 Vanessa Granger
Vanessa Granger Marine Peyneau1,2
Marine Peyneau1,2 Sylvie Chollet-Martin
Sylvie Chollet-Martin Luc de Chaisemartin
Luc de Chaisemartin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 03 December 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02824
This article is part of the Research Topic Role of Neutrophils in Inflammatory Diseases View all 11 articles
Neutrophil extracellular traps (NETs) have been initially described as main actors in host defense owing to their ability to immobilize and sometimes kill microorganisms. Subsequent studies have demonstrated their implication in the pathophysiology of various diseases, due to the toxic effects of their main components on surrounding tissues. Several distinct NETosis pathways have been described in response to various triggers. Among these triggers, IgG immune complexes (IC) play an important role since they induce robust NET release upon binding to activating FcγRs on neutrophils. Few in vitro studies have documented the mechanisms of IC-induced NET release and evidence about the partners involved is controversial. In vivo, animal models and clinical studies have strongly suggested the importance of IgG IC-induced NET release for autoimmunity and anaphylaxis. In this review, we will focus on two autoimmune diseases in which NETs are undoubtedly major players, systemic lupus erythematosus (SLE), and rheumatoid arthritis (RA). We will also discuss anaphylaxis as another example of disease recently associated with IC-induced NET release. Understanding the role of IC-induced NETs in these settings will pave the way for new diagnostic tools and therapeutic strategies.
Neutrophil extracellular traps (NETs) are extracellular chromatin filaments produced upon cell activation and decorated with many proteins normally confined to neutrophil cytoplasm and granules. This process was first described in 2004 as a new mechanism to catch, immobilize, and potentially kill bacteria (1). Subsequently, NETs have been shown to kill several species of bacteria, and rapidly limit the extent of the infection in some models (2–4). However, NET contribution to infectious diseases is double-edged. On the one hand, they may play a major role in defense against pathogens but on the other hand, collateral damage in infected host tissues can be significant, due to proteolytic enzymes release or histone toxicity (5–7). For instance, a deleterious role for NETs have been described in life-threatening infectious conditions such as sepsis or pneumonia-associated acute respiratory distress syndrome (8–12). Interestingly, besides their major pro-inflammatory role, NETs might also be able to downregulate dendritic cell activation and promote Th2 response, thus participating in the resolution of inflammation (13).
In addition to their role during infection, increasing evidence shows that NETosis also happens in a large number of non-infectious inflammation-associated diseases, including various lung diseases, thrombosis, cancer, and auto-immune diseases (14). In the lung, we and others reported that NETs are found in high concentrations in patients with chronic obstruction pulmonary disease or asthma (15–20). In thrombosis, a major interplay between platelets, components of the coagulation system and NETs have been unraveled, leading to the emergence of a new concept named immunothrombosis (21). Finally, as NETs are both pro-inflammatory and composed of many potential autoimmune targets, it has been hypothesized that NETs could be strong inducers of autoimmunity. Indeed, a crucial role for NETs has been described in various autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), vasculitis or diabetes (22–24).
In the initial studies about NETosis, the authors described a relatively long process (several hours) leading to neutrophil lysis and dependent on NADPH oxidase 2 (NOX2) activation (25–27). Fifteen years later, several distinct pathways in response to various triggers have been described making the definition of NETosis even more complex (28). The lytic or suicidal form of NETosis relies on NOX2-derived reactive oxygen species (ROS) release, allowing the liberation of neutrophil elastase (NE) and myeloperoxidase (MPO) from azurophilic granules. Both histone cleavage by NE and their citrullination by peptidyl deaminase 4 (PAD4) have been initially described as required for chromatin decondensation and extrusion out of the cell. However, several intracellular pathways have been described, and some NET release could be NE- or PAD4-independent (29–31). Autophagy is also probably involved, even if there is yet no consensus as conflicting results were obtained (28, 32–34). Very interestingly, quick non-suicidal pathways of NET release have also been described, where neutrophils remain viable, and can still perform functions such as phagocytosis, chemotaxis or dendritic cell activation (14, 35–37). Some studies have also shown NETs composed of mitochondrial DNA (38) but this mechanism requires more investigation. In addition, NOX2–independent pathways have been described, where calcium influx could triggers the activation of mitochondrial ROS (39, 40). This mechanism has been shown in chronic granulomatous disease patients, who have impaired NOX2 activation (41, 42).
The mechanisms of NET formation and release may vary depending upon the initial trigger (23, 43, 44). A large number of triggers have been described, both artificial and physiological. Among them, IgG and IgA immune complexes (IC) have been shown to trigger NETosis in different situations (38, 45–48).
In this review, we will focus on the role of IC on NET formation. To date, few in vitro studies have documented the mechanisms of NET release in response to pre-formed IC in both murine (49, 50) and human neutrophils (49, 51). Moreover, these studies provided conflicting results especially regarding type of FcγR involved or NOX2 requirement. Using soluble bovine serum albumin (BSA)-IgG IC, Chen et al. highlighted some discrepancies between the results obtained in human FcγRIIA transgenic mice and those using blocking antibodies on purified human neutrophils. The first model suggested the importance of human FcγRIIA during in vivo NET release whereas the latter supported a role of FcγRIIIB in vitro. The differences between the models could explain this discrepancy. Indeed, in transgenic FcγRIIIB+/γ−/− mice which do not express FcγRIIA, FcγRIIIB engagement lead to IC clearance. Moreover, blocking FcγRIIA on human neutrophils does not affect receptor intracellular signaling domain, allowing FcγRIIA to transduce signal mediated by FcγRIIIB engagement. Hence, these results support a cooperative role of FcγRIIA and FcγRIIIB, the two activating receptors constitutively expressed on neutrophils, during IC-induced NET-release (49). However, other studies have pointed out an exclusive role of FcγRIIIB in NET release in response to immobilized ICs (51) or by direct receptor aggregation (52, 53). Similarly, the requirement of NOX2-generated ROS in IC-induced NET release is controversial. Although most studies supported a pivotal role of NOX2 (38, 45, 51, 52), two of them provided opposite results despite similar pharmacological inhibitor [diphenyleneiodonium (DPI)] but different concentrations (49, 54). As DPI inhibits a wide variety of NADP-dependent enzymes as well as mitochondrial flavoenzymes (55), DPI-related NET release inhibition only means that ROS (whatever their cellular origin) are important in this process. Additionally, a role of actin cytoskeleton (49), Syk/Src (49–51), and MAPK (49, 51, 52) have been suggested by a few studies whereas the implication of NE/MPO (49, 51) and PI3K/AKT (49, 51, 52) are debated and need confirmation. All these conflicting results regarding the molecular pathways implicated in IC-induced NET release could be explained at least in part by the heterogeneity of the protocols: type of IC (antigenic system, spatial configuration, antigen, and antibody concentrations), pharmacological inhibitors (type and concentration), blocking antibodies [clone, type (Fab or full Ab)], use of human transgenic mice, and method to quantify NETs. Thus, there is a need for recapitulative studies comparing side by side the different triggers and inhibition strategies to obtain a definitive and clear view of IC-induced NET release pathways.
