 Tiziana Vaisitti
Tiziana Vaisitti Francesca Arruga
Francesca Arruga Giulia Guerra
Giulia Guerra Silvia Deaglio
Silvia Deaglio
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 04 October 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02301
This article is part of the Research Topic The Role of Ectonucleotidases in Acute and Chronic Inflammation View all 6 articles
Leukemia develops as the result of intrinsic features of the transformed cell, such as gene mutations and derived oncogenic signaling, and extrinsic factors, such as a tumor-friendly, immunosuppressed microenvironment, predominantly in the lymph nodes and the bone marrow. There, high extracellular levels of nucleotides, mainly NAD+ and ATP, are catabolized by different ectonucleotidases, which can be divided in two families according to substrate specificity: on one side those that metabolize NAD+, including CD38, CD157, and CD203a; on the other, those that convert ATP, namely CD39 (and other ENTPDases) and CD73. They generate products that modulate intracellular calcium levels and that activate purinergic receptors. They can also converge on adenosine generation with profound effects, both on leukemic cells, enhancing chemoresistance and homing, and on non-malignant immune cells, polarizing them toward tolerance. This review will first provide an overview of ectonucleotidases expression within the immune system, in physiological and pathological conditions. We will then focus on different hematological malignancies, discussing their role as disease markers and possibly pathogenic agents. Lastly, we will describe current efforts aimed at therapeutic targeting of this family of enzymes.
Adenosine triphosphate (ATP) and nicotinamide dinucleotide (NAD+) are present at millimolar concentrations inside the cell, where they represent the currency for energy metabolism. In the extracellular space their concentrations are usually low, but can transiently increase through active or passive mechanisms. Specifically, active release can occur via exocytosis or can be mediated by selective transmembrane proteins (e.g., pannexin, connexin). Otherwise, extracellular ATP can derive from passive leakage from necrotic or injured cells. Both mechanisms are enhanced during inflammation, hypoxia and cancer (1–3). High levels of extracellular ATP and NAD+ behave as danger signals, alerting the immune system to possible tissue damage. To prevent protracted reactions leading to chronic inflammation, homeostasis is rapidly restored through a scavenging circuit operated by nucleotide-catabolizing enzymes that produce the immunosuppressant adenosine and inosine, which can re-enter the cell, reconstituting the nucleotide pool (4).
Extracellular ATP influences cell metabolism, adhesion, and migration in settings of acute inflammation (5). Likewise, extracellular NAD+ is critical in immune system homeostasis by regulating homing processes of different subsets of immune cells (6). Both nucleotides exert these effects through binding surface purinergic receptors of the P2X and P2Y families. An extensive overview of their expression in different cell types and their role in healthy and pathological conditions can be found in Burnstock (7) and Di Virgilio et al. (8). Beside receptor-mediated signaling, which will be not addressed in the current review, extracellular ATP and NAD+ can be degraded by an integrated network of ectonucleotidases, which generate intermediates that modulate signaling and activate immunoregulatory circuits. To date, eight different ectonucleoside triphosphate diphosphohydrolase (ENTPDase) subtypes have been identified (ENTPD1-8, each with distinct catalytic properties and tissue-specific distribution) (9, 10). The best characterized ATP degrading pathway proceeds through the sequential action of CD39 (ectonucleoside triphosphate diphosphohydrolase-1, ENTPD-1), which converts extracellular ATP (or ADP) to adenosine monophosphate (AMP), and CD73 (5'-nucleotidase) that converts AMP to adenosine (ADO). As an alternative, ADO can be generated from NAD+ through to the coordinated action of CD38 (NAD glycohydrolase), which generates ADP ribose (ADPR), and PC-1 (ectonucleotide pyrophosphatase/phosphodiesterase family member 1), which generates AMP. The removal of the final phosphate occurs via CD73, which consequently acts as bottleneck enzyme for both cascades (11). It is noteworthy to mention that lack of other key nucleotide-metabolizing enzymes, including NTPDase or CD38, can globally impact on ATP/NAD dismantling. Furthermore, not all extracellular NAD+ is converted to ADO: ADP ribose (ADPR) and—to a lesser extent—cyclic ADPR (cAPDR), generated by CD38, elicit signal transduction by modulating intracellular calcium levels (Figure 1).
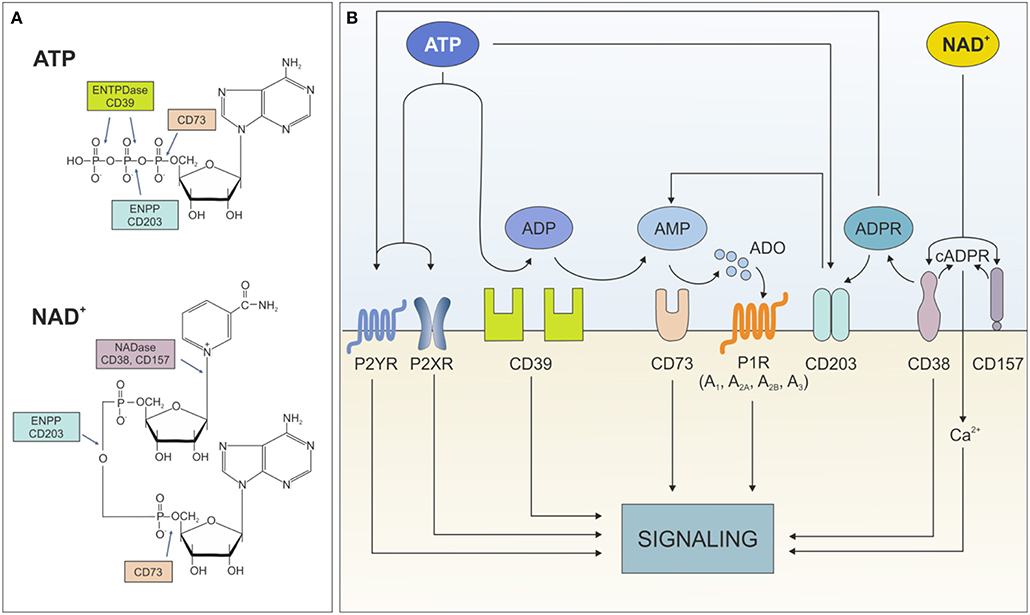
Figure 1. Ectonucleotidases network. (A) Structure of ATP and NAD+ with indication of cleavage sites by the different ectonucleotidases. (B) Schematic representation of the fate of extracellular ATP and NAD+ dismantling operated by the different ectonucleotidases and the functional consequences of the generated metabolites. ENTPDase, ecto-nucleoside triphosphate diphosphohydrolase; ENPP, ecto-phosphodiesterase/nucleotide phosphohydrolase; ATP, adenine-triphosphate; ADP, adenine-diphosphate; AMP, adenine-monophosphate; ADO, adenosine; P2XR, ATP-gated P2X receptor cation channel; P2YR, G protein-coupled P2Y receptor; P1R, adenosine purinergic receptor; NAD, nicotinamide adenine dinucleotide; ADPR, ADP-ribose; cADPR, cyclic-ADPR; Ca2+, calcium ions.
Despite being a physiological way to prevent excessive inflammation and tissue damage, in tumor biology ectonucleotidases can contribute to generate immunosuppressive conditions (4).
Similar to other mechanisms that are hijacked to serve pro-tumor purposes, tumor cells take advantage of the expression of these molecules. Levels of extracellular nucleotides rise in response to hypoxia and ischemia, two defining feature of the tumor environment (12, 13). In cancer, including hematological malignancies, over-expression of ectonucleotidases has been linked to increased homing to protected niches, increased survival and proliferation, as well as modulation of immune responses toward tolerance. In some instances, ectonucleotidases have become reliable markers to monitor disease and to stratify patient subsets or molecular targets for novel treatment strategies (14).
The aim of this review is to provide an overview of the structure and function of these enzymes in physiological conditions and then to focus on their expression and role in several hematological diseases. The reader should take in consideration that while CD38 has been extensively studied from the biological standpoint over the years and is now widely adopted as a marker in the clinical practice, the enzymes of the ATP-dismantling cascade gained visibility in the field of hematological malignancies only recently. For this reason, most of the notions concerning CD39 and CD73 reported in the present review are based on experimental rather than clinical experience.
