 Ana Batista-Gonzalez
Ana Batista-Gonzalez Roberto Vidal
Roberto Vidal Alfredo Criollo
Alfredo Criollo Leandro J. Carreño
Leandro J. Carreño
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 10 January 2020
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02993
This article is part of the Research TopicMacrophage Metabolism and Immune ResponsesView all 9 articles
Macrophage activation is intimately linked to metabolic reprogramming. Inflammatory (M1) macrophages are able to sustain inflammatory responses and to kill pathogens, mostly by relying on aerobic glycolysis and fatty acid biosynthesis. Glycolysis is a fast way of producing ATP, and fatty acids serve as precursors for the synthesis of inflammatory mediators. On the opposite side, anti-inflammatory (M2) macrophages mediate the resolution of inflammation and tissue repair, switching their metabolism to fatty acid oxidation and oxidative phosphorylation. Over the years, this classical view has been challenged by recent discoveries pointing to a more complex metabolic network during macrophage activation. Lipid metabolism plays a critical role in the activation of both M1 and M2 macrophages. Recent evidence shows that fatty acid oxidation is also essential for inflammasome activation in M1 macrophages, and glycolysis is now known to fuel fatty acid oxidation in M2 macrophages. Ultimately, targeting lipid metabolism in macrophages can improve the outcome of metabolic diseases. Here, we review the main aspects of macrophage immunometabolism from the perspective of the metabolism of lipids. Building a reliable metabolic network during macrophage activation will bring us closer to targeting macrophages for improving human health.
Macrophages are a heterogeneous population of immune cells found in all tissues of the organism (1, 2). They were first discovered and described as phagocytic cells by the Russian zoologist Élie Metchnikoff. Since then, macrophages have emerged not only as mediators of the first line of immune defense but also as key players in tissue homeostasis, development, and pathology (3). One of the main characteristics of macrophages is their plasticity, as they respond to different stimuli by rapidly changing their functional profile in a process called polarization. Classically activated macrophages (M1) are induced by lipopolysaccharide (LPS), toll-like receptors (TLR) ligands, or interferon-gamma (4). Their function is to kill pathogens and to present antigens to T cells to initiate the immune response, and they do so by secreting proinflammatory cytokines such as tumor necrosis factor-alpha, interleukin (IL) 1β (IL1β), IL6, IL12, and IL23 (4). M1 macrophages also express high levels of inducible nitric oxide synthase (4, 5). This enzyme plays an essential role in pathogen killing using arginine to synthesize nitric oxide (NO), which can form reactive oxygen species (ROS) with microbicidal properties (6). Alternatively activated macrophages (M2), on the other hand, are induced by products secreted by innate and adaptive immune cells as a result of parasitic infections, such as IL4 and IL13 (7, 8). These anti-inflammatory macrophages act mainly to resolve inflammation and in tissue remodeling through the secretion of insulin-like growth factor 1 (9), transforming growth factor-beta, and vascular endothelial growth factor (10). In contrast to M1, in M2 macrophages, arginase 1 is induced, and thus, arginine is used to produce polyamines precursors for collagen synthesis, used for tissue repair (11). In addition, different stimuli can induce different subsets of M2 macrophages, such as M2b (activated by TLR ligands or by IL1R agonists), M2c (induced by glucocorticoids or IL10), and M2d (induced by TLR ligands and A2 adenosine receptor agonists) (12).
The notion that M1 and M2 macrophages also differ in their metabolism has been around for more than a decade now, and metabolic adaptations of each type of macrophage intimately respond to their primary function. M1 macrophages are known to rely on aerobic glycolysis and to have impaired oxidative phosphorylation (OXPHOS). This metabolic adaptation favors rapid ATP production to sustain their phagocytic function and provides metabolic precursors to feed the pentose phosphate pathway. In these macrophages, the tricarboxylic acid (TCA) cycle is broken into two parts to provide precursors needed for the synthesis of several lipids (3) and the stabilization of transcription factors such as hypoxia inducible factor 1α, a key player in the activation of glycolysis (13). On the contrary, M2 macrophages have an intact TCA cycle and enhanced fatty acid oxidation (FAO) and OXPHOS (3). Although it was initially thought that proinflammatory macrophages were solely glycolytic and that FAO and OXPHOS were characteristic of anti-inflammatory macrophages, through the years, it has become clear that this equation is not that simple, and recent findings support the need of glycolysis for M2 macrophages, and FAO has been found to occur also in M1 macrophages (14, 15). Here, we present a summary of how lipid metabolism is differentially regulated in M1 and M2 macrophages to promote unique activation and cell functions. Controversial findings are discussed, as well as the potential of using lipid metabolism in macrophages as a target for the treatment of metabolic diseases. Finally, the M1/M2 classification serves us to simplify the rather complex phenomena of macrophage polarization.
