 Malte Roerden
Malte Roerden Annika Nelde
Annika Nelde Juliane S. Walz
Juliane S. Walz
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 20 December 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.03004
This article is part of the Research Topic Genetic Variants as Targets for Immunotherapy of Hematological Tumors View all 12 articles
Neoantigens derive from non-synonymous somatic mutations in malignant cells. Recognition of neoantigens presented via human leukocyte antigen (HLA) molecules on the tumor cell surface by T cells holds promise to enable highly specific and effective anti-cancer immune responses and thus neoantigens provide an exceptionally attractive target for immunotherapy. While genome sequencing approaches already enable the reliable identification of somatic mutations in tumor samples, the identification of mutation-derived, naturally HLA-presented neoepitopes as targets for immunotherapy remains challenging, particularly in low mutational burden cancer entities, including hematological malignancies. Several approaches have been utilized to identify neoepitopes from primary tumor samples. Besides whole genome sequencing with subsequent in silico prediction of potential mutation-derived HLA ligands, mass spectrometry (MS) allows for the only unbiased identification of naturally presented mutation-derived HLA ligands. The feasibility of characterizing and targeting these novel antigens has recently been demonstrated in acute myeloid leukemia (AML). Several immunogenic, HLA-presented peptides derived from mutated Nucleophosmin 1 (NPM1) were identified, allowing for the generation of T-cell receptor-transduced NPM1mut-specific T cells with anti-leukemic activity in a xenograft mouse model. Neoantigen-specific T-cell responses have also been identified for peptides derived from mutated isocitrate dehydrogenase (IDHmut), and specific T-cell responses could be induced by IDHmut peptide vaccination. In this review, we give a comprehensive overview on known neoantigens in hematological malignancies, present possible prediction and discovery tools and discuss their role as targets for immunotherapy approaches.
Recognition of tumor-associated antigens via human leukocyte antigen (HLA) molecules is pivotal for T cell-mediated tumor control and the induction of anti-tumor responses by immunotherapy (1). Neoantigens derive from non-synonymous somatic mutations and are of special interest, as they entail optimal tumor-specificity and lack central T-cell tolerance (2). These potentially highly immunogenic antigens are therefore considered prime targets for immunotherapy, particularly since neoantigens were described as targets of immune checkpoint inhibitor-induced anti-tumor T-cell responses (3, 4, 4–6). Recent advances in mass spectrometry (MS) (7, 8) and HLA antigen prediction algorithms (9–14) as well as the broad availability of whole genome sequencing (WGS) portrayed milestones in the field of cancer immunotherapy and hold promise to enable a robust and personalized identification of neoantigens in the future. The identification of spontaneous, neoantigen-specific T-cell responses in patients with long-lasting remissions suggests that neoantigen-specific targeting of tumor cells might enable durable anti-tumor responses (15–17). Long-lasting remissions could also be observed after personalized neoantigen-based peptide vaccination therapy in melanoma patients. Keeping the small sample size in these studies in consideration, these reports further indicate toward a therapeutic potential of neoantigens (18, 19). Following the success of checkpoint inhibitor therapy and the uncovering of the specificities of respective T-cell responses, a multitude of HLA-presented neoantigens have been identified in high mutational burden diseases such as melanoma (1, 3, 20). Hematological malignancies (HM) on the other hand are characterized by a low mutational burden and the role of neoantigens for immune-mediated tumor control and immunotherapeutic approaches in these entities remains to be elucidated. The immunogenicity of acute myeloid leukemia (AML) and other HM is demonstrated by the graft-vs.-leukemia/lymphoma effect and despite the typically low mutational burden, there has been steady progress in the identification of targetable neoantigens in these diseases in recent years. In this review we give an overview on known neoantigens in HM, their means of identification as well as the current state of efforts regarding the translation of these discoveries into the clinic.
While genome sequencing approaches already allow for the reliable identification of patient-individual, tumor-specific mutations (1, 21), the subsequent identification of mutation-derived neoantigens remains challenging (1, 22). These novel targets can be present as mutated membrane proteins or, more frequently, as HLA-presented peptide fragments derived from intracellular proteins comprising the mutated sequence (1, 22). Frequently, identification of these HLA-presented neoepitopes is performed by in silico prediction of potential HLA binding motifs based on identified somatic mutations and subsequent confirmation of immunogenicity in in vitro T-cell assays by priming of naïve T cells or demonstration of pre-existing memory T-cell responses (1). However, as there is no direct correlation between genome, transcriptome, and immunopeptidome (23–25), this “reverse immunology approach” based on gene expression data and in vitro analyses can provide several “false positive” neoantigens lacking natural presentation on the tumor cell surface (18). This lack of correlation between gene expression and the immunopeptidome can be explained by the complex process of HLA ligand formation, which is furthermore frequently altered in tumor cells (26–29). Thus, only a very small fraction of predicted neoantigens is actually naturally processed and presented via HLA molecules on the tumor cell, calling for direct identification methods of HLA-presented neoepitopes to identify suitable targets for immunotherapy. This can be achieved by MS-based immunopeptidomics, which enables the only unbiased, in-depth analysis of the naturally presented HLA immunopeptidome (8, 30). Recent reports estimate, that only approximately one mutation-derived HLA-presented neoepitope arises from about 1,000 non-synonymous mutations (18, 22, 31–34). In HM, which are typically low mutational burden diseases with only a handful to a few hundred mutations (20), this implicates a low abundance or even absence of HLA-presented neoepitopes. Considering further that these can derive from passenger mutations, which are sensitive to immune escape mechanisms (1, 22) and are mainly patient-specific, the presence of broadly targetable neoantigens cannot be taken for granted in these diseases. Nevertheless, identification and successful targeting of recurrent and mainly driver mutation-derived neoantigens has recently been demonstrated in various HM (35–49) (Table 1), thereby expanding the prospects for immunotherapy in these entities.