Human CD38 and CD157/BST-1 are part of the same family of NADase/ADPR cyclase enzymes and share several features that reflect their origin from a common ancestor, a soluble enzyme found in the sea mollusk Aplysia californica (15, 16). Consistently, analysis of the coding genes shows a high degree of similarity in terms of exon-intron structure to the Aplysia ADP Ribosyl Cyclase (ADPRC) gene. Moreover, phylogenic analysis highlighted that CD38 and CD157 clearly derived from gene duplication, an event happened millions of years ago (17). During evolution from the original ancestral gene, CD38 and CD157 molecules acquired novel characteristics, including cell surface localization (18).
CD38 is a surface glycoprotein characterized by a relatively large extracellular domain that harbors the catalytic site, a single transmembrane pass and a short cytoplasmic tail (19). CD157 on the contrary, is attached to the membrane via a glycosylphosphatidylinositol (GPI) anchor (20). The extracellular domain of both molecules contains conserved critical residues that are essential for the enzymatic activity (21–24).
CD38 and CD157 pattern of expression is distinct in most tissues, including the hematopoietic system, suggesting that they regulate different cellular functions. Specifically, within the immune system, CD38 expression is high in immature hematopoietic cells, as well in activated T, B, dendritic and natural killer cells, but it is down-modulated in mature lymphocytes (20). CD157 on the other hand is mainly expressed by cells of the myelomonocytic lineage, including neutrophils, eosinophils, basophils, monocytes, macrophages, and plasmacytoid dendritic cells (20) (Table 1).
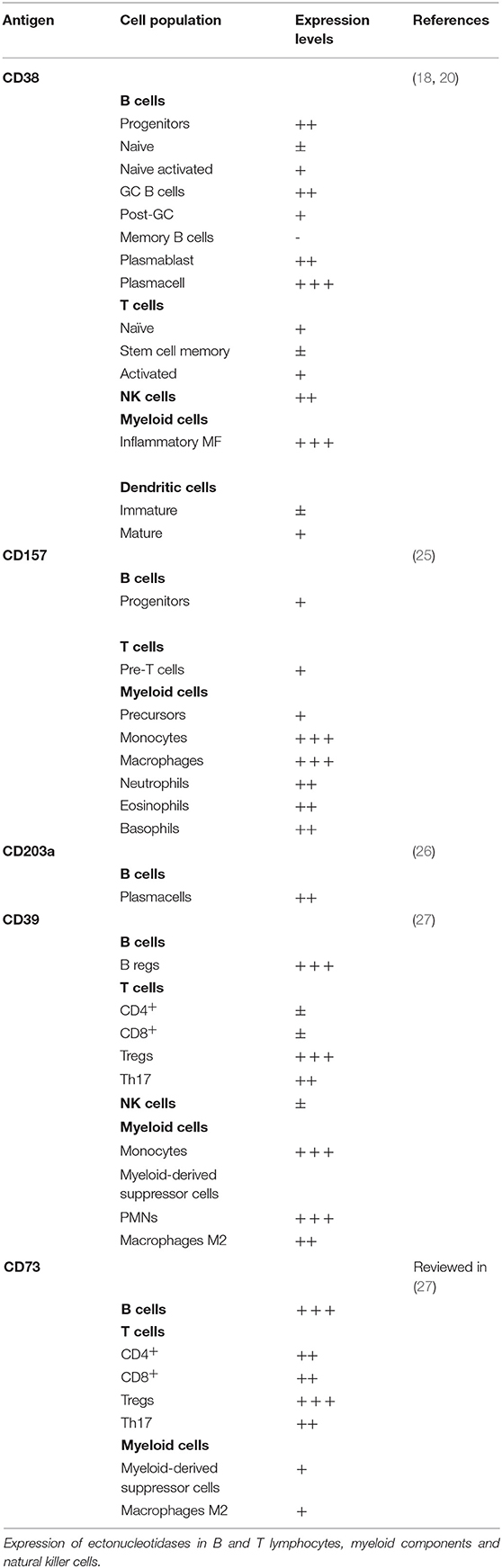
Table 1. Pattern of ectonucleotidases expression in non-malignant blood cells.
CD38, the main mammalian member of the ADPRC family, is a multifunctional ectoenzyme, involved in the catabolism and degradation of extracellular NAD+ (under normal pH) and NADP+ (under acidic pH). The main catalytic activity is that of NAD+ glycohydrolase that generates nicotinamide and ADPR. A minor activity of CD38 is that of NAD cyclase, which generates cADPR, then hydrolyzed to ADPR. Lastly, in the presence of NADP+ as substrate and of nicotinic acid and under acidic pH levels, CD38 can generate NAADP [revised in (28)]. ADPR, the main product of all these activities, undergoes different fates: (i) it can be covalently attached to target proteins modifying their functions, a process mediated by ADP-ribosyltransferases (ARTs) and known as ADP-ribosylation; (ii) it may bind purinergic receptors, specifically P2Y1, serving as a signaling molecule (29, 30); (iii) alternatively, it can enters the cells, where it can activate Ca2+ channels, activating calcium-induced calcium release. The calcium mobilizing property of ADPR is shared with the other products originated by CD38 activities, including cADPR and NAADP (31, 32). cADPR binds to ryanodine receptors (RyR) expressed on the endoplasmic reticulum, ADPR binds to membrane melastatin-related transient receptor potential cation channels TRPM2 (33) and NAADP binds to receptors expressed by acidic organelles, such as lysosomes, suggesting a role as calcium messenger in the endocytic pathway (32). Therefore, the final result of CD38 enzymatic activities is the modulation of intracellular calcium levels, which contribute to cell homeostasis and signaling (34). Similar to CD38, CD157 is able to generate cADPR and subsequently ADPR when incubated with NAD+, suggesting that this molecule has ADPRC and cADPR hydrolase activities (35, 36). However, its catalytic efficiency is markedly lower than that of CD38 (37).
The position of CD38 and CD157 on the cell surface suggests that their enzymatic activities can be modulated through protein-protein interactions. For example, CD38 can interact with CD31, a non-substrate ligand (38) and can form strong lateral association within lipid rafts. In T lymphocytes CD38 localizes to the immune synapse in close contact with the T cell receptor (39). Similar observations of a critical localization of CD38 in the “center of action” were reported for B lymphocytes, monocytes and NK cells (39–42). Although the functional implications of these interactions remain largely unknown, it is conceivable that CD38 may somehow act as a signaling booster, providing the additional increase in intracellular Ca2+ levels that may be necessary to induce or maintain receptor-mediated signaling.
CD38 has been implicated in T and B cell maturation, proliferation and cytokine production, mainly through the activation of intracellular signaling cascades that include phosphorylation of Zap70, c-Cbl, PLC-gamma, PI-3K, and MAPK (43). In addition, CD38 appears a critical mediator of chemotaxis and homing of immune cells. Genetic evidence was obtained from experiments performed using CD38−/− mice, showing that dendritic cells and neutrophils of these animals have defective ability to migrate toward inflammation sites in response to specific chemokines, mainly CCL2, CCL19, CCL21, and N-Formylmethionine-leucyl-phenylalanine (fMLP). The underlying mechanism resides in the loss of cADPR production by CD38, thereby limiting calcium mobilization upon engagement of the chemokine receptors (41, 44, 45). Similar to CD38, also CD157 is involved in chemotaxis and trans-endothelial migration of neutrophils (46).
Plasma cell antigen 1 (PC-1) is a class II transmembrane glycoprotein, belonging to the ecto-nucleotide pyrophosphatases/phosphodiesterases (E-NPPs) family that comprises seven different members. It is expressed on the cell surface and in different cellular compartments, including the endoplasmic reticulum. It is a homodimer characterized by a short amino-terminal intracellular portion, a single transmembrane sequence and a large extracellular domain, containing the catalytic site of the molecule. PC-1 has been detected in almost all tissues. In the immune system, its expression is restricted to plasma B cells, where it has been considered for several years as a marker (47).
