 Elena Martin-Orozco
Elena Martin-Orozco Ana Sanchez-Fernandez
Ana Sanchez-Fernandez Irene Ortiz-Parra
Irene Ortiz-Parra Maria Ayala-San Nicolas
Maria Ayala-San Nicolas- 1Department of Biochemistry and Molecular Biology (B) and Immunology, School of Medicine, University of Murcia, Murcia, Spain
- 2Biomedical Research Institute of Murcia (IMIB), ARADyAL, Murcia, Spain
WNT/β-catenin signaling is involved in many physiological processes. Its implication in embryonic development, cell migration, and polarization has been shown. Nevertheless, alterations in this signaling have also been related with pathological events such as sustaining and proliferating the cancer stem cell (CSC) subset present in the tumor bulk. Related with this, WNT signaling has been associated with the maintenance, expansion, and epithelial-mesenchymal transition of stem cells, and furthermore with two distinctive features of this tumor population: therapeutic resistance (MDR, multidrug resistance) and immune escape. These mechanisms are developed and maintained by WNT activation through the transcriptional control of the genes involved in such processes. This review focuses on the description of the best known WNT pathways and the molecules involved in them. Special attention is given to the WNT cascade proteins deregulated in tumors, which have a decisive role in tumor survival. Some of these proteins function as extrusion pumps that, in the course of chemotherapy, expel the drugs from the cells; others help the tumoral cells hide from the immune effector mechanisms. Among the WNT targets involved in drug resistance, the drug extrusion pump MDR-1 (P-GP, ABCB1) and the cell adhesion molecules from the CD44 family are highlighted. The chemokine CCL4 and the immune checkpoint proteins CD47 and PD-L1 are included in the list of WNT target molecules with a role in immunity escape. This pathway should be a main target in cancer therapy as WNT signaling activation is essential for tumor progression and survival, even in the presence of the anti-tumoral immune response and/or antineoplastic drugs. The appropriate design and combination of anti-tumoral strategies, based on the modulation of WNT mediators and/or protein targets, could negatively affect the growth of tumoral cells, improving the efficacy of these types of therapies.
Introduction
Tumors consist of a heterogeneous mix of cellular populations composed of a small number of cancer stem cells, stromal cells, and tumor infiltrating immune cells, among others. Supporting this idea, several observations have indicated that each specific tumor contains subclones with a wide assortment of gene mutations and promoter hypermethylation in all the cells that constitute the tumor and its microenvironment (1–3).
Currently, two proposals seek to explain tumor heterogeneity. The clonal evolution model proposes that arbitrary mutations in each tumor could generate clones with acquired advantages under adverse selection requirements such as oxygen and/or nutrients. Such clones will expand while the others, less adapted to these conditions, will disappear. Different requirements for tumor growth may be present in different areas of the tumor or at different times or, furthermore, clones with better resistance mechanisms could became advantageous after therapy application. With this proposal, all the cells within a tumor could regenerate the tumor in a different location. The cancer stem cell (CSC) model assumes that only the subset of cancer stem cells on each tumor possesses the capacity to initiate and maintain tumor growth (4). Thus, CSCs are responsible for tumor heterogeneity by generating a variety of cell types that can be reverted, since the terminally differentiated cells can go back and gain CSCs properties under specific conditions. This concept of cellular plasticity unifies the two current models proposed (5–10).
Normal stem or differentiated cells could originate CSCs through a process of consecutive mutations that lead to the acquisition of characteristic cancer stem cell properties of self-renewal, pluripotency, tumor reconstitution capacity, chemo-resistance, low immunogenicity, and/or immune escape capacity. Drug resistance could be mediated by several mechanisms such as the acquisition of quiescence, improved DNA repair, drug efflux capacity, decreased apoptosis, and interaction with the microenvironment (11). Low immunogenicity and/or immunity evasion could be acquired by different strategies such as the production of immunosuppressive molecules; the recruitment of regulatory cells of the immune system; the expression of inhibitory molecules of the anti-tumoral immune response; the loss of tumor antigen expression, the downregulation of major histocompatibility complex I and II (MHC-I and MHC-II) expression, and/or the inhibition of co-stimulatory molecules on antigen presenting cells. These characteristics, mean that the CSCs are the main players in tumor initiation, progression, invasion, metastasis, drug-resistance, and recurrence (11).
The oncogenic properties of CSCs require a number of developmental pathways previously associated to the regulation of normal stem cells (12). Among them, the WNT pathway is deregulated in epithelial-mesenchymal transition (EMT) and CSCs (13). Mutations in WNT signaling components, such as APC, AXIN, β-catenin, and WNT ligands were first observed in colorectal cancers, but have also been reported in other solid tumors (14–21) and hematological malignancies, including leukemia and Multiple Myeloma (MM) (22–24).
There are different WNT pathways interconnected with each other which have been classified as canonical (β-catenin-dependent) and non-canonical signaling pathways. In general, it is assumed that the canonical WNT cascade is responsible for self-renewal, proliferation, or differentiation of progenitor cells, and that the non-canonical cascade participates in the maintenance of stem cells, cell movement, or inhibition of the canonical pathway (25).
WNT Signaling Components and Pathways
WNT Ligands, Receptors, and Co-receptors
The WNT ligands family is constituted by 19 secreted glycoproteins that can participate in one or several WNT signaling pathways in a manner that can be autocrine or paracrine. They exhibit specific expression patterns and functions and are highly conserved from invertebrates to mammals (26, 27). All WNT ligands become glycosylated in the endoplasmic reticulum (ER); and also acylated by the O-acetyltransferase Porcupine (PRCN) (28–31). Next, lipid-modified WNT ligands bind to the transmembrane protein Evenness interrupted WNTless (Evi/WIs) and are transported to the plasma membrane via the Golgi apparatus with the assistance of the p24 proteins (32–34). Finally, the transportation of WNT ligands on the extracellular space occurs in membrane enclosed vesicles such as exosomes (28, 31, 35).
The family of Frizzled (FZD) receptors interacts with WNT ligands and with the co-receptor's low-density lipoprotein receptor-related proteins 5,6 (LRP5/6). While the complex consisting of WNT, FZD, and LRP proteins activates the canonical WNT/β-catenin signaling cascade, the complex formed by FZD and/or ROR1/ROR2/RYK (Receptor tyrosine kinase-like orphan receptor) receptors activates non-canonical WNT signaling cascades (WNT/PCP or planar cell polarity and the WNT/Ca2+ signaling cascades). The complex WNT-FZD-LRP also activates the WNT/STOP (stabilization of proteins) route which is a subtype of the non-canonical WNT signaling pathway which decelerates protein degradation when cells prepare to divide during mitosis (36–38).
WNT Canonical Pathway: On and Off
The central point of this pathway is the activation of the protein β-catenin, which can be found in the cell in different forms and locations. Thus, at the cytoplasmic membrane, β-catenin remains associated with E-cadherin and, through α-catenin, connects actin filaments to form the cytoskeleton (Figure 1A, left panel); in the cytoplasm, β-catenin levels are strictly controlled; and in the nucleus this protein regulates transcriptional activation and chromatin remodeling.
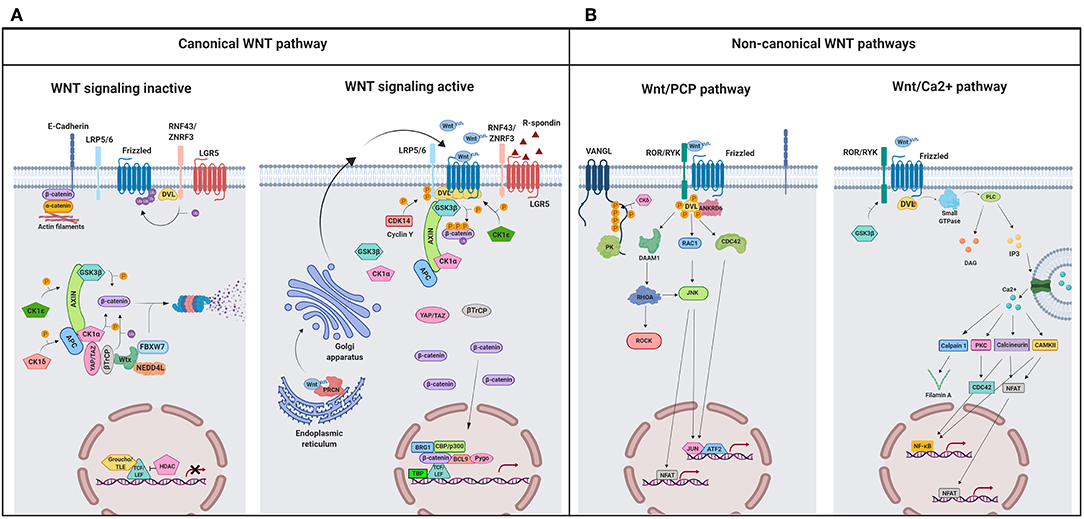
Figure 1. A schematic illustration representing different WNT signaling pathways. (A) Canonical WNT signaling. Left panel shows inactive pathway. In the absence of WNT ligands, β-catenin is phosphorylated by the destruction complex, constituted by the scaffolding proteins APC and AXIN and the kinases GSK3β and CK1α. Then, β-catenin is ubiquitinated and targeted for proteasomal degradation by the complex containing β-TrCP, FBXW7, NEDDL4, and WTX proteins. Thus, β-catenin degradation prevents its presence in the nucleus where a complex formed by TCF/LEF and TLE/Groucho binds HDACs to inhibit transcription of target genes. Right panel shows canonical WNT signaling active. The binding of WNT ligands to FZD receptors and LRP co-receptors activates WNT signaling. LRP receptors are phosphorylated by CK1α and GSK3β. Then, DVL proteins polymerize and are activated at the plasma membrane inhibiting the destruction complex. This results in stabilization and accumulation of β-catenin in the cytosol and its subsequent translocation into the nucleus where it displaces TLE/Groucho repressors forming an active complex with TCF/LEF proteins that bind co-activators such as CBP/p300, BRG1, BCL9, and PYGO. An alternative way of β-catenin signaling includes the disruption of epithelial E-cadherin interactions, which breaks the binding of β-catenin to the cytoplasmic domain of cadherin and leads to the accumulation of β-catenin first in the cytosol, and later in the nucleus. (B) Schematic illustration representing the main non-canonical WNT pathways. Left panel shows the WNT/PCP pathway. WNT ligands bind to the FZD receptor and the co-receptors ROR 1/2 (or RYK). Then, DVL is recruited and activated followed by VANGL activation. Then DVL binds to the small GTPase RHO A with the collaboration of the cytoplasmic protein DAAM1. The small GTPases RAC1 and RHO activate ROCK and JNK. This leads to rearrangements of the cytoskeleton and/or transcriptional responses via for example, ATF2 and/or NFAT. Right panel shows the WNT/Ca2+ pathway. The signaling is initiated when WNT ligands bind to the FZD receptor and the co-receptor ROR 1/2 (or RYK). Then, DVL is recruited and activated and binds to the small GTPase which activates phospholipase C leading to intracellular calcium fluxes and downstream calcium dependent cytoskeletal and/or transcriptional responses. APC, adenomatous polyposis coli; BCL9, B-cell CLL/lymphoma 9 protein; β-TrCP, β-Transducin repeat-containing protein; BRG1, Brahma related gene 1; CAMKII, calmodulin-dependent protein kinase II; CBP, CREB-binding protein; CDC42, cell division control protein 42; CELSR, cadherin EGF LAG seven-pass G-type receptor; CK1α,ε,δ, casein kinase 1α,ε,δ; DAAM1, DVL associated activator of morphogenesis; DAG, diacylglycerol; DVL, disheveled; FBXW7, F box/WD repeat-containing protein 7; FZD, Frizzled; GSK3β, glycogen synthase kinase 3β; IP3, inositol 1,4,5 triphosphate; JNK, JUN kinase; LGR5, Leucine-rich repeat-containing G-protein-coupled receptor 5; LRP5/6, low-density lipoprotein receptor-related protein 5/6; NEDD4L, neural precursor cell expressed, developmentally downregulated 4-like; NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor kappa B; PK, Prickle; PKC, protein kinase C; PLC, Phospholipase C; p300, E1A Binding Protein p300; RAC, Ras-related C3 botulinum toxin substrate; RHOA, Ras homolog gene family member A; ROCK, Rho kinase; ROR1/2, bind tyrosine kinase-like orphan receptor 1 or 2; RYK, receptor-like tyrosine kinase; TBP, TATA-binding protein; PRCN, Porcupine; PYGO, Pygopus; RNF43, Ring finger protein 43; RSPO, R-spondin; TCF/LEF, T-cell factor/lymphoid enhancer factor; TLE, Transducin-Like Enhancer of Split proteins; VANGL, Van Gogh-like; WTX, Wilms tumor suppressor protein complex; YAP/TAZ, Yes-associated protein/Transcriptional co-activator with a PDZ-binding domain; ZNRF3, Zinc and Ring Finger 3. Created with BioRender.com.
In the absence of ligands, the WNT pathway is inactive (Figure 1A, left panel) and β-catenin is continuously synthesized, ubiquitinated, and degraded in the cytosol by a destruction complex constituted by the two scaffold proteins, adenomatous polyposis coli (APC) and axis inhibition protein 1 (AXIN1), Ser/Thr kinases such as casein kinase 1 (CK1α, ε y δ) and glycogen synthase kinase 3β (GSK3β), and two transcriptional regulators (YAP/TAZ; Yes-associated protein/transcriptional co-activator with a PDZ-binding domain) of the Hippo pathway (37–39). Thus, β-catenin will not be available for nuclear transport and transcriptional regulation.
On the contrary, when WNT binds to the FZD receptor, the canonical WNT pathway becomes active (Figure 1A, right panel) and the recruitment of co-receptors LRP5/6 changes AXIN conformation, impairing its interaction with β-catenin and preventing the phosphorylation of this protein (40, 41). Alternatively, Li et al. (42) propose a model in which the destruction complex can bind and phosphorylate β-catenin, but the absence of β-TrCP within the complex prevents ubiquitination and degradation of the protein (38, 43), so that β-catenin accumulates in the cytosol and travels to the nucleus, where it induces the transcription of specific genes (Figure 1A, right panel) (40, 43–46) (listed in http://www.stanford.edu/Brnusse/WNTwindow.html).
Non-canonical or Alternative WNT Signaling Pathways
Non-canonical WNT pathways do not require β-catenin stabilization and the signal initiates through the binding of WNT to the FZD receptor without LRP co-receptor participation. Although several non-canonical WNT pathways have been identified, the best known are the WNT/PCP and the WNT/Ca2+ pathways (Figure 1B).
WNT/PCP signaling is implicated in the establishment of cell polarity and cell migration (31). In this pathway (Figure 1B, left panel), the binding of the WNT ligand initiates a cascade in which ROR/RYK helps to transmit the signal to VANGL2 and induces its phosphorylation (47–50). Additionally, RHOA and RAC activate JUN kinase (JNK), which can induce gene transcription through the activation of AP1 (jun-ATF-2) and the nuclear factor of activated T cells (NFAT) (Figure 1B, left panel). A crosstalk exists between WNT/PCP and the canonical WNT pathway as it has been shown that VANG-1/VANGL negatively regulates the WNT/β-catenin signaling by a mechanism dependent on DVL (51).
The WNT/Ca2+ signaling pathway (Figure 1B, right panel) is activated by the binding of the WNT ligands to the FZD family of proteins, which in turn activates phospholipase C (PLC). The secondary messengers induce the release of intracellular calcium and then calcium dependent kinases such as calpain-1 and calcineurin (Cn) are activated. These kinases activate the expression of transcription factors such as NFAT and nuclear factor kappa B (NFκB). This pathway is fundamental in the regulation of cell adhesion, cell migration, and embryonic development (25).
