 Madison R. Rogan
Madison R. Rogan LaNisha L. Patterson
LaNisha L. Patterson Jennifer Y. Wang
Jennifer Y. Wang Jere W. McBride
Jere W. McBride
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 17 October 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02390
This article is part of the Research Topic Wnt Signaling in Immune Cell Regulation During Microbial Infection and Cancer View all 10 articles
The host-pathogen interface is a crucial battleground during bacterial infection in which host defenses are met with an array of bacterial counter-mechanisms whereby the invader aims to make the host environment more favorable to survival and dissemination. Interestingly, the eukaryotic Wnt signaling pathway has emerged as a key player in the host and pathogen tug-of-war. Although studied for decades as a regulator of embryogenesis, stem cell maintenance, bone formation, and organogenesis, Wnt signaling has recently been shown to control processes related to bacterial infection in the human host. Wnt signaling pathways contribute to cell cycle control, cytoskeleton reorganization during phagocytosis and cell migration, autophagy, apoptosis, and a number of inflammation-related events. Unsurprisingly, bacterial pathogens have evolved strategies to manipulate these Wnt-associated processes in order to enhance infection and survival within the human host. In this review, we examine the different ways human bacterial pathogens with distinct host cell tropisms and lifestyles exploit Wnt signaling for infection and address the potential of harnessing Wnt-related mechanisms to combat infectious disease.
The innate immune response is the first, and in many successful cases, the primary barrier between bacterial invader and human host. Fine-tuned through co-evolution with microbial insults and largely evolutionarily conserved across the Metazoa, the innate immune system specializes in both pathogen recognition and the formation of a rapid response (1). At the foundation of the innate cellular antimicrobial response are families of germline-encoded pattern recognition receptors (PRRs) expressed by professional phagocytes, including Toll-like, C-type lectin, NOD-like, and RIG-I-like receptors (2, 3). Interaction of receptor and ligand, which include lipoproteins, polysaccharides, nucleic acids, and other conserved microbial molecular patterns, results in an inflammatory response involving cytokine, and chemokine gene transcription, pathogen clearance through various mechanisms such as lysosomes, antimicrobial peptides, or membrane attack complexes, and coordination of the adaptive immune response (2, 4). Unsurprisingly, bacterial pathogens have evolved an armament of immunoevasion mechanisms as carefully selected for as the immune system that defends the host. The very foundation of bacterial virulence is the ability to subvert host defenses in order to establish a replicative niche. This involves mechanisms beneficial to pathogens that replicate in the extracellular niche, such as serum resistance, tissue adherence, and motility; mechanisms beneficial to the pathogens that replicate in an intracellular niche, such as controlling host cell fate and avoiding lysosomal destruction; and mechanisms employed by pathogens of both niches including competitive nutrient acquisition systems and secretion of effector proteins to modulate the host.
The means by which pathogens subvert the host innate immune responses have helped expand our knowledge regarding how eukaryotic cellular signaling pathways cooperate to modulate innate immunity. Often, these pathways moonlight as branches of the immune system, as they were originally discovered in the context of cell development or cancer. For example, eukaryotic Notch signaling is a well-characterized regulator of cell fate that is highly active in development and tissue homeostasis (5). This pathway has been shown to function within innate immunity through regulation of PRR expression. The obligately intracellular pathogen Ehrlichia chaffeensis utilizes a type 1 secretion system (T1SS) effector to activate Notch signaling which indirectly downregulates PU.1, a transcriptional activator of TLR2 and 4 (6). The discovery of xenophagy as a type of autophagy is another example of a moonlighting innate immune pathway. Years of research have demonstrated that a process originally thought only to function as a cellular starvation and stress response also functions within the innate immune system (7). Xenophagy is deployed against Group A Streptococcus as a method of bacterial clearance, while other pathogens express effector proteins to escape xenophagy, as is the case with Listeria monocytogenes expression of xenophagy evasion protein ActA (8, 9). As evidence linking conserved eukaryotic cell pathways to the immunosubversion of human pathogens continues to grow, as does our model of the innate immune system and the networks that comprise it.
Mounting research within the last two decades has demonstrated that the conserved eukaryotic signaling pathway Wnt is a significant part of the interplay between the human host and both extracellular and intracellular bacterial pathogens. The discovery of the Wnt pathway began with the identification of the murine oncogene int-1 that was found to be homologous with a Drosophila gene that controlled body segmentation during development (10, 11). Later renamed Wnt proteins, these gene products are a family of 19 highly conserved, secreted, lipidated glycoproteins that regulate metazoan development and tissue homeostasis (12). Wnt proteins participate in paracrine and autocrine signaling through binding of 1 of 10 homologs of the seven-pass transmembrane receptor Frizzled (Fzd1-10) and a cognate coreceptor expressed on the surface of the signal-receiving cell. The signal is transduced through the intracellular mediator Disheveled (Dvl) which, depending on its phosphorylation state, activates either canonical or non-canonical pathways (13). Canonical Wnt signaling, also known as β-catenin-dependent signaling, is the most well-studied Wnt pathway (Figure 1) (14). In the pathway off state, the β-catenin destruction complex consisting of Axin, adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK3β), and casein kinase 1 (CK1) facilitates the phosphorylation of β-catenin by GSK3β which induces ubiquitination of β-catenin by the β-TrCP-SCF E3 RING-type ubiquitin ligase complex (β-TrCP) and subsequent proteasomal degradation. When Dvl is activated through interaction of a Wnt ligand with a Fzd receptor and the canonical pathway coreceptor lipoprotein receptor-related protein 5/6 (LRP5/6), the destruction complex is recruited to the Frizzled-Dvl complex at the plasma membrane, freeing β-catenin from degradation. Accumulation of the cytoplasmic pool of β-catenin induces its translocation into the nucleus where it binds with T-cell factor (TCF) transcription factor at the Wnt response element (WRE) DNA sequence and activates transcription of target genes involved in processes such as development (SNAIL, ENGRAILED, SLUG) and cell proliferation (CMYC, CCND1, and MMP7).
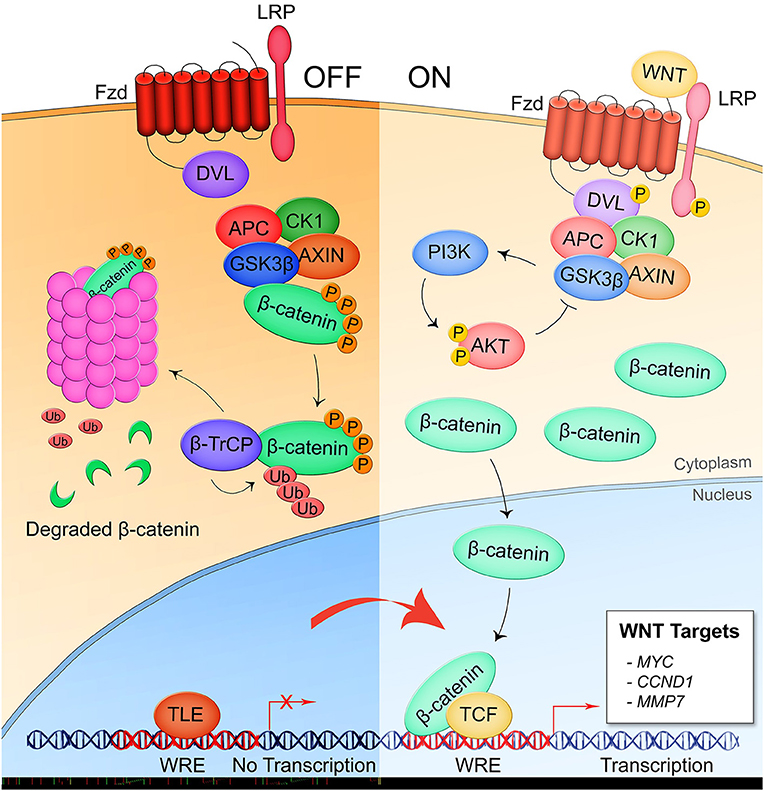
Figure 1. Canonical/β-catenin-dependent Wnt signaling. In the pathway off state, the β-catenin destruction complex consisting of APC, CK1, GSK3β, and AXIN binds β-catenin leading to its phosphorylation by GSK3β and subsequent ubiquitination by the E3 ubiquitin ligase complex β-TrCP which targets β-catenin for proteasomal degradation. Thus, the Wnt response element (WRE) located in the promoter region of Wnt pathway target genes remains bound by transcriptional repressor TLE. When a secreted Wnt ligand originating from the same or a nearby cell binds Fzd and the coreceptor LRP5/6, Dvl is phosphorylated and sequesters the destruction complex, preventing the phosphorylation of β-catenin. Accumulation of β-catenin is the cytoplasm leads to nuclear translocation of the protein where it binds with co-activator TCF at the WRE and drives expression of Wnt target genes.
Non-canonical, β-catenin-independent signaling can be divided into two pathways: the Wnt/Ca2+ pathway and the planar cell polarity (Wnt/PCP) pathway. In the Wnt/Ca2+ pathway, Wnt ligands signal through Fzd and the coreceptor receptor tyrosine kinase-like orphan receptor 1/2 (ROR1/2) to induce Dvl-dependent phospholipase C (PLC) cleavage of phosphatidylinositol 4,5-biphosphate (PIP2), producing inositol triphosphate (IP3) and diacyl glycerol (DAG) (Figure 2A) (15). IP3 acts on Ca2+ channels at the endoplasmic reticulum resulting in a wave of cytosolic Ca2+ that drives protein kinase C (PKC) and Ca2+/calmodulin-dependent protein kinase II (CAMKII) activity. This controls nuclear translocation of nuclear factor of activated T cells (NFAT) for target gene transcription and actin polymerization through the Rho GTPase CDC42. NFAT target genes have been most thoroughly researched in the context of osteoclast formation and T cell regulation. In the transcription-independent Wnt/PCP pathway, cell polarity and migration are regulated through the direct interaction of Dvl and Dvl-associated activator of morphogenesis (DAAM1), G protein activation of the small GTPase Rac, and Dvl activation of phosphoinositide 3-kinase (PI3K) (Figure 2B) (16). Non-canonical pathways demonstrate a high amount of crosstalk and together regulate events such as filopodia formation, cell movement, and establishment of cell polarity.
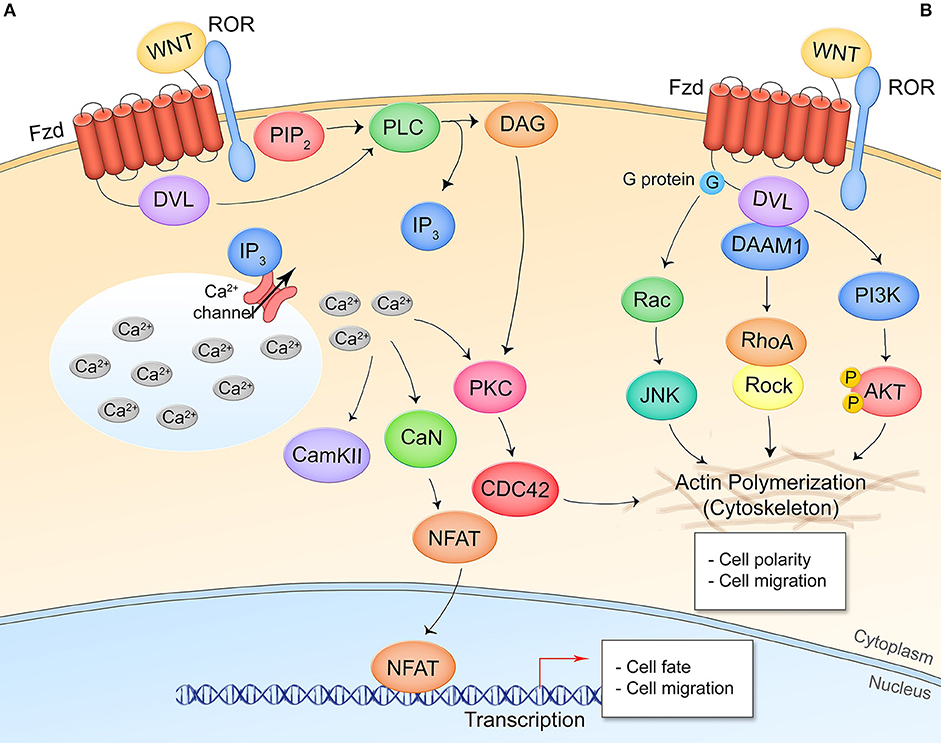
Figure 2. Non-canonical/β-catenin-independent Wnt signaling pathways. (A) Wnt/Ca2+ signaling. A Wnt ligand interaction with Fzd and the non-canonical coreceptors ROR1/2 activates Dvl which activates PLC, leading to production of IP3 and opening of cytosolic Ca2+ stores. CAMKII, CaN, and PKC are activated by the increased cytosolic Ca2+ levels. CaN dephosphorylates NFAT leading to its nuclear translocation and expression of NFAT target genes. PKC crosstalks with Wnt/PCP signaling. (B) Wnt/planar cell polarity signaling. Interaction of Wnt and respective receptor/coreceptor leads to cytoskeletal reorganization through G-protein activation of Ras-JNK signaling, Dvl-DAAM1 association and Rho activation, and Dvl activation of PI3K and Akt. This pathway is transcription-independent.
Pathogen manipulation of Wnt signaling as a mechanism of innate immune subversion takes advantage of three main outcomes of the signal cascade: cell fate determination (including maintenance of epithelia and endothelia), anti-inflammatory effects, and phagolysosome formation. In this review, we highlight mechanisms of Wnt pathway manipulation by several extracellular, and obligate intracellular pathogens, with a specific focus on the virulence factors involved and the consequences on innate immune subversion. Table 1 summarizes the role of Wnt signaling in the pathogenesis of the bacteria reviewed herein. Ultimately, our understanding of host-pathogen interaction at the cellular and molecular levels will highlight potential targets for therapeutic intervention and expand our model of the many roles of Wnt signaling within the eukaryotic cell.
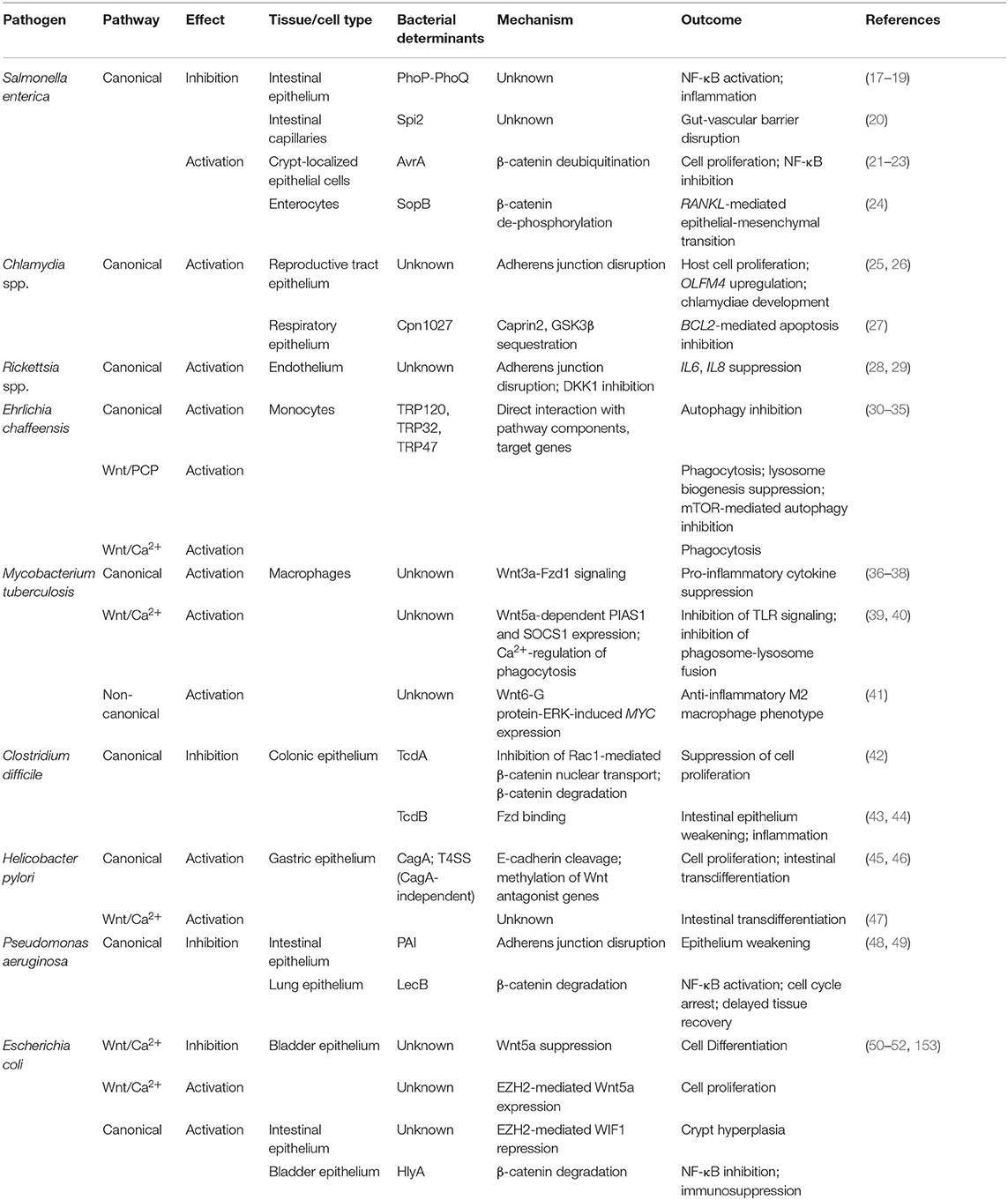
Table 1. Summary of the role of Wnt signaling pathways and respective bacterial factors involved in the pathogenesis of representative bacteria.
