 Ayesha Zahid
Ayesha Zahid Bofeng Li
Bofeng Li Arnaud John Kombe Kombe2
Arnaud John Kombe Kombe2 Tengchuan Jin
Tengchuan Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 25 October 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02538
This article is part of the Research Topic Role of NOD-like Receptors in Infectious and Immunological Diseases View all 9 articles
Inflammasomes play a crucial role in innate immunity by serving as signaling platforms which deal with a plethora of pathogenic products and cellular products associated with stress and damage. By far, the best studied and most characterized inflammasome is NLRP3 inflammasome, which consists of NLRP3 (nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3), ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), and procaspase-1. Activation of NLRP3 inflammasome is mediated by highly diverse stimuli. Upon activation, NLRP3 protein recruits the adapter ASC protein, which recruits the procaspase-1 resulting in its cleavage and activation, inducing the maturation, and secretion of inflammatory cytokines and pyroptosis. However, aberrant activation of the NLRP3 inflammasome is implicated in various diseases including diabetes, atherosclerosis, metabolic syndrome, cardiovascular, and neurodegenerative diseases; raising a tremendous clinical interest in exploring the potential inhibitors of NLRP3 inflammasome. Recent investigations have disclosed various inhibitors of the NLRP3 inflammasome pathway which were validated through in vitro studies and in vivo experiments in animal models of NLRP3-associated disorders. Some of these inhibitors directly target the NLRP3 protein whereas some are aimed at other components and products of the inflammasome. Direct targeting of NLRP3 protein can be a better choice because it can prevent off target immunosuppressive effects, thus restrain tissue destruction. This paper will review the various pharmacological inhibitors of the NLRP3 inflammasome and will also discuss their mechanism of action.
In mammals, the immune system relies on innate immunity and adaptive immunity to protect the host from any external or internal danger (1). The innate immune response utilizes pattern-recognition receptors (PRRs) to sense endogenous or exogenous pathogens (2). A newly identified PRR, which was reported in detail for the first time in 2002, is the inflammasome. It is a high molecular weight protein complex which elicits the activation of inflammatory caspases and processing of pro-interleukin-1β (pro-IL-1β). Inflammasomes are of vital importance in innate immunity because they serve as signaling platforms which are capable of dealing with a plethora of pathogenic products and cellular products associated with stress and damage (3, 4). At present, there are five inflammasomes which are clearly identified, including nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 1 (NLRP1), NLRP3, and NLR and caspase recruitment domain containing receptor 4 (NLRC4) and the AIM2-like receptors (ALR) family including absent in melanoma 2 (AIM2) (5, 6). This review will describe NLRP3 inflammasome and some reported pharmacological inhibitors targeting this most important inflammasome complex.
NLRP3 inflammasome is the best characterized inflammasome at present, named after the NLRP3 protein in the complex which belongs to the NLR family and is also termed as NALP3, CIASI or pyrin domain-containing protein 3 (7). Besides NLRP3 protein, the adapter protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and procaspase-1 are also part of this inflammasome (8, 9). NLRP3 is a 115 kDa cytosolic protein expressed in monocytes, neutrophils, dendritic cells, lymphocytes, osteoblasts, and epithelial cells (10). It contains three domains which are: a leucine-rich repeat (LRR) at the C-terminal, a central nucleotide-binding and oligomerization domain NACHT which possesses ATPase activity, and a pyrin domain (PYD) at the N-terminal which recruits ASC (11). The danger signal is sensed by the LRR domain which leads to the oligomerization of NLRP3 monomers through their NACHT domains. This is followed by the interaction between the PYD domains of NLRP3 and ASC. Finally, procaspase-1 is recruited into the complex through its CARD domain by ASC which acts as an adaptor protein (12). Recently, Sharif et al. determined the structure of recombinant complex of maltose binding protein (MBP)-tagged NLRP3 protein without pyrin domain and mitotic Ser/Thr kinase NEK7. The cryo EM map showed an earring shape structure composed of curved LRR and globular NACHT domains. The C-terminal lobe of NEK7 interacts with multiple NLRP3 domains including the LRR, HD2 (helical domain 2), and NBD (nucleotide-binding domain). This structure suggests the possibility that NEK7 joins adjacent NLRP3 subunits into bipartite interactions to bring about the activation of NLRP3 inflammasome (13).
NLRP3 Inflammasome recognizes a wide range of stimuli which include various protozoans, e.g., Plasmodium, ameba, viruses such as adenoviruses, influenza, and Sendai virus, fungi such as Saccharomyces cerevisiae and Candida albicans, different bacteria such as Listeria monocytogenes, Escherichia coli, and Staphylococcus aureus (14). NLRP3 Inflammasome can also respond to damage-associated endogenous factors such as drusen (15), uric acid crystals (16), extracellular adenosine triphosphate (ATP) (17), β-amyloid plaques (11), and islet amyloid polypeptide (18).
Activation of NLRP3 inflammasome signaling pathway needs two independent yet parallel steps i.e., priming and activation (19–21). Basal expression of NLRP3 protein and the precursor pro-form of IL-1β is very low, therefore a priming step or “signal 1” initiates the transcription of these targets. Priming step is induced by toll-like receptors (TLRs), myeloid differentiation primary response 88 (MyD88) and/or cytokine receptors, e.g., TNF receptor, which recognize PAMPs or DAMPs and activate the transcription of NLRP3 and pro-IL-1β (14, 22, 23) as illustrated in Figure 1. Recently, many studies have provided strong evidences that priming step is not limited to transcriptional upregulation, post-translational modifications (PTMs) such as ubiquitination and phosphorylation of NLRP3 protein also play critical roles in NLRP3 inflammasome activation (24–26). The second activation step occurs as the primed cell recognizes another stimulus (usually a DAMP) (27, 28).