Modulators of WNT Signaling: RNF43/ZNRF3, RSPO, YAP/TAZ, and VANGL
RNF43 (Ring finger protein 43) and its homolog ZNRF3 (Zinc and Ring Finger 3) promote poly-ubiquitination of lysines in the cytoplasmic sequence of FZDs proteins (52, 53) inducing endocytosis and the destruction of these receptors at the lysosome (Figure 1A) (28).
Another WNT regulator is Leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), a protein that binds to the R-spondin ligand family members (RSPO) and maintains WNT signaling through the neutralization of the RNF43/ZNF3 ligases (Figure 1A) (36, 37, 54, 55). YAP/TAZ are also WNT regulators and compete with LRP5/6 for the same binding domain of AXIN. Thus, the association of AXIN to YAP/TAZ is incompatible with its association to LRP5/6 (Figure 1A, right panel) (56). Finally, Van Gogh-like (VANGL) and Prickle (PK) are two crucial components of the planar cell polarity pathway and antagonists to the canonical WNT/β-catenin pathway (Figure 1B, left panel). Therefore, the absence of VANGL1/2 decreases the response threshold to WNT3a, without initiating activation of canonical WNT signaling however (51).
WNT Signaling Activation in Healthy Immune Response and in Tumor Cell Survival
WNT signaling should be considered a multifactorial pathway which regulates the transcription of genes that participate in most of the mechanisms used by healthy cells to differentiate and survive. Supporting this, WNT signaling plays a main role in cellular homeostasis, regulating immune cell development and function. Thus, the non-canonical WNT pathway inhibits the differentiation of quiescent HSCs by controlling β-catenin activation (57, 58). Conversely, canonical WNT signaling promotes T cell lymphopoiesis (5, 59–61). The role of WNT signaling in immunity also includes the regulation of peripheral T cell activation and differentiation. As evidence of this, TCF activity has been shown to play crucial roles in the differentiation of memory CD8+ T cells as well as in inducing specific TH subset responses (59, 61). In this sense, the non-canonical WNT signaling by WNT5A induces the secretion of IL-12, by dendritic cells favoring TH1 responses (59, 61–63). However, activation of the canonical pathway promotes TH2 polarization. Specifically, TCF1 inhibits differentiation of naive CD4+ T cells into TH1 and TH17 cells and promotes differentiation into the TH2 and TFH (T follicular helper cells) subsets. Regulatory T (Tr) cell survival is also promoted by the canonical WNT pathway, whilst the effector function and development of TH17 cells are inhibited. Nevertheless, TCF1 also limits the suppressive activity exerted by regulatory T cells in the effector T cell population. Finally, TCF1 is essential for the development of innate lymphoid cells (ILCs) natural killer (NK) cells and DC differentiation (Figure 2) (59, 61, 64, 65).
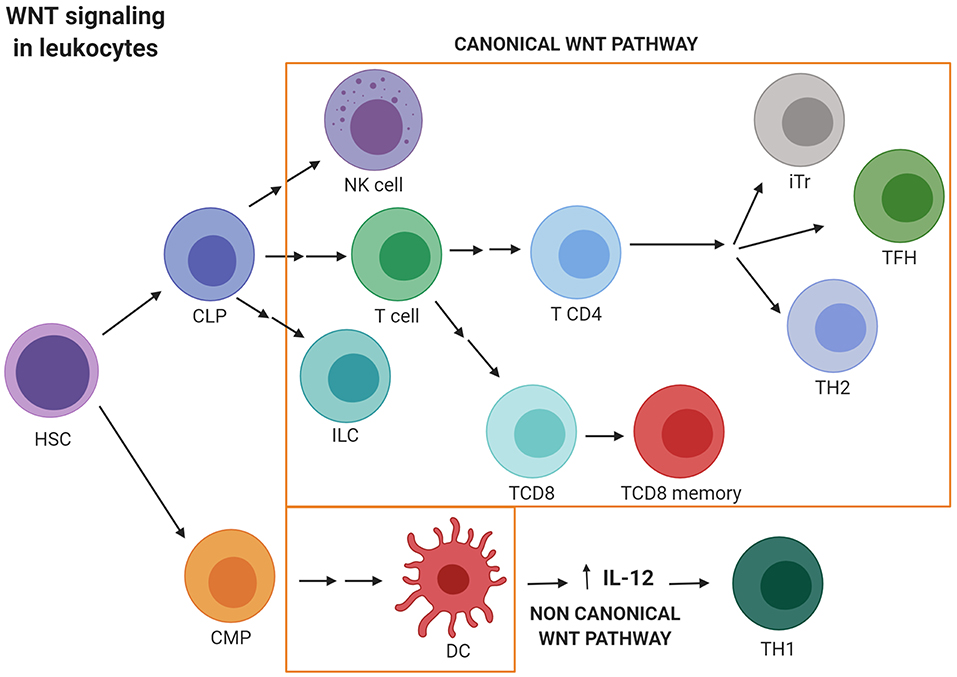
Figure 2. WNT signaling activation in leukocytes. Canonical WNT signaling promotes T cell lymphopoiesis and regulates peripheral T cell activation and differentiation. Thus, canonical WNT activity promotes differentiation into the TH2 and TFH subsets. Regulatory T (Tr) cells survival is also promoted by the canonical WNT pathway. Nevertheless, TCF1 limits the suppressive activity exerted by Tr in the effector T cell population. Finally, WNT/β-catenin activation is essential for the development of innate lymphoid cells (ILCs) natural killer (NK) cells and DC differentiation. Conversely, non-canonical WNT signaling induces the secretion of IL-12 by dendritic cells favoring TH1 responses. CLP, Common lymphoid progenitor; CMP, Common myeloid progenitor; DC, Dendritic cell; ILC, Innate lymphoid cell; iTr, induced regulatory T cell; HSC, hematopoietic stem cell; NK, Natural killer; TFH, T follicular helper cell; TH, T helper. Created with BioRender.com.
Since WNT signaling is crucial to immune homeostasis, alterations of this pathway in pathologies such as cancer can also be associated with the profound dysregulation of protective anti-tumoral immune responses [reviewed in (45, 66, 67)]. In fact, WNT signaling is associated with the onset of many types of solid and liquid tumors. As an example, mutations involved in the WNT signaling pathway were found in more than 90% of carcinomas of the colon and rectum [reviewed in (68)]. Related to that, it is broadly accepted that the first step in the progression to CRC frequently consists of APC gene mutations associated with the activation of the canonical WNT pathway. Supporting this, adenoma regression was observed in a murine model when APC function was recovered. Additionally, activating mutations in β-catenin, mediated or not by APC mutations, were observed in 80% of cases of CRC (69). RNF43 mutations causing WNT signaling activation were found in over 18% of colorectal and endometrial cancers (70, 71) which were extremely dependent on the WNT pathway. Since RNF43 mutations make the PDAC cell lines sensitive to treatment with Porcupine inhibitors, the growth of these tumors also relies on WNT ligand secretion. Conversely, induction of DKK1, a WNT antagonist, or treatment with an FZD-specific antibody delays PDAC development (72). Additionally, RSPO translocations are described in 4–18% of patients with gastric, ovarian, and endometrial cancer and about 9% of colorectal cancers (70). Recently, ROR1 and ROR2 co-receptor upregulation was associated to the two different melanoma states: proliferative and invasive. These phenotypes are in part the result of the balance between canonical and non-canonical WNT activation. The treatment of proliferative melanoma cells, expressing ROR1, with WNT5a induced ROR1 degradation, increased ROR2 expression and high invasiveness in vivo. (65). WNT signaling activation is also observed in ~50% of patients with breast cancer and is associated with a decrease in overall survival. Although only a small percentage of tumors carry mutations of the main WNT pathway regulators, canonical WNT ligands and receptors are frequently overexpressed, whereas antagonists are downregulated. Among all the WNT regulators, high LGR4 expression was correlated with low patient survival. In Lgr4-deficient mouse models, researchers found a significant delay in mammary tumor development, proliferation, and the occurrence of metastasis. Further molecular analysis demonstrated that LGR4 knockdown inhibited WNT/β-catenin signaling, and the expression of epithelial–mesenchymal transition (EMT) mediators. Finally, there was a 90% decline in the number of CSCs in human breast-cancer cells and in the mouse mammary tumor virus (MMTV)-WNT1 transgenic mouse model. Thus, WNT signaling is implicated in the maintenance of mammary stem cells and breast-cancer stem cells (73–76). Supporting these results, in non-small cell lung cancer it has been shown that maintaining the lung cancer stem cell population requires the degradation of the WNT negative regulators, increasing β-catenin mediated WNT activity (77). Furthermore, WNT signaling plays a critical role in the maintenance and propagation of ovarian cancer stem cells (78); it is critical in desmoid tumor formation (79), and in head and neck squamous carcinomas (HNSCC), where researchers have shown CSC elimination after WNT/β-catenin signaling was downregulated.
WNT signaling deregulation also plays an important role in the development of hematological tumors. WNT ligands and receptors are expressed in the hematopoietic stem cells (HSC) and are present in the bone marrow (59). Thus, the WNT/β-catenin pathway is highly activated in AML (80–82), CML (83, 84), Multiple myeloma (MM), and other types of leukemia. In fact, in mouse models of AML, the WNT/β-catenin signaling pathway is required for leukemia stem cells (LSCs) self-renewal (85). Similarly, a high expression of β-catenin and WNT pathway associated genes has been observed in LSCs in CML (86). Deregulation of the WNT pathway is also associated with abnormal expression of LGR4 in cells isolated from MM patients (87, 88).
WNT signaling has been involved not only in tumor development but also in the capacity of tumoral cells to escape different types of cellular stresses, such as drug treatments and the host immune response. In this sense, many attempts have been made to find the connection between drug resistance and immune evasion (89), and common pathways controlling both processes have been explored. Within the WNT signaling pathways, many molecules have been described as being the main drivers of the transcriptional activation of the genes involved in the two mechanisms (90–94).
WNT Signaling in Tumors Drug Resistance
Cancer stem cells develop several resistance mechanisms, which protect the cells from drug damage and make them resistant to chemotherapeutic drugs (11, 12). In parallel with the models explaining tumor heterogeneity, two different proposals account for the existence of cancer drug resistance. According to the clonal evolution model, a population of tumor cells can acquire drug resistance by sequential genetic modifications. After chemotherapy, only the drug-resistant cells within the tumor survive and proliferate, regenerating the tumor bulk made up of the drug-resistant cells progeny. Thus, all tumoral cells became drug resistant. By contrast, the CSC model postulates that after drug exposure, only CSCs (which harbor intrinsic resistance mechanisms) survive. These stem cells then divide and regenerate the tumor mass that will be constituted by stem and differentiated cells, reestablishing the tumoral heterogeneity. The acquisition of drug resistance seems to have features from both models.
Generally, cancer drug resistance involves the participation of a variety of cellular mechanisms, including: drug target mutations; oncogene/onco-suppressor deregulations; activation of pathways blocking the drug action; increased DNA damage repair; overexpression of drug efflux pumps (ABC-transporters); and the induction of cell adhesion-mediated drug resistance, originating due to the crosstalk between tumor and stromal cells. Some of these resistance mechanisms, specifically the over-expression of ABCB1 and CD44 molecules among others, develop because of WNT signaling pathway deregulation in the cancer stem cell subset (Table 1) (104).

Table 1. Role of WNT components in drug resistance.
WNT Signaling and Extrusion of Chemotherapeutic Drugs: ABCB1
The superfamily of ABC transporters mediates the efflux or uptake of specific substrates through cellular membranes (plasma membrane, endoplasmic reticulum, Golgi apparatus, peroxisomes, and mitochondria). The number and nature of the substrates is varied and includes physiological and xenobiotic molecules. These transporters are highly conserved and present in prokaryotes and eukaryotes. The ABC transporters are characterized by two transmembrane (TMs) and two nucleotide-binding domains (NBs) or ATP binding cassettes. Human ABC transporters include 48 functional genes classified into seven subfamilies (from A to G) based on different characteristics: similarity in gene structure, order, and sequence homology in the NB and TM domains.
ABCB1 (ATP-binding cassette B1, also known as MDR-1 or P-GP) was the first cloned human ABC transporter. Physiologically, this protein, and other members of the family, transports hydrophobic, and hydrophilic compounds across the placenta, intestine, and other locations and contribute to the blood-brain barrier. Furthermore, this and other transporters actively efflux xenobiotics to protect the cells from cytotoxic agents (11). This includes the capacity to expel a variety of drugs outside the cancer cells, thus inducing chemoresistance in numerous solid tumors and hematological malignancies (13, 109, 110). In that way, ABCB1 contributes to the acquisition of a multidrug resistant (MDR) phenotype since it can bind and extrude a huge repertoire of drugs, thus leading to treatment failure and tumor relapse. Other roles attributed to these proteins include the transport of ABC proteins to intracellular (e.g., cytoplasmic vesicles) or extracellular (e.g., exosomes) compartments to improve drug sequestration (111, 112); tumor cell proliferation, invasion, and deregulation of the pathways involved in apoptosis or complement-mediated cytotoxicity (109, 110, 113–116).
Stem cells, including CSC, exhibit high expression levels of the two main MDR genes, ABCB1 and ABCG2 (ATP-binding cassette G2) (11). In addition, since the promoter of the ABCB1 gene in humans contains several TCF4/LEF binding motifs, this protein is a target gene of the β-catenin/TCF4 transcriptional regulators. In fact, activation of β-catenin augments ABCB1 expression, which confirms the direct connection between the WNT/β-catenin pathway and chemoresistance. In fact, several studies have demonstrated the existence of this connection in tumor cells which have, in this way, acquired a multidrug resistant phenotype (Figure 3) (117, 118).
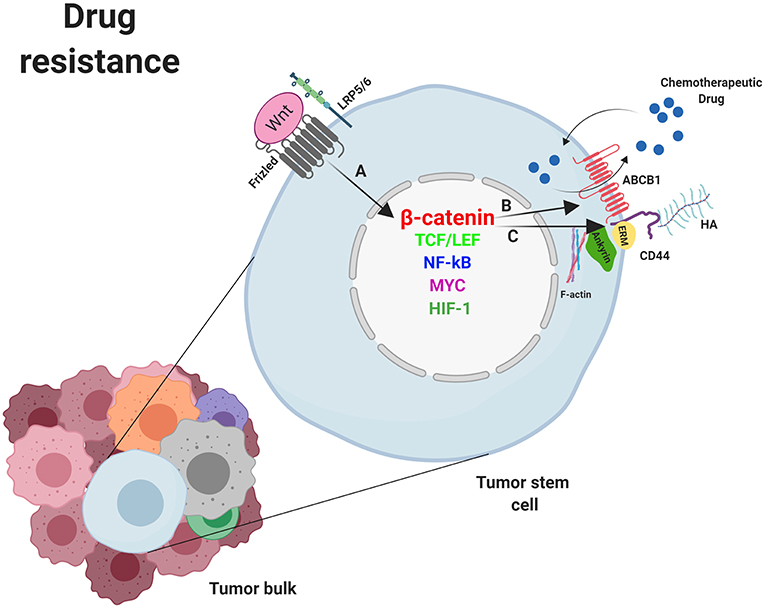
Figure 3. Mechanisms of drug resistance induced by WNT/β-catenin signaling in cancer stem cells. (A) Nuclear β-catenin, by means of MYC, TCF/LEF, NF-κB, SNAI1, HIF-1, and other transcription factors, promotes the transcription of genes involved in mechanisms supporting tumor cells survival such as deregulation of molecules involved in drug resistance. (B) ABCB-1 and (C) CD44. Overexpression of ABCB1 and/or CD44 favor the extrusion of chemotherapeutic drugs out of the cell. ABCB1, ATP-binding cassette B1; ERM, Ezrin, Radixin, Moesin; HA, Hyaluronan; HIF-1, Hypoxia inducible factor 1; LRP5/6, Low-density lipoprotein receptor-related protein 5/6; NF-κB, nuclear factor kappa B; MYC, Myelocytomatosis oncogene cellular homolog; SNAIL, Zinc finger protein SNAI1; TCF/LEF, T-cell factor/lymphoid enhancer factor. Created with BioRender.com.