The Gram-negative, facultative intracellular bacillus Salmonella enterica causes typhoid fever or non-typhoidal salmonellosis in humans. The bacteria are commonly foodborne pathogens but can also be transmitted fecal-orally (53). Salmonella establish infection in the gut where they replicate within the lumen until sufficient numbers induce their entry into M cells which is triggered by T3SS effectors. S. enterica are also phagocytosed by various phagocytic cells but can survive phagolysosome acidification and replicate within the intracellular vacuole. Dissemination to other organs is accompanied by a robust immune response and the potential for persistent infection within various cell types. Crossing of the gut barrier and infection of infiltrating immune cells including neutrophils, monocytes, and macrophages are essential to dissemination.
Nearly two decades of research have created the model of Salmonella enterica serovar Typhimurium manipulation of canonical Wnt signaling to promote infection (Figure 3A). Curiously, S. Typhimurium suppresses Wnt signaling in intestinal epithelium and the underlying capillary endothelium but activates the pathway in intestinal stem cells in what appears to be a cell type- or temporally-specific mechanism. In transformed T84 colon carcinoma cells, wild type S. Typhimurium represses pathway activation as identified by significant suppression of β-catenin levels that correlates with decreased formation of the TCF-β-catenin complex in the nucleus, dampened expression of the Wnt target gene CMYC, and suppression of cellular proliferation (17). This is contrasted with colonization by non-pathogenic S. Typhimurium strain PhoPC which possesses an attenuating, constitutively active PhoP-PhoQ two-component system and does not suppress β-catenin-TCF complex formation, implicating a role for this response regulator in suppression of Wnt signaling by pathogenic salmonellae in the colonic epithelium, both in cell culture and in a mouse model (18).
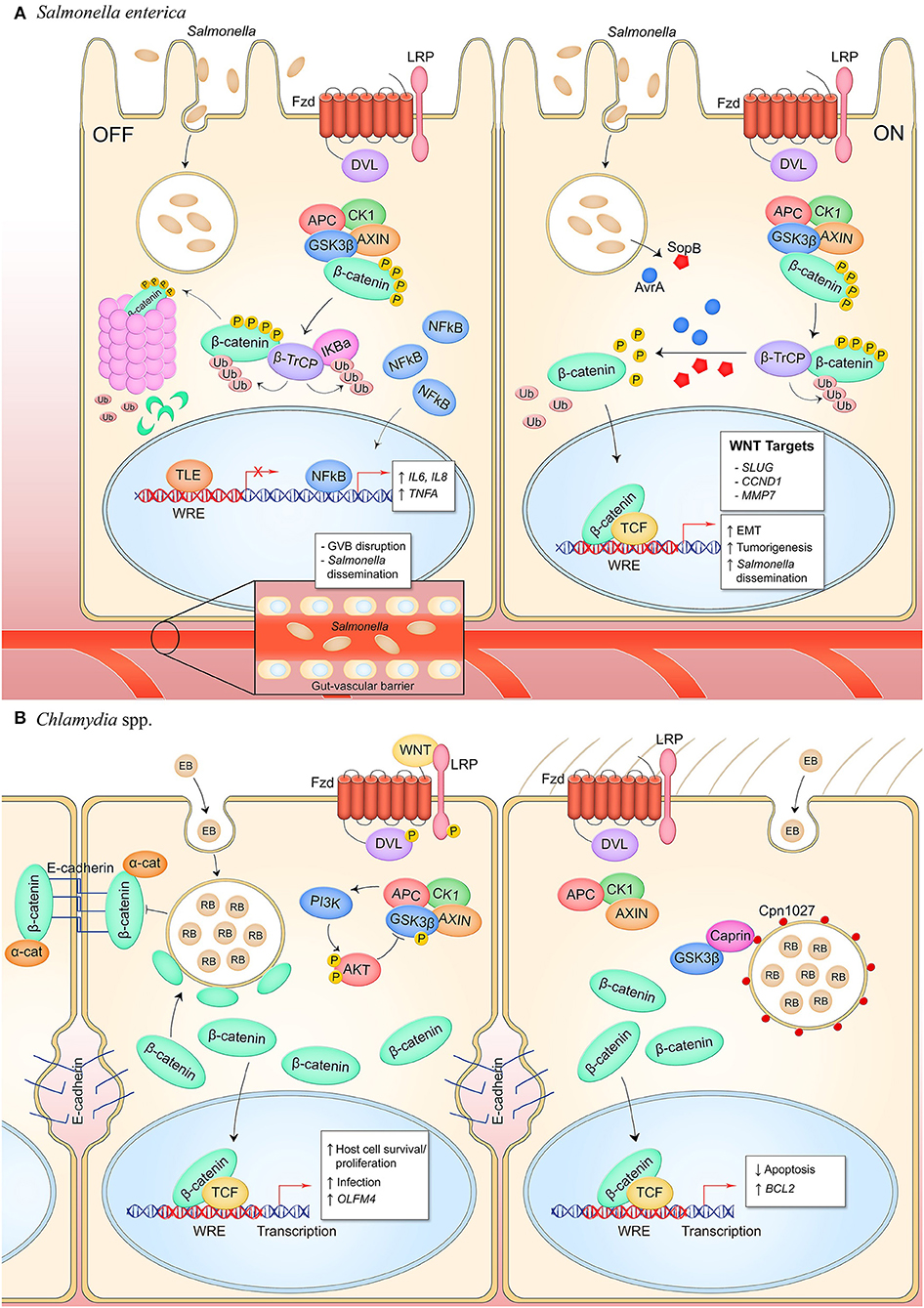
Figure 3. Canonical Wnt signaling manipulation during bacterial infection of epithelial cells. (A) S. enterica infection of intestinal epithelial cells inhibits Wnt signaling through an unknown mechanism (left). Activity of the E3 ubiquitin ligase β-TrCP, which targets both β-catenin and IκBα for proteasomal degradation, stabilizes NF-κB levels which causes nuclear translocation of the protein and expression of pro-inflammatory target genes. Wnt signaling is also inhibited in the GVB during infection, promoting bacterial access to vasculature by increasing vascular permeability. S. enterica secretes the T3SS effectors AvrA and SopB into intestinal M cells and crypt-localized epithelial cells which causes activation of canonical Wnt signaling (right). Deubiquitinase AvrA and phosphatase SopB induce pathway activation through reversal of β-catenin posttranslational modifications, promoting expression of β-catenin-dependent genes that drive EMT in M cells, induce intestinal stem cell proliferation, and inhibit NF-κB activity in stem cells. (B) C. trachomatis infection of reproductive tract epithelium induces breakdown of adherens junctions and accumulation of β-catenin in the cytoplasm (left). β-catenin localizes to the chlamydial inclusion and translocates into the nucleus to activate transcription. Pathway activity promotes the bacterial developmental cycle through unknown mechanisms as well as drives expression of OLFM4 which is known to inhibit NF-κB signaling. C. pneumoniae expresses inclusion membrane protein Cpn1027 during infection of respiratory epithelium (right). Cpn1027 recruits Caprin2, a scaffold protein of the β-catenin destruction complex, as well as GSK3β, thereby reducing β-catenin turnover and allowing nuclear translocation for expression of target gene BCL2 to inhibit host cell apoptosis.
The effects of S. Typhimurium suppression of Wnt signaling in the intestines have created two models for bacterial host manipulation by Wnt signaling modulation. First, Salmonella studies have revealed a mechanism behind the immunosuppressive effects of β-catenin signaling. IκBα negatively regulates NF-κB transcriptional activity through cytosolic sequestration of NF-κB. It has been shown that β-catenin associates with this complex, indirectly stabilizing IκBα through an unknown mechanism (19). Activation of the NF-κB pathway causes ubiquitination of IκBα by the ubiquitin E3 ligase β-TrCP, the same ligase that targets β-catenin for degradation to suppress Wnt pathway activity, which results in ubiquitin-dependent degradation of IκBα (54). Activation of this E3 ligase therefore results in silencing of Wnt signaling through loss of β-catenin and activation of NF-κB signaling through loss of IκBα repression, while suppression of β-TrCP has the opposite effect. During S. Typhimurium infection of colonic epithelial cells, the NF-κB target genes IL6, IL8, and TNFA are induced, corresponding with enhanced ubiquitination of IκBα as well as β-catenin degradation which demonstrates the reciprocal activation of these two pathways by infection (18, 19). Infection in the presence of lithium chloride, a β-catenin stabilizing agent, or a constitutively active β-catenin mutant CTNNB1−/Δ45 attenuates this effect. Activation of NF- κB signaling through direct inhibition of β-catenin-dependent Wnt signaling induces the proinflammatory response which recruits dendritic cells and macrophages. While TNF-α is important for controlling replication and spread of bacteria, the cells producing it are also host cells for both replicative and persistent salmonellae, thereby supporting survival and dissemination of the bacteria (53, 55, 56).
Recently, another pathogenic mechanism was discovered by which salmonellae inhibit canonical Wnt signaling at the gut-vascular barrier (GVB), a system of tight and adherens junctions in the capillaries underlying the gut epithelium that functions as a size-selective barrier to molecules traversing the gut barrier (20). Similar to the blood-brain barrier, the GVB is regulated by canonical Wnt signaling (20, 57). This protective barrier is impermeable to bacteria, but infection by S. Typhimurium results in downregulation of Wnt signaling in the endothelium which enhances vascular leakiness and promotes bacterial dissemination (20). In the presence of recombinant canonical pathway ligand Wnt3a, canonical signaling is activated and S. Typhimurium-induced leakiness is reduced. S. Typhimurium possess two pathogenicity islands (Spi1 and Spi2) that together encode the T3SS. A strain lacking Spi2 is unable to induce vascular permeability, indicating this pathogenic mechanism is Spi2-dependent. This model demonstrates how salmonellae permeabilize endothelial layers utilizing T3SS factors without directly manipulating cell-cell contacts, a mechanism that is employed by other endothelial pathogens such as Rickettsia. Whether this implicates activation of Wnt signaling as a potential therapeutic for S. enterica infection, and whether specific S. enterica pathogenicity island effectors are responsible for Wnt pathway inhibition remains to be investigated.
Studies using a Salmonella colitis mouse model have demonstrated that S. Typhimurium is able to stimulate Wnt signaling in intestinal stem cells in contrast to the inhibitory mechanism deployed in the epithelium and endothelium. The T3SS effector AvrA, a deubiquitinase, has been shown to activate canonical Wnt signaling in intestinal stem cells through deubiquitination and subsequent stabilization of β-catenin (21, 58). Additionally, AvrA alone, in the absence of infection, can activate pathway activity. In the colon of the Salmonella colitis mouse model, stem cells demonstrate β-catenin nuclear localization and expression of Wnt target genes MMP7 and CCND1 (22). Hyperactivation mediates pathological effects, and studies have shown that in mice with induced inflammation, infection with AvrA+ S. Typhimurium drives tumorigenesis (22). In addition to AvrA, T3SS effector SopB has tumorigenic properties through inducing cellular transformation of follicular-associated epithelial enterocytes into microfold cells (24). This has also been linked to a canonical Wnt-dependent mechanism. The phosphatase dephosphorylates β-catenin and Akt in primary rectal epithelial cells, resulting in β-catenin-dependent signaling and expression of target gene SLUG which activates RANKL expression, a critical cytokine for M cell development (24, 59). Indeed, increased M cells is a phenotype of S. enterica infection and proliferation of this cell type promotes S. Typhimurium invasion of the intestines for enhanced survival and dissemination.
S. Typhimurium has proven to be a model pathogen for understanding the role of canonical Wnt signaling in both gut tissue maintenance and suppression of inflammatory pathway signaling, physiological processes that are applicable to a range of pathogens that occupy a similar niche. Insight to Salmonella inhibition of signaling to perturb the GVB brings to light the therapeutic potential of Wnt ligands to maintain endothelial barriers. R-spondin3, a Wnt homolog, has been shown to exhibit an anti-inflammatory effect in an ischemia/reperfusion mouse model and induces tightening of endothelial junctions and loss of vascular leakiness (60). Furthermore, understanding how effectors SopB and AvrA contribute to pathway manipulation during infection highlights the potential of such mechanisms as therapeutics for infection or other disease states mediated by dysregulated Wnt signaling.
Chlamydia trachomatis and Chlamydia pneumoniae are obligately intracellular, anaerobic pathogens that typically target the human genital and respiratory tract mucosal epithelial cells, respectively. Chlamydiae undergo a developmental cycle in which they transition between two ultrastructural forms at different phases of infection (61). The elementary body (EB) is the infectious form of the bacteria that triggers entry into the host cell. Once intracellular, the chlamydiae transition into the replicative reticulate body (RB) form within the inclusion, the membrane-bound microcolony. The intracellular chlamydiae interact with the host cell through both T3SS effector proteins as well as inclusion membrane-localized proteins that interface with the host cell cytoplasm. Environmental cues such as drug presence or immunological stress can induce the EB to enter a persistent form in which they evade immune detection by entering a dormant state within the inclusion (62). At the end of the infection cycle, the RB transition back into and EB and leave the cell through lysis or exocytosis to infect a neighboring cell.
C. trachomatis targets epithelial cells of the genital tract, including the endometrial and fallopian epithelium. Wnt/β-catenin signaling is known to maintain epithelial cell homeostasis through regulating tissue renewal and cell proliferation, and through facilitating epithelial barrier integrity by the β-catenin-E-cadherin complex that constitutes adherens junctions (63–65). C. trachomatis infection in the fallopian tube has been shown to disrupt adherens junctions and cause redistribution of β-catenin from the plasma membrane to the chlamydial inclusion (25) (Figure 3B). It is unclear if disruption of these junctions amplifies β-catenin nuclear localization and target gene expression, but infected epithelium does demonstrate increased Wnt pathway activity evidenced by phosphorylation-dependent inactivation of GSK3β and redistribution of APC which indicates inactivation of the β-catenin destruction complex (25). Additionally, inhibition of Wnt signaling through either RNA silencing of β-catenin or a small molecule inhibitor reduces infectivity of the chlamydiae and impairs chlamydiae intracellular development (25, 26). Thus, signaling is beneficial to chlamydiae and may be synergistically activated through inhibition of the β-catenin destruction complex and disruption of adherens junctions. The known beneficial phenotypes of Wnt signaling for the chlamydial niche are 2-fold. First, infection causes Wnt signaling-dependent host cell proliferation, a critical survival strategy for an obligately intracellular pathogen that can maintain persistent infection (25). Second, Wnt signaling upregulates the stem cell marker OLFM4 which is a suppressor of NOD1/2 and NF-κB-dependent pro-inflammatory cytokine expression (23). Therefore, Wnt signaling appears to be an active mechanism of pathogenesis by which Chlamydia establishes infection and suppresses NF-kB-mediated innate immune mechanisms.
