 Erkan Demirkaya
Erkan Demirkaya Zehra Serap Arici
Zehra Serap Arici Micol Romano
Micol Romano Roberta Audrey Berard1
Roberta Audrey Berard1 Ivona Aksentijevich
Ivona Aksentijevich- 1Division of Paediatric Rheumatology, Department of Paediatrics, Schulich School of Medicine & Dentistry, University of Western Ontario, London, ON, Canada
- 2Department of Paediatric Rheumatology, Sanliurfa Training and Research Hospital, Sanliurfa, Turkey
- 3Department of Pediatric Rheumatology, Istituto Ortopedico Gaetano Pini, Milan, Italy
- 4Inflammatory Disease Section, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
Precision medicine (PM) is an emerging data-driven health care approach that integrates phenotypic, genomic, epigenetic, and environmental factors unique to an individual. The goal of PM is to facilitate diagnosis, predict effective therapy, and avoid adverse reactions specific for each patient. The forefront of PM is in oncology; nonetheless, it is developing in other fields of medicine, including rheumatology. Recent studies on elucidating the genetic architecture of polygenic and monogenic rheumatological diseases have made PM possible by enabling physicians to customize medical treatment through the incorporation of clinical features and genetic data. For complex inflammatory disorders, the prevailing paradigm is that disease susceptibility is due to additive effects of common reduced-penetrance gene variants and environmental factors. Efforts have been made to calculate cumulative genetic risk score (GRS) and to relate specific susceptibility alleles for use of target therapies. The discovery of rare patients with single-gene high-penetrance mutations informed our understanding of pathways driving systemic inflammation. Here, we review the advances in practicing PM in patients with primary systemic vasculitides (PSVs). We summarize recent genetic studies and discuss current knowledge on the contribution of epigenetic factors and extracellular vesicles (EVs) in disease progression and treatment response. Implementation of PM in PSVs is a developing field that will require analysis of a large cohort of patients to validate data from genomics, transcriptomics, metabolomics, proteomics, and epigenomics studies for accurate disease profiling. This multi-omics approach to study disease pathogeneses should ultimately provide a powerful tool for stratification of patients to receive tailored optimal therapies and for monitoring their disease activity.
Introduction
Precision medicine (PM) can be defined as tailored medical care that is primarily based upon understanding the molecular sequence of biologic events causal to disease and critical for diagnosis and therapy. The term precision medicine should be distinguished from the term personalized medicine whereby prevention and treatment are being developed uniquely for each patient, although these two terms are used interchangeably (1). Both communicate a concept of individualized medicine, although in PM, the focus is on identifying health care approaches that will be effective for a group of patients who share similar genetic, environmental, and lifestyle disease susceptibility factors. Recognizing subgroups of patients with similar disease risk factors will ensure that they will receive optimal treatment to improve their quality of life and health outcomes.
Primary systemic vasculitides (PSVs) (Table 1) are a heterogeneous group of diseases in their etiology, clinical presentation, and response to therapy. Severity and location of symptoms vary greatly, and most classification schemes that attempt to advance clinicians the ability to diagnose and treat patients, are based on blood vessel size, autoantibodies profile [e.g., anti-neutrophil cytoplasmic antibody (ANCA)], and histopathological findings. In addition to genetic predisposition, there is a role for environmental factors, including exposure to drugs and infectious agents, in the pathogenesis, and prognosis of PSVs. The past decade has witnessed major advances in genetics research that have improved our understanding on molecular mechanisms in PSVs. New sequencing technologies and high-throughput genotyping arrays used for genome-wide association studies (GWAS) generated vast amount of data that will require validation by meta-analysis in large cohorts of patients. Discovery of monogenic diseases mimicking PSVs offered additional insights on biochemical pathways that are shared between rare and more common forms of vasculitis. Recent advances in epigenetic research support the role of extracellular vesicles (EVs) in the pathogenesis of PSVs, which could be used for developing novel biomarkers to monitor disease activity, prognosis, and treatment outcomes.

Table 1. Chapel Hill Consensus Conference Nomenclature (CHCC 2012) of vasculitides (2).
Herein, we discuss the up-to-date knowledge on the genetic predisposition to PSVs and review recent clinical research studies aimed to further our understanding of the pathogeneses and outcomes of vasculitides.
GWAS in PSVs
GWAS have been used, following the completion of the Human Genome project in 2003 and the International HapMap Project in 2005, to identify association of common and reduced-penetrance variants, termed single nucleotide polymorphisms (SNPs), with human traits or diseases (3). The primary goal of GWAS has been to elucidate the biology of polygenic and complex human diseases, which can be translated toward the development of novel therapeutics. GWAS have also been used to study gene-environment interactions, response to therapies, and more recently for risk prediction. GWAS are based on the genotyping of large cohorts of patients and ancestry-matched controls for SNPs in the entire genome. These associations are tested with gene variants that have a minor allele frequency (MAF) higher than 5% in the general population and are considered significant at the p-value genome-wide threshold of 5 × 10−8 (4). For a number of autoimmune disorders, SNPs in the human leukocyte antigen (HLA) [also known as major histocompatibility complex (MHC)] and genes encoded within the locus have been shown to play a major role in susceptibility to disease (4). In primary vasculitides (PSVs), GWAS have been performed primarily in Kawasaki disease (KD) and Behçet's disease (BD) (Table 2) (5–42, 44–46).

Table 2. Genome-wide association studies (GWAS) in primary systemic vasculitides (PSVs).
Kawasaki Disease
KD is an acute, self-limited vasculitis that typically affects infants and children under the age of 5 years. Coronary artery aneurysms (CAAs) occur in 25% of untreated patients and may lead to ischemic heart disease, myocardial infarction, and sudden death at a young age. The pathogenesis of KD remains unknown; however, it is thought that host genetics play an important role in susceptibility and disease outcome. Interestingly, the incidence of KD is up to 50-fold higher in children of Asian descent. Epidemiologic and clinical features of KD also strongly support an infectious etiology in genetically predisposed children (47).
GWAS in KD have identified a number of susceptibility SNPs/genes that contribute to the risk of KD (HLA, FCGR2A, SLC8A1, ZFHX3, NMNAT2, BLK, DAB1, HCP5, COPB2, ERAP1, NAALADL2, CD40, NFKBIL1, IGHV, EBF2, CACNB2, LTA, and LEF1) (5–16) to the risk of cardiovascular disease in KD (TIAM1, NEBL, PLCB4/PLCB1, TUBA3C, SLC8A1, PELI1, KCNN2, TIFAB, and AGT) (8, 12, 17–22) and to the risk of intravenous immunoglobulin (IVIG) resistance (BCL2L11 and SAMD9L) (23, 24). Involvement of the HLA region in susceptibility to KD has been controversial and has not been replicated across different ancestral groups.
Shimizu et al. (12) identified three functional SNPs in the solute carrier family 8, member 1 (SLC8A1) gene to be associated with susceptibility to KD in Japanese and European cohorts (meta analysis p = 0.0001). SLC8A1 encodes NCX1 (a sodium/calcium exchanger) that functions as a bidirectional sodium/calcium channel. Patients homozygous for the risk allele (rs13017968) have higher rates of coronary artery abnormalities. Homozygosity for rs13017968 is associated with an increase in Ca2+ flux in EBV-transformed B cells of healthy individuals. The NCX1 protein expression was detected in the postmortem coronary artery tissue of a young KD patient. Another study by Onouchi et al. (48) found a coding SNP (rs3741596) in the ORAI Calcium Release-Activated Calcium Modulator 1 (ORAI1) gene to be significantly associated with KD in Japanese patients (meta analysis p = 0.00041). Interestingly, frequency of the risk allele is more than 20 times higher in Japanese compared to Europeans, which may account for higher prevalence of KD in the Japanese population. Together, these genetic and functional data provide evidence for the role of Ca2+-mediated signaling pathways in the pathogenesis of KD and for the use of calcineurin inhibitors (49).
Lv et al. (46) used statistically significant candidate variants from multiple GWAS and other gene association studies for pathways analysis. This investigation showed that KD susceptibility genes are enriched in functional networks for calcium ion homeostasis and immune responses and highlighted the role of nuclear transcription factor of activated T cells (NF-AT) and nuclear factor (NF) kappa light chain enhancer of activated B cells (NF-κB) in the pathogenesis of KD.
Another indication from GWAS for the use of new therapies in KD has come from the study by Chang et al. (44). The promoter variant, rs2736340, in the B lymphoid tyrosine kinase (BLK) gene was significantly associated with susceptibility to KD in multiple Asian populations [odds ratio (OR) = 1.498, 1.354–1.657; p = 4.74 × 10(−31)]. The transformed and primary B cells with the risk allele express significantly lower levels of BLK and have reduced signaling downstream of B cell receptors. These data suggest a role for humoral immunity in the pathogenesis of the acute stage of KD (44). Although B cells and autoantibodies have been found in blood samples of KD patients, their role, whether they are causal or bystanders of an activated immune system or specific to an infectious agent in the etiology of KD, is currently unknown (50).
Standard treatment for KD consists of a single infusion of high-dose IVIGs and high-dose aspirin (until the fever has resolved); however, the mechanism of action remains elusive. Because IVIG therapy is the mainstay of treatment for KD, there has been interest to study the effect of SNPs and copy number variants (CNVs) on the function of receptors for IgG, the Fc-gamma receptors (FcγRs). There are five genes encoding the low-affinity FcγRs (FCGR2A, FCGR2B, FCGR2C, FCGR3A, and FCGR3B) that are clustered in the FCGR2/3 locus. Recently, Nagelkerke et al. published a study that evaluated the previously reported genetic association with the rs1801274 variant in this gene locus. The FCGR2A coding SNP rs1801274 has been validated in a number of KD ancestry cohorts and was shown to cause a difference in the ability of FcγRIIa to bind the human IgG2 immunoglobulin isotype. This new study defined haplotype blocks (set of closely linked alleles/markers that are inherited together) across the FCGR2/3 gene locus in over 4,000 individuals from different populations. The FCGR2C-ORF haplotype has stronger susceptibility to KD in the European cohort than the previously reported risk allele rs1801274. However, the FCGR2C-ORF haplotype is extremely rare in Asians and thus cannot explain the high prevalence of KD in Asian populations. This research points out the importance of proper interpretation of genetic association studies in the context of patients' ancestry (51).
Kuo et al. developed a genetic model to predict IVIG resistance in KD. They identified 11 candidate SNPs in ADTRP, KLF6, EX0C2, ZNF438-ZEB1, and MIR548A3-ZPLD1 genes to calculate a weighted genetic risk score (wGRS) in 126 IVIG responders and 24 IVIG non-responders (52). Patients with IVIG resistance had higher wGRS scores than individuals who were IVIG responsive; however, the wide confident intervals (CIs) for the OR suggest that these findings should be replicated in larger cohorts of patients.
Kim et al. (24) recently identified a new gene locus, SAMD9L, that is associated with IVIG resistance in Korean and Japanese KD cohorts. The meta-analysis of IVIG non-responders (n = 317) and IVIG responders (n = 1221) showed the association of the common intronic variant (rs28662; MAF = 13% in gnomAD) with IVIG resistance [(OR) = 4.30, p = 5.3 × 10−6]. This SNP was predicted to change the motif for transcription factor binding site and was also associated with differential expression of the SAMD9L gene in lymphoblastoid cell lines. Previously, variants in SAMD9L have been associated with susceptibility for systemic lupus erythematosus, systemic sclerosis, and other immune-related diseases, such as juvenile myelomonocytic leukemia, acute myeloid leukemia, myelodysplastic syndrome, hepatitis-B-related hepatocellular carcinoma, and ataxia-pancytopenia syndrome. However, the role of SAMD9L in immune signaling pathway is unclear (24).
Behçet's Disease
BD is a chronic multisystem inflammatory disorder characterized by recurrent oral/genital ulcers, ocular involvement, skin lesions/pathergy, presenting with remissions and exacerbations. The disease is common in populations living along the ancient Silk Road: Turkish, Iranian, Chinese, Korean, and Japanese. Classic BD is a complex disease with a strong genetic predisposition. Although the etiology of BD remains unclear, recent immunogenetic findings from GWAS provide clues to its pathogenesis (53, 54).
HLA-B*51 remains the most significant genetic susceptibility factor for BD in multiple populations; however, it accounts for <20% of the genetic risk (55, 56). The ongoing controversy is about whether the disease association with HLA-B*51 is attributed to a role of this variant itself or is due to its linkage disequilibrium (LD) with another variant in the region. There are also other BD disease-associated amino acids variants in the HLA-B locus that are thought to reside within the antigen-binding grove of the molecule (57). The association between specific MHC class I alleles and certain disease manifestations have been reported (58–61). A meta-analysis based on 72 studies in 74 study populations revealed moderate association of HLA-B*51/B5 with male gender and high prevalence of eye and mucocutaneous involvement (60).
GWAS have identified a number of population-specific associations in the following genes: IL23R-IL12RB2, IL10, TNFAIP3, STAT4, CCR1-CCR3, KLRC4, ERAP1, and FUT2. Variants in some genes, including KIAA1529, UBASH3B, GIMAP, IL12A, NAALADL2, YIPF7, CPVL, LOC100129342, and UBAC2, did not reach genome-wide significance (p < 5.0 × 10−8) (25–34). Most of these alleles act as susceptibility factors, while some variants appear to be protective. BD-risk alleles in the IL10 susceptibility gene locus were the first variants identified outside of the HLA region by GWAS in Turkish and Japanese populations (29, 30). The SNP rs1518111 was subsequently replicated in Middle Eastern Arab, Greek, British, Korean, Iranian, and Han-Chinese samples and is today the only variant consistently associated with susceptibility to BD across all ancestries (57).
More recent studies investigated whether there is evidence for interaction (epistasis) among BD susceptibility genes. The endoplasmic reticulum (ER)-associated protease (ERAP1) haplotype tagged by the coding variant, rs17482078 (p.Arg725Gln), has a large effect with an OR of 3.78 in patient carriers for HLA-B*51. Interestingly, the same variant rs17482078 is protective for psoriasis and ankylosing spondylitis (AS). ERAP1 is an enzyme that clips proteasome-processed peptides in the ER before these peptides are loaded on class I HLA molecules (56). The epistasis observed between HLA-B*51 and the risk haplotype of ERAP1 suggests that an HLA class I-peptide presentation contributes to BD. The difference between risk and protection conferred by the ERAP1 haplotype may be explained by altered binding affinities among specific peptides, trimmed or not trimmed by the ERAP1 variant, for different HLA class I molecules (62).