Frizzled receptors and ABCB1-mediated multidrug resistance
FZD1 was reported to upregulate ABCB1 expression through WNT/β-catenin signaling in solid tumors and hematological malignancies. Both FZD1 and ABCB1 locate in the same region within the 7q21 chromosome. Knockdown of FZD1 induces a significant decrease of ABCB1 protein expression in a human myelogenous leukemia cell line and the recovery of chemosensitivity to antineoplastic drugs by those cells. Either way, the knockdown of ABCB1 using antisense oligonucleotides restored drug sensitivity in the previously resistant cell line (95). Downregulation of FZD1 also resulted in β-catenin degradation and therefore the absence of this protein in the nucleus. Moreover, it has been shown that the knockdown of FZD1 triggers also a decrease in the proliferative capacity of MDR leukemic cells through the WNT/β-catenin cascade. In fact, in cells from patients with acute myeloid leukemia (AML), elevated FZD1 expression at diagnosis was associated with more problems in achieving remission, and a tendency to recurrence. Thus, in patients with relapse, increased FZD1, rather than ABCB1 expression, induced high proliferation and chemoresistance in leukemic cells (96).
WNT/β-catenin signaling deregulation, specifically FZD1 overexpression, has also been observed in neuroblastoma multidrug resistant cell lines. After silencing FZD1 in MDR cells there was inhibition of β-catenin translocation to the nucleus. Consequently, ABCB1 expression and efflux activity were reduced, causing the reversal of the MDR phenotype in those cancer cells (97, 98). Another study explored the role of miR-27a in the modulation of ABCB1-mediated MDR in hepatocellular carcinoma (HCC) using the sensitive cell line BEL-7402 and its resistant counterpart, BEL-7402/5-fluorouracil. The increase in miR-27a levels was associated with enhanced sensitivity of these cells to 5-fluorouracil (5-FU), higher 5-FU-induced apoptosis and downregulation of MDR1/P-glycoprotein expression. That study showed that miR-27a reverses the ABCB1-mediated MDR by downregulation of FZD7 and the inhibition of the WNT/β-catenin pathway (99).
Furthermore, methylation of SFRPs (secreted frizzle-related protein), which inhibits WNT/β-catenin signaling, was reported in several types of leukemia, and associated with resistance to cisplatin in ovarian cancer and with a poor prognosis in cervical and breast cancers. In fact, overexpression of β-catenin was observed in leukemia cells from patients with methylated SFRP5, and a demethylation compound recovered SFRP5 protein expression and reduced ABCB1 protein levels. Thus, altered SFRP5 gene methylation may participate in the development and/or progression of different types of cancers through WNT signaling pathway activation (100).
LGR5 and ABCB1-mediated drug resistance
LGR5 is another target gene of the WNT pathway as well as a marker of colorectal carcinoma CSCs. Several studies have associated higher LGR5 levels with a poor response of colorectal cancer patients to 5-fluoracil-based treatment. In addition, LGR5 positively modulates the ABCB1 expression in CRC cells. That study, as well as others, demonstrated that in cancer cells, LGR5 promotes stem cell properties such as chemoresistance through the positive regulation of the extrusion pump ABCB1 (101).
YAP/TAZ and ABCB1-mediated drug resistance
Like other WNT regulators, the activation of YAP/TAZ supports the survival of CSCs treated with conventional chemotherapy, protecting cancer cells against DNA damage. Thus, YAP/TAZ activation protects breast CSCs from paclitaxel, doxorubicin, cisplatin, and radiations and favors resistance to therapies targeting certain molecules in tumor cells with specific oncogenic alterations. Lin et al. reported that YAP confers resistance to inhibitors of RAF and MEK signaling pathways in tumoral cell lines containing activating mutations in BRAF, KRAS or NRAS genes (102). Conversely, the decrease of YAP enhanced RAF and MEK inhibitor efficacy in mutant cells resistant to monotherapy with those inhibitors. Furthermore, the YAP mechano-transduction pathway is involved in resistance in melanoma and breast cancer (103).
Pygopus and ABCB1-mediated drug resistance
PYGO2 is overexpressed in several types of cancer (breast, ovarian, lung, glioma, and esophageal squamous cell carcinoma), and plays a decisive role in the carcinogenesis of these tumors. In fact, PYGO2 was the most upregulated gene in chemo-resistant breast cancer cells. Experimental results indicated that PYGO2 upregulated ABCB1 expression in resistant cells through the WNT/β-catenin cascade. As expected, PYGO2 inhibition restored drug sensitivity in MDR cells by decreasing ABCB1 expression, reducing the breast cancer stem cell subset following chemotherapy. Furthermore, RNA samples from tumors extracted by surgery from 64 paired patients significantly increased PYGO2 and/or ABCB1 expression after chemotherapy, thus underlining a crucial role for the WNT/β-catenin pathway mediated by PYGO2 in the clinical chemoresistance of breast cancer (104).
WNT and Activation of Cell Adhesion-Mediated Drug Resistance (CAM-DR): CD44
The constant interaction between the tumor and the surrounding microenvironment influences the cellular fate of CSCs. The protein CD44 is especially important in this communication process since it constitutes a platform for signaling that incorporates cellular microenvironmental information. This molecule integrates signals from growth factors, cytokines and others, and sends them to cytoskeletal proteins associated to the membrane or the nucleus where gene expression is regulated. CD44 upregulates the expression of cell cycle proteins inhibitors, anti-apoptotic proteins, and ABC transporters, and others, thus inducing a form of drug resistance known as cell adhesion-mediated drug resistance (CAM-DR).
The CD44 family is constituted by single-pass, glycosylated class-I transmembrane proteins of 85–90 kDa. The extracellular N-terminal portion of CD44 proteins binds to the glycosaminoglycan hyaluronan (HA), among other ligands, while the C-terminal cytoplasmic tail binds the cytoskeletal linker proteins ezrin, radixin, moesin (ERM), and ankyrin (119), and F-actin (120–122).
The different members of the CD44 family are originated by the alternative splicing of 10 exons which codified for the extracellular domain of the protein. This domain can be completely deleted, generating CD44s or giving rise to various combinations encoding CD44 variant members (CD44v). Although CD44 is found in many normal cell types, it is used as a surface marker for CSCs from several types of tumors (11).
CD44 is a positive regulator of the WNT/β-catenin signaling pathway through the control of LRP5/6 phosphorylation and its location at the plasma membrane (105). Therefore, the ability of CD44 to bind to the cytoskeleton might provide a platform necessary for the interaction between LRP5/6 and kinases such as GSK3β and CK1. Furthermore, the shuttle of LRP5/6-charged vesicles from the Golgi to the membrane might use the F-actin tracks anchored to the plasma membrane through the complex CD44-ERM (123).
Several articles have been published supporting a role for CD44 in CAM-DR. Thus, overexpression of CD44v6 associates with colorectal cancer in advanced stages and is characterized by mutations in the WNT pathway (e.g., APC mutations) (105). Additionally, it was reported that the degree of cell adhesion in MM showed a negative correlation with the sensitivity of these cells to doxorubicin (106). Furthermore, long term exposure of human multiple myeloma cell lines (HMCLs) to lenalidomide increased the level of resistance to this molecule (107) and the expression and activity of β-catenin with enhanced transcription of the WNT target genes cyclin-D1 and myelocytomatosis oncogene cellular homolog (MYC). These effects were the consequence of CK1α suppression, and GSK3α/β inactivation, since an increase in the phosphorylation of the inhibitory residues of this protein was observed. In another study in MM, CD44 was identified as the main effector molecule of lenalidomide resistance mediated by the WNT cascade. Overexpression of this protein was observed in lenalidomide-resistant human multiple myeloma cell lines (HMCLs), and in correlation with that, increased adhesion to bone marrow (BM) stromal cells was detected, indicative of CAM-DR. Inhibition of CD44 reduced the adhesion of MM cells and reversed the resistance to lenalidomide (108).
CD44 associates with ABC pumps (124, 125) such as the ABCB1 protein and regulates its gene expression (Figure 3). The process requires the activation of CD44 by HA binding, which promotes PI3K activation and stimulates HA production and ABCB1 expression (126). Alternatively, the binding of HA to CD44 induces the expression and activation of the transcription factor p300 which, together with β-catenin and NFκB-p65, sustains ABCB1 transcription (127). Engagement of CD44 and activation of specific transcription factors in EMT can also induce apoptosis resistance. Thus, it was demonstrated that the CD44–HA interaction to PKCε induced transport of Nanog to the nucleus, leading to miR-21 synthesis and upregulation of ABCB1 and apoptosis inhibitors (128, 129).
The association between ABCB1/P-GP and CD44 has also been demonstrated by experiments of co-immunoprecipitation and co-localization within the plasma membrane, and further confirmed in a yeast two-hybrid system. In another study, the examination of primary and metastatic tumor samples from osteosarcoma patients showed that a high expression of CD44 was associated with osteosarcoma metastasis and recurrence and CD44 was considered as a solid predictor for chemotherapy response and overall survival in osteosarcoma patients. The results in osteosarcoma cell lines with constitutive knockout of CD44 gene by CRISPR/Cas9 system verified that CD44 mediates migration, invasion, proliferation and drug resistance to doxorubicin in osteosarcoma cells. In another study, the downregulation of CD44 protein by siRNA in cancer cells from patients with ovarian carcinoma confirmed that the mRNA levels of CD44 and ABCB1 correlate positively. In summary, numerous studies support the hypothesis that CD44 may contribute to tumor drug resistance by regulating ABCB1 expression (130).
WNT Signaling in Immune Escape by Tumors
The host immune system constitutes a wall against tumor formation through the innate and the adaptive immune response. The primary role of immune effectors such as macrophages, dendritic cells (DCs), and T cells is to discriminate healthy cells from pathogens or tumoral cells through receptors on their cell surface. These molecules integrate all the signals received from microorganisms and/or cells and switch the balance to activation or inhibition of the immune response. Nevertheless, cancer cells can evade detection by the immune cells through the expression of surface molecules that mimic the signals released by healthy cells. Thus, tumoral cells prevent the arrival of immune effectors to the tumor area or, in the event that effector cells infiltrate the tumor, induce their inactivation and death. These molecules expressed by healthy or tumoral cells to keep the immune system under control and their receptors are collectively called checkpoint proteins. These “immune checkpoints” normally function to control excessive immune activation but are also used by tumors to evade the immune system. Because of this, tumors can be classified based on the existence or lack of a T-cell-inflamed tumor microenvironment, and this phenotype correlates with the response to an immune-checkpoint blockade. An inverse correlation has been observed between WNT/β-catenin pathway activation and T-cell infiltration across most human cancers (131, 132). The connection between immune exclusion and the WNT/β-catenin pathway was identified for the first time in metastatic melanoma. When comparing samples categorized as either T cell–inflamed or non-T cell–inflamed it was observed that almost half of the non-T cell–inflamed tumor subset showed increased activation of the WNT/β-catenin signaling pathway. Mechanistic studies using genetically engineered mouse models confirmed that melanomas with increased WNT/β-catenin activation lacked tumor-infiltrating T cells, mimicking the non-inflamed phenotype observed in patients with melanoma. This effect is due to failed recruitment of specific dendritic cells into tumors, leading to impaired recruitment of T cells to the tumor microenvironment (131, 132). Some of the mechanisms involved in the escape of immunity develop after the activation of WNT in tumor cells. Among them, decrease of secretion by tumoral and/or stromal cells of immune cell-attracting chemokines, such as CC-chemokine ligand 4 (CCL4), should be highlighted (61). Other escape mechanisms involve the expression of checkpoint inhibitors such as programmed death ligand 1 (PD-L1) and CD47 which mainly control the activity of tumor-specific T cells and macrophages (61, 133, 134). PD-L1 and CD47 transcription is controlled by MYC, a proto-oncogene identified as a WNT target gene. Thus, mutations in components of the WNT/β-catenin signaling pathway induce aberrant MYC expression and, because of that, increased expression of the immune checkpoint proteins PD-L1 and CD47 and a non-T cell-inflamed tumor phenotype. Conversely, MYC inactivation led to a decrease in the expression of the above proteins, favoring the accumulation of tumor-associated T cells and macrophages (133–135).
Impaired DCs Recruitment: CCL4 Inhibition
Anti-tumoral immune cells such as DCs and T cells need to reach the tumor bulk to perform their anti-tumoral activity. Nevertheless, tumors develop mechanisms to avoid immunity by disrupting chemokine secretion. It has been observed that the secretion of chemokines implicated in effector T-cell recruitment is significantly reduced in several types of tumors lacking dendritic cells and CD8+ T-cell infiltrate. Among the chemokines that act on the recruitment of DCs and whose secretion is reduced in many non-T cell inflamed tumors, CC-chemokine ligand 4 (CCL4/MIP-1beta) should be highlighted (61, 136). As an example, immune exclusion mediated by activation of the WNT–β-catenin pathway was observed in melanoma when expression of CCL4 was inhibited and DCs were no longer recruited into the tumor. Consequently, no T cell priming occurs, and effector T cells were not present at the tumor area (137). Supporting the previous results, a study involving 266 cases of metastatic melanoma found that approximately one third of the melanoma metastatic lesions were non-T cell inflamed and half of them showed activation of WNT–β-catenin signaling in tumor cells. Further support that activation of β-catenin in tumors was related to T cell exclusion was obtained in a genetically engineered mouse model of melanoma which conditionally expressed a dominant stable form of β-catenin. Although all the mice developed melanoma, β-catenin-positive tumors had minimal T cell infiltration and were resistant to therapy based on checkpoint blockade. In β-catenin-positive melanoma cells, secretion of CCL4 and other chemokines was reduced, with insufficient recruitment of specific DCs into the tumor area and defective host priming of antigen-specific T cells (138, 139). Another study in bladder cancer demonstrated the WNT7/β-catenin pathway activation in non-T cell-inflamed tumors (140). Studies in melanoma cells also showed that the CCL4 chemokine and the BATF3 transcription factor were linked to the recruitment of the dendritic cells necessary for T-cell activation (141). Additionally, gene expression of WNT7B was explored in urothelial bladder cancer and an inverse correlation with the presence of CD8 cells was found, further supporting a link between the absence of intratumoral T cells and the activation of WNT signaling. This result was also confirmed by immunohistochemistry. Indeed, detection of CD8 transcripts and BATF3 and CCL4 expression within the tumor area inversely correlated with WNT7B expression (Figure 4) (140).