Table 1. Overview of neoantigens in hematological malignancies.
In AML, the mutational landscape is well-characterized (64) and several novel antigens derived from recurrent genetic alterations have been identified recently. Neoantigens derived from Nucleophosmin 1 mutations (NPM1mut), which occur in about 35% of AML patients (65), are arguably the most prominent targets in this regard. In most cases of NPM1mut AML, a frameshift mutation in exon 12 leads to an altered c-terminal protein sequence, which can specifically be recognized by cytotoxic CD8+ T cells (66). It has been proposed, that the immunogenicity of NPM1mut neoepitopes might add to the favorable prognosis of AML patients with NPM1 mutations (39). Several NPM1mut-derived HLA class I neoepitopes were identified by MS analysis in two recent studies (46, 50) and specific T-cell responses could be demonstrated in respective patients. Furthermore, isolation and transfer of a T-cell receptor (TCR) gene with an NPM1mut neoepitope specificity was performed in one of these studies. TCR-transduced T cells subsequently showed anti-tumor efficacy in an AML xenograft mouse model, thereby emphasizing the potential of NPM1mut-specific T-cell-based immunotherapy approaches for the treatment of NPM1mut AML (46). Missense mutations of isocitrate dehydrogenase (IDH) 1 or 2 can be detected in about 20% of AML patients, resulting in an altered, leukemogenesis promoting function of the enzymes (67). A study in glioma patients, where IDH mutations occur particularly frequent, identified an IDH1mut-derived HLA class II neoepitope and demonstrated its natural presentation and immunogenicity by detection of spontaneous CD4+ T-cell responses and mutation-specific antibodies in respective patients. CD4+ T-cell responses and mutation-specific antibody formation were subsequently induced by peptide vaccination in an HLA-humanized mouse model and led to IDH1mut-specific immune responses (44). Our own data support NPM1mut- and IDHmut-derived neoepitopes as promising targets in AML. Using our MS-based immunopeptidomics approach (24, 68–70), we were able to identify naturally presented HLA class I and II neoepitopes derived from mutated NPM1 and IDH2 in primary AML samples. Further analysis revealed multifunctional T-cell responses, and peptide-specific target cell killing was proven for one naturally presented NPM1mut neoepitope (51). Additionally, AML-specific neoantigens can arise from internal tandem duplications (ITD) of the FMS like tyrosine kinase 3 (FLT3) gene, which occur in up to 30% of AML patients (71). While these duplications vary in length, the same protein domain is affected in the majority of cases (72). These mutations can yield immunogenic peptides, as described for a FLT3-ITD-derived HLA-A*01:01-restricted neoepitope showing specific T-cell responses in vitro as well as ex vivo (38, 43). A further source of HLA-presented neoepitopes are fusion proteins. In AML, in vitro T-cell recognition of fusion protein-derived HLA-presented peptides has been demonstrated for PML-RARα (52), DEK-CAN (53), and ETV6–AML1 (54). While these reports arouse interest in these potential targets, the clinical significance of these in vitro analyses remains to be elucidated as natural presentation and spontaneous immune responses against respective HLA-presented neoepitopes have not been demonstrated.
In chronic myeloid leukemia (CML), peptides encompassing the BCR-ABL fusion site in theory represent optimal targets for immunotherapy, as this fusion protein is essential for the malignant transformation, is present in virtually all CML cells and patients, and potentially provides HLA binding motifs. One major throwback however is the occurrence of several different fusion sites resulting in diverse mutation-derived peptides in distinct patients. The t(9;22) translocation mainly leads to the formation of an exon junction between exon 2 or 3 of BCR and exon 2 of ABL (b2a2 and b3a2, respectively) (73). Several groups have described specific T-cell responses against HLA-presented peptides derived from b3a2 (41, 42, 55, 56). Furthermore, it has been demonstrated that BCR-ABL-specific T-cell responses can be induced with peptide vaccination in CML patients (57). One study reported the direct identification of a HLA-presented b3a2-derived neoepitope by MS on primary CML cells (58). However, in our recently performed extensive MS-based analysis of the primary CML immunopeptidome, we could not identify any naturally HLA-presented peptides encompassing BCR-ABL- or ABL-BCR-derived neoepitopes (68), keeping in mind that especially for MS-based immunopeptidomics, absence of evidence does not equal evidence of absence. Notably, neoantigens can also arise under therapy with tyrosine kinase inhibitors. In patients with imatinib-resistant CML, drug resistance-mediating mutations outside the BCR-ABL fusion site have been identified (59) and specific T-cell responses against neoepitopes derived from these mutations have been demonstrated and were linked with clinical response (59). Myeloproliferative disorders (MPN) are characterized by a homogenous mutational landscape with recurrent driver mutations, which in theory represent shared and therefore broadly applicable targets for immunotherapy. A single nucleotide mutation of the janus kinase 2 gene (JAK2 V416F) is the most frequent among MPN driver mutations, occurring in more than 90% of patients with Polycythemia vera (PV) and about 50% of patients with Essential thrombocytosis (ET) and Primary myelofibrosis (PMF), respectively (74). While experimental recognition of JAK2mut-derived neoepitopes has been demonstrated after in vitro priming of healthy donor T cells, thereby indicating the immunogenic potential, no spontaneous T-cell responses have been identified in JAK2mut MPN patients (48). Direct identification of naturally presented JAK2mut neoepitopes has not been reported so far. In MPN with Calreticulin mutations (CALRmut)—the most common driver mutation in JAK2 