C. pneumoniae inclusion protein Cpn1027 is the only chlamydial protein known to directly interface with the canonical Wnt pathway. During C. pneumoniae infection of respiratory epithelium, Cpn1027 directly binds the Caprin2, an adaptor protein within the β-catenin destruction complex (27, 66). GSK3β also localizes to the Cpn1027-Caprin2 complex and demonstrates decreased kinase activity. Consequently, β-catenin translocates to the nucleus and drives expression of the anti-apoptotic BCL2 gene, linking the infection phenotype of apoptosis inhibition to manipulation of Wnt signaling for enhanced intracellular survival. This mechanism of pathway manipulation is unique to C. pneumoniae as Cpn1027 is not expressed by other species of the Chlamydia genus (67).
Several questions remain regarding the role of Wnt signaling during Chlamydia spp. infection. The consequence of β-catenin localization to the C. trachomatis inclusion concurrent with adherens junction disruption is unclear, as signaling is not inhibited in the host cell, and β-catenin is necessary for the chlamydial intracellular life cycle. The chlamydial deubiquitinase ChlaDub 1 has been shown to deubiquitinate NF-κB inhibitor IκBα in order to suppress the NF-κB-dependent expression of proinflammatory cytokines (68). β-catenin and IκBα are both substrates of the E3 ligase SCFβ−TrCP, raising the question of whether β-catenin localizes to the chlamydial inclusion to also serve as a ChlaDub 1 substrate which would lead to pathway activation (69). Identifying interactions between chlamydial secreted effectors or inclusion proteins and components of the Wnt pathway will define novel host-bacterial pathogenic interactions that can be targeted by therapeutics. In addition to understanding pathogenic mechanisms, further research will shed light on chlamydial mechanisms of cellular transformation. Chlamydia trachomatis is associated with cancer development through infection-induced degradation of p53 and dysregulated ROS production (70–72). It is well-known that canonical Wnt signaling is a driver of tumorigenesis in multiple human cancers, including cervical cancer (73). However, a model for cervical cancer development through C. trachomatis-induced hyperactivation of Wnt signaling has not been investigated.
The Rickettsia genus comprises 27 species of obligately intracellular Gram-negative bacteria, over half of which are human pathogens primarily transmitted by various arthropod vectors (74). R. conorii and R. rickettsii are both members of the spotted fever group of Rickettsia and causative agents of the human diseases Mediterranean spotted fever and Rocky Mountain spotted fever, respectively. These pathogens establish infection in the endothelium which induces an inflammatory response consisting of increased vascular permeability, recruitment and activation of natural killer cells and macrophages, and ROS- and cytokine-mediated vascular damage. The bacteria escape from their endocytic vesicle and replicate in the cytosol where they utilize actin-based motility for cell-to-cell spread. Robust production of cytokines IL-6 and IL-8 correlate with infection lethality, and clearance of the bacteria is typically mediated by PRR engagement (75).
Wnt signaling has a complex role in the endothelium, and activation of the pathway induces endothelial cell proliferation and enhanced interaction between endothelial cells and monocytes (76, 77). Additionally, β-catenin is present at endothelial adherens junctions, regulating cell-cell contacts (78). The Wnt signaling pathway controls neovascularization during development but demonstrates decreased activity in adult vasculature (79). However, activation in certain disease states including infection and cancer can induce Wnt-dependent vascular endothelial growth factor A (VEGF-A) expression followed by angiogenesis (76). Activated endothelial cells also release DKK1, a member of the Dickkopf family of secreted Wnt signaling antagonists that exert their effect by outcompeting Wnt ligands for binding of coreceptor LRP5/6 (80). DKK1 is a β-catenin target gene that functions through feedback inhibition of Wnt signaling, thereby decreasing neovascularization induced by VEGF-A (81, 82).
In a HUVEC model of R. conorii infection, β-catenin rapidly localizes to the nucleus within 2 hpi, indicating activation of Wnt signaling early in infection (Figure 4) (28). It is unclear how the pathway is activated by rickettsiae, as other markers of signaling activity have not been investigated. In a HUVEC model of R. rickettsii infection, adherens junctions are disrupted and β-catenin is redistributed from primarily membrane-localized to diffuse localization throughout the cell (29). A correlation was identified between vascular permeability through loss of adherens junction integrity as well as increased expression of NF-κB-dependent inflammatory cytokines, but whether this is related to canonical Wnt signaling reciprocal regulation of the NF-κB signaling pathway is unknown. Increased nuclear entry of β-catenin during rickettsial infections has not been specifically investigated, but consistent with other models of infection-induced adherens junctions remodeling, it is likely that Rickettsia-induced cell contact disruption can activate canonical Wnt signaling through increasing the cytoplasmic pool of β-catenin.
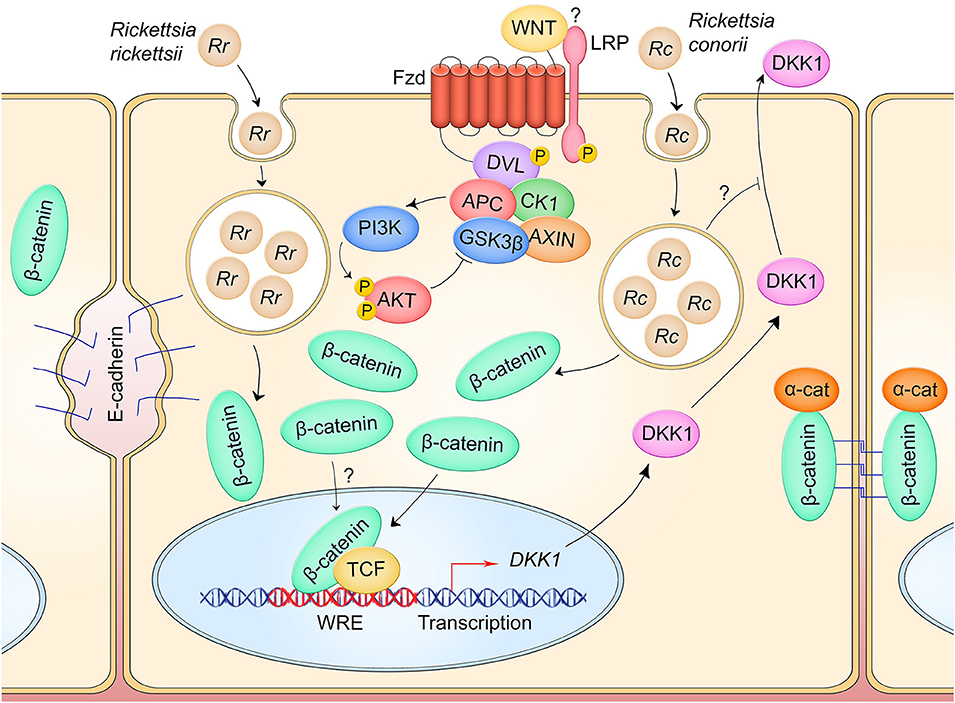
Figure 4. Canonical Wnt signaling manipulation during Rickettsia spp. infection of endothelium. R. rickettsii (Rr) infection of endothelial cells induces breakdown of adherens junctions and accumulation of cytoplasmic β-catenin which may drive pathway activity (left). R. conorii (Rc) infection leads to a suppression of secreted Wnt antagonist DKK1, which may facilitate activation of Wnt signaling to induce an anti-inflammatory environment during infection. IL-6 and IL-8 secretion are suppressed during infection in a DKK1-dependent manner.
In accordance with Wnt signaling activation, DKK1 secreted protein level is significantly reduced in a HUVEC model of R. conorii infection relative to uninfected controls from 5 to 180 hpi (28). Infection activates endothelial cells and causes significant increase in secreted protein levels of inflammatory cytokines IL-6 and IL-8. However, RNA-mediated silencing of DKK1 in infected HUVECs significantly reduces production of these cytokines, suggesting that DKK1 plays a role in modulating the inflammatory response. It is hypothesized that R. conorii suppresses DKK1 to prevent negative regulation of Wnt signaling, thereby keeping proinflammatory cytokine expression relatively low during infection.
These studies highlight a unique role of Wnt signaling feedback inhibitors in pathogenic mechanisms of infection. In seeking to augment Wnt signaling for its anti-inflammatory effect, Rickettsia may possess active mechanisms for reducing the activity of Wnt pathway antagonists like DKK1. A critical gap in this model is whether DKK1 is inhibited at the transcriptional, translational, or post-translation level. Evidence suggests Rickettsia may be inhibiting DKK secretion, although further studies are needed to confirm this. This research also presents the therapeutic potential of harnessing the Wnt pathway to mitigate disease. Recombinant DKK1 has been shown to reduce pathological Wnt signaling activation in a mouse model of neovascularization-induced blindness (81). DKK1 levels in R. conorii-infected patient sera indicate a decline in secreted DKK1 throughout infection, but whether this is a direct result of pathogen manipulation has not been investigated (28). Altogether, these results propose a complex model in which intracellular pathogens act on feedback inhibition mechanisms of Wnt signaling to interfere with pathway activity, and they propose a unique role for DKK1 in influencing the proinflammatory response to Rickettsia.
Ehrlichia chaffeensis is the causative agent of the tick-transmitted disease human monocytic ehrlichiosis (HME). The pathogen is an obligately intracellular bacterium and infects mononuclear phagocytes including monocytes. E. chaffeensis enters the host cell through phagocytosis and replicates within a membrane-bound vacuole to form a microcolony known as a morula (83). The morula resembles an autophagosome, but through pathogenic mechanisms never fuses with the lysosome (31). Similar tochlamydiae, ehrlichiae undergo a biphasic developmental cycle within the host cell (84). The infectious dense-cored cell (DC) ehrlichiae invade the host cell and transition into replicating reticulate cells (RC) that divide through binary fission. The ehrlichiae complete their infection cycle by transitioning back into DC, rupturing the host cell, and spreading hematogenously to the next host cell. Throughout infection, ehrlichiae secrete a variety of T1SS and T4SS effectors, including the TISS tandem repeat protein (TRP) effectors TRP32, TRP47, and TRP120 that induce pathogenesis through direct interactions with numerous host proteins and host DNA (34, 35, 85, 86). Through unknown mechanisms, the TRPs also localize to the surface of the ehrlichiae and decorate the outer membrane, facilitating interactions with the host cell leading to invasion (87, 88).
E. chaffeensis has emerged as a model organism for pathogenesis mediated by the hijacking of conserved cell signaling pathways including Wnt signaling (87). Both canonical and non-canonical Wnt signaling are active early during infection and are necessary for enhancement of ehrlichial infection, as inhibitors or gene silencing of canonical and non-canonical pathway components significantly reduce E. chaffeensis survival within the monocyte (Figure 5) (30, 32). Of note, RNA silencing of antagonist DKK3 results in increased infection, while RNA silencing of canonical and non-canonical pathway components such as CK1, CAMKII, NFAT, and β-catenin significantly reduces infection. E. chaffeensis enters the host cell via phagocytic pathways regulated by Ca2+ signaling and actin filamentation, and the morula labels with autophagosomal markers LC3 and beclin-1 even though autophagy appears to be inhibited and lysosomal marker LAMP2 never localizes to the morula (31, 89, 90). The Wnt pathway has been shown to involved in E. chaffeensis phagocytosis because microspheres coated in ehrlichial surface TRPs can stimulate phagocytosis by monocytes but are unable to do so in the presence of a small molecule inhibitor of Wnt signaling (30). Additionally, RNA silencing of Wnt receptors Fzd5 and Fzd9, and Wnt coreceptor LRP6, significantly reduces the number of intracellular bacteria, indicating a potential role for Fzds as E. chaffeensis receptors. The hypothesis that E. chaffeensis uses activation of non-canonical Wnt signaling to drive actin filamentation and bacterial uptake is supported by evidence demonstrating Wnt5-Fzd5-PI3K non-canonical Wnt signaling induces uptake of non-pathogenic E. coli without leading to bacterial killing, as well as the role of non-canonical Wnt signaling in cytoskeletal control (91–94). E. chaffeensis avoidance of lysosomal fusion was also found to be dependent on ehrlichial activation of Wnt-PI3K signaling, mediated by TRP32 and TRP120, to stimulate mTOR inhibition of autophagy as well as inhibition of TFEB-dependent lysosome synthesis genes (31). Accordingly, inhibition of Wnt signaling following E. chaffeensis infection in monocytes promotes colocalization of lysosomal marker LAMP2 with the ehrlichial cytoplasmic vacuole (31). β-catenin activation is known to repress the autophagy protein p62, indicating another potential mechanism by which ehrlichial activation of canonical Wnt signaling drives intracellular survival (95).
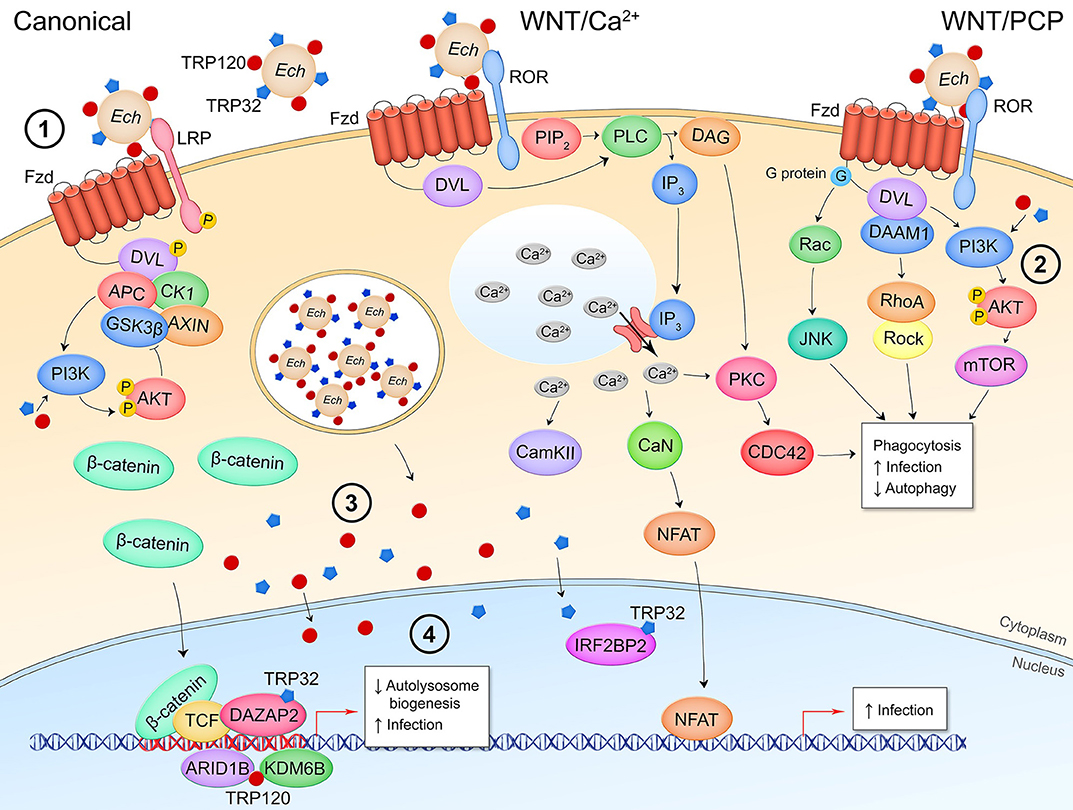
Figure 5. Canonical and non-canonical Wnt signaling manipulation during E. chaffeensis infection of monocytes. (1) E. chaffeensis dense-cored cells express surface proteins TRP32 and TRP120 that stimulate phagocytosis in a Wnt signaling-dependent manner, potentially through interaction with the Wnt receptor complex and stimulation of non-canonical Wnt pathways to induce cytosolic Ca2+ flux and cytoskeletal reorganization. (2) Pathway activity during bacterial entry as well as T1SS secreted effectors TRP32 and TRP120 direct stimulation of PI3K-AKT signaling facilitates mTOR activation which prevents fusion of the bacterial replicative autophagosome with the lysosome. During infection, the secreted TRP effectors localize to the host cytoplasm where they interact with proteins involved in Wnt signaling (3) as well as translocate to the nucleus where they bind host DNA within the promoter regions of Wnt target genes or directly interact with transcriptional regulators of Wnt target genes (4).