As they expose intracellular endogenous components to the immune system, NETs have been very soon suspected to participate in the initiation of the autoimmune response (Figure 1). Indeed, autoantibodies against several NET components such as DNA, MPO, elastase, citrullinated histones, or proteinase 3 (PR3) are hallmarks of several systemic autoimmune diseases. It has been speculated for several years that anti-ribonucleoprotein (RNP) and anti-DNA antibodies found in the serum of patients with SLE could be produced in response to NET constituents and thus participate to the high level of circulating IC in lupus (56–58). ICs containing self-antigens, in particular RNP/anti-RNP ICs, have been shown to induce NET release, creating an amplification loop where NET components induce autoantibodies leading to ICs, which subsequently trigger NET formation and perpetuate the phenomenon. The role of these IC-induced NETs in the pathophysiology of lupus is not obvious, as the high levels of NETs released during infection does not usually lead to autoimmune response. This suggests that some additional mechanisms must lead to a break of tolerance to NETs. Interestingly, RNP-containing ICs were shown to induce mitochondrial hyperpolarization, increased mitochondrial ROS production and extracellular release of oxidized mitochondrial DNA, a potent proinflammatory compound able to activate type 1 interferon pathway (38). Additionally, self-DNA ICs have been shown to activate plasmacytoid dendritic cells via TLR9, also leading to type 1 interferon release, which has been linked to loss of tolerance (56, 57). An impairment of NETs regulatory mechanisms could also favor loss of tolerance. Some patients with active lupus or lupus nephritis have a deficiency in DNase 1 activity and/or anti-NET antibodies that inhibit DNase effect (23, 59, 60), leading to abnormal NET accumulation. This prolonged presence of NETs could favor rupture of tolerance as well as increase tissue damage (61). Another interesting element is that the composition of NETs from SLE patients is different from that of healthy controls. NETs from SLE patients are richer in toxic compounds (e.g., oxidized alpha-enolase) leading to tissue damage, especially in lupus nephritis (62). Recent observations suggest that several PAD4 polymorphisms are associated to SLE and lupus nephritis, reinforcing the link between NETs and SLE pathophysiology (63, 64). Additionally, in lupus nephritis, circulating IC deposit in the glomerular basement membrane, giving an additional pathogenic role for these IC. Taken together these findings emphasize a major role for IC in the different NETosis pathways involved in SLE, particularly in lupus nephritis (65).
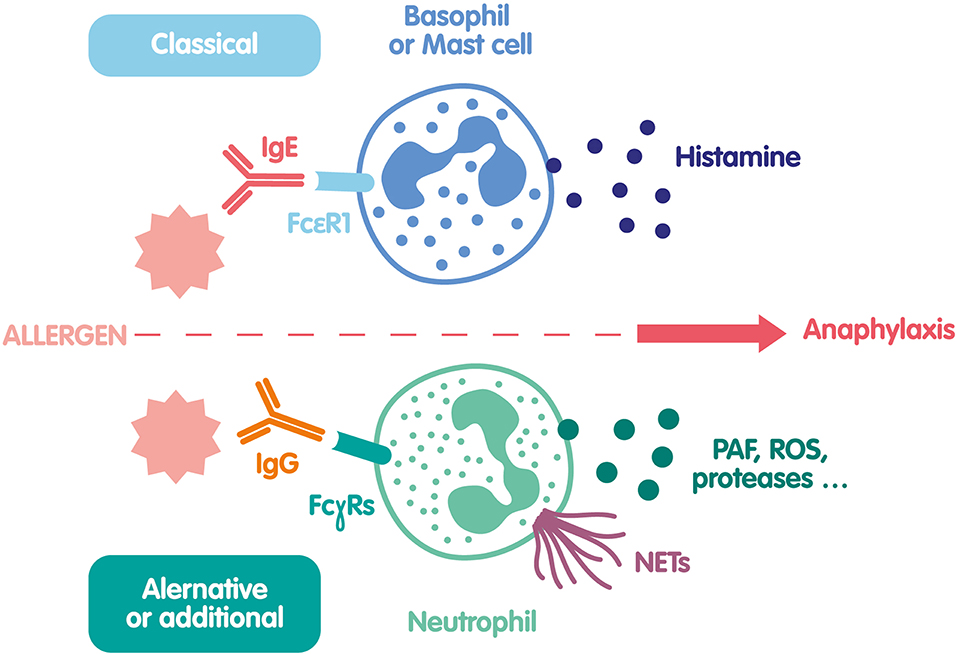
Figure 1. Mechanisms of NET formation in autoimmunity. Autoantigen/IgG IC can bind to several FcγRs expressed at the neutrophil surface and induce their activation. In particular, NOX2 is activated and produce ROS that can in turn activate PAD4 leading to protein citrullination and chromatin decondensation. In parallel, ROS can also help MPO and NE degranulation and translocation to the nucleus contributing to chromatin unfolding. The nuclear membrane breaks down, the decondensed chromatin is released in the cytosol and becomes decorated with various cytosolic and granule-derived proteins. Finally, NETs are released exposing to the immune system a large number of autoantigens that can amplify this mechanism called lytic NETosis. In some conditions, in particular in SLE, these IC can also induce a non-lytic NOX2-independent NETosis via the production of mitochondria-derived ROS and/or DNA; in that case, neutrophils are still alive.
RA is another example where IC-induced NETs are of importance (66, 67). Even if the pathogenesis of RA is not fully understood, many studies have shown that in genetically predisposed patients, anti-citrullinated protein antibodies (ACPAs) play a major role. These autoantibodies target citrullinated self-proteins like histones, vimentin, enolase, collagen, filaggrin, fibrinogen, or calreticulin. Citrullination is a physiological process that occurs in inflammation and during NETosis, due to the activation of PAD4 (66–69). PAD4 activity contributes to RA development, since PAD4-deficient mice have reduced autoantibodies and joint damage in arthritis models (70, 71), and a single nucleotide polymorphism of PAD4 is associated to an increased risk to develop RA in humans (72). Neutrophils from RA patients are activated and produce spontaneously more NETs than healthy donors (73). Additionally, NETosis can be activated by ACPA IgG and IgA ICs (46, 69, 74). Circulating NETs and netting neutrophils in joints are found in patients with RA (69, 75, 76), demonstrating active and widespread NETosis in this context. Thus, IC-induced NETs together with inflammation and synoviocyte activation can enhance the production of citrullinated autoantigens and fuel the autoimmune response, which will in turn produce more ICs (24, 77). Furthermore, synovial fibrocytes can internalize NETs via a RAGE-TLR9 pathway leading to MHC-class II upregulation and presentation to specific T cells of NET-associated citrullinated peptides (78).
The breaking of tolerance to citrullinated proteins is suspected to occur in the airway, in particular in smoker's airway (79–81). Identical citrullinated proteins are present in the joints and in the lung of patients with RA, and high levels of NETs can be found in the sputum of ACPA-positive RA patients, and even in at-risk patients' relatives (positive for HLA-DRB1 allele and ACPA) (82). Nicotine could induce NETs via PAD4 activation (83), and smoking triggered NETosis in several experimental models (84, 85). The link between nicotine, NETs, and loss of tolerance to citrullinated proteins is not fully elucidated yet, but the more recent studies point to an intense lung citrullination process related to high levels of PAD4 and neutrophil activation (22, 86). These NETs can then induce dendritic cell maturation, type 1 IFN release, Th1 expansion, and B cell activation. Furthermore, ectopic lymphoid tissue and high levels of ACPA are observed in the lung of patients with RA reinforcing the idea of a local autoimmune response (82, 87). Finally, the microenvironment, in particular the microbial agents, might themselves play a role in breaking the tolerance; it was for instance recently demonstrated that PAD from Porphyromonas gingivalis is able to produce citrullinated proteins and participate to RA pathogenesis (23, 88). Thus, NETs produced in response to infection could constitute in some instances a bridge between infection and autoimmunity. To summarize, NETs are an important source of citrullinated autoantigens in RA, fueling the production of ACPAs in predisposed individuals. They also maintain an inflammatory environment in the lung and in the joints, facilitating neutrophil activation and NET production by the ACPA/citrullinated peptides ICs.