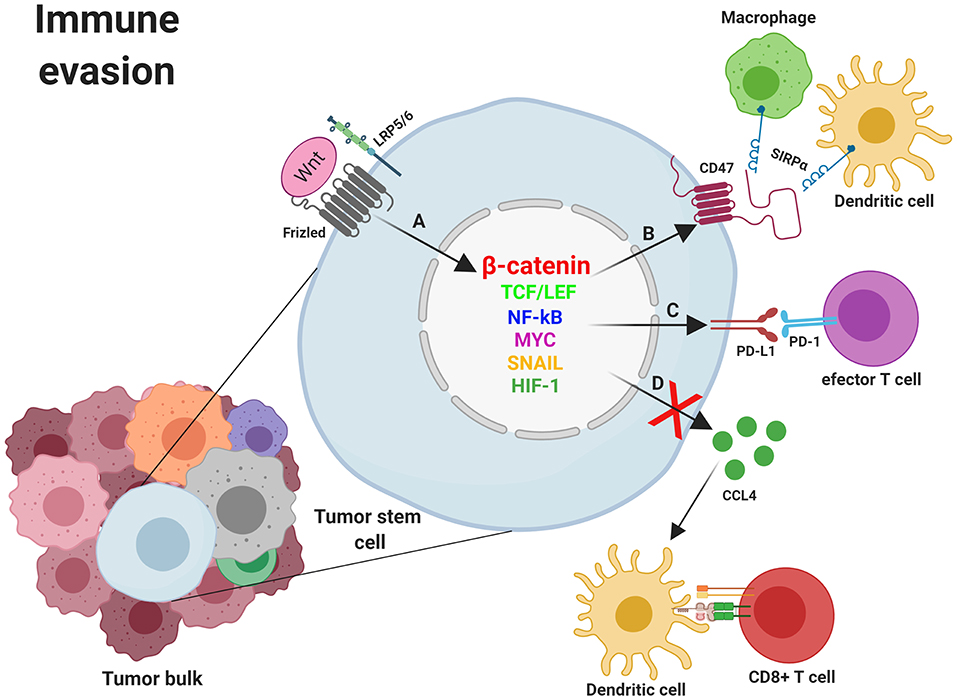
Figure 4. Mechanisms of immune evasion induced by WNT/β-catenin signaling in cancer stem cells. (A) Nuclear β-catenin, by means of MYC, TCF/LEF, NF-κB, SNAI1, and other transcription factors, promotes the transcription of genes involved in mechanisms supporting tumor cells survival such as the deregulation of molecules involved in immune evasion (CCL4, CD47, and PD-L1). (B) The binding of CD47 to SIRPα might prevent phagocytosis of tumor cells (and/or fragments derived from them) by macrophages and dendritic cells with the subsequent absence of T cell activation. (C) Expression of PD-L1 might directly impair effector T cells function within the tumor microenvironment. (D) Inhibition of CCL4 secretion by tumor cells avoids Dendritic cells recruitment to the tumor microenvironment and the subsequent T cell priming and activation. CCL4, CC-chemokine ligand 4; LRP5/6, Low-density lipoprotein receptor-related protein 5/6; NF-κB, Nuclear factor kappa B; MYC, Myelocytomatosis oncogene cellular homolog; PD-1, Programmed cell death 1; PD-L1, programmed death ligand 1; SIRPα, Signal regulatory protein Alpha; SNAIL, Zinc finger protein SNAI1; TCF/LEF, T-cell factor/lymphoid enhancer factor. Created with BioRender.com.
Inhibition of Tumor-Associate Macrophages (TAM) and DCs Phagocytosis Through “Don't Eat Me” Signals: CD47
CD47 (Integrin-Associated Protein, IAP) is a transmembrane glycoprotein, member of the Ig superfamily, with an IgV-like extracellular domain and a short cytoplasmic tail. CD47 expression is ubiquitous in human cells where it is a “marker of self” functioning as a “don't eat me” signal. CD47 has been shown to interact in cis with αvβ3, αIIbβ3, and α2β1 integrins and in trans with thrombospondins (TSPs) and with regulatory molecules belonging to the signal regulatory protein (SIRP) family. SIRPα and SIRPγ are ligands for CD47. SIRPα is highly expressed on myeloid cells (dendritic cells, macrophages, and neutrophils) and smooth muscle cells and discretely expressed by cultured murine and human endothelium. SIRPγ is expressed by T-cells, NK cells, and some B-cells (142).
Interaction of CD47 to SIRP-α promotes the phosphorylation of the immunoreceptor tyrosine-based inhibitory motif (ITIM) on SIRP-α, and the recruitment to the membrane of Src homology region 2 domain-containing phosphatases (SHP-1 and SHP-2), with the subsequent inhibition of myosin-IIA accumulation at the phagocytic area, blocking phagocytosis (143, 144).
CD47 can be expressed on many healthy cells in mice and humans and is highly expressed on a variety of CSCs, including both hematopoietic and solid tumors (145). Expression of CD47 in tumor cells avoid their recognition and elimination by macrophages, dendritic cells, and T cells and induces epithelial-mesenchymal transition (EMT) through modulation of N-cadherin and E-cadherin (146). In fact, CD47 protein expression was significantly high in ovarian cancer and associated with patient stage, chemotherapy resistance, and prognosis. Similar results have been observed in glioma cells, breast cancer cells (147), not small-cells lung carcinoma (NSCLC), PDAC, and others (147–150). The presence of CD47 in AML was also associated with a high self-renewal potential of cancer stem cells and with low patient survival (151, 152). CD47 overexpression also correlates with poor prognosis in head and neck squamous cell carcinoma, melanoma, and osteosarcoma (153–156).
Various transcription factors have been proposed to bind to the promoter of CD47 and explain its upregulated expression in different tumors. Some of them like NF-κB, MYC, SNAI1, ZEB1, HIF-1 and the PKM2–β-catenin–BRG1-TCF4 complex (134, 148, 157, 158) have been implicated in CD47 expression, which strongly suggests that the WNT/β-catenin pathway is involved (Figure 4). Supporting this idea, it has been shown that a constant activation of β-catenin is needed for glioma progression (159), and increased levels of its target genes, such as CD47, have been associated with high-grade GBM (16). Conversely, the inhibition of β-catenin in mutant glioma cells abrogated CD47 expression as well as the interaction between β-catenin and TCF4. The reverse effect was observed in the same cells upon the pharmacological elevation of nuclear β-catenin levels. As expected, the CD47 transcriptional downregulation negatively affected the phagocytosis of cancer cells by microglia (158).
In addition, miRNAs have been described as regulators of stem cells and related with the overexpression of CD47 in cancers. miR-133a acts as a tumor suppressor gene, and is downregulated in many types of tumors (160). miR-133a also regulates the transcription factor TCF7, which is essential in the activation of canonical WNT signaling (161).
In another study, miR-708 induced repression of the WNT/β-catenin signaling pathway in BCSCs, causing inhibition of self-renewal and chemoresistance in these cells. miR-708 was shown to directly bind the CD47 gene regulating the expression of this molecule and the tumor-associated macrophage-mediated phagocytosis (162).
Finally, a role for CD47 in drug resistance has been described. In hepatocellular carcinoma cells (HCC), it was found that clones resistant to sorafenib exhibited increased cancer stem cell characteristics, such as tumorigenicity, self-renewal, and invasiveness. Moreover, an increase in CD47 expression, dependent on nuclear factor kappa B (NF-κB) activation, was found. The knockdown or blocking of CD47 in sorafenib-resistant HCC cells consistently demonstrated an increased sensitization to sorafenib by these cells, suggesting that CD47 signaling might be involved in the sensitization to this drug. Therefore, CD47 may have a role through many as yet unknown pathways in drug resistance (163).
Elimination of Activated T Cells Through “Don't Find Me” Signals: PDL-1
PD-L1 is a type I single-pass transmembrane protein, member of the B7 family, organized in an IgV-like domain, an IgC-like domain, and a cytoplasmic tail (163, 164). PD-L1 interacts with the receptor, programmed cell death 1 (PD-1), on activated cytotoxic T cells through the IgV domain. Next, PD-1 forms aggregates with TCR and costimulatory receptor CD28 and recruits the SHP2 phosphatase, leading to its dephosphorylation and inactivation (165, 166). Last, effector T cells become exhausted by the decreased phosphorylation of crucial signaling molecules which regulate activation and proliferation mediated by NFAT (165–167). PD-L1 is expressed in many cell types including macrophages and dendritic cells (164), and tissues such as heart, lung, and placenta (168), and is also overexpressed with immune activation (169). The PD-L1/PD-1 interaction keeps the balance between tolerance and autoimmunity and its deficient or excess functioning can trigger several diseases, including auto-immune diseases such as arthritis and lupus (170). PD-L1 expression has been found to be positive in 5–40% tumor cells such as lung, colon, melanoma, bladder and renal and hepatocellular carcinomas, head and neck cancers, ovarian cancer, and hematologic malignancies (171), inducing elimination of effector cells through interaction of PD-L1 on the surface of cancer cells with PD-1 on the T cells plasma membrane (171–174). It has also been shown that PD-L1 expression is modulated by the WNT pathway (Figure 4). Thus, Triple Negative Breast Cancer Stem Cells (TNBCSCs) exhibit PD-L1 overexpression through the WNT cascade and the upregulation or downregulation of this cascade significantly affects the expression of this molecule (175). In testicular germ tumors β-catenin was described as a marker of poor outcome and a positive correlation with PD-L1 expression was observed, as was a decrease in immune infiltration (176). In melanoma cells, the activation of WNT/β-catenin results in the absence of T cell infiltration in the tumor microenvironment. This effect appears to be a consequence of the action of the negative regulatory pathway PD-L1-PD-1 (141). Finally, an increase in PD-L1 in the population of breast mesenchymal-like cancer cells, but specially in breast CSCs, has been observed during EMT by the EMT/β-catenin/STT3/PD-L1 signaling axis. Specifically, it has been shown that the ER-associated N-glycosyltransferases STT3A and STT3B are required for PD-L1 induction through regulating its glycosylation. This produces PD-L1 stabilization by antagonizing β-TrCP-dependent proteasome degradation of this molecule. In this study, the authors also identified that etoposide suppressed the EMT/β-catenin/STT3/PD-L1 axis through TOP2B degradation-dependent nuclear β-catenin reduction, leading to PD-L1 downregulation of CSCs and non-CSCs, and sensitization of cancer cells to anti-Tim-3 therapy. (177). In summary, several studies support an association between WNT/β-catenin signaling and immune evasion through the regulation of the “don't find me signal” PD-L1 and other molecules involved in immune control.
New Strategies for Effective Antitumoral Therapy
Most of the anticancer chemotherapies currently being used work by killing highly proliferating cells, which, in many tumors, are mostly non-CSCs, thus decreasing the tumor size. However, the small CSC population present in the tumor bulk is constituted by relatively slow cycling quiescent cells with a higher repair mechanism and are innately resistant to therapy. Furthermore, radiation and chemotherapy may trigger cellular stress response mechanisms enhancing stemness characteristics in non-CSCs and thereby increasing their capacity for adaptation and survival. Thus, conventional chemotherapy can increase the fraction of CSCs within a cancer, making them resistant to treatment, and re-establishing tumor growth in the same or in a distant location from the primary tumor, leading to the formation of metastases which are usually far more resistant to chemotherapy than primary tumors. This has been shown following the treatment of different tumor types, including brain, head and neck, lung, breast, and liver, amongst others (178). Therefore, in contrast to previous therapeutic approaches, new treatments need to consider strategic combinations that could kill CSCs and non-CSCs at once as well as preventing the transition from non-CSCs to CSCs.
Several anti-neoplastic drugs targeting specific molecules are currently in clinical use. One type includes inhibitors of signaling molecules such as inhibitors of tyrosine kinases, Raf, MEK, PI3K, mTOR (mammalian target of rapamycin), and WNT/β-catenin signaling pathways, which are essential for the proliferation of tumoral cells. Another type of therapy is based on obtaining and using monoclonal antibodies (mAbs) to surface proteins highly expressed in cancer cells, such as CD20 in lymphoma (rituximab), HER2 in breast cancer (trastuzumab), and epidermal growth factor receptor (EGFR) in colon cancer (cetuximab). Another type of therapy includes compounds and/or mAbs targeting molecules with a high expression in CSCs and with an essential role in drug resistance and/or tumor immune escape. Inhibitors and antibodies (Abs) specific for ABCB1, CD44, CD47, and/or PD-L1 belong to this group. The great heterogeneity of tumor cellular composition and the complex mechanisms involved in the tumorgenicity in CSCs and non-CSCs subsets requires the use of many different therapeutic approaches and targets, based on the characteristics of the tumor and the patient. Many of these approaches developed to eliminate tumor bulk are used in combination and are currently under clinical evaluation.
Targeting Molecules Involved in WNT Pathway
Targeting the WNT signaling pathway in CSCs is an interesting and promising approach for anti-tumoral therapy. Although this signaling pathway is expressed in normal cells, deregulation is found in CSCs. Compounds with WNT inhibitory properties can target the CSCs but also normal cells, inducing adverse effects. In this regard, therapeutic molecules should be modified or combined with other therapies to improve their specificity and efficiency. Thus, current chemotherapeutic strategies focus on targeting WNT signaling in specific tumor subclasses or with specific mutational characteristics. Ideally, this tumor-targeted therapy should have at least, three main effects: first, recruitment of dendritic cells, macrophages, and effector T cells in the tumor area; second, reversal of resistance to tumor drugs; and third, inhibition of tumor evasion. Since canonical WNT signaling promotes not only T cell lymphopoiesis but also regulation of peripheral immune cells activation and differentiation (61, 72), ongoing therapy should also have a minimal incidence on immune cells, specifically those infiltrated in the tumor area. Otherwise, mechanisms of anti-tumoral immunity will not function properly and the effectiveness of the immune cells fighting the tumor will be seriously compromised. Finally, given the crucial role of WNT signaling in the maintenance of stem cells and the regeneration of tissues and organs in homeostasis, unwanted side effects should be carefully evaluated. In fact, although diverse types of WNT/β-catenin pathway inhibitors are under development as anti-neoplastic therapies in many hematologic and solid malignancies, none have been approved for clinical use (Figure 5A) (36).
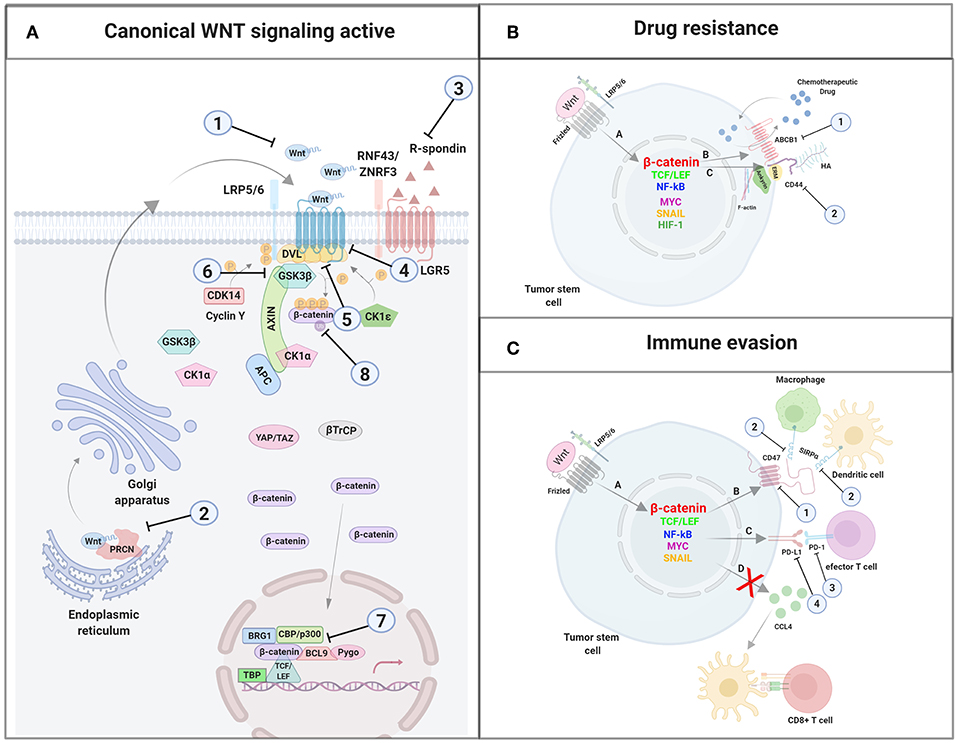
Figure 5. Inhibitors used for antitumoral therapy. (A) Agents targeting molecules of the WNT canonical pathway: (1) Targeting WNT ligands or agonists; (2) Porcupine specific inhibitors; (3) Blocking antibody targeting RSPO3; (4) Frizzled blocking agents; (5) Agents targeting LGR5; (6) GSK3β inhibitors; (7) Transcription complex inhibitors; (8) Agents targeting β-catenin expression. (B) Agents targeting molecules involved in drug resistance: (1) Inhibitors of the extrusion pump ABCB1; (2) Blocking antibodies targeting CD44. (C) Agents targeting molecules involved in immune evasion: (1) CD47 blocking agents; (2) Agents targeting SIRPα; (3) Agents targeting PD-1; (4) Agents targeting PD-L1. Created with BioRender.com.
