1463
39. Machner MP, Isberg RR. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev Cell. (2006) 11:47–56. doi: 10.1016/j.devcel.2006.05.013
40. Schoebel S, Blankenfeldt W, Goody RS, Itzen A. High-affinity binding of phosphatidylinositol 4-phosphate by Legionella pneumophila DrrA. EMBO Rep. (2010) 11:598–604. doi: 10.1038/embor.2010.97
41. Itzen A, Goody RS. Covalent coercion by Legionella pneumophila. Cell Host Microbe. (2011) 10:89–91. doi: 10.1016/j.chom.2011.08.002
42. Simon S, Wagner MA, Rothmeier E, Müller-Taubenberger A, Hilbi H. Icm/Dot-dependent inhibition of phagocyte migration by Legionella is antagonized by a translocated Ran GTPase activator. Cell Microbiol. (2014) 16:977–92. doi: 10.1111/cmi.12258
43. Bärlocher K, Hutter CAJ, Swart AL, Steiner B, Welin A, Hohl M, et al. Structural insights into Legionella RidL-Vps29 retromer subunit interaction reveal displacement of the regulator TBC1D5. Nat Commun. (2017) 8:1543. doi: 10.1038/s41467-017-01512-5
44. Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, van Rahden VA, et al. The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell Host Microbe. (2013) 14:38–50. doi: 10.1016/j.chom.2013.06.001
45. Romano-Moreno M, Rojas AL, Williamson CD, Gershlick DC, Lucas M, Isupov MN, et al. Molecular mechanism for the subversion of the retromer coat by the Legionella effector RidL. Proc Natl Acad Sci USA. (2017) 114:E11151–60. doi: 10.1073/pnas.1715361115
46. Yao J, Yang F, Sun X, Wang S, Gan N, Liu Q, et al. Mechanism of inhibition of retromer transport by the bacterial effector RidL. Proc Natl Acad Sci USA. (2018) 115:E1446–E1454. doi: 10.1073/pnas.1717383115
47. Xu L, Shen X, Bryan A, Banga S, Swanson MS, Luo ZQ. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. (2010) 6:e1000822. doi: 10.1371/journal.ppat.1000822
48. Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. (2012) 338:1072–6. doi: 10.1126/science.1227026
49. Horenkamp FA, Kauffman KJ, Kohler LJ, Sherwood RK, Krueger KP, Shteyn V, et al. The Legionella anti-autophagy effector RavZ targets the autophagosome via PI3P- and curvature-sensing motifs. Dev Cell. (2015) 34:569–76. doi: 10.1016/j.devcel.2015.08.010
50. Yang A, Pantoom S, Wu YW. Elucidation of the anti-autophagy mechanism of the Legionella effector RavZ using semisynthetic LC3 proteins. Elife. (2017) 6:e23905. doi: 10.7554/eLife.23905
51. Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. (2006) 2:e46. doi: 10.1371/journal.ppat.0020046
52. Weber SS, Ragaz C, Hilbi H. Pathogen trafficking pathways and host phosphoinositide metabolism. Mol Microbiol. (2009) 71:1341–52. doi: 10.1111/j.1365-2958.2009.06608.x
53. Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev. (2013) 93:1019–137. doi: 10.1152/physrev.00028.2012
54. Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. (2006) 443:651–7. doi: 10.1038/nature05185
55. Michell RH. Inositol derivatives: evolution and functions. Nat Rev Mol Cell Biol. (2008) 9:151–61. doi: 10.1038/nrm2334
56. Payrastre B, Missy K, Giuriato S, Bodin S, Plantavid M, Gratacap M. Phosphoinositides: key players in cell signalling, in time and space. Cell Signal. (2001) 13:377–87. doi: 10.1016/S0898-6568(01)00158-9
57. De Matteis MA, Godi A. PI-loting membrane traffic. Nat Cell Biol. (2004) 6:487–92. doi: 10.1038/ncb0604-487
58. Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, et al. Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res. (2009) 48:307–43. doi: 10.1016/j.plipres.2009.06.001
59. Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. (2008) 9:99–111. doi: 10.1038/nrm2328
60. Behnia R, Munro S. Organelle identity and the signposts for membrane traffic. Nature. (2005) 438:597–604. doi: 10.1038/nature04397
61. Schink KO, Tan KW, Stenmark H. Phosphoinositides in control of membrane dynamics. Annu Rev Cell Dev Biol. (2016) 32:143–71. doi: 10.1146/annurev-cellbio-111315-125349
62. Saarikangas J, Zhao H, Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol Rev. (2010) 90:259–89. doi: 10.1152/physrev.00036.2009
63. Jeschke A, Zehethofer N, Lindner B, Krupp J, Schwudke D, Haneburger I, et al. Phosphatidylinositol 4-phosphate and phosphatidylinositol 3-phosphate regulate phagolysosome biogenesis. Proc Natl Acad Sci USA. (2015) 112:4636–41. doi: 10.1073/pnas.1423456112
64. Peracino B, Balest A, Bozzaro S. Phosphoinositides differentially regulate bacterial uptake and Nramp1-induced resistance to Legionella infection in Dictyostelium. J Cell Sci. (2010) 123:4039–51. doi: 10.1242/jcs.072124
65. Weber S, Hilbi H. Live cell imaging of phosphoinositide dynamics during Legionella infection. Methods Mol Biol. (2014) 1197:153–67. doi: 10.1007/978-1-4939-1261-2_9
66. Khelef N, Shuman HA, Maxfield FR. Phagocytosis of wild-type Legionella pneumophila occurs through a wortmannin-insensitive pathway. Infect Immun. (2001) 69:5157–61. doi: 10.1128/IAI.69.8.5157-5161.2001
67. Harada T, Tanikawa T, Iwasaki Y, Yamada M, Imai Y, Miyake M. Phagocytic entry of Legionella pneumophila into macrophages through phosphatidylinositol 3,4,5-trisphosphate-independent pathway. Biol Pharm Bull. (2012) 35:1460–8. doi: 10.1248/bpb.b11-00011
68. Hilbi H, Segal G, Shuman HA. Icm/Dot-dependent upregulation of phagocytosis by Legionella pneumophila. Mol Microbiol. (2001) 42:603–17. doi: 10.1046/j.1365-2958.2001.02645.x
69. Watarai M, Derre I, Kirby J, Growney JD, Dietrich WF, Isberg RR. Legionella pneumophila is internalized by a macropinocytotic uptake pathway controlled by the Dot/Icm system and the mouse lgn1 locus. J Exp Med. (2001) 194:1081–96. doi: 10.1084/jem.194.8.1081
70. Buckley CM, Heath VL, Gueho A, Bosmani C, Knobloch P, Sikakana P, et al. PIKfyve/Fab1 is required for efficient V-ATPase and hydrolase delivery to phagosomes, phagosomal killing, and restriction of Legionella infection. PLoS Pathog. (2019) 15:e1007551. doi: 10.1371/journal.ppat.1007551
71. Weber S, Wagner M, Hilbi H. Live-cell imaging of phosphoinositide dynamics and membrane architecture during Legionella infection. MBio. (2014) 5:e00839–e00813. doi: 10.1128/mBio.00839-13
72. Weber S, Steiner B, Welin A, Hilbi H. Legionella-containing vacuoles capture PtdIns(4)P-rich vesicles derived from the Golgi apparatus. MBio. (2018) 9:e02420–e02418. doi: 10.1128/mBio.02420-18
73. Hilbi H, Weber S, Finsel I. Anchors for effectors: subversion of phosphoinositide lipids by Legionella. Front Microbiol. (2011) 2:91. doi: 10.3389/fmicb.2011.00091
74. Del Campo CM, Mishra AK, Wang YH, Roy CR, Janmey PA, Lambright DG. Structural basis for PI(4)P-specific membrane recruitment of the Legionella pneumophila effector DrrA/SidM. Structure. (2014) 22:397–408. doi: 10.1016/j.str.2013.12.018
75. Dolinsky S, Haneburger I, Cichy A, Hannemann M, Itzen A, Hilbi H. The Legionella longbeachae Icm/Dot substrate SidC selectively binds phosphatidylinositol 4-phosphate with nanomolar affinity and promotes pathogen vacuole-endoplasmic reticulum interactions. Infect Immun. (2014) 82:4021–33. doi: 10.1128/IAI.01685-14