Dense genotyping of immune-related loci extended the list of susceptibility genes to include risk alleles in IL1A-IL1B, IRF8, CEBPB-PTPN1, and ADO-EGR2 genes in the Turkish set of 1,900 cases and 1,779 controls. This study implicated the innate immune response to microbial exposure in susceptibility to BD (63). Bakir-Gungor et al. reported new BD-associated common variants in pathways, including focal adhesion, complement and coagulation cascades, mitogen-activated protein kinases (MAPK) signaling, proteasome pathways, extracellular matrix (ECM)–receptor interaction, and transforming growth factor-β (TGFβ) signaling both in Japanese and Turkish populations (64).
Targeted sequencing of candidate immunological genes has revealed the contribution of rare non-synonymous variants of NOD2, TLR4, IL23, and MEFV in the BD pathogenesis. In particular, heterozygosity for a common high-penetrance familial Mediterranean fever (FMF)-associated mutation p.Met694Val is significantly associated with BD risk in the Turkish population, although not in Japanese patients due to its absence in the Asian populations (65). MEFV encodes pyrin, which regulates caspase-1-mediated interleukin (IL)-1β production.
Nakano et al. investigated the functional effect of GWAS-identified BD risk alleles in IL-10 and CCR1. They observed a differential protein expression of IL10 and CCR1 in M1 vs. M2 macrophages (Mϕ) derived from BD patients. BD skin lesions showed predominance of M1 Mϕ. This study suggests that targeting pathways and genes that play a role in the Mϕ polarization should be explored in the treatment of BD (66).
Taken together, GWAS have implicated a number of genes of the innate and adaptive immunity with increased susceptibility to BD. Whether inhibition of these molecular pathways will be effective in the treatment of BD remains to be seen.
ANCA-Associated Vasculitis (AAV)
AAV is a heterogeneous group of disorders characterized by necrotizing inflammation of small vessels and by the presence of ANCA. The two most common ANCAs are the autoantibodies that target the proteins: myeloperoxidase (MPO) and proteinase 3 (PR3). AAV includes the clinical syndromes of microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA), and eosinophilic granulomatosis with polyangiitis (EGPA). ANCA directed against PR3 are preferentially associated with GPA, and anti-MPO is associated mainly with MPA and EGPA. Although the role of ANCA in the pathogenesis of AAV is appreciated, the mechanism for ANCA-mediated development of AAV is not clear. Genetic predisposition and exposure to environmental factors, such as drugs and infectious agents, are possible triggers of excessive activation of neutrophils and injury to small blood vessels. The clinical signs vary and may affect several organs, such as the kidney, intestine, lung, and skin.
Currently, three GWAS have been performed in AAV. These studies identified risk alleles in HLA-DQ, HLA-DP, HLA-DR, SERPINA1, PTPN22, PRTN3, and SEMA6A genes as contributing to AAV (35–37). A meta-analysis of 140 reported genetic variants associated with AAV distinguished the most significant 33 alleles in 15 genes (67). Twenty of these 33 genetic variants were present in the HLA region, suggesting an important role for the antigen presentation in the disease pathogenesis. Two GWAS showed that the SNP association signal in the HLA region was fully accounted for by HLA-DPB1*04 allele (35, 37).
Lyons et al. established that MPA and GPA are two distinct diseases based on their genetic profile (35). Anti-MPO positive AAV (MPA) is strongly associated with the HLA-DQ, whereas anti-PR3 AAV (GPA) is associated with the HLA-DP region and the genes encoding α(1)-antitrypsin (SERPINA1) and serine proteinase 3 (PRTN3) (35, 68). SERPINA1 is a major inhibitor of PR3, and lower levels of α (1)-antitrypsin can lead to higher levels of circulating PR3 and higher production of anti-PR3 autoantibodies. Population difference in HLA-class II genotype frequency plays a role in the epidemiology of this disease. Among AAVs, GPA and PR3-AAV are prevalent in European populations, whereas MPA and MPO-AAV are predominant in the Japanese population. In the North American population, the HLA- DPB1*04 has the strongest association with GPA, whereas in the Japanese population, HLA- DRB1*09:01 is strongly associated with MPA (68–70).
Other notable variants outside the HLA region include alleles in protein tyrosine phosphatase non-receptor type 2 (PTPN22) gene, which negatively regulates the production of an immunosuppressive cytokine interleukin (IL-10) (71). The gain-of-function risk allele, p.Arg260Trp, is linked to GPA-AAV in addition to a number of other autoimmune diseases (72, 73). A non-coding SNP in the vicinity of SEMA6A is linked to GPA in the GWAS study of Caucasian cohort of 528 patients and at a genome-wide signficance (p = 2.09 × 10−8). The SEMA6A gene codes for semaphorin 6A and is highly expressed in dendritic cells; however, its function is unclear (37).
Lower and higher copy number variants in the FCGR3B gene and a higher DEFB4 gene copy number have been associated with risk for AAV (74–76). A possible relationship between the FCGR3B copy number and susceptibility to AAV needs to be clarified experimentally (77).
Lee et al. performed gene-ontology (GO) enrichment and protein–protein interaction (PPI) networks analysis of significant risk alleles to identify the key pathophysiological pathways in the pathogenesis of AAV. The most significant pathways include the processing of antigens via HLA class II, T-cell receptor signaling, and interferon (IFN)-γ mediated signaling pathways (78).
Takayasu Arteritis (TAK)
TAK is a large-vessel vasculitis that predominantly affects young women and can cause organ-threatening ischemia, such as pulseless limbs, aortic regurgitation, pulmonary infarct, and kidney failure by occluding the aorta and/or its branches. TAK is seen all over the world, but Eastern Asia, especially Japan, has the highest incidence. The etiology of TAK is elusive, but genetic predisposition is likely. Corticosteroids, anti-tumor necrosis factor (TNF)-α, anti-IL-6, and abatacept are used for induction of remission and with aim to prevent relapse.
GWAS have shown that HLA-B/MICA, HLA-DQB1/HLA-DRB1, FCGR2A/FCGR3A, IL12B, RPS9/LILRB3, IL6, PTK2B, LILRA3/LILRB2, DUSP22, KLHL33, and HSPA6/FCGR3A and the intergenic locus on chromosomes 21q.22, MICB, and HLA-B*52 are susceptibility gene loci in TAK (38–41). Risk alleles in HLA-B/MICA and HLA-DQB1/HLA-DRB1 were identified in Turkish and European cohorts of patients. Other notable associations are in PSMG1, IL12, and IL23 gene loci but not at the level of genome-wide significance (39). HLA-B*52 allele is related to poor prognosis in TAK (79, 80). Risk alleles in the RPS9/LILRB3 locus and IL6 may have immunoregulatory effects (38). The disease risk variant in RPS9/LILRB3, rs11666543 (p = 2.34 × 10−8), correlates with reduced mRNA expression of the inhibitory leukocyte immunoglobulin-like receptor gene LILRB3. One study proposed that risk alleles in the IL6 gene and serum level of IL-6 can be used to stratify patients for anti-IL6 treatment (81). Another risk allele, rs6871626, in IL12B is shown to influence the expression of IL-12p70 and IL-12p40 genes, resulting in enhanced IL-12 signaling that might further contribute to the pathophysiology of TAK (82). Terao et al. showed that interaction of HLA-B*52 and LILRB1 may be responsible for activation of natural killer (NK) cells by a way of suppression of inhibitory pathways (41).
FCGR2A/FCGR3A is a common susceptibility gene locus for TAK and KD. The Fc-gamma receptors (FcγRs) are responsible for interactive relationship between adaptive and innate immune systems. All immune cells possess these receptors. FcγRs can be classified into one high-affinity receptor (FcγRI) and five low-affinity FcγRs (the different isoforms of FcγRII and FcγRIII) (83). Genetic variants of these receptors have been associated with susceptibility to autoimmune, autoinflammatory, and infectious diseases and response to immunotherapy in cancer patients (6, 84–91). Nagelkerke et al. (51) showed that activation of IgG receptors on monocytes and neutrophils plays a role in the pathophysiology of KD. Inhibition of this activation may be a targeted treatment for patients who carry the FCGR2A/FCGR3A risk alleles.
Immunoglobulin A Vasculitis/Henoch-Schönlein Purpura (IgAV/HSP)
IgAV/HSP is a small-vessel vasculitis of the skin, kidney, gastrointestinal tract, and joints. IgAV/HSP has typically a good prognosis because of the self-limiting disease course; however, renal and gastrointestinal involvement can cause morbidity. IgAV/HSP is the most common childhood vasculitis. Diagnosis of IgAV/HSP is based on clinical features; however, skin and kidney biopsy are useful for diagnosis in selected cases.
The first GWAS of IgAV/HSP revealed that the HLA class II region (mainly HLA-DRB1*01 allele) in the European population is associated with the risk for IgAV/HSP, although this finding did not reach genome-wide significance (42). A meta-analysis confirmed that genetic susceptibility to IgAV/HSP is associated with HLA-DRB1*01 and HLA-DRB1*07 variants and suggested that IgAV may be an HLA class II disease (92). Immunoglobulin A vasculitis with nephritis (IgAVN) and IgA nephropathy (IgAN; also known as Berger's disease) are considered related diseases. The chromosome 1q32 locus that contains the complement factor H (CFH) was identified as an IgAN-susceptible locus. Jia et al. showed the contribution of rs6677604 allele in CFH to the regulation of the complement activation in both IgAN and IgAVN (93).
Giant Cell Arteritis (GCA)
GCA is a systemic vasculitis characterized by granulomatous inflammation of medium- and large-sized vessels and with particular predilection for the aorta and its extracranial branches.
GCA is the most common vasculitis in the elderly (mostly women) population from western countries (94). GCA can cause headaches, visual loss, jaw claudication, painful scalp, impairment of limb arteries, systemic inflammation, and polymyalgia rheumatica. GCA is a potentially devastating disease, with up to 25% of patients developing aneurysmal disease and stroke. First-line treatment of GCA is a high-dose glucocorticoid (GC), and it should be maintained until symptoms are resolved.
GC resistance and adverse effects are main concerns for treatment of patients with GCA (95). Other agents, such as methotrexate and TNF inhibitors, have been used with limited or no evidence of benefit. Recent studies implicated important roles for TH1- and TH17-driven inflammatory cascades. Results from new therapies with abatacept and ustekinumab have also been promising.
In 2017, the first GWAS in GCA identified the HLA-DRB1*04 as the strongest risk allele (43). This finding implies that GCA is a predominantly HLA class II disease and is distinct from the other large-vessel vasculitis, TAK, which is considered an HLA class I disease. Carmona et al. (43) reported susceptibility alleles in plasminogen (PLG) and prolyl 4-hydroxylase subunit alpha 2 (P4HA2) genes in 10 different populations of European ancestry at a genome-wide level of significance (rs4252134, p = 1.23 × 10−10, OR = 1.28; and rs128738, p = 4.60 × 10−9, OR = 1.32) (43). Both PLG and P4HA2 play a role in the regulation of angiogenesis and vascular remodeling. The plasminogen system is known to regulate several pathways possibly related to the GCA pathogenesis, such as lymphocyte recruitment and production of TNF-α and IL-6 cytokines (96, 97). The P4HA2 gene is a hypoxia-responsive gene with a role in collagen biosynthesis and composition of ECM. Hypoxia-inducible factor-1 (HIF-1) regulates the expression of P4HA2 and other genes, such as VEGF, IL6, and MMP9 (98).
Epigenetics And Epigenome-Wide Association Studies (Ewas) In Primary Systemic Vasculitides
Epigenetic mechanisms including DNA methylation, histone modifications, and non-coding RNAs regulate gene expression, cellular development, differentiation, and activity (99). Epigenomic alterations are not the result of genetic mutations, and they are reversible; however, they can act as integrators of genetic and environmental disease risk factors. Recent work has suggested that epigenome modifications, for example, histone modifications and micro RNA (miRNA) expression, may be used as a biomarker for disease activity and for monitoring disease progression (100). EWAS aim to identify epigenetic variations associated with disease, for example, differentially methylated CpG sites, by performing a hypothesis-free testing across the whole genome (101). Thus far, EWAS have mainly investigated DNA methylation and its association with human diseases. In general, hypermethylation of promoters typically acts to repress gene transcription.
Kawasaki Disease
FCGR2A (IgG receptor gene) encodes the low-affinity immunoglobulin gamma Fc region receptor II-a protein that is expressed on the surface of macrophages, neutrophils, monocytes, and dendritic cells. This protein leads to increased phagocytosis and production of inflammatory mediators. Genetic variants in FCGR2A gene have been linked to a genetic risk for KD (100, 102). The first study to examine the DNA methylation array in KD showed that CpG sites within the promoter region of FCGR2A were hypomethylated in whole blood cells from KD patients compared with controls, and especially in patients resistant to IVIG treatment (103). Li et al. demonstrated that DNA methylation patterns changed after IVIG treatment in KD, including reversal of the disease-associated hypomethylation in FCGR2A (104). Furthermore, interaction of FCGR2A and Toll-like receptors (TLR) can induce a proinflammatory response (105, 106). DNA hypomethylation on Toll-like receptors (TLRs) was detected in KD patients, and the methylation level of the TLRs genes was rescued 3 weeks post-IVIG therapy (107).
FoxP3 acts as both transcriptional activator and repressor in regulatory T cells (Tregs), and FoxP3-dependent transcriptional program is epigenetically controlled by histone modifications at its binding site (108). FoxP3 binds to ~700 genes and a number of miRNAs, including miRNA-155, which upregulation likely contributes to proliferative potential of TR cells. FoxP3 expression in Tregs of patients with KD is affected by miR-155 and miR-31. In patients with acute KD, decrease in FoxP3+ Treg might be associated with reduced expression of miR-155 and overexpression of miR-31. These effects were partially reversed following the IVIG treatment (109).
Huang et al. (110) showed that the expression of a neutrophil surface molecule, CD177, is higher during acute stage of KD and correlates with epigenetic hypomethylation. They observed that the CD177 mRNA level diminished sharply after IVIG treatment and were higher in patients with atypical presentation or KD or IVIG-resistant patients.
Chang et al. (111) recently published that gene expression of PHLPP1 and MAPK14 is significantly high in KD and is influenced by methylation. They proposed that hypomethylation status and upregulated expressions of MAPK14 and PHLPP1 could be candidate biomarkers for the diagnosis of KD.