wildtype (JAK2wt) MPN, occurring in about 25% of ET and PMF patients (74)—a frameshift mutation leads to an altered c-terminus of the protein. Recent reports evaluated CALRmut-derived HLA-presented neoepitopes as targets for immunotherapy (45, 47, 49, 61, 62) as these peptides were predicted to bind HLA-A*03:01 and HLA-B*07:02. While natural presentation of these HLA class I neoepitopes could not be demonstrated upon MS analysis (45), spontaneous, primarily CD4+ T-cell responses against several CALRmut-derived neoepitopes could be identified in CALRmut MPN patients (49). Further, CALRmut-dependent killing of autologous CALRmut cells was demonstrated in an HLA-DR-restricted manner (47). Of note, while the observed CALRmut-specific T-cell responses in CALRmut MPN patients were often weak (45, 49, 61), these could be restored by immune checkpoint blockade both in vitro and in vivo (61). These findings indicate that CALRmut is naturally presented but respective T-cell responses are suppressed by immune checkpoint receptor signaling (61), illustrating the potential of combining distinct immunotherapeutic approaches for the treatment of HM. Taken together, the results revealed CALRmut-derived neoepitopes as shared MPN-specific neoantigens, prompting their further evaluation for therapeutic targeting (47, 49, 61, 62). Interestingly, CALRmut-specific memory T-cell responses were frequently detectable in healthy individuals in a subsequent study, suggesting a previous clearance of CALRmut cells by immunosurveillant T cells and thereby further highlighting the immunogenic potential of CALRmut neoantigens (62). Mutations in the myeloproliferative leukemia virus oncogene (MPL) are further recurrent driver mutations in JAK2wt MPN (75). A recent study performing an in silico analysis based on whole transcriptome sequencing of MPN patients predicted several MPLmut HLA class I binding neoepitopes (63). However, demonstration of natural presentation of MPLmut neoepitopes by MS has not been performed thus far.
FBXW7 is a tumor suppressor gene with mutations occurring in various HM, most frequently in T-ALL (76). Specific CD8+ T-cell responses against a recurrent FBXW7mut-derived neoepitope have been demonstrated, suggesting that this recurrent mutation might represent another neoantigen applicable for immunotherapy in HM (45). Mutation-derived neoantigens have also been identified in chronic lymphocytic leukemia (CLL). In a study, implementing a reverse immunology approach, immune responses were detected against HLA antigens derived from somatic mutations in ALMS1, C6ORF89, and FNDC3B (17). However, it should be kept in mind that these mutations are not considered driver mutations, thus theoretically making them suboptimal targets for immunotherapy. The identification of T-cell responses against these mutated neoantigens nevertheless demonstrated the applicability of neoantigen-specific targeting of CLL. Evaluation of neoantigens in mantle cell lymphoma patients applying a combined approach of whole exome sequencing and direct HLA ligand identification by MS revealed the presence of naturally presented HLA class II neoepitopes derived from the lymphoma immunoglobulin heavy- or light-chain variable regions (60). Spontaneous CD4+ T-cell responses could be identified against these neoepitopes and mediated tumor-specific killing of autologous lymphoma cells (60). MYD88 (L265P) is a recurrent driver mutation in Waldenstom's macroglobulinemia, CLL and other Non-Hodgkin lymphomas (77). We previously evaluated HLA class I neoepitopes derived from MYD88mut as targets for immunotherapy in lymphoma patients (40). Based on in silico HLA motif prediction, further immunogenicity evaluation of possible MYD88mut-derived HLA class I neoepitopes was performed. In vitro priming of naïve T cells from MYD88mut CLL patients and healthy donors was successful for several HLA-B:07- and HLA-B:15-restricted neoepitopes. While further analysis revealed that spontaneous MYD88mut-specific T-cell responses are infrequent in lymphoma patients, these de novo induced MYD88mut-specific T cells were multifunctional and elicited mutation-restricted cytotoxicity (40), highlighting the potential of MYD88mut neoepitopes as targets for immunotherapy.
While the term neoantigen is mostly used referring to mutation-derived HLA-presented neoepitopes, the following section will discuss further sources of neoantigens that might also represent promising targets for immunotherapy. In distinction to mutations of the target's primary genome sequence, non-canonical neoantigens or cryptic peptides arise among others from tumor-specific alterations of the HLA antigen presentation machinery, DNA methylation, RNA editing or protein biosynthesis, proteasomal splicing, or non-canonical translation products (Figure 1) (29, 78–83). Further, tumor-specific splice variant proteins can result from splice-site creating mutations or mutations of spliceosome proteins and can lead to the formation of tumor-specific HLA-presented neoepitopes (84). Mutations directly affecting the spliceosome, such as SF3B1 or SRSF2 mutations, have been shown to occur in up to 20% of de novo AML (64) and 15% of PMF (63). In AML, frequently occurring splice variants have been identified for NOTCH2 (35), FLT3 (35), and CD44 (36), each leading to the occurrence of an altered cell surface protein. Specific targeting of a CD44 splice variant has been demonstrated by a CD44v6 CAR T cell in a mouse model (36), thereby highlighting the potential of targeting this class of neoantigens with immunotherapeutic approaches. Post-translational protein modifications, which are preserved in HLA-presented peptides, can portray another source of neoantigens as these tumor-specific alterations can lead to the formation of novel epitopes (29). In this regard, neoantigens resulting from tumor-specific phosphorylation as well as glycosylation have been identified in AML and immunogenicity of this class of neoantigens has been demonstrated (28, 37, 85, 86).