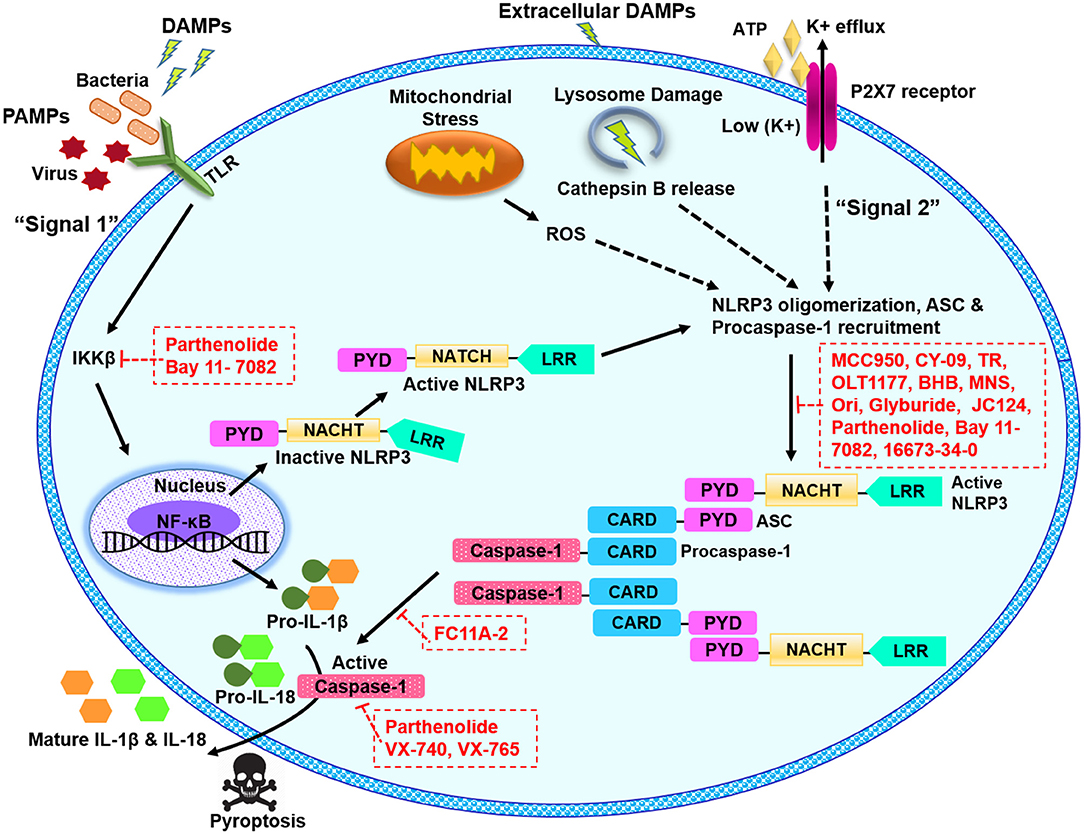
Figure 1. Schematic illustration of NLRP3 inflammasome pathway and potential blockade sites of various pharmacological inhibitors. The signal 1 or the priming signal is mediated by pathogenic PAMPs from bacteria or virus, or sterile DAMPs resulting in NF-κB-dependent upregulation of NLRP3 and pro-IL-1β expression. The signal 2 or activation signal mediated by numerous PAMP or DAMP stimulation, promotes the NLRP3 oligomerization, and recruitment of ASC and pro-caspase-1, leading to the activation of NLRP3 inflammasome complex. NLRP3 can be activated in response to extracellular ATP and K+ efflux through the ATP-gated P2X7 channel, in response to cathepsin B release from damaged lysosomes or in response to reactive oxygen species (ROS) released from damaged mitochondria. NLRP3 inflammasome activation results in active caspase-1, which cleaves the proforms of IL-1β and IL-18 into their mature forms. ASC, apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain; ATP, adenosine triphosphate; BHB, β-Hydroxybutyrate; CARD, caspase recruitment domain; DAMPS, danger or damage associated molecular patterns; IL, interleukin; LRR, leucine-rich repeat; MNS, methylenedioxy-β-nitrostyrene; NACHT, central nucleotide-binding and oligomerization; NF-κB, nuclear factor kappa B; Ori, oridonin; P2X7, P2X purinergic receptor 7; PAMPS, pathogen associated molecular patterns; PYD, pyrin domain; ROS, reactive oxygen species; TLR, toll-like receptor; TR, tranilast.
As a result of the second step, caspase-1 is activated and carries out resultant processing and secretion of IL-1β and IL-18 (29). Various molecular mechanisms to explain the activation of NLRP3 inflammasome have been proposed which include mitochondrial reactive oxygen species (ROS) generation (30, 31), pore formation and potassium (K+) efflux (32, 33) and lysosomal destabilization and rupture (30, 34).
Anomalous NLRP3 inflammasome activation is linked with the development of many diseases, especially age-associated ailments for example various metabolic syndromes and metabolic disorders including gout (16), atherosclerosis (35), Alzheimer's disease (AD) (11), and type II diabetes (T2D) (36). Enhanced secretion of IL-1β and IL-18 by NLRP3 inflammasome is associated with the progression of atherosclerotic plaque in atherosclerotic patients and animal models (37–39). NLRP3 inflammasome is involved in experimental autoimmune encephalomyelitis (EAE) in animal models and multiple sclerosis (MS) in humans (40, 41). Inappropriate NLRP3 inflammasome activation is also implicated in Crohn's disease, inflammatory bowel disease (IBD), and ulcerative colitis (42–44). NLRP3 inflammasome is also linked with various cancers, such as colon cancer, breast cancer, melanoma, hepatitis C virus-associated hepatocellular carcinoma, and gastrointestinal cancers (45, 46). In addition to NLRP3 activation anomalies, there are also NLRP3 genetic abnormalities collectively termed as cryopyrin-associated periodic syndromes (CAPS). Gain of function mutations in NLRP3 gene give rise CAPS disorders, resulting in enhanced IL-1β secretion, and other CAPS specific symptoms (47).
The association of NLRP3 inflammasome with the plethora of diseases evokes a substantial interest in the scientific community to discover the effective NLRP3 inflammasome inhibitors. By taking advantage of complex signaling cascade of NLRP3 inflammasome, a diverse range of targets can be used for its inhibition. For example, inhibition of NLRP3 inflammasome activation, suppression of upstream signals, blockade of inflammasome assembly, caspase-1 activation inhibition, blockade of pore-forming protein gasdermin D (GSDMD) cleavage, and neutralizing the inflammatory cytokines produced by the NLRP3 inflammasome can be targeted for potential inhibition of NLRP3 inflammasome. Different mechanisms can be opted to achieve these outcomes for example inhibition of NLRP3 inflammasome assembly, inhibition of P2X7 receptor, inhibition of K+ efflux, and ROS scavengers can be used (48–51). Furthermore, NLRP3-NLRP3 interactions or NLRP3-ASC interactions can be disrupted. Inhibitors can be directed at the ATP-binding domain of NLRP3 resulting in blockade of its ATPase activity (52, 53). PTMs of NLRP3 protein and other constituents of the NLRP3 inflammasome are reported as an important step to control its assembly. It can be anticipated that future studies may target the biological components which are involved in these PTMs to inhibit NLRP3 inflammasome. In the recent years, several inhibitors of NLRP3 inflammasome pathway have been reported. Here, we describe some recent pharmacological inhibitors of NLRP3 inflammasome pathway, their proposed mode of action and therapeutic potential (Table 1). Figure 1 depicts the proposed sites of action of these inhibitors provided by in vitro and in vivo experimental data.