Hepcidin, encoded by the HAMP gene, has a main role in the pathogenesis of inflammation-associated anemia. Hepcidin levels are high in KD patients, and they decrease after IVIG treatment. Huang et al. demonstrated that KD patients had a considerably higher epigenetic hypomethylation of HAMP promoter than controls, which was reversed following therapy with IVIG. They proposed that HAMP promoter hypomethylation and increased hepcidin levels may serve as a biomarker of KD (112). Huang et al. (113) performed the first genome-wide DNA methylation and gene expression assays in blood samples of KD patients. They found that specific CpG markers were hypomethylated in samples from patients with active disease compared to controls and hypermethylated in samples from patients in convalescent phase compared with samples from patients in acute phase. In particular, methylation changes of CpG markers correlated with the expression S100A genes. S100A8 and S100A9 are inflammatory biomarkers that are usually highly expressed in acute and chronic inflammation. Using in vitro model, they showed that S100A proteins enhanced leukocyte transendothelial migration of neutrophils. In summary, this study implicates the role of S100A proteins in the pathogenesis of KD (113).
Behçet's Disease
The first EWAS in BD showed that CD4+ T cells and monocytes extracted from the peripheral blood of BD patients were hypomethylated in genes associated with cytoskeletal remodeling, cell migration, and tissue invasion, such as RAC1, RGS14, and FSCN2. Some methylation deficiencies return to normal level during disease remission. These findings suggest that epigenetic modifications of cytoskeleton-related genes are important in the pathogenesis of BD (114). Hypermethylation of the IL-4, TGF-β, and GATA binding protein 3 (GATA3) in CD4+ cells was noted in patients with active BD, but this may not be disease-specific (115, 116). Conversely, treatment with corticosteroids and cyclosporine (CsA) has beneficial effects by decreasing the methylation level of TGF-β and GATA3 (115). A systematic analysis of miRNA expression profiles and pathway analyses in BD identified a specific miRNA signatures in active disease that regulated the IFNγ, TNF, and VEGF-VEGFR signaling cascades (117). Woo et al. (118) observed differential expressions of miRNAs miR-638 and miR-4488 to be associated with elevated production of IL-6. Activation of the Notch pathway in active BD disease and its association with decreased expression of mir-23 and higher Th17 response have been observed (119). Two functional variants, rs2910164 (miR-146a) and rs11614913 (miR-196a2; Ets-1), confer the risk for BD in Han-Chinese by regulating production of proinflammatory cytokines (120, 121).
The differentially expressed genes identified by EWAS might be good candidate biomarkers for monitoring the disease activity and might represent promising candidates for design of novel therapeutic strategies in BD.
ANCA-Associated Vasculitis
Normally, the expression of MPO and PRTN3 genes (encoding MPO and PR3 autoantigenes) in neutrophils occurs only in the early stages of cell maturation; however, MPO and PRTN3 are found continuously expressed in neutrophils and monocytes of patients with AAV (122). This deregulation in gene expression is postulated to be related to epigenetic mechanisms (123–125). Jones et al. (124) showed that the gene-specific DNA methylation changes may regulate autoantigen expression, and they correlate with disease activity in AAV. In remission, DNA methylation is generally increased. Patients with active disease had hypomethylation and increased expression of MPO and PRTN3. The findings suggest that the reactivation of once-silenced genes can lead to increased antigen availability. Patients with decreased DNA methylation at the PRTN3 promoter have a significantly greater risk of relapse. Therefore, fluctuations in the DNA methylation of the PRTN3 promoter may be associated with stable remission. Methylation status may prove to be a potential biomarker for prognosis in AVV patients (124).
Immunoglobulin A Vasculitis/Henoch-Schönlein Purpura
Luo et al. (126) observed that global histone H3 acetylation and H3K4 methylation are increased in peripheral blood mononuclear cells (PBMCs) isolated from IgAV patients. These epigenetic modifications had positive correlations with disease activity and were more common in IgAV patients with renal involvement compared with IgAV patients without renal involvement and healthy controls. They observed increased mRNA expression of GATA3, IL-4, IL-6, and IL-13 genes in IgAV patients. Serum protein levels of IL-4, IL-6, and IL-13 were significantly increased in HSP with kidney damage patients compared to healthy controls. Abnormal histone modifications of transcription factors such as GATA3 may lead to the Th1/Th2 cytokine imbalance in HSP and other immune diseases (126).
Giant Cell Arteritis
Only two epigenetic studies evaluated DNA methylation status and miRNA expression in temporal artery biopsies (TABs) of patients with GCA (127, 128). Coit et al. (127) identified 1,555 hypomethylated CpG sites in 853 genes by comparing 12 patients and 12 healthy age-, gender-, and ancrestry-matched controls. DNA was extracted from the affected temporal artery tissues in patients with GCA and from histologically confirmed normal arteries in controls.
Most of these genes have roles in T-cell activation and differentiation pathways, especially Th1 and Th17 cells. TNF, LTA, LTB, CCR7, RUNX3, CD6, CD40LG, IL2, IL6, NLRP1, IL1B, IL18, IL21, IL23R, and IFNG were proinflammatory hypomethylated genes that were found in this study. Interestingly, the significant hypomethylation of genes in the calcineurin (CaN)/NFAT pathway was shown within the temporal artery of GCA patients.
Increased activity in NFAT is one of the key factors in production of proinflammatory cytokines. This study provided evidence for use of NFAT inhibitors (e.g., dipyridamole) in the treatment of GCA (127, 129).
Croci et al. showed that miR-146b-5p,−146a,−21,−150,−155, and−299-5p are expressed at higher levels in affected vessels of TAB-positive GCA patients compared to non-inflamed TABs from GCA patients and to non-inflamed TABs from non-GCA patients. Interestingly, these miRNAs were mainly deregulated at the tissue level, whereas no difference in their expression was observed in peripheral blood mononuclear and polymorphonuclear cells from these three groups of patients and controls. Especially, miR-146b-5p expression was the most promising diagnostic biomarker for the presence of inflammation in TABs of GCA patients and the follow up of the disease activity. Further research is necessary to optimize their use in medical practice (128).
Extracellular Vesicles In Primary Systemic Vasculitides
EVs were defined in 1967 as “dust” from platelets (130). More recently, it has been shown that EVs are membrane vesicles that are released by almost all eukaryotic cells during cell activation and programmed cell death (131–134). Heterogeneity of EVs is essential. They are classified into three groups with regard to biological features and their size: exosomes, microvesicles (MVs), and apoptotic bodies (132, 133, 135, 136). EVs are responsible for immune regulation, cell-to-cell interaction, and signal transmission by transporting bioactive molecules including proteins and lipids, DNA, and various RNAs, such as mRNAs, small-interfering RNAs (siRNAs), microRNAs (miRNAs), and long non-coding RNAs (lncRNAs) and other molecules produced by cells (132, 137–139). EVs can be detected in many organs, tissues, and all body fluids, such as urine, blood, and synovial fluid at low levels in physiological conditions (140, 141). The increased levels of EVs are noted in cardiovascular disease, cancer, and pathological conditions that are associated with vasculitis: inflammation, autoimmunity, endothelial damage, angiogenesis, procoagulation, and intimal hyperplasia (131–133, 136, 140–142). EVs are known to be released from injured endothelial cells and are found increased in many diseases associated with endothelial dysfunction. It is thought that EVs from platelets, lymphocytes, and monocyte/macrophages contribute to the pathogenesis of systemic vasculitis (143–170) (Table 3).
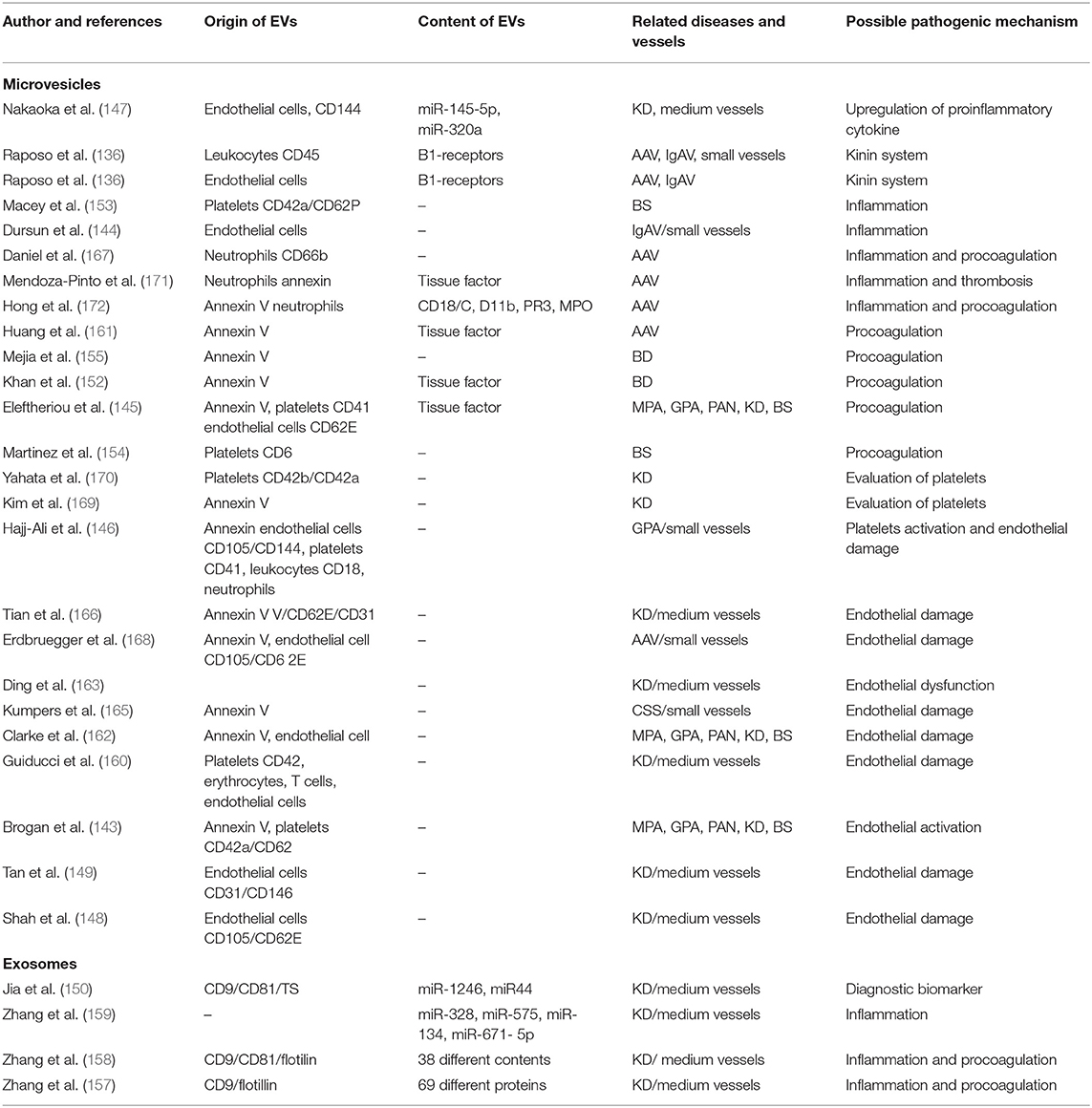
Table 3. The role of extracellular vesicles in vasculitis. This table is adapted from Wu et al. (133).
In 2004, Brogan et al. (143) reported that the endothelial microparticles expressing E-selectin-positive or CD105 and the platelet MVs expressing CD42a were significantly increased in patients with active vasculitis compared to controls or children with inactive vasculitis or other febrile illnesses. They found that endothelial microparticle levels correlate with the Birmingham Vasculitis Activity Score (BVAS) and acute-phase reactant levels (143).
Activation of the contact system has proinflammatory and vasoactive properties. The contact system triggers the kallikrein-kinin cascade releasing bradykinin from high-molecular-weight kininogen. Bradykinin concentrations were higher in the patients' plasma than in plasma from controls, and kinins were present in lesional biopsy tissues of kidney and skin of children with PSVs (173, 174). The kinin B1 receptor is induced by inflammatory stimuli, particularly in response to the cytokine IL-1β, and plays a major role in neutrophil recruitment. Kahn et al. (151) found that leukocyte-derived MVs transfer functional kinin B1-receptors and in this way may further promote kinin-mediated inflammation in vasculitis. Mossberg et al. (156) showed that patients with acute vasculitis had significantly higher levels of circulating endothelial MVs and more MVs carrying B1-receptors. These MVs can induce the inflammation by increasing neutrophil chemotaxis, whereas addition of a C1-inhibitor decreased the release of B1R+MVs. Kinin-B1 receptor activation of CXCL5 can further amplify neutrophil chemotaxis and chronic inflammation (175). Exploring inhibition of the kallikrein-kinin pathway is potentially a novel target for reducing neutrophil-mediated inflammation in PSVs.
Kawasaki Disease
Endothelial cell-derived MVs were detected in higher levels in children with KD (147–149, 163, 166) and HSP (144). These MVs may serve as biomarkers to predict coronary artery aneurysms in KD and to detect subclinical inflammation in HSP (144, 148).
The role of platelet activation dynamics in acute-phase KD patients was explored by assaying platelet-derived MVs (PDMVs). Prior to aspirin treatment, PDMV level was significantly higher in the acute-phase KD patients in comparison to patients with common febrile diseases. Guiducci et al. observed that MVs derived from platelets, endothelial cells, erythrocytes, and T cells are significantly elevated in plasma samples of patients with KD. Endothelial and T cells were the major source of MVs, and the levels were reduced by IVIG treatment (160). Platelet activation is important in the pathogenesis of KD, thus PDMVs may serve as a biomarker to evaluate the antiplatelet therapy response in KD (169, 170).
Measuring microRNAs (miR-4436b-5p, miR-1246, miR-671-5p, and miR-197-3p) in serum exosomes has been proposed as a diagnostic biomarker for the prediction of IVIG response in KD (149, 159). Serum exosomal microRNAs may have a role in the pathogenesis of KD by regulating the expression of inflammatory genes (CX chemokine receptor types 1 and 2, IL-8, and its receptor) in mononuclear cells (159). Serum exosomes from patients with KD contain many proteins, such as apolipoprotein A-IV, insulin-like growth factor-binding protein complex, acid-labile subunit and complement C3, and their proteomic profile correlates with IVIG therapy (157). Zhang et al. performed proteomic analyses of serum exosomes from KD patients with coronary artery dilatation. They found 38 differentially expressed proteins, and the majority are involved in inflammatory and coagulation pathways (158). More recent proteomic analysis validated TN, APOA4, LRG1, and RBP4 proteins as differentially expressed in patients with coronary artery aneurysms (CAA) (176). These finding provide additional insights in the pathogenesis of CAA in patients with KD.
ANCA-Associated Vasculitis
Endothelium-derived MVs have been reported in active AAV patients (168). Plasma levels of MVs derived from platelet and neutrophil are high in acute-phase vasculitis (167). The neutrophil-derived MVs cause increased production of proinflammatory cytokines IL-6 and IL-8, increased expression of intercellular adhesion molecule-1, and production of reactive oxygen radicals by binding to endothelial cells in a CD18-dependent manner (172). Furthermore, endothelial MVs carry PR3 and MPO and may contribute to the extensive endothelial damage and inflammation seen in AAV (177, 178).