E-NNPs, including PC-1, are part of the network of surface ectonucleotidases involved in several physiological functions, including nucleotide recycling, tuning of purinergic receptor signaling, modulation of pyrophosphate levels (critical for physiological calcification) and regulation of chemotaxis (10, 48). They exert their enzymatic functions by hydrolyzing the 5′-phosphodiester bonds in nucleotides and their derivatives, leading to the formation of nucleoside mono- or di-phosphate while releasing pyrophosphate (PPi) (Figure 1). Recent data prompted the idea that E-NNPs are part of the enzymatic cascade that produces ADO starting from ATP and NAD+, through the generation of AMP which is in turn the substrate for CD73, releasing ADO (10).
The cascade starting from ATP and leading to ADO production is governed by CD39 and CD73 and affects purinergic signaling by modulating ligand availability.
CD39, also known as NTPDase1, belongs to the (ecto-)nucleotide triphosphate diphosphohydrolase [(E)NTPDases] family of ectoenzymes that includes eight members differing for their catalytic properties and for their cellular localization. It is encoded by the ETNPD1 gene and it was the first NTPDase to be cloned and sequenced. Different splicing products have also been identified.
Together with NTPDase2, 3, and 8, CD39 has the active site facing the extracellular space. This site contains the “apyrase conserved regions,” highly conserved sequence domains, which are required for the phosphohydrolysis of extracellular nucleotides. Distinct phosphohydrolytic activities among ENTPDase family members are due to substantial differences in their sequences, which reflect in secondary, tertiary and quaternary structural differences (49). Consequently, they have distinct preferences for substrates and divalent cations, hydrolyze nucleoside triphosphates at varying rates, and generate different products. Micromolar levels of Ca2+ or Mg2+ ions are absolutely required for these four cell-surface-located ectoenzymes to exert maximal activity. CD39/ENTPD1, with a preference of Mg2+ over Ca2+, equally degrades ATP and ADP.
Other NTPDase are located inside the cells or toward the lumen of intracellular organelles. At variance with other NTPDases, CD39 can hydrolyze both ATP and ADP thus representing the rate-limiting enzyme in AMP production. A recent general description of CD39 is reviewed in Allard et al. (27).
Several structural requirements control the activity of this enzyme. First, two transmembrane domains are essential to anchor the protein to the cell membrane and to maintain the catalytic activity, as well as substrate specificity (50). Second, post-translational modifications, such as proteolysis and glycosylation, make the enzyme fully functional. Third, palmitoylation of the N-terminal intracytoplasmic domain enables association of CD39 with the lipid rafts, another requirement for full CD39 activity (51, 52).
Whereas, CD39 catalyzes the hydrolysis of ATP to AMP, CD73 is the rate-limiting enzyme in ADO generation pathways and it represents the step where NAD+ and ATP degradation cascades can converge. CD73 belongs to the ecto-5′-nucleotidase family that catalyzes the hydrolysis of 5′-AMP to ADO and inorganic phosphate (53, 54). It is encoded by the NT5E gene and is a GPI-anchored protein of ~70 kDa. This enzyme also exists in a soluble form derived from shedding of the GPI anchor and maintaining a similar enzymatic activity (55). The structure of CD73 is organized in three domains: a N-terminal domain with metal-binding sites, a C-terminal domain where the catalytic site is located, and a bridge alpha helix domain. Post-translational glycosylation, resulting in different molecular weight glycoforms, has also been reported (56). Full catalytic activity requires CD73 homodimerization, stabilized by non-covalent hydrophobic interactions between adjacent C-terminal domains, as well as the binding of two zinc ions. CD73 homodimers cycle from through open and closed conformation for efficient AMP hydrolysis (57). Other forms of 5'- nucleotidase exist in the cytoplasm and lysosomes and can be distinguished from CD73 by their substrate affinities, requirement for Mg2+ ion, activation by ATP, and inhibition by inorganic phosphate.
Furthermore, both CD39 and CD73 are characterized by specific structural features important for their potential activity as signal transducers, independent of their role as ATP-dismantling enzymes. For instance, CD39 shows sequence motifs, as well as secondary and tertiary structure similarities, with members of the actin/HSP70/sugar kinase superfamily (58) and can directly interact with Ran-binding protein M (RanBPM), a membrane scaffolding protein with GTPase activity (59). Likewise, CD73 can act as a cell adhesion molecule (59, 60) and can interact with and activate specific G-protein coupled receptor (GPCR) to increase intracellular cAMP levels, with reported effects on cancer cell aggressiveness, metastasis and angiogenesis [reviewed in (61, 62)].
In human peripheral blood, the majority of B cells and monocytes express CD39, as roughly one third of CD4+ T cells and, in a very small proportion, CD8+ T cells and NK cells. On the contrary, CD73 is expressed by B cells and by ~50% of CD8+ T cells, but only by ~10% of CD4+ T cells and <5% of NK cells [reviewed in (27)]. NK cells can acquire CD73 expression upon contact with mesenchymal stem cells and can then contribute to immune response modulation by converting AMP to ADO (63).
CD39 was initially described as a B cell activation marker (64), although, functionally, CD39 deletion did not impair global B cell number, but rather affected B cell memory responses to T-dependent antigens. This suggested that CD39 contributes to affinity maturation of antibody response and to post-germinal center B cell differentiation (65). Human B cells co-express CD39 and CD73, as well as adenosine receptors and they benefit from the generated ADO through an autocrine mechanism. The activation of adenosine receptors favors expansion and functions of adenosine-producing B cells. Furthermore, ADO generated by B cells influences the behavior of neighboring T cells (66). It was also observed that ectonucleotidases expression is associated with high proliferative capacity of regulatory B cells (Bregs) and with increased secretion of IL-10 and IL-6, which in turn potentiate the immunosuppressive activity toward T cells (67). Moreover, CD73 expression and adenosine production are involved in class-switch recombination. Indeed, CD73 lacking B cells presented impaired maturation of antibodies production (68).
Within the T cell compartment, CD39 is mainly expressed by CD4+ cells and principally by Tregs and Th17 even though it was reported on effector memory cells as well. Generally, expression of CD39 associates with the acquisition of an immunosuppressive phenotype and increased susceptibility to apoptosis and metabolic stress, therefore representing a mechanism to control expansion of cytotoxic T lymphocytes (69–71). On the other hand, CD73 expression marks selected T cell lineages, being found mainly on naïve T cells and in a smaller proportion of memory CD8+ cells. While decreasing upon T cell activation, CD73 is high in conditions of chronic inflammation where it associates with a memory T cell phenotype (72).
The finding that CD39 and CD73 are co-expressed on Treg cells (73), highlighted the central role of these ectonucleotidases in driving immune suppression through ADO generation, as witnessed by the observation that CD39−/− mice show significantly impaired suppressive properties in a model of allograft rejection in vivo (73). Several studies also suggested that CD39 expression on Tregs suppresses IL-17 production, either preventing trans-differentiation of Tregs into Th17 or endowing already differentiated Th17 cells with an immunosuppressive phenotype (74, 75). At variance with CD39, CD73 is present only in a small proportion of circulating Tregs, but it can be induced upon activation and in conditions of hypoxia. In a study from Stagg et al., it was shown that CD73−/− Tregs are less tumor-supportive than the WT counterpart (76), indicating a role for CD73 and suggesting that its expression is possibly modulated and induced upon Tregs activation. It was also proposed that Tregs are provided with ADO by paracrine interactions with neighboring CD73-expressing cells or through exosome-mediated exchanges (77). ADO, in turn, modulates Treg functions by promoting their proliferation and by increasing CTLA-4 and PD-1 expression, thus ultimately enhancing immunosuppressive potential (78) (Table 1). Increasing evidence suggests that exosome-mediated enzyme exchange could be a more general mechanism, exploited also by tumor cells to provide bystander cells with enzymes creating local conditions of immunosuppression and a tumor-friendly environment (79, 80).
CD39 and CD73 exert their pro-tolerogenic effects on the myeloid compartment as well. For instance, whereas extracellular ATP favors dendritic cells (DC) maturation and activity, by upregulating co-stimulatory molecules as CD38, CD83, CD86, and IL-12 secretion (81), ADO promotes tolerance by increasing IL-6 and IL-10 secretion and expression of immunosuppressive proteins such as indoleamine 2,3-dioxygenase (IDO), TGFβ, and arginase-2 (82). Consistently, anti-inflammatory macrophages with an M2 phenotype co-express CD39 and CD73 and show a higher ATP/AMP hydrolysis compared to pro-inflammatory M1 macrophages, in contrast characterized by low levels of both enzymes (83). This finding suggests that M2 population is accompanied by increase ATP conversion to ADO, which further modulates macrophage functions toward tolerance as witnessed by ADO-mediated increase of M2 markers and enhancement of IL-10 driven responses (84).