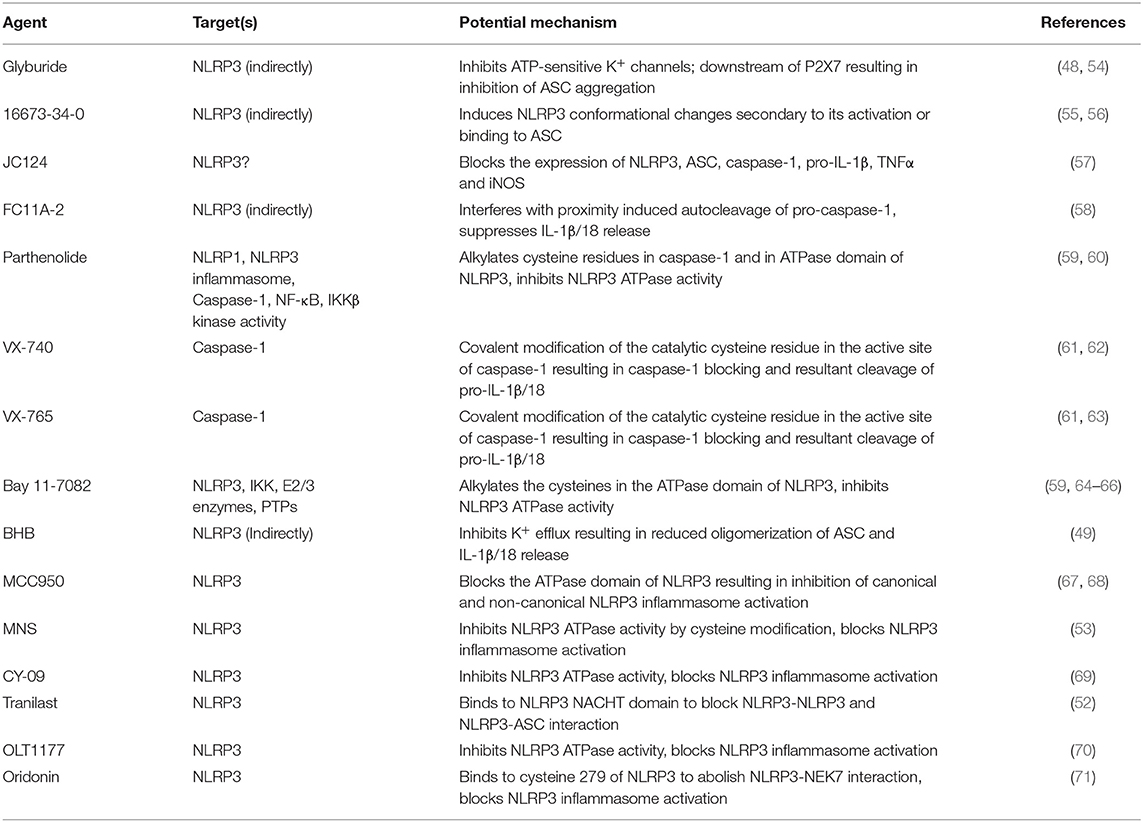
Table 1. Potential inhibitors of NLRP3 inflammasome and their targets.
Glyburide is a sulfonylurea drug which is widely used in the United States for the treatment of T2D (72). It inhibits ATP-sensitive K+ (KATP) channels in pancreatic β cells (73). One study conducted by Lamkanfi et al. showed that glyburide prevents PAMP-, DAMP-, and crystal-induced NLRP3 inflammasome activation in bone marrow-derived macrophages (BMDMs). Its inhibitory potential seems to be specific for NLRP3 inflammasome, since it did not prevent the IL-1β release from activated NLRC4 or NLRP1 pathway (48). When tested in response to stimuli which work independent of the P2X7 receptor but require TLR4 signaling, glyburide effectively prevented the activation of caspase-1 and 1L-1β secretion, suggesting that it works downstream of the P2X7 receptor (48). It did not block the caspase-1 activation in S. typhimurium-infected BMDMs which do not require NLRP3 for caspase-1 activation (74), suggesting that it works upstream of NLRP3 (48). Furthermore, glyburide showed inhibitory activity in vitro (48, 75) or in vivo (76) during NLRP3 inflammasome activation. However, the in vivo doses of glyburide to exert its inhibitory affect are quite high, which cause hypoglycemia, therefore its usage is limited to T2D only (21).
16673-34-0 is an intermediate substrate produced during glyburide synthesis, however, it lacks the cyclohexylurea moiety of glyburide which is involved in insulin release, therefore, it does not affect glucose metabolism. A study carried out by Marchetti et al. in J774A.1 murine macrophages and primary adult rat cardiomyocytes showed that 16673-34-0 inhibits NLRP3 inflammasome formation, but shows no effect on AIM2 or NLRC4 inflammasome. When tested in vivo, it showed positive outcome in mouse models of non-reperfused and reperfused acute myocardial infarction. 16673-34-0 was tested with multiple diverse stimuli of NLRP3 inflammasome, independent of which stimuli is used, inhibitory effects of 16673-34-0 remained the same suggesting that it interferes with downstream events involved in either NLRP3 conformational changes secondary to activation or binding to ASC (55, 56). However, the exact mechanism of inhibition is not completely clear and additional studies are needed to fully determine its inhibitory potential.
Kuwar et al. recently developed a novel small molecule JC124, through structural optimization of glyburide. JC124 was rationally designed to remove the potential hypoglycemic effects of glyburide. They explored the potential of JC124 for traumatic brain injury (TBI) therapy and it was demonstrated to exert significant anti-inflammatory effect to protect the injured brain following TBI. JC124 treatment significantly reduced the expression of NLRP3, ASC, caspase-1, pro-IL-1β, TNFα, and inducible nitric oxide synthase (iNOS). This targeting of NLRP3 inflammasome activation and its downstream neuroinflammatory cascade is suggested to confer JC124 its protective effect for TBI (57). It blocked ASC aggregation, caspase-1 activation, and IL-1β secretion. JC124 showed protective effects in a mouse model of acute myocardial infarction (77) and in transgenic AD models (77, 78). Further studies aiming at determining the efficacy of JC124 will render more information for its translational value.
Liu et al. investigated a synthetic small molecular compound, 1-ethyl-5-methyl-2-phenyl-1H-benzo[d]imidazole, which is also known as FC11A-2, for its inhibitory potential of NLRP3 inflammasome. FC11A-2 was examined in THP-1 cells and in mouse model of dextran sulfate sodium (DSS)-induced experimental colitis, and it showed highly effective outcomes by repressing IL-1β/18 release. FC11A-2 hindered the proximity-induced autocleavage of procaspase-1, eventually resulting in reduced amount of activated caspase-1, by a pathway which is independent of activation of NF-κB (58).
Parthenolide is a plant sesquiterpene lactone and has numerous anti-inflammatory properties, therefore, it is utilized in herbal medicines of various inflammatory diseases (79). It inhibited caspase-1 activation in response to NLRP1, NLRC4, and NLRP3 stimulation by alkylating many cysteine residues of caspase-1. Parthenolide can also target ATPase activity of NLRP3 protein directly, probably through cysteine modification (59). However, it had poor solubility and bioavailability, therefore now its water soluble analogs are being evaluated (80, 81).