Lipogenesis comprehends a series of enzymatic reactions where fatty acids and triglycerides are synthesized. Acetyl-Coenzyme A (acetyl-CoA) serves as a building block for the synthesis of cholesterol, isoprenoids, and fatty acids (16). At the same time, fatty acids are used for the synthesis of triglycerides and complex lipids (16). Lipid biosynthesis is essential for membrane remodeling and the synthesis of inflammatory mediators in M1 macrophages. In these cells, glycolysis is upregulated not only to provide ATP in a faster way, but to fuel the TCA cycle to obtain acetyl-CoA from citrate (Figure 1). In agreement with this, the levels of ATP-citrate lyase (ACLY), the enzyme that converts citrate in acetyl-CoA quickly increases in activated macrophages, and ACLY silencing or inhibition is sufficient to reduce the expression of inflammatory mediators such as NO and ROS (17). Lipogenesis is regulated at the transcriptional level by the sterol regulatory element-binding proteins (SREBPs), which are key elements in the synthesis of fatty acids and cholesterol (18). Srebp1-a, one of the three SREBP isoforms, is abundantly expressed in macrophages and positively regulates their inflammatory response. Im et al. showed that LPS induces Srebp1-a expression in macrophages and that mice with Srebp1-a deficiency have a defective innate immune response (19). Interestingly, macrophages isolated from these mice not only were unable to induce lipid biosynthesis in response to LPS but also secreted lower cytokine levels due to a defective inflammasome. Lipidomic studies have shed light on the importance of lipid metabolism for macrophage polarization toward an inflammatory phenotype. For instance, it is now known that, upon selective TLR4 stimulation, remodeling of glycerolipids, glycerophospholipids, and prenols happens in macrophages (20), which is accompanied by an increase in the synthesis of eicosanoid, sphingolipids, and sterols. Mechanistically, this happens through the activation of the signal transducer and activator of transcription 3 (STAT3) through nuclear factor kappa light-chain enhancer of activated B cells (21).
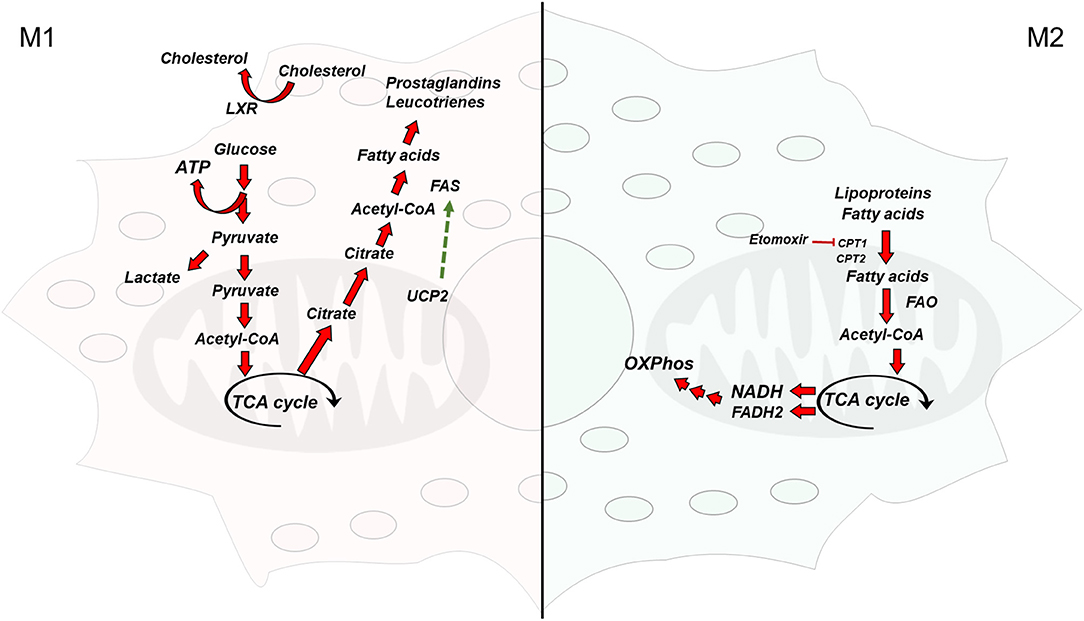
Figure 1. Overview of the metabolic pathways differentially activated in macrophages. (Left) Classically activated macrophages are glycolytic and synthesize fatty acids from acetyl-CoA to obtain inflammatory mediators (Right) Alternatively activated macrophages rely on fatty acid oxidation and have a functional electron transport chain and oxidative phosphorylation. TCA cycle, tricarboxylic acid cycle; FAS, fatty acid synthase; acetyl-CoA, acetyl-coenzyme A; ATP, adenosine triphosphate; FAO, fatty acid oxidation; NADH, nicotinamide adenine dinucleotide (reduced form); FADH2, flavin adenine dinucleotide (reduced form); UCP2, uncoupling protein 2; LXR, liver X receptor; CPT1/2, carnitine palmitoyltransferase 1/2.