Taken together, these observations highlight the fine tuning underneath the regulation of immune system responses. Similar to other biological mechanisms that are derailed in tumors, expression of CD39 and CD73 is often deregulated in neoplastic cells, which take advantage of an immunosuppressive environment that facilitates tumor growth.
The main aim of this review is to discuss the role of ectonucleotidases in hematological malignancies. Wandering around literature, it comes to light that these molecules may have a role as simple markers to distinguish specific cellular populations or subset of patients characterized by a different prognosis, while in other cases they are deeply involved in the pathogenesis of the disease and in molding the tumor microenvironment, critically contributing to leukemic cells proliferation and survival. Similar to solid tumors, leukemia, lymphoma, and myeloma cells establish critical interactions with surrounding non-tumor cells. These interactions take place both in the blood circulation and in protected niches and provide extra stimuli that influence cell behavior. Those occurring in the environmental niches are mostly critical to drive disease progression and chemoresistance, and leukemic cells themselves progressively re-shape the environment to further support their growth while eluding immune surveillance.
Independently on their role, ectonucleotidases have been explored as therapeutic targets, representing in some instances, a winning strategy. In the following sections, we will go through the mechanisms through which NAD- and ATP-dismantling enzymes shape leukemic cells intrinsic features as well as tumor microenvironment in hematological malignancies, exploring also their applications useful for the clinical practice. Targeting strategies will be discussed in a dedicated paragraph.
CD38 is expressed in several hematological malignancies, including acute B lymphoblastic leukemia (B-ALL) (85), acute myeloid leukemia (AML) (85–87), mantle cell lymphoma (MCL) (88), chronic lymphocytic leukemia (CLL) (89, 90), multiple myeloma (MM) (91) and NK/T cell leukemia (T-ALL) (92, 93) (Figure 2).
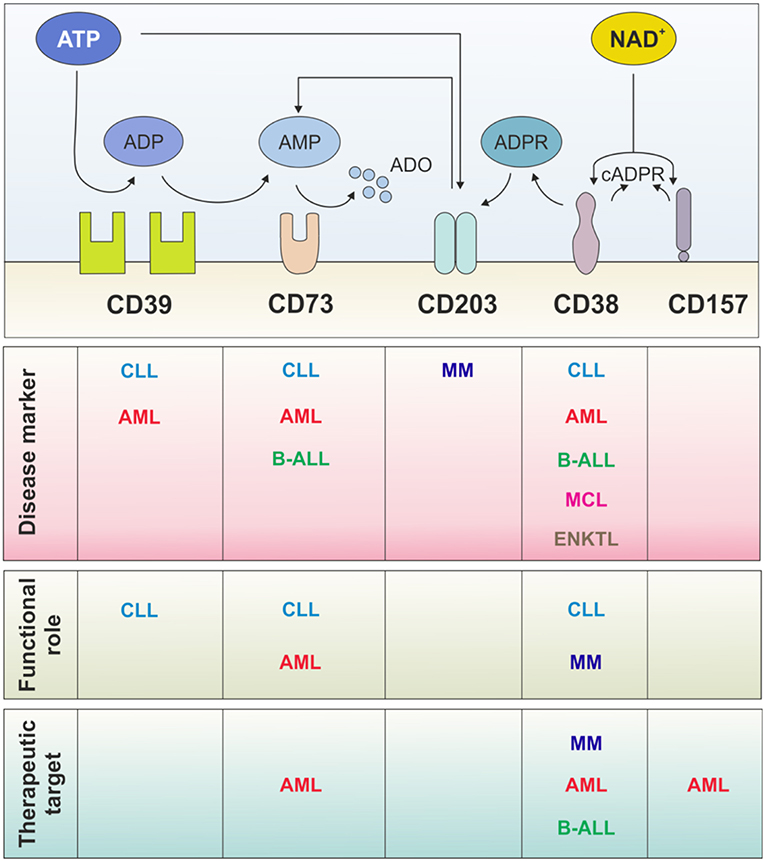
Figure 2. Multiple roles of ectonucleotidases in hematological malignancies. Surface NAD+- and ATP-dismantling enzymes have been widely exploited as disease markers for patient stratification and with prognostic relevance. In contrast, few studies, mainly in CLL and AML, address their functional role in the disease biology. Independently of their role, they may represent therapeutic targets: CD38 is a leading example with monoclonal antibodies already in the clinical practice. Several strategies have been designed to target ATP-dismantling enzymes, mainly in solid tumors, and conceivably should be exploited in hematological malignancies in the near future. CLL, chronic lymphocytic leukemia; MCL, mantle-cell lymphoma; B-ALL, B-acute lymphoblastic leukemia; AML, acute myeloid leukemia; ENKTL; extranodal natural killer/T cell lymphoma; MM, multiple myeloma.
The role of CD38 has been widely explored and defined in CLL, and to a lesser extent in MM, whereas little is known about its molecular and biological significance in other malignancies, where it is mainly reported as a negative or positive prognostic indicator.
Several years ago, Keyhani and colleagues reported CD38 expression as a positive predictor of disease outcome in previously untreated adult AML and ALL, where CD38 levels correlate with prolonged complete remission and overall survival and represent and independent positive prognostic marker (85). In line with these data, more recently, several papers reported that relapse of Philadephia chromosome positive ALL (Ph+ ALL) patients is due to the persistence of leukemia-initiating cells (LICs), a self-renewing CD34+CD38−CD58− population, capable of initiating human leukemia in immune-deficient mice (94–97). Higher LICs frequencies at diagnosis are predictive of unfavorable prognosis and clinical outcome in AML (98–100) and are considered an independent risk factor for relapse (101). This holds true also in childhood ALL, where a high proportion of CD34+CD38− cells negatively correlates with disease outcome and represents a useful marker for patients' risk stratification (102).
At variance with CD38, CD157 presents a more restricted pattern of expression in hematological malignancies, being present only in B-ALL and AML, where it serves as target for selective monoclonal antibodies (103). Specifically, in ALL CD157 was reported as an informative surface marker to discriminate leukemic from normal cells, suggesting its potential application to monitor residual leukemia cell during treatment regimens (104).
In contrast, despite being occasionally reported in solid tumors (105), little is known about pattern of expression of CD203a in hematological malignancies.
Twenty years ago, in the attempt to find reliable markers to distinguish among different subset of patients, the association between CD38 expression by CLL cells and a more aggressive behavior was reported for the first time (106). Several independent studies have subsequently confirmed the role of CD38 as a negative prognosticator for this disease and correlated its elevated expression with several adverse prognostic factors such as advanced disease stage, higher incidence of lymphadenopathy, high-risk cytogenetics, shorter lymphocytes doubling time (LDT), shorter time to initiation of first treatment (TFT) and poorer response to therapy (107–111).
Combining CD38 with other prognostic markers, such as Zap70 and CD49d, provides complementary prognostic information. Indeed, flow cytometry analyses on large cohorts of CLL patients indicated that co-expression of CD38 (30% cut-off) with Zap70 (20% cut-off) or with CD49d (30% cut-off) is associated with an unfavorable clinical course (112, 113). A close association between CD38 expression and elevated percentages of Zap70 and Ki-67 has also been highlighted, confirming the attitude of CD38+ clones to enter cell cycle (89). The finding of deleterious chromosomal abnormalities such as 11q and 17p deletions or trisomy 12 in patients with a CD38+ clone is also consistent with an enhanced proliferative potential (114–116) and a higher degree of clonal evolution (117, 118).
CD38 is highly expressed by MM cells although the characterization of its pathophysiological role in the disease is still an open field of research. Immunophenotyping data indicate that the combination of CD38 and CD138, another known marker of fully differentiated plasma cells, is a validated strategy to identify CD45+ myeloma and separate CD20+ myeloma from B-cell lymphoma. Moreover, plasma cells immunophenotyping might be useful for detecting minimal residual disease. Given its constituitve expression on plasma cells, it is not surprising that CD38 became an attractive target for CD38+ diseases.