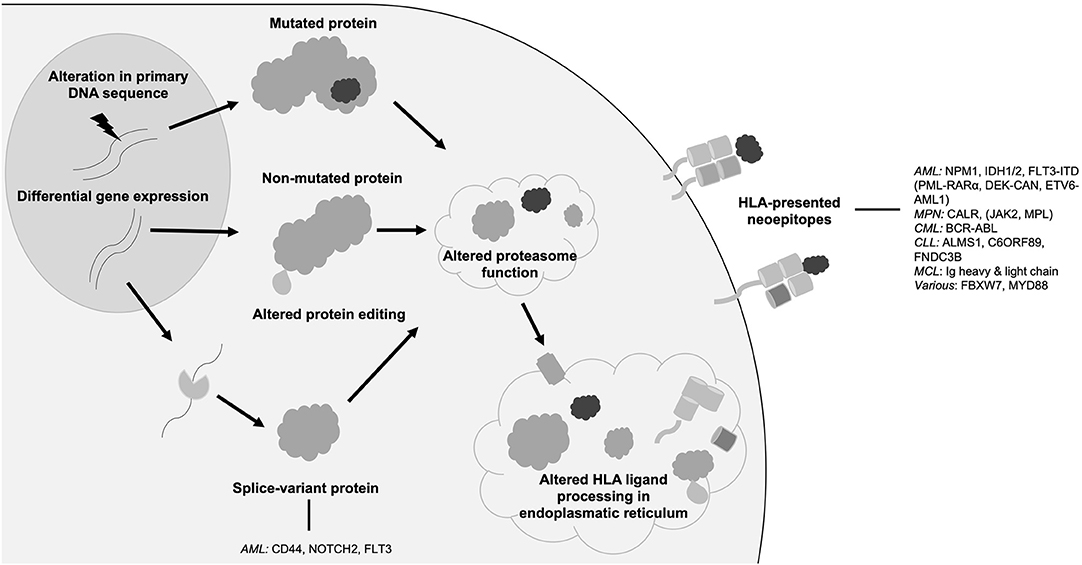
Figure 1. Origins of neoantigens. Schematic overview showing different origins of neoantigens with prominent examples in hematological malignancies. AML, acute myeloid leukemia; FLT3, FMS like tyrosine kinase 3; HLA, human leukocyte antigen; NPM1, nucleophosmin 1; IDH, isocitrate dehydrogenase; MPN, myeloproliferative neoplasia; CALR, calreticulin; JAK2 janus like kinase 2; CML, chronic myeloid leukemia; CLL, chronic lymphocytic leukemia; MCL, mantle cell lymphoma; Ig, immunoglobulin.
Neoantigen targeting holds promise to enable highly specific and durable anti-tumor immune responses (1, 22). Although HM are typically low mutational burden diseases (20), there has recently been remarkable progress in the uncovering of neoantigens in these entities. These discoveries were fueled by an immense progress in the field of WGS, steadily improved HLA motif prediction algorithms as well as technical advances in MS in recent years (1, 21, 31). While these advances already facilitate neoantigen identification from primary tumor samples, we are likely only seeing the beginning of personalized target evaluation. In this progress, a standard approach for target identification has yet to be defined (1, 22). As optimal target selection is a prerequisite for effective immunotherapy, we consider the direct identification of HLA-presented neoantigens by MS as the optimal approach. The direct identification of potential targets with MS harbors essential advantages when compared to the reverse immunology approach, which relies on neoantigen prediction, experimental HLA-binding and further immunogenicity confirmation in T-cell assays (1, 22). Here, “false targets” might be identified if HLA binding is demonstrated for predicted neoantigens in vitro, but these antigens are not naturally presented via HLA antigens in vivo. Furthermore, while the ex vivo identification of spontaneous T-cell responses against neoantigens can be regarded as evidence for natural HLA-presentation, potential targets without pre-existing responses might be missed. This is concerning as antigen-specific immunotherapy, including antibody strategies and peptide vaccines in particular aim to induce de novo anti-tumor responses. HM have been the first entities where immunotherapy—in form of allogenic stem cell transplantation (87)—has been performed and the immunogenicity of HM is long known (88, 89). While targeting of mutated membrane proteins by antibodies or CAR T cells has already been established for the treatment of HM, but is restricted to very few suitable surface antigens (90–94), HLA-presented neoantigens derived from intracellular proteins are of particular interest for immunotherapy. Hence, recent highly noted reports on the identification of neoantigens in HM have raised hopes that these novel targets might bring along new therapeutic options. In AML, NPM1mut- and IDHmut-derived neoantigens thus far represent the most promising targets, as these mutations occur frequently and successful as well as specific targeting has already been demonstrated in preclinical studies (44, 46, 51). Neoepitopes derived from fusion proteins are equally interesting targets, but natural presentation via HLA molecules has not been demonstrated so far (41, 42, 52–54, 56, 57, 95). While adoptive T-cell transfer and peptide vaccination approaches using non-mutated antigens are already under clinical evaluation in AML and other HM (96–98), the identification of these neoantigens will allow for an even more targeted approach in the future. However, it should be kept in mind that a personalized target selection remains challenging, as MS analysis is elaborate and not universally available. Furthermore, completely personalized immunotherapy approaches at the same time bring the difficulty of manufacturing an individualized product, e.g., a peptide vaccine, for each patient. A “warehouse” model, where a patient-specific selection of therapeutics targeting frequently occurring neoantigens can be made, might represent an elegant solution to this problem. This is particular true for malignancies with a well-characterized mutational landscape and a narrow spectrum of recurrent mutations, such as AML and MPN (20). At the same time and despite the recent progress in neoantigen identification in HM, the infrequency of individual mutations and HLA allotype restrictions still limit specific neoantigen targeting to a subset of patients. To overcome this issue, combined targeting of both mutated and non-mutated, tumor-exclusive antigens might be a suitable approach. Tumor-exclusive non-mutated neoepitopes can arise as a consequence of differential gene expression or tumor-specific alterations of RNA- and protein-processing, as described for splice variant proteins in AML (35, 36, 64) and have been shown to possess equally immunogenic properties as mutated neoepitopes (99). With mutations of spliceosome proteins, transcription factors and DNA methylation related proteins occurring frequently in AML and other HM (20, 64), additional non-canonical neoantigens are likely awaiting uncovering (35, 36, 84). Recent neoantigen discoveries have created novel and promising prospects for immunotherapy in HM. With currently ongoing endeavors, additional neoantigens might be uncovered and personalized target evaluation will be taken another step further. Considering the paucity of targetable mutated neoantigens in the individual patient due to HLA allotype restrictions and patient-individual mutations, combined approaches targeting both mutated and non-mutated tumor-exclusive antigens are likely warranted in patients with HM. Harnessing “the best of both worlds” might then enable immunotherapy to unfold its full potential in hematological malignancies.