Anaphylaxis is an acute systemic hypersensitivity reaction that can be life-threatening. Because of its extremely fast and unpredictable onset, it is difficult to obtain data on its mechanisms in human, and animal models have been developed to better understand this complex disease (89). The classical pathway is based on the triad IgE/basophil-mastocyte/histamine. During anaphylaxis, cell-surface bound specific IgE on basophils and mast cells react with the allergen and induce the release of preformed mediators such as histamine and proteases, leading to clinical signs of anaphylaxis. However, anaphylaxis can be triggered in mice lacking IgE or their receptor (90, 91), and we reported that up to 30% of patients with neuromuscular blocking agent (NMBA) perioperative anaphylaxis do not have any sign of the IgE pathway (92, 93). An IgE-independent anaphylaxis mechanism has thus been proposed and demonstrated in mice, mediated by neutrophils, specific IgG and FcγRs (94). Specific IgG-IC can bind to various activating FcγRs at the surface of cells such as neutrophils and induce their activation. High circulating levels of several neutrophil-related components and platelet activating factor (PAF) have been described in mice models of anaphylaxis, and in patients experiencing anaphylaxis as markers of neutrophil activation (95–97). The mechanisms of IgG-mediated neutrophil activation during anaphylaxis were first demonstrated in mice models of BSA-induced anaphylaxis. Using depletion and inhibition strategies it was shown that specific IgG-IC binding to neutrophil FcγRIIIA or FcγRIV was sufficient to induce fatal anaphylaxis (94). As human neutrophils do not express these two activating receptors but FcγRIIA, transgenic mice expressing the human FcγRIIA were used to demonstrate a major role for this receptor during anaphylaxis (98, 99). Very recently, these findings were confirmed in an elegant humanized mouse model where the human low-affinity IgG receptor locus, comprising both activating and inhibitory FcγR genes was inserted into the equivalent murine locus (100, 101). The implication of such an IC-mediated anaphylaxis via a new IgG/neutrophil/PAF triad is thus well-demonstrated in animal models and suggested to be relevant in humans by the studies on humanized mice. The existence of this alternative or additional mechanism in humans has been very recently demonstrated in a cohort of 86 patients experiencing NMBA anaphylaxis (93). Blood neutrophils were activated in patients as shown by the upregulation of CD11b, CD18, CD66b, and high levels of circulating elastase. NETosis was also triggered and patients had high levels of circulating NETs remnants (DNA-MPO complexes). Interestingly, a decreased expression of neutrophil FcγRIIA and FCγRIIIB was observed 30 min after anaphylaxis onset. This negative modulation is consistent with the engagement of FcγRs by circulating IC. Moreover, purified anti-rocuronium IgG isolated from a patient could form IC in vitro with a rocuronium bioconjugate. These IC were able to activate human neutrophils in vitro, and induce NET release (93). Concentration of anti-NMBA IgG and neutrophil activation markers correlated with anaphylaxis severity. This mechanism could be observed in patients lacking any evidence of IgE-dependent anaphylaxis, suggesting that IgG and IgE pathways could be independent, at least in some instances.
Besides the role of IgG/IC in NET release during anaphylaxis, one could speculate that other mechanisms exist that could modulate neutrophil activation. Some mediators released both in the acute and the late inflammatory phase of anaphylaxis such as pro-inflammatory cytokines, PAF, or C5a seem able to activate NET release in some conditions, even if there is still conflicting results on the subject (102–105). Recently, a major role of platelets in anaphylaxis has been suggested both in hFcγRIIA transgenic mice model and in humans (106, 107). In the mouse model, the interaction of platelet FcγRIIA with IgG-ICs induced platelet activation/aggregation, whose intensity correlated with the severity of anaphylaxis. Moreover, platelets depletion substantially attenuated symptomatology in mice. As platelets are much more abundant in bloodstream than neutrophils, it seems likely that IgG-ICs interact with platelet FcγRIIA first, before interacting with neutrophils. Activated platelets can aggregate on neutrophils to form platelet-neutrophil complexes detectable in vivo during several inflammatory conditions such as sepsis, pulmonary diseases, atherosclerosis (108), and recently allergic shock (106). The formation of these complexes involves the GP1b (glycoprotein 1b)/MAC-1 (macrophage 1 antigen) interaction (108), which is able to induce NET release. Platelets also release several soluble mediators known to activate NETosis (Von Willebrand Factor, platelet factor 4, HMGB1, PAF) (4, 109). Thus, besides direct neutrophil activation by IgG-ICs, other mechanisms involving released mediators or/and activated platelets may contribute to NET release during the acute phase of anaphylaxis.
To date, only one study showed NET formation during anaphylaxis in human (93). Therefore, the pathogenic role of NET in this context is still unclear. However, some hypotheses could be raised according to well-established NET component properties. NET cytotoxicity on vascular endothelium and epithelia has already been shown to be responsible for organ failure in mouse models of sepsis and acute lung injury (4, 5, 110) and may therefore contribute to the pulmonary and vascular symptomatology of anaphylaxis. Whether NETs are formed in the lungs during anaphylaxis has not been directly investigated so far, but interstitial accumulation of neutrophil associated with pulmonary congestion has been demonstrated in a model of casein-induced active anaphylaxis (111). It is also possible that circulating NETs reach lung microcirculation and damage the alveolar-capillary interface as observed in mouse models of acute lung injury (112).
Complement activation is one of the mechanisms implicated in alternative routes of anaphylaxis and in worsening classical IgE-mediated anaphylaxis through C3a and C5a production (89). Along with direct toxicity, NETs could amplify mast cell degranulation by activating the alternative complement pathway (113, 114). Similarly, NETs could amplify bradykinin-mediated circulatory complications through their capacity to activate contact coagulation system (115).
To summarize, very recent human studies have shown that allergen/specific IgG IC are able to activate neutrophils and induce NETs. This new anaphylaxis pathway could participate to clinical manifestations of anaphylaxis (Figure 2), and should be considered in future investigations of diagnostic markers or therapeutic interventions.