Another critical player regulating lipid biosynthesis in M1 macrophages is fatty acid synthase (FAS), a key enzyme for fatty acid biosynthesis (22). FAS has proven to be essential for M1 induction. Wei et al. showed that FAS deletion in macrophages prevented adipose macrophage recruitment and inflammation in mice (23). In addition, these mice were resistant to diet-induced insulin insensitivity. The authors showed that FAS is necessary for membrane remodeling in macrophages and that deficiency in FAS led to changes in the composition of the plasma membrane and Rho GTPase trafficking, which blunted the inflammatory signaling in macrophages. The activation of FAS by the mitochondrial uncoupling protein 2 in macrophages also mediates the induction of the NLR family pyrin domain containing 3 (NLRP3) inflammasome and the consequent secretion of IL1β and IL18 in response to an LPS challenge (24). Consistent with this, uncoupling protein 2 deficiency improved survival in a mouse model of polymicrobial sepsis, which was associated with a decrease in FAS-mediated free fatty acid synthesis. In addition to the de novo synthesis of fatty acids, the NLRP3 inflammasome can also be activated in macrophages by exogenous lipids, such as palmitate (25), a saturated fatty acid. An increase in the levels of circulating saturated fatty acids occurs during obesity, which is thought to trigger inflammation in adipose tissue in part by activating macrophages inflammasome (15). However, the mechanisms by which endogenous and exogenous fatty acids induce inflammasome activation in macrophages are not fully understood. Recently, Gianfrancesco et al. showed that saturated but not unsaturated fatty acids induced inflammasome activation by increasing the level of saturated phosphatidylcholine. This led to a loss in membrane fluidity and the consequent disruption of the Na+/K+ ATPase, which caused a K+ efflux (25). In addition, Wen et al. showed that palmitic acid induction of inflammasome activation in macrophages requires LPS (26). In summary, in M1 macrophages, lipids serve as precursors for the synthesis of inflammatory molecules, as well as to potentiate inflammasome activation.
In sharp contrast to M1 macrophages, M2 macrophages are characterized by an intact TCA cycle and an enhanced mitochondrial OXPHOS that sustain constant energy production. OXPHOS in M2 macrophages is fueled by fatty acid uptake (27), which are oxidized via FAO (Figure 1). Fatty acid uptake happens through lipolysis of circulating lipoproteins (28) and fatty acids, which are internalized through CD36 (29). In a few cases, lipoproteins are internalized by endocytosis (30, 31). In this sense, IL4 treatment induces triglyceride lipolysis in macrophages (29). The importance of FAO for M2 polarization is highlighted in several studies. Using etomoxir, an inhibitor of the mitochondrial carnitine palmitoyl-transferase 1, Malandrino et al. showed that blocking FAO inhibits the activation of M2 macrophages induced by IL4. In the same note, the expression of a constitutively active carnitine palmitoyl-transferase 1 in cultured macrophages prevented the palmitic acid induction of a proinflammatory phenotype (32). Mechanistically, the activation of the oxidative program in M2 macrophages happens through the activation of the peroxisome proliferator activated receptor-γ (PPARγ) (33) and the proliferator-activated receptor-coactivator 1β (34). PPARγ can sense fatty acids, and it has been shown to mediate the transcription of M2 signature genes upon oleic acid and IL4 stimulation (35). Interestingly, PPARγ was recently found to mediate M2 polarization by promoting the oxidation of glutamine (36), an amino acid that fuels OXPHOS (37). In spite of these advances, it is still not fully understood how FAO and OXPHOS are mechanistically linked to the anti-inflammatory phenotype of M2 macrophages.