Thromboembolic disease complicating primary systemic vasculitis is associated with significant morbidity and mortality. The mechanisms of hypercoagulability in PSV remain poorly defined. Several studies attempted to identify risk factors of thrombosis to stratify patients who could benefit from prophylaxis with antiplatelet or anticoagulant agents. One study showed that stimulation with ANCAs causes C5a-primed neutrophils to release neutrophil tissue factor (TF)-expressing MVs and neutrophil extracellular traps (NETs) that might promote hypercoagulability in AAV (161). Eleftheriou at al. have demonstrated that thromboembolic disease in children with systemic vasculitis is linked to increased level of MVs-mediated thrombin (145).
Behçet's Disease
Thrombosis is common in BD patients, and there is a need for better assessment of risk factors. BD patients with a history of thrombosis have high serum levels of MVs expressing tissue factor (TF). The ratio of TF pathway inhibitor (TFPI)-positive MVs to TF-positive MVs was significantly lower in patients with thrombosis (152). In contrast, platelet-derived MVs and procoagulant MVs did not differentiate between BD patients with or without thrombosis (155). Macey et al. (153) showed that high-level platelet-derived CD62P+ MVs correlate with active disease in patients in a younger age group, whereas lower levels of MVs correlate with decreased disease activity in patients older than 50 years.
Insights From Studies of Monogenic Systemic Autoinflammatory Diseases
Rare Mendelian diseases of systemic inflammation often present with severe-like phenotypes of polygenic diseases and can share underlying biochemical pathways with more common rheumatic diseases. Identification of patients with monogenic disease can point to genes and pathways that could be investigated in patients with polygenic disorders. For instance, polyarteritis nodosa (PAN) can present either as early-onset monogenic or late-onset polygenic disease. PAN is a systemic necrotizing vasculitis that predominantly affects medium-sized arteries, causing tissue ischemia and organ damage. The disease commonly affects the skin, gastrointestinal tract, and kidneys, and patients are at an increased risk of stroke. Most cases of PAN occur in the fourth or fifth decade, and men are more likely to be affected. A subset of patients with childhood-onset PAN are found to carry biallelic loss of function mutations in adenosine deaminase 2 gene. This disease is named deficiency of ADA2 (DADA2)(179, 180). Among many features of DADA2, subcortical ischemic strokes, hypertension, aneurysms, renal infarcts, and peripheral amputations have been reported in up to 50% of patients. DADA2 patients are highly responsive to treatment with TNF inhibitors (181). Although pathogenic variants in the ADA2 gene may not account for a large number of sporadic adult-onset PAN patients, TNF inhibitors should be explored in the treatment of those patients as well (181).
Inflammasomopathies
Inflammasomopathies are rare monogenic autoinflammatory diseases caused by gain-of -function mutations in the multiprotein complexes termed the inflammasome. The inflammasome functions as cytosolic pathogen and danger-associated molecular patterns (PAMPs/DAMPs) recognition receptors (PRRs). The core of inflammasome is one of the nucleotide-binding domain leucine-rich repeat (NLR) proteins (NLRP1, NLRP3, AIM2, NLRC4) or pyrin. Upon stimulation, the inflammasome interacts with an inflammasome adaptor protein, apoptosis-associated speck-like protein with a caspase recruitment domain (ASC), or with pro-caspase 1 to form platform for caspase-1-mediated production of IL-1β and IL-18 cytokines. The best known inflammasomopathies are familial Mediterranean fever (FMF) and cryopyrinopathities (CAPS). FMF is caused by recessively inherited hypomorphic mutations in pyrin, whereas CAPS is linked to dominantly inherited gain-of-function mutations in NLRP3. The net effect of these pathogenic variants is increased production of IL-1β cytokine and systemic inflammation. FMF is a common disease in many Eastern Mediterranean countries where the frequency of pathogenic mutations is very high (182–205). Although vasculitis is not a primary feature of FMF, IgAV and PAN have been reported in about 3 and 1% of FMF patients, respectively (206). FMF-associated mutations predispose to the development of BD in the Turkish population (207–210). Small and medium vessel skin vasculitis and CNS vasculitis were reported in other inflammasomopathies, including CAPS and other pyrin-mediated autoinflammatory diseases mevalonate kinase deficiency/hyperimmunoglobulin D (MKD/HIDS); pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA); and pyoderma gangrenosum, acne, hidradenitis suppurativa (PASH) syndromes (211–221). NLRP3 inflammasome activation was demonstrated in two Kawasaki mouse models (222, 223). Underlying mechanisms of vasculitis in inflammasomopathies are unclear, although it is assumed that high systemic IL-1β production may cause endothelial cell inflammation and damage. Patients with BD may respond to colchicine and anti-IL1 therapies like patients with inflammasomopathies (224).
Relopathies
Relopathies are NF kappa-B (NF-κB)-mediated monogenic systemic autoinflammatory diseases driven by multiple cytokines including TNF and IL-6 (225). They result from pathogenic variants in proteins that regulate posttranslational ubiquitin modifications in the NF-κB pathway. Up to now, haploinsufficency of A20 (HA20) and OTULIN deficiency have been defined as relopathies (226). HA20 is a dominantly inherited disease caused by heterozygous loss-of-function mutations in TNFAIP3, which encodes the K-63 deubiquitinase protein A20 (227). Common variants in TNFAIP3 have been linked to BD in the Chinese-Han population (228). Clinically, HA20 resembles early-onset BD. Two patients with HA20 were diagnosed with CNS vasculitis based on brain imaging and a frontal lobe punctate (229). OTULIN deficiency is caused by recessively inherited loss-of-function mutations in the enzyme that regulates linear deubiquitination. There are only few patients reported, and they present with early-onset severe systemic inflammation. One patient was described with vasculitis of small and medium vessels on skin biopsy (230). In most patients with HA20 and OTULIN deficiency, anti-cytokine therapies targeting TNF and IL-1 are efficient in suppressing the disease activity. Ubiquitin pathway has been implicated in the pathogenesis of BD by multiple association studies. The rs9517723 variant in the 3' region of ubiquitin-associated domain containing ubiquitin-associated domain containing 2 (UBAC2) gene is significantly associated with ocular and central nervous system (CNS) lesions under the recessive model (32, 231). UBAC2 encodes a protein that plays a role in ubiquitination and proteasomal degradation. Homozygous risk allele (TT) of the rs9517723 correlates with increased UBAC2 expression. BD has been also associated with other ubiquitination pathway-related genes, including ubiquitin associated and SH3 domain containing B (UBASH3B) and small ubiquitin-like modifier 4 (SUMO4) (232, 233).
Interferonopathies
Type I interferonopathies consist of Aicardi–Goutières syndrome (AGS), STING-associated vasculopathy with onset in infancy (SAVI), chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome, and COPA syndrome (226). Intracerebral vasculitis is common in patients with AGS (234). High levels of IFN activities were demonstrated in the cerebrospinal fluid and serum of AGS patients (234). In SAVI, gain-of-function mutations in the TMEM173 gene lead to a constitutively active STING protein, and a high expression of type I IFN-induced genes (235, 236). Clinically, the SAVI phenotype mimics the AAV with cutaneous rashes, interstitial lung disease, peripheral ulcerations/gangrene, and ANCA positivity in some patients (237). Patients with CANDLE have dysfunction in the protesome-mediated degradation pathway. Skin biopsies from CANDLE patients showed evidence for perivascular infiltration prominent for myeloid cells and leukocytoclasis but not for the typical vasculitis with fibrinoid necrosis of the vessel walls (238). JAK1/2 inhibition with baricitinib has been successful in treating clinical disease manifestations and in suppression of the IFN signature (239). The role of the IFN pathway in the pathogenesis of polygenic/complex vasculitides has been recently postulated by analysis of data from multiple GWAS (78).
Reports of vasculitis in other rare autoinflammatory diseases are scarce. One patient with the deficiency of IL-1 receptor antagonist (DIRA) had histopathological evidence of vasculitis in the connective tissue and cerebral vasculitis or vasculopathy was detected on magnetic resonance imaging (240). IL-1Ra-deficient mice frequently develop aortitis at the root of the aorta, which can be suppressed by a cross with TNF-α-deficient mice. Bone marrow transplantation of T cells from IL-1Ra−/− mice induced aortitis in recipient nu/nu mice or in irradiated wild-type recipient mice (241). This study demonstrates that IL-1Ra deficiency in T cells is responsible for the development of aortitis.
Expert Commentary
i) The main limitations of current genetic datasets in PSVs are the lack of multiple meta-analyses of GWAS and inability to confirm functional significance of identified risk alleles. Validation of disease-associated variants in cohorts of patients of different ancestry would give more creditability to the reported findings; however, some genetic variants may not be present in all populations.
ii) As epigenetic modifications evolve and change over the life span, the data from EWAS will require validation in longitudinal cohort studies that are costly to maintain. Epigenomic investigations maybe accomplished in a shorter time period if they focus on studies in monozygotic twin pairs (MZ pairs) or if they would investigate differences in young vs. old MZ pairs. In addition, it would be interesting to explore cell-specific epigenomic modifications.
iii) Use of extracellular vesicles (MVs, PDMVs, EMVs) as potential biomarkers of disease activity in patients with primary systemic vasculitis is important for diagnostic and treatment monitoring. At present time, there are no standardized methods for the sampling process and detection of EVs.
5-Year Forward View
Multicenter collaborations and studies in large cohorts of patients would increase the power of detection and confirmation of PSV disease-associated variants. These meta studies would also help estimate a size effect of identified risk alleles, which is important for calculation of genetic risk score (GRS) and genetic counseling. Considering a significant decrease in the cost of whole exome and whole genome sequencing (WES/WGS), these new sequencing technologies should capture rare germ-line variants, structural variants, and mosaic mutations that may explain the missing heritability in these diseases. At this time, it is still unclear whether SNPs captured by GWAS are true disease-associated variants or in linkage disequilibrium with a risk allele. Performing WGS of patient cohorts would avoid a need for imputation studies. Effects of most common disease-associated variants on protein function are unclear, in particular for non-coding variants. Disease-associated common variants may have very small effect sizes but could lead to important and targetable pathways. Studies in cell culture systems and model organisms and experiments in primary patient cells are needed to answer this question.
Conclusion
Management of PSVs and other human diseases is beginning to be more directed with increased use of molecular data to improve diagnosis and to guide optimal treatment options. This new clinical paradigm has premise to apply knowledge of genomic, proteomic, and metabolic variants and epigenomic biomarkers to generate “omic” profiles. The translation into clinical practice can be achieved by integrating the “omic” information into a unique algorithm that will be able to make accurate diagnosis and to optimize therapeutic decisions to maximize benefit and minimize harm. Proper implementation of PM will require multidisciplinary teams of clinicians, bioinformaticians, geneticists, genetic counselors, and research scientists that can address multiple challenges in interpretation and integration of the multi-omics results. Along these lines, successful transition toward personalized care will also necessitate updating medical curriculums to facilitate training of a new generation of “omic”-literate physicians.
Author Contributions
ED and ZA drafted the manuscript. MR and RB revised the manuscript and designed the tables. IA revised and finalized the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MR was the recipient of ARG-ITALIA Onlus (http://www.argitalia.it/) bursary for international research fellowship.
References
1. Berman JJ. Precision Medicine and the Reinvention of Human Disease. London: Elsevier (2018). doi: 10.1016/B978-0-12-814393-3.00008-1
2. Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthr Rheumat. (2013) 65:1–11. doi: 10.1002/art.37715
3. Institute. NNHGR. Genome-Wide Association Studies Fact Sheet. (2019). Available online at: https://www.genome.gov/about-genomics/fact-sheets/Genome-Wide-Association-Studies-Fact-Sheet (accessed May 01, 2019).
4. Gutierrez-Arcelus M, Rich SS, Raychaudhuri S. Autoimmune diseases - connecting risk alleles with molecular traits of the immune system. Nat Rev Genet. (2016) 17:160–74. doi: 10.1038/nrg.2015.33
5. Burgner D, Davila S, Breunis WB, Ng SB, Li Y, Bonnard C, et al. A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. (2009) 5:e1000319. doi: 10.1371/journal.pgen.1000319
6. Khor CC, Davila S, Breunis WB, Lee YC, Shimizu C, Wright VJ, et al. Genome-wide association study identifies FCGR2A as a susceptibility locus for Kawasaki disease. Nat Genet. (2011) 43:1241–6. doi: 10.1038/ng.981
7. Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. A genome-wide association analysis identifies NMNAT2 and HCP5 as susceptibility loci for Kawasaki disease. J Hum Genet. (2017) 62:1023–9. doi: 10.1038/jhg.2017.87
8. Kuo HC, Li SC, Guo MM, Huang YH, Yu HR, Huang FC, et al. Genome-wide association study identifies novel susceptibility genes associated with coronary artery aneurysm formation in Kawasaki disease. PLoS ONE. (2016) 11:e0154943. doi: 10.1371/journal.pone.0154943
9. Kwon YC, Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, et al. Male-specific association of the FCGR2A His167Arg polymorphism with Kawasaki disease. PLoS ONE. (2017) 12:e0184248. doi: 10.1371/journal.pone.0184248
10. Lee YC, Kuo HC, Chang JS, Chang LY, Huang LM, Chen MR, et al. Two new susceptibility loci for Kawasaki disease identified through genome-wide association analysis. Nat Genet. (2012) 44:522–5. doi: 10.1038/ng.2227
11. Onouchi Y, Ozaki K, Burns JC, Shimizu C, Terai M, Hamada H, et al. A genome-wide association study identifies three new risk loci for Kawasaki disease. Nat Genet. (2012) 44:517–21. doi: 10.1038/ng.2220
12. Shimizu C, Eleftherohorinou H, Wright VJ, Kim J, Alphonse MP, Perry JC, et al. Genetic variation in the SLC8A1 calcium signaling pathway is associated with susceptibility to Kawasaki disease and coronary artery abnormalities. Circ Cardiovasc Genet. (2016) 9:559–68. doi: 10.1161/CIRCGENETICS.116.001533
13. Tsai FJ, Lee YC, Chang JS, Huang LM, Huang FY, Chiu NC, et al. Identification of novel susceptibility Loci for Kawasaki disease in a Han chinese population by a genome-wide association study. PLoS ONE. (2011) 6:e16853. doi: 10.1371/journal.pone.0016853
14. Kim HJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. Identification of LEF1 as a susceptibility locus for Kawasaki disease in patients younger than 6 months of age. Genomics Inform. (2018) 16:36–41. doi: 10.5808/GI.2018.16.2.36
15. Bae Y, Shin D, Nam J, Lee HR, Kim JS, Kim KY, et al. Variants in the gene EBF2 are associated with Kawasaki disease in a korean population. Yonsei Med J. (2018) 59:519–23. doi: 10.3349/ymj.2018.59.4.519
16. Oyamada J, Shimizu C, Kim J, Williams MR, Png E, Hibberd ML, et al. Bifid T waves on the ECG and genetic variation in calcium channel voltage-dependent beta 2 subunit gene (CACNB2) in acute Kawasaki disease. Congenit Heart Dis. (2019) 14:213–20. doi: 10.1111/chd.12696
17. Kim JJ, Hong YM, Sohn S, Jang GY, Ha KS, Yun SW, et al. A genome-wide association analysis reveals 1p31 and 2p13.3 as susceptibility loci for Kawasaki disease. Hum Genet. (2011) 129:487–95. doi: 10.1007/s00439-010-0937-x
18. Kim JJ, Park YM, Yoon D, Lee KY, Seob Song M, Doo Lee H, et al. Identification of KCNN2 as a susceptibility locus for coronary artery aneurysms in Kawasaki disease using genome-wide association analysis. J Hum Genet. (2013) 58:521–5. doi: 10.1038/jhg.2013.43
19. Lin MT, Hsu CL, Taiwan Pediatric CVG, Chen PL, Yang WS, Wang JK, et al. A genome-wide association analysis identifies novel susceptibility loci for coronary arterial lesions in patients with Kawasaki disease. Transl Res. (2013) 161:513–5. doi: 10.1016/j.trsl.2013.02.002
20. Lin YJ, Chang JS, Liu X, Tsang H, Chien WK, Chen JH, et al. Genetic variants in PLCB4/PLCB1 as susceptibility loci for coronary artery aneurysm formation in Kawasaki disease in Han Chinese in Taiwan. Sci Rep. (2015) 5:14762. doi: 10.1038/srep14762
21. Liu Y, Fu L, Pi L, Che D, Xu Y, Zheng H, et al. An angiotensinogen gene polymorphism (rs5050) is associated with the risk of coronary artery aneurysm in southern chinese children with Kawasaki disease. Dis Markers. (2019) 2019:2849695. doi: 10.1155/2019/2849695
22. Kwon YC, Kim JJ, Yu JJ, Yun SW, Yoon KL, Lee KY, et al. Identification of the TIFAB gene as a susceptibility locus for coronary artery aneurysm in patients with Kawasaki disease. Pediatr Cardiol. (2019) 40:483–8. doi: 10.1007/s00246-018-1992-7
23. Kwon YC, Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, et al. BCL2L11 is associated with Kawasaki disease in intravenous immunoglobulin responder patients. Circ Genom Precis Med. (2018) 11:e002020. doi: 10.1161/CIRCGEN.117.002020
24. Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. Identification of SAMD9L as a susceptibility locus for intravenous immunoglobulin resistance in Kawasaki disease by genome-wide association analysis. Pharmacogenomics J. (2019). doi: 10.1038/s41397-019-0085-1. [Epub ahead of print].