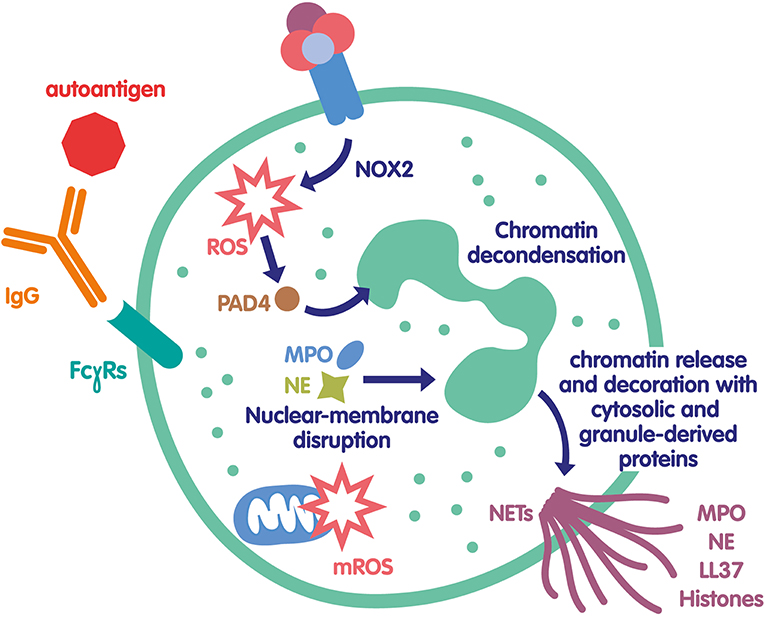
Figure 2. Mechanisms of NET formation during anaphylaxis. The classical pathway of anaphylaxis is based on histamine release by mast cells and basophils activated by the engagement of FcεRI after interaction of specific IgE with an allergen. A second pathway was recently demonstrated both in mice and human. In this pathway the allergen reacts with specific IgG and form an IC that binds to several FcγRs at the neutrophil surface and activate them. In addition to ROS and protease release, neutrophils release PAF and NETs, that could be also involved in anaphylaxis clinical manifestations.
The implication of NETs in pathology has been prompting several studies investigating its potential as a diagnostic marker or a therapeutic target.
Circulating NETs have been detected in patient's serum in many diseases. Accordingly, NET concentrations have been studied as diagnostic or prognostic markers. Many studies have focused on the concentrations of cell-free DNA or circulating nucleosomes as NET surrogates. However, those are a poor reflect of NETosis since they will be released by any dying cell (116). Some other works focused on citrullinated H3 (H3citr) quantification in serum or tissues. For example, serum H3citr levels predicted the risk of venous thromboembolism in a cohort of 946 cancer patients. Additionally, Jin et al. showed in an interesting study that intratumoral NETs identified by H3citr staining could predict poor survival in post-surgery pancreatic cancer patients (117). The most specific marker of NETs to date are DNA-MPO complexes (116), but fewer studies have investigated their diagnostic relevance. Concentration of DNA-MPO complexes in serum are associated with poor control in asthma (20), severity in anaphylaxis (93), and development of extra-articular nodules in rheumatoid arthritis (118). It could also predict organ dysfunction and 28-day mortality in septic shock (119). High levels of circulating NETs were also associated with poor prognosis in community-acquired pneumonia, though it's not clear exactly which NETs surrogate marker was used in this study (120).
As for therapeutic intervention, two main approaches have been investigated: destruction of NETs or inhibition of their production. NETs can be dismantled by DNAse I treatment or by heparin (121), while their production has been blocked with PAD-4 inhibitors, mostly chloramidine (Cl-Amidine). Disruption of NETs with DNAse I have proven efficient in mouse models of stroke (122), ischemia reperfusion (123, 124), heparin-induced thrombocytopenia (125), and deep vein thrombosis (126). Furthermore, mice treated with DNAse I show less metastasis in mammary tumor models (127, 128). Treatment with chloramidine has shown efficacy in murine models of abdominal aortic aneurysm (122), arterial thrombosis (129), photothrombotic stroke (122), and sepsis (130).
However, studies in pathologies involving IC-induced NETs are scarce. The most relevant works show that pristane-induced lupus is reduced after inhalation of DNAse 1 (131), and arthritis symptoms are reduced by chloramidine treatment in collagen-induced arthritis model (132). Specific inhibition strategies using FcR blocking antibodies represent an interesting possibility, but no study has tested this approach so far, despite the existence of broadly available efficient antibodies.
Globally, while several potential clinical uses of NETs have been described, most results come from mouse models, and large scale clinical trials results are missing.
ICs can be formed in several clinical conditions. In autoimmunity they can be continuously present in circulation or in tissues, depending on the accessibility of the self-antigen. In contrast, during anaphylaxis they are formed as soon as the allergen enters the body and encounters pre-existing IgGs. As neutrophils express high levels of FcγRs, they can be activated by ICs and release NETs. Beside their tissue toxicity and proinflammatory properties, NETs contain autoantigen and can thus perpetuate autoimmunity. In anaphylaxis, IC-induced NETs release represents a new pathway that may participate in symptoms and severity of the disease. New fundamental and clinical investigations are needed to better elucidate the intracellular mechanisms of IC-induced NET release and evaluate the potential clinical applications of NETs as a biomarker and a therapeutic target.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would like to thank Prof. Marc Pallardy for helpful discussions about the manuscript.
1. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
2. Honda M, Kubes P. Neutrophils and neutrophil extracellular traps in the liver and gastrointestinal system. Nat Rev Gastroenterol Hepatol. (2018) 15:206–21. doi: 10.1038/nrgastro.2017.183
3. Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med. (2010) 207:1853–62. doi: 10.1084/jem.20100239
4. Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
5. Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, et al. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE. (2012) 7:e32366. doi: 10.1371/journal.pone.0032366
6. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E, et al. Neutrophil extracellular traps induce trypsin activation, inflammation, and tissue damage in mice with severe acute pancreatitis. Gastroenterology. (2015) 149:1920–31.e8. doi: 10.1053/j.gastro.2015.08.026
7. Daniel C, Leppkes M, Muñoz LE, Schley G, Schett G, Herrmann M. Extracellular DNA traps in inflammation, injury and healing. Nat Rev Nephrol. (2019) 15:559–75. doi: 10.1038/s41581-019-0163-2
8. Sakurai K, Miyashita T, Okazaki M, Yamaguchi T, Ohbatake Y, Nakanuma S, et al. Role for neutrophil extracellular traps (NETs) and platelet aggregation in early sepsis-induced hepatic dysfunction. Vivo Athens Greece. (2017) 31:1051–8. doi: 10.21873/invivo.11169
9. McDonald B, Davis RP, Kim S-J, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. (2017) 129:1357–67. doi: 10.1182/blood-2016-09-741298
10. Lefrançais E, Mallavia B, Zhuo H, Calfee CS, Looney MR. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. JCI Insight. (2018) 3:98178. doi: 10.1172/jci.insight.98178
11. Li H, Zhou X, Tan H, Hu Y, Zhang L, Liu S, et al. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget. (2018) 9:1772–84. doi: 10.18632/oncotarget.22744
12. Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep. (2016) 6:37252. doi: 10.1038/srep37252
13. Barrientos L, Bignon A, Gueguen C, de Chaisemartin L, Gorges R, Sandré C, et al. Neutrophil extracellular traps downregulate lipopolysaccharide-induced activation of monocyte-derived dendritic cells. J Immunol. (2014) 193:5689–98. doi: 10.4049/jimmunol.1400586
14. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. (2019) 133:2178–85. doi: 10.1182/blood-2018-11-844530
15. Wright TK, Gibson PG, Simpson JL, McDonald VM, Wood LG, Baines KJ. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirol Carlton Vic. (2016) 21:467–75. doi: 10.1111/resp.12730
16. Lachowicz-Scroggins ME, Dunican EM, Charbit AR, Raymond W, Looney MR, Peters MC, et al. Extracellular DNA, neutrophil extracellular traps, and inflammasome activation in severe asthma. Am J Respir Crit Care Med. (2019) 199:1076–85. doi: 10.1164/rccm.201810-1869OC
17. Dicker AJ, Crichton ML, Pumphrey EG, Cassidy AJ, Suarez-Cuartin G, Sibila O, et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol. (2017) 141:117–27. doi: 10.1016/j.jaci.2017.04.022
18. Grabcanovic-Musija F, Obermayer A, Stoiber W, Krautgartner W-D, Steinbacher P, Winterberg N, et al. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir Res. (2015) 16:59. doi: 10.1186/s12931-015-0221-7
19. Twaddell SH, Baines KJ, Grainge C, Gibson PG. The emerging role of neutrophil extracellular traps in respiratory disease. Chest. (2019) 156:774–82. doi: 10.1016/j.chest.2019.06.012
20. Granger V, Taillé C, Roach D, Letuvé S, Dupin C, Hamidi F, et al. Circulating neutrophil and eosinophil extracellular traps are markers of severe asthma. Allergy. (2019). doi: 10.1111/all.14059. [Epub ahead of print].