Although the consensus is that aerobic glycolysis defines M1 macrophages, and FAO is characteristic of M2 macrophages, several pieces of evidence challenge this paradigm and open questions about the metabolic programming during macrophage activation. For instance, it has been shown that FAO is necessary for inflammasome activation in bone-marrow-derived macrophages (BMDM). Pharmacological and genetic inhibition of NADPH oxidase 4, an enzyme that induces inflammasome activation through CPT1-mediated FAO, inhibits NLRP3 activation, and the consequent secretion of IL1b and IL18 (38). Other studies show that FAO is required for palmitate-induced NLRP3 inflammasome activation, which happens through the mitochondrial oxidation of palmitate and the generation of ROS (26, 39). The notion that M2 macrophages are completely independent of glycolysis has also been questioned. Huang et al. showed that a source of external fatty acids is dispensable for M2 activation as long as glucose is present in the media (29). In agreement with this, the same authors showed that inhibiting glycolysis in IL4-stimulated BMDM blunted the expression of M2 activation markers (14). The authors suggested that glycolysis in M2 macrophages fuels the TCA cycle for fatty acid biosynthesis through FAS, which are used later for FAO. This was further supported by Wang et al. by showing that 2-deoxyglucose, an inhibitor of glycolysis that also impairs OXPHOS indirectly, blunted M2 macrophage activation. However, galactose, a glucose analog that inhibits glycolysis but not OXPHOS, did not affect M2 polarization (40). These data reinforce the idea that in M2 macrophages the function of glycolysis could be to fuel OXPHOS, and inhibiting glycolysis will not affect M2 polarization unless OXPHOS is affected.
Liver X receptors (LXRs) are a family of nuclear receptors that act as cholesterol sensors to control cholesterol efflux and lipogenesis (41, 42). The role of LXRs in macrophage polarization seems to be complex. For instance, it has been shown that LXR-deficient mice are more susceptible to infections with the intracellular bacteria Listeria monocytogenes (43) and Mycobacterium tuberculosis (44). However, the LXR agonist GW3965, which targets both LRXα and LXRβ, is able to inhibit the LPS-induced expression of inflammatory genes such as inducible nitric oxide synthase, cyclooxygenase 2, and IL6 in macrophages (45). In addition, Bruhn et al. showed that LXR deficiency in mice confers protection against Leishmania infection (46). Furthermore, expressing a constitutively active LXR in macrophages in vivo not only drives the synthesis of genes involved in cholesterol efflux but also inhibits LPS-induced expression of inflammatory genes (47).
Much controversy also exists on whether FAO is obligatory for macrophage polarization into the M2 phenotype. Although it was initially demonstrated that FAO is essential for M2 polarization in murine macrophages (14, 29, 32), later, several authors used the CPT1 inhibitor etomoxir to demonstrate that FAO is dispensable for IL4-induced M2 polarization in mouse and human macrophages (48, 49). However, proteomic data revealed that human macrophages seem not to depend on FAO for M2 polarization, but they instead use gluconeogenesis as an energy source (50). This piece of data could explain why in human macrophages M2 polarization is unaffected when FAO is inhibited. More recently, Nomura et al. used a genetic approach to directly assess the role of FAO in macrophage polarization. By deleting Cpt2 specifically in the myeloid lineage of mice, the authors demonstrated that BMDM lacking Cpt2 were unable to oxidase fatty acids, as expected. However, they did not find differences in IL4-induced expression of M2 markers between control and Cpt2-deficient BMDM (51). Interestingly, the authors also found that etomoxir was able to inhibit M2 polarization in both control and Cpt2-deficient BMDM, which suggests that the data based on the use of etomoxir must be taken with caution, since it is possible that CPT1 has other functions outside FAO or that etomoxir is not specific for CPT1 (51). Divakaruni et al. further supported this by showing that etomoxir has off-target effects indeed and that the inhibition of M2 polarization by this compound happens through the alteration of CoA levels rather than the inhibition of fatty acid oxidation (52). Overall, the available data suggest that the role of FAO in macrophage polarization is more complicated than initially thought. Future experiments using genetic approaches rather than pharmacological inhibitors, which have the drawback of potentially being unspecific, will help elucidate not only the role of FAO in macrophage polarization but the molecular mechanism behind this process.
Macrophages infiltrate healthy tissue, where they play a critical role in maintaining tissue homeostasis during aseptic conditions (53). Under normal circumstances, M2 polarized macrophages are thought to constitute the majority of macrophages in adipose tissue and are necessary for maintaining insulin sensitivity through the secretion of IL10 (54). However, infiltration of inflammatory macrophages in adipose tissue happens during obesity (55). In addition, an increase in circulating saturated fatty acids is known to activate TLR4 in adipocytes (56). Fatty acid-binding protein 1, an intracellular protein that mediates the uptake of fatty acids, is important in mediating macrophage switch between inflammatory and anti-inflammatory and has been proposed as an interesting target to limit adipose tissue inflammation during obesity (57). Boutens et al. studied the metabolic programming of adipose tissue-resident macrophages in lean and obese mice using transcriptomic analysis and found that adipose tissue macrophages in obese mice have an increase in glycolysis and OXPHOS (58). Interestingly, cytokine release in these macrophages not only depends on glycolysis but also fatty acid oxidation. Future approaches to control adipose tissue inflammation in obese individuals might include the reprogramming of adipose tissue-resident macrophages into an anti-inflammatory phenotype.