MR wrote the first draft of the manuscript. AN and JW wrote further sections of the manuscript. All authors contributed to manuscript revision, read, and approved the final version.
We acknowledge support by Deutsche Forschungsgemeinschaft and Open Access Publishing Fund of University of Tübingen.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
2. Gilboa E. The makings of a tumor rejection antigen. Immunity. (1999) 11:263–70. doi: 10.1016/S1074-7613(00)80101-6
3. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. (2016) 351:1463–9. doi: 10.1126/science.aaf1490
4. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. (2017) 7:264–76. doi: 10.1158/2159-8290.CD-16-0828
5. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. (2017) 16:2598–608. doi: 10.1158/1535-7163.MCT-17-0386
6. Hellmann MD, Ciuleanu T-E, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Eng J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
7. Marino F, Chong C, Michaux J, Bassani-Sternberg M. High-throughput, fast, and sensitive immunopeptidomics sample processing for mass spectrometry. Methods Mol Biol. (2019) 1913:67–79. doi: 10.1007/978-1-4939-8979-9_5
8. Bassani-Sternberg M. Mass spectrometry based immunopeptidomics for the discovery of cancer neoantigens. Methods Mol Biol. (2018) 1719:209–21. doi: 10.1007/978-1-4939-7537-2_14
9. Bulik-Sullivan B, Busby J, Palmer CD, Davis MJ, Murphy T, Clark A, et al. Deep learning using tumor HLA peptide mass spectrometry datasets improves neoantigen identification. Nat Biotechnol. (2019) 37:55. doi: 10.1038/nbt.4313
10. Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol. (2017) 199:3360–8. doi: 10.4049/jimmunol.1700893
11. Martins J, Magalhães C, Rocha M, Osório NS. Machine learning-enhanced T cell neoepitope discovery for immunotherapy design. Cancer Inform. (2019) 18:1176935119852081. doi: 10.1177/1176935119852081
12. Mellacheruvu D, Phillips N, Bartha G, Harris J, Power R, McClory R, et al. Applying Immunopeptidomics and Machine Learning to Improve Neoantigen Prediction for Therapeutic and Diagnostic Use. AACR (2019).
13. O'Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. (2018) 7:129–32.e4. doi: 10.1016/j.cels.2018.05.014
14. Phloyphisut P, Pornputtapong N, Sriswasdi S, Chuangsuwanich E. MHCSeqNet: a deep neural network model for universal MHC binding prediction. BMC Bioinformatics. (2019) 20:270. doi: 10.1186/s12859-019-2892-4
15. Melero I, Navarro B, Teijeira A, Coukos G. Cancer immunotherapy full speed ahead. Ann Oncol. (2017) 28(Suppl. 12):xii1–xii2. doi: 10.1093/annonc/mdx739
16. Müller-Schmah C, Solari L, Weis R, Pfeifer D, Scheibenbogen C, Trepel M, et al. Immune response as a possible mechanism of long-lasting disease control in spontaneous remission of MLL/AF9-positive acute myeloid leukemia. Ann Hematol. (2012) 91:27–32. doi: 10.1007/s00277-011-1332-y
17. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. (2014) 124:453–62. doi: 10.1182/blood-2014-04-567933
18. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. (2017) 547:217. doi: 10.1038/nature22991
19. Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222. doi: 10.1038/nature23003
20. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. (2017) 9:34. doi: 10.1186/s13073-017-0424-2
21. Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. (2010) 11:31. doi: 10.1038/nrg2626
22. Finn OJ, Rammensee H-G. Is it possible to develop cancer vaccines to neoantigens, what are the major challenges, and how can these be overcome? Neoantigens: nothing new in spite of the name. Cold Spring Harb Perspect Biol. (2018) 10:a028829. doi: 10.1101/cshperspect.a028829
23. Bassani-Sternberg M, Pletscher-Frankild S, Jensen LJ, Mann M. Mass spectrometry of human leukocyte antigen class I peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol Cell Proteomics. (2015) 14:658–73. doi: 10.1074/mcp.M114.042812
24. Berlin C, Kowalewski D, Schuster H, Mirza N, Walz S, Handel M, et al. Mapping the HLA ligandome landscape of acute myeloid leukemia: a targeted approach toward peptide-based immunotherapy. Leukemia. (2015) 29:647. doi: 10.1038/leu.2014.233
25. Weinzierl AO, Lemmel C, Schoor O, Müller M, Krüger T, Wernet D, et al. Distorted relation between mRNA copy number and corresponding major histocompatibility complex ligand density on the cell surface. Mol Cell Proteomics. (2007) 6:102–13. doi: 10.1074/mcp.M600310-MCP200
26. Hoyos LE, Abdel-Wahab O. Cancer-specific splicing changes and the potential for splicing-derived neoantigens. Cancer Cell. (2018) 34:181–3. doi: 10.1016/j.ccell.2018.07.008
27. Mani A, Gelmann EP. The ubiquitin-proteasome pathway and its role in cancer. J Clin Oncol. (2005) 23:4776–89. doi: 10.1200/JCO.2005.05.081
28. Depontieu FR, Qian J, Zarling AL, McMiller TL, Salay TM, Norris A, et al. Identification of tumor-associated, MHC class II-restricted phosphopeptides as targets for immunotherapy. Proc Natl Acad Sci USA. (2009) 106:12073–8. doi: 10.1073/pnas.0903852106
29. Janeway CA, Travers P, Walport M, Shlomchik M. Immunobiology: The Immune System in Health and Disease. London: Current Biology (1996).