In MCL, expression of CD38, together with CD20 and CD23, represents a robust marker to identify leukemic cells (88, 119). In addition, CD38 represents a marker to distinguish nodal (~94% of CD38+ cases) and non-nodal (~48% of CD38+ cases) MCL, with expression correlating with a shorter median survival in several independent series of MCL cases (120, 121).
A similar role for CD38 as prognostic marker was reported for extranodal natural killer/T cell lymphoma (ENKTL), an Epstein-Barr virus associated lymphoma relatively rare in western countries, but more diffuse in the Asian area. The majority of ENKTL cases are CD38+ and a stronger expression of this molecule significantly correlates with an inferior outcome, while a weak expression is associated with an increased complete response rate, opening to the possibility of using CD38 as a therapeutic target (93, 122).
A still open and controversial point concerns the role of CD38 in acute promyelocytic leukemia (APL), the M3 subtype of AML, which is characterized by the presence of the chimeric gene promyelocytic leukemia-retinoic acid receptor α (PML-RARA) (123) and by an arrest of leukocyte differentiation at the promyelocyte stage. Several clinical regimens combining anthracyclines, arsenic trioxide and retinoic acid (RA) induce differentiation and apoptosis of leukemic cells, resulting in high rates of durable remission in most patients (123, 124). CD38 is one of the earliest markers of RA response: its levels increase exponentially, starting 2–3 h after RA administration (125, 126). Hence, granulocytes differentiated from APL blasts express very high levels of CD38, at variance with their normal counterpart. A recent paper investigated the potential role and impact of CD38 in APL concluding that this molecule is not necessary for RA-induced differentiation. By using selective blocking substrates of the enzymatic activity and CRISPR/Cas9-mediated CD38 truncation, the authors showed that the molecule and its enzymatic activities are dispensable to the differentiation process (127). In conclusion, while the physiologic significance of this aberrant expression remains unclear, it was suggested that CD38 can play a role in a complication of RA therapy known as retinoic acid syndrome (RAS). This complication, which arises soon after RA administration, is characterized by fever, dyspnea and pulmonary edema. One hypothesis to explain this phenomenon involves adhesion of newly differentiated CD38+ granulocytes to lung endothelial cells expressing CD31, the CD38 ligand, and resulting in the local production of inflammatory cytokines, apoptosis of endothelial cells and eventually contributing to the development of RAS (128) (Figure 2).
Possibly due to intrinsic advantages, such as the high frequency of the disease and the presence of leukemic cells in the peripheral blood, CLL is the leukemia where the functions of CD38 have been studied more in depth.
The first evidence linking CD38 expression to functional responses in leukemic cells came from the observation that CD38+ clones showed a more robust response to IgM stimulation compared to negative counterparts (129–131). CD38 ligation with antibodies induced a signaling cascade, including phosphorylation of Zap70 and the MAP kinases, and leading to a more pronounced proliferation and the acquisition of immunoblast like features (132, 133).
Analysis of gene expression profiling after CD38 engagement highlighted migration as one of the most activated biological process (134). Consistently, when exposed to CXCL12, the main chemokine involved in CLL re-circulation from blood to lymph nodes through the binding to CXCR4 (135), CD38+ clones exhibited a more pronounced ability to migrate and to activate a selective signaling pathway that the counterpart (136). Lentiviral-induced expression of CD38 in negative primary CLL cells restored their ability to actively respond to the chemokine stimulus (136, 137). In addition to migration, CD38+ CLL cells showed more efficient integrin mediated adhesion and matrix digestion. The molecular mechanism behind this functional cooperation resides in their closed physical proximity on the CLL membrane, indicating the existence of a supra-molecular hub that tunes chemotactic responses, adhesion and ability to invade tissues (138, 139).
Within this hub, the likely functions of CD38 is to provide Ca2+-mobilizing compounds, mainly ADPR, through NAD+ hydrolysis. The implication is that inhibition of the enzymatic activity, impairs CLL chemotaxis, adhesion and in vivo homing, by trapping leukemic cells in the blood, preventing their localization in growth-permissive niches and increasing their sensitivity to chemotherapy (140).
MM is a tumor highly dependent on the BM microenvironment, which functions in a supportive way to promote proliferation, survival, and migration (141). One mechanism through which MM cells communicate with the microenvironment is via microvesiscles trafficking. Immunophenotypic characterization of these particles showed that they express high levels of CD38, together with other ectoenzymes, including CD203a, CD73, and CD39, which show functional activities thereby increasing ADO levels in the niche, ultimately contributing to microenvironment remodeling (142).
Recent data indicate that besides microvesicles, stromal cells from the environment can also deliver mitochondria to MM, contributing to the metabolic adaptation necessary to sustain the high proliferation rate of cancer cells and their homing capability. This organelle transfer has been reported also in MM, where it relies on CD38 and on non-malignant BM stromal cells acting as donors. The use of blocking anti-CD38 antibodies significantly impairs mitochondrial transfer from stromal to MM cells, in turn impacting on their viability (143).
In addition, CD38 is highly expressed also on the Treg fraction of MM patients compared to healthy subjects, and its levels correlate with the suppressive function of Tregs contributing to disease progression and expansion (144).
Similar to solid tumors, deregulation of CD39 and CD73 was also described in hematological malignancies, both of lymphoid and myeloid series. Expression of CD39 and CD73 is generally associated to the development of an immune tolerant environment, contributing to disease expansion. For this reason, levels of these ectonucleotidases, both on leukemic and bystander cells, are often exploited as disease markers or prognosticators. In some instances, the role of CD39 and CD73 has been functionally characterized unraveling a direct contribution in disease pathogenesis and outcome.
Monitoring the expression of CD39 and/or CD73 is a useful marker also in B-ALL and AML, although the role of nucleotide dismantling in these diseases is far less characterized than in CLL.
In a genome-wide analysis that compared 270 newly diagnosed ALL patients to normal B cell progenitors, CD73 was identified as a differentially expressed molecule, both at the transcript and at the protein levels (145). Flow cytometry validation identified CD73 as a useful marker to monitor minimal residual disease (MRD) as its expression correlated quite precisely to that of molecular markers already in use to predict ALL relapse. Specifically, CD73 is upregulated in B cell precursors of ALL patients and its expression is not down-modulated upon treatment thus making it a reliable marker to identify residual ALL cells resistant to therapies and potentially responsible for future relapses (146, 147).
AML is mainly characterized by aberrant differentiation of myeloid progenitors and impaired control of tumor growth likely due to the immunosuppressive environment created by leukemic blasts, leading to Tregs expansion (148). A study from Dulphy et al. reported that both Tregs and AML blasts express CD39 at higher levels compared to healthy subjects. The authors suggest that Tregs expansion and delocalization, together with ectonucleotidase expression and activity both on Tregs and on AML blasts, could contribute to immune escape of AML ultimately favoring leukemia growth (149). In support of a role for ATP dismantling in eliciting immune suppression in AML, another paper from Lecciso and colleagues reported that AML cells release ATP in response to chemotherapeutics and that through this they modulate dendritic cells and Tregs functions toward tolerance by upregulating CD39 expression on these subsets (150). In contrast, a very recent paper reported that CD73 expression on CD8 T cells favors a reawakening of immune response at variance with CD73−/CD8+ cells that exhibit a more exhausted profile (151). Beside its role in modulating immune system, CD73 may directly impact on leukemic cell proliferation under the control of CEBPA. In CEBPA mutant AML, CD73 is among the most upregulated genes suggesting that the adenosinergic axis may contribute to AML aggressiveness. Accordingly, silencing of CD73 significantly delays leukemia development in mice thus establishing a tumor-promoting role of this enzyme in leukemia progression in vivo (152).
It is therefore becoming clear that upregulation of CD39 and CD73 marks leukemic cells characterized by a more aggressive behavior likely resulting from a more permissive environment or a more efficient adaptation. The pathogenic role of nucleotide dismantling and its contribution to disease evolution has been characterized in relatively few disease models, on top of which there is CLL (Figure 2).