In addition to manipulation of Wnt pathway activation early during infection to facilitate phagocytosis and intracellular survival, E. chaffeensis maintains pathway activity throughout infection of the monocyte (30). Yeast-two-hybrid data identified multiple interactions between TRP32 or TRP120 and Wnt signaling pathway components and regulators in the host cytoplasm and nucleus, implicating intracellular pathway manipulation through direct interference with Wnt signaling regulators (85, 86). These interacting partners include pathway negative regulators CEP164 (96), KLHL12 (97), ILF3 (98), and LMO2 (99); and positive regulators PPP3R1 (100) and VPS29 (101). RNA-mediated knockdown of these interacting targets results in significant, differential regulation of THP-1 monocyte infection which demonstrates these TRP-host protein interactions are relevant to E. chaffeensis establishment of infection (32). TRPs also interact with nuclear proteins involved in Wnt target gene epigenetic modification and expression, including the Wnt response element transcriptional repressor TLE4 (102), co-activator DAZAP2 (103), and histone remodeling proteins ARID1B, KDM6B, and IRF2BP2 (104–106). Furthermore, TRP32, TRP47, and TRP120 DNA-binding motifs are within the promoter region of numerous Wnt target genes, indicating the TRPs may directly influence Wnt gene transcription through nucleomodulin activity (33–35). Although these specific interactions have not yet been investigated, it is likely that the nucleomodulin activity of TRP effectors or the recently discovered ubiquitin ligase activity of TRP120, plays a role in manipulation of these target proteins, facilitating Wnt pathway activity and ultimately enhancing E. chaffeensis intracellular survival (107).
E. chaffeensis stands out among human pathogens as a bacterium that targets Wnt signaling at both canonical and non-canonical levels and takes advantage of a wide range of pathway outcomes. Gaps that remain to be filled in the E. chaffeensis monocyte infection model include how both signaling branches are activated. The studies reviewed demonstrate that E. chaffeensis utilizes an extracellular mechanism of pathway activation, potentially through surface TRPs interaction with the Wnt receptor complex. While many bacteria use secreted effectors to activate Wnt signaling through manipulation of midstream signaling components, ehrlichial TRPs are the only bacterial factors that are known to trigger entry into a cell in a Wnt-dependent mechanism which suggests the surface proteins mimic Wnt ligands and utilize Wnt control of the cytoskeleton for phagocytosis. E. chaffeensis demonstrates a potential for intracellular activation of signaling through the secretion of TRP effectors that interact with Wnt pathway-related proteins and DNA in the cytosol and nucleus. These dual modes of extra/intracellular pathway activation may be necessary for temporal regulation of signal activity to facilitate the bacterium's complex intracellular developmental cycle. Understanding the relevance of these vast interactions will shed light ehrlichial pathogenesis while also serving as a model for other intracellular bacteria that reprogram the host cell to establish a replicative niche.
The causative agent of one of the most prevalent infectious diseases in the world, Mycobacterium tuberculosis establishes infection in the lower respiratory tract and infects alveolar macrophages as well as epithelial cells. The outcome of infection involves a delicate balance between the innate immune response control of intracellular bacterial killing early in infection and containment of infection by the adaptive immune response in lung granulomas to prevent dissemination. However, this can result in persistent infection with the ability to transition into an acute infection even decades after the initial exposure.
Canonical Wnt signaling in infected macrophages in the lung has been shown to be part of the innate immune response to M. tuberculosis and is regulated by the Wnt3a-Fzd1 signaling axis, with significant crosstalk from other Wnt ligands as well as TLR signaling. Although canonical signaling is involved in activation of macrophages, it has also been shown to lead to suppression of pro-inflammatory cytokine expression (36–38). It is hypothesized this anti-inflammatory effect contributes to tissue renewal following activation of the inflammatory response, but canonical pathway activation prior to complete clearance of the bacteria can also facilitate persistent infection. Several non-canonical pathways also play a part in M. tuberculosis infection, including Wnt/Ca2+ signaling which may be responsible for Ca2+ signaling that is necessary for recruitment of phagosome coat protein TACO which allows the bacterium to avoid the lysosomal pathway (39). Wnt5a signaling drives expression of PIAS1 and SOCS1 which inhibit TLR signaling, thereby controlling innate immune coordination (40). A non-canonical signaling pathway in which Wnt6 signals through G proteins to activate ERK and drive MYC expression has also been identified as a mechanism of induction of an anti-inflammatory M2 macrophage phenotype which may cooperate with canonical Wnt signaling suppression of inflammation to not only permit tissue renewal but also allow pathogen persistence.
Wnt signaling plays a large role in the processes that both permit and control mycobacterium survival. For further detail, the authors would like to direct the reader to a publication from 2017 that thoroughly reviews the mechanisms controlled by various Wnt ligands during M. tuberculosis infection (108).
The anaerobic, Gram-positive bacterium Clostridium difficile is part of the human intestinal normal flora. It is the causative agent of pseudomembranous colitis (PMC), a severe inflammatory disease of the colon that arises from opportunistic infection by C. difficile typically following antibiotic-induced disturbances in the normal flora population proportions (109). The bacteria are transmitted fecal-orally in the form of spores and upon environmental triggers, such as exposure to bile acids, enter a vegetative state in the intestines. C. difficile pathogenesis is highly dependent on the expression of a family of exotoxins that includes toxin A (TcdA) and toxin B (TcdB) (110). The toxins are secreted from C. difficile through non-canonical secretion mechanisms and are endocytosed by target intestinal epithelial cells (111). These toxins are glucosyltransferases that glucosylate host small GTPases including Rho and Ras. Initially, TcdA, and TcdB together were thought to be responsible for symptoms of PMC. Alone, TcdA, but not TcdB, in small animal models can induce clinical signs of PMC including tissue damage, intestinal endothelial leakage, and inflammation (112). However, TcdA−/TcdB+ clinically relevant strains have been isolated, demonstrating TcdB is sufficient for induction of PMC in humans (113).
TcdA and TcdB exert enterotoxigenic effects including weakening of the epithelial barrier through disruption of tight junctions caused by the glucosylation of small GTPases that regulate actin filamentation, and stimulation of epithelial cells to recruit immune cells and program a pro-inflammatory response (110). Additionally, C. difficile is known to inhibit cell proliferation within the intestinal epithelium, a process typically regulated by canonical Wnt signaling, through the direct inhibitory effect of TcdA and TcdB on the Wnt pathway (Figure 6A) (114). TcdA has been shown to induce β-catenin degradation in the presence of pathway stimulation by Wnt3a, as well as prevent stimulation of pathway activity following pretreatment of intestinal epithelial cells with Wnt3a. Consequently, CMYC transcription is suppressed resulting in cell cycle arrest (42). TcdA-mediated degradation of β-catenin is independent of the β-catenin destruction complex, as TcdA stimulation of cells expressing a GSK3β phosphorylation-resistant β-catenin mutant cannot rescue pathway inhibition. It is speculated that TcdA glucosylation of Rac1 also inhibits canonical Wnt signaling as Rac1 expression is necessary for β-catenin nuclear translocation in certain cell types (115).
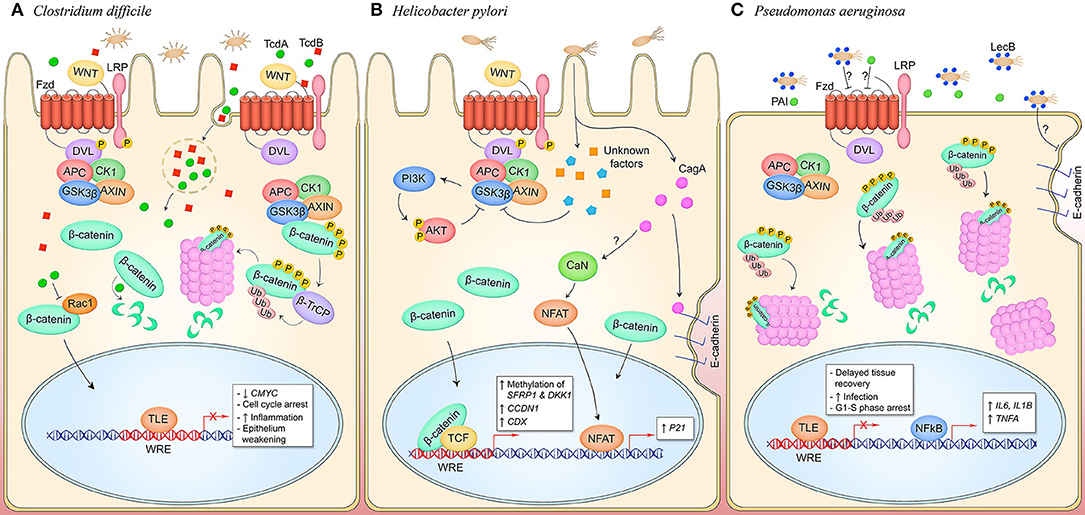
Figure 6. Canonical and non-canonical Wnt signaling manipulation by pathogens occupying an extracellular niche. (A) C. difficile secretes toxins TcdA and TcdB which are phagocytosed by host cells in the intestinal and colonic epithelia. TcdA glucosyltransferase activity inhibits small GTPases like RacA which may inhibit pathway activity through inhibiting nuclear translocation of β-catenin. TcdB interacts with Wnt pathway components LRP6, Wnt5a, and GSK3β, and Fzd1, 2, and 7. Direct binding of Fzds prevents binding of endogenous Wnt ligands and silences pathway activity. This promotes epithelium permeability and a pro-inflammatory state. (B) During H. pylori infection of gastric mucosa, CagA activates canonical Wnt signaling through the breakdown of adherens junctions, and non-canonical Wnt signaling through stimulation of CaN and NFAT nuclear translocation by an unknown mechanism. Canonical pathway signaling is also activated through unknown T4SS effectors that induce phosphorylation of LRP and DVL and inhibit GSK3β, promoting cytosolic accumulation of β-catenin and subsequent nuclear translocation to activate genes that drive cell proliferation. Furthermore, unknown mechanisms induce methylation of Wnt pathway inhibitory genes SFRP1 and DKK1 to hypothetically facilitate Wnt signaling activity during infection. (C) P. aeruginosa is decorated with lectin LecB which induces β-catenin degradation through an unknown extracellular mechanism, resulting in decreased cell proliferation. Coincidentally, NF-κB signaling is activated and inflammatory target genes IL6, IL1B, and TNFA are expressed. LecB as well as the quorum-sensing molecule PAI both induce disruption of adherens junction in infected epithelia, contributing to prolonged tissue damage in the host.
Recent studies have identified a unique role for TcdB in inhibition of Wnt signaling. Through sgRNA screening, TcdB has been shown to directly bind Fzd receptors in the intestinal epithelium, including Fzd1, Fzd2, and Fzd7 (43). This results in inhibition of LRP and Dvl phosphorylation, indicating a TcdB mechanism of Wnt inhibition through direct manipulation of the Wnt receptor complex. Wnt ligands interact with Fzd through two distinct binding sites, as well as hydrophobic interactions between a groove on the Fzd-CRD and the palmitoleic acid moiety (PAM) of Wnt (116). TcdB interacts with the Fzd-CRD as well, although the binding site is distinct from the Wnt binding sites as revealed by the co-crystallization of TcdB Fzd-binding domain and Fzd-CRD (44). In the pathway off state under normal physiological conditions, Fzd-CRD binds an endogenous lipid which becomes displaced by the Wnt PAM when the signal-initiating ligand binds. The endogenous lipid acts as coreceptor for TcdB that TcdB directly engages and locks in place, preventing displacement by the Wnt PAM and inhibiting pathway activation. It was also demonstrated that TcdB can bind Fzd when a Wnt ligand has already bound, with the Wnt PAM acting as a TcdB coreceptor, suggesting TcdB may have other inhibitory mechanisms including inhibition of receptor dimerization which has been shown to be stimulated through Wnt binding and necessary for signal transduction to occur (44, 117). This blockade of Wnt signaling is hypothesized to be a major underlying cause of weakening of the intestinal epithelium, tissue damage, and inflammation, hallmarks of PMC and C. difficile infection (44).
The unique inhibitory mechanisms of toxins TcdA and TcdB on Wnt signaling in host cells has expanded the model of pathogen manipulation of Wnt signaling through the identification of both intra- and extra-cellular molecular interactions that contribute to C. difficile pathogenesis. It remains to be discovered if TcdA glucosyltransferase activity modulates non-canonical Wnt signaling in the intestinal epithelium due to the major role of small GTPases in non-canonical pathways (118). Further investigation of TcdB interference with Fzd receptors will also lend insight into the critical interactions for Wnt pathway signaling initiation under normal physiological conditions. Of note, the canonical Wnt pathway components APC, GSK3β, Wnt5a, and LRP6 were also identified as potential targets of TcdB in an sgRNA screen (43). Whether the pool of TcdB that is endocytosed has a relevant role in Wnt signaling manipulation through interactions with these intracellular components remains to be determined. Of particular interest is the exploitation of TcdB-Fzd interaction for rational drug design. A recombinant TcdB mutant lacking Fzd-binding ability can induce significantly less swelling, epithelial damage, and infiltration of immune cells in a mouse cecum injection model, demonstrating that modulation of this toxin activity in the intestines has strong potential to reduce disease severity (44).
Helicobacter pylori is a Gram-negative bacilli and most common infectious agent found in both adults and children worldwide. The bacterium is transmitted fecal-orally and oral-orally and replicates within the mucin layer in the stomach adhered to gastric epithelium. Pathogenic H. pylori strains possess the cag pathogenicity island, a genetic locus that consists of 31 genes including the effector CagA and the T4SS that secretes it. CagA is injected into gastric epithelial cells and directly interferes with cellular signaling pathways such as NF-κB and MAPK signaling, alters cell-cell contacts, and stimulates a proinflammatory response, thereby promoting the H. pylori replicative niche (119). CagA induction of the cell cycle and host survival pathways including canonical β-catenin-dependent Wnt signaling induce cellular transformation and replication which is why Cag+ strains of H. pylori increase host predisposition to tumorigenesis and gastric carcinoma, and H. pylori is classified as a type I carcinogen (120, 121).
While it is well-documented that H. pylori infection activates β-catenin-dependent Wnt signaling in the gastric epithelium, the role of CagA in Wnt activation is controversial. An early observation in the field determined that during infection of human gastric epithelia, β-catenin is stabilized. This was associated with infection by CagA+ strains (Figure 6B) (122). The mechanism for activation of β-catenin has been shown to be in part mediated by CagA disruption of adherens junctions. CagA directly interacts with E-cadherin which causes β-catenin dissociation from the adherens junctions complex and subsequent translocation to the nucleus (45). CagA induction of β-catenin target gene CDX and transactivation of p21 links disruption of adherens junctions and Wnt pathway activation with H. pylori CagA carcinogenesis as these gene products are required for transformation of gastric epithelium to intestinal epithelium which is a critical event in the development of intestinal metaplasia. An earlier finding connected activation of non-canonical NFAT-dependent Wnt signaling with CagA induction of p21, demonstrating regulation of Wnt signaling by CagA at multiple levels during infection (47). In a mammary epithelium model of infection, adherens junctions proteins were shown to dissociate upon infection in a CagA-independent mechanism resulting in loss of junctional integrity (123). While this group demonstrates β-catenin is stabilized, they do not detect nuclear localization of the transcription factor, but rather propose the hypothesis some other factor is able to cleave E-cadherin and manipulation junctions (123). This could indicate CagA− strains of H. pylori stimulate β-catenin signaling through other mechanisms, or these results could suggest host factor-dependent predisposition to pathway hyperactivation.