VX-740 (Pralnacasan) and its analog VX-765 are peptidomimetic inhibitor of caspase-1. They are prodrugs which are metabolized by plasma esterases to their corresponding aldo-acids (63, 82). Both compound act by covalent modification of the catalytic cysteine residue in the active site of caspase-1, hence they block caspase-1 and resultant cleavage of pro-IL-1β/18 (61). VX-740 showed good results for the treatment of rheumatoid arthritis (RA) and osteoarthritis (OA) in mice models (62). In phase I and II clinical trials in RA patients, it exhibited significant anti-inflammatory effects with good pharmacokinetics profile (83, 84). However, hepatic toxicity in animals after its long-term exposure led to discontinuation of further development (85). VX-765 showed even higher potency for RA and also showed reduction in IL-1β/18 in mouse model of dermatitis. It also had positive outcomes for treatment of epilepsy and psoriasis in mice and was announced to undergo clinical trial (63, 86). Some recent findings have reported that VX-765 helped in alleviating the cognitive impairment and severity of AD in mice (87). It also lowered myocardial infarction and preserved ventricular function in mice (88).
Bay 11-7082 is a phenyl vinyl sulfone, it inhibits NF-κB pathway through blockade of kinase activity of IKKβ. It inhibits its target proteins using alkylation of essential nucleophilic residues, for example cysteines. Studies with NG5 cells and mouse primary BMDMs showed that bay 11-7082 prevents the organization of ASC pyroptosome and NLRP3 inflammasome function through alkylation of cysteine residues of NLRP3 ATPase region. Importantly, it showed selective inhibition of NLRP3 inflammasome as compare to other inflammasomes (59). Recently, vinyl sulfone derivatives were used as antiparasitic agents in dogs and mice (89), these preclinical trials revealed that these compounds are well-tolerated, non-mutagenic and have suitable pharmacokinetic profiles. They also permeate cell membrane easily (59). Bay 11-7082 and other vinyl sulfone/sulfonate compounds provides an applicable framework for the future design.
ß-hydroxy butyrate (BHB) is a ketone metabolite, which was tested by Youm et al. for NLRP3 inflammasome blockade. It affectively lowered the production of IL-1ß and IL-18 in human monocytes in response to activated NLRP3 inflammasome, without interfering with activated AIM2 or NLRC4 inflammasome. Treatment of BMDMs from mouse models of familial cold auto inflammatory syndrome (FCAS) and Muckle–Wells syndrome (MWS) with BHB dose-dependently inhibited constitutive NLRP3 inflammasome activation. BHB is effective only for canonical activation during which it inhibits K+ efflux and reduces the oligomerization and speck formation of ASC. It blocks the activation of NLRP3 inflammasome independent of ROS, AMP-activated protein kinase, glycolytic inhibition, or autophagy (49). From these findings it can be anticipated that pharmacological or dietary attempts to raise BHB level may reduce the severity of NLRP3-mediated chronic inflammatory diseases.
A diarylsulfonylurea-containing compound termed as MCC950, is considered one of the most potent and selective inhibitor of NLRP3 inflammasome. There is an extensive consideration in the development of MCC950 as a treatment for the NLRP3-driven disorders. MCC950 was previously reported to block the processing of IL-1β by caspase-1 (54), later it was described by Coll et al. that in mouse and human macrophages, MCC950 has the potential to block both canonical and non-canonical NLRP3 inflammasome activation and IL-1β production by abrogating ASC oligomerization. Notably, MCC950 had no effect on AIM2, NLRC4, or NLRP1 inflammasome activation (67, 90, 91). Another latest study has reported that MCC950 directly binds to the NLRP3 NACHT domain's Walker B motif, and blocks the hydrolysis of ATP and formation of NLRP3 inflammasome (68). Very recently, a preprint paper at BioRxiv has reported by utilizing photoaffinity labeling and iBody technology that MCC950 interacts with the NACHT domain of wild type NLRP3. The binding was lessened in most of CAPS-related NLRP3 mutants, moreover, in two mouse models of CAPS, MCC950 did not inhibit the NLRP3-driven inflammatory pathology. This study implies that MCC950 may only be effective in inflammation driven by wild type NLRP3 protein, but not in ailments driven by CAPS-related NLRP3 mutants (92).
MCC950 was reported to lower skin and pulmonary inflammation in mice (93) and some other in vivo experiments in mouse model of human MS showed that MCC950 alleviates the severity of EAE (67). Oral treatment of MCC950 rescued the dopaminergic degeneration in a mouse model of Parkinson's disease (PD) (94). Future studies are needed to warrant the exact potential of MCC950.
A potent NLRP3 inhibitor, 3,4-Methylenedioxy-β-nitrostyrene (MNS) was found through screening a kinase inhibitory library by He et al. By utilizing immunoprecipitation, mass spectrometry, and mutational studies, it was demonstrated that MNS binds to the LRR and NACHT domains and suppresses ATPase activity of NLRP3, while the activation of AIM2 or NLRC4 inflammasomes was unaffected by it. MNS may directly target the cysteine(s) of NLRP3 as implicated by its inhibition of ATPase activity of NLRP3 (53). Future studies on MNS may confer additional insights on this potential inhibitor.
Jiang et al. identified an effective and direct inhibitor of NLRP3 which showed significant inhibition of NLRP3 inflammasome in vivo in mice models and ex vivo in human cells (52). CY-09 is an analog of CFTR(inh)-172 (C172), which inhibits the cystic fibrosis transmembrane conductance regulator (CFTR) channel (95). CY-09 lacks CFTR-inhibitory activity (96). In BMDMs primed with LPS, CY-09 dose-dependently blocked the ATP, monosodium urate (MSU), and nigericin-induced activation of caspase-1 and resultant release of IL-1β. Its inhibitory effect is not dependent on signal 1 and NLRP3 post-translational modification (ubiquitination). Mechanistically, it directly interacts with the NLRP3 Walker A motif to eliminate the ATP binding of NLRP3, however, it does not affect NLRP1, NLRC4, RIG-1, or NOD2 (52).
CY-09 demonstrated outstanding preventive or therapeutic properties in the mice models of gout, T2D, and CAPS. Most importantly, it exhibited a promising pharmacokinetic profile and showed good oral bioavailability, safety, and stability. Nonetheless, more studies are required to broaden its full potential (52).
Tranilast (N-[3′,4′-dimethoxycinnamoyl]-anthranilic acid, TR) is a tryptophan metabolite analog which showed inhibitory potential for homologous passive cutaneous anaphylaxis (97). TR is a fairly safe compound and its high doses showed appropriate tolerance levels when tested in patients (98, 99). It showed inhibitory effect for NLRP3 inflammasome but not for NLRC4 or AIM2 inflammasome. TR impaired the endogenous NLRP3-ASC interaction but did not affect the NLRP3-NEK7 interaction, raising the possibility that it targets NLRP3 directly. Indeed, it was demonstrated to bind to the NLRP3 NACHT domain and, abolish the direct NLRP3-NLRP3 interaction (52). Moreover, TR does not impede with the upstream signaling events of NLRP3 inflammasome, e.g., expression of NLRP3 and pro-IL-1β, ROS production, K+ efflux, chloride efflux, and mitochondrial damage. TR has demonstrated significant therapeutic and preventive outcomes in gout, CAPS, and T2D mice models (69). Considering the high safety of TR in clinic, it can be of significant importance for treating NLRP3-driven diseases.