Atherosclerosis is an inflammatory disease where cholesterol-rich lipoproteins accumulate in arterial walls leading to their oxidation and the consequent recruitment of several subtypes of immune cells (59). Macrophages are known to internalize oxidized low-density lipoproteins (oxLDL) and lipids within atherosclerotic plaques, which leads to the formation of foam cells with an inflammatory phenotype, all of which contributes to artery occlusion. In addition, lipid uptake causes an increase in ROS in macrophages, which leads to mitochondrial dysfunction and an impaired OXPHOS. This is thought to prevent polarization into the M2 phenotype and to contribute to chronic inflammation and atherosclerosis progress (60). Several authors have shown that targeting lipid metabolism in macrophages can improve atherosclerosis outcomes in mice models. The overexpression of LXRα in macrophages in a mouse model of atherosclerosis had an antiatherogenic effect by increasing cholesterol efflux in macrophages (61). The same effect is seeing using LXR agonists in mice (62). Recently, it was shown that individuals with a specific variant of Perilipin-2, a lipid droplet-associated protein, are less susceptible to the development of atherosclerosis (63). Interestingly, the mechanism behind this protection involves the upregulation of LXR in primary monocyte-derived macrophages from these individuals. In recent years, trained innate immunity has also emerged as a potential mechanism of atherogenesis (64). It is known that macrophages exposed to oxLDL have an enhanced response to restimulation with TLR ligands and have an increased propensity to form foam cells (65, 66). In addition, during the induction of trained immunity, there is an activation of the cholesterol synthesis pathway in macrophages (67).
CCAAT enhancer-binding proteins (C/EBP) are a family of transcription factors involved in adipocyte differentiation with key roles during macrophage polarization (68). In general, it has been proposed that C/EBPα promotes M1 polarization, whereas C/EBPβ participates in M2 activation (69, 70), although several studies suggest that this distinction is more complicated (71, 72). Interestingly, a recent study showed that C/EBPβ was enriched in open chromatin regions in primary human macrophages exposed to oxLDL (73), establishing a link between this transcription factor and the development of atherosclerosis. Overall, redirecting macrophage polarization toward an anti-inflammatory phenotype could have a significant impact on the treatment of metabolic diseases where inflammation plays critical roles.
Lipids are critical metabolites during macrophage polarization. M1 macrophages synthesize fatty acids to use them as precursors for the synthesis of inflammatory mediators while at the same time obtaining most of the ATP from aerobic glycolysis. This adaptation is most likely a result of the rapid activation of M1 macrophages during inflammatory responses. M2 macrophages, on the other hand, are involved in the resolution of inflammation and thus do not need to produce energy in a fast way. As a consequence, M2 macrophages have a functional mitochondrial respiratory chain fueled by the oxidation of fatty acids. Despite these advances, many questions remain on the role of lipid metabolism for macrophage polarization. During the past decade, several pieces of data have questioned the classical view that M1 macrophages rely only on aerobic glycolysis and M2 depend solely on FAO. The evidence suggests that the metabolism of macrophages during activation is more complex, and more studies are needed to unravel the metabolic signature of macrophages. Furthermore, this task gets complicated by the fact that most of the data on macrophage polarization come from murine studies. However, several differences exist between human and murine macrophages in terms of gene expression (74) and the metabolic pathways activated during polarization (48, 50, 75, 76). This makes difficult the extrapolation from mouse to humans, especially whether the reprogramming of macrophage polarization by metabolic interventions will be helpful in the treatment of human diseases. More importantly, the question of whether metabolic adaptations are the cause or the consequence of macrophage polarization needs to be addressed. Answering these questions will take us one step further to understand the relation between macrophage activation and metabolism.
AB-G and LC wrote the article and made figures. AB-G, RV, AC, and LC reviewed the article.