Thus, a CagA-independent mechanism of Wnt activation may be active during H. pylori infection in addition to manipulation of the signaling pathway through interference with adherens junctions. LRP5/6 and Dvl are both phosphorylated in a Cag-independent, T4SS-dependent manner in a gastric epithelial infection model (124). H. pylori has also been shown to inhibit GSK3β activity which suppresses β-catenin degradation and is shown to induce Wnt pathway target gene CCND1 (125, 126). Intracellularly, β-catenin activation and Wnt target gene expression is linked to activation of PI3K and Akt which inhibit GSK3β during infection, but the role of the Wnt receptor complex protein LRP6 in this mechanism remains to be investigated. Ultimately, there may be cell type- and temporally-specific deregulation of Wnt signaling during infection, as well as diverse effects of different effectors, that promote a tightly controlled differentiation vs. proliferation profile during H. pylori infection that ultimately serves to facilitate bacterial survival and dissemination.
H. pylori is well-known to modify target cell epigenetics which can also promote infection and tumorigenesis (127). A recent report linked H. pylori infection with modulation of Wnt signaling through epigenetic modification of Wnt pathway-related genes. Human H. pylori-positive gastric carcinoma samples display significantly increased methylation at the host genes SFRP1 and DKK1, inhibitors of Wnt signaling (46). This corresponded with slight to moderate increases in β-catenin nuclear localization. Patient samples that demonstrated H. pylori eradication at a one-year follow-up did not have significantly altered epigenetic footprints, indicating H. pylori can induce long-lasting changes to host genomics and impact Wnt signaling activity in hosts post-eradication (46).
Understanding the connection between H. pylori secreted effectors and changes to host cell signaling pathways provides insight into potential avenues for therapeutics, infection or gastric cancer biomarkers, and pathogen-mediated tumorigenesis. Both CagA-dependent and -independent mechanisms are at play during infection to manipulate Wnt signaling and facilitate an environment within the host that supports bacterial replication. The role of LRP in activation of signaling and CagA cleavage of E-cadherin demonstrates H. pylori may utilize secreted proteins to manipulate Wnt signaling at the level of the receptor complex to induce pathway activity, a model of pathway activation applicable to intracellular and extracellular pathogens alike. Interestingly, pathways active during infection including PI3K-AKT and Wnt signaling both converge at the inactivation of GSK3β, indicating redundant mechanisms in place to ensure the different outcomes that are mediated by inhibition of the kinase. Further understanding of how the kinase activity of GSK3β is a crux for infection may lead to the development of therapeutics that act on cellular signaling pathways and have the potential to delay pathogen-induced tumorigenesis.
Pseudomonas aeruginosa is a Gram-negative, opportunistic pathogen and a causative agent of microbial keratitis, burn wound infections, and hospital-acquired pneumonia. The bacteria is found in biofilms that are essential for host attachment and survival (128). P. aeruginosa utilizes quorum-sensing (QS) systems to sense bacterial population density and regulate biofilm development, mammalian host defense, host-microbe interactions, virulence and metabolite acquisition (129, 130) P. aeruginosa pathogenesis also encompasses disruption of epithelial barrier function, increased inflammatory response, and bacterial virulence factors.
Canonical Wnt signaling plays a major role in epithelial barrier integrity and proliferation during P. aeruginosa infection. Host response to P. aeruginosa infection leads to activation of the Wnt signaling pathway to regulate the pro-inflammatory response and promote bacterial clearance; therefore, inhibition of Wnt signaling is advantageous for persistent infection and survival. A recent study has shown that macrophages stimulated with Wnt3a conditioned media inhibits production of pro-inflammatory cytokines IL-6, IL-1β, MIP2, and TNF-α during P. aeruginosa infection. Wnt3a treatment also promotes apoptosis of macrophages at 12 and 24 hpi and enhances intracellular bacterial killing via upregulation of anti-microbial peptides CRAMP and BD1 (131). During P. aeruginosa infection inhibition of Wnt3a upregulates expression of pro-inflammatory cytokines, likely to delay tissue recovery and promote P. aeruginosa colonization (Figure 6C) (49). In a P. aeruginosa keratitis model of infection, melting of the cornea occurs due to bacterial proteases, activation of matrix metalloproteases, and deregulated immune response (132). β-catenin inhibits pro-inflammatory cytokines and decreases bacterial burden in the corneal stroma to reduce the severity of P. aeruginosa keratitis (133). Overexpression of β-catenin in B6 corneas results in suppression of IL1B, MIP2, and TNFA in phagocytes, but not in corneal epithelial cells, for host resistance of P. aeruginosa infection. Bacterial load is also decreased during overexpression of β-catenin in B6 corneas infected with P. aeruginosa. Collectively these studies suggest inhibition of Wnt signaling, specifically by downregulation of Wnt3a and β-catenin, increases bacterial burden and the inflammatory response during P. aeruginosa infection. Along with these findings, lithium chloride (LiCl) was shown to promote bacterial clearance. LiCl promotes canonical Wnt pathway activity through the inhibition of GSK3β (134). Various studies have determined that GSK3β is important in regulating inflammatory responses during bacterial infections (135). For example, Klebsiella pneumoniae-infected mice provided with LiCl-treated drinking water display increased survival and decreased liver injury due to a decrease in bacterial burden and cytokine production in blood and liver tissues (136). During P. aeruginosa infection, LiCl has been shown to inhibit proinflammatory cytokine TNF-α, enhance production of anti-inflammatory cytokine IL-10, and ultimately lead to host resistance against P. aeruginosa infection in the cornea (137). In correlation with previous listed studies, a reduction in inflammatory infiltration is observed in LiCl-treated cells, and increased apoptosis of phagocytes occurs. Taken together, P. aeruginosa-mediated inhibition of Wnt/β-catenin signaling is important for recruitment of infiltrating phagocytes and increased inflammatory response to promote dissemination and survival of the bacteria.
As previously described, β-catenin is a major contributor to the integrity of adherens junctions and may play an indirect role in the integrity of tight junctions and epithelial barrier during P. aeruginosa infection. P. aeruginosa contains two quorum-sensing systems which regulate specific signaling molecules that contribute to infection and survival (138). Adherens junctions integrity is decreased by a P. aeruginosa acyl-homoserine lactone (PAI) quorum-sensing molecule (48, 139). Treatment of CaCO-2 cells with the PAI results in a decrease in adherens junction-associated proteins E-cadherin and β-catenin expression during early infection. Hyperphosphorylation of occludin, ZO-1, E-cadherin and β-catenin are detected on tyrosine residues during early infection; however, occludin and β-catenin are dephosphorylated on serine and threonine residues. These outcomes are responsible for dissociation of E-cadherin-β-catenin complexes and association of ZO-1-occludin complexes ultimately leading to disassembly of adherens junctions (48). Of note, the expression levels of E-cadherin and β-catenin increase back to normal levels at later time points of infection (5–24 h), demonstrating a reversible effect of PAI during P. aeruginosa infection. Taken together, distinct phosphorylation events on adherens junction proteins, including β-catenin, play an important role in P. aeruginosa PAI-mediated tissue damage.
Along with P. aeruginosa PAI, soluble carbohydrate-binding lectin LecB is regulated by both las and rhl QS systems and reprograms the Wnt signaling pathway for infection and survival. LecB is one of two lectins expressed on the outer bacterial membrane of P. aeruginosa and is important for adhesion, biofilm formation, pilus biogenesis, and protease activity (140). LecB acts antagonistically to Wnt signaling through proteasomal degradation of β-catenin in a GSK3β-dependent manner, resulting in a G1-S phase cell cycle arrest in lung epithelial cells (49). LecB reduces β-catenin at adherens junctions and causes accumulation of the protein at centrosomes expressing high levels of proteasomes. Furthermore, co-incubation of Wnt3a and LecB repressed nuclear translocation of β-catenin, indicating infection actively suppresses canonical Wnt signaling activation. In addition, Ser536 phosphorylation of p65, a marker for increased transcriptional activity and acetylation of NF-κB, increases during P. aeruginosa infection, resulting in activation of NF-κB and subsequent elevation of downstream target genes TNFA, IL6, and IL1B which are linked to delayed tissue recovery (49). Taken together, these results indicate that P. aeruginosa manipulates the Wnt/β-catenin signaling pathway via β-catenin degradation and NF-κB activation to delay tissue recovery and promote infection.
P. aeruginosa is an opportunistic pathogen that utilizes various approaches to cross the epithelium at cell-cell junctions, including disruption of the epithelial cell barrier via PAI induction of phosphorylation of E-cadherin-β-catenin complexes (141). P. aeruginosa PAI also modulates mechanisms of immune tolerance to provide a niche for bacterial survival (142). Targeting the quorum sensing abilities of P. aeruginosa has been explored as an antimicrobial strategy, so understanding the extent of PAI in manipulating the host cell may identify mechanisms by which anti-quorum sensing drugs can also target mechanisms involving Wnt signaling. As previously described, S. typhimurium infection results in simultaneous β-catenin degradation and increased NF-κB activity in a murine model (18). P. aeruginosa LecB degrades β-catenin to decrease proliferation of lung epithelial cells, ultimately leading to delayed tissue recovery and persistent infection. Further investigation of the role of LecB-mediated β-catenin degradation inducing activation of NF-κB should also be explored. LecB is localized to the outer membrane of P. aeruginosa and binds to host cell plasma membrane receptors stimulating changes to epithelial barrier function and increased inflammatory response (143). Therefore, LecB-dependent β-catenin degradation may be stimulated by direct binding to extracellular portions of the Wnt receptor complex or extracellular adherens junction components. Understanding the mechanisms utilized by LecB to manipulate Wnt signaling to decrease proliferation and tissue recovery of lung epithelial cells may serve as a potential therapeutic approach to P. aeruginosa infection.
Uropathogenic Escherichia coli (UPEC) is an extraintestinal pathogenic isolate of Escherichia coli and the causative agent of 40% of hospital- and 80% of community-acquired urinary tract infections (144). UPEC infects the urothelium, a highly specialized epithelium lining the lower urinary tract that is composed of basal, intermediate, and superficial cell layers (145). The superficial apical cell layer provides the primary urinary barrier and is composed of large, hexagonal cells known as umbrella cells which are exfoliated and regenerated upon infection (145, 146). Upon infection, UPEC enters and ascends into the urinary tract, enters the urethra, and migrates into the bladder lumen where it engages with bladder umbrella cells by binding to mannose-containing glycoprotein receptors via the adhesin FimH (50). Engulfment into the host cell is followed by the formation of intracellular bacterial communities (IBCs) that demonstrate biofilm-like properties (147, 148). UPEC detach from the intracellular biofilm and establish intracellular colonization leading to eruption of bladder epithelial cells. Dissemination and detachment lead to UPEC mobilization to the bladder lumen which increases the risk of bacteremia and septicemia (148).
UPEC has evolved several molecular-based strategies to subvert the innate immune response for efficient colonization and persistent infection in the urinary tract. One mechanism of host response to UPEC infection is the process of exfoliation of bladder epithelial cells that are overwhelmed with bacteria (146, 147). To renew exfoliated superficial epithelium, genetic programs are activated for proliferation and differentiation of urothelial cells (149). Despite the role of exfoliation in bacterial clearance, this outcome also leads to exposure of immature bladder epithelial cells which are more susceptible to UPEC infection (50). Within these compartments, bacteria are in a quiescent state; however, as the immature urothelial cells differentiate into mature urothelial cells, UPEC replicates for recurrent infection (147). Modulation of Wnt signaling has been linked to differentiation and proliferation of basal/intermediate cells during UPEC infection. More specifically, expression of Wnt5a during UPEC infection leads to proliferation of basal/intermediate cells while suppression of Wnt5a expression results in differentiation (50). A recent study demonstrated suppression of Wnt5a/Ca2+ signaling promotes differentiation of basal/intermediate cells prior to exfoliation (50). Infection by FimH+ UPEC results in a decrease in Wnt5a expression and subsequent decrease of non-canonical Wnt pathway targets PKCδ and CamKIIδ up to relatively late infection timepoints (50). This suggests that prior to exfoliation, non-canonical Wnt signaling is suppressed to promote differentiation of umbrella cells upon initiation of UPEC infection. However, it has also been shown that UPEC induces infected cells to secrete paracrine factors that cause alterations in the expression of the epigenetic writer EZH2 which enhances Wnt5a expression, promoting proliferation of infected cells at relatively early infection time points (51). Further studies must be done to determine if there is temporal regulation of Wnt5a expression during UPEC infection. Despite these discrepancies, research suggests that modulation of Wnt signaling during UPEC infection promotes exfoliation and cell proliferation for dissemination and survival, respectively. Consistent with these findings, EZH2 has been shown to be involved in Wnt-mediated proliferation in Citrobacter rodentium infection (52). C. rodentium is a Gram-negative, murine enteric bacterial pathogen and closely related to enteropathogenic (EPEC) and enterohaemorrhagic E. coli (EHEC) (150–152). EPEC, EHEC, and C. rodentium are members of the attaching and effacing (A/E) family of bacterial pathogens due to the destructive effect of colonization of the intestinal epithelium and enterocytes (150, 151). The natural host range and genetic make-up of C. rodentium makes it a good in vivo model for A/E pathogens. EZH2 represses Wnt inhibitory factor WIF1 resulting in activation of Wnt/β-catenin signaling and ultimately C. rodentium-induced crypt hyperplasia and tumorigenesis (52). Therefore, modulation of the Wnt signaling pathway by A/E pathogens would appear to be a useful regulatory mechanism by providing a niche for adaptation and survival in the host.
Along with a diverse range of adhesin molecules, UPEC utilizes a variety of toxins to increase invasiveness and facilitate virulence. Of these secreted toxins is a pore-forming, T1SS toxin α-hemolysin (HlyA). HlyA is inserted into epithelial and macrophage membranes and triggers rapid degradation of various host proteins involved in the proinflammatory response and cell-cell and cell-matrix interactions, including β-catenin (153). HlyA mediated degradation of β-catenin in infected BECs occurs simultaneously with loss of IκBα. Additionally, the NF-κB subunit RelA (p65) is also degraded in HlyA-intoxicated bladder epithelial cells. HlyA-mediated degradation of RelA (p65) correlates with reduced IL6 expression (153). This study suggests a novel mechanism by which UPEC inhibits NF-κB-mediated inflammatory response via β-catenin degradation, and independent of IκBα-mediated proteasomal degradation. Similar results were shown with macrophages, demonstrating a significant role of HlyA in modulating phagocytes and epithelial cell function for UPEC survival. Investigation of the cross-regulation of β-catenin and NF-κB signaling pathways during UPEC infection would demonstrate if UPEC utilizes Wnt signaling as a means of immunosuppression.
Several gaps in knowledge remain in reference to the role of Wnt signaling in subversion of innate immunity during UPEC infection. While exfoliation and proliferation are mechanisms of host response to UPEC infection, this response is beneficial for establishing a niche for persistent infection (153, 154). Identifying the role of Wnt signaling and possible UPEC effectors involved in pathway activation to induce exfoliation and proliferation for survival may allow for better understanding of how UPEC infections can persist in the face of host defenses. Inhibition of Wnt5a-induced non-canonical signaling and EZH2 may potentially inhibit proliferative processes necessary for UPEC infection and may therefore serve as potential therapeutic targets.