OLT1177 is an active β-sulfonyl nitrile compound, which cleared phase I clinical trial for the treatment of degenerative arthritis successfully, and now being evaluated under phase II clinical trial (100).
A study in mice model of MSU- and zymosan-induced arthritis by Marchetti et al. demonstrated that OLT1177 has the potential to lower the neutrophil infiltration and joint swelling, as well as to inhibit the secretion of IL-1β and IL-6. In in vitro studies, OLT1177 blocked both canonical and non-canonical activation of NLRP3 inflammasome and showed direct binding with NLRP3 to block its ATPase activity. Moreover, in monocytes from CAPS patients, it lowered caspase-1 activity and resultant IL-1β secretion, and reduced LPS-induced systemic inflammation in mice. Significantly, OLT1177 did not inhibit NLRC4 or AIM2 inflammasome. OLT1177 was given orally to the healthy subjects in phase 1 trials, and it showed good safety and tolerance levels. Additionally, it had long half-life and did not show any organ or hematological toxicity at various doses (70). Thus, OLT1177 seems to have a significant potential to treat NLRP3-related diseases.
Oridonin (Ori) is a bioactive ent-kaurane diterpenoid, a main component of herbal plant Rabdosia rubescens, which is extensively utilized in traditional Chinese medicine (101, 102). There are a number of anticancer activities which have been associated with Ori, such as cell cycle arrest, angiogenesis suppression and apoptosis induction (103, 104). It is reported to inhibit the NF-κB or MAPK activation and repress the release of inflammasome-independent proinflammatory cytokines release (105–107). Furthermore, it has good therapeutic effects on neuroinflammation, sepsis and colitis (108–110). He et al. reported that Ori interacts with the cysteine 279 of NLRP3 NACHT domain through a covalent bond, abolishes NLRP3-NEK7 interaction, and inhibits consequent activation of NLRP3 inflammasome. The inhibitory effects of Ori are limited to NLRP3 inflammasome only and it does not inhibit AIM2 or NLRC4 inflammasome activation. When used in mice models of T2D, peritonitis and gouty arthritis, Ori exhibited significant preventive, and therapeutic effects (71). Thus, it can be anticipated that future studies may establish Ori as a clinically applicable inhibitor of NLRP3 inflammasome.
NLRP3-induced pyroptosis and IL-1β/18 secretion is linked to various diseases. The extent to which NLRP3 inflammasome activation contributes to the pyroptosis is still unclear, however, NLRP3 activation does results in pyroptosis which in turn can cause serious injury to vital organs (111). At present, to treat NLRP3-associated diseases, many drugs are available which block IL-1β such as neutralizing IL-1β antibody canakinumab, recombinant IL-1 receptor antagonist anakinra, and the soluble decoy IL-1 receptor rilonacept. These biological agents are being used to treat CAPS and other diseases associated with IL-1β (112). However, activated NLRP3 inflammasome does not produce only IL-1β, there are other cytokines such as IL-18 which may also contribute to the NLRP3-associated disorders (113, 114). Moreover, IL-1β production can be mediated by other inflammasomes or by inflammasome-independent pathways; thus inhibitors aimed at IL-1β can result in unintentional immunosuppressive effects. Therefore, pharmacological inhibitors which specifically target the NLRP3 inflammasome only could be a better option for treatment of NLRP3-associated diseases. NLRP3-induced pyroptosis has been reported by many recent studies as a critical mechanism contributing to the NLRP3 inflammasome related pathologies (115, 116). Emerging evidences have reported GSDMD as an executive protein responsible for pyroptosis (117, 118), making it an attractive therapeutic target for curing NLRP3-induced pyroptosis associated diseases. Future studies should take advantage of now available structure of NLRP3 and focus on the development of structure-guided direct inhibitors with improved specificity and potency. Furthermore, nanobodies (Nbs) are now being explored extensively as therapeutics due to their high specificity, stability, and low toxicity (119, 120). It can be anticipated that Nbs may also be evaluated for NLRP3 inflammasome inhibition. In the past decade, great leaps forward were made to determine the structure of NLRP3 inflammasome, its activation mechanisms and its contribution to the initiation and progression of different diseases. Moreover, many small molecule inhibitors for NLRP3 inflammasome have been reported and some of them have shown remarkable therapeutic potential. However, none of them is currently approved by food and drug administration (FDA) or other agents. Current research should focus on the development of specific, small-molecular inhibitors of NLRP3 inflammasome which have improved pharmacokinetic properties, can penetrate the blood brain barrier more readily and be more cost-effective.
AZ prepared the draft. All authors revised the draft.