In CLL, several studies observed that CD39 expression is markedly increased in circulating leukemic cells compared to matched healthy subjects, and levels are further increased when looking at cells residing in the LN, representing a CLL sanctuary. Specifically, the proliferating Ki-67+ CLL fraction expresses CD39 at higher levels compared to the resting population that showed consistently weaker expression, suggesting a role in disease aggressiveness (153, 154). Besides leukemic cells, CLL LNs are rich of stromal components that are intensely CD39+, including T lymphocytes. The comparative analysis of T cell subsets highlighted that, overall, CLL patients have a higher percentage of CD39+ T lymphocytes than controls, both in the CD4+ and in the CD8+ compartment (155). Similar to CD39, CD73 expression is also associated with the proliferating CLL fraction that is in close contact with CD4+CD25+ Tregs although with more variability (156). CD73 expression characterize roughly one third of CLL patients, showing also intra-leukemic heterogeneity. This means that, despite virtually all CLL cells could degrade ADP to AMP, as they constitutively express CD39, not all of them can produce ADO at appreciable levels. It was observed that a cut-off of ≥30% CD73+ cells, is needed and ectoenzyme expression is in turn tightly regulated by local conditions, most important hypoxia (155, 157). Both CD39 and CD73 were investigated for CLL patients' prognostic classification. CD39 expression on the T cells fraction strongly associates with a more advanced disease stage and is considered predictive of treatment requirement, as significantly shorter TTFT was observed in patients with CD39high T cells compared to CD39low (158).
CD73 on leukemic B cells associates with other negative prognostic markers, such as CD38 and Zap70. It was proposed that CD73 expression identifies a leukemic subset of cells characterized by (i) an increased recirculation to and from the lymphoid niche, (ii) a more aggressive clinical behavior, (iii) a higher cellular turnover and (iv) associates with time to disease progression after fludarabine treatment (155, 159).
In the tumor microenvironment, ATP is abundantly released in the extracellular space, likely as the result of metabolic stress due to hypoxia and pro-inflammatory signals. Even if it could potentially recruit immune cells to fight against the tumor, ectonucleotidases ensure a rapid conversion of ATP to ADO, which accumulates in the extracellular space, contributing to skewing immune cells toward tolerance, supporting tumor growth instead (160). The role of ATP ectonucleotidases is mostly connected to the control of immunomodulatory activities and has been inferred by using drugs that modulate ADO production and signaling. Overexpression of both ATP-dismantling ectoenzymes and ADO receptors might carry negative prognostic relevance in hematological malignancies, marking aggressive disease and immunosuppression (161).
In a model of FL, hypo-responsiveness of infiltrating T cells could be partially reversed by blockade of adenosine receptors or inhibitors of CD39 activity. Accordingly, CD39 was upregulated in T cells residing in FL lymph nodes suggesting that ATP-dismantling and ADO signaling could be responsible for T cell anergy favoring FL expansion (162).
In CLL there is an intense cross-talk between leukemic and non-tumor bystander cells in microenvironment-associated structures (e.g., LN and BM), where adenosinergic signaling is integrated in the complex network of cell-cell contacts and soluble mediators driving leukemia (14, 163). CLL cells are equipped with both CD39/CD73 and A2A ADO receptors, indicating that adenosinergic signaling operates through both autocrine and paracrine effects. On the CLL side, high ADO levels contribute to keep leukemic cells in the growth-supportive environment of the LN, where they are recruited by chemokine signals (155). Furthermore, ADO up-regulates anti-apoptotic mediators, limiting the efficacy of chemotherapy. Consequently, in the LN environment, leukemic cells are protected from spontaneous and drug-induced apoptosis, maintaining a leukemia reservoir that fuels disease progression (164).
On the other hand, ADO acts on immune system cells, polarizing them toward tolerance, further sustaining leukemic cell expansion. The molecular mechanisms behind this effect involve the activation of the HIF1α genetic program (165), which directly upregulates ectonucleotidases expression and ADO signaling (166), as observed in other disease conditions (167, 168). Experimental evidence confirms that CD73 expression is significantly increased under in vitro hypoxic conditions, both at the mRNA level (NT5E) and on the surface of nurse-like cells (157), a type 2 (M2) macrophage population. When derived in vitro under low oxygen tension, NLCs significantly upregulated ADO production, fueling A2A signaling of surrounding cells and activating autocrine signaling through MAPK, PI3K, and NF-κB (157). ADO receptor signaling further contributes to M2 macrophage polarization by upregulating the transcription factor IRF4 and the tryptophan-metabolizing enzyme IDO, both M2 macrophages markers (169), and increasing synthesis of IL-6 (157).
Moreover, hypoxia-mediated upregulation of CD73 and of ADO generation also influences metabolic adaptation of the T lymphocytes surrounding the tumor, inducing a switch toward glycolysis, with robust upregulation of glucose and lactate transporters and of lactate dehydrogenase (LDHA) and pyruvate kinase 2 (PKM2). This metabolic conversion skews T cells functions to a regulatory phenotype, as indicated by increased expression of FOXP3, PD-1, IL-10, and VEGFA and the down-modulation of interferon γ (IFNG) mRNA. These effects can be recapitulated upon pharmacological modulation of A2A activation (157). Similar outcomes are observed on the leukemic fraction, in line with evidence that metabolic adaptation of tumor cells upon oxygen deprivation rapidly fosters energy production via glycolysis through HIF1α-mediated transcriptional control (170). In keeping with the immunomodulatory effects registered in the T cell compartment, metabolic adaptation induced by the hypoxia-ADO axis on leukemic B cells tunes CLL cytokine production and release and favors the acquisition of a B-regulatory phenotype, as defined by a marked upregulation of IL-10 (171). These findings link hypoxia and CD73-generated ADO in a common axis aimed at re-shaping a tumor-favorable niche with tumor-supportive and immunosuppressive features (157) (Figure 2).
Interrupting deregulated circuits promoting leukemia expansion, while awakening the immune surveillance against tumor cells, represents the “fil rouge” of target therapy approaches. In this scenario, nucleotide-dismantling enzymes represent good candidates as they play multifaceted roles, thus offering multiple levels of targeting. First, given their specific pattern of expression, they may serve as markers to selectively tag leukemic cells and deliver therapeutic agents while limiting off-target effects. Second, since they operate in a coordinated cascade of events, inhibition of one of them is sufficient to block the downstream processes. Lastly, they play several roles in tumor biology, so interfering with them opens the possibility to modulate immunosuppression. However, at least in hematological malignancies, clinical trials have just reached the first level, i.e., using antibodies to ectonucleotidases to specifically target a disease population.
Daratumumab, a humanized anti-CD38 IgG1 mAb, is the forefather and the most advanced immunotherapeutic agent, which was approved for clinical use in 2015 (172). Its primary application is in multiple myeloma (MM) where CD38 is highly and uniformly expressed, but it is also being evaluated in lymphomas, including diffuse large B cell lymphoma, FL and mantle cell lymphoma, in AML and in ALL (87, 173). Generally well-tolerated, daratumumab induces partial or complete responses in ~30% of heavily pretreated MM patients, when used as monotherapy and in this setting received its first approval in 2015. However, based on its distinct mechanisms of action, favorable toxicity profile and single-agent activity, daratumumab is an attractive partner in combination regimens. Significant responses and prolonged progression-free survival can be achieved in combination with immunomodulatory agents or proteasome inhibitors (174, 175). For these reasons, daratumumab is now approved by the FDA and EMA in combination with lenalidomide or bortezomib in MM patients who have received at least one prior therapy. Several clinical trials are ongoing both in the United States and Europe, evaluating the efficacy of this mAb with other drug combinations or as frontline therapy (175). The documented mechanisms of action include antibody-dependent cell cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), inhibition of CD38 enzymatic activities and induction of apoptosis in a caspase-dependent manner (176–178).
More recently new anti-CD38 antibodies are being developed, including isatuximab, a chimeric mouse/human Ab (179), and MOR202, a fully human Ab (180), underlying the interest of the medical community.
Besides mAbs, an alternative approach to target CD38 is the use of enzyme inhibitors. For example, flavonoids, such as kuromanin, bind to critical residues in the catalytic site competing with the substrates (140, 181). However, these strategies are still very far from clinical applications.