Pathogens' respective niches and modes of dissemination shape how they avoid innate immune pressure during infection. Wnt signaling is a host cell pathway targeted by both obligately intracellular and extracellular bacterial pathogens alike, demonstrating the breadth of the pathway's control over eukaryotic cellular processes. A consistent theme among the intracellular pathogens discussed in this review is activation of the pathway in the host cell. Evidence from Salmonella, Chlamydia, Rickettsia, and Mycobacteria demonstrates canonical pathway activation contributes to an anti-inflammatory state which mediates immunosuppression and may prolong infection. The former two pathogens also appear to stimulate canonical Wnt signaling to prolong host cell survival by inducing either proliferation or differentiation of the host cell. Ehrlichia is unique in that studies have shown the bacterium activates non-canonical pathways which govern cytoskeletal reorganization and autophagy regulation, permitting phagocytosis of the bacterium without destruction by the lysosome. Inhibition of the canonical Wnt pathway by Salmonella and C. difficile occurs in intestinal epithelium and the intestinal capillary endothelium for the former and in the colonic epithelium for the latter, and contributes to pathogen manipulation of tissue barriers, facilitating dissemination into the bloodstream. In the case of Salmonella, inhibition of Wnt signaling may also lead to an inflammatory response that recruits target cells for Salmonella and promotes dissemination. Studies have shown that C. difficile and P. aeruginosa utilize multiple effector molecules to interfere with canonical Wnt signaling activity of the surrounding tissue during infection. In both cases, viability of the cell within the infected tissue is reduced which may contribute to nutrient accumulation or dissemination for the bacteria. E. coli and H. pylori, contrastingly, activate canonical and non-canonical Wnt signaling from their extracellular niches, resulting in increased proliferation or differentiation of cells in the surrounding epithelium. Although evidence is yet to demonstrate a clear phenotype, it is likely that pathway activation also contributes to an anti-inflammatory state in the bacterial replicative niche due to reciprocal regulation of the transcription factors NF-κB and β-catenin.
Studies have implicated a range of bacterial effectors with the capability to manipulate canonical and non-canonical Wnt signaling, including T1,3,4SS effector proteins of S. enterica, C. pneumoniae, E. chaffeensis, E. coli, and H. pylori; lectins and quorum-sensing molecules of P. aeruginosa; two-component regulatory systems of S. enterica; and likely many more unknown factors. Shared mechanisms involve disassembly of adherens junctions as is the case for C. trachomatis, R. rickettsii, H. pylori, and P. aeruginosa which may be a means for increasing the amount of cytosolic β-catenin available to activate the Wnt response element, or a means of inactivating the protein and facilitating its degradation. Cpn1027 of C. pneumoniae and TcdB of C. difficile both inactivate pathway components through direct binding. Such may be the case for E. chaffeensis TRP32, 47, and 120, as many host-bacterial interactions between negative regulators of Wnt signaling and the TRPs have been identified for which mechanisms have yet to be described. Pathogens also deploy enzymes during infection to manipulate Wnt signaling components at the post-translation modification level, as is the case with C. difficile TcdA and S. enterica AvrA and SopB. Research identifying Wnt pathway regulatory mechanisms and foreign molecules that can interfere with the pathway has applications ranging from research tools to potential therapeutics.
As research detailing mechanisms of pathogenesis and bacteria-host interaction accumulates, our knowledge of how various cellular mechanisms participate in innate immune signaling expands. Wnt signaling regulates stem cell renewal, cell proliferation, and cellular morphology, but the ability of pathogens to usurp these processes to establish infection demonstrates that Wnt signaling is also a target for bacterial immunoevasion strategies. By drawing a parallel between pathway dysregulation during infection and dysregulation in “classical” diseases of Wnt signaling, we can identify targetable pathway components for drug intervention of infectious disease. One such strategy involves the use of the existing armaments of small molecule inhibitors and activators that target various components of canonical Wnt signaling thereby overriding the pathogen reprogramming strategy. Another approach is the use of antivirulence therapy to target the pathogen-host molecular interactions that are beneficial to bacterial virulence, thereby weakening the pathogen through impairing its ability to manipulate the Wnt pathway.
Multiple small molecules have been identified that modulate Wnt signaling activity through activation or inhibition of pathway components or regulators (14). Use of these drugs as an approach for infection control is most suitable for bacteria for which stimulation or repression of pathway activity significantly impairs establishment of infection or a damaging inflammatory response. GSK3 promotes Wnt signaling through inactivation of the destruction complex and a number of small molecules target GSK3 activity. These inhibitors have been used in mouse infection models for Streptococcus and Porphyromonas gingivalis and demonstrate efficacy in reducing bacterial burden or excessive inflammation (155–157). GSK3 represents an attractive target for drug design because of its suppressive effect on NF-κB signaling, and upregulation of its kinase activity by different pathogens makes it a potential point for therapeutic intervention during infection (158). Modulators of extracellular components of Wnt signaling include pathway agonists that mimic Wnt ligands and neutralizing antibodies that target secreted pathway inhibitors (159–161). These types of drugs may be utilized when infection causes suppression of Wnt signaling which leads to tissue damage. Salmonella perturbation of the GVB represents a case in point—stimulation of Wnt signaling reverses infection-induced endothelial weakening (20). The effectiveness of these potential Wnt-modulating therapeutics will be impacted by the particular points within the Wnt signal cascade that different pathogens exert manipulation, which prospective mechanism-oriented studies should address. Ultimately, the vast amount of signaling proteins within the Wnt pathway suggests that while this cellular signal cascade can be targeted by drugs, such intervention should be thoroughly researched to anticipate off-target and adverse side effects (162).
Studies of how pathogens interact with the host signalosome allows us to exploit this knowledge to target crucial interactions between bacterial and host factors that enable bacterial virulence (163). Antivirulence therapy has risen as a strategy to combat pathogenic bacteria by disarming them of virulence factors, and an in-depth understanding of the interplay between virulence factors and Wnt signaling enables the development of such therapeutic strategies. For example, C. difficile infection has been shown to be controlled by small molecules and monoclonal antibodies that target critical enzymatic or protein-protein interaction-mediating domains within TcdB (164, 165). The identification of a Fzd-binding domain within TcdB uncovers a target against which small molecules or neutralizing antibodies can be directed, thereby reducing the pathological effects of bacterial inhibition of Wnt signaling within the intestines. A similar strategy could be directed toward E. chaffeensis infection, as surface TRPs including TRP120 mediate phagocytosis of the bacterium through stimulation of Wnt pathway activity. As further research identifies the molecular determinants of such pathological events, interference with TRP-Wnt receptor complex could reduce the ability of E. chaffeensis to establish infection within monocytes. P. aeruginosa utilizes surface protein LecB to extracellularly modulate β-catenin activity. Antivirulence drugs in preclinical development that target LecB demonstrate how this protein can be manipulated to reduce infection, and suggest that the efficacy of these drugs is, at least in part, due to the disruption of P. aeruginosa modulation of Wnt signaling (166).
The era of Wnt signaling research has shifted from understanding the basic molecular biology of the pathway to identifying how the pathway is manipulated by infectious agents. The ultimate goal is to use this knowledge to repurpose or develop novel therapeutics for infectious disease that are relevant in the age of antibiotic resistance. Pathogens have coevolved with the human host and are master manipulators of signaling pathways that have been in place throughout the evolution of metazoans. Understanding the interactions that dictate how pathogens can usurp the Wnt signal cascade to benefit their survival and dissemination facilitates the development of counter-defense strategies that enable the host innate immune system to control and clear infection.
MR and JM conceived the work. MR gathered information and contributed all sections except for P. aeruginosa and E. coli which were contributed by LP. JW performed artwork. All authors participated in editing of the final draft.
This work was supported by the National Institutes of Health grants AI126144, AI123610, and AI106859; by UTMB James B. McLaughlin Endowment Predoctoral Fellowship to MR and JW; and by Gates Millennium Foundation funding to LP.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all current and former laboratory members for their contributions to understanding the role of Wnt signaling in the molecular pathogenesis of E. chaffeensis.
1. Christophides GK, Zdobnov E, Barillas-Mury C, Birney E, Blandin S, Blass C, et al. Immunity-related genes and gene families in Anopheles gambiae. Science. (2002) 298:159–65. doi: 10.1126/science.1077136
2. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
3. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
4. Riera Romo M, Pérez-Martínez D, Castillo Ferrer C. Innate immunity in vertebrates: an overview. Immunology. (2016) 148:125–39. doi: 10.1111/imm.12597
5. Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. (2017) 97:1235–94. doi: 10.1152/physrev.00005.2017
6. Lina TT, Dunphy PS, Luo T, McBride JW. Ehrlichia chaffeensis TRP120 activates canonical notch signaling to downregulate TLR2/4 expression and promote intracellular survival. MBio. (2016) 7:e00672–16. doi: 10.1128/mBio.00672-16
7. Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. (2005) 120:159–62. doi: 10.1016/S0092-8674(05)00043-7
8. Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, et al. Autophagy defends cells against invading group A Streptococcus. Science. (2004) 306:1037–40. doi: 10.1126/science.1103966
9. Cheng MI, Chen C, Engström P, Portnoy DA, Mitchell G. Actin-based motility allows Listeria monocytogenes to avoid autophagy in the macrophage cytosol. Cell Microbiol. (2018) 20:1–13. doi: 10.1111/cmi.12854
10. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. (1982) 31:99–109. doi: 10.1016/0092-8674(82)90409-3
11. Sharma RP, Chopra VL. Effect of the wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev Biol. (1976) 48:461–5. doi: 10.1016/0012-1606(76)90108-1
12. Ng LF, Kaur P, Bunnag N, Suresh J, Sung ICH, Tan QH, et al. WNT signaling in disease. Cells. (2019) 8:826. doi: 10.3390/cells8080826
13. Gao C, Chen YG. Dishevelled: the hub of Wnt signaling. Cell Signal. (2010) 22:717–27. doi: 10.1016/j.cellsig.2009.11.021
14. Nusse R, Clevers H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. (2017) 169:985–99. doi: 10.1016/j.cell.2017.05.016
15. De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin. (2011) 43:745–56. doi: 10.1093/abbs/gmr079
16. Yang Y, Mlodzik M. Wnt-frizzled/planar cell polarity signaling: cellular orientation by facing the wind (Wnt). Annu Rev Cell Dev Biol. (2015) 31:623–46. doi: 10.1146/annurev-cellbio-100814-125315
17. Sun J, Hobert ME, Neish AS, Madara JL. Bacterial activation of β-catenin signaling in human epithelia. AJP Gastrointest Liver Physiol. (2004) 287:G220–7. doi: 10.1152/ajpgi.00498.2003
18. Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. β-Catenin activity negatively regulates bacteria-induced inflammation. Lab Investig. (2007) 87:613–24. doi: 10.1038/labinvest.3700545
19. Sun J, Hobert ME, Duan Y, Rao AS, HE T-C, Change EB, et al. Crosstalk between NFκB and β-catenin pathways in bacterial-colonized intestinal epithelial cells. AJP Gastrointest Liver Physiol. (2005) 289:G129–37. doi: 10.1152/ajpgi.00515.2004
20. Spadoni I, Zagato E, Bertocchi A, Paolinelli R, Hot E, Di Sabatino A, et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science. (2015) 350:830–4. doi: 10.1126/science.aad0135
21. Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol. (2007) 171:882–92. doi: 10.2353/ajpath.2007.070220
22. Lu R, Wu S, Zhang Y, Xia Y, Liu X, Zheng Y, et al. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic β-catenin signaling pathway. Oncogenesis. (2014) 3:e105–5. doi: 10.1038/oncsis.2014.20
23. Liu W, Yan M, Liu Y, Wang R, Li C, Deng C, et al. Olfactomedin 4 down-regulates innate immunity against Helicobacter pylori infection. Proc Natl Acad Sci USA. (2010) 107:11056–61. doi: 10.1073/pnas.1001269107
24. Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS, Wang D, et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe. (2012) 12:645–56. doi: 10.1016/j.chom.2012.10.009
25. Kessler M, Zielecki J, Thieck O, Mollenkopf HJ, Fotopoulou C, Meyer TF. Chlamydia trachomatis disturbs epithelial tissue homeostasis in fallopian tubes via paracrine Wnt signaling. Am J Pathol. (2012) 180:186–98. doi: 10.1016/j.ajpath.2011.09.015
26. Kintner J, Moore CG, Whittimore JD, Butler M, Hall JV. Inhibition of Wnt signaling pathways impairs chlamydia trachomatis infection in endometrial epithelial cells. Front Cell Infect Microbiol. (2017) 7:501. doi: 10.3389/fcimb.2017.00501
27. Flores R, Zhong G. The Chlamydia pneumoniae inclusion membrane protein Cpn1027 interacts with host cell Wnt signaling pathway regulator cytoplasmic activation/ proliferation-associated protein 2 (Caprin2). PLoS ONE. (2015) 10:e127909. doi: 10.1371/journal.pone.0127909
28. Astrup E, Lekva T, Davì G, Otterdal K, Santilli F, Øie E, et al. A complex interaction between Rickettsia conorii and dickkopf-1 - potential role in immune evasion mechanisms in endothelial cells. PLoS ONE. (2012).7:e43638. doi: 10.1371/journal.pone.0043638
29. Woods ME, Olano JP. Host defenses to Rickettsia rickettsii infection contribute to increased microvascular permeability in human cerebral endothelial cells. J Clin Immunol. (2008) 28:174–85. doi: 10.1007/s10875-007-9140-9
30. Luo T, Dunphy PS, Lina TT, Mcbride JW. Ehrlichia chaffeensis exploits canonical and noncanonical host wnt signaling pathways to stimulate phagocytosis and promote intracellular survival. Infect Immun. (2016) 84:686–700. doi: 10.1128/IAI.01289-15
31. Lina TT, Luo T, Velayutham T, Das S, McBride JW. Ehrlichia activation of Wnt-PI3K-mTOR signaling inhibits autolysosome generation and autophagic destruction by the mononuclear phagocyte. Infect Immun. (2017) 85:e00690–17. doi: 10.1128/IAI.00690-17
32. Luo T, Dunphy PS, McBride JW. Ehrlichia chaffeensis tandem repeat effector targets differentially influence infection. Front Cell Infect Microbiol. (2017) 7:178. doi: 10.3389/fcimb.2017.00178
33. Kibler CE, Milligan SL, Farris TR, Zhu B, Mitra S, McBride JW. Ehrlichia chaffeensis TRP47 enters the nucleus via a MYND-binding domain-dependent mechanism and predominantly binds enhancers of host genes associated with signal transduction, cytoskeletal organization, and immune response. PLoS ONE. (2018) 13:e205983. doi: 10.1371/journal.pone.0205983
34. Farris TR, Dunphy PS, Zhu B, Kibler CE, McBride JW. Ehrlichia chaffeensis TRP32 Is a nucleomodulin that directly regulates expression of host genes governing differentiation and proliferation. Infect Immun. (2016) 84:3182–94. doi: 10.1128/IAI.00657-16
35. Zhu B, Kuriakose JA, Luo T, Ballesteros E, Gupta S, Fofanov Y, et al. Ehrlichia chaffeensis TRP120 binds a G+C-rich motif in host cell DNA and exhibits eukaryotic transcriptional activator function. Infect Immun. (2011) 79:4370–81. doi: 10.1128/IAI.05422-11