21. Kimball AS, Obi AT, Diaz JA, Henke PK. The emerging role of NETs in venous thrombosis and immunothrombosis. Front Immunol. (2016) 7:236. doi: 10.3389/fimmu.2016.00236
22. Skopelja-Gardner S, Jones JD, Rigby WFC. “NETtling” the host: breaking of tolerance in chronic inflammation and chronic infection. J Autoimmun. (2018) 88:1–10. doi: 10.1016/j.jaut.2017.10.008
23. Dwivedi N, Radic M. Burning controversies in NETs and autoimmunity: the mysteries of cell death and autoimmune disease. Autoimmunity. (2018) 51:267–80. doi: 10.1080/08916934.2018.1523395
24. Lee KH, Kronbichler A, Park DD-Y, Park Y, Moon H, Kim H, et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: a comprehensive review. Autoimmun Rev. (2017) 16:1160–73. doi: 10.1016/j.autrev.2017.09.012
25. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
26. Kirchner T, Möller S, Klinger M, Solbach W, Laskay T, Behnen M. The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators Inflamm. (2012) 2012:849136. doi: 10.1155/2012/849136
27. Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. (2011) 21:290–304. doi: 10.1038/cr.2010.150
28. Boeltz S, Amini P, Anders H-J, Andrade F, Bilyy R, Chatfield S, et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. (2019) 26:395–408. doi: 10.1038/s41418-018-0261-x
29. Kenny EF, Herzig A, Krüger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. eLife. (2017) 6:24437. doi: 10.7554/eLife.24437
30. Silva JC, Rodrigues NC, Thompson-Souza GA, Valdirene de Muniz S, Neves JS, Figueiredo RT. Mac-1 triggers neutrophil DNA extracellular trap formation to Aspergillus fumigatus independently of PAD4 histone citrullination. J Leukoc Biol. (2019). doi: 10.1002/JLB.4A0119-009RR. [Epub ahead of print].
31. Guiducci E, Lemberg C, Küng N, Schraner E, Theocharides APA, LeibundGut-Landmann S. Candida albicans-Induced NETosis Is independent of peptidylarginine deiminase 4. Front Immunol. (2018) 9:1573. doi: 10.3389/fimmu.2018.01573
32. Kenno S, Perito S, Mosci P, Vecchiarelli A, Monari C. Autophagy and reactive oxygen species are involved in neutrophil extracellular traps release induced by C. albicans morphotypes. Front Microbiol. (2016) 7:879. doi: 10.3389/fmicb.2016.00879
33. Xu F, Zhang C, Zou Z, Fan EKY, Chen L, Li Y, et al. Aging-related Atg5 defect impairs neutrophil extracellular traps formation. Immunology. (2017) 151:417–32. doi: 10.1111/imm.12740
34. Germic N, Stojkov D, Oberson K, Yousefi S, Simon H-U. Neither eosinophils nor neutrophils require ATG5-dependent autophagy for extracellular DNA trap formation. Immunology. (2017) 152:517–25. doi: 10.1111/imm.12790
35. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
36. Yipp BG, Petri B, Salina D, Jenne CN, Scott BNV, Zbytnuik LD, et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat Med. (2012) 18:1386–93. doi: 10.1038/nm.2847
37. Pilsczek FH, Salina D, Poon KKH, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol. (2010) 185:7413–25. doi: 10.4049/jimmunol.1000675
38. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
39. Douda DN, Khan MA, Grasemann H, Palaniyar N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc Natl Acad Sci USA. (2015) 112:2817–22. doi: 10.1073/pnas.1414055112
40. Naffah de Souza C, Breda LCD, Khan MA, de Almeida SR, Câmara NOS, Sweezey N, et al. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: a matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front Immunol. (2017) 8:1849. doi: 10.3389/fimmu.2017.01849
41. Flament H, Granger V, Vezinet C, Marzaioli V, Kannengiesser C, de Chaisemartin L, et al. Aspergillus-induced pneumonia in adult without obvious immunodeficiency: test the burst! Eur Respir J. (2018) 51:1702711. doi: 10.1183/13993003.02711-2017
42. Dahlgren C, Karlsson A, Bylund J. Intracellular neutrophil oxidants: from laboratory curiosity to clinical reality. J Immunol. (2019) 202:3127–34. doi: 10.4049/jimmunol.1900235
43. Petretto A, Bruschi M, Pratesi F, Croia C, Candiano G, Ghiggeri G, et al. Neutrophil extracellular traps (NET) induced by different stimuli: a comparative proteomic analysis. PLoS ONE. (2019) 14:e0218946. doi: 10.1371/journal.pone.0218946
44. van der Linden M, Westerlaken GHA, van der Vlist M, van Montfrans J, Meyaard L. Differential signalling and kinetics of neutrophil extracellular trap release revealed by quantitative live imaging. Sci Rep. (2017) 7:6529. doi: 10.1038/s41598-017-06901-w
45. Aleyd E, van Hout MWM, Ganzevles SH, Hoeben KA, Everts V, Bakema JE, et al. IgA enhances NETosis and release of neutrophil extracellular traps by polymorphonuclear cells via Fcα receptor I. J Immunol. (2014) 192:2374–83. doi: 10.4049/jimmunol.1300261
46. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via FcαRI. J Immunol. (2016) 197:4552–9. doi: 10.4049/jimmunol.1502353
47. Carmona-Rivera C, Zhao W, Yalavarthi S, Kaplan MJ. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann Rheum Dis. (2015) 74:1417–24. doi: 10.1136/annrheumdis-2013-204837
48. Kelley JM, Monach PA, Ji C, Zhou Y, Wu J, Tanaka S, et al. IgA and IgG antineutrophil cytoplasmic antibody engagement of Fc receptor genetic variants influences granulomatosis with polyangiitis. Proc Natl Acad Sci USA. (2011) 108:20736–41. doi: 10.1073/pnas.1109227109
49. Chen K, Nishi H, Travers R, Tsuboi N, Martinod K, Wagner DD, et al. Endocytosis of soluble immune complexes leads to their clearance by FcγRIIIB but induces neutrophil extracellular traps via FcγRIIA in vivo. Blood. (2012) 120:4421–31. doi: 10.1182/blood-2011-12-401133
50. Ozaki N, Suzuki S, Ishida M, Harada Y, Tanaka K, Sato Y, et al. Syk-dependent signaling pathways in neutrophils and macrophages are indispensable in the pathogenesis of anti-collagen antibody-induced arthritis. Int Immunol. (2012) 24:539–50. doi: 10.1093/intimm/dxs078
51. Behnen M, Leschczyk C, Möller S, Batel T, Klinger M, Solbach W, et al. Immobilized immune complexes induce neutrophil extracellular trap release by human neutrophil granulocytes via FcγRIIIB and Mac-1. J Immunol. (2014) 193:1954–65. doi: 10.4049/jimmunol.1400478
52. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Differential use of human neutrophil Fcγ receptors for inducing neutrophil extracellular trap formation. J Immunol Res. (2016) 2016:2908034. doi: 10.1155/2016/2908034
53. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Transforming growth factor-β-activated kinase 1 is required for human FcγRIIIb-induced neutrophil extracellular trap formation. Front Immunol. (2016) 7:277. doi: 10.3389/fimmu.2016.00277
54. Kraaij T, Tengström FC, Kamerling SWA, Pusey CD, Scherer HU, Toes REM, et al. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmun Rev. (2016) 15:577–84. doi: 10.1016/j.autrev.2016.02.018
55. Stoiber W, Obermayer A, Steinbacher P, Krautgartner W-D. The role of reactive oxygen species (ROS) in the formation of extracellular traps (ETs) in humans. Biomolecules. (2015) 5:702–23. doi: 10.3390/biom5020702
56. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
57. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
58. Jeremic I, Djuric O, Nikolic M, Vlajnic M, Nikolic A, Radojkovic D, et al. Neutrophil extracellular traps-associated markers are elevated in patients with systemic lupus erythematosus. Rheumatol Int. (2019) 39:1849–57. doi: 10.1007/s00296-019-04426-1
59. Leffler J, Martin M, Gullstrand B, Tydén H, Lood C, Truedsson L, et al. Neutrophil extracellular traps that are not degraded in systemic lupus erythematosus activate complement exacerbating the disease. J Immunol. (2012) 188:3522–31. doi: 10.4049/jimmunol.1102404
60. Bruschi M, Bonanni A, Petretto A, Vaglio A, Pratesi F, Santucci L, et al. Neutrophil extracellular traps (NETs) profiles in patients with incident SLE and lupus nephritis. J Rheumatol. (2019) 46:181232. doi: 10.3899/jrheum.181232
61. Pruchniak MP, Ostafin M, Wachowska M, Jakubaszek M, Kwiatkowska B, Olesinska M, et al. Neutrophil extracellular traps generation and degradation in patients with granulomatosis with polyangiitis and systemic lupus erythematosus. Autoimmunity. (2019) 52:126–35. doi: 10.1080/08916934.2019.1631812
62. Bruschi M, Petretto A, Santucci L, Vaglio A, Pratesi F, Migliorini P, et al. Neutrophil Extracellular Traps protein composition is specific for patients with Lupus nephritis and includes methyl-oxidized αenolase (methionine sulfoxide 93). Sci Rep. (2019) 9:7934. doi: 10.1038/s41598-019-44379-w
63. Massarenti L, Enevold C, Damgaard D, Ødum N, Nielsen CH, Jacobsen S. Peptidylarginine deiminase-4 gene polymorphisms are associated with systemic lupus erythematosus and lupus nephritis. Scand J Rheumatol. (2019) 48:133–40. doi: 10.1080/03009742.2018.1488273
64. Odqvist L, Jevnikar Z, Riise R, Öberg L, Rhedin M, Leonard D, et al. Genetic variations in A20 DUB domain provide a genetic link to citrullination and neutrophil extracellular traps in systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:1363–70. doi: 10.1136/annrheumdis-2019-215434
65. Nishi H, Mayadas TN. Neutrophils in lupus nephritis. Curr Opin Rheumatol. (2019) 31:193–200. doi: 10.1097/BOR.0000000000000577
66. Apel F, Zychlinsky A, Kenny EF. The role of neutrophil extracellular traps in rheumatic diseases. Nat Rev Rheumatol. (2018) 14:467–75. doi: 10.1038/s41584-018-0039-z
67. Podolska MJ, Mahajan A, Knopf J, Hahn J, Boeltz S, Munoz L, et al. Autoimmune, rheumatic, chronic inflammatory diseases: neutrophil extracellular traps on parade. Autoimmunity. (2018) 51:281–7. doi: 10.1080/08916934.2018.1519804
68. Pratesi F, Dioni I, Tommasi C, Alcaro MC, Paolini I, Barbetti F, et al. Antibodies from patients with rheumatoid arthritis target citrullinated histone 4 contained in neutrophils extracellular traps. Ann Rheum Dis. (2014) 73:1414–22. doi: 10.1136/annrheumdis-2012-202765
69. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 5:178ra40. doi: 10.1126/scitranslmed.3005580
70. Seri Y, Shoda H, Suzuki A, Matsumoto I, Sumida T, Fujio K, et al. Peptidylarginine deiminase type 4 deficiency reduced arthritis severity in a glucose-6-phosphate isomerase-induced arthritis model. Sci Rep. (2015) 5:13041. doi: 10.1038/srep13041
71. Suzuki A, Kochi Y, Shoda H, Seri Y, Fujio K, Sawada T, et al. Decreased severity of experimental autoimmune arthritis in peptidylarginine deiminase type 4 knockout mice. BMC Musculoskelet Disord. (2016) 17:205. doi: 10.1186/s12891-016-1055-2
72. Mergaert AM, Bawadekar M, Nguyen TQ, Massarenti L, Holmes CL, Rebernick R, et al. Reduced anti-histone antibodies and increased risk of rheumatoid arthritis associated with a single nucleotide polymorphism in PADI4 in North Americans. Int J Mol Sci. (2019) 20:E3093. doi: 10.3390/ijms20123093
73. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. (2014) 16:R122. doi: 10.1186/ar4579
74. Ribon M, Seninet S, Mussard J, Sebbag M, Clavel C, Serre G, et al. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J Autoimmun. (2019) 98:122–31. doi: 10.1016/j.jaut.2019.01.003
75. Wang W, Peng W, Ning X. Increased levels of neutrophil extracellular trap remnants in the serum of patients with rheumatoid arthritis. Int J Rheum Dis. (2017) 21:415–21. doi: 10.1111/1756-185X.13226
76. Pérez-Sánchez C, Ruiz-Limón P, Aguirre MA, Jiménez-Gómez Y, Arias-de la Rosa I, Ábalos-Aguilera MC, et al. Diagnostic potential of NETosis-derived products for disease activity, atherosclerosis and therapeutic effectiveness in rheumatoid arthritis patients. J Autoimmun. (2017) 82:31–40. doi: 10.1016/j.jaut.2017.04.007
77. Chen W, Wang Q, Ke Y, Lin J. Neutrophil function in an inflammatory milieu of rheumatoid arthritis. J Immunol Res. (2018) 2018:8549329. doi: 10.1155/2018/8549329
78. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. (2017) 2:eaag3358. doi: 10.1126/sciimmunol.aag3358
79. O'Neil LJ, Kaplan MJ. Neutrophils in rheumatoid arthritis: breaking immune tolerance and fueling disease. Trends Mol Med. (2019) 25:215–27. doi: 10.1016/j.molmed.2018.12.008