30. Freudenmann LK, Marcu A, Stevanović S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology. (2018) 154:331–45. doi: 10.1111/imm.12936
31. Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. (2014) 515:572. doi: 10.1038/nature14001
32. Bassani-Sternberg M, Braunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat Commun. (2016) 7:13404. doi: 10.1038/ncomms13404
33. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. (2014) 515:577. doi: 10.1038/nature13988
34. Löffler MW, Chandran PA, Laske K, Schroeder C, Bonzheim I, Walzer M, et al. Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J Hepatol. (2016) 65:849–55. doi: 10.1016/j.jhep.2016.06.027
35. Adamia S, Bar-Natan M, Haibe-Kains B, Pilarski PM, Bach C, Pevzner S, et al. NOTCH2 and FLT3 gene mis-splicings are common events in patients with acute myeloid leukemia (AML): new potential targets in AML. Blood. (2014) 123:2816–25. doi: 10.1182/blood-2013-02-481507
36. Casucci M, di Robilant BN, Falcone L, Camisa B, Norelli M, Genovese P, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. (2013) 122:3461–72. doi: 10.1182/blood-2013-04-493361
37. Cobbold M, De La Peña H, Norris A, Polefrone JM, Qian J, English AM, et al. MHC class I–associated phosphopeptides are the targets of memory-like immunity in leukemia. Sci Transl Med. (2013) 5:203ra125. doi: 10.1126/scitranslmed.3006061
38. Graf C, Heidel F, Tenzer S, Radsak MP, Solem FK, Britten CM, et al. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood. (2007) 109:2985–8. doi: 10.1182/blood-2006-07-032839
39. Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Götz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4+ and CD8+ T-cell responses in patients with acute myeloid leukemia. Blood. (2012) 120:1282–9. doi: 10.1182/blood-2011-11-394395
40. Nelde A, Walz JS, Kowalewski DJ, Schuster H, Wolz O-O, Peper JK, et al. HLA class I-restricted MYD88 L265P-derived peptides as specific targets for lymphoma immunotherapy. Oncoimmunology. (2017) 6:e1219825. doi: 10.1080/2162402X.2016.1219825
41. Norbury LC, Clark RE, Christmas SE. b3a2 BCR–ABL fusion peptides as targets for cytotoxic T cells in chronic myeloid leukaemia. Br J Haematol. (2000) 109:616–21. doi: 10.1046/j.1365-2141.2000.02090.x
42. Pawelec G, Max H, Halder T, Bruserud O, Merl A, da Silva P, et al. BCR/ABL leukemia oncogene fusion peptides selectively bind to certain HLA-DR alleles and can be recognized by T cells found at low frequency in the repertoire of normal donors. Blood. (1996) 88:2118–24. doi: 10.1182/blood.V88.6.2118.bloodjournal8862118
43. Scholl S, Salzmann S, Kaufmann AM, Höffken K. Flt3-ITD mutations can generate leukaemia specific neoepitopes: potential role for immunotherapeutic approaches. Leukemia Lymph. (2006) 47:307–12. doi: 10.1080/10428190500301306
44. Schumacher T, Bunse L, Pusch S, Sahm F, Wiestler B, Quandt J, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. (2014) 512:324. doi: 10.1038/nature13387
45. Tubb VM, Schrikkema DS, Croft NP, Purcell AW, Linnemann C, Freriks MR, et al. Isolation of T cell receptors targeting recurrent neoantigens in hematological malignancies. J Immunother Cancer. (2018) 6:70. doi: 10.1186/s40425-018-0386-y
46. van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RC, Kester MG, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Investig. (2019) 129:774–78. doi: 10.1172/JCI97482
47. Holmström M, Martinenaite E, Ahmad S, Met Ö, Friese C, Kjaer L, et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia. (2018) 32:429. doi: 10.1038/leu.2017.214
48. Holmström MO, Hjortsø M, Ahmad S, Met Ö, Martinenaite E, Riley C, et al. The JAK2 V617F mutation is a target for specific T cells in the JAK2 V617F-positive myeloproliferative neoplasms. Leukemia. (2017) 31:495–8. doi: 10.1038/leu.2016.290
49. Holmström MO, Riley CH, Svane IM, Hasselbalch HC, Andersen MH. The CALR exon 9 mutations are shared neoantigens in patients with CALR mutant chronic myeloproliferative neoplasms. Leukemia. (2016) 30:2413. doi: 10.1038/leu.2016.233
50. Narayan R, Olsson N, Wagar LE, Medeiros BC, Meyer E, Czerwinski D, et al. Acute myeloid leukemia immunopeptidome reveals HLA presentation of mutated nucleophosmin. PloS ONE. (2019) 14:e0219547. doi: 10.1371/journal.pone.0219547
51. Nelde A, Kowalewski DJ, Salih HR, Marcu A, Bilich T, Neidert MC, et al. Identification of naturally presented HLA ligands of CD34+CD38- acute myeloid leukemia (AML) progenitor cells for T cell-based immunotherapy – neoepitopes and self-peptides as novel targets for anti-cancer therapy. In: Abstract CIMT 2019 Annual Meeting, (Mainz) (2019).