Targeting WNT Ligands
Monoclonal antibodies, recombinant proteins, and other inhibitors that neutralize WNT ligands or agonists of these molecules are under clinical investigation and have demonstrated WNT inhibition and tumor reduction in melanoma, sarcoma, colorectal cancers, non-small cell lung carcinoma, and mesothelioma. For example, a hexapeptide has been synthesized which can imitate the characteristics of the WNT5a ligand and inhibit tumor cell migration in vitro. That compound is being tested in a phase 1 study of patients with breast, colon, and prostate metastatic cancers (36, 37). Moreover, a WNT inhibitor which binds and sequesters WNT, blocking the signaling cascade, is in Phase 1b clinical trials (clinicaltrials.gov). Additionally, specific inhibitors have been found that neutralize Porcupine, preventing acylation, and WNT secretion (179). These inhibitors have been tested in phase I trials in melanoma, breast cancer, and pancreatic cancer, metastatic colorectal and head and neck cancers. Promising therapeutic results have also been obtained in pre-clinical models of CRC with another treatment, based on the use of a blocking antibody targeting RSPO3 which produces a loss of stemness and differentiation.
Targeting Frizzled Receptors and Co-receptors
Several trials testing Abs specific for receptors and co-receptors of the WNT signaling pathway are currently in process. For example, an anti-Fz10 radiolabeled mAb is being evaluated for the treatment of synovial sarcoma in a phase I clinical trial. Also, a monoclonal antibody specific to five human FZD receptors inhibits the growth of human tumor xenografts in mouse models. This antibody is currently included in phase I clinical trials used as monotherapy or in combination with taxanes in breast and pancreatic cancer and non-small cell lung cancer. It is also being used in combination with conventional chemotherapy in phase 1b clinical trials with breast, liver, ovarian, pancreatic, and lung cancer patients (180). Additionally, the use of Abs conjugated to cytotoxic drugs targeting LGR5 has demonstrated therapeutic effectiveness in xenografts and in genetic mouse models of CRC.
Disheveled Inhibitors
A non-steroidal anti-inflammatory drug (NSAID) which targets DVL is currently undergoing testing in phase II trials, although multiple non-NSAID inhibitors of DVL have also been developed. Specifically, these agents block the binding between FZD and DVL at the membrane, inhibiting DVL activation. As a consequence of this, the destruction complex is stabilized, thus promoting β-catenin degradation (12, 151, 181).
GSK3β Inhibitors
The effect of an inductor of GSK3β degradation was tested in combination with chemotherapy on cholangiocarcinoma (CCA) cells and was shown to enhance the effect of classical chemotherapy. This agent downregulated ABCB1 expression in a GSK3β-dependent manner (182). In addition, lawsone derivatives are compounds which induce collateral sensitivity in MDR ABCB1-overexpressing cells. These compounds decrease β-catenin activity in a reporter cell line and downregulate c-MYC, ABCB1/P-GP, and FZD7 protein expression. Furthermore, WNT/β-catenin signaling was selectively inhibited in resistant cell lines (108).
TCF/CBP/β-Catenin Transcription Complex Inhibitors
Another inhibitor of WNT/β-catenin signaling that has been synthesized specifically binds to the N-terminus of CBP and not p300, blocking only the interaction between CBP and β-catenin. The compound leads to differentiation and loss of self-renewal capacity in pre-B ALL cells. Also, this drug downregulated survivin, an inhibitor-of-apoptosis protein, (IAP) in primary ALL and, in combination with conventional therapy and independently of the mutational status of CBP and chromosomal aberration, eradicates drug-resistant primary leukemia in vitro and prolongs the survival of NOD/SCID mice inoculated with primary ALL (183–185).
Additionally, the use of RNA interference technology to knock down the expression of β-catenin results in the reduction of many CSC properties, such as proliferation, migration, drug resistance, and expression of transcription factors such as OCT-4, amongst others (77).
Also, CRISPR/Cas9 genome editing technology has been used to delete β-catenin. Thus, results have shown reduced tumorigenesis in mixed lineage leukemia and regression of epidermal tumors by depleting CSCs.
Another experimental strategy involves the activation of quiescent CSCs subsets which increases their sensitivity to chemotherapeutic drugs. Quiescence has been associated with the activity of the ubiquitin ligase FBXW7, which downregulates MYC levels. Thus, in experimental models of chronic myeloid leukemia (CML), genetic ablation of FBXW7 in quiescent leukemic stem cells reprograms them to re-enter the cell cycle and the cells become susceptible to imatinib (186).
Targeting Molecules Involved in Drug Resistance: ABCB1 and CD44
Targeting ABCB1
Several attempts have been made to efficiently counteract the action of the extrusion pump ABCB1/P-GP and although many pump extrusion inhibitors have been assayed in clinical trials, none of them have been approved for use in patients. Currently, the third and fourth generations of ABCB1/P-GP inhibitors are being designed and/or tested. Third generation inhibitors use nanomolar concentrations to increase their effectiveness at reversing MDR compared to first- and second-generation compounds. Thus, elacridar and zosuquidar significantly inhibited ABCB1 with low toxicity in phase I clinical trials. More specifically, zosuquidar, an oral ABCB1 inhibitor, has been used in the treatment of acute myeloid leukemia, and improves the uptake of daunorubicin, idarubicin, and mitoxantrone. Another inhibitor, tariquidar, has elevated ABCB1 affinity and reduces the ATPase activity of the extrusion pump. Nevertheless, phase III assays of tariquidar with carboplatin/paclitaxel or with vinorelbine were closed due to toxicity issues. Tetrandrine, an alkaloid isolated from Stephania tetrandra, modulates the activity of ABCB1 and has also been used with doxorubicin in phase I clinical trials (www.clinicaltrials.gov) in the treatment of multidrug resistant cancers (187).
More recently, researchers have been working on the fourth generation of ABCB1 inhibitors, which are natural compounds derived from plant extracts, marine organisms, fungi and other sources, and exhibit modulatory properties on ABCB1, less cytotoxicity, and better oral bioavailability. The catalog of these compounds includes alkaloids, coumarins, flavonoids, and terpenoids, many of which are ABCB1 inhibitors. Thus, trabectedin, cytarabine, and Halaven have been approved for use with patients due to their powerful ABC drug transport reversal activity (188). Other phytochemicals such as curcumin and quercetin prevent ABCB1 function reversing MDR in human cancer cell lines (189, 190). Another example is piperine, an alkaloid and major component of black pepper (Piper nigrum) and long pepper (Piper longum). This compound exhibits P-GP inhibitory activity, and also antitumoral, antioxidant, antimicrobial, and hepatoprotective functions (191). Other flavonoids such as luteolin and casticin have been shown to be active on glioma stem-like cells (192, 193). 8-Bromo-7-methoxychrysin (BrMC) induces apoptotic cell death on hepatocellular carcinoma (Hep-G2 cell line) by generating reactive oxygen species (ROS) (194). Additionally, LY294002 is a compound with a PI3K inhibitory activity that induces apoptosis in osteosarcoma CSCs by blocking the cell cycle. This compound also inhibits ABCG2/BCRP, ABCB1/MDR1/P-GP, and ABCC1/MRP1, three transporters with a high expression in stem cells. In addition, salinomycin, an antibiotic isolated from Streptomyces albus, interferes with ABC transporters and CSC pathways in many solid tumors and hematological cancers (187, 195).
Thus, novel compounds are being considered as new extrusion pumps inhibitors. Nevertheless, many adverse effects must be considered. Inhibition of efflux pumps can cause adverse effects in normal stem cells, since they exhibit high expression of several extrusion pumps which they need for their physiological function. In addition, ABCB1, and other pumps, play an essential role in keeping the integrity of the blood brain barrier, and interfering with their normal function could have negative consequences for the patient's health (Figure 5B) (187, 196, 197).
Targeting CD44
Another promising approach to blocking essential CSC signaling pathways is the use of therapy based on Abs. Thus, several anti-CD44 monoclonal Abs have been obtained with very promising results, since some of them selectively eliminate the self-renewal properties of CSCs in several malignancies and cancer cell lines. Specifically, the antibody H90 can discern between the stem cells from conventional hematopoietic progenitor cells and from AML cells. Also, the antibody P245 inhibits estrogen and progesterone receptor and HER2 in TNBCC and H4C4 blocks tumor sphere formation in human pancreatic carcinoma cells and inhibits tumorigenesis in a murine xenograft model. Notably, another recombinant and humanized anti-CD44 monoclonal antibody (RO5429083/RG7356) blocks tumorigenesis in head-and-neck carcinoma cells in mice by the cytolytic action of natural killer (NK) cells. Moreover, this antibody can eliminate triple-negative MDA-MB-231 breast cancer and CLL cells (Figure 5B) (198).
Targeting Molecules Involved in Immune Evasion: CD47 and PD-L1
Targeting CD47
The interaction CD47-SIRPα has been intensively investigated as potential cancer therapy (199–201). Several humanized monoclonal Abs have been obtained with encouraging therapeutic results. For example, Hu5F9-G4, the first humanized mAb to human CD47 increases the phagocytosis of tumor cells by macrophages in vitro and eliminates tumors in xenograft mouse models. The humanized mAb to human CD47, Hu5F9-G4, and CC-90002, are being tested in phase I or I/II clinical trials for solid tumors and hematological malignancies (202). Treatment with another anti-CD47 antibody, B6H12.2, has been shown to improve CD8+ T-cell cytotoxicity and macrophages phagocytosis and decrease tumorigenesis in AML CSCs in animals. Additionally, promising results have been obtained in many types of cancer cells. Therapy based on the use of B6H12 also decreased tumor formation in a leiomyosarcoma mouse model and the reduction of CSCs in pediatric brain carcinomas (151).
Moreover, another therapeutic approach is the design and synthesis of engineered recombinant SIRPα proteins with improved affinity for the CD47 molecule blocking endogenous CD47-SIRPα interaction. Specifically, TTI-621 is formed by the Ig-V-like domain of human SIRPα linked to the Fc region of human IgG1 (203). Similarly, the recombinant protein ALX148 is constituted by a variant of the Ig-V-like domain of human SIRPα anchored to an inactive Fc domain (203, 204). Both treatments are being used in hematological malignancies or solid tumors; as single medication or with Abs specific for tumoral antigens, with radiotherapy or with immune checkpoint inhibitors. CD47 blockade promotes antibody-dependent cellular phagocytosis (ADCP) of tumor cells by macrophages and tumor killing by cytotoxic T lymphocytes. In contrast, since many cell types express CD47, therapy based on targeting that molecule could induce adverse effects and, in fact, treatment with anti-CD47 Abs induced the appearance of anemia in monkeys (Figure 5C) (204).
Targeting SIRPα
Another potentially successful strategy for cancer therapy is the combination of a molecule that blocks SIRPα with Abs specific for tumoral antigens. In fact, the combined use of anti-SIRPα murine Abs and rituximab, eliminated human Raji cells grafted into non-obese diabetic (NOD)/SCID mice (205). Moreover, blocking CD47-SIRPα interaction with an antibody specific for human SIRPα enhanced killing in vitro by macrophages of HER2+ breast cancer cells previously opsonized with the anti-HER2 monoclonal antibody, trastuzumab (206).
The therapeutic success of anti-SIRPα blocking Abs in tumors SIRPα-positives, might be based on the activation of the ADCP mechanism against tumor cells by macrophages together with the elimination of the phagocytosis blockade exerted by the CD47-SIRPα interaction. In fact, the use of a murine Ab anti-SIRPα reduced tumorigenesis in mice inoculated with cells from renal cell carcinoma or melanoma (205). Cytotoxic T cells and NK cells could also participate in the antitumor mechanisms triggered by the anti-SIRPα Abs (205). Such Abs could favor activation of cytotoxic T lymphocytes by the macrophages and DCs infiltrated in the tumor area and induce antibody-dependent cellular cytotoxicity (ADCC) by NK cells toward tumor cells (206–208). It is of great importance that this type of treatment has thus far not induced adverse effects such as anemia or neurotoxicity in mice (206). In addition, recombinant CD47 proteins blocking the interaction between CD47 and SIRPα may also contribute to the elimination of tumor cells. Indeed, a variant of CD47 with improved affinity for SIRPα had a synergistic effect when used in combination with tumor-specific monoclonal Abs to ameliorate phagocytosis of tumor cells in vitro (209), although its antitumoral effects in vivo have not been evaluated (Figure 5C).
Targeting PD-L1
Therapies based on anti-PD-1 and anti-PD-L1 Abs have been designed to augment the cytotoxic T-cell attack to tumor cells. However, these treatments sometimes fail because of the emergence of resistance in patients. Furthermore, these Abs sometimes do not target all the CSCs present in the tumor bulk or induce insufficient signaling responses. To solve these problems, researchers have obtained multi-specific Abs with the capacity to bind more than one membrane receptor. These chimeric Abs are very efficient at blocking ligand binding and inhibiting downstream signaling. Several inhibitors of PD-L1 have been obtained such as atezolizumab, durvalumab, avelumab, and inhibitors of PD-1 include nivolumab and pembrolizumab (168). Treatment of patients with PD-1 and PD-L1 inhibitors, alone or in combination with standard chemotherapy, has demonstrated preliminary positive results in advanced TNBC (151, 175).
Finally, CRISPR/Cas9 is another method to cleave DNA, allowing edition in any cell. A clinical trial based on this technology was initiated to combat non-small-cell lung cancer by deleting the PD-1 receptor (Figure 5C) (151).
Combination of Treatments to Target Several Immune Checkpoint Inhibitors
Combinations of therapies based on targeting immune molecules with standard chemotherapeutic drugs and novel therapeutic agents could improve the efficacy of immune checkpoint inhibition in unresponsive patients. The strategy is based on the joint use of Abs and/or recombinant proteins targeting checkpoint inhibitors, chemotherapeutic drugs, cancer vaccines, and immune-stimulatory molecules. These combination therapies are currently being tested in pre-clinical models and patients. The joint blockade of the CD47-SIRPα and PD-1-PD-L1 interactions might have a synergistic effect in the elimination of tumor cells. Indeed, an anti-CD47 nanobody that inhibits the CD47-SIRPα interaction synergized with a PD-L1 antagonist significantly reducing the growth of tumors in mice previously injected with melanoma cells (210, 211). The efficacy of therapy based on the combined use of anti-PD-1 and anti-CTLA-4 antibodies was enhanced by using anti-CD47 blocking Abs in a murine model of esophageal squamous cell cancer (210). Moreover, combined therapy with SIRPα and PD-1 blocking agents had a synergistic antitumoral effect in a murine model of colon cancer (205). The combination of agents with capacity to inhibit the CD47-SIRPα interaction with Abs that block the PD-1-PD-L1 binding is a promising therapeutic approach for the treatment of a broad range of cancers (144, 210, 211).
Conclusions
WNT/β-catenin signaling is a highly conserved pathway involved in multiple essential homeostatic functions. Nevertheless, deregulation of this pathway is involved in cancer progression, drug resistance, and immunity escape. Thus, at the same time the WNT pathway is activated in CSCs, a complex transcriptional program is initiated, resulting in the expression of genes such as ABCB1 and CD44, which are involved in drug resistance, and PD-L1 and CD47, which are well-known in immunity. This increased WNT activation in tumor cells is responsible for the start of different survival programs which have made tumor cells highly resistant to the anti-tumoral response and/or the drug treatment. For a long time, inhibitors and/or blocking Abs have been used to counteract the action of ABCB1 as an extrusion pump or to block PD-L1 and/or CD47, and combined therapy has been relatively successful. Additionally, many components of the WNT pathway have been the focus of the scientific community as promising therapeutic targets to eliminate tumor cells. Nevertheless, although the combination of several therapeutic approaches has been promising, many challenges remain to be solved in order to design an effective anti-tumoral treatment which targets tumoral cells, including the small population of CSCs that constitutes a critical subset of the tumor bulk.
Author Contributions
EM-O wrote and revised the manuscript and supervised the figures and references. AS-F assisted in the elaboration of the figures and references list. IO-P organized the references list. All authors assisted in the conception of this review, acquisition of relevant literature, and gave their approval of the last version to be published.