80. Makrygiannakis D, Hermansson M, Ulfgren A-K, Nicholas AP, Zendman AJW, Eklund A, et al. Smoking increases peptidylarginine deiminase 2 enzyme expression in human lungs and increases citrullination in BAL cells. Ann Rheum Dis. (2008) 67:1488–92. doi: 10.1136/ard.2007.075192
81. van Wesemael TJ, Ajeganova S, Humphreys J, Terao C, Muhammad A, Symmons DPM, et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: a multicenter cohort study. Arthritis Res Ther. (2016) 18:285. doi: 10.1186/s13075-016-1177-9
82. Demoruelle MK, Harrall KK, Ho L, Purmalek MM, Seto NL, Rothfuss HM, et al. Anti-citrullinated protein antibodies are associated with neutrophil extracellular traps in the sputum in relatives of rheumatoid arthritis patients. Arthritis Rheumatol. (2017) 69:1165–75. doi: 10.1002/art.40066
83. Hosseinzadeh A, Thompson PR, Segal BH, Urban CF. Nicotine induces neutrophil extracellular traps. J Leukoc Biol. (2016) 100:1105–12. doi: 10.1189/jlb.3AB0815-379RR
84. Lee J, Luria A, Rhodes C, Raghu H, Lingampalli N, Sharpe O, et al. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatol Oxf Engl. (2017) 56:644–53. doi: 10.1093/rheumatology/kew449
85. Chrysanthopoulou A, Mitroulis I, Apostolidou E, Arelaki S, Mikroulis D, Konstantinidis T, et al. Neutrophil extracellular traps promote differentiation and function of fibroblasts. J Pathol. (2014) 233:294–307. doi: 10.1002/path.4359
86. Corsiero E, Pratesi F, Prediletto E, Bombardieri M, Migliorini P. NETosis as source of autoantigens in rheumatoid arthritis. Front Immunol. (2016) 7:485. doi: 10.3389/fimmu.2016.00485
87. Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest. (2006) 116:3183–94. doi: 10.1172/JCI28756
88. Montgomery AB, Kopec J, Shrestha L, Thezenas M-L, Burgess-Brown NA, Fischer R, et al. Crystal structure of Porphyromonas gingivalis peptidylarginine deiminase: implications for autoimmunity in rheumatoid arthritis. Ann Rheum Dis. (2016) 75:1255–61. doi: 10.1136/annrheumdis-2015-207656
89. Reber LL, Hernandez JD, Galli SJ. The pathophysiology of anaphylaxis. J Allergy Clin Immunol. (2017) 140:335–48. doi: 10.1016/j.jaci.2017.06.003
90. Oettgen HC, Martin TR, Wynshaw-Boris A, Deng C, Drazen JM, Leder P. Active anaphylaxis in IgE-deficient mice. Nature. (1994) 370:367–70. doi: 10.1038/370367a0
91. Miyajima I, Dombrowicz D, Martin TR, Ravetch JV, Kinet JP, Galli SJ. Systemic anaphylaxis in the mouse can be mediated largely through IgG1 and Fc gammaRIII. Assessment of the cardiopulmonary changes, mast cell degranulation, and death associated with active or IgE- or IgG1-dependent passive anaphylaxis. J Clin Invest. (1997) 99:901–14. doi: 10.1172/JCI119255
92. Laroche D, Chollet-Martin S, Léturgie P, Malzac L, Vergnaud M-C, Neukirch C, et al. Evaluation of a new routine diagnostic test for immunoglobulin E sensitization to neuromuscular blocking agents. Anesthesiology. (2011) 114:91–7. doi: 10.1097/ALN.0b013e31820164d2
93. Jönsson F, de Chaisemartin L, Granger V, Gouel-Chéron A, Gillis CM, Zhu Q, et al. An IgG-induced neutrophil activation pathway contributes to human drug-induced anaphylaxis. Sci Transl Med. (2019) 11:eaat1479. doi: 10.1126/scitranslmed.aat1479
94. Jönsson F, Mancardi DA, Kita Y, Karasuyama H, Iannascoli B, Van Rooijen N, et al. Mouse and human neutrophils induce anaphylaxis. J Clin Invest. (2011) 121:1484–96. doi: 10.1172/JCI45232
95. Francis A, Bosio E, Stone SF, Fatovich DM, Arendts G, Nagree Y, et al. Neutrophil activation during acute human anaphylaxis: analysis of MPO and sCD62L. Clin Exp Allergy J Br Soc Allergy Clin Immunol. (2017) 47:361–70. doi: 10.1111/cea.12868
96. Vadas P, Gold M, Perelman B, Liss GM, Lack G, Blyth T, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med. (2008) 358:28–35. doi: 10.1056/NEJMoa070030
97. Francis A, Bosio E, Stone SF, Fatovich DM, Arendts G, MacDonald SPJ, et al. Markers involved in innate immunity and neutrophil activation are elevated during acute human anaphylaxis: validation of a microarray study. J Innate Immun. (2019) 11:63–73. doi: 10.1159/000492301
98. Jönsson F, Mancardi DA, Zhao W, Kita Y, Iannascoli B, Khun H, et al. Human FcγRIIA induces anaphylactic and allergic reactions. Blood. (2012) 119:2533–44. doi: 10.1182/blood-2011-07-367334
99. Jönsson F, Mancardi DA, Albanesi M, Bruhns P. Neutrophils in local and systemic antibody-dependent inflammatory and anaphylactic reactions. J Leukoc Biol. (2013) 94:643–56. doi: 10.1189/jlb.1212623
100. Gillis CM, Jönsson F, Mancardi DA, Tu N, Beutier H, Van Rooijen N, et al. Mechanisms of anaphylaxis in human low-affinity IgG receptor locus knock-in mice. J Allergy Clin Immunol. (2017) 139:1253–65.e14. doi: 10.1016/j.jaci.2016.06.058
101. Gillis CM, Zenatti PP, Mancardi DA, Beutier H, Fiette L, Macdonald LE, et al. In vivo effector functions of high-affinity mouse IgG receptor FcγRI in disease and therapy models. J Autoimmun. (2017) 80:95–102. doi: 10.1016/j.jaut.2016.09.009
102. Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, et al. Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS ONE. (2012) 7:e48111. doi: 10.1371/journal.pone.0048111
103. Hoppenbrouwers T, Autar ASA, Sultan AR, Abraham TE, van Cappellen WA, Houtsmuller AB, et al. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE. (2017) 12:e0176472. doi: 10.1371/journal.pone.0176472
104. Guimarães-Costa AB, Nascimento MTC, Wardini AB, Pinto-da-Silva LH, Saraiva EM. ETosis: a microbicidal mechanism beyond cell death. J Parasitol Res. (2012) 2012:929743. doi: 10.1155/2012/929743
105. Granger V, Faille D, Marani V, Noël B, Gallais Y, Szely N, et al. Human blood monocytes are able to form extracellular traps. J Leukoc Biol. (2017) 102:775–81. doi: 10.1189/jlb.3MA0916-411R
106. Beutier H, Hechler B, Godon O, Wang Y, Gillis CM, de Chaisemartin L, et al. Platelets expressing IgG receptor FcγRIIA/CD32A determine the severity of experimental anaphylaxis. Sci Immunol. (2018) 3:eaan5997. doi: 10.1126/sciimmunol.aan5997