76. Peurois F, Veyron S, Ferrandez Y, Ladid I, Benabdi S, Zeghouf M, et al. Characterization of the activation of small GTPases by their GEFs on membranes using artificial membrane tethering. Biochem J. (2017) 474:1259–72. doi: 10.1042/BCJ20170015
77. Alix E, Chesnel L, Bowzard BJ, Tucker AM, Delprato A, Cherfils J, et al. The capping domain in RalF regulates effector functions. PLoS Pathog. (2012) 8:e1003012. doi: 10.1371/journal.ppat.1003012
78. Folly-Klan M, Alix E, Stalder D, Ray P, Duarte LV, Delprato A, et al. A novel membrane sensor controls the localization and ArfGEF activity of bacterial RalF. PLoS Pathog. (2013) 9:e1003747. doi: 10.1371/journal.ppat.1003747
79. Isaac DT, Laguna RK, Valtz N, Isberg RR. MavN is a Legionella pneumophila vacuole-associated protein required for efficient iron acquisition during intracellular growth. Proc Natl Acad Sci USA. (2015) 112:E5208–E5217. doi: 10.1073/pnas.1511389112
80. Christenson ET, Isaac DT, Yoshida K, Lipo E, Kim JS, Ghirlando R, et al. The iron-regulated vacuolar Legionella pneumophila MavN protein is a transition-metal transporter. Proc Natl Acad Sci USA. (2019) 116:17775–85. doi: 10.1073/pnas.1902806116
81. Ivanov SS, Charron G, Hang HC, Roy CR. Lipidation by the host prenyltransferase machinery facilitates membrane localization of Legionella pneumophila effector proteins. J Biol Chem. (2010) 285:34686–98. doi: 10.1074/jbc.M110.170746
82. Price CT, Al-Quadan T, Santic M, Jones SC, Abu Kwaik Y. Exploitation of conserved eukaryotic host cell farnesylation machinery by an F-box effector of Legionella pneumophila. J Exp Med. (2010) 207:1713–26. doi: 10.1084/jem.20100771
83. Schroeder GN, Aurass P, Oates CV, Tate EW, Hartland EL, Flieger A, et al. Legionella pneumophila effector LpdA is a palmitoylated phospholipase D virulence factor. Infect Immun. (2015) 83:3989–4002. doi: 10.1128/IAI.00785-15
84. Brombacher E, Urwyler S, Ragaz C, Weber SS, Kami K, Overduin M, et al. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J Biol Chem. (2009) 284:4846–56. doi: 10.1074/jbc.M807505200
85. Levanova N, Mattheis C, Carson D, To KN, Jank T, Frankel G, et al. The Legionella effector LtpM is a new type of phosphoinositide-activated glucosyltransferase. J Biol Chem. (2019) 294:2862–79. doi: 10.1074/jbc.RA118.005952
86. Campanacci V, Mukherjee S, Roy CR, Cherfils J. Structure of the Legionella effector AnkX reveals the mechanism of phosphocholine transfer by the FIC domain. EMBO J. (2013) 32:1469–77. doi: 10.1038/emboj.2013.82
87. Mukherjee S, Liu X, Arasaki K, McDonough J, Galan JE, Roy CR. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature. (2011) 477:103–6. doi: 10.1038/nature10335
88. Tan Y, Arnold RJ, Luo ZQ. Legionella pneumophila regulates the small GTPase Rab1 activity by reversible phosphorylcholination. Proc Natl Acad Sci USA. (2011) 108:21212–7. doi: 10.1073/pnas.1114023109
89. Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science. (2008) 320:1651–4. doi: 10.1126/science.1158160
90. Oesterlin LK, Goody RS, Itzen A. Posttranslational modifications of Rab proteins cause effective displacement of GDP dissociation inhibitor. Proc Natl Acad Sci USA. (2012) 109:5621–6. doi: 10.1073/pnas.1121161109
91. Gavriljuk K, Schartner J, Seidel H, Dickhut C, Zahedi RP, Hedberg C, et al. Unraveling the phosphocholination mechanism of the Legionella pneumophila enzyme AnkX. Biochemistry. (2016) 55:4375–85. doi: 10.1021/acs.biochem.6b00524