52. Gambacorti-Passerini C, Grignani F, Arienti F, Pandolfi PP, Pelicci PG, Parmiani G. Human CD4 lymphocytes specifically recognize a peptide representing the fusion region of the hybrid protein pml/RAR alpha present in acute promyelocytic leukemia cells. Blood. (1993) 81:1369–75. doi: 10.1182/blood.V81.5.1369.1369
53. Makita M, Azuma T, Hamaguchi H, Niiya H, Kojima K, Fujita S, et al. Leukemia-associated fusion proteins, dek-can and bcr-abl, represent immunogenic HLA-DR-restricted epitopes recognized by fusion peptide-specific CD4+ T lymphocytes. Leukemia. (2002) 16:2400. doi: 10.1038/sj.leu.2402742
54. Yotnda P, Garcia F, Peuchmaur M, Grandchamp B, Duval M, Lemonnier F, et al. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J Clin Investig. (1998) 102:455–62. doi: 10.1172/JCI3126
55. Bocchia M, Korontsvit T, Xu Q, Mackinnon S, Yang S, Sette A, et al. Specific human cellular immunity to bcr-abl oncogene-derived peptides. Blood. (1996) 87:3587–92. doi: 10.1182/blood.V87.9.3587.bloodjournal8793587
56. Yasukawa M, Ohminami H, Kaneko S, Yakushijin Y, Nishimura Y, Inokuchi K, et al. CD4+ Cytotoxic T-cell clones specific for bcr-abl b3a2 fusion peptide augment colony formation by chronic myelogenous leukemia cells in a b3a2-specific and HLA-DR–restricted manner. Blood. (1998) 92:3355–61.
57. Pinilla-Ibarz J, Cathcart K, Korontsvit T, Soignet S, Bocchia M, Caggiano J, et al. Vaccination of patients with chronic myelogenous leukemia with bcr-abl oncogene breakpoint fusion peptides generates specific immune responses. Blood. (2000) 95:1781–7. doi: 10.1182/blood.V95.5.1781.005k46_1781_1787
58. Clark RE, Dodi IA, Hill SC, Lill JR, Aubert G, Macintyre AR, et al. Direct evidence that leukemic cells present HLA-associated immunogenic peptides derived from the BCR-ABL b3a2 fusion protein. Blood. (2001) 98:2887–93. doi: 10.1182/blood.V98.10.2887
59. Cai A, Keskin DB, DeLuca DS, Alonso A, Zhang W, Zhang GL, et al. Mutated BCR-ABL generates immunogenic T-cell epitopes in CML patients. Clin Cancer Res. (2012) 18:5761–72. doi: 10.1158/1078-0432.CCR-12-1182
60. Khodadoust MS, Olsson N, Wagar LE, Haabeth OA, Chen B, Swaminathan K, et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature. (2017) 543:723. doi: 10.1038/nature21433
61. Bozkus CC, Roudko V, Finnigan JP, Mascarenhas J, Hoffman R, Iancu-Rubin C, et al. Immune checkpoint blockade enhances shared neoantigen-induced T cell immunity directed against mutated calreticulin in myeloproliferative neoplasms. Cancer Discov. (2019) 9:1192–207. doi: 10.1158/2159-8290.CD-18-1356
62. Holmström MO, Ahmad SM, Klausen U, Bendtsen SK, Martinenaite E, Riley CH, et al. High frequencies of circulating memory T cells specific for calreticulin exon 9 mutations in healthy individuals. Blood Cancer J. (2019) 9:8. doi: 10.1038/s41408-018-0166-4
63. Schischlik F, Jäger R, Rosebrock F, Hug E, Schuster M, Holly R, et al. Mutational landscape of the transcriptome offers a rich neoantigen resource for immunotherapy of myeloproliferative neoplasms. Blood. (2018) 132(Suppl. 1):3058. doi: 10.1182/blood-2018-99-118049
64. Eisfeld A-K, Mrózek K, Kohlschmidt J, Nicolet D, Orwick S, Walker CJ, et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia. (2017) 31:2211. doi: 10.1038/leu.2017.86
65. Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Eng J Med. (2005) 352:254–66. doi: 10.1056/NEJMoa041974
66. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Eng J Med. (2016) 374:2209–21. doi: 10.1056/NEJMoa1516192
67. Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Krönke J, Bullinger L, et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol. (2010) 28:3636–43. doi: 10.1200/JCO.2010.28.3762
68. Bilich T, Nelde A, Bichmann L, Roerden M, Salih HR, Kowalewski DJ, et al. The HLA ligandome landscape of chronic myeloid leukemia delineates novel T-cell epitopes for immunotherapy. Blood. (2019) 133:550–65. doi: 10.1182/blood-2018-07-866830
69. Kowalewski DJ, Schuster H, Backert L, Berlin C, Kahn S, Kanz L, et al. HLA ligandome analysis identifies the underlying specificities of spontaneous antileukemia immune responses in chronic lymphocytic leukemia (CLL). Proc Natl Acad Sci USA. (2015) 112:E166–75. doi: 10.1073/pnas.1416389112
70. Walz S, Stickel JS, Kowalewski DJ, Schuster H, Weisel K, Backert L, et al. The antigenic landscape of multiple myeloma: mass spectrometry (re) defines targets for T-cell–based immunotherapy. Blood. (2015) 126:1203–13. doi: 10.1182/blood-2015-04-640532
71. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. (2003) 3:650. doi: 10.1038/nrc1169
72. Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. (2002) 21:2555. doi: 10.1038/sj.onc.1205332
73. Ren R. Mechanisms of BCR–ABL in the pathogenesis of chronic myelogenous leukaemia. Nat Rev Cancer. (2005) 5:172. doi: 10.1038/nrc1567
74. Nangalia J, Green TR. The evolving genomic landscape of myeloproliferative neoplasms. Hematology Am Soc Hematol Educ Program. (2014) 2014:287–96. doi: 10.1182/asheducation-2014.1.287