25. Hou S, Yang Z, Du L, Jiang Z, Shu Q, Chen Y, et al. Identification of a susceptibility locus in STAT4 for Behcet's disease in Han Chinese in a genome-wide association study. Arthritis Rheum. (2012) 64:4104–13. doi: 10.1002/art.37708
26. Kappen JH, Medina-Gomez C, van Hagen PM, Stolk L, Estrada K, Rivadeneira F, et al. Genome-wide association study in an admixed case series reveals IL12A as a new candidate in Behcet disease. PLoS ONE. (2015) 10:e0119085. doi: 10.1371/journal.pone.0119085
27. Kim SW, Jung YS, Ahn JB, Shin ES, Jang HW, Lee HJ, et al. Identification of genetic susceptibility loci for intestinal Behcet's disease. Sci Rep. (2017) 7:39850. doi: 10.1038/srep39850
28. Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Genome-wide association analysis identifies new susceptibility loci for Behcet's disease and epistasis between HLA-B*51 and ERAP1. Nat Genet. (2013) 45:202–7. doi: 10.1038/ng.2520
29. Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, et al. Genome-wide association studies identify IL23R-IL12RB2 and IL10 as Behcet's disease susceptibility loci. Nat Genet. (2010) 42:703–6. doi: 10.1038/ng.624
30. Remmers EF, Cosan F, Kirino Y, Ombrello MJ, Abaci N, Satorius C, et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet's disease. Nature genetics. (2010) 42:698–702. doi: 10.1038/ng.625
31. Xavier JM, Shahram F, Sousa I, Davatchi F, Matos M, Abdollahi BS, et al. FUT2: filling the gap between genes and environment in Behcet's disease? Ann Rheum Dis. (2015) 74:618–24. doi: 10.1136/annrheumdis-2013-204475
32. Fei Y, Webb R, Cobb BL, Direskeneli H, Saruhan-Direskeneli G, Sawalha AH. Identification of novel genetic susceptibility loci for Behcet's disease using a genome-wide association study. Arthritis Res Ther. (2009) 11:R66. doi: 10.1186/ar2695
33. Lee YJ, Horie Y, Wallace GR, Choi YS, Park JA, Choi JY, et al. Genome-wide association study identifies GIMAP as a novel susceptibility locus for Behcet's disease. Ann Rheum Dis. (2013) 72:1510–6. doi: 10.1136/annrheumdis-2011-200288
34. Yu H, Zheng M, Zhang L, Li H, Zhu Y, Cheng L, et al. Identification of susceptibility SNPs in IL10 and IL23R-IL12RB2 for Behcet's disease in Han Chinese. J Allergy Clin Immunol. (2017) 139:621–7. doi: 10.1016/j.jaci.2016.05.024
35. Lyons PA, Rayner TF, Trivedi S, Holle JU, Watts RA, Jayne DR, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med. (2012) 367:214–23.
36. Merkel PA, Xie G, Monach PA, Ji X, Ciavatta DJ, Byun J, et al. Identification of functional and expression polymorphisms associated with risk for antineutrophil cytoplasmic autoantibody-associated vasculitis. Arthritis Rheumatol. (2017) 69:1054–66. doi: 10.1002/art.40034
37. Xie G, Roshandel D, Sherva R, Monach PA, Lu EY, Kung T, et al. Association of granulomatosis with polyangiitis (Wegener's) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis Rheum. (2013) 65:2457–68. doi: 10.1002/art.38036
38. Renauer PA, Saruhan-Direskeneli G, Coit P, Adler A, Aksu K, Keser G, et al. Identification of susceptibility loci in IL6, RPS9/LILRB3, and an intergenic locus on chromosome 21q22 in takayasu arteritis in a genome-wide association study. Arthritis Rheumatol. (2015) 67:1361–8. doi: 10.1002/art.39035
39. Saruhan-Direskeneli G, Hughes T, Aksu K, Keser G, Coit P, Aydin SZ, et al. Identification of multiple genetic susceptibility loci in Takayasu arteritis. Am J Hum Genet. (2013) 93:298–305. 10.1016/j.ajhg.2013.05.026
40. Terao C, Yoshifuji H, Kimura A, Matsumura T, Ohmura K, Takahashi M, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet. (2013) 93:289–97. doi: 10.1016/j.ajhg.2013.05.024
41. Terao C, Yoshifuji H, Matsumura T, Naruse TK, Ishii T, Nakaoka Y, et al. Genetic determinants and an epistasis of LILRA3 and HLA-B*52 in Takayasu arteritis. Proc Natl Acad Sci USA. (2018) 115:13045–50. doi: 10.1073/pnas.1808850115
42. Lopez-Mejias R, Carmona FD, Castaneda S, Genre F, Remuzgo-Martinez S, Sevilla-Perez B, et al. A genome-wide association study suggests the HLA Class II region as the major susceptibility locus for IgA vasculitis. Sci Rep. (2017) 7:5088. doi: 10.1038/s41598-017-03915-2
43. Carmona FD, Vaglio A, Mackie SL, Hernandez-Rodriguez J, Monach PA, Castaneda S, et al. A Genome-wide association study identifies risk alleles in plasminogen and P4HA2 associated with giant cell arteritis. Am J Hum Genet. (2017) 100:64–74. doi: 10.1016/j.ajhg.2016.11.013
44. Chang CJ, Kuo HC, Chang JS, Lee JK, Tsai FJ, Khor CC, et al. Replication and meta-analysis of GWAS identified susceptibility loci in Kawasaki disease confirm the importance of B lymphoid tyrosine kinase (BLK) in disease susceptibility. PLoS ONE. (2013) 8:e72037. doi: 10.1371/journal.pone.0072037
45. Kim JJ, Yun SW, Yu JJ, Yoon KL, Lee KY, Kil HR, et al. Common variants in the CRP promoter are associated with a high C-reactive protein level in Kawasaki disease. Pediatr Cardiol. (2015) 36:438–44. doi: 10.1007/s00246-014-1032-1
46. Lv YW, Wang J, Sun L, Zhang JM, Cao L, Ding YY, et al. Understanding the pathogenesis of Kawasaki disease by network and pathway analysis. Comput Math Methods Med. (2013) 2013:989307. doi: 10.1155/2013/989307
47. Rowley AH, Shulman ST. The Epidemiology and pathogenesis of Kawasaki disease. Front Pediatr. (2018) 6:374. doi: 10.3389/fped.2018.00374
48. Onouchi Y, Fukazawa R, Yamamura K, Suzuki H, Kakimoto N, Suenaga T, et al. Variations in ORAI1 gene associated with Kawasaki disease. PLoS ONE. (2016) 11:e0145486. doi: 10.1371/journal.pone.0145486
49. Hamada H, Suzuki H, Onouchi Y, Ebata R, Terai M, Fuse S, et al. Efficacy of primary treatment with immunoglobulin plus ciclosporin for prevention of coronary artery abnormalities in patients with Kawasaki disease predicted to be at increased risk of non-response to intravenous immunoglobulin (KAICA): a randomised controlled, open-label, blinded-endpoints, phase 3 trial. Lancet. (2019) 393:1128–37. doi: 10.1093/rheumatology/kez063.031
50. Lindquist ME, Hicar MD. B cells and antibodies in Kawasaki disease. Int J Mol Sci. (2019) 20:E1834. doi: 10.3390/ijms20081834
51. Nagelkerke SQ, Tacke CE, Breunis WB, Tanck MWT, Geissler J, Png E, et al. Extensive ethnic variation and linkage disequilibrium at the FCGR2/3 locus: different genetic associations revealed in Kawasaki disease. Front Immunol. (2019) 10:185. doi: 10.3389/fimmu.2019.00185
52. Kuo HC, Wong HS, Chang WP, Chen BK, Wu MS, Yang KD, et al. Prediction for intravenous immunoglobulin resistance by using weighted genetic risk score identified from genome-wide association study in Kawasaki disease. Circ Cardiovasc Genet. (2017) 10:e001625. doi: 10.1161/CIRCGENETICS.116.001625
53. Gul A. Genetics of Behcet's disease: lessons learned from genomewide association studies. Curr Opin Rheumatol. (2014) 26:56–63. doi: 10.1097/BOR.0000000000000003
54. Deng Y, Zhu W, Zhou X. Immune regulatory genes are major genetic factors to behcet disease: systematic review. Open Rheumatol J. (2018) 12:70–85. doi: 10.2174/1874312901812010070
55. Hatemi G, Karatemiz G, Yazici H. Behcet's disease: an MHC-I-opathy? Clin Exp Rheumatol. (2017) 35 Suppl 104:5.
56. Takeuchi M, Ombrello MJ, Kirino Y, Erer B, Tugal-Tutkun I, Seyahi E, et al. A single endoplasmic reticulum aminopeptidase-1 protein allotype is a strong risk factor for Behcet's disease in HLA-B*51 carriers. Ann Rheum Dis. (2016) 75:2208–11. doi: 10.1136/annrheumdis-2015-209059
57. Takeuchi M, Kastner DL, Remmers EF. The immunogenetics of Behcet's disease: A comprehensive review. J Autoimmun. (2015) 64:137–48. doi: 10.1016/j.jaut.2015.08.013
58. Chang HK, Kim JU, Cheon KS, Chung HR, Lee KW, Lee IH. HLA-B51 and its allelic types in association with Behcet's disease and recurrent aphthous stomatitis in Korea. Clin Exp Rheumatol. (2001) 19(5 Suppl. 24):S31–5.
59. Hamzaoui A, Houman MH, Massouadia M, Ben Salem T, Khanfir MS, Ben Ghorbel I, et al. Contribution of Hla-B51 in the susceptibility and specific clinical features of Behcet's disease in Tunisian patients. Eur J Intern Med. (2012) 23:347–9. doi: 10.1016/j.ejim.2011.12.011
60. Maldini C, Lavalley MP, Cheminant M, de Menthon M, Mahr A. Relationships of HLA-B51 or B5 genotype with Behcet's disease clinical characteristics: systematic review and meta-analyses of observational studies. Rheumatology. (2012) 51:887–900. doi: 10.1093/rheumatology/ker428
61. Wallace GR. HLA-B*51 the primary risk in Behcet disease. Proc Natl Acad Sci USA. (2014) 111:8706–7. doi: 10.1073/pnas.1407307111
62. Pepelyayeva Y, Amalfitano A. The role of ERAP1 in autoinflammation and autoimmunity. Hum Immunol. (2019) 80:302–9. doi: 10.1016/j.humimm.2019.02.013
63. Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, et al. Dense genotyping of immune-related loci implicates host responses to microbial exposure in Behcet's disease susceptibility. Nat Genet. (2017) 49:438–43. doi: 10.1038/ng.3786
64. Bakir-Gungor B, Remmers EF, Meguro A, Mizuki N, Kastner DL, Gul A, et al. Identification of possible pathogenic pathways in Behcet's disease using genome-wide association study data from two different populations. Eur J Hum Genet. (2015) 23:678–87. doi: 10.1038/ejhg.2014.158
65. Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, et al. Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll-like receptor 4 gene TLR4 in Behcet disease. Proc Natl Acad Sci USA. (2013) 110:8134–9. doi: 10.1073/pnas.1306352110
66. Nakano H, Kirino Y, Takeno M, Higashitani K, Nagai H, Yoshimi R, et al. GWAS-identified CCR1 and IL10 loci contribute to M1 macrophage-predominant inflammation in Behcet's disease. Arthritis Res Ther. (2018) 20:124. doi: 10.1186/s13075-018-1613-0
67. Rahmattulla C, Mooyaart AL, van Hooven D, Schoones JW, Bruijn JA, Dekkers OM, et al. Genetic variants in ANCA-associated vasculitis: a meta-analysis. Ann Rheum Dis. (2016) 75:1687–92. doi: 10.1136/annrheumdis-2015-207601
68. Nakazawa D, Masuda S, Tomaru U, Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol. (2019) 15:91–101. doi: 10.1038/s41584-018-0145-y
69. Fujimoto S, Watts RA, Kobayashi S, Suzuki K, Jayne DR, Scott DG, et al. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatology. (2011) 50:1916–20. doi: 10.1093/rheumatology/ker205
70. Kawasaki A, Hasebe N, Hidaka M, Hirano F, Sada KE, Kobayashi S, et al. Protective role of HLA-DRB1*13:02 against Microscopic Polyangiitis and MPO-ANCA-Positive vasculitides in a japanese population: a case-control study. PLoS ONE. (2016) 11:e0154393. doi: 10.1371/journal.pone.0154393
71. Cao Y, Yang J, Colby K, Hogan SL, Hu Y, Jennette CE, et al. High basal activity of the PTPN22 gain-of-function variant blunts leukocyte responsiveness negatively affecting IL-10 production in ANCA vasculitis. PLoS ONE. (2012) 7:e42783. doi: 10.1371/journal.pone.0042783
72. Carr EJ, Niederer HA, Williams J, Harper L, Watts RA, Lyons PA, et al. Confirmation of the genetic association of CTLA4 and PTPN22 with ANCA-associated vasculitis. BMC Med Genet. (2009) 10:121. doi: 10.1186/1471-2350-10-121
73. Jagiello P, Aries P, Arning L, Wagenleiter SE, Csernok E, Hellmich B, et al. The PTPN22 620W allele is a risk factor for Wegener's granulomatosis. Arthritis Rheum. (2005) 52:4039–43. doi: 10.1002/art.21487
74. Alberici F, Martorana D, Bonatti F, Gioffredi A, Lyons PA, Vaglio A. Genetics of ANCA-associated vasculitides: HLA and beyond. Clin Exp Rheumatol. (2014) 32(3 Suppl. 82):S90–7.