Funding
This work was supported by a grant from the University of Murcia (Plan Propio de Fomento de la Investigación y la Transferencia de la Universidad de Murcia 2019).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to those authors whose work we could not cite due to space constraints. We thank Dr. Manuel Sánchez-Angulo for the critical reading of this manuscript and helpful suggestions.
References
1. Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. (2011) 469:356–61. doi: 10.1038/nature09650
2. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) 366:883–92. doi: 10.1056/NEJMoa1113205
3. Anderson ARA, Weaver AM, Cummings PT, Quaranta V. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell. (2006) 127:905–15. doi: 10.1016/j.cell.2006.09.042
4. Sottoriva A, Verhoeff JJC, Borovski T, McWeeney SK, Naumov L, Medema JP, et al. Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Cancer Res. (2010) 70:46–56. doi: 10.1158/0008-5472.CAN-09-3663
5. Waclaw B, Bozic I, Pittman ME, Hruban RH, Nowak MA, Guthrie P, et al. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. (2015) 525:261–4. doi: 10.1038/nature14971.A
6. Medema JP. Cancer stem cells: the challenges ahead. Nat Cell Biol. (2013) 15:338–44. doi: 10.1038/ncb2717
7. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. (2013) 501:328–37. doi: 10.1038/nature12624
8. Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci USA. (2008) 105:13427–32. doi: 10.1073/pnas.0805706105
9. Campbell LL, Polyak K. Breast tumor heterogeneity: cancer stem cells or clonal evolution? Cell Cycle. (2007) 6:2332–8. doi: 10.4161/cc.6.19.4914
10. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. (2017) 16:41. doi: 10.1186/s12943-017-0600-4
11. Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by cancer stem cells. Semin Cancer Biol. (2018) 53:156–67. doi: 10.1016/j.semcancer.2018.11.006
12. Ramos EK, Hoffman AD, Gerson SL, Liu H. New opportunities and challenges to defeat cancer stem cells. Trends Cancer. (2017) 3:780–96. doi: 10.1016/j.trecan.2017.08.007
13. Singh A, Settleman J. EMT cancer stem cells and drug resistance. Oncogene. (2010) 29:4741–51. doi: 10.1038/onc.2010.215
14. Flanagan DJ, Barker N, Di Costanzo NS, Mason EA, Gurney A, Meniel VS, et al. Frizzled-7 is required for Wnt signaling in gastric tumors with and without APC mutations. Cancer Res. (2019) 79:970–81. doi: 10.1158/0008-5472.CAN-18-2095
15. Zhang Y, Morris JP IV, Yan W, Schofield HK, Gurney A, Simeone DM, et al. Canonical Wnt signaling is required for pancreatic carcinogenesis. Cancer Res. (2013) 73:4909–22. doi: 10.1158/0008-5472.CAN-12-4384
16. Sinnberg T, Levesque MP, Krochmann J, Cheng PF, Ikenberg K, Meraz-Torres F, et al. Wnt-signaling enhances neural crest migration of melanoma cells and induces an invasive phenotype. Mol Cancer. (2018) 17:59. doi: 10.1186/s12943-018-0773-5
17. Vesel M, Rapp J, Feller D, Kiss E, Jaromi L, Meggyes M, et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir Res. (2017) 18:52. doi: 10.1186/s12931-017-0537-6
18. Liu Y, Chang Y, Lu S, Xiang YY. Downregulation of long noncoding RNA DGCR5 contributes to the proliferation, migration, and invasion of cervical cancer by activating Wnt signaling pathway. J Cell Physiol. (2019) 234:11662–9. doi: 10.1002/jcp.27825
19. Veeck J, Bektas N, Hartmann A, Kristiansen G, Heindrichs U, Knüchel R, et al. Wnt signalling in human breast cancer: expression of the putative Wnt inhibitor Dickkopf-3 (DKK3) is frequently suppressed by promoter hypermethylation in mammary tumours. Breast Cancer Res. (2008) 10:R82. doi: 10.1186/bcr2151
20. Murillo-Garzón V, Kypta R. WNT signalling in prostate cancer. Nat Rev Urol. (2017) 14:683–96. doi: 10.1038/nrurol.2017.144
21. McCord M, Mukouyama Y, Gilbert MR, Jackson S. Targeting WNT signaling for multifaceted glioblastoma therapy. Front Cell Neurosci. (2017) 11:318. doi: 10.3389/fncel.2017.00318
22. van Andel H, Kocemba KA, Spaargaren M, Pals ST. Aberrant Wnt signaling in multiple myeloma: molecular mechanisms and targeting options. Leukemia. (2019) 33:1063–75. doi: 10.1038/s41375-019-0404-1
23. Ashihara E, Takada T, Maekawa T. Targeting the canonical Wnt/β-catenin pathway in hematological malignancies. Cancer Sci. (2015) 106:665–71. doi: 10.1111/cas.12655
24. Yadav AK, Desai NS. Cancer stem cells: acquisition, characteristics, therapeutic implications, targeting strategies and future prospects. Stem Cell Rev Rep. (2019) 15:331–55. doi: 10.1007/s12015-019-09887-2
25. Duchartre Y, Kim Y-M, Kahn M. The Wnt signaling pathway in cancer. Crit Rev Oncol Hematol. (2016) 99:141–9. doi: 10.1016/j.critrevonc.2015.12.005
26. Mikels AJ, Nusse R. Wnts as ligands: processing, secretion and reception. Oncogene. (2006) 25:7461–8. doi: 10.1038/sj.onc.1210053
27. Tao Q, Yokota C, Puck H, Kofron M, Birsoy B, Yan D, et al. Maternal Wnt11 activates the canonical Wnt signaling pathway required for axis formation in Xenopus embryos. Cell. (2005) 120:857–71. doi: 10.1016/j.cell.2005.01.013
28. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
29. Coudreuse D, Korswagen HC. The making of Wnt: new insights into Wnt maturation, sorting and secretion. Development. (2007) 134:3–12. doi: 10.1242/dev.02699
30. Willert K, Nusse R. Wnt proteins. Cold Spring Harb Perspect Biol. (2012) 4:a007864. doi: 10.1101/cshperspect.a007864
31. Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. (2012) 151:1542–56. doi: 10.1016/j.cell.2012.11.024
32. Buechling T, Chaudhary V, Spirohn K, Weiss M, Boutros M. P24 proteins are required for secretion of Wnt ligands. EMBO Rep. (2011) 12:1265–72. doi: 10.1038/embor.2011.212
33. Port F, Hausmann G, Basler K. A genome-wide RNA interference screen uncovers two p24 proteins as regulators of Wingless secretion. EMBO Rep. (2011) 12:1144–52. doi: 10.1038/embor.2011.165
34. Nile AH, Hannoush RN. Fatty acylation of Wnt proteins. Nat Chem Biol. (2016) 12:60–9. doi: 10.1038/nchembio.2005
35. Gross JC, Chaudhary V, Bartscherer K, Boutros M. Active Wnt proteins are secreted on exosomes. Nat Cell Biol. (2012) 14:1036–45. doi: 10.1038/ncb2574
36. Krishnamurthy N, Kurzrock R. Targeting the Wnt/beta-catenin pathway in cancer: update on effectors and inhibitors. Cancer Treat Rev. (2018) 62:50–60. doi: 10.1016/j.ctrv.2017.11.002
37. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. (2017) 36:1461–73. doi: 10.1038/onc.2016.304
38. Acebron SP, Niehrs C. β-catenin-independent roles of Wnt/LRP6 signaling. Trends Cell Biol. (2016) 26:956–67. doi: 10.1016/j.tcb.2016.07.009
39. Kimelman D, Xu W. β-Catenin destruction complex: insights and questions from a structural perspective. Oncogene. (2006) 25:7482–91. doi: 10.1038/sj.onc.1210055
40. Davidson G, Niehrs C. Emerging links between CDK cell cycle regulators and Wnt signaling. Trends Cell Biol. (2010) 20:453–60. doi: 10.1016/j.tcb.2010.05.002
41. Wang X, Jia Y, Fei C, Song X, Li L. Activation/proliferation-associated protein 2 (Caprin-2) positively regulates CDK14/cyclin Y-mediated lipoprotein. J Biol Chem. (2016) 291:26427–34. doi: 10.1074/jbc.M116.744607
42. Li VSW, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, et al. Wnt signaling through inhibition of β-catenin degradation in an intact axin1 complex. Cell. (2012) 149:1245–56. doi: 10.1016/j.cell.2012.05.002
43. Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, et al. Wnt signaling requires sequestration of Glycogen Synthase Kinase 3 inside multivesicular endosomes. Cell. (2010) 143:1136–48. doi: 10.1016/j.cell.2010.11.034
44. Kim N-G, Xu C, Gumbiner BM. Identification of targets of the Wnt pathway destruction complex in addition to β-catenin. Proc Natl Acad Sci USA. (2009) 106:5165–70. doi: 10.1073/pnas.0810185106
45. Gattinoni L, Ji Y, Restifo NP. Wnt/β-catenin signaling in T cell immunity and cancer immunotherapy. Clin Cancer Res. (2010) 16:4695–701. doi: 10.1158/1078-0432.CCR-10-0356.
46. Sineva GS, Pospelov VA. β-catenin in pluripotency. Adhering to self-renewal or wnting to differentiate? In: International Review of Cell and Molecular Biology. Elsevier Inc. (2014), p. 53–78. doi: 10.1016/B978-0-12-800178-3.00002-6
47. Yang Y, Mlodzik M. Wnt-frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu Rev Cell Dev Biol. (2015) 31:623–46. doi: 10.1146/annurev-cellbio-100814-125315
48. Gao B, Song H, Bishop K, Elliot G, Garrett L, English M, et al. Wnt signaling gradients establish planar cell polarity by inducing Vangl2 phosphorylation through Ror2. Dev Cell. (2011) 20:163–76. doi: 10.1016/j.devcel.2011.01.001.Wnt
49. Hatakeyama J, Wald JH, Printsev I, Ho H-YH, Carraway III KL. Vangl1 and Vangl2: planar cell polarity components with a developing role in cancer. Endocr Relat Cancer. (2014) 21:R345–56. doi: 10.1530/ERC-14-0141
50. Yanfeng WA, Berhane H, Mola M, Singh J, Jenny A, Mlodzik M. Functional dissection of phosphorylation of dishevelled in Drosophila. Dev Biol. (2011) 360:132–42. doi: 10.1016/j.ydbio.2011.09.017
51. Mentink RA, Rella L, Radaszkiewicz TW, Gybel T, Betist MC, Bryja V, et al. The planar cell polarity protein VANG-1/Vangl negatively regulates Wnt/β-catenin signaling through a Dvl dependent mechanism. PLoS Genet. (2018) 14:e1007840. doi: 10.1371/journal.pgen.1007840
52. Hao HX, Xie Y, Zhang Y, Zhang O, Oster E, Avello M, et al. ZNRF3 promotes Wnt receptor turnover in an R-spondin-sensitive manner. Nature. (2012) 485:195–202. doi: 10.1038/nature11019
53. Koo BK, Spit M, Jordens I, Low TY, Stange DE, Van De Wetering M, et al. Tumour suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. (2012) 488:665–9. doi: 10.1038/nature11308
54. Morgan RG, Mortensson E, Williams AC. Targeting LGR5 in colorectal cancer: therapeutic gold or too plastic? Br J Cancer. (2018) 118:1410–8. doi: 10.1038/s41416-018-0118-6
55. Li X, Xiang Y, Li F, Ying C, Li B, Ke X. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol. (2019) 10:2293. doi: 10.3389/fimmu.2019.02293
56. De Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. (2014) 28:305–16. doi: 10.1101/gad.235473.113
57. Azzolin L, Panciera T, Soligo S, Enzo E, Bicciato S, Dupont S, et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell. (2014) 158:157–70. doi: 10.1016/j.cell.2014.06.013
58. Sugimura R, He XC, Venkatraman A, Arai F, Box A, Semerad C, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. (2012) 150:351–65. doi: 10.1016/j.cell.2012.05.041.
59. Galluzzi L, Spranger S, Fuchs E, López-Soto A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. (2019) 29:44–65. doi: 10.1016/j.tcb.2018.08.005
60. Nemeth MJ, Topol L, Anderson SM, Yang Y, Bodine DM. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation PNAS. (2007) 104:15436–41. doi: 10.1073/pnas.0704747104
61. Staal FJ, Luis TC, Tiemessen MM. Wnt signalling in the immune system: Wnt is spreading its wings. Nature Rev Immunol. (2008) 8:581–93. doi: 10.1038/nri2360
62. Xu Y, Banerjee D, Huelsken J, Birchmeier W, Sen JM. Deletion of beta-catenin impairs T cell development. Nat Immunol. (2003) 4:1177–82. doi: 10.1038/ni1008
63. Valencia J, Martínez VG, Hidalgo L, Hernández-López C, Canseco NM, Vicente A, et al. Wnt5a signaling increases IL-12 secretion by human dendritic cells and enhances IFN-γ production by CD4+ T cells. Immunol Lett. (2014) 162(1 Pt A):188–99. doi: 10.1016/j.imlet.2014.08.015
64. Sato A, Kayama H, Shojima K, Matsumoto S, Koyama H, Minami Y, et al. The Wnt5a-Ror2 axis promotes the signaling circuit between interleukin-12 and interferon-γ in colitis. Sci Repl. (2015) 5:10536. doi: 10.1038/srep10536
65. Zhou J, Cheng P, Youn JI, Cotter MJ, Gabrilovich DI. Notch and wingless signaling cooperate in regulation of dendritic cell differentiation. Immunity. (2009) 30:845–59. doi: 10.1016/j.immuni.2009.03.021
66. van Loosdregt J, Coffer PJ. The Role of WNT signaling in mature T cells: T cell factor is coming home. J Immunol. (2018) 201:2193–200. doi: 10.4049/jimmunol.1800633
67. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. (2012) 4:a008052. doi: 10.1101/cshperspect.a008052
68. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. (2014) 26:570–9. doi: 10.1016/j.cellsig.2013.11.032
69. Giannakis M, Hodis E, Mu XJ, Yamauchi M, Rosenbluh J, Cibulskis K, et al. RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. (2014) 46:1264–6. doi: 10.1038/ng.3127
70. Loregger A, Grandl M, Mejías-Luque R, Allgäuer M, Degenhart K, Haselmann V, et al. The E3 ligase RNF43 inhibits Wnt signaling downstream of mutated b-catenin by sequestering TCF4 to the nuclear membrane. Sci Signal. (2015) 8:393. doi: 10.1126/scisignal.aac6757
71. Zhong Y, Wang Z, Fu B, Pan F, Yachida S, Dhara M, et al. GATA6 activates Wnt signaling in pancreatic cancer by negatively regulating the Wnt antagonist Dickkopf-1. PLoS ONE. (2011) 6:e22129. doi: 10.1371/journal.pone.0022129
72. Fernández NB, Lorenzo D, Picco ME, Barbero G, Dergan-Dylon LS, Marks MP, et al. ROR1 contributes to melanoma cell growth and migration by regulating N-cadherin expression via the PI3K/Akt pathway. Mol Carcinog. (2016) 55:1772–85. doi: 10.1002/mc.22426
73. Yue Z, Yuan Z, Zeng L, Wang Y, Lai L, Li J, et al. LGR4 modulates breast cancer initiation, metastasis, and cancer stem cells. FASEB J. (2018) 32:2422–37. doi: 10.1096/fj.201700897R.