107. Peppers BP, Vatsayan A, Dalal J, Bonfield T, Tcheurekdjian H, Hostoffer R. A case series: association of anaphylaxis with a significant decrease in platelet levels and possible secondary risk of thrombosis. Immun Inflamm Dis. (2018) 6:377–81. doi: 10.1002/iid3.224
108. Lisman T. Platelet-neutrophil interactions as drivers of inflammatory and thrombotic disease. Cell Tissue Res. (2018) 371:567–76. doi: 10.1007/s00441-017-2727-4
109. Carestia A, Kaufman T, Schattner M. Platelets: new bricks in the building of neutrophil extracellular traps. Front Immunol. (2016) 7:271. doi: 10.3389/fimmu.2016.00271
110. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. (2012) 122:2661–71. doi: 10.1172/JCI61303
111. Krishnamurthy D, Starkl P, Szalai K, Roth-Walter F, Diesner SC, Mittlboeck M, et al. Monitoring neutrophils and platelets during casein-induced anaphylaxis in an experimental BALB/c mouse model. Clin Exp Allergy J Br Soc Allergy Clin Immunol. (2012) 42:1119–28. doi: 10.1111/j.1365-2222.2012.04012.x
112. Cortjens B, van Woensel JBM, Bem RA. Neutrophil extracellular traps in respiratory disease: guided anti-microbial traps or toxic webs? Paediatr Respir Rev. (2017) 21:54–61. doi: 10.1016/j.prrv.2016.03.007
113. Yuen J, Pluthero FG, Douda DN, Riedl M, Cherry A, Ulanova M, et al. NETosing neutrophils activate complement both on their own NETs and bacteria via alternative and non-alternative pathways. Front Immunol. (2016) 7:137. doi: 10.3389/fimmu.2016.00137
114. Wang H, Wang C, Zhao M-H, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. (2015) 181:518–27. doi: 10.1111/cei.12654
115. Oehmcke S, Mörgelin M, Herwald H. Activation of the human contact system on neutrophil extracellular traps. J Innate Immun. (2009) 1:225–30. doi: 10.1159/000203700
116. Masuda S, Nakazawa D, Shida H, Miyoshi A, Kusunoki Y, Tomaru U, et al. NETosis markers: quest for specific, objective, and quantitative markers. Clin Chim Acta. (2016) 459:89–93. doi: 10.1016/j.cca.2016.05.029
117. Jin W, Xu H-X, Zhang S-R, Li H, Wang W-Q, Gao H-L, et al. Tumor-infiltrating NETs predict postsurgical survival in patients with pancreatic ductal adenocarcinoma. Ann Surg Oncol. (2019) 26:635–43. doi: 10.1245/s10434-018-6941-4
118. Bach M, Moon J, Moore R, Pan T, Nelson JL, Lood C. A neutrophil activation biomarker panel in prognosis and monitoring of patients with rheumatoid arthritis. Arthritis Rheumatol Hoboken NJ. (2019). doi: 10.1002/art.41062. [Epub ahead of print].
119. Maruchi Y, Tsuda M, Mori H, Takenaka N, Gocho T, Huq MA, et al. Plasma myeloperoxidase-conjugated DNA level predicts outcomes and organ dysfunction in patients with septic shock. Crit Care Lond Engl. (2018) 22:176. doi: 10.1186/s13054-018-2109-7
120. Ebrahimi F, Giaglis S, Hahn S, Blum CA, Baumgartner C, Kutz A, et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: secondary analysis of a randomised controlled trial. Eur Respir J. (2018) 51:1701389. doi: 10.1183/13993003.01389-2017
121. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
122. Peña-Martínez C, Durán-Laforet V, García-Culebras A, Ostos F, Hernández-Jiménez M, Bravo-Ferrer I, et al. Pharmacological modulation of neutrophil extracellular traps reverses thrombotic stroke tPA (tissue-type plasminogen activator) resistance. Stroke. (2019) 50:3228–37. doi: 10.1161/STROKEAHA.119.026848
123. Wang S, Xie T, Sun S, Wang K, Liu B, Wu X, et al. DNase-1 treatment exerts protective effects in a rat model of intestinal ischemia-reperfusion injury. Sci Rep. (2018) 8:17788. doi: 10.1038/s41598-018-36198-2
124. Albadawi H, Oklu R, Raacke Malley RE, O'Keefe RM, Uong TP, Cormier NR, et al. Effect of DNase I treatment and neutrophil depletion on acute limb ischemia-reperfusion injury in mice. J Vasc Surg. (2016) 64:484–93. doi: 10.1016/j.jvs.2015.01.031
125. Gollomp K, Kim M, Johnston I, Hayes V, Welsh J, Arepally GM, et al. Neutrophil accumulation and NET release contribute to thrombosis in HIT. JCI Insight. (2018) 3:99445. doi: 10.1172/jci.insight.99445
126. Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
127. Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. (2016) 8:361ra138. doi: 10.1126/scitranslmed.aag1711
128. Rocks N, Vanwinge C, Radermecker C, Blacher S, Gilles C, Marée R, et al. Ozone-primed neutrophils promote early steps of tumour cell metastasis to lungs by enhancing their NET production. Thorax. (2019) 74:768–79. doi: 10.1136/thoraxjnl-2018-211990
129. Novotny J, Chandraratne S, Weinberger T, Philippi V, Stark K, Ehrlich A, et al. Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS ONE. (2018) 13:e0190728. doi: 10.1371/journal.pone.0190728
130. Biron BM, Chung C-S, O'Brien XM, Chen Y, Reichner JS, Ayala A. Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun. (2017) 9:22–32. doi: 10.1159/000448808
131. Jarrot P-A, Tellier E, Plantureux L, Crescence L, Robert S, Chareyre C, et al. Neutrophil extracellular traps are associated with the pathogenesis of diffuse alveolar hemorrhage in murine lupus. J Autoimmun. (2019) 100:120–30. doi: 10.1016/j.jaut.2019.03.009
Keywords: NETs, autoimmunity, anaphylaxis, immune complexes, neutrophils
Citation: Granger V, Peyneau M, Chollet-Martin S and de Chaisemartin L (2019) Neutrophil Extracellular Traps in Autoimmunity and Allergy: Immune Complexes at Work. Front. Immunol. 10:2824. doi: 10.3389/fimmu.2019.02824
Received: 31 July 2019; Accepted: 15 November 2019;
Published: 03 December 2019.
Edited by:
Thomas Marichal, University of Liège, BelgiumReviewed by:
Dipyaman Ganguly, Indian Institute of Chemical Biology (CSIR), IndiaCopyright © 2019 Granger, Peyneau, Chollet-Martin and de Chaisemartin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Luc de Chaisemartin, bHVjLmRlLWNoYWlzZW1hcnRpbkB1LXBzdWQuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.