92. Ingmundson A, Delprato A, Lambright DG, Roy CR. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature. (2007) 450:365–9. doi: 10.1038/nature06336
93. Hardiman CA, Roy CR. AMPylation is critical for Rab1 localization to vacuoles containing Legionella pneumophila. MBio. (2014) 5:e01035-13. doi: 10.1128/mBio.01035-13
94. Gazdag EM, Streller A, Haneburger I, Hilbi H, Vetter IR, Goody RS, et al. Mechanism of Rab1b deactivation by the Legionella pneumophila GAP LepB. EMBO Rep. (2013) 14:199–205. doi: 10.1038/embor.2012.211
95. Nachmias N, Zusman T, Segal G. Study of Legionella effector domains revealed novel and prevalent phosphatidylinositol 3-phosphate binding domains. Infect Immun. (2019) 87:e00153–e00119. doi: 10.1128/IAI.00153-19
96. Dong N, Niu M, Hu L, Yao Q, Zhou R, Shao F. Modulation of membrane phosphoinositide dynamics by the phosphatidylinositide 4-kinase activity of the Legionella LepB effector. Nat Microbiol. (2016) 2:16236. doi: 10.1038/nmicrobiol.2016.236
97. Chen J, de Felipe KS, Clarke M, Lu H, Anderson OR, Segal G, et al. Legionella effectors that promote nonlytic release from protozoa. Science. (2004) 303:1358–61. doi: 10.1126/science.1094226
98. Mishra AK, Del Campo CM, Collins RE, Roy CR, Lambright DG. The Legionella pneumophila GTPase activating protein LepB accelerates Rab1 deactivation by a non-canonical hydrolytic mechanism. J Biol Chem. (2013) 288:24000–11. doi: 10.1074/jbc.M113.470625
99. Viner R, Chetrit D, Ehrlich M, Segal G. Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathog. (2012) 8:e1002988. doi: 10.1371/journal.ppat.1002988
100. Zhu W, Banga S, Tan Y, Zheng C, Stephenson R, Gately J, et al. Comprehensive identification of protein substrates of the Dot/Icm type IV transporter of Legionella pneumophila. PLoS ONE. (2011) 6:e17638. doi: 10.1371/journal.pone.0017638
101. Ledvina HE, Kelly KA, Eshraghi A, Plemel RL, Peterson SB, Lee B, et al. A phosphatidylinositol 3-kinase effector alters phagosomal maturation to promote intracellular growth of Francisella. Cell Host Microbe. (2018) 24:285–95. doi: 10.1016/j.chom.2018.07.003
102. Hubber A, Arasaki K, Nakatsu F, Hardiman C, Lambright D, De Camilli P, et al. The machinery at endoplasmic reticulum-plasma membrane contact sites contributes to spatial regulation of multiple Legionella effector proteins. PLoS Pathog. (2014) 10:e1004222. doi: 10.1371/journal.ppat.1004222
103. Neunuebel MR, Mohammadi S, Jarnik M, Machner MP. Legionella pneumophila LidA affects nucleotide binding and activity of the host GTPase Rab1. J Bacteriol. (2012) 194:1389–400. doi: 10.1128/JB.06306-11
104. Cheng W, Yin K, Lu D, Li B, Zhu D, Chen Y, et al. Structural insights into a unique Legionella pneumophila effector LidA recognizing both GDP and GTP bound Rab1 in their active state. PLoS Pathog. (2012) 8:e1002528. doi: 10.1371/journal.ppat.1002528
105. Schoebel S, Cichy AL, Goody RS, Itzen A. Protein LidA from Legionella is a Rab GTPase supereffector. Proc Natl Acad Sci USA. (2011) 108:17945–50. doi: 10.1073/pnas.1113133108
106. Conover GM, Derre I, Vogel JP, Isberg RR. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol Microbiol. (2003) 48:305–21. doi: 10.1046/j.1365-2958.2003.03400.x
107. Derre I, Isberg RR. LidA, a translocated substrate of the Legionella pneumophila type IV secretion system, interferes with the early secretory pathway. Infect Immun. (2005) 73:4370–80. doi: 10.1128/IAI.73.7.4370-4380.2005