TJ was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB29030104), the National Natural Science Fund (Grant No. 31870731 and U1732109), the Fundamental Research Funds for the Central Universities, and the 100 Talents Programme of the Chinese Academy of Sciences. AZ was supported by CAS-TWAS fellowship. AK was supported by Chinese Government Scholarship. JT was supported by the National Natural Science Foundation of China (81771774), the Anhui Provincial Natural Science Foundation (1708085MH191), and the Key Projects of Research and Development Program of Anhui Provence (201904a07020103).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. (2010) 464:1367–70. doi: 10.1038/nature08900
2. Fullard N, O'Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol. (2015) 37:511–7. doi: 10.1007/s00281-015-0503-7
3. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
4. Sanders M, Parsons M, Howard A, Liu J, Fassio S, Martinez J, et al. Single-cell imaging of inflammatory caspase dimerization reveals differential recruitment to inflammasomes. Cell Death Dis. (2015) 6:e1813. doi: 10.1038/cddis.2015.186
5. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
6. Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. (2013) 61:1113–21. doi: 10.1002/glia.22499
7. Inoue M, Shinohara ML. The role of interferon-β in the treatment of multiple sclerosis and experimental autoimmune encephalomyelitis–in the perspective of inflammasomes. Immunology. (2013) 139:11–8. doi: 10.1111/imm.12081
8. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton's tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun. (2015) 6:7360. doi: 10.1038/ncomms8360
9. Inoue M, Shinohara ML. Nlrp3 inflammasome and MS/EAE. Autoimmune Dis. (2013) 2013:859145. doi: 10.1155/2013/859145
10. Zhong Y, Kinio A, Saleh M. Functions of NOD-like receptors in human diseases. Front Immunol. (2013) 4:333. doi: 10.3389/fimmu.2013.00333
11. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat Immunol. (2008) 9:857–65. doi: 10.1038/ni.1636
12. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci USA. (2007) 104:8041–6. doi: 10.1073/pnas.0611496104
13. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. (2019) 570:338–43. doi: 10.1038/s41586-019-1295-z
14. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. (2012) 13:325–32. doi: 10.1038/ni.2231
15. Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. (2012) 18:791–8. doi: 10.1038/nm.2717
16. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. (2006) 440:237–41. doi: 10.1038/nature04516
17. Mariathasan S, Weiss DS, Newton K, McBride J, O'rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. (2006) 440:228–32. doi: 10.1038/nature04515
18. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat Immunol. (2010) 11:897–904. doi: 10.1038/ni.1935
19. Martín-Sánchez F, Diamond C, Zeitler M, Gomez A, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Different. (2016) 23:1219–31. doi: 10.1038/cdd.2015.176
20. Gaidt MM, Hornung V. The NLRP3 inflammasome renders cell death pro-inflammatory. J Mol Biol. (2018) 430:133–41. doi: 10.1016/j.jmb.2017.11.013
21. Mangan MS, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov. (2018) 17:588–606. doi: 10.1038/nrd.2018.97
22. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, Macdonald K, Speert D, et al. Cutting edge: NF-κB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
23. Franchi L, Eigenbrod T, Muñozplanillo R, Ozkurede U, Kim YG, Arindam C, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol. (2014) 193:4214–22. doi: 10.4049/jimmunol.1400582
24. Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. (2012) 287:36617–22. doi: 10.1074/jbc.M112.407130
25. Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandesalnemri T, Alnemri ES, et al. Inflammasome priming by LPS is dependent upon ERK signaling and proteasome function. J Immunol. (2014) 192:3881–8. doi: 10.4049/jimmunol.1301974
26. Py BF, Kim MS, Vakifahmetoglunorberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. (2013) 49:331–8. doi: 10.1016/j.molcel.2012.11.009
27. Kim EH, Park MJ, Sun P, Lee ES. Increased expression of the NLRP3 inflammasome components in patients with Behçet's disease. Journal of Inflammation. (2015) 12:41. doi: 10.1186/s12950-015-0086-z
28. Ozaki E, Campbell M, Doyle SL. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res. (2015) 8:15–27. doi: 10.2147/JIR.S51250
29. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann NY Acad Sci. (2014) 1319:82–95. doi: 10.1111/nyas.12458
30. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol. (2013) 191:5230–8. doi: 10.4049/jimmunol.1301490
31. Gurung P, Lukens JR, Kanneganti T-D. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. (2015) 21:193–201. doi: 10.1016/j.molmed.2014.11.008
32. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem. (1994) 269:15195–203.
33. Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Different. (2007) 14:1583–9. doi: 10.1038/sj.cdd.4402195
34. Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The lysosome rupture-activated TAK1-JNK pathway regulates NLRP3 inflammasome activation. J Biol Chem. (2014) 289:32926–36. doi: 10.1074/jbc.M114.579961
35. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
36. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. (2013) 62:194–204. doi: 10.2337/db12-0420
37. Altaf A, Qu P, Zhao Y, Wang H, Lou D, Niu N. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron Artery Dis. (2015) 26:409–21. doi: 10.1097/MCA.0000000000000255
38. Patel MN, Bernard WG, Milev NB, Cawthorn WP, Figg N, Hart D, et al. Hematopoietic IKBKE limits the chronicity of inflammasome priming and metaflammation. Proc Natl Acad Sci USA. (2015) 112:506–11. doi: 10.1073/pnas.1414536112
39. Peng K, Liu L, Wei D, Lv Y, Wang G, Xiong W, et al. P2X7R is involved in the progression of atherosclerosis by promoting NLRP3 inflammasome activation. Int J Mol Med. (2015) 35:1179–88. doi: 10.3892/ijmm.2015.2129
40. Shao BZ, Wei W, Ke P, Xu ZQ, Zhou JX, Liu C. Activating cannabinoid receptor 2 alleviates pathogenesis of experimental autoimmune encephalomyelitis via activation of autophagy and inhibiting NLRP 3 inflammasome. CNS Neurosci Ther. (2014) 20:1021–8. doi: 10.1111/cns.12349
41. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. (2012) 109:10480–5. doi: 10.1073/pnas.1201836109
42. Cheng YL, Song LQ, Huang YM, Xiong YW, Zhang XA, Sun H, et al. Effect of enterohaemorrhagic Escherichia coli O 157: H 7-specific enterohaemolysin on interleukin-1β production differs between human and mouse macrophages due to the different sensitivity of NLRP 3 activation. Immunology. (2015) 145:258–67. doi: 10.1111/imm.12442
43. Guo W, Liu W, Jin B, Geng J, Li J, Ding H, et al. Asiatic acid ameliorates dextran sulfate sodium-induced murine experimental colitis via suppressing mitochondria-mediated NLRP3 inflammasome activation. Int Immunopharmacol. (2015) 24:232–8. doi: 10.1016/j.intimp.2014.12.009
44. Sun Y, Zhao Y, Yao J, Zhao L, Wu Z, Wang Y, et al. Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-κB and NLRP3 inflammasome activation. Biochem pharmacol. (2015) 94:142–54. doi: 10.1016/j.bcp.2015.02.002
45. Kolb R, Liu G-H, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. (2014) 5:12–20. doi: 10.1007/s13238-013-0001-4
46. Karki R, Man S, Kanneganti T. Inflammasomes and cancer. Cancer Immunol Res. 5:94–99. doi: 10.1158/2326-6066.CIR-16-0269
47. Ozkurede V, Franchi L. Immunology in clinic review series; focus on autoinflammatory diseases: role of inflammasomes in autoinflammatory syndromes. Clin Exp Immunol. (2012) 167:382–90. doi: 10.1111/j.1365-2249.2011.04535.x
48. Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. (2009) 187:61–70. doi: 10.1083/jcb.200903124
49. Youm Y-H, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D, et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome–mediated inflammatory disease. Nat Med. (2015) 21:263–9. doi: 10.1038/nm.3804
50. Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T, et al. Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS ONE. (2014) 9:e85765. doi: 10.1371/journal.pone.0085765
51. Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C, et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. (2014) 109:415. doi: 10.1007/s00395-014-0415-z
52. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. (2017) 214:3219–38. doi: 10.1084/jem.20171419
53. He Y, Varadarajan S, Muñoz-Planillo R, Burberry A, Nakamura Y, Núñez G. 3, 4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem. (2014) 289:1142–50. doi: 10.1074/jbc.M113.515080
54. Perregaux DG, McNiff P, Laliberte R, Hawryluk N, Peurano H, Stam E, et al. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J Pharmacol Exp Ther. (2001) 299:187–97.