74. Wang D, Cai C, Dong X, Yu QC, Zhang XO, Yang L, et al. Identification of multipotent mammary stem cells by protein C receptor expression. Nature. (2015) 517:81–4. doi: 10.1038/nature13851
75. Monteiro J, Gaspar C, Richer W, Franken PF, Sacchetti A, Joosten R, et al. Cancer stemness in Wnt-driven mammary tumorigenesis. Carcinogenesis. (2014) 35:2–13. doi: 10.1093/carcin/bgt279
76. Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, et al. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol. (2014) 16:1004–15. doi: 10.1038/ncb3040
77. Teng Y, Wang X, Wang Y, Ma D. Wnt/beta-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem Biophys Res Commun. (2010) 392:373–9. doi: 10.1016/j.bbrc.2010.01.028
78. Yamamoto TM, McMellen A, Watson ZL, Aguilera J, Ferguson R, Nurmemmedov E, et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol Carcinog. (2019) 58:1770–82. doi: 10.1002/mc.23064
79. Huss S, Nehles J, Binot E, Wardelmann E, Mittler J, Kleine MA, et al. β-catenin (CTNNB1) mutations and clinicopathological features of mesenteric desmoid-type fibromatosis. Histopathology. (2013) 62:294–304. doi: 10.1111/j.1365-2559.2012.04355.x
80. Gandillet A, Park S, Lassailly F, Griessinger E, Vargaftig J, Filby A, et al. Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia. (2011) 25:770–80. doi: 10.1038/leu.2011.17
81. Siapati EK, Papadaki M, Kozaou Z, Rouka E, Michali E, Savvidou I, Gogos D, et al. Proliferation and bone marrow engraftment of AML blasts is dependent on beta-catenin signalling. Br J Haematol. (2011) 152:164–74. doi: 10.1111/j.1365-2141.2010.08471.x
82. Griffiths EA, Gore SD, Hooker C, McDevitt MA, Karp JE, Smith BD, et al. Acute myeloid leukemia is characterized by Wnt pathway inhibitor promoter hypermethylation. Leukemia Lymphoma. (2010) 51:1711–9. doi: 10.3109/10428194.2010.496505
83. Hu Y, Chen Y, Douglas L, Li S. β-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia. (2009) 23:109–16. doi: 10.1038/leu.2008.262
84. Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. (2007) 12:528–41. doi: 10.1016/j.ccr.2007.11.003
85. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. (2010) 327:1650–3. doi: 10.1126/science.1186624
86. Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, et al. β-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. (2010) 18:606–18. doi: 10.1016/j.ccr.2010.10.032
87. Kumar SK, Rajkumar V, Kyle RA, van Duin M, Sonneveld P, Mateos MV, et al. Multiple myeloma. Nat Rev Dis Primers. (2017) 3:17046. doi: 10.1038/nrdp.2017.46
88. Rollig C, Knop S, Bornhauser M. Multiple myeloma. Lancet. (2015) 385:2197–208. doi: 10.1016/S0140-6736(14)60493-1
89. Martín-Orozco E, Ferragut JA, Garcia-Peñarrubia P, Ferrer-Montiel A. Acquisition of multidrug resistance by L1210 leukemia cells decreases their tumorigenicity and enhances their susceptibility to the host immune response. Cancer Immunol Immunother. (2005) 54:328–36. doi: 10.1007/s00262-004-0588-7
90. Chen W, Qin Y, Liu S. Cytokines, breast cancer stem cells (BCSCs) and chemoresistance. Clin Transl Med. (2018) 7:27. doi: 10.1186/s40169-018-0205-6
91. Turato C, Scarpa M, Kotsafti A, Cappon A, Quarta S, Biasiolo A, et al. Squamous cell carcinoma antigen 1 is associated to poor prognosis in esophageal cancer through immune surveillance impairment and reduced chemosensitivity. Cancer Sci. (2019) 110:1552–63. doi: 10.1111/cas.13986
92. Showalter LE, Oechsle C, Ghimirey N, Steele C, Czerniecki BJ, Koski GK. Th1 cytokines sensitize HER-expressing breast cancer cells to lapatinib. PLoS ONE. (2019) 14:e0210209. doi: 10.1371/journal.pone.0210209
93. Patel SA, Minn AJ. Combination cancer therapy with immune checkpoint blockade: mechanisms and strategies. Immunity. (2018) 48:417–33. doi: 10.1016/j.immuni.2018.03.007
94. Jaiswal R, Johnson MS, Pokharel D, Krishnan SR, Bebawy M. Microparticles shed from multidrug resistant breast cancer cells provide a parallel survival pathway through immune evasion. BMC Cancer. (2017) 17:104. doi: 10.1186/s12885-017-3102-2
95. Corrêa S, Binato R, Du Rocher B, Castelo-Branco MTL, Pizzatti L, Abdelhay E. Wnt/β-catenin pathway regulates ABCB1 transcription in chronic myeloid leukemia. BMC Cancer. (2012) 12:303. doi: 10.1186/1471-2407-12-303
96. Wang YH, Imai Y, Shiseki M, Tanaka J, Motoji T. Knockdown of the Wnt receptor Frizzled-1 (FZD1) reduces MDR1/P-glycoprotein expression in multidrug resistant leukemic cells and inhibits leukemic cell proliferation. Leuk Res. (2018) 67:99–108. doi: 10.1016/j.leukres.2018.01.020
97. Zhang H, Zhang X, Wu X, Li W, Su P, Cheng H, et al. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/β-catenin pathway. Cancer Lett. (2012) 323:106–13. doi: 10.1016/j.canlet.2012.03.039
98. Flahaut M, Meier R, Coulon A, Nardou KA, Niggli FK, Martinet D, et al. The Wnt receptor FZD1 mediates chemoresistance in neuroblastoma through activation of the Wntβ-catenin pathway. Oncogene. (2009) 28:2245–56. doi: 10.1038/onc.2009.80
99. Chen Z, Ma T, Huang C, Zhang L, Lv X, Xu T, et al. MiR-27a modulates the MDR1/P-glycoprotein expression by inhibiting FZD7/β-catenin pathway in hepatocellular carcinoma cells. Cell Signal. (2013) 25:2693–701. doi: 10.1016/j.cellsig.2013.08.032
100. Wang H, Wang X, Hu R, Yang W, Liao A, Zhao C, et al. Methylation of SFRP5 is related to multidrug-resistance in leukemia cells. Cancer Gene Therapy. (2014) 21:83–9. doi: 10.1038/cgt.2013.87
101. Liu YS, Hsu HC, Tseng KC, Chen HC, Chen SJ. Lgr5 promotes cancer stemness and confers chemoresistance through ABCB1 in colorectal cancer. Biomed Pharmacother. (2013) 67:791–9. doi: 10.1016/j.biopha.2013.08.001
102. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, et al. The hippo effector YAP promotes resistance to RAF- and MEK- targeted cancer therapies. Nat Genet. (2015) 47:250–6. doi: 10.1038/ng.3218
103. Zanconato F, Battilana G, Cordenonsi M, Piccolo S. YAP/TAZ as therapeutic targets in cancer. Curr Opin Pharmacol. (2016) 29:26–33. doi: 10.1016/j.coph.2016.05.002
104. Zhang ZM, Wu JF, Luo QC, Liu QF, Wu QW, Ye GD, et al. Pygo2 activates MDR1 expression and mediates chemoresistance in breast cancer via the Wnt/β-catenin pathway. Oncogene. (2016) 35:4787–97. doi: 10.1038/onc.2016.10
105. Schmitt M, Metzger M, Gradl D, Davidson G, Orian-Rousseau V. CD44 functions in Wnt signaling by regulating LRP6 localization and activation. Cell Death Differ. (2015) 22:677–89. doi: 10.1038/cdd.2014.156
106. Kobune M, Chiba H, Kato J, Kato K, Nakamura K, Kawano Y, et al. Wnt3/RhoA/ROCK signaling pathway is involved in adhesion-mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol Cancer Ther. (2007) 6:1774–84. doi: 10.1158/1535-7163.mct-06-0684
107. Bjorklund CC, Baladandayuthapani V, Lin HY, Jones RJ, Kuiatse I, Wang H, et al. Evidence of a role for CD44 and cell adhesion in mediating resistance to lenalidomide in multiple myeloma: therapeutic implications. Leukemia. (2014) 28:373–83. doi: 10.1038/leu.2013.174
108. Spaan I, Raymakers RA, van de Stolpe A, Peperzak V. Wnt signaling in multiple myeloma: a central player in disease with therapeutic potential. J Hematol Oncol. (2018) 11:67. doi: 10.1186/s13045-018-0615-3
109. Cerezo D, Lencina M, Ruiz-Alcaraz AJ, Ferragut JA, Saceda M, Sanchez M, et al. Acquisition of MDR phenotype by leukemic cells is associated with increased caspase-3 activity and a collateral sensitivity to cold stress. J Cell Biochem. (2012) 113:1416–25. doi: 10.1002/jcb.24016
110. Cerezo D, Ruiz-Alcaraz AJ, Lencina-Guardiola M, Cánovas M, García-Peñarrubia P, Martínez-López I, et al. Attenuated JNK signaling in multidrug-resistant leukemic cells. Dual role of MAPK in cell survival. Cell Signal. (2017) 30:162–70. doi: 10.1016/j.cellsig.2016.12.003
111. Goler-Baron V, Assaraf YG. Structure and function of ABCG2-rich extracellular vesicles mediating multidrug resistance. PLoS ONE. (2011) 6:e16007. doi: 10.1371/journal.pone.0016007
112. Horio M, Gottesman MM, Pastan I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc Natl Acad Sci USA. (1988) 85:3580–4. doi: 10.1073/pnas.85.10.3580
113. Pallis M, Russell N. P-glycoprotein plays a drug-efflux-independent role in augmenting cell survival in acute myeloblastic leukemia and is associated with modulation of a sphingomyelin-ceramide apoptotic pathway. Blood. (2000) 95:2897–904. doi: 10.1182/blood.V95.9.2897.009k14_2897_2904
114. Weisburg JH, Roepe PD, Dzekunov S, Scheinberg DA. Intracellular ph and multidrug resistance regulate complement-mediated cytotoxicity of nucleated human cells. J Biol Chem. (1999) 274:10877–88. doi: 10.1074/jbc.274.16.10877
115. Ding S, Chamberlain M, McLaren A, Goh L-b, Duncan I, Wolf CR. Cross-talk between signalling pathways and the multidrug resistant protein MDR-1. Br J Cancer. (2001) 85:1175–84. doi: 10.1054/bjoc.2001.2044
116. Cerezo D, Cánovas M, García-Peñarrubia P, Martín-Orozco E. Collateral sensitivity to cold stress and differential BCL-2 family expression in new daunomycin-resistant lynphoblastoide cell lines. Exp Cell Res. (2015) 331:11–20. doi: 10.1016/j.yexcr.2014.11.017
117. Jaiswal R, Luk F, Dalla PV, Grau GER, Bebawy M. Breast cancer-derived microparticles display tissue selectivity in the transfer of resistance proteins to cells. PLoS ONE. (2013) 8:e61515. doi: 10.1371/journal.pone.0061515
118. Jaiswal R, Sedger LM. Intercellular vesicular transfer by exosomes, microparticles and oncosomes - implications for cancer biology and treatments. Front Oncol. (2019) 9:125. doi: 10.3389/fonc.2019.00125
119. Lokeshwar VB, Fregien N, Bourguignon LYW. Ankyrin-binding domain of CD44(GP85) is required for the expression of hyaluronic acid-mediated adhesion function. J Cell Biol. (1994) 126:1099–109. doi: 10.1083/jcb.126.4.1099
120. Mori T, Kitano K, Terawaki SI, Maesaki R, Fukami Y, Hakoshima T. Structural basis for CD44 recognition by ERM proteins. J Biol Chem. (2008) 283:29602–12. doi: 10.1074/jbc.M803606200
121. Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. (2010) 11:276–87. doi: 10.1038/nrm2866
122. Bourguignon LYW, Shiina M, Li JJ. Hyaluronan-CD44 interaction promotes oncogenic signaling, microRNA functions, chemoresistance, and radiation resistance in cancer stem cells leading to tumor progression. Adv Cancer Res. (2014) 123:255–75. doi: 10.1016/B978-0-12-800092-2.00010-1
123. Orian-Rousseau V, Schmitt M. CD44 regulates Wnt signaling at the level of LRP6. Mol Cell Oncol. (2015) 2:e995046. doi: 10.4161/23723556.2014.995046
124. Grass GD, Dai L, Qin Z, Parsons C, Toole BP. CD147: Regulator of hyaluronan signaling in invasiveness and chemoresistance. Adv Cancer Res. 123:351–73. doi: 10.1016/B978-0-12-800092-2.00013-7
125. Kozovska Z, Gabrisova V, Kucerova L. Colon cancer: cancer stem cells markers, drug resistance and treatment. Biomed Pharmacother. (2014) 68:911–6. doi: 10.1016/j.biopha.2014.10.019
126. Misra S, Ghatak S, Toole BP. Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J Biol Chem. (2005) 280:20310–5. doi: 10.1074/jbc.M500737200
127. Wang Z, Zhao K, Hackert T, Zöller M. CD44/CD44v6 a reliable companion in cancer-initiating cell maintenance and tumor progression. Front Cell Dev Biol. (2018) 6:97. doi: 10.3389/fcell.2018.00097
128. Bourguignon LYW, Spevak CC, Wong G, Xia W, Gilad E. Hyaluronan-CD44 interaction with protein kinase Cε promotes oncogenic signaling by the stem cell marker nanog and the production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance. J Biol Chem. (2009) 284:26533–46. doi: 10.1074/jbc.M109.027466
129. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/β-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. (2019) 25:3074–83. doi: 10.1158/1078-0432.CCR-18-1942
130. Xiao Z, Wan J, Nur AA, Dou P, Mankin H, Liu T, et al. Targeting CD44 by CRISPR-Cas9 in multi-drug resistant osteosarcoma cells. Cell Physiol Biochem. (2018) 51:1879–93. doi: 10.1159/000495714
131. Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/β-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. (2018) 39:648–58. doi: 10.1016/j.tips.2018.03.008
132. Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell–inflamed versus non-T cell–inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. (2018) 6:990–1000. doi: 10.1158/2326-6066.CIR-18-0277
133. Casey SC, Baylot V, Felsher DW. The MYC oncogene is a global regulator of the immune response. Blood. (2018) 131:2007–15. doi: 10.1182/blood-2017-11-742577
134. Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the anti-tumor immune response through CD47 and PD-L1. Science. (2016) 352:227–31. doi: 10.1126/science.aac9935
135. Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. β-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. (2019) 9:1124–41. doi: 10.1158/2159-8290.CD-19-0074
136. Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. (2017) 10:101. doi: 10.1186/s13045-017-0471-6
137. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. (2018) 18:139–47. doi: 10.1038/nrc.2017.117
138. Wang Q, Wu X. Primary and acquired resistance to PD-1/PD-L1 blockade in cancer treatment. Int Immunopharmacol. (2017) 46:210–9. doi: 10.1016/j.intimp.2017.03.015
139. Spranger S, Gajewski TF. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J Immunother Cancer. (2015) 3:43. doi: 10.1186/s40425-015-0089-6
140. Sweis RF, Spranger S, Bao R, Paner GP, Stadler WM, Steinberg G, et al. Molecular drivers of the non-T cell-inflamed tumor microenvironment in urothelial bladder cancer randy. Cancer Immunol Res. (2016) 4:563–8. doi: 10.1158/2326-6066.CIR-15-0274
141. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
142. Azcutia V, Stefanidakis M, Tsuboi N, Mayadas T, Croce KJ, Fukuda D, et al. Endothelial CD47 promotes vascular endothelial-cadherin tyrosine phosphorylation and participates in T cell recruitment at sites of inflammation in vivo. J Immunol. (2012) 189:2553–62. doi: 10.4049/jimmunol.1103606
143. Fujioka Y, Matozaki T, Noguchi T, Iwamatsu A, Yamao T, Takahashi N, et al. A novel membrane glycoprotein, SHPS-1, that binds the SH2-domain-containing protein tyrosine phosphatase SHP-2 in response to mitogens and cell adhesion. Mol Cell Biol. (1996) 16:6887–99.