This work was supported by Millennium Institute on Immunology and Immunotherapy P09/016-F, FONDECYT grant 1160336, ICGEB grant CRP-CHL17-06-EC, and COPEC-UC grant 2017.J.924.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
2. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic induction of trained immunity through the mevalonate pathway. Cell. (2018) 172:135–46.e9. doi: 10.1016/j.cell.2017.11.025
3. Van den Bossche J, O'Neill LA, Menon D. Macrophage immunometabolism: where are we (going)? Trends Immunol. (2017) 38:395–406. doi: 10.1016/j.it.2017.03.001
4. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. (2008) 8:958–69. doi: 10.1038/nri2448
5. Munder M, Eichmann K, Modolell M. Alternative metabolic states in murine macrophages reflected by the nitric oxide synthase/arginase balance: competitive regulation by CD4+ T cells correlates with Th1/Th2 phenotype. J Immunol. (1998) 160:5347–54.
6. Schairer DO, Chouake JS, Nosanchuk JD, Friedman AJ. The potential of nitric oxide releasing therapies as antimicrobial agents. Virulence. (2012) 3:271–9. doi: 10.4161/viru.20328
7. Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, Ferrara P, et al. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur J Immunol. (1994) 24:1441–5. doi: 10.1002/eji.1830240630
8. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. (1992) 176:287–92. doi: 10.1084/jem.176.1.287
9. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. (2013) 229:176–85. doi: 10.1002/path.4133
10. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. (2014) 6:716–26. doi: 10.1159/000364945
11. Corraliza IM, Soler G, Eichmann K, Modolell M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem Biophys Res Commun. (1995) 206:667–73. doi: 10.1006/bbrc.1995.1094
12. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
13. Wang T, Liu H, Lian G, Zhang SY, Wang X, Jiang C. HIF1α-induced glycolysis metabolism is essential to the activation of inflammatory macrophages. Mediators Inflamm. (2017) 2017:9029327. doi: 10.1155/2017/9029327
14. Huang SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45:817–30. doi: 10.1016/j.immuni.2016.09.016
15. Korbecki J, Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflamm Res. (2019) 68:915–32. doi: 10.1007/s00011-019-01273-5
16. Song Z, Xiaoli AM, Yang F. Regulation and metabolic significance of de novo lipogenesis in adipose tissues. Nutrients. (2018) 10:E1383. doi: 10.3390/nu10101383
17. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem Biophys Res Commun. (2013) 440:105–11. doi: 10.1016/j.bbrc.2013.09.037
18. Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. (2002) 109:1125–31. doi: 10.1172/JCI15593
19. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
20. Dennis EA, Deems RA, Harkewicz R, Quehenberger O, Brown HA, Milne SB, et al. A mouse macrophage lipidome. J Biol Chem. (2010) 285:39976–85. doi: 10.1074/jbc.M110.182915
21. Dinasarapu AR, Gupta S, Ram Maurya M, Fahy E, Min J, Sud M, et al. A combined omics study on activated macrophages–enhanced role of STATs in apoptosis, immunity and lipid metabolism. Bioinformatics. (2013) 29:2735–43. doi: 10.1093/bioinformatics/btt469
22. Beld J, Lee DJ, Burkart MD. Fatty acid biosynthesis revisited: structure elucidation and metabolic engineering. Mol Biosyst. (2015) 11:38–59. doi: 10.1039/C4MB00443D
23. Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, et al. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. (2016) 539:294–8. doi: 10.1038/nature20117
24. Moon J-S, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. (2015) 125:665–80. doi: 10.1172/JCI78253
25. Gianfrancesco MA, Dehairs J, L'homme L, Herinckx G, Esser N, Jansen O, et al. Saturated fatty acids induce NLRP3 activation in human macrophages through K(+) efflux resulting from phospholipid saturation and Na, K-ATPase disruption. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:1017–30. doi: 10.1016/j.bbalip.2019.04.001
26. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
27. Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. (2011) 6:275–97. doi: 10.1146/annurev-pathol-011110-130138
28. Evans AJ, Sawyez CG, Wolfe BM, Connelly PW, Maguire GF, Huff MW. Evidence that cholesteryl ester and triglyceride accumulation in J774 macrophages induced by very low density lipoprotein subfractions occurs by different mechanisms. J Lipid Res. (1993) 34:703–17.
29. Huang SC, Everts B, Ivanova Y, O'Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
30. Huff MW, Evans AJ, Sawyez CG, Wolfe BM, Nestel PJ. Cholesterol accumulation in J774 macrophages induced by triglyceride-rich lipoproteins. Comparison of very low density lipoprotein from subjects with type III, IV, and V hyperlipoproteinemias. Arterioscler Thromb. (1991) 11:221–33. doi: 10.1161/01.ATV.11.2.221
31. Lindqvist P, Ostlund-Lindqvist AM, Witztum JL, Steinberg D, Little JA. The role of lipoprotein lipase in the metabolism of triglyceride-rich lipoproteins by macrophages. J Biol Chem. (1983) 258:9086–92.
32. Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab. (2015) 308:E756–69. doi: 10.1152/ajpendo.00362.2014
33. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. (2010) 106:1559–69. doi: 10.1161/CIRCRESAHA.110.216523
34. Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. (2006) 4:13–24. doi: 10.1016/j.cmet.2006.05.011
35. Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. (2008) 7:496–507. doi: 10.1016/j.cmet.2008.04.003
36. Nelson VL, Nguyen HCB, Garcìa-Cañaveras JC, Briggs ER, Ho WY, DiSpirito JR, et al. PPARγ is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev. (2018) 32:1035–44. doi: 10.1101/gad.312355.118
37. Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. (2013) 9:712. doi: 10.1038/msb.2013.65
38. Moon J-S, Nakahira K, Chung KP, DeNicola GM, Koo MJ, Pabón MA, et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat Med. (2016) 22:1002–12. doi: 10.1038/nm.4153
39. Hall CJ, Boyle RH, Astin JW, Flores MV, Oehlers SH, Sanderson LE, et al. Immunoresponsive gene 1 augments bactericidal activity of macrophage-lineage cells by regulating beta-oxidation-dependent mitochondrial ROS production. Cell Metab. (2013) 18:265–78. doi: 10.1016/j.cmet.2013.06.018
40. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–75.e4. doi: 10.1016/j.cmet.2018.08.012
41. Repa JJ, Mangelsdorf DJ. The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol. (2000) 16:459–81. doi: 10.1146/annurev.cellbio.16.1.459
42. Yang C, McDonald JG, Patel A, Zhang Y, Umetani M, Xu F, et al. Sterol intermediates from cholesterol biosynthetic pathway as liver X receptor ligands. J Biol Chem. (2006) 281:27816–26. doi: 10.1074/jbc.M603781200
43. Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, et al. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell. (2004) 119:299–309. doi: 10.1016/j.cell.2004.09.032
44. Korf H, Vander Beken S, Romano M, Steffensen KR, Stijlemans B, Gustafsson JA, et al. Liver X receptors contribute to the protective immune response against Mycobacterium tuberculosis in mice. J Clin Invest. (2009) 119:1626–37. doi: 10.1172/JCI35288
45. Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med. (2003) 9:213–9. doi: 10.1038/nm820
46. Bruhn KW, Marathe C, Maretti-Mira AC, Nguyen H, Haskell J, Tran TA, et al. LXR deficiency confers increased protection against visceral Leishmania infection in mice. PLoS Negl Trop Dis. (2010) 4:e886. doi: 10.1371/journal.pntd.0000886
47. Hong C, Walczak R, Dhamko H, Bradley MN, Marathe C, Boyadjian R, et al. Constitutive activation of LXR in macrophages regulates metabolic and inflammatory gene expression: identification of ARL7 as a direct target. J Lipid Res. (2011) 52:531–9. doi: 10.1194/jlr.M010686
48. Namgaladze D, Brune B. Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim Biophys Acta. (2014) 1841:1329–35. doi: 10.1016/j.bbalip.2014.06.007
49. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
50. Reales-Calderón JA, Aguilera-Montilla N, Corbí ÁL, Molero G, Gil C. Proteomic characterization of human proinflammatory M1 and anti-inflammatory M2 macrophages and their response to Candida albicans. Proteomics. (2014) 14:1503–18. doi: 10.1002/pmic.201300508
51. Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, et al. Fatty acid oxidation in macrophage polarization. Nat Immunol. (2016) 17:216–7. doi: 10.1038/ni.3366
52. Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, et al. Etomoxir inhibits macrophage polarization by disrupting CoA homeostasis. Cell Metab. (2018) 28:490–503.e7. doi: 10.1016/j.cmet.2018.06.001
53. Thapa B, Lee K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. (2019) 52:360–72. doi: 10.5483/BMBRep.2019.52.6.140
54. Odegaard JI, Chawla A. The immune system as a sensor of the metabolic state. Immunity. (2013) 38:644–54. doi: 10.1016/j.immuni.2013.04.001
55. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. (2007) 117:175–84. doi: 10.1172/JCI29881
56. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
57. Johnson AR, Qin Y, Cozzo AJ, Freemerman AJ, Huang MJ, Zhao L, et al. Metabolic reprogramming through fatty acid transport protein 1 (FATP1) regulates macrophage inflammatory potential and adipose inflammation. Mol Metab. (2016) 5:506–26. doi: 10.1016/j.molmet.2016.04.005