55. Marchetti C, Toldo S, Chojnacki J, Mezzaroma E, Liu K, Salloum FN, et al. Pharmacologic inhibition of the NLRP3 inflammasome preserves cardiac function after ischemic and non-ischemic injury in the mouse. J Cardiovasc Pharmacol. (2015) 66:1–8. doi: 10.1097/FJC.0000000000000247
56. Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury following ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol. (2014) 63:316–22. doi: 10.1097/FJC.0000000000000053
57. Kuwar R, Rolfe A, Di L, Xu H, He L, Jiang Y, et al. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J Neuroinflamm. (2019) 16:81. doi: 10.1186/s12974-019-1471-y
58. Liu W, Guo W, Wu J, Luo Q, Tao F, Gu Y, et al. A novel benzo[d]imidazole derivate prevents the development of dextran sulfate sodium-induced murine experimental colitis via inhibition of NLRP3 inflammasome. Biochem Pharmacol. (2013) 85:1504–12. doi: 10.1016/j.bcp.2013.03.008
59. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu J-W, et al. Anti-inflammatory compounds parthenolide and Bay 11–7082 are direct inhibitors of the inflammasome. J Biol Chem. (2010) 285:9792–802. doi: 10.1074/jbc.M109.082305
60. Saadane A, Masters S, Didonato J, Li J, Berger M. Parthenolide inhibits IκB Kinase, NF-κB activation, and inflammatory response in cystic fibrosis cells and mice. Am J Respir Cell Mol Biol. (2007) 36:728–36. doi: 10.1165/rcmb.2006-0323OC
61. Boxer MB, Shen M, Auld DS, Wells JA, Thomas CJ. A Small Molecule Inhibitor of Caspase 1. Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda, MD: National Center for Biotechnology Information (2010).
62. Rudolphi K, Gerwin N, Verzijl N, Kraan PVD, Berg WVD. Pralnacasan, an inhibitor of interleukin-1β converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage. (2003) 11:738–46. doi: 10.1016/S1063-4584(03)00153-5
63. Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. (S)-1-((S)-2-{[1-(4-Amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoyl)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an Orally Available Selective Interleukin (IL)-Converting Enzyme/Caspase-1 Inhi. J Pharmacol Exp Ther. (2007) 321:509–16. doi: 10.1124/jpet.106.111344
64. Strickson S, Campbell DG, Emmerich CH, Knebel A, Plater L, Ritorto MS, et al. The anti-inflammatory drug BAY 11–7082 suppresses the MyD88-dependent signalling network by targeting the ubiquitin system. Biochem J. (2013) 451(Pt 3):427–37. doi: 10.1042/BJ20121651
65. Meng X, Martinez MA, Raymond-Stintz MA, Winter SS, Wilson BS. IKK inhibitor bay 11-7082 induces necroptotic cell death in precursor-B acute lymphoblastic leukaemic blasts. Br J Haematol. (2010) 148:487–90. doi: 10.1111/j.1365-2141.2009.07988.x
66. Krishnan N, Bencze G, Cohen P, Tonks NK. The anti-inflammatory compound BAY 11–7082 is a potent inhibitor of Protein Tyrosine Phosphatases. FEBS J. (2013) 280:2830–41. doi: 10.1111/febs.12283
67. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. (2015) 21:248–55. doi: 10.1038/nm.3806
68. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
69. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med. (2018) 10:e8689. doi: 10.15252/emmm.201708689
70. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci USA. (2018) 115:E1530–9. doi: 10.1073/pnas.1716095115
71. He H, Jiang H, Chen Y, Ye J, Wang A, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. (2018) 9:2550. doi: 10.1038/s41467-018-04947-6
72. Riddle MC. Editorial: sulfonylureas differ in effects on ischemic preconditioning–is it time to retire glyburide? J Clin Endocrinol Metab. (2003) 88:528–30. doi: 10.1210/jc.2002-021971
73. Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest. (2005) 115:2047–58. doi: 10.1172/JCI25495
74. Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature. (2004) 430:213–8. doi: 10.1038/nature02664
75. Hamon Y, Luciani MF, Becq F, Verrier B, Rubartelli A, Chimini G. Interleukin-1beta secretion is impaired by inhibitors of the Atp binding cassette transporter, ABC1. Blood. (1997) 90:2911–5. doi: 10.1182/blood.V90.8.2911
76. Lottaz D, Beleznay Z, Bickel M. Inhibition of ATP-binding cassette transporter downregulates interleukin-1beta-mediated autocrine activation of human dermal fibroblasts. J Invest Dermatol. (2001) 117:871–6. doi: 10.1046/j.0022-202x.2001.01451.x
77. Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, et al. Structural insights of benzenesulfonamide analogues as NLRP3 inflammasome inhibitors: design, synthesis, and biological characterization. J Med Chem. (2018) 61:5412–23. doi: 10.1021/acs.jmedchem.8b00733
78. Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, et al. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer's disease. Mol Neurobiol. (2018) 55:1977–87. doi: 10.1007/s12035-017-0467-9
79. Heinrich M, Robles M, West JE, Ortiz de Montellano BR, Rodriguez E. Ethnopharmacology of Mexican asteraceae (compositae). Ann Rev Pharmacol Toxicol. (1998) 38:539–65. doi: 10.1146/annurev.pharmtox.38.1.539
80. Guzman ML, Rossi RM, Neelakantan S, Li X, Corbett CA, Hassane DC, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. (2007) 110:4427–35. doi: 10.1182/blood-2007-05-090621
81. D'anneo A, Carlisi D, Lauricella M, Puleio R, Martinez R, Di Bella S, et al. Parthenolide generates reactive oxygen species and autophagy in MDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. (2013) 4:e891. doi: 10.1038/cddis.2013.415
82. Mackenzie SH, Schipper JL, Clark AC. The potential for caspases in drug discovery. Curr Opin Drug Discov Devel. (2010) 13:568–76. doi: 10.4049/jimmunol.168.6.3024
84. Strand V, Sokolove J. Randomized controlled trial design in rheumatoid arthritis: the past decade. Arthritis Res Ther. (2009) 11:205. doi: 10.1186/ar2555
85. Fischer U, Schulzeosthoff K. Apoptosis-based therapies and drug targets. Cell Death Different. (2005) 12(Suppl 1):942–61. doi: 10.1038/sj.cdd.4401556
86. Maroso M, Balosso S, Ravizza T, Liu J, Bianchi ME, Vezzani A. Interleukin-1 type 1 receptor/Toll-like receptor signalling in epilepsy: the importance of IL-1beta and high-mobility group box 1. J Int Med. (2011) 270:319–26. doi: 10.1111/j.1365-2796.2011.02431.x
87. Flores J, Noël A, Foveau B, Lynham J, Lecrux C, Leblanc AC. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer's disease mouse model. Nat Commun. (2018) 9:3916. doi: 10.1038/s41467-018-06449-x