36. Wu X, Deng G, Hao X, Li Y, Zeng J, Ma C, et al. A caspase-dependent pathway is involved in Wnt/β-catenin signaling promoted apoptosis in bacillus Calmette-Guerin infected RAW264.7 macrophages. Int J Mol Sci. (2014) 15:5045–62. doi: 10.3390/ijms15035045
37. Schaale K, Neumann J, Schneider D, Ehlers S, Reiling N. Wnt signaling in macrophages: Augmenting and inhibiting mycobacteria-induced inflammatory responses. Eur J Cell Biol. (2011) 90:553–9. doi: 10.1016/j.ejcb.2010.11.004
38. Neumann J, Schaale K, Farhat K, Endermann T, Ulmer AJ, Ehlers S, et al. Frizzled1 is a marker of inflammatory macrophages, and its ligand Wnt3a is involved in reprogramming Mycobacterium tuberculosis-infected macrophages. FASEB J. (2010) 24:4599–612. doi: 10.1096/fj.10-160994
39. Ferrari G, Langen H, Naito M, Pieters J. A coat protein on phagosomes involved in the intracellular survival of Mycobacteria. Cell. (1999) 97:435–47. doi: 10.1016/S0092-8674(00)80754-0
40. Trinath J, Holla S, Mahadik K, Prakhar P, Singh V, Balaji KN. The WNT signaling pathway contributes to dectin-1-dependent inhibition of toll-like receptor-induced inflammatory signature. Mol Cell Biol. (2014) 34:4301–14. doi: 10.1128/MCB.00641-14
41. Schaale K, Reiling N, Leitges M, Brandenburg J, Kispert A, Ehlers S. Wnt6 is expressed in granulomatous lesions of Mycobacterium tuberculosis-infected mice and is involved in macrophage differentiation and proliferation. J Immunol. (2013) 191:5182–95. doi: 10.4049/jimmunol.1201819
42. Bezerra Lima B, Faria Fonseca B, da Graça Amado N, Moreira Lima D, Albuquerque Ribeiro R, Garcia Abreu J, et al. Clostridium difficile toxin A attenuates Wnt/β-catenin Signaling in intestinal epithelial cells. Infect Immun. (2014) 82:2680–7. doi: 10.1128/IAI.00567-13
43. Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, et al. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature. (2016) 538:350–5. doi: 10.1038/nature19799
44. Chen P, Tao L, Wang T, Zhang J, He A, Lam K, et al. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science. (2018) 360:664–9. doi: 10.1126/science.aar1999
45. Murata-Kamiya N, Kurashima Y, Teishikata Y, Yamahashi Y, Saito Y, Higashi H, et al. Helicobacter pylori CagA interacts with E-cadherin and deregulates the β-catenin signal that promotes intestinal transdifferentiation in gastric epithelial cells. Oncogene. (2007) 26:4617–26. doi: 10.1038/sj.onc.1210251
46. Yang H-J, Kim SG, Lim JH, Choi JM, Kim WH, Jung HC. Helicobacter pylori-induced modulation of the promoter methylation of Wnt antagonist genes in gastric carcinogenesis. Gastric Cancer. (2018) 21:237–48. doi: 10.1007/s10120-017-0741-6
47. Yokoyama K, Higashi H, Ishikawa S, Fujii Y, Kondo S, Kato H, et al. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signaling pathway in gastric epithelial cells. Proc Natl Acad Sci USA. (2005) 102:9661–6. doi: 10.1073/pnas.0502529102
48. Vikström E, Bui L, Konradsson P, Magnusson KE. The junctional integrity of epithelial cells is modulated by Pseudomonas aeruginosa quorum sensing molecule through phosphorylation-dependent mechanisms. Exp Cell Res. (2009) 315:313–26. doi: 10.1016/j.yexcr.2008.10.044
49. Cott C, Thuenauer R, Landi A, Kühn K, Juillot S, Imberty A, et al. Pseudomonas aeruginosa lectin LecB inhibits tissue repair processes by triggering β-catenin degradation. Biochim Biophys Acta Mol Cell Res. (2016) 1863:1106–18. doi: 10.1016/j.bbamcr.2016.02.004
50. Mysorekar IU, Mulvey MA, Hultgren SJ, Gordon JI. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J Biol Chem. (2002) 277:7412–9. doi: 10.1074/jbc.M110560200
51. Ting K, Aitken KJ, Penna F, Samiei AN, Sidler M, Jiang JX, et al. Uropathogenic E. coli (UPEC) infection induces proliferation through enhancer of zeste homologue 2 (EZH2). PLoS ONE. (2016) 11:e0149118. doi: 10.1371/journal.pone.0149118
52. Roy BC, Subramaniam D, Ahmed I, Jala VR, Hester CM, Greiner KA, et al. Role of bacterial infection in the epigenetic regulation of Wnt antagonist WIF1 by PRC2 protein EZH2. Oncogene. (2015) 34:4519–30. doi: 10.1038/onc.2014.386
53. Kurtz JR, Goggins JA, McLachlan JB. Salmonella infection: interplay between the bacteria and host immune system. Immunol Lett. (2017) 190:42–50. doi: 10.1016/j.imlet.2017.07.006
54. Wertz IE, Dixit VM. Signaling to NF-κB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. (2010) 2:a003350. doi: 10.1101/cshperspect.a003350
55. Nauciel C, Espinasse-Maes F. Role of gamma interferon and tumor necrosis factor alpha in resistance to Salmonella typhimurium infection. Infect Immun. (1992) 60:450–4. doi: 10.1007/978-1-4615-2854-8_28
56. Nix RN, Altschuler SE, Henson PM, Detweiler CS. Hemophagocytic macrophages harbor Salmonella enterica during persistent infection. PLoS Pathog. (2007) 3:1982–92. doi: 10.1371/journal.ppat.0030193
57. Engelhardt B, Liebner S. Novel insights into the development and maintenance of the blood–brain barrier. Cell Tissue Res. (2014) 355:687–99. doi: 10.1007/s00441-014-1811-2
58. Liu X, Lu R, Wu S, Sun J. Salmonella regulation of intestinal stem cells through the Wnt/β-catenin pathway. FEBS Lett. (2010) 584:911–6. doi: 10.1016/j.febslet.2010.01.024
59. Knoop KA, Kumar N, Butler BR, Sakthivel SK, Taylor RT, Nochi T, et al. RANKL is necessary and sufficient to initiate development of antigen-sampling m cells in the intestinal epithelium. J Immunol. (2009) 183:5738–47. doi: 10.4049/jimmunol.0901563
60. Kannan L, Kis-Toth K, Yoshiya K, Thai T-H, Sehrawat S, Mayadas TN, et al. R-spondin3 prevents mesenteric ischemia/reperfusion-induced tissue damage by tightening endothelium and preventing vascular leakage. Proc Natl Acad Sci USA. (2013) 110:14348–53. doi: 10.1073/pnas.1309393110
61. Elwell C, Mirrashidi K, Engel J. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol. (2016) 14:385–400. doi: 10.1038/nrmicro.2016.30
62. Hogan RJ, Mathews SA, Mukhopadhyay S, Summersgill JT, Timms P. Chlamydial persistence: beyond the biphasic paradigm. Infect Immun. (2004) 72:1843–55. doi: 10.1128/IAI.72.4.1843-1855.2004
63. Clevers H, Loh KM, Nusse R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science. (2014) 346:1248012. doi: 10.1126/science.1248012
64. Brembeck FH, Rosário M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of β-catenin. Curr Opin Genet Dev. (2006) 16:51–9. doi: 10.1016/j.gde.2005.12.007
65. Vlad-Fiegen A, Langerak A, Eberth S, Müller O. The Wnt pathway destabilizes adherens junctions and promotes cell migration via β-catenin and its target gene cyclin D1. FEBS Open Bio. (2012) 2:26–31. doi: 10.1016/j.fob.2012.02.004
66. Ding Y, Xi Y, Chen T, Wang J, Tao D, Wu Z-L, et al. Caprin-2 enhances canonical Wnt signaling through regulating LRP5/6 phosphorylation. J Cell Biol. (2008) 182:865–72. doi: 10.1083/jcb.200803147
67. Flores R, Luo J, Chen D, Sturgeon G, Shivshankar P, Zhong Y, et al. Characterization of the hypothetical protein Cpn1027, a newly identified inclusion membrane protein unique to Chlamydia pneumoniae. Microbiology. (2007) 153:777–86. doi: 10.1099/mic.0.2006/002956-0
68. Le Negrate G, Krieg A, Faustin B, Loeffler M, Godzik A, Krajewski S, et al. ChlaDub1 of Chlamydia trachomatis suppresses NF-κB activation and inhibits IκBα ubiquitination and degradation. Cell Microbiol. (2008) 10:1879–92. doi: 10.1111/j.1462-5822.2008.01178.x
69. Latres E, Chiaur DS, Pagano M. The human F box protein β-Trcp associates with the Cul1/Skp1 complex and regulates the stability of β-catenin. Oncogene. (1999) 18:849–54. doi: 10.1038/sj.onc.1202653
70. Nadel B, Gorvel J-P, Neefjes J, Gagnaire A, Raoult D. Collateral damage: insights into bacterial mechanisms that predispose host cells to cancer. Nat Rev Microbiol. (2017) 15:109–28. doi: 10.1038/nrmicro.2016.171
71. Eluf-Neto J, Herrero R, van den Brule AJC, Smith JS, Franceschi S, Peeling RW, et al. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer. (2004) 111:431–9. doi: 10.1002/ijc.20257
72. Zhu H, Shen Z, Luo H, Zhang W, Zhu X. Chlamydia trachomatis infection-associated risk of cervical cancer. Medicine. (2016) 95:e3077. doi: 10.1097/MD.0000000000003077
73. Bahrami A, Hasanzadeh M, ShahidSales S, Yousefi Z, Kadkhodayan S, Farazestanian M, et al. Clinical significance and prognosis value of wnt signaling pathway in cervical cancer. J Cell Biochem. (2017) 118:3028–33. doi: 10.1002/jcb.25992
74. Fang R, Blanton LS, Walker DH. Rickettsiae as emerging infectious agents. Clin Lab Med. (2017) 37:383–400. doi: 10.1016/j.cll.2017.01.009
75. Sahni A, Fang R, Sahni SK, Walker DH. Pathogenesis of Rickettsial diseases: pathogenic and immune mechanisms of an endotheliotropic infection. Annu Rev Pathol Mech Dis. (2019) 14:127–52. doi: 10.1146/annurev-pathmechdis-012418-012800
76. Masckauchán TNH, Shawber CJ, Funahashi Y, Li CM, Kitajewski J. Wnt/β-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis. (2005) 8:43–51. doi: 10.1007/s10456-005-5612-9
77. Lee DK, Nathan Grantham R, Trachte AL, Mannion JD, Wilson CL. Activation of the canonical Wnt/β-catenin pathway enhances monocyte adhesion to endothelial cells. Biochem Biophys Res Commun. (2006) 347:109–16. doi: 10.1016/j.bbrc.2006.06.082
78. Dejana E, Orsenigo F. Endothelial adherens junctions at a glance. J Cell Sci. (2013) 126:2545–9. doi: 10.1242/jcs.124529
79. Choi H-J, Park H, Lee H-W, Kwon Y-G. The Wnt pathway and the roles for its antagonists, DKKS, in angiogenesis. IUBMB Life. (2012) 64:724–31. doi: 10.1002/iub.1062
80. Ueland T, Otterdal K, Lekva T, Halvorsen B, Gabrielsen A, Sandberg WJ, et al. Dickkopf-1 enhances inflammatory interaction between platelets and endothelial cells and shows increased expression in atherosclerosis. Arterioscler Thromb Vasc Biol. (2009) 29:1228–34. doi: 10.1161/ATVBAHA.109.189761
81. Chen Y, Hu Y, Lu K, Flannery JG, Ma J-X. Very low density lipoprotein receptor, a negative regulator of the Wnt signaling pathway and choroidal neovascularization. J Biol Chem. (2007) 282:34420–8. doi: 10.1074/jbc.M611289200
82. Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, et al. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene. (2004) 23:8520–6. doi: 10.1038/sj.onc.1207892
83. Rikihisa Y. Molecular pathogenesis of Ehrlichia chaffeensis infection. Annu Rev Microbiol. (2015) 69:283–304. doi: 10.1146/annurev-micro-091014-104411
84. Zhang JZ, Popov VL, Gao S, Walker DH, Yu XJ. The developmental cycle of Ehrlichia chaffeensis in vertebrate cells. Cell Microbiol. (2007) 9:610–8. doi: 10.1111/j.1462-5822.2006.00812.x
85. Luo T, McBride JW. Ehrlichia chaffeensis TRP32 interacts with host cell targets that influence intracellular survival. Infect Immun. (2012) 80:2297–306. doi: 10.1128/IAI.00154-12
86. Luo T, Kuriakose JA, Zhu B, Wakeel A, McBride JW. Ehrlichia chaffeensis TRP120 interacts with a diverse array of eukaryotic proteins involved in transcription, signaling, and cytoskeleton organization. Infect. Immun. (2011) 79:4382–91. doi: 10.1128/IAI.05608-11
87. Lina TT, Farris T, Luo T, Mitra S, Zhu B, McBride JW. Hacker within! Ehrlichia chaffeensis effector driven phagocyte reprogramming strategy. Front Cell Infect Microbiol. (2016) 6:58. doi: 10.3389/fcimb.2016.00058
88. Mohan Kumar D, Yamaguchi M, Miura K, Lin M, Los M, Coy JF, et al. Ehrlichia chaffeensis uses its surface protein EtpE to bind GPI-anchored protein DNase X and trigger entry into mammalian cells. PLoS Pathog. (2013) 9:e1003666. doi: 10.1371/journal.ppat.1003666
89. Lin M, Zhu MX, Rikihisa Y. Rapid activation of protein tyrosine kinase and phospholipase cγ2 and increase in cytosolic free calcium are required by Ehrlichia chaffeensis for internalization and growth in THP-1 cells. Infect Immun. (2002) 70:889–98. doi: 10.1128/IAI.70.2.889-898.2002
90. Kumar DM, Lin M, Xiong Q, Webber MJ, Kural C, Rikihisa Y. EtpE binding to DNase X induces ehrlichial entry via CD147 and hnRNP-K recruitment, followed by mobilization of N-WASP and actin. MBio. (2015) 6:1–12. doi: 10.1128/mBio.01541-15
91. Maiti G, Naskar D, Sen M. The Wingless homolog Wnt5a stimulates phagocytosis but not bacterial killing. Proc Natl Acad Sci USA. (2012) 109:16600–5. doi: 10.1073/pnas.1207789109
92. Winter CG, Wang B, Ballew A, Royou A, Karess R, Axelrod JD, et al. Drosophila Rho-associated kinase (Drok) links Frizzled-mediated planar cell polarity signaling to the actin cytoskeleton. Cell. (2001) 105:81–91. doi: 10.1016/S0092-8674(01)00298-7
93. Wang Q, Symes AJ, Kane CA, Freeman A, Nariculam J, Munson P, et al. A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS ONE. (2010) 5:e10456. doi: 10.1371/journal.pone.0010456
94. Witze ES, Litman ES, Argast GM, Moon RT, Ahn NG. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science. (2008) 320:365–9. doi: 10.1126/science.1151250
95. Petherick KJ, Williams AC, Lane JD, Ordóñez-Morán P, Huelsken J, Collard TJ, et al. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. (2013) 32:1903–16. doi: 10.1038/emboj.2013.123
96. Steere N, Chae V, Burke M, Li FQ, Takemaru KI, Kuriyama R. A Wnt/β-catenin pathway antagonist Chibby binds Cenexin at the distal end of mother centrioles and functions in primary cilia formation. PLoS ONE. (2012) 7:e41077. doi: 10.1371/journal.pone.0041077
97. Angers S, Thorpe CJ, Biechele TL, Goldenberg SJ, Zheng N, MacCoss MJ, et al. The KLHL12–Cullin-3 ubiquitin ligase negatively regulates the Wnt–β-catenin pathway by targeting Dishevelled for degradation. Nat Cell Biol. (2006) 8:348–57. doi: 10.1038/ncb1381
98. Nakadai T, Fukuda A, Shimada M, Nishimura K, Hisatake K. The RNA binding complexes NF45-NF90 and NF45-NF110 associate dynamically with the c-fos gene and function as transcriptional coactivators. J Biol Chem. (2015) 290:26832–45. doi: 10.1074/jbc.M115.688317
99. Liu Y, Huang D, Wang Z, Wu C, Zhang Z, Wang D, et al. LMO2 attenuates tumor growth by targeting the Wnt signaling pathway in breast and colorectal cancer. Sci Rep. (2016) 6:36050. doi: 10.1038/srep36050
100. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. (2000) 80:1483–521. doi: 10.1152/physrev.2000.80.4.1483
101. Lorenowicz MJ, Macurkova M, Harterink M, Middelkoop TC, de Groot R, Betist MC, et al. Inhibition of late endosomal maturation restores Wnt secretion in Caenorhabditis elegans vps-29 retromer mutants. Cell Signal. (2014) 26:19–31. doi: 10.1016/j.cellsig.2013.09.013
102. Gasperowicz M, Otto F. Mammalian groucho homologs: redundancy or specificity? J Cell Biochem. (2005) 95:670–87. doi: 10.1002/jcb.20476
103. Lukas J, Mazna P, Valenta T, Doubravska L, Pospichalova V, Vojtechova M, et al. Dazap2 modulates transcription driven by the Wnt effector TCF-4. Nucleic Acids Res. (2009) 37:3007–20. doi: 10.1093/nar/gkp179
104. Vasileiou G, Ekici AB, Uebe S, Zweier C, Hoyer J, Engels H, et al. Chromatin-remodeling-factor ARID1B represses Wnt/β-catenin signaling. Am J Hum Genet. (2015) 97:445–56. doi: 10.1016/j.ajhg.2015.08.002
105. Jiang W, Wang J, Zhang Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res. (2013) 23:122–30. doi: 10.1038/cr.2012.119
106. Carneiro FRG, Ramalho-Oliveira R, Mognol GP, Viola JPB. Interferon regulatory factor 2 binding protein 2 is a new NFAT1 partner and represses its transcriptional activity. Mol Cell Biol. (2011) 31:2889–901. doi: 10.1128/MCB.00974-10
107. Zhu B, Das S, Mitra S, Farris TR, McBride JW. Ehrlichia chaffeensis TRP120 moonlights as a HECT E3 ligase involved in self- and host ubiquitination to influence protein interactions and stability for intracellular survival. Infect Immun. (2017) 85:e00290–17. doi: 10.1128/IAI.00290-17
108. Villaseñor T, Madrid-Paulino E, Maldonado-Bravo R, Urbán-Aragón A, Pérez-Martínez L, Pedraza-Alva G. Activation of the Wnt pathway by Mycobacterium tuberculosis: a Wnt-Wnt situation. Front Immunol. (2017) 8:50. doi: 10.3389/fimmu.2017.00050
109. Farooq PD, Urrunaga NH, Tang DM, von Rosenvinge EC. Pseudomembranous colitis. Dis Mon. (2015) 61:181–206. doi: 10.1016/j.disamonth.2015.01.006
110. Chandrasekaran R, Lacy DB. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev. (2017) 41:723–50. doi: 10.1093/femsre/fux048
111. Govind R, Dupuy B. Secretion of Clostridium difficile toxins A and B requires the holin-like protein TcdE. PLoS Pathog. (2012) 8:e1002727. doi: 10.1371/journal.ppat.1002727
112. Lyerly DM, Saum KE, MacDonald DK, Wilkins TD. Effects of Clostridium difficile toxins given intragastrically to animals. Infect. Immun. (1985) 47:349–52.