75. Fanciulli M, Norsworthy PJ, Petretto E, Dong R, Harper L, Kamesh L, et al. FCGR3B copy number variation is associated with susceptibility to systemic, but not organ-specific, autoimmunity. Nat Genet. (2007) 39:721–3. doi: 10.1038/ng2046
76. Zhou XJ, Cheng FJ, Lv JC, Luo H, Yu F, Chen M, et al. Higher DEFB4 genomic copy number in SLE and ANCA-associated small vasculitis. Rheumatology. (2012) 51:992–5. doi: 10.1093/rheumatology/ker419
77. Qi Y, Zhou X, Bu D, Hou P, Lv J, Zhang H. Low copy numbers of FCGR3A and FCGR3B associated with Chinese patients with SLE and AASV. Lupus. (2017) 26:1383–9. doi: 10.1177/0961203317700485
78. Lee KS, Kronbichler A, Pereira Vasconcelos DF, Pereira da Silva FR, Ko Y, Oh YS, et al. Genetic variants in antineutrophil cytoplasmic antibody-associated vasculitis: a bayesian approach and systematic review. J Clin Med. (2019) 8:E266. doi: 10.3390/jcm8020266
79. Harky A, Fok M, Balmforth D, Bashir M. Pathogenesis of large vessel vasculitis: Implications for disease classification and future therapies. Vasc Med. (2019) 24:79–88. doi: 10.1177/1358863X18802989
80. Sahin Z, Bicakcigil M, Aksu K, Kamali S, Akar S, Onen F, et al. Takayasu's arteritis is associated with HLA-B*52, but not with HLA-B*51, in Turkey. Arthritis Res Ther. (2012) 14:R27. doi: 10.1186/ar3730
81. Ozen S, Batu ED. Vasculitis pathogenesis: can we talk about precision medicine? Front Immunol. (2018) 9:1892. doi: 10.3389/fimmu.2018.01892
82. Nakajima T, Yoshifuji H, Shimizu M, Kitagori K, Murakami K, Nakashima R, et al. A novel susceptibility locus in the IL12B region is associated with the pathophysiology of Takayasu arteritis through IL-12p40 and IL-12p70 production. Arthritis Res Ther. (2017) 19:197. doi: 10.1186/s13075-017-1408-8
83. Nagelkerke SQ, Kuijpers TW. Immunomodulation by IVIg and the role of Fc-gamma receptors: classic mechanisms of action after all? Front Immunol. (2014) 5:674. doi: 10.3389/fimmu.2014.00674
84. Adu B, Dodoo D, Adukpo S, Hedley PL, Arthur FK, Gerds TA, et al. Fc gamma receptor IIIB (FcgammaRIIIB) polymorphisms are associated with clinical malaria in Ghanaian children. PLoS ONE. (2012) 7:e46197. doi: 10.1371/journal.pone.0046197
85. Asano K, Matsushita T, Umeno J, Hosono N, Takahashi A, Kawaguchi T, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. (2009) 41:1325–9. doi: 10.1038/ng.482
86. Breunis WB, van Mirre E, Bruin M, Geissler J, de Boer M, Peters M, et al. Copy number variation of the activating FCGR2C gene predisposes to idiopathic thrombocytopenic purpura. Blood. (2008) 111:1029–38. doi: 10.1182/blood-2007-03-079913
87. Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. (2002) 99:754–8. doi: 10.1182/blood.V99.3.754
88. Lee YH, Ji JD, Song GG. Associations between FCGR3A polymorphisms and susceptibility to rheumatoid arthritis: a metaanalysis. J Rheumatol. (2008) 35:2129–35. doi: 10.3899/jrheum.080186
89. Lee YH, Ji JD, Song GG. Fcgamma receptor IIB and IIIB polymorphisms and susceptibility to systemic lupus erythematosus and lupus nephritis: a meta-analysis. Lupus. (2009) 18:727–34. doi: 10.1177/0961203309104020
90. Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. FcgammaR2A and 3A polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with HER2-positive breast cancer. Ann Oncol. (2011) 22:1302–7. doi: 10.1093/annonc/mdq585
91. Treon SP, Yang G, Hanzis C, Ioakimidis L, Verselis SJ, Fox EA, et al. Attainment of complete/very good partial response following rituximab-based therapy is an important determinant to progression-free survival, and is impacted by polymorphisms in FCGR3A in Waldenstrom macroglobulinaemia. Br J Haematol. (2011) 154:223–8. doi: 10.1111/j.1365-2141.2011.08726.x
92. Carmona FD, Martin J, Gonzalez-Gay MA. Genetics of vasculitis. Curr Opin Rheumatol. (2015) 27:10–7. doi: 10.1097/BOR.0000000000000124
93. Jia M, Zhu L, Zhai YL, Chen P, Xu BY, Guo WY, et al. Variation in complement factor H affects complement activation in immunoglobulin A vasculitis with nephritis. Nephrology. (2019). doi: 10.1111/nep.13580. [Epub ahead of print].
94. Watts RA, Scott DG. Epidemiology of the vasculitides. Semin Respir Crit Care Med. (2004) 25:455–64. doi: 10.1055/s-2004-836139
95. Smutny T, Barvik I, Veleta T, Pavek P, Soukup T. Genetic predispositions of glucocorticoid resistance and therapeutic outcomes in polymyalgia rheumatica and giant cell arteritis. J Clin Med. (2019) 8:582. doi: 10.3390/jcm8050582
96. Acosta-Herrera M, Gonzalez-Gay MA, Martin J, Marquez A. Leveraging genetic findings for precision medicine in vasculitis. Front Immunol. (2019) 10:1796. doi: 10.3389/fimmu.2019.01796
97. Ly KH, Regent A, Tamby MC, Mouthon L. Pathogenesis of giant cell arteritis: more than just an inflammatory condition? Autoimmun Rev. (2010) 9:635–45. doi: 10.1016/j.autrev.2010.05.002
98. Gilkes DM, Bajpai S, Chaturvedi P, Wirtz D, Semenza GL. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. (2013) 288:10819–29. doi: 10.1074/jbc.M112.442939
99. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet. (2016) 17:487–500. doi: 10.1038/nrg.2016.59
100. Coit P, Direskeneli H, Sawalha AH. An update on the role of epigenetics in systemic vasculitis. Curr Opin Rheumatol. (2018) 30:4–15. doi: 10.1097/BOR.0000000000000451
101. Carnero-Montoro E, Alarcon-Riquelme ME. Epigenome-wide association studies for systemic autoimmune diseases: the road behind and the road ahead. Clin Immunol. (2018) 196:21–33. doi: 10.1016/j.clim.2018.03.014
102. Duan J, Lou J, Zhang Q, Ke J, Qi Y, Shen N, et al. A genetic variant rs1801274 in FCGR2A as a potential risk marker for Kawasaki disease: a case-control study and meta-analysis. PLoS ONE. (2014) 9:e103329. doi: 10.1371/journal.pone.0103329
103. Kuo HC, Chang JC, Kuo HC, Yu HR, Wang CL, Lee CP, et al. Identification of an association between genomic hypomethylation of FCGR2A and susceptibility to Kawasaki disease and intravenous immunoglobulin resistance by DNA methylation array. Arthritis Rheumatol. (2015) 67:828–36. doi: 10.1002/art.38976
104. Li SC, Chan WC, Huang YH, Guo MM, Yu HR, Huang FC, et al. Major methylation alterations on the CpG markers of inflammatory immune associated genes after IVIG treatment in Kawasaki disease. BMC Med Genomics. (2016) 9 (Suppl. 1):37. doi: 10.1186/s12920-016-0197-2
105. Ellerman JE, Brown CK, de Vera M, Zeh HJ, Billiar T, Rubartelli A, et al. Masquerader: high mobility group box-1 and cancer. Clin Cancer Res. (2007) 13:2836–48. doi: 10.1158/1078-0432.CCR-06-1953
106. Vogelpoel LT, Hansen IS, Visser MW, Nagelkerke SQ, Kuijpers TW, Kapsenberg ML, et al. FcgammaRIIa cross-talk with TLRs, IL-1R, and IFNgammaR selectively modulates cytokine production in human myeloid cells. Immunobiology. (2015) 220:193–9. doi: 10.1016/j.imbio.2014.07.016
107. Huang YH, Li SC, Huang LH, Chen PC, Lin YY, Lin CC, et al. Identifying genetic hypomethylation and upregulation of Toll-like receptors in Kawasaki disease. Oncotarget. (2017) 8:11249–58. doi: 10.18632/oncotarget.14497
108. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev. (2011) 241:260–8. doi: 10.1111/j.1600-065X.2011.01018.x
109. Ni FF, Li CR, Li Q, Xia Y, Wang GB, Yang J. Regulatory T cell microRNA expression changes in children with acute Kawasaki disease. Clin Exp Immunol. (2014) 178:384–93. doi: 10.1111/cei.12418
110. Huang YH, Lo MH, Cai XY, Liu SF, Kuo HC. Increase expression of CD177 in Kawasaki disease. Pediatr Rheumatol Online J. (2019) 17:13. doi: 10.1186/s12969-019-0315-8
111. Chang D, Qian C, Li H, Feng H. Comprehensive analyses of DNA methylation and gene expression profiles of Kawasaki disease. J Cell Biochem. (2019) 120:13001–11. doi: 10.1002/jcb.28571
112. Huang YH, Kuo HC, Li SC, Cai XY, Liu SF, Kuo HC. HAMP promoter hypomethylation and increased hepcidin levels as biomarkers for Kawasaki disease. J Mol Cell Cardiol. (2018) 117:82–7. doi: 10.1016/j.yjmcc.2018.02.017
113. Huang LH, Kuo HC, Pan CT, Lin YS, Huang YH, Li SC. Multiomics analyses identified epigenetic modulation of the S100A gene family in Kawasaki disease and their significant involvement in neutrophil transendothelial migration. Clin Epigenetics. (2018) 10:135. doi: 10.1186/s13148-018-0557-1
114. Hughes T, Ture-Ozdemir F, Alibaz-Oner F, Coit P, Direskeneli H, Sawalha AH. Epigenome-wide scan identifies a treatment-responsive pattern of altered DNA methylation among cytoskeletal remodeling genes in monocytes and CD4+ T cells from patients with Behcet's disease. Arthritis Rheumatol. (2014) 66:1648–58. doi: 10.1002/art.38409
115. Zhu Y, Qiu Y, Yu H, Yi S, Su W, Kijlstra A, et al. Aberrant DNA methylation of GATA binding protein 3 (GATA3), interleukin-4 (IL-4), and transforming growth factor-beta (TGF-beta) promoters in Behcet's disease. Oncotarget. (2017) 8:64263–72. doi: 10.18632/oncotarget.19500
116. Zhu Y, Yu H, Qiu Y, Ye Z, Su W, Deng J, et al. Promoter hypermethylation of GATA3, IL-4, and TGF-beta confers susceptibility to vogt-koyanagi-harada disease in Han Chinese. Invest Ophthalmol Vis Sci. (2017) 58:1529–36. doi: 10.1167/iovs.16-21188
117. Puccetti A, Pelosi A, Fiore PF, Patuzzo G, Lunardi C, Dolcino M. MicroRNA expression profiling in Behcet's disease. J Immunol Res. (2018) 2018:2405150. doi: 10.1155/2018/2405150
118. Woo MY, Yun SJ, Cho O, Kim K, Lee ES, Park S. MicroRNAs differentially expressed in Behcet disease are involved in interleukin-6 production. J Inflamm. (2016) 13:22. doi: 10.1186/s12950-016-0130-7
119. Qi J, Yang Y, Hou S, Qiao Y, Wang Q, Yu H, et al. Increased Notch pathway activation in Behcet's disease. Rheumatology. (2014) 53:810–20. doi: 10.1093/rheumatology/ket438
120. Qi J, Hou S, Zhang Q, Liao D, Wei L, Fang J, et al. A functional variant of pre-miRNA-196a2 confers risk for Behcet's disease but not for Vogt-Koyanagi-Harada syndrome or AAU in ankylosing spondylitis. Hum Genet. (2013) 132:1395–404. doi: 10.1007/s00439-013-1346-8
121. Zhou Q, Hou S, Liang L, Li X, Tan X, Wei L, et al. MicroRNA-146a and Ets-1 gene polymorphisms in ocular Behcet's disease and Vogt-Koyanagi-Harada syndrome. Ann Rheum Dis. (2014) 73:170–6. doi: 10.1136/annrheumdis-2012-201627
122. Yang JJ, Pendergraft WF, Alcorta DA, Nachman PH, Hogan SL, Thomas RP, et al. Circumvention of normal constraints on granule protein gene expression in peripheral blood neutrophils and monocytes of patients with antineutrophil cytoplasmic autoantibody-associated glomerulonephritis. J Am Soc Nephrol. (2004) 15:2103–14. doi: 10.1097/01.ASN.0000135058.46193.72
123. Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, et al. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. (2010) 120:3209–19. doi: 10.1172/JCI40034
124. Jones BE, Yang J, Muthigi A, Hogan SL, Hu Y, Starmer J, et al. Gene-specific DNA methylation changes predict remission in patients with ANCA-associated vasculitis. J Am Soc Nephrol. (2017) 28:1175–87. doi: 10.1681/ASN.2016050548
125. Yang J, Ge H, Poulton CJ, Hogan SL, Hu Y, Jones BE, et al. Histone modification signature at myeloperoxidase and proteinase 3 in patients with anti-neutrophil cytoplasmic autoantibody-associated vasculitis. Clin Epigenetics. (2016) 8:85. doi: 10.1186/s13148-016-0251-0
126. Luo S, Liang G, Zhang P, Zhao M, Lu Q. Aberrant histone modifications in peripheral blood mononuclear cells from patients with Henoch-Schonlein purpura. Clin Immunol. (2013) 146:165–75. doi: 10.1016/j.clim.2012.12.009
127. Coit P, De Lott LB, Nan B, Elner VM, Sawalha AH. DNA methylation analysis of the temporal artery microenvironment in giant cell arteritis. Ann Rheum Dis. (2016) 75:1196–202. doi: 10.1136/annrheumdis-2014-207116
128. Croci S, Zerbini A, Boiardi L, Muratore F, Bisagni A, Nicoli D, et al. MicroRNA markers of inflammation and remodelling in temporal arteries from patients with giant cell arteritis. Ann Rheum Dis. (2016) 75:1527–33. doi: 10.1136/annrheumdis-2015-207846
129. Kuret T, Burja B, Feichtinger J, Thallinger GG, Frank-Bertoncelj M, Lakota K, et al. Gene and miRNA expression in giant cell arteritis-a concise systematic review of significantly modified studies. Clin Rheumatol. (2019) 38:307–16. doi: 10.1007/s10067-018-4231-y
130. Wolf P. The nature and significance of platelet products in human plasma. Br J Haematol. (1967) 13:269–88. doi: 10.1111/j.1365-2141.1967.tb08741.x
131. Chen Y, Li G, Liu ML. Microvesicles as emerging biomarkers and therapeutic targets in cardiometabolic diseases. Genom Proteom Bioinform. (2018) 16:50–62. doi: 10.1016/j.gpb.2017.03.006
132. Liu ML, Williams KJ, Werth VP. Microvesicles in autoimmune diseases. Adv Clin Chem. (2016) 77:125–75. doi: 10.1016/bs.acc.2016.06.005
133. Wu X, Liu Y, Wei W, Liu ML. Extracellular vesicles in autoimmune vasculitis - Little dirts light the fire in blood vessels. Autoimmun Rev. (2019) 18:593–606. doi: 10.1016/j.autrev.2018.12.007.
134. Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the Protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. (2017) 47:51–65 e7. doi: 10.1016/j.immuni.2017.06.001
135. Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. (2014) 30:255–89. doi: 10.1146/annurev-cellbio-101512-122326
136. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. (2013) 200:373–83. doi: 10.1083/jcb.201211138
137. Chaput N, Thery C. Exosomes: immune properties and potential clinical implementations. Semin Immunopathol. (2011) 33:419–40. doi: 10.1007/s00281-010-0233-9
138. Record M, Subra C, Silvente-Poirot S, Poirot M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem Pharmacol. (2011) 81:1171–82. doi: 10.1016/j.bcp.2011.02.011
139. Sluijter JP, Verhage V, Deddens JC, van den Akker F, Doevendans PA. Microvesicles and exosomes for intracardiac communication. Cardiovasc Res. (2014) 102:302–11. doi: 10.1093/cvr/cvu022
140. Park KS, Lee J, Lee C, Park HT, Kim JW, Kim OY, et al. Sepsis-like systemic inflammation induced by nano-sized extracellular vesicles from feces. Front Microbiol. (2018) 9:1735. doi: 10.3389/fmicb.2018.01735
141. van der Pol E, Boing AN, Harrison P, Sturk A, Nieuwland R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. (2012) 64:676–705. doi: 10.1124/pr.112.005983
142. Buzas EI, Gyorgy B, Nagy G, Falus A, Gay S. Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol. (2014) 10:356–64. doi: 10.1038/nrrheum.2014.19
143. Brogan PA, Shah V, Brachet C, Harnden A, Mant D, Klein N, et al. Endothelial and platelet microparticles in vasculitis of the young. Arthritis Rheum. (2004) 50:927–36. doi: 10.1002/art.20199
144. Dursun I, Dusunsel R, Poyrazoglu HM, Gunduz Z, Patiroglu T, Ulger H, et al. Circulating endothelial microparticles in children with Henoch-Schonlein purpura; preliminary results. Rheumatol Int. (2011) 31:1595–600. doi: 10.1007/s00296-010-1528-9
145. Eleftheriou D, Hong Y, Klein NJ, Brogan PA. Thromboembolic disease in systemic vasculitis is associated with enhanced microparticle-mediated thrombin generation. J Thromb Haemost. (2011) 9:1864–7. doi: 10.1111/j.1538-7836.2011.04434.x
146. Hajj-Ali RA, Major J, Langford C, Hoffman GS, Clark T, Zhang L, et al. The interface of inflammation and subclinical atherosclerosis in granulomatosis with polyangiitis (Wegener's): a preliminary study. Transl Res. (2015) 166:366–74. doi: 10.1016/j.trsl.2015.04.001
147. Nakaoka H, Hirono K, Yamamoto S, Takasaki I, Takahashi K, Kinoshita K, et al. MicroRNA-145-5p and microRNA-320a encapsulated in endothelial microparticles contribute to the progression of vasculitis in acute Kawasaki Disease. Sci Rep. (2018) 8:1016. doi: 10.1038/s41598-018-19310-4
148. Shah V, Christov G, Mukasa T, Brogan KS, Wade A, Eleftheriou D, et al. Cardiovascular status after Kawasaki disease in the UK. Heart. (2015) 101:1646–55. doi: 10.1136/heartjnl-2015-307734
149. Tan Z, Yuan Y, Chen S, Chen Y, Chen TX. Plasma endothelial microparticles, TNF-a and IL-6 in Kawasaki disease. Indian Pediatr. (2013) 50:501–3. doi: 10.1007/s13312-013-0152-7
150. Jia HL, Liu CW, Zhang L, Xu WJ, Gao XJ, Bai J, et al. Sets of serum exosomal microRNAs as candidate diagnostic biomarkers for Kawasaki disease. Sci Rep. (2017) 7:44706. doi: 10.1038/srep44706
151. Kahn R, Mossberg M, Stahl AL, Johansson K, Lopatko Lindman I, Heijl C, et al. Microvesicle transfer of kinin B1-receptors is a novel inflammatory mechanism in vasculitis. Kidney Int. (2017) 91:96–105. doi: 10.1016/j.kint.2016.09.023
152. Khan E, Ambrose NL, Ahnstrom J, Kiprianos AP, Stanford MR, Eleftheriou D, et al. A low balance between microparticles expressing tissue factor pathway inhibitor and tissue factor is associated with thrombosis in Behcet's syndrome. Sci Rep. (2016) 6:38104. doi: 10.1038/srep38104
153. Macey M, Hagi-Pavli E, Stewart J, Wallace GR, Stanford M, Shirlaw P, et al. Age, gender and disease-related platelet and neutrophil activation ex vivo in whole blood samples from patients with Behcet's disease. Rheumatology. (2011) 50:1849–59. doi: 10.1093/rheumatology/ker177
154. Martinez M, Ricart JM, Ruiz-Aja S, Rus A, Todoli J, Calvo J, et al. Platelet activation and red blood cell phosphatidylserine exposure evaluated by flow cytometry in patients with Behcet's disease: are they related to thrombotic events? Pathophysiol Haemost Thromb. (2007) 36:18–22. doi: 10.1159/000112635
155. Mejia JC, Ortiz T, Tassies D, Solanich X, Vidaller A, Cervera R, et al. Procoagulant microparticles are increased in patients with Behcet's disease but do not define a specific subset of clinical manifestations. Clin Rheumatol. (2016) 35:695–9. doi: 10.1007/s10067-015-2903-4
156. Mossberg M, Stahl AL, Kahn R, Kristoffersson AC, Tati R, Heijl C, et al. C1-inhibitor decreases the release of vasculitis-like chemotactic endothelial microvesicles. J Am Soc Nephrol. (2017) 28:2472–81. doi: 10.1681/ASN.2016060637
157. Zhang L, Song QF, Jin JJ, Huang P, Wang ZP, Xie XF, et al. Differential protein analysis of serum exosomes post-intravenous immunoglobulin therapy in patients with Kawasaki disease. Cardiol Young. (2017) 27:1786–96. doi: 10.1017/S1047951117001433
158. Zhang L, Wang W, Bai J, Xu YF, Li LQ, Hua L, et al. Proteomic analysis associated with coronary artery dilatation caused by Kawasaki disease using serum exosomes. Rev Port Cardiol. (2016) 35:265–73. doi: 10.1016/j.repce.2015.11.024
159. Zhang X, Xin G, Sun D. Serum exosomal miR-328, miR-575, miR-134 and miR-671-5p as potential biomarkers for the diagnosis of Kawasaki disease and the prediction of therapeutic outcomes of intravenous immunoglobulin therapy. Exp Ther Med. (2018) 16:2420–32. doi: 10.3892/etm.2018.6458
160. Guiducci S, Ricci L, Romano E, Ceccarelli C, Distler JH, Miniati I, et al. Microparticles and Kawasaki disease: a marker of vascular damage? Clin Exp Rheumatol. (2011) 29(1 Suppl. 64):S121–5.
161. Huang YM, Wang H, Wang C, Chen M, Zhao MH. Promotion of hypercoagulability in antineutrophil cytoplasmic antibody-associated vasculitis by C5a-induced tissue factor-expressing microparticles and neutrophil extracellular traps. Arthritis Rheumatol. (2015) 67:2780–90. doi: 10.1002/art.39239
162. Clarke LA, Hong Y, Eleftheriou D, Shah V, Arrigoni F, Klein NJ, et al. Endothelial injury and repair in systemic vasculitis of the young. Arthritis Rheum. (2010) 62:1770–80. doi: 10.1002/art.27418
163. Ding YY, Ren Y, Feng X, Xu QQ, Sun L, Zhang JM, et al. Correlation between brachial artery flow-mediated dilation and endothelial microparticle levels for identifying endothelial dysfunction in children with Kawasaki disease. Pediatr Res. (2014) 75:453–8. doi: 10.1038/pr.2013.240
164. Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. (2014) 73:1854–63. doi: 10.1136/annrheumdis-2013-203430
165. Kumpers P, Erdbrugger U, Grossheim M, Meyer GP, Hiss M, Gwinner W, et al. Endothelial microparticles as a diagnostic aid in Churg-Strauss vasculitis-induced cardiomyopathy. Clin Exp Rheumatol. (2008) 26(3. Suppl 49):S86–9.
166. Tian J, Lv HT, An XJ, Ling N, Xu F. Endothelial microparticles induce vascular endothelial cell injury in children with Kawasaki disease. Eur Rev Med Pharmacol Sci. (2016) 20:1814–8.
167. Daniel L, Fakhouri F, Joly D, Mouthon L, Nusbaum P, Grunfeld JP, et al. Increase of circulating neutrophil and platelet microparticles during acute vasculitis and hemodialysis. Kidney Int. (2006) 69:1416–23. doi: 10.1038/sj.ki.5000306
168. Erdbruegger U, Grossheim M, Hertel B, Wyss K, Kirsch T, Woywodt A, et al. Diagnostic role of endothelial microparticles in vasculitis. Rheumatology. (2008) 47:1820–5. doi: 10.1093/rheumatology/ken373
169. Kim HJ, Choi EH, Lim YJ, Kil HR. The usefulness of platelet-derived microparticle as biomarker of antiplatelet therapy in Kawasaki disease. J Korean Med Sci. (2017) 32:1147–53. doi: 10.3346/jkms.2017.32.7.1147
170. Yahata T, Suzuki C, Yoshioka A, Hamaoka A, Ikeda K. Platelet activation dynamics evaluated using platelet-derived microparticles in Kawasaki disease. Circ J. (2014) 78:188–93. doi: 10.1253/circj.CJ-12-1037
171. Mendoza-Pinto C, Garcia-Carrasco M, Jimenez-Hernandez M, Jimenez Hernandez C, Riebeling-Navarro C, Nava Zavala A, et al. Etiopathogenesis of Behcet's disease. Autoimmun Rev. (2010) 9:241–5. doi: 10.1016/j.autrev.2009.10.005
172. Hong Y, Eleftheriou D, Hussain AA, Price-Kuehne FE, Savage CO, Jayne D, et al. Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J Am Soc Nephrol. (2012) 23:49–62. doi: 10.1681/ASN.2011030298
173. Kahn R, Hellmark T, Leeb-Lundberg LM, Akbari N, Todiras M, Olofsson T, et al. Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J Immunol. (2009) 182:7906–15. doi: 10.4049/jimmunol.0803624
174. Kahn R, Herwald H, Muller-Esterl W, Schmitt R, Sjogren AC, Truedsson L, et al. Contact-system activation in children with vasculitis. Lancet. (2002) 360:535–41. doi: 10.1016/S0140-6736(02)09743-X
175. Duchene J, Lecomte F, Ahmed S, Cayla C, Pesquero J, Bader M, et al. A novel inflammatory pathway involved in leukocyte recruitment: role for the kinin B1 receptor and the chemokine CXCL5. J Immunol. (2007) 179:4849–56. doi: 10.4049/jimmunol.179.7.4849
176. Xie XF, Chu HJ, Xu YF, Hua L, Wang ZP, Huang P, et al. Proteomics study of serum exosomes in Kawasaki disease patients with coronary artery aneurysms. Cardiol J. (2018) 26:584–93. doi: 10.5603/CJ.a2018.0032
177. Pitanga TN, de Aragao Franca L, Rocha VC, Meirelles T, Borges VM, Goncalves MS, et al. Neutrophil-derived microparticles induce myeloperoxidase-mediated damage of vascular endothelial cells. BMC Cell Biol. (2014) 15:21. doi: 10.1186/1471-2121-15-21
178. Slater TW, Finkielsztein A, Mascarenhas LA, Mehl LC, Butin-Israeli V, Sumagin R. Neutrophil microparticles deliver active myeloperoxidase to injured mucosa to inhibit epithelial wound healing. J Immunol. (2017) 198:2886–97. doi: 10.4049/jimmunol.1601810
179. Kastner DL, Zhou Q, Aksentijevich I. Mutant ADA2 in vasculopathies. N Engl J Med. (2014) 371:480–1. doi: 10.1056/NEJMc1405506
180. Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. (2014) 370:911–20. doi: 10.1056/NEJMoa1307361
181. Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment strategies for deficiency of adenosine deaminase 2. N Engl J Med. (2019) 380:1582–4. doi: 10.1056/NEJMc1801927
182. Glikson M, Galun E, Schlesinger M, Cohen D, Haskell L, Rubinow A, et al. Polyarteritis nodosa and familial Mediterranean fever: a report of 2 cases and review of the literature. J Rheumatol. (1989) 16:536–9.