144. Murata Y, Saito Y, Kotani T, Matozaki T. CD47-signal regulatory protein α signaling system and its application to cancer immunotherapy. Cancer Sci. (2018) 109:2349–57. doi: 10.1111/cas.13663
145. Schürch CM, Forster S, Brühl F, Yang SH, Felley-Bosco E, Hewer E. The “don't eat me” signal CD47 is a novel diagnostic biomarker and potential therapeutic target for diffuse malignant mesothelioma. Oncoimmunology. (2018) 7:e1373235. doi: 10.1080/2162402X.2017.1373235
146. Li F, Lv B, Liu Y, Hua T, Han J, Sun C, et al. Blocking the CD47-SIRPα axis by delivery of anti-CD47 antibody induces antitumor effects in glioma and glioma stem cells. Oncoimmunology. (2018) 7:e1391973. doi: 10.1080/2162402X.2017.1391973
147. Zhao H, Wang J, Kong X, Li E, Liu Y, Du X, et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci Rep. (2016) 6:29719. doi: 10.1038/srep29719
148. Noman MZ, Van Moer K, Marani V, Gemmill RM, Tranchevent LC, Azuaje F, et al. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. Oncoimmunology. (2018) 7:e1345415. doi: 10.1080/2162402X.2017.1345415
149. Barrera L, Montes-Servín E, Hernandez-Martinez JM, Garciá-Vicente MDLÁ, Montes-Servín E, Herrera-Martínez M, et al. CD47 overexpression is associated with decreased neutrophil apoptosis/phagocytosis and poor prognosis in non-small-cell lung cancer patients. Br J Cancer. (2017) 117:385–97. doi: 10.1038/bjc.2017.173
150. Michaels AD, Newhook TE, Adair SJ, Morioka S, Goudreau BJ, Nagdas S, et al. CD47 blockade as an adjuvant immunotherapy for resectable pancreatic cancer. Clin Cancer Res. (2018) 24:1415–25. doi: 10.1158/1078-0432.CCR-17-2283
151. Marquardt S, Solanki M, Spitschak A, Vera J, Pützer BM. Emerging functional markers for cancer stem cell-based therapies: Understanding signaling networks for targeting metastasis. Semin Cancer Biol. (2018) 53:90–109. doi: 10.1016/j.semcancer.2018.06.006
152. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. (2009) 138:286–99. doi: 10.1016/j.cell.2009.05.045
153. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. (2012) 109:6662–7. doi: 10.1073/pnas.1121623109
154. Sakakura K, Takahashi H, Kaira K, Toyoda M, Murata T, Ohnishi H, et al. Relationship between tumor-associated macrophage subsets and CD47 expression in squamous cell carcinoma of the head and neck in the tumor microenvironment. Lab Invest. (2016) 96:994–1003. doi: 10.1038/labinvest.2016.70.
155. Fu W, Li J, Zhang W, Li P. High expression of CD47 predicts adverse prognosis in Chinese patients and suppresses immune response in melanoma. Biomed Pharmacother. (2017) 93:1190–6. doi: 10.1016/j.biopha.2017.06.030
156. Dheilly E, Moine V, Broyer L, Salgado-Pires S, Johnson Z, Papaioannou A, et al. Selective blockade of the ubiquitous checkpoint receptor CD47 is enabled by dual-targeting bispecific antibodies. Mol Ther. (2017) 25:523–33. doi: 10.1016/j.ymthe.2016.11.006
157. Betancur PA, Abraham BJ, Yiu YY, Willingham SB, Khameneh F, Zarnegar M, et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat Commun. (2017) 8:14802. doi: 10.1038/ncomms14802
158. Gowda P, Patrick S, Singh A, Sheikh T, Sen E. Mutant isocitrate dehydrogenase 1 disrupts PKM2-β-catenin-BRG1 transcriptional network- driven CD47 expression. Mol Cell Biol. (2018) 38:e00001–18. doi: 10.1128/MCB.00001-18
159. Zheng H, Ying H, Wiedemeyer R, Yan H, Quayle SN, Ivanova EV, et al. PLAGL2 regulates Wnt signaling to impede differentiation in neural stem cells and gliomas. Cancer Cell. (2010) 17:497–509. doi: 10.1016/j.ccr.2010.03.020
160. Yang H, Shao R, Huang H, Wang X, Rong Z, Lin Y. Engineering macrophages to phagocytose cancer cells by blocking the CD47/SIRP? axis. Cancer Med. (2019) 8:4245–53. doi: 10.1002/cam4.2332
161. Waterman ML. Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev. (2004) 1–2:41–52. doi: 10.1023/A:1025858928620
162. Tan W, Tang H, Jiang X, Ye F, Huang L, Shi D, et al. Metformin mediates induction of miR-708 to inhibit self-renewal and chemoresistance of breast cancer stem cells through targeting CD47. J Cell Mol Med. (2019) 23:5994–6004. doi: 10.1111/jcmm.14462
163. Lo J, Lau EY, Ching RH, Cheng BY, Ma MK, Ng IO, et al. Nuclear factor kappa B–mediated CD47 up-regulation promotes sorafenib resistance and its blockade synergizes the effect of sorafenib in hepatocellular carcinoma in mice. Hepatology. (2015) 62:534–45. doi: 10.1002/hep.27859
164. Shi L, Chen S, Yang L, Li Y. The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J Hematol Oncol. (2013) 6:74. doi: 10.1186/1756-8722-6-74
165. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. (2012) 209:1201–17. doi: 10.1084/jem.20112741
166. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Heidi A, et al. T cell costimulatory receptor CD28 is a primary target for PD-1– mediated inhibition. Science. (2017) 355:1428–33. doi: 10.1126/science.aaf1292.T
167. Gong B, Kiyotani K, Sakata S, Nagano S, Kumehara S, Baba S, et al. Secreted PD-L1 variants mediate resistance to PD-L1 blockade therapy in non–small cell lung cancer. J Exp Med. (2019) 216:982–1000. doi: 10.1084/jem.20180870
168. Bellmunt J, Powles T, Vogelzang NJ. A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: the future is now. Cancer Treat Rev. (2017) 54:58–67. doi: 10.1016/j.ctrv.2017.01.007
169. Ritprajak P, Azuma M. Intrinsic and extrinsic control of expression of the immunoregulatory molecule PD-L1 in epithelial cells and squamous cell carcinoma. Oral Oncol. (2015) 51:221–8. doi: 10.1016/j.oraloncology.2014.11.014
170. Xie QK, Zhao YJ, Pan T, Lyu N, Mu LW, Li SL, et al. Programmed death ligand 1 as an indicator of pre-existing adaptive immune responses in human hepatocellular carcinoma. Oncoimmunology. (2016) 5:e1181252. doi: 10.1080/2162402X.2016.1181252
171. Wang Y, Wang H, Yao H, Li C, Fang JY, Xu J. Regulation of PD-L1: emerging routes for targeting tumor immune evasion. Front Pharmacol. (2018) 9:536. doi: 10.3389/fphar.2018.00536
172. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
173. Barach YS, Lee JS, Zang X. T cell coinhibition in prostate cancer: new immune evasion pathways and emerging therapeutics. Trends Mol Med. (2011) 17:47–55. doi: 10.1016/j.molmed.2010.09.006
174. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
175. Castagnoli L, Cancila V, Cordoba-Romero SL, Faraci S, Talarico G, Belmonte B, et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene. (2019) 38:4047–60. doi: 10.1038/s41388-019-0700-2
176. Chovanec M, Cierna Z, Miskovska V, Machalekova K, Kalavska K, Rejlekova K, et al. βcatenin is a marker of poor clinical characteristics and suppressed immune infiltration in testicular germ cell tumors. BMC Cancer. (2018) 18:1062. doi: 10.1186/s12885-018-4929-x
177. Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. (2018) 9:1908. doi: 10.1038/s41467-018-04313-6
178. Lathia JD, Liu H. Overview of cancer stem cells and stemness for community oncologists. Target Oncol. (2017) 12:387–99. doi: 10.1007/s11523-017-0508-3
179. Ho SY, Keller TH. The use of porcupine inhibitors to target Wnt-driven cancers. Bioorganic Med Chem Lett. (2015) 25:5472–6. doi: 10.1016/j.bmcl.2015.10.032
180. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting notch, hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
181. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
182. Huang GL, Shen DY, Cai CF, Zhang QY, Ren HY, Chen QX. β-escin reverses multidrug resistance through inhibition of the GSK3β/β-catenin pathway in cholangiocarcinoma. World J Gastroenterol. (2015) 21:1148–57. doi: 10.3748/wjg.v21.i4.1148
183. Zhao Y, Masiello D, McMillian M, Nguyen C, Yongfeng W, Melendez E, et al. CBP/catenin antagonist safely eliminates drug resistant leukemia initiating cells. Oncogene. (2016) 35:3705–17. doi: 10.1038/onc.2015.438
184. Han L, Shi S, Gong T, Zhang Z, Sun X. Cancer stem cells: therapeutic implications and perspectives in cancer therapy. Acta Pharm Sin B. (2013) 3:65–75. doi: 10.1016/j.apsb.2013.02.006
185. Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. (2010) 16:3153–62. doi: 10.1158/1078-0432.CCR-09-2943
186. Takeishi S, Matsumoto A, Onoyama I, Naka K, Hirao A, Nakayama KI. Ablation of Fbxw7 eliminates leukemia-initiating cells by preventing quiescence. Cancer cell. (2013) 23:347–61. doi: 10.1016/j.ccr.2013.01.026
187. Mollazadeh S, Sahebkar A, Hadizadeh F, Behravan J, Arabzadeh S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. (2018) 214:118–23. doi: 10.1016/j.lfs.2018.10.048
188. Li W, Zhang H, Assaraf YG, Zhao K, Xu X, Xie J, et al. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist Updat. (2016) 27:14–29. doi: 10.1016/j.drup.2016.05.001
189. El-Araby ME, Omar AM, Khayat MT, Assiri HA, Al-Abd AM. Molecular mimics of classic p-glycoprotein inhibitors as multidrug resistance suppressors and their synergistic effect on paclitaxel. PLoS ONE. (2017) 12:e0168938. doi: 10.1371/journal.pone.0168938
190. Mohana S, Ganesan M, Rajendra Prasad N, Ananthakrishnan D, Velmurugan D. Flavonoids modulate multidrug resistance through Wnt signaling in P-glycoprotein overexpressing cell lines. BMC Cancer. (2018) 18:1168. doi: 10.1186/s12885-018-5103-1
191. Syed SB, Arya H, Fu IH, Yeh TK, Periyasamy L, Hsieh HP, et al. Targeting P-glycoprotein: investigation of piperine analogs for overcoming drug resistance in cancer. Sci Rep. (2017) 7:7972. doi: 10.1038/s41598-017-08062-2
192. Chakrabarti M, Ray SK. Anti-tumor activities of luteolin and silibinin in glioblastoma cells: overexpression of miR-7-1-3p augmented luteolin and silibinin to inhibit autophagy and induce apoptosis in glioblastoma in vivo. Apoptosis. (2016) 21:312–28. doi: 10.1007/s10495-015-1198-x
193. Liu E, Kuang Y, He W, Xing X, Gu J. Casticin induces human glioma cell death through apoptosis and mitotic arrest. Cell Physiol Biochem. (2013) 31:805–14. doi: 10.1159/000350098
194. Yang XH, Zheng X, Cao JG, Xiang HL, Liu F, Lv Y. 8-bromo-7-methoxychrysin-induced apoptosis of hepatocellular carcinoma cells involves ROS and JNK. World J Gastroenterol. (2010) 16:3385–93. doi: 10.3748/wjg.v16.i27.3385
195. Zinzi L, Contino M, Cantore M, Capparelli E, Leopoldo M, Colabufo NA. ABC transporters in CSCs membranes as a novel target for treating tumor relapse. Front Pharmacol. (2014) 5:163. doi: 10.3389/fphar.2014.00163
196. Deshmukh A, Deshpande K, Arfuso F, Newsholme P, Dharmarajan A. Cancer stem cell metabolism: a potential target for cancer therapy. Mol Cancer. (2016) 15:69. doi: 10.1186/s12943-016-0555-x
197. Hamed AR, Abdel-Azim NS, Shams KA, Hammouda FM. Targeting multidrug resistance in cancer by natural chemosensitizers. Bull Natl Res Cent. (2019) 43:8. doi: 10.1186/s42269-019-0043-8
198. Yan Y, Zao X, Wei D. Concise review: emerging role of CD44 in cancer stem cells: a promising biomarker and therapeutic target. Stem Cells Transl Med. (2015) 4:1033–43. doi: 10.5966/sctm.2015-0048
199. Ho CCM, Guo N, Sockolosky JT, Ring AM, Weiskopf K, Özkan E, et al. “Velcro” engineering of high affinity CD47 Ectodomain as signal regulatory protein α (SIRPα) antagonists that enhance antibody-dependent cellular phagocytosis. J Biol Chem. (2015) 290:12650–63. doi: 10.1074/jbc.M115.648220
200. Tseng D, Volkmer J-P, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci USA. (2013) 110:11103–8. doi: 10.1073/pnas.1305569110
201. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Blood Rev. (2018) 21:1209–15. doi: 10.1038/nm.3931
202. Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, et al. Blocking ‘don't eat me' signal of CD47-SIRPα in hematological malignancies, an in-depth review Nat Med. (2015) 32:480–9. doi: 10.1016/j.blre.2018.04.005
203. Petrova PS, Viller NN, Wong M, Pang X, Lin GHY, Dodge K, et al. TTI-621 (SIRPaFc): a CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin Cancer Res. (2017) 23:1068–79. doi: 10.1158/1078-0432.CCR-16-1700
204. Kauder SE, Kuo TC, Harrabi O, Chen A, Sangaland E, Doyle L, et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS ONE. (2015) 13:e0201832. doi: 10.1371/journal.pone.0201832
205. Murata Y, Tanaka D, Hazama D, Yanagita T, Saito Y, Kotani T, et al. Anti-human SIRPα antibody is a new tool for cancer immunotherapy. Cancer Sci. (2018) 109:1300–8. doi: 10.1111/cas.13548
206. Zhao XW, van Beek EM, Schornagel K, Van der Maaden H, Van Houdt M, Otten MA, et al. CD47-signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc Natl Acad Sci USA. (2011) 108:18342–7. doi: 10.1073/pnas.1106550108
207. Mattiola I, Pesant M, Tentorio PF, Molgora M, Marcenaro E, Lugli E, et al. Priming of human resting NK cells by autologous M1 macrophages via the engagement of IL-1β, IFN-β, and IL-15 pathways. J Immunol. (2015) 195:2818–28. doi: 10.4049/jimmunol.1500325
208. Bodduluru LN, Kasala ER, Madhana RMR, Sriram CS. Natural killer cells: the journey from puzzles in biology to treatment of cancer. Cancer Lett. (2015) 357:454–67. doi: 10.1016/j.canlet.2014.12.020
209. Sockolosky JT, Dougan M, Ingram JR, Ho CCM, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci USA. (2016) 113:E2646–54. doi: 10.1073/pnas.1604268113
210. Ingram JR, Blomberg OS, Sockolosky JT, Ali L, Schmidt FI, Pishesha N, et al. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc Natl Acad Sci USA. (2017) 114:10184–9. doi: 10.1073/pnas.1710776114
Keywords: WNT, β-catenin, ABCB1, PD-L1, CD47, multidrug resistance (MDR), immunity escape, cancer
Citation: Martin-Orozco E, Sanchez-Fernandez A, Ortiz-Parra I and Ayala-San Nicolas M (2019) WNT Signaling in Tumors: The Way to Evade Drugs and Immunity. Front. Immunol. 10:2854. doi: 10.3389/fimmu.2019.02854
Received: 01 July 2019; Accepted: 20 November 2019;
Published: 20 December 2019.
Edited by:
Nicola Giuliani, University of Parma, ItalyReviewed by:
Magdalena Joanna Lorenowicz, University Medical Center Utrecht, NetherlandsAlessandro Poggi, San Martino Hospital (IRCCS), Italy
Copyright © 2019 Martin-Orozco, Sanchez-Fernandez, Ortiz-Parra and Ayala-San Nicolas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Martin-Orozco, ZW1hcnRpbkB1bS5lcw==