108. Weber S, Stirnimann CU, Wieser M, Frey D, Meier R, Engelhardt S, et al. A type IV translocated Legionella cysteine phytase counteracts intracellular growth restriction by phytate. J Biol Chem. (2014) 289:34175–88. doi: 10.1074/jbc.M114.592568
109. Harding CR, Mattheis C, Mousnier A, Oates CV, Hartland EL, Frankel G, et al. LtpD is a novel Legionella pneumophila effector that binds phosphatidylinositol 3-phosphate and inositol monophosphatase IMPA1. Infect Immun. (2013) 81:4261–70. doi: 10.1128/IAI.01054-13
110. Heidtman M, Chen EJ, Moy MY, Isberg RR. Large-scale identification of Legionella pneumophila Dot/Icm substrates that modulate host cell vesicle trafficking pathways. Cell Microbiol. (2009) 11:230–48. doi: 10.1111/j.1462-5822.2008.01249.x
111. Aurass P, Schlegel M, Metwally O, Harding CR, Schroeder GN, Frankel G, et al. The Legionella pneumophila Dot/Icm-secreted effector PlcC/CegC1 together with PlcA and PlcB promotes virulence and belongs to a novel zinc metallophospholipase C family present in bacteria and fungi. J Biol Chem. (2013) 288:11080–92. doi: 10.1074/jbc.M112.426049
112. Amor JC, Swails J, Zhu X, Roy CR, Nagai H, Ingmundson A, et al. The structure of RalF, an ADP-ribosylation factor guanine nucleotide exchange factor from Legionella pneumophila, reveals the presence of a cap over the active site. J Biol Chem. (2005) 280:1392–400. doi: 10.1074/jbc.M410820200
113. Nagai H, Cambronne ED, Kagan JC, Amor JC, Kahn RA, Roy CR. A C-terminal translocation signal required for Dot/Icm-dependent delivery of the Legionella RalF protein to host cells. Proc Natl Acad Sci USA. (2005) 102:826–31. doi: 10.1073/pnas.0406239101
114. Pike CM, Boyer-Andersen R, Kinch LN, Caplan JL, Neunuebel MR. The Legionella effector RavD binds phosphatidylinositol-3-phosphate and helps suppress endolysosomal maturation of the Legionella-containing vacuole. J Biol Chem. (2019) 294:6405–15. doi: 10.1074/jbc.RA118.007086
115. Luo ZQ, Isberg RR. Multiple substrates of the Legionella pneumophila Dot/Icm system identified by interbacterial protein transfer. Proc Natl Acad Sci USA. (2004) 101:841–6. doi: 10.1073/pnas.0304916101
116. Ragaz C, Pietsch H, Urwyler S, Tiaden A, Weber SS, Hilbi H. The Legionella pneumophila phosphatidylinositol 4-phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol. (2008) 10:2416–33. doi: 10.1111/j.1462-5822.2008.01219.x
117. Horenkamp FA, Mukherjee S, Alix E, Schauder CM, Hubber AM, Roy CR, et al. Legionella pneumophila subversion of host vesicular transport by SidC effector proteins. Traffic. (2014) 15:488–99. doi: 10.1111/tra.12158
118. Hsu F, Luo X, Qiu J, Teng YB, Jin J, Smolka MB, et al. The Legionella effector SidC defines a unique family of ubiquitin ligases important for bacterial phagosomal remodeling. Proc Natl Acad Sci USA. (2014) 111:10538–43. doi: 10.1073/pnas.1402605111
119. Wasilko DJ, Huang Q, Mao Y. Insights into the ubiquitin transfer cascade catalyzed by the Legionella effector SidC. Elife. (2018) 7:e36154. doi: 10.7554/eLife.36154
120. Jank T, Bohmer KE, Tzivelekidis T, Schwan C, Belyi Y, Aktories K. Domain organization of Legionella effector SetA. Cell Microbiol. (2012) 14:852–68. doi: 10.1111/j.1462-5822.2012.01761.x
121. Gazdag EM, Schoebel S, Shkumatov AV, Goody RS, Itzen A. The structure of the N-terminal domain of the Legionella protein SidC. J Struct Biol. (2014) 186:188–94. doi: 10.1016/j.jsb.2014.02.003
122. Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, et al. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci USA. (2007) 104:5121–6. doi: 10.1073/pnas.0611030104
123. Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, Mao Y. Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc Natl Acad Sci USA. (2012) 109:13567–72. doi: 10.1073/pnas.1207903109
124. Machner MP, Isberg RR. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science. (2007) 318:974–7. doi: 10.1126/science.1149121
125. Schoebel S, Oesterlin LK, Blankenfeldt W, Goody RS, Itzen A. RabGDI displacement by DrrA from Legionella is a consequence of its guanine nucleotide exchange activity. Mol Cell. (2009) 36:1060–72. doi: 10.1016/j.molcel.2009.11.014
126. Suh HY, Lee DW, Lee KH, Ku B, Choi SJ, Woo JS, et al. Structural insights into the dual nucleotide exchange and GDI displacement activity of SidM/DrrA. EMBO J. (2010) 29:496–504. doi: 10.1038/emboj.2009.347
127. Zhu Y, Hu L, Zhou Y, Yao Q, Liu L, Shao F. Structural mechanism of host Rab1 activation by the bifunctional Legionella type IV effector SidM/DrrA. Proc Natl Acad Sci USA. (2010) 107:4699–704. doi: 10.1073/pnas.0914231107
128. Muller MP, Peters H, Blumer J, Blankenfeldt W, Goody RS, Itzen A. The Legionella effector protein DrrA AMPylates the membrane traffic regulator Rab1b. Science. (2010) 329:946–9. doi: 10.1126/science.1192276
129. Toulabi L, Wu X, Cheng Y, Mao Y. Identification and structural characterization of a Legionella phosphoinositide phosphatase. J Biol Chem. (2013) 288:24518–27. doi: 10.1074/jbc.M113.474239
130. Shohdy N, Efe JA, Emr SD, Shuman HA. Pathogen effector protein screening in yeast identifies Legionella factors that interfere with membrane trafficking. Proc Natl Acad Sci USA. (2005) 102:4866–71. doi: 10.1073/pnas.0501315102
131. VanRheenen SM, Luo ZQ, O'Connor T, Isberg RR. Members of a Legionella pneumophila family of proteins with ExoU (phospholipase A) active sites are translocated to target cells. Infect Immun. (2006) 74:3597–606. doi: 10.1128/IAI.02060-05
132. Zhu W, Hammad LA, Hsu F, Mao Y, Luo ZQ. Induction of caspase 3 activation by multiple Legionella pneumophila Dot/Icm substrates. Cell Microbiol. (2013) 15:1783–95. doi: 10.1111/cmi.12157
133. Gaspar AH, Machner MP. VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci USA. (2014) 111:4560–5. doi: 10.1073/pnas.1316376111
134. Ku B, Lee KH, Park WS, Yang CS, Ge J, Lee SG, et al. VipD of Legionella pneumophila targets activated Rab5 and Rab22 to interfere with endosomal trafficking in macrophages. PLoS Pathog. (2012) 8:e1003082. doi: 10.1371/journal.ppat.1003082
135. Lucas M, Gaspar AH, Pallara C, Rojas AL, Fernandez-Recio J, Machner MP, et al. Structural basis for the recruitment and activation of the Legionella phospholipase VipD by the host GTPase Rab5. Proc Natl Acad Sci USA. (2014) 111:E3514–E3523. doi: 10.1073/pnas.1405391111
136. Weber S, Dolinsky S, Hilbi H. Interactions of Legionella effector proteins with host phosphoinositide lipids. Methods Mol Biol. (2013) 954:367–80. doi: 10.1007/978-1-62703-161-5_23
137. Luo X, Wasilko DJ, Liu Y, Sun J, Wu X, Luo ZQ, et al. Structure of the Legionella virulence factor, SidC reveals a unique PI(4)P-specific binding domain essential for its targeting to the bacterial phagosome. PLoS Pathog. (2015) 11:e1004965. doi: 10.1371/journal.ppat.1004965