183. Henckes M, Roskams T, Vanneste S, Van Damme B, Vanrenterghem Y. Polyarteritis nodosa type vasculitis in a patient with familial Mediterranean fever treated with cyclosporin A. Transpl Int. (1994) 7:292–6. doi: 10.1111/j.1432-2277.1994.tb01577.x
184. Kocak H, Cakar N, Hekimoglu B, Atakan C, Akkok N, Unal S. The coexistence of familial Mediterranean fever and polyarteritis nodosa; report of a case. Pediatr Nephrol. (1996) 10:631–3. doi: 10.1007/s004670050176
185. Ozdogan H, Arisoy N, Kasapcapur O, Sever L, Caliskan S, Tuzuner N, et al. Vasculitis in familial Mediterranean fever. J Rheumatol. (1997) 24:323–7.
186. Sachs D, Langevitz P, Morag B, Pras M. Polyarteritis nodosa and familial Mediterranean fever. Br J Rheumatol. (1987) 26:139–41. doi: 10.1093/rheumatology/26.2.139
187. Schlesinger M, Kopolovic J, Viskoper RJ, Ron N. A case of familial Mediterranean fever with cutaneous vasculitis and immune complex nephritis: light, electron, and immunofluorescent study of renal biopsy. Am J Clin Pathol. (1983) 80:511–4. doi: 10.1093/ajcp/80.4.511
188. Serrano R, Martinez MA, Andres A, Morales JM, Samartin R. Familial mediterranean fever and acute myocardial infarction secondary to coronary vasculitis. Histopathology. (1998) 33:163–7. doi: 10.1046/j.1365-2559.1998.00462.x
189. Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. (1967) 43:227–53. doi: 10.1016/0002-9343(67)90167-2
190. Tekin M, Yalcinkaya F, Tumer N, Akar N, Misirlioglu M, Cakar N. Clinical, laboratory and molecular characteristics of children with Familial Mediterranean Fever-associated vasculitis. Acta Paediatr. (2000) 89:177–82. doi: 10.1080/080352500750028799
191. Balbir-Gurman A, Nahir AM, Braun-Moscovici Y. Vasculitis in siblings with familial Mediterranean fever: a report of three cases and review of the literature. Clin Rheumatol. (2007) 26:1183–5. doi: 10.1007/s10067-006-0323-1
192. Bosacki C, Richard O, Freycon F, Mosnier JF, Cathebras P. [The association of polyarteritis nodosa and familial Mediterranean fever]. Presse Med. (2003) 32:24–6.
193. Braun E, Schapira D, Guralnik L, Azzam ZS. Acute vasculitis with multiorgan involvement in a patient with familial Mediterranean fever. Am J Med Sci. (2003) 325:363–4. doi: 10.1097/00000441-200306000-00007
194. Hatemi G, Masatlioglu S, Gogus F, Ozdogan H. Necrotizing vasculitis associated with familial Mediterranean fever. Am J Med. (2004) 117:516–9. doi: 10.1016/j.amjmed.2004.02.050
195. Kone-Paut I, Hentgen V, Guillaume-Czitrom S, Compeyrot-Lacassagne S, Tran TA, Touitou I. The clinical spectrum of 94 patients carrying a single mutated MEFV allele. Rheumatology. (2009) 48:840–2. doi: 10.1093/rheumatology/kep121
196. Luger S, Harter PN, Mittelbronn M, Wagner M, Foerch C. Brain stem infarction associated with familial Mediterranean fever and central nervous system vasculitis. Clin Exp Rheumatol. (2013) 31(3 Suppl. 77):93–5.
197. Ozen S, Bakkaloglu A, Yilmaz E, Duzova A, Balci B, Topaloglu R, et al. Mutations in the gene for familial Mediterranean fever: do they predispose to inflammation? J Rheumatol. (2003) 30:2014–8.
198. Ozkaya O, Bek K, Alaca N, Ceyhan M, Acikgoz Y, Tasdemir HA. Cerebral vasculitis in a child with Henoch-Schonlein purpura and familial Mediterranean fever. Clin Rheumatol. (2007) 26:1729–32. doi: 10.1007/s10067-006-0485-x
199. Topaloglu R, Ozaltin F, Yilmaz E, Ozen S, Balci B, Besbas N, et al. E148Q is a disease-causing MEFV mutation: a phenotypic evaluation in patients with familial Mediterranean fever. Ann. Rheumat. Dis. (2005) 64:750–2. doi: 10.1136/ard.2004.026963
200. Zihni FY, Kalfa M, Ocakci PT, Tarhan F, Parildar M, Keser G, et al. Coexistence of Takayasu's arteritis with familial Mediterranean fever. Rheumatol Int. (2012) 32:1675–8. doi: 10.1007/s00296-011-1853-7
201. Jarjour RA, Al-Berrawi S. Familial Mediterranean fever in Syrian children: phenotype-genotype correlation. Rheumatol Int. (2015) 35:629–34. doi: 10.1007/s00296-014-3116-x
202. Komatsu S, Honma M, Igawa S, Tsuji H, Ishida-Yamamoto A, Migita K, et al. Cutaneous necrotizing vasculitis as a manifestation of familial Mediterranean fever. J Dermatol. (2014) 41:827–9. doi: 10.1111/1346-8138.12588
203. Taylan A, Yildiz Y, Sari I, Ozkok G. Vasculitis and long standing ankylosing spondylitis in a patient with familial Mediterranean fever. J Res Med Sci. (2014) 19:1009–11.
204. Ozcakar ZB, Cakar N, Uncu N, Celikel BA, Yalcinkaya F. Familial Mediterranean fever-associated diseases in children. QJM. (2017) 110:287–90. doi: 10.1093/qjmed/hcw230
205. Ugan Y, Dogru A, Sencan H, Sahin M, Ercan Tunc S. Sacroiliitis and polyarteritis nodosa in a patient with familial mediterranean fever. Case Rep Med. (2016) 2016:5134546. doi: 10.1155/2016/5134546
206. Jain A, Misra DP, Sharma A, Wakhlu A, Agarwal V, Negi VS. Vasculitis and vasculitis-like manifestations in monogenic autoinflammatory syndromes. Rheumatol Int. (2018) 38:13–24. doi: 10.1007/s00296-017-3839-6
207. Imirzalioglu N, Dursun A, Tastan B, Soysal Y, Yakicier MC. MEFV gene is a probable susceptibility gene for Behcet's disease. Scand J Rheumatol. (2005) 34:56–8. doi: 10.1080/03009740510017931
208. Schwartz T, Langevitz P, Zemer D, Gazit E, Pras M, Livneh A. Behcet's disease in Familial Mediterranean fever: characterization of the association between the two diseases. Semin Arthritis Rheum. (2000) 29:286–95. doi: 10.1016/S0049-0172(00)80015-3
209. Touitou I, Magne X, Molinari N, Navarro A, Quellec AL, Picco P, et al. MEFV mutations in Behcet's disease. Hum Mutat. (2000) 16:271–2. doi: 10.1002/1098-1004(200009)16:3<271::AID-HUMU16>3.0.CO;2-A
210. Wu Z, Zhang S, Li J, Chen S, Li P, Sun F, et al. Association between MEFV mutations M694V and M680I and behcet's disease: A Meta-Analysis. PLoS ONE. (2015) 10:e0132704. doi: 10.1371/journal.pone.0132704
211. Ebrahimi-Fakhari D, Wahlster L, Mackensen F, Blank N. Clinical manifestations and longterm followup of a patient with CINCA/NOMID syndrome. J Rheumatol. (2010) 37:2196–7. doi: 10.3899/jrheum.100290
212. Kolivras A, Theunis A, Ferster A, Lipsker D, Sass U, Dussart A, et al. Cryopyrin-associated periodic syndrome: an autoinflammatory disease manifested as neutrophilic urticarial dermatosis with additional perieccrine involvement. J Cutan Pathol. (2011) 38:202–8. doi: 10.1111/j.1600-0560.2010.01638.x
213. Russo RA, Katsicas MM. Chronic infantile neurological cutaneous and articular syndrome: two new cases with rare manifestations. Acta Paediatr. (2001) 90:1076–9. doi: 10.1080/080352501316978192
215. Boom BW, Daha MR, Vermeer BJ, van der Meer JW. IgD immune complex vasculitis in a patient with hyperimmunoglobulinemia D and periodic fever. Arch Dermatol. (1990) 126:1621–4. doi: 10.1001/archderm.126.12.1621
216. Drenth JP, Boom BW, Toonstra J, Van der Meer JW. Cutaneous manifestations and histologic findings in the hyperimmunoglobulinemia D syndrome. International Hyper IgD Study Group. Arch Dermatol. (1994) 130:59–65. doi: 10.1001/archderm.130.1.59
217. Topaloglu R, Saatci U. Hyperimmunoglobulinaemia D and periodic fever mimicking familial Mediterranean fever in the Mediterranean. Postgrad Med J. (1991) 67:490–1. doi: 10.1136/pgmj.67.787.490-a
218. van der Meer JW, Vossen JM, Radl J, van Nieuwkoop JA, Meyer CJ, Lobatto S, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet. (1984) 1:1087–90. doi: 10.1016/S0140-6736(84)92505-4
219. Toro JR, Aksentijevich I, Hull K, Dean J, Kastner DL. Tumor necrosis factor receptor-associated periodic syndrome: a novel syndrome with cutaneous manifestations. Arch Dermatol. (2000) 136:1487–94. doi: 10.1001/archderm.136.12.1487
220. Khatibi K, Heit JJ, Telischak NA, Elbers JM, Do HM. Cerebral vascular findings in PAPA syndrome: cerebral arterial vasculopathy or vasculitis and a posterior cerebral artery dissecting aneurysm. J Neurointerv Surg. (2016) 8:e29. doi: 10.1136/neurintsurg-2015-011753.rep
221. Niv D, Ramirez JA, Fivenson DP. Pyoderma gangrenosum, acne, and hidradenitis suppurativa (PASH) syndrome with recurrent vasculitis. JAAD Case Rep. (2017) 3:70–3. doi: 10.1016/j.jdcr.2016.11.006
222. Chen Y, Li X, Boini KM, Pitzer AL, Gulbins E, Zhang Y, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta. (2015) 1853:396–408. doi: 10.1016/j.bbamcr.2014.11.012
223. Xia M, Boini KM, Abais JM, Xu M, Zhang Y, Li PL. Endothelial NLRP3 inflammasome activation and enhanced neointima formation in mice by adipokine visfatin. Am J Pathol. (2014) 184:1617–28. doi: 10.1016/j.ajpath.2014.01.032
224. Bettiol A, Silvestri E, Di Scala G, Amedei A, Becatti M, Fiorillo C, et al. The right place of interleukin-1 inhibitors in the treatment of Behcet's syndrome: a systematic review. Rheumatol Int. (2019) 39:971–90. doi: 10.1007/s00296-019-04259-y
225. Moghaddas F, Masters SL. Monogenic autoinflammatory diseases: cytokinopathies. Cytokine. (2015) 74:237–46. doi: 10.1016/j.cyto.2015.02.012
226. Demir S, Sag E, Dedeoglu F, Ozen S. Vasculitis in systemic autoinflammatory diseases. Front Pediatr. (2018) 6:377. doi: 10.3389/fped.2018.00377
227. Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet. (2016) 48:67–73. doi: 10.1038/ng.3459
228. Li H, Liu Q, Hou S, Du L, Zhou Q, Zhou Y, et al. TNFAIP3 gene polymorphisms confer risk for Behcet's disease in a Chinese Han population. Hum Genet. (2013) 132:293–300. doi: 10.1007/s00439-012-1250-7
229. Aeschlimann FA, Batu ED, Canna SW, Go E, Gul A, Hoffmann P, et al. A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheumat Dis. (2018) 77:728–35. doi: 10.1136/annrheumdis-2017-212403
230. Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, et al. Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc Natl Acad Sci USA. (2016) 113:10127–32. doi: 10.1073/pnas.1612594113
231. Hou S, Shu Q, Jiang Z, Chen Y, Li F, Chen F, et al. Replication study confirms the association between UBAC2 and Behcet's disease in two independent Chinese sets of patients and controls. Arthritis Res Ther. (2012) 14:R70. doi: 10.1186/ar3789
232. Hou S, Kijlstra A, Yang P. The genetics of Behcet's disease in a Chinese population. Front Med. (2012) 6:354–9. doi: 10.1007/s11684-012-0234-2
233. Park G, Kim HS, Choe JY, Kim SK. SUMO4 C438T polymorphism is associated with papulopustular skin lesion in Korean patients with Behcet's disease. Rheumatol Int. (2012) 32:3031–7. doi: 10.1007/s00296-011-2086-5
234. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A. (2015) 167A:296–312. doi: 10.1055/s-0036-1592307
235. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
236. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
237. Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, et al. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol. (2015) 151:872–7. doi: 10.1001/jamadermatol.2015.0251
238. Torrelo A, Colmenero I, Requena L, Paller AS, Ramot Y, Richard Lee CC, et al. Histologic and Immunohistochemical Features of the Skin Lesions in CANDLE Syndrome. Am J Dermatopathol. (2015) 37:517–22. doi: 10.1097/DAD.0000000000000340
239. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest. (2018) 128:3041–52. doi: 10.1172/JCI98814
240. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. (2009) 360:2426–37. doi: 10.1056/NEJMoa0807865
Keywords: precision medicine, vasculitis, vasculitides, genome-wide association studies, epigenetics, extracellular vesicles, monogenic systemic autoinflammatory diseases
Citation: Demirkaya E, Arici ZS, Romano M, Berard RA and Aksentijevich I (2019) Current State of Precision Medicine in Primary Systemic Vasculitides. Front. Immunol. 10:2813. doi: 10.3389/fimmu.2019.02813
Received: 02 August 2019; Accepted: 15 November 2019;
Published: 17 December 2019.
Edited by:
Andreas Kronbichler, Innsbruck Medical University, AustriaReviewed by:
Davide Martorana, University Hospital of Parma, ItalyMd Yuzaiful Md Yusof, University of Leeds, United Kingdom
Copyright © 2019 Demirkaya, Arici, Romano, Berard and Aksentijevich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erkan Demirkaya, ZXJrYW4uZGVtaXJrYXlhQGxoc2Mub24uY2E=