88. Audia JP, Yang XM, Crockett ES, Housley N, Haq EU, O'Donnell K, et al. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res Cardiol. (2018) 113:32. doi: 10.1007/s00395-018-0692-z
89. Kerr ID, Lee JH, Farady CJ, Marion R, Rickert M, Sajid M, et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J Biol Chem. (2009) 284:25697–703. doi: 10.1074/jbc.M109.014340
90. Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer's disease in rodent models. Nat Commun. (2016) 7:12504. doi: 10.1038/ncomms12504
91. Dempsey C, Araiz AR, Bryson K, Finucane O, Larkin C, Mills E, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun. (2017) 61:306–16. doi: 10.1016/j.bbi.2016.12.014
92. Vande Walle L, Stowe IB, Šácha P, Lee BL, Demon D, Fossoul A, et al. MCC950/CRID3 potently targets the NACHT domain of wildtype NLRP3 but not disease-associated mutants for inflammasome inhibition. bioRxiv. (2019) 634493. doi: 10.1101/634493
93. Primiano MJ, Lefker BA, Bowman MR, Bree AG, Hubeau C, Bonin PD, et al. Efficacy and pharmacology of the NLRP3 inflammasome inhibitor CP-456,773 (CRID3) in murine models of dermal and pulmonary inflammation. J Immunol. (2016) 197:2421–33. doi: 10.4049/jimmunol.1600035
94. Gordon R, Albornoz EA, Christie DC, Langley MR, Kumar V, Mantovani S, et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Trans Med. (2018) 10:eaah4066. doi: 10.1126/scitranslmed.aah4066
95. Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, et al. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin–induced intestinal fluid secretion. J Clin Invest. (2002) 110:1651–8. doi: 10.1172/JCI16112
96. Sonawane N, Verkman A. Thiazolidinone CFTR inhibitors with improved water solubility identified by structure–activity analysis. Bioorgan Med Chem. (2008) 16:8187–95. doi: 10.1016/j.bmc.2008.07.044
97. Darakhshan S, Pour AB. Tranilast: a review of its therapeutic applications. Pharmacol Res. (2015) 91:15–28. doi: 10.1016/j.phrs.2014.10.009
99. Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. (2005) 310:850–5. doi: 10.1126/science.1117634
100. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
101. Kadota S, Basnet P, Ishii E, Tamura T, Namba T. Antibacterial activity of trichorabdal A from Rabdosia trichocarpa against Helicobacter pylori. Zentralblatt Bakteriol. (1997) 286:63–7. doi: 10.1016/S0934-8840(97)80076-X
102. Kuo L-M, Kuo C-Y, Lin C-Y, Hung M-F, Shen J-J, Hwang T-L. Intracellular glutathione depletion by oridonin leads to apoptosis in hepatic stellate cells. Molecules. (2014) 19:3327–44. doi: 10.3390/molecules19033327
103. Ding Y, Ding C, Ye N, Liu Z, Wold EA, Chen H, et al. Discovery and development of natural product oridonin-inspired anticancer agents. Eur J Med Chem. (2016) 122:102–17. doi: 10.1016/j.ejmech.2016.06.015
104. Fujita E, Nagao Y, Kaneko K, Nakazawa S, Kuroda H. The antitumor and antibacterial activity of the Isodon diterpenoids. Chem Pharm Bull. (1976) 24:2118–27. doi: 10.1248/cpb.24.2118
105. Huang J, Wu L, Tashiro S-i, Onodera S, Ikejima T. A comparison of the signal pathways between the TNFα-and oridonin-induced Murine L929 fibrosarcoma cell death. Acta Med Okayama. (2005) 59:261–70. doi: 10.18926/AMO/31960
106. Xu Y, Xue Y, Wang Y, Feng D, Lin S, Xu L. Multiple-modulation effects of Oridonin on the production of proinflammatory cytokines and neurotrophic factors in LPS-activated microglia. Int Immunopharmacol. (2009) 9:360–5. doi: 10.1016/j.intimp.2009.01.002
107. Zhao G, Zhang T, Ma X, Jiang K, Wu H, Qiu C, et al. Oridonin attenuates the release of pro-inflammatory cytokines in lipopolysaccharide-induced RAW264. 7 cells and acute lung injury. Oncotarget. (2017) 8:68153. doi: 10.18632/oncotarget.19249
108. Wang S, Zhang Y, Saas P, Wang H, Xu Y, Chen K, et al. Oridonin's therapeutic effect: Suppressing T h1/T h17 simultaneously in a mouse model of C rohn's disease. J Gastroenterol Hepatol. (2015) 30:504–12. doi: 10.1111/jgh.12710
109. Wang S, Yang H, Yu L, Jin J, Qian L, Zhao H, et al. Oridonin attenuates Aβ1–42-induced neuroinflammation and inhibits NF-κB pathway. PLoS ONE. (2014) 9:e104745. doi: 10.1371/journal.pone.0104745
110. Zhang ZY, Daniels R, Schluesener HJ. Oridonin ameliorates neuropathological changes and behavioural deficits in a mouse model of cerebral amyloidosis. J Cell Mol Med. (2013) 17:1566–76. doi: 10.1111/jcmm.12124
111. Jiang D, Chen S, Sun R, Zhang X, Wang D. The NLRP3 inflammasome: role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. (2018) 419:8–19. doi: 10.1016/j.canlet.2018.01.034
112. Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. (2013) 25:469–84. doi: 10.1016/j.smim.2013.10.008
113. Nowarski R, Jackson R, Gagliani N, de Zoete MR, Palm NW, Bailis W, et al. Epithelial IL-18 equilibrium controls barrier function in colitis. Cell. (2015) 163:1444–56. doi: 10.1016/j.cell.2015.10.072
114. Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundbäck P, et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. (2012) 488:670–4. doi: 10.1038/nature11290
115. Kanneganti A, Malireddi RS, Saavedra PH, Walle LV, Van Gorp H, Kambara H, et al. GSDMD is critical for autoinflammatory pathology in a mouse model of Familial Mediterranean Fever. Journal of Experimental Medicine. (2018) 215:1519–29. doi: 10.1084/jem.20172060
116. Xiao J, Wang C, Yao J-C, Alippe Y, Xu C, Kress D, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. (2018) 16:e3000047. doi: 10.1371/journal.pbio.3000047
117. Gaidt MM, Hornung V. Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J. (2016) 35:2167–9. doi: 10.15252/embj.201695415
118. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
119. Salvador JP, Vilaplana L, Marco MP. Nanobody: outstanding features for diagnostic and therapeutic applications. Anal Bioanal Chem. (2019) 411:1703–13. doi: 10.1007/s00216-019-01633-4
Keywords: NLRP3 inflammasome, inhibitors, MCC950, drug screening, IL-1β
Citation: Zahid A, Li B, Kombe AJK, Jin T and Tao J (2019) Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 10:2538. doi: 10.3389/fimmu.2019.02538
Received: 27 July 2019; Accepted: 14 October 2019;
Published: 25 October 2019.
Edited by:
Christopher Lupfer, Missouri State University, United StatesReviewed by:
Mark Wewers, The Ohio State University, United StatesCopyright © 2019 Zahid, Li, Kombe, Jin and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tengchuan Jin, amludEB1c3RjLmVkdS5jbg==; Jinhui Tao, dGFvamluaHVpQHVzdGMuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.