138. Urwyler S, Finsel I, Ragaz C, Hilbi H. Isolation of Legionella-containing vacuoles by immuno-magnetic separation. Curr Protoc Cell Biol. (2010) Chapter 3:Unit 3.34. doi: 10.1002/0471143030.cb0334s46
139. Hoffmann C, Finsel I, Hilbi H. Pathogen vacuole purification from Legionella-infected amoeba and macrophages. Methods Mol Biol. (2013) 954:309–21. doi: 10.1007/978-1-62703-161-5_18
140. Urwyler S, Nyfeler Y, Ragaz C, Lee H, Mueller LN, Aebersold R, et al. Proteome analysis of Legionella vacuoles purified by magnetic immunoseparation reveals secretory and endosomal GTPases. Traffic. (2009) 10:76–87. doi: 10.1111/j.1600-0854.2008.00851.x
141. Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, et al. IFNs modify the proteome of Legionella-containing vacuoles and restrict infection via IRG1-derived itaconic acid. PLoS Pathog. (2016) 12:e1005408. doi: 10.1371/journal.ppat.1005408
142. Finsel I, Hoffmann C, Hilbi H. Immunomagnetic purification of fluorescent Legionella-containing vacuoles. Methods Mol Biol. (2013) 983:431–43. doi: 10.1007/978-1-62703-302-2_24
143. Hammond GR, Machner MP, Balla T. A novel probe for phosphatidylinositol 4-phosphate reveals multiple pools beyond the Golgi. J Cell Biol. (2014) 205:113–26. doi: 10.1083/jcb.201312072
144. Hardie RC, Liu CH, Randall AS, Sengupta S. In vivo tracking of phosphoinositides in Drosophila photoreceptors. J Cell Sci. (2015) 128:4328–40. doi: 10.1242/jcs.180364
145. Rigden DJ. Identification and modelling of a PPM protein phosphatase fold in the Legionella pneumophila deAMPylase SidD. FEBS Lett. (2011) 585:2749–54. doi: 10.1016/j.febslet.2011.08.006
146. Tan Y, Luo ZQ. Legionella pneumophila SidD is a deAMPylase that modifies Rab1. Nature. (2011) 475:506–9. doi: 10.1038/nature10307
147. Neunuebel MR, Chen Y, Gaspar AH, Backlund PS Jr, Yergey A, Machner MP. De-AMPylation of the small GTPase Rab1 by the pathogen Legionella pneumophila. Science. (2011) 333:453–6. doi: 10.1126/science.1207193
148. Arasaki K, Roy CR. Legionella pneumophila promotes functional interactions between plasma membrane syntaxins and Sec22b. Traffic. (2010) 11:587–600. doi: 10.1111/j.1600-0854.2010.01050.x
149. Arasaki K, Toomre DK, Roy CR. The Legionella pneumophila effector DrrA is sufficient to stimulate SNARE-dependent membrane fusion. Cell Host Microbe. (2012) 11:46–57. doi: 10.1016/j.chom.2011.11.009
150. Derre I, Isberg RR. Legionella pneumophila replication vacuole formation involves rapid recruitment of proteins of the early secretory system. Infect Immun. (2004) 72:3048–53. doi: 10.1128/IAI.72.5.3048-3053.2004
151. Kagan JC, Stein MP, Pypaert M, Roy CR. Legionella subvert the functions of rab1 and sec22b to create a replicative organelle. J Exp Med. (2004) 199:1201–11. doi: 10.1084/jem.20031706
152. Chen Y, Machner MP. Targeting of the small GTPase Rab6A by the Legionella pneumophila effector LidA. Infect Immun. (2013) 81:2226–35. doi: 10.1128/IAI.00157-13
153. So EC, Schroeder GN, Carson D, Mattheis C, Mousnier A, Broncel M, et al. The Rab-binding profiles of bacterial virulence factors during infection. J Biol Chem. (2016) 291:5832–43. doi: 10.1074/jbc.M115.700930
154. Allgood SC, Romero Duenas BP, Noll RR, Pike C, Lein S, Neunuebel MR. Legionella effector AnkX disrupts host cell endocytic recycling in a phosphocholination-dependent manner. Front Cell Infect Microbiol. (2017) 7:397. doi: 10.3389/fcimb.2017.00397