113. King AM, Mackin KE, Lyras D. Emergence of toxin A-negative, toxin B-positive Clostridium difficile strains: epidemiological and clinical considerations. Fut Microbiol. (2015) 10:1–4. doi: 10.2217/fmb.14.115
114. van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. (2009) 71:241–60. doi: 10.1146/annurev.physiol.010908.163145
115. Wu X, Tu X, Joeng KS, Hilton MJ, Williams DA, Long F. Rac1 activation controls nuclear localization of β-catenin during canonical Wnt signaling. Cell. (2008) 133:340–53. doi: 10.1016/j.cell.2008.01.052
116. Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC. Structural basis of Wnt recognition by frizzled. Science. (2012) 337:59–64. doi: 10.1126/science.1222879
117. Nile AH, Mukund S, Stanger K, Wang W, Hannoush RN. Unsaturated fatty acyl recognition by Frizzled receptors mediates dimerization upon Wnt ligand binding. Proc Natl Acad Sci USA. (2017) 114:4147–52. doi: 10.1073/pnas.1618293114
118. Schlessinger K, Hall A, Tolwinski N. Wnt signaling pathways meet Rho GTPases. Genes Dev. (2009) 23:265–77. doi: 10.1101/gad.1760809
119. Tohidpour A. CagA-mediated pathogenesis of Helicobacter pylori. Microb Pathog. (2016) 93:44–55. doi: 10.1016/j.micpath.2016.01.005
120. IARC. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr. Eval Carcinog Risks Hum. (1994) 61:1–241.
121. Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. (2002) 2:28–37. doi: 10.1038/nrc703
122. Franco AT, Israel DA, Washington MK, Krishna U, Fox JG, Rogers AB, et al. Activation of β-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA. (2005) 102:10646–51. doi: 10.1073/pnas.0504927102
123. Weydig C, Starzinski-Powitz A, Carra G, Löwer J, Wessler S. CagA-independent disruption of adherence junction complexes involves E-cadherin shedding and implies multiple steps in Helicobacter pylori pathogenicity. Exp Cell Res. (2007) 313:3459–71. doi: 10.1016/j.yexcr.2007.07.015
124. Gnad T, Feoktistova M, Leverkus M, Lendeckel U, Naumann M. Helicobacter pylori-induced activation of β-catenin involves low density lipoprotein receptor-related protein 6 and Dishevelled. Mol Cancer. (2010) 9:31. doi: 10.1186/1476-4598-9-31
125. Sokolova O, Bozko PM, Naumann M. Helicobacter pylori suppresses glycogen synthase kinase 3β to promote β-catenin activity. J Biol Chem. (2008) 283:29367–74. doi: 10.1074/jbc.M801818200
126. Lee D-G, Kim HS, Lee YS, Kim S, Cha SY, Ota I, et al. Helicobacter pylori CagA promotes Snail-mediated epithelial–mesenchymal transition by reducing GSK-3 activity. Nat Commun. (2014) 5:4423. doi: 10.1038/ncomms5423
127. Gorrell R, Kwok T. The Helicobacter pylori methylome: roles in gene regulation and virulence. Curr Top Microbiol Immunol. (2017) 400:105–27. doi: 10.1007/978-3-319-50520-6_5
128. Huang JJ, Han JI, Zhang LH, Leadbetter JR. Utilization of Acyl-homoserine lactone quorum signals for growth by a soil pseudomonad and Pseudomonas aeruginosa PAO1. Appl Environ Microbiol. (2003) 69:5941–9. doi: 10.1128/AEM.69.10.5941-5949.2003
129. Turkina MV, Vikström E. Bacteria-host crosstalk: sensing of the quorum in the context of Pseudomonas aeruginosa infections. J Innate Immun. (2019) 11:263–79. doi: 10.1159/000494069
130. Smith RS, Iglewski BH. Pseudomonas aeruginosa quorum sensing as a potential antimicrobial target. J Clin Invest. (2003) 112:1460–5. doi: 10.1172/JCI200320364
131. Chen K, Fu Q, Li D, Wu Y, Sun S, Zhang X. Wnt3a suppresses Pseudomonas aeruginosa-induced inflammation and promotes bacterial killing in macrophages. Mol Med Rep. (2016) 13:2439–46. doi: 10.3892/mmr.2016.4869
132. Marquart ME, O'Callaghan RJ. Infectious keratitis: secreted bacterial proteins that mediate corneal damage. J Ophthalmol. (2013) 2013:1–9. doi: 10.1155/2013/369094
133. Chen K, Yin L, Nie X, Deng Q, Wu Y, Zhu M, et al. β-Catenin promotes host resistance against Pseudomonas aeruginosa keratitis. J Infect. (2013) 67:584–94. doi: 10.1016/j.jinf.2013.07.025
134. Voronkov A, Krauss S. Wnt/β-catenin signaling and small molecule inhibitors. Curr Pharm Des. (2013) 19:634–64. doi: 10.2174/138161213804581837
135. Wang H, Kumar A, Lamont RJ, Scott DA. GSK3β and the control of infectious bacterial diseases. Trends Microbiol. (2014) 22:208–17. doi: 10.1016/j.tim.2014.01.009
136. Tsao N, Kuo CF, Chiu CC, Lin WC, Huang WH, Chen LY. Protection against Klebsiella pneumoniae using lithium chloride in an intragastric infection model. Antimicrob Agents Chemother. (2015) 59:1525–33. doi: 10.1128/AAC.04261-14
137. Chen K, Wu Y, Zhu M, Deng Q, Nie X, Li M, et al. Lithium chloride promotes host resistance against Pseudomonas aeruginosa keratitis. Mol Vis. (2013) 19:1502–14. Available online at: http://www.molvis.org/molvis/v19/1502
138. Chemani C, Imberty A, De Bentzmann S, Pierre M, Wimmerová M, Guery BP, et al. Role of LecA and LecB lectins in Pseudomonas aeruginosa-induced lung injury and effect of carbohydrate ligands. Infect Immun. (2009) 77:2065–75. doi: 10.1128/IAI.01204-08
139. Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. J Lab Autom. (2015) 20:107–26. doi: 10.1177/2211068214561025
140. Sonawane A, Jyot J, Ramphal R. Pseudomonas aeruginosa LecB is involved in pilus biogenesis and protease IV activity but not in adhesion to respiratory mucins. Infect Immun. (2006) 74:7035–9. doi: 10.1128/IAI.00551-06
141. Golovkine G, Faudry E, Bouillot S, Elsen S, Attrée I, Huber P. Pseudomonas aeruginosa transmigrates at epithelial cell-cell junctions, exploiting sites of cell division and senescent cell extrusion. PLoS Pathog. (2016) 12:1–22. doi: 10.1371/journal.ppat.1005377
142. Smith RS, Harris SG, Phipps R, Iglewski B. The Pseudomonas aeruginosa quorum-sensing molecule N-(3-Oxododecanoyl)homoserine lactone contributes to virulence and induces inflammation in vivo. J Bacteriol. (2002) 184:1132–9. doi: 10.1128/jb.184.4.1132-1139.2002
143. Imberty A, Wimmerová M, Mitchell EP, Gilboa-Garber N. Structures of the lectins from Pseudomonas aeruginosa: Insights into the molecular basis for host glycan recognition. Microbes Infect. (2004) 6:221–8. doi: 10.1016/j.micinf.2003.10.016
144. Kulkarni R, Dhakal BK, Slechta ES, Kurtz Z, Mulvey MA, Thanassi DG. Roles of putative type II secretion and type IV pilus systems in the virulence of uropathogenic Escherichia coli. PLoS ONE. (2009) 4:e4752. doi: 10.1371/journal.pone.0004752
145. Birder LA. Urothelial signaling. Auton Neurosci Basic Clin. (2010) 153:33–40. doi: 10.1016/j.autneu.2009.07.005
146. Zhang Y, Atala A. Urothelial cell culture: stratified urothelial sheet and three-dimensional growth of urothelial structure. In: Randell S, Fulcher M, editors. Epithelial Cell Culture Protocols. Methods in Molecular Biology (Methods and Protocols), Vol. 945. Totowa, NJ: Humana Press (2012).
147. Lewis AJ, Richards AC, Mulvey MA. Invasion of host cells and tissues by uropathogenic bacteria. Microbiol Spectr. (2017) 4:1–18. doi: 10.1128/microbiolspec.UTI-0026-2016
148. Terlizzi ME, Gribaudo G, Maffei ME. UroPathogenic Escherichia coli (UPEC) infections: Virulence factors, bladder responses, antibiotic, and non-antibiotic antimicrobial strategies. Front Microbiol. (2017) 8:e01566. doi: 10.3389/fmicb.2017.01566
149. Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. (2003) 301:105–7. doi: 10.1126/science.1084550
150. Maaser C, Housley MP, Iimura M, Smith JR, Vallance BA, Finlay BB, et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect Immun. (2004) 72:3315–24. doi: 10.1128/IAI.72.6.3315-3324.2004
151. Alipour M, Lou Y, Zimmerman D, Bording-Jorgensen MW, Sergi C, Liu JJ, et al. A balanced IL-1β activity is required for host response to Citrobacter rodentium infection. PLoS One. (2013) 8:e080656. doi: 10.1371/journal.pone.0080656
152. Petty NK, Bulgin R, Crepin VF, Cerdeño-Tárraga AM, Schroeder GN, Quail MA, et al. The Citrobacter rodentium genome sequence reveals convergent evolution with human pathogenic Escherichia coli. J Bacteriol. (2010) 192:525–38. doi: 10.1128/JB.01144-09
153. Dhakal BK, Mulvey MA. The UPEC pore-forming toxin α-hemolysin triggers proteolysis of host proteins to disrupt cell adhesion, inflammatory, and survival pathways. Cell Host Microbe. (2012) 11:58–69. doi: 10.1016/j.chom.2011.12.003
154. Bower JM, Eto DS, Mulvey MA. Covert operations of uropathogenic Escherichia coli within the urinary tract. Traffic. (2005) 6:18–31. doi: 10.1111/j.1600-0854.2004.00251.x
155. Chang Y-T, Chen C-L, Lin C-F, Lu S-L, Cheng M-H, Kuo C-F, et al. Regulatory role of GSK-3β on NF-κB, nitric oxide, and TNF-α in group A streptococcal infection. Mediators Inflamm. (2013) 2013:1–10. doi: 10.1155/2013/720689
156. Boren J, Shryock G, Fergis A, Jeffers A, Owens S, Qin W, et al. Inhibition of glycogen synthase kinase 3β blocks mesomesenchymal transition and attenuates Streptococcus pneumonia–mediated pleural injury in mice. Am J Pathol. (2017) 187:2461–72. doi: 10.1016/j.ajpath.2017.07.007
157. Adamowicz K, Wang H, Jotwani R, Zeller I, Potempa J, Scott DA. Inhibition of GSK3 abolishes bacterial-induced periodontal bone loss in mice. Mol Med. (2012) 18:1190–6. doi: 10.2119/molmed.2012.00180
158. Dugo L, Collin M, Thiemermann C. Glycogen synthase kinase 3β as a target for the therapy of shock and inflammation. Shock. (2007) 27:113–23. doi: 10.1097/01.shk.0000238059.23837.68
159. Hoeppner LH, Secreto FJ, Westendorf JJ. Wnt signaling as a therapeutic target for bone diseases. Expert Opin Ther Targets. (2009) 13:485–96. doi: 10.1517/14728220902841961
160. Janda CY, Dang LT, You C, Chang J, de Lau W, Zhong ZA, et al. Surrogate Wnt agonists that phenocopy canonical Wnt and β-catenin signalling. Nature. (2017) 545:234–7. doi: 10.1038/nature22306
161. Brogi S, Maramai S, Brindisi M, Chemi G, Porcari V, Corallo C, et al. Activation of the Wnt pathway by small peptides: rational design, synthesis and biological evaluation. ChemMedChem. (2017) 12:2074–85. doi: 10.1002/cmdc.201700551
162. Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. (2014) 13:513–32. doi: 10.1038/nrd4233
163. Clatworthy AE, Pierson E, Hung DT. Targeting virulence: a new paradigm for antimicrobial therapy. Nat Chem Biol. (2007) 3:541–8. doi: 10.1038/nchembio.2007.24
164. Bender KO, Garland M, Ferreyra JA, Hryckowian AJ, Child MA, Puri AW, et al. A small-molecule antivirulence agent for treating Clostridium difficile infection. Sci Transl Med. (2015) 7:306ra148. doi: 10.1126/scitranslmed.aac9103
165. Davies NL, Compson JE, MacKenzie B, O'Dowd VL, Oxbrow AKF, Heads JT, et al. A mixture of functionally oligoclonal humanized monoclonal antibodies that neutralize Clostridium difficile TcdA and TcdB with high levels of in vitro potency shows in vivo protection in a hamster infection model. Clin Vaccine Immunol. (2013) 20:377–90. doi: 10.1128/CVI.00625-12
Keywords: Wnt, β-catenin, bacteria, pathogen, innate immunity, immunoevasion
Citation: Rogan MR, Patterson LL, Wang JY and McBride JW (2019) Bacterial Manipulation of Wnt Signaling: A Host-Pathogen Tug-of-Wnt. Front. Immunol. 10:2390. doi: 10.3389/fimmu.2019.02390
Received: 01 July 2019; Accepted: 23 September 2019;
Published: 17 October 2019.
Edited by:
Wanderley De Souza, Federal University of Rio de Janeiro, BrazilReviewed by:
Emile Santos Barrias, National Institute of Metrology, Quality and Technology, BrazilCopyright © 2019 Rogan, Patterson, Wang and McBride. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jere W. McBride, amVtY2JyaWRAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.