58. Boutens L, Hooiveld GJ, Dhingra S, Cramer RA, Netea MG, Stienstra R. Unique metabolic activation of adipose tissue macrophages in obesity promotes inflammatory responses. Diabetologia. (2018) 61:942–53. doi: 10.1007/s00125-017-4526-6
59. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
60. Yang X, Li Y, Li Y, Ren X, Zhang X, Hu D, et al. Oxidative stress-mediated atherosclerosis: mechanisms and therapies. Front Physiol. (2017) 8:600. doi: 10.3389/fphys.2017.00600
61. Teupser D, Kretzschmar D, Tennert C, Burkhardt R, Wilfert W, Fengler D, et al. Effect of macrophage overexpression of murine liver X receptor-alpha (LXR-alpha) on atherosclerosis in LDL-receptor deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:2009–15. doi: 10.1161/ATVBAHA.108.175257
62. Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. (2010) 30:1513–8. doi: 10.1161/ATVBAHA.109.191197
63. Saliba-Gustafsson P, Pedrelli M, Gertow K, Werngren O, Janas V, Pourteymour S, et al. Subclinical atherosclerosis and its progression are modulated by PLIN2 through a feed-forward loop between LXR and autophagy. J Intern Med. (2019) 286:660–75. doi: 10.1111/joim.12951
64. Groh L, Keating ST, Joosten LAB, Netea MG, Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. (2018) 40:203–14. doi: 10.1007/s00281-017-0656-7
65. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
66. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134:611–24. doi: 10.1161/CIRCULATIONAHA.116.020838
67. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345:1251086. doi: 10.1126/science.1251086
68. Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. (2002) 365(Pt 3):561–75. doi: 10.1042/bj20020508
69. El Kasmi KC, Qualls JE, Pesce JT, Smith AM, Thompson RW, Henao-Tamayo M, et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat Immunol. (2008) 9:1399–406. doi: 10.1038/ni.1671
70. Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, et al. A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci USA. (2009) 106:17475–80. doi: 10.1073/pnas.0908641106
71. Gorgoni B, Maritano D, Marthyn P, Righi M, Poli V. C/EBP beta gene inactivation causes both impaired and enhanced gene expression and inverse regulation of IL-12 p40 and p35 mRNAs in macrophages. J Immunol. (2002) 168:4055–62. doi: 10.4049/jimmunol.168.8.4055
72. Lee B, Qiao L, Lu M, Yoo HS, Cheung W, Mak R, et al. C/EBPalpha regulates macrophage activation and systemic metabolism. Am J Physiol Endocrinol Metab. (2014) 306:E1144–54. doi: 10.1152/ajpendo.00002.2014
73. Reschen ME, Gaulton KJ, Lin D, Soilleux EJ, Morris AJ, Smyth SS, et al. Lipid-induced epigenomic changes in human macrophages identify a coronary artery disease-associated variant that regulates PPAP2B Expression through Altered C/EBP-beta binding. PLoS Genet. (2015) 11:e1005061. doi: 10.1371/journal.pgen.1005061
74. Raes G, Van den Bergh R, De Baetselier P, Ghassabeh GH, Scotton C, Locati M, et al. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J Immunol. (2005) 174:6561; author reply 6561–2. doi: 10.4049/jimmunol.174.11.6561
75. Cameron ML, Granger DL, Weinberg JB, Kozumbo WJ, Koren HS. Human alveolar and peritoneal macrophages mediate fungistasis independently of L-arginine oxidation to nitrite or nitrate. Am Rev Respir Dis. (1990) 142(6 Pt 1):1313–9. doi: 10.1164/ajrccm/142.6_Pt_1.1313
Keywords: macrophages, lipid metabolism, metabolic reprogramming, oxidative phosphorylation, fatty acid oxidation
Citation: Batista-Gonzalez A, Vidal R, Criollo A and Carreño LJ (2020) New Insights on the Role of Lipid Metabolism in the Metabolic Reprogramming of Macrophages. Front. Immunol. 10:2993. doi: 10.3389/fimmu.2019.02993
Received: 30 September 2019; Accepted: 05 December 2019;
Published: 10 January 2020.
Edited by:
Rafael Prados-Rosales, Albert Einstein College of Medicine, United StatesReviewed by:
Aize Pellón Rodríguez, Dental Institute, King's College London, United KingdomCopyright © 2020 Batista-Gonzalez, Vidal, Criollo and Carreño. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leandro J. Carreño, bGVhbmRyb2NhcnJlbm9AdWNoaWxlLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.