155. Goody PR, Heller K, Oesterlin LK, Muller MP, Itzen A, Goody RS. Reversible phosphocholination of Rab proteins by Legionella pneumophila effector proteins. EMBO J. (2012) 31:1774–84. doi: 10.1038/emboj.2012.16
156. Weber SS, Ragaz C, Hilbi H. The inositol polyphosphate 5-phosphatase OCRL1 restricts intracellular growth of Legionella, localizes to the replicative vacuole and binds to the bacterial effector LpnE. Cell Microbiol. (2009) 11:442–60. doi: 10.1111/j.1462-5822.2008.01266.x
157. Newton HJ, Sansom FM, Bennett-Wood V, Hartland EL. Identification of Legionella pneumophila-specific genes by genomic subtractive hybridization with Legionella micdadei and identification of lpnE, a gene required for efficient host cell entry. Infect Immun. (2006) 74:1683–91. doi: 10.1128/IAI.74.3.1683-1691.2006
158. Newton HJ, Sansom FM, Dao J, McAlister AD, Sloan J, Cianciotto NP, et al. Sel1 repeat protein LpnE is a Legionella pneumophila virulence determinant that influences vacuolar trafficking. Infect Immun. (2007) 75:5575–85. doi: 10.1128/IAI.00443-07
159. de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, et al. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. (2008) 4:e1000117. doi: 10.1371/journal.ppat.1000117
160. Weigele BA, Orchard RC, Jimenez A, Cox GW, Alto NM. A systematic exploration of the interactions between bacterial effector proteins and host cell membranes. Nat Commun. (2017) 8:532. doi: 10.1038/s41467-017-00700-7
161. Voth KA, Chung IYW, van Straaten K, Li L, Boniecki MT, Cygler M. The structure of Legionella effector protein LpnE provides insights into its interaction with Oculocerebrorenal syndrome of Lowe (OCRL) protein. FEBS J. (2019) 286:710–25. doi: 10.1111/febs.14710
162. Loovers HM, Kortholt A, de Groote H, Whitty L, Nussbaum RL, van Haastert PJ. Regulation of phagocytosis in Dictyostelium by the inositol 5-phosphatase OCRL homolog Dd5P4. Traffic. (2007) 8:618–28. doi: 10.1111/j.1600-0854.2007.00546.x
163. Zhang X, Jefferson AB, Auethavekiat V, Majerus PW. The protein deficient in Lowe syndrome is a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. Proc Natl Acad Sci USA. (1995) 92:4853–6. doi: 10.1073/pnas.92.11.4853
164. Welin A, Weber S, Hilbi H. Quantitative imaging flow cytometry of Legionella-infected Dictyostelium amoebae reveals the impact of retrograde trafficking on pathogen vacuole composition. Appl Environ Microbiol. (2018) 84:e00158–e00118. doi: 10.1128/AEM.00158-18
165. Godi A, Pertile P, Meyers R, Marra P, Di Tullio G, Iurisci C, et al. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. (1999) 1:280–7. doi: 10.1038/12993
166. Hyvola N, Diao A, McKenzie E, Skippen A, Cockcroft S, Lowe M. Membrane targeting and activation of the Lowe syndrome protein OCRL1 by rab GTPases. EMBO J. (2006) 25:3750–61. doi: 10.1038/sj.emboj.7601274
Keywords: Dictyostelium discoideum, effector protein, endoplasmic reticulum, host-pathogen interaction, macrophage, pathogen vacuole, type IV secretion, vesicle trafficking
Citation: Swart AL and Hilbi H (2020) Phosphoinositides and the Fate of Legionella in Phagocytes. Front. Immunol. 11:25. doi: 10.3389/fimmu.2020.00025
Received: 14 November 2019; Accepted: 08 January 2020;
Published: 30 January 2020.
Edited by:
Carlos Rosales, National Autonomous University of Mexico, MexicoReviewed by:
Hayley J. Newton, The University of Melbourne, AustraliaYuxin Mao, Cornell University, United States
Copyright © 2020 Swart and Hilbi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hubert Hilbi, aGlsYmlAaW1tLnV6aC5jaA==