75. Beer PA, Campbell PJ, Scott LM, Bench AJ, Erber WN, Bareford D, et al. MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood. (2008) 112:141–9. doi: 10.1182/blood-2008-01-131664
76. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. (2007) 67:9006–12. doi: 10.1158/0008-5472.CAN-07-1320
77. Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim K-H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. (2011) 470:115. doi: 10.1038/nature09671
78. Chong C, Müller M, Pak H, Harnett D, Huber F, Grun D, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. bioRxiv. (2019). doi: 10.1101/758680
79. Laumont CM, Daouda T, Laverdure J-P, Bonneil É, Caron-Lizotte O, Hardy M-P, et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat Commun. (2016) 7:10238. doi: 10.1038/ncomms10238
80. Liepe J, Marino F, Sidney J, Jeko A, Bunting DE, Sette A, et al. A large fraction of HLA class I ligands are proteasome-generated spliced peptides. Science. (2016) 354:354–8. doi: 10.1126/science.aaf4384
81. Liepe J, Sidney J, Lorenz FK, Sette A, Mishto M. Mapping the MHC Class I–Spliced Immunopeptidome of Cancer Cells. Cancer Immunol Res. (2019) 7:62–76. doi: 10.1158/2326-6066.CIR-18-0424
82. Mishto M, Liepe J. Post-translational peptide splicing and T cell responses. Trends Immunol. (2017) 38:904–15. doi: 10.1016/j.it.2017.07.011
83. Starck SR, Shastri N. Non-conventional sources of peptides presented by MHC class I. Cell Mol Life Sci. (2011) 68:1471–9. doi: 10.1007/s00018-011-0655-0
84. Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. (2018) 23:270–81. e3. doi: 10.1016/j.celrep.2018.03.052
85. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
86. Malaker SA, Penny SA, Steadman LG, Myers PT, Loke JC, Raghavan M, et al. Identification of glycopeptides as posttranslationally modified neoantigens in leukemia. Cancer Immunol Res. (2017) 5:376–84. doi: 10.1158/2326-6066.CIR-16-0280
87. Thomas ED, Lochte HL Jr, Lu WC, Ferrebee JW. Intravenous infusion of bone marrow in patients receiving radiation and chemotherapy. N Eng J Med. (1957) 257:491–6. doi: 10.1056/NEJM195709122571102
88. Greiner J, Ringhoffer M, Simikopinko O, Szmaragowska A, Huebsch S, Maurer U, et al. Simultaneous expression of different immunogenic antigens in acute myeloid leukemia. Exp Hematol. (2000) 28:1413–22. doi: 10.1016/S0301-472X(00)00550-6
89. Heslop HE, Stevenson FK, Molldrem JJ. Immunotherapy of hematologic malignancy. Hematology Am Soc Hematol Educ Program. (2003) 2003:331–49. doi: 10.1182/asheducation-2003.1.331
90. DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Eng J Med. (2018) 378:2386–98. doi: 10.1056/NEJMoa1716984
91. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Eng J Med. (2017) 377:454–64. doi: 10.1056/NEJMoa1614359
92. Katz J, Janik JE, Younes A. Brentuximab vedotin (SGN-35). Clin Cancer Res. (2011) 17:6428–36. doi: 10.1158/1078-0432.CCR-11-0488
93. Plosker GL, Figgitt DP. Rituximab. Drugs. (2003) 63:803–43. doi: 10.2165/00003495-200363080-00005
94. Kochenderfer JN, Rosenberg SA. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat Rev Clin Oncol. (2013) 10:267. doi: 10.1038/nrclinonc.2013.46
95. Yang W, Lee K-W, Srivastava RM, Kuo F, Krishna C, Chowell D, et al. Immunogenic neoantigens derived from gene fusions stimulate T cell responses. Nat Med. (2019) 25:767. doi: 10.1038/s41591-019-0434-2
96. ClinicalTrials.gov. Identifier NCT02802943, iVAC-CLL01: Patient-Individualized Peptide Vaccination After First Line Therapy of CLL. Bethesda, MD: National Library of Medicine (2016). Available online at: https://clinicaltrials.gov/ct2/show/NCT02802943 (accessed September 22, 2019).
97. ClinicalTrials.gov. Identifier NCT00153582, WT1 Peptide Vaccination in Acute Myeloid Leukemia (AML). Bethesda, MD: National Library of Medicine (2005). Available online at: https://clinicaltrials.gov/ct2/show/NCT00153582 (accessed September 22, 2019).
98. ClinicalTrials.gov. Identifier NCT02550535, A Phase I/II Study of Gene-Modified WT1 TCR Therapy in MDS & AML Patients. Bethesda, MD: National Library of Medicine (2015) Available online at: https://clinicaltrials.gov/ct2/show/NCT02550535 (accessed September 22, 2019).
Keywords: neoantigens, hematological malignancies, mass spectrometry, immunopeptidomics, HLA antigens, NPM1 mutations
Citation: Roerden M, Nelde A and Walz JS (2019) Neoantigens in Hematological Malignancies—Ultimate Targets for Immunotherapy? Front. Immunol. 10:3004. doi: 10.3389/fimmu.2019.03004
Received: 28 September 2019; Accepted: 06 December 2019;
Published: 20 December 2019.
Edited by:
Marieke Griffioen, Leiden University Medical Center, NetherlandsReviewed by:
Per Thor Straten, Herlev Hospital, DenmarkCopyright © 2019 Roerden, Nelde and Walz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juliane S. Walz, anVsaWFuZS53YWx6QG1lZC51bmktdHVlYmluZ2VuLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.