Targeting leukemic cells exploiting ectonucleotidases as surface markers is an approach pursued also with CD157. In vitro and ex vivo preclinical studies in AML and ALL show that anti-CD157 antibody, namely OBT357/MEN1112, has highly specific cytotoxicity against leukemic cells (103, 182). The antibody is currently being tested in a clinical trial for patients with relapsed or refractory AML.
In contrast, targeting of CD39 and CD73 in hematological malignancies remains an unexplored field despite promising results obtained in solid tumors, where modulation of their enzymatic activities is achieved by multiple strategies. Several CD39 inhibitors, including ARL67156 and POM-1, have shown efficacy in animal models of follicular lymphoma, sarcoma, or murine melanoma, resulting in a partial overcome of T-cell hypo-responsiveness to stimulation (162), increased therapeutic response to chemotherapeutic agents (183), or inhibition of tumor growth (184), respectively. Anti-CD39 antibodies, which also block the enzymatic activity, seem to be effective in different tumors, such as sarcoma and ovarian cancer (185–187). The efficacy of targeting ATP-dismantling enzymes as an anti-tumor therapy derives also from studies in breast cancer models where treatment of mice with an anti-CD73 antibody partially prevented lung metastases (188). Accordingly, a therapeutic anti-CD73 antibody, MEDI9447 is currently in clinical trials in solid cancer patients (NCT02503774; NCT03611556) (189). Potentially promising in a translational perspective is the compound a,b-methylene-ADP (APCP), the most potent competitive CD73 inhibitor, although no clinical trials are currently ongoing (190, 191).
More recently, nucleotide metabolizing enzymes have been explored as targets for the generation of nanobodies, soluble antibodies that correspond to the variable domain fragments derived from the heavy chain. A pool of nanobodies targeting CD38 is currently undergoing selection and functional characterization. Stemming from these results, nanobodies against CD39, CD73, and CD203 are likely to be designed (192) (Figure 2).
In this review, we have discussed basic knowledge on ectonucleotidases, addressing their role in hematological malignancies where they can be surface markers and, in some instances, they can directly contribute to leukemia expansion. The contributing role of ATP and NAD-dismantling enzymes is underlined by their involvement in the regulation of homing processes and in favoring local conditions of immunosuppression.
The emerging picture is that they represent good targets for novel therapeutic strategies. However, several unaddressed questions remain, including the convergence and cross-talk of these pathways toward the production of adenosine, the functional characterization and impact in leukemia other than CLL and the feasibility and relevance of their inhibition. This holds true mainly for the adenosinergic axis for which most of the current knowledge on therapeutic strategies comes from solid tumors. Efforts are being made to design inhibitors of adenosine signaling, which include compounds targeting A2A (CPI-444, PBF-509 and AZD4635) currently in Phase I/II clinical trials (193–195).
At the light of the major effects obtained with Daratumumab in MM, it is conceivable that in the near future additional “bullets” targeting ectonucleotidases will reach the market as adjunctive tools to conventional chemotherapy.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the European Union H2020 funds (INTEGRATA project #813284 to SD), by the GILEAD Fellowship Program (Gilead Italia 2018 to SD), by the Cassa di Risparmio di Torino Foundation (Fondazione CRT RF2017.0979 to TV), by the Italian Ministry of Health (GR-2016-02364298 to TV), and by the Ministry of Education, University and Research-MIUR Progetto strategico di Eccellenza Dipartimentale #D15D18000410001 to the Department of Medical Sciences, University of Turin.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kepp O, Loos F, Liu P, Kroemer G. Extracellular nucleosides and nucleotides as immunomodulators. Immunol Rev. (2017) 280:83–92. doi: 10.1111/imr.12571
2. Dosch M, Gerber J, Jebbawi F, Beldi G. Mechanisms of ATP release by inflammatory cells. Int J Mol Sci. (2018) 19:E1222. doi: 10.3390/ijms19041222
3. Principi E, Raffaghello L. The role of the P2X7 receptor in myeloid-derived suppressor cells and immunosuppression. Curr Opin Pharmacol. (2019) 47:82–9. doi: 10.1016/j.coph.2019.02.010
4. Antonioli L, Blandizzi C, Pacher P, Hasko G. Immunity, inflammation and cancer: a leading role for adenosine. Nat Rev Cancer. (2013) 13:842–57. doi: 10.1038/nrc3613
5. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. (2014) 509:310–7. doi: 10.1038/nature13085
6. Salmi M, Jalkanen S. Ectoenzymes in leukocyte migration and their therapeutic potential. Semin Immunopathol. (2014) 36:163–76. doi: 10.1007/s00281-014-0417-9
7. Burnstock G. Purinergic signalling: therapeutic developments. Front Pharmacol. (2017) 8:661. doi: 10.3389/fphar.2017.00661
8. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
9. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: important modulators of purinergic signalling cascade. Biochim Biophys Acta. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
10. Zimmermann H, Zebisch M, Strater N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. (2012) 8:437–502. doi: 10.1007/s11302-012-9309-4
11. Longhi MS, Robson SC, Bernstein SH, Serra S, Deaglio S. Biological functions of ecto-enzymes in regulating extracellular adenosine levels in neoplastic and inflammatory disease states. J Mol Med. (2013) 91:165–72. doi: 10.1007/s00109-012-0991-z
12. Kumar V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: where to go? Purinergic Signal. (2013) 9:145–65. doi: 10.1007/s11302-012-9349-9
13. Petrova V, Annicchiarico-Petruzzelli M, Melino G, Amelio I. The hypoxic tumour microenvironment. Oncogenesis. (2018) 7:10. doi: 10.1038/s41389-017-0011-9
14. Vaisitti T, Arruga F, Deaglio S. Targeting the adenosinergic axis in chronic lymphocytic leukemia: a way to disrupt the tumor niche? Int J Mol Sci. (2018) 19:E1167. doi: 10.3390/ijms19041167
15. States DJ, Walseth TF, Lee HC. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem Sci. (1992) 17:495. doi: 10.1016/0968-0004(92)90337-9
16. Itoh M, Ishihara K, Tomizawa H, Tanaka H, Kobune Y, Ishikawa J, et al. Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem Biophys Res Commun. (1994) 203:1309–17. doi: 10.1006/bbrc.1994.2325
17. Ferrero E, Lo Buono N, Horenstein AL, Funaro A, Malavasi F. The ADP-ribosyl cyclases–the current evolutionary state of the ARCs. Front Biosci. (2014) 19:986–1002. doi: 10.2741/4262
18. Malavasi F, Deaglio S, Ferrero E, Funaro A, Sancho J, Ausiello CM, et al. CD38 and CD157 as receptors of the immune system: a bridge between innate and adaptive immunity. Mol Med. (2006) 12:334–41. doi: 10.2119/2006-00094.Malavasi
19. Alessio M, Roggero S, Funaro A, De Monte LB, Peruzzi L, Geuna M, et al. CD38 molecule: structural and biochemical analysis on human T lymphocytes, thymocytes, and plasma cells. J Immunol. (1990) 145:878–84.
20. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
21. Prasad GS, McRee DE, Stura EA, Levitt DG, Lee HC, Stout CD. Crystal structure of Aplysia ADP ribosyl cyclase, a homologue of the bifunctional ectozyme CD38. Nat Struct Biol. (1996) 3:957–64. doi: 10.1038/nsb1196-957
22. Liu Q, Kriksunov IA, Graeff R, Munshi C, Lee HC, Hao Q. Crystal structure of human CD38 extracellular domain. Structure. (2005) 13:1331–9. doi: 10.1016/j.str.2005.05.012
23. Liu Q, Kriksunov IA, Graeff R, Lee HC, Hao Q. Structural basis for formation and hydrolysis of the calcium messenger cyclic ADP-ribose by human CD38. J Biol Chem. (2007) 282:5853–61. doi: 10.1074/jbc.M609093200
24. Liu Q, Kriksunov IA, Moreau C, Graeff R, Potter BV, Lee HC, et al. Catalysis-associated conformational changes revealed by human CD38 complexed with a non-hydrolyzable substrate analog. J Biol Chem. (2007) 282:24825–32. doi: 10.1074/jbc.M701653200
25. Ortolan E, Augeri S, Fissolo G, Musso I, Funaro A. CD157: from immunoregulatory protein to potential therapeutic target. Immunol Lett. (2019) 205:59–64. doi: 10.1016/j.imlet.2018.06.007
26. Horenstein AL, Bracci C, Morandi F, Malavasi F. CD38 in adenosinergic pathways and metabolic re-programming in human multiple myeloma cells: in-tandem insights from basic science to therapy. Front Immunol. (2019) 10:760. doi: 10.3389/fimmu.2019.00760
27. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
28. Lee HC. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J Biol Chem. (2012) 287:31633–40. doi: 10.1074/jbc.R112.349464
29. Moreschi I, Bruzzone S, Nicholas RA, Fruscione F, Sturla L, Benvenuto F, et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J Biol Chem. (2006) 281:31419–29. doi: 10.1074/jbc.M606625200
30. Klein C, Grahnert A, Abdelrahman A, Muller CE, Hauschildt S. Extracellular NAD(+) induces a rise in [Ca(2+)](i) in activated human monocytes via engagement of P2Y(1) and P2Y(11) receptors. Cell Calcium. (2009) 46:263–72. doi: 10.1016/j.ceca.2009.08.004
31. Adebanjo OA, Anandatheerthavarada HK, Koval AP, Moonga BS, Biswas G, Sun L, et al. A new function for CD38/ADP-ribosyl cyclase in nuclear Ca2+ homeostasis. Nat Cell Biol. (1999) 1:409–14. doi: 10.1038/15640
32. Lee HC. Structure and enzymatic functions of human CD38. Mol Med. (2006) 12:317–23. doi: 10.2119/2006-00086.Lee
33. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. (2001) 411:590–5. doi: 10.1038/35079092
34. Guse AH. Cyclic ADP-ribose: a novel Ca2+-mobilising second messenger. Cell Signal. (1999) 11:309–16. doi: 10.1016/S0898-6568(99)00004-2
35. Hirata Y, Kimura N, Sato K, Ohsugi Y, Takasawa S, Okamoto H, et al. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. (1994) 356:244–8. doi: 10.1016/0014-5793(94)01279-2
36. Kajimoto Y, Miyagawa J, Ishihara K, Okuyama Y, Fujitani Y, Itoh M, et al. Pancreatic islet cells express BST-1, a CD38-like surface molecule having ADP-ribosyl cyclase activity. Biochem Biophys Res Commun. (1996) 219:941–6. doi: 10.1006/bbrc.1996.0327
37. Hussain AM, Lee HC, Chang CF. Functional expression of secreted mouse BST-1 in yeast. Protein Expr Purif. (1998) 12:133–7. doi: 10.1006/prep.1997.0811
38. Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, et al. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol. (1998) 160:395–402.
39. Zubiaur M, Fernandez O, Ferrero E, Salmeron J, Malissen B, Malavasi F, et al. CD38 is associated with lipid rafts and upon receptor stimulation leads to Akt/protein kinase B and Erk activation in the absence of the CD3-zeta immune receptor tyrosine-based activation motifs. J Biol Chem. (2002) 277:13–22. doi: 10.1074/jbc.M107474200
40. Deaglio S, Zubiaur M, Gregorini A, Bottarel F, Ausiello CM, Dianzani U, et al. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood. (2002) 99:2490–8. doi: 10.1182/blood.V99.7.2490
41. Frasca L, Fedele G, Deaglio S, Capuano C, Palazzo R, Vaisitti T, et al. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood. (2006) 107:2392–9. doi: 10.1182/blood-2005-07-2913
42. Deaglio S, Vaisitti T, Billington R, Bergui L, Omede P, Genazzani AA, et al. CD38/CD19: a lipid raft-dependent signaling complex in human B cells. Blood. (2007) 109:5390–8. doi: 10.1182/blood-2006-12-061812
43. Romero-Ramirez H, Morales-Guadarrama MT, Pelayo R, Lopez-Santiago R, Santos-Argumedo L. CD38 expression in early B-cell precursors contributes to extracellular signal-regulated kinase-mediated apoptosis. Immunology. (2015) 144:271–81. doi: 10.1111/imm.12370
44. Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med. (2001) 7:1209–16. doi: 10.1038/nm1101-1209
45. Partida-Sanchez S, Goodrich S, Kusser K, Oppenheimer N, Randall TD, Lund FE. Regulation of dendritic cell trafficking by the ADP-ribosyl cyclase CD38: impact on the development of humoral immunity. Immunity. (2004) 20:279–91. doi: 10.1016/S1074-7613(04)00048-2
46. Lo Buono N, Parrotta R, Morone S, Bovino P, Nacci G, Ortolan E, et al. The CD157-integrin partnership controls transendothelial migration and adhesion of human monocytes. J Biol Chem. (2011) 286:18681–91. doi: 10.1074/jbc.M111.227876
47. Ferrero E, Faini AC, Malavasi F. A phylogenetic view of the leukocyte ectonucleotidases. Immunol Lett. (2019) 205:51–8. doi: 10.1016/j.imlet.2018.06.008
48. Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci. (2005) 30:542–50. doi: 10.1016/j.tibs.2005.08.005
49. Heine P, Braun N, Sevigny J, Robson SC, Servos J, Zimmermann H. The C-terminal cysteine-rich region dictates specific catalytic properties in chimeras of the ectonucleotidases NTPDase1 and NTPDase2. Eur J Biochem. (2001) 268:364–73. doi: 10.1046/j.1432-1033.2001.01896.x
50. Grinthal A, Guidotti G. Transmembrane domains confer different substrate specificities and adenosine diphosphate hydrolysis mechanisms on CD39, CD39L1, and chimeras. Biochemistry. (2002) 41:1947–56. doi: 10.1021/bi015563h
51. Koziak K, Kaczmarek E, Kittel A, Sevigny J, Blusztajn JK, Schulte Am Esch J II, et al. Palmitoylation targets CD39/endothelial ATP diphosphohydrolase to caveolae. J Biol Chem. (2000) 275:2057–62. doi: 10.1074/jbc.275.3.2057
52. Zhong X, Malhotra R, Woodruff R, Guidotti G. Mammalian plasma membrane ecto-nucleoside triphosphate diphosphohydrolase 1, CD39, is not active intracellularly. The N-glycosylation state of CD39 correlates with surface activity and localization. J Biol Chem. (2001) 276:41518–25. doi: 10.1074/jbc.M104415200
53. Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Physiological roles for ecto-5'-nucleotidase (CD73). Purinergic Signal. (2006) 2:351–60. doi: 10.1007/s11302-005-5302-5
54. Robson SC, Sevigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: structure function relationships and pathophysiological significance. Purinergic Signal. (2006) 2:409–30. doi: 10.1007/s11302-006-9003-5
55. Yegutkin G, Bodin P, Burnstock G. Effect of shear stress on the release of soluble ecto-enzymes ATPase and 5′-nucleotidase along with endogenous ATP from vascular endothelial cells. Br J Pharmacol. (2000) 129:921–6. doi: 10.1038/sj.bjp.0703136
56. Fini C, Talamo F, Cherri S, Coli M, Floridi A, Ferrara L, et al. Biochemical and mass spectrometric characterization of soluble ecto-5'-nucleotidase from bull seminal plasma. Biochem J. (2003) 372(Pt 2):443–51. doi: 10.1042/bj20021687
57. Knapp K, Zebisch M, Pippel J, El-Tayeb A, Muller CE, Strater N. Crystal structure of the human ecto-5′-nucleotidase (CD73): insights into the regulation of purinergic signaling. Structure. (2012) 20:2161–73. doi: 10.1016/j.str.2012.10.001
58. Kirley TL, Crawford PA, Smith TM. The structure of the nucleoside triphosphate diphosphohydrolases (NTPDases) as revealed by mutagenic and computational modeling analyses. Purinergic Signal. (2006) 2:379–89. doi: 10.1007/s11302-005-5301-6
59. Wu Y, Sun X, Kaczmarek E, Dwyer KM, Bianchi E, Usheva A, et al. RanBPM associates with CD39 and modulates ecto-nucleotidase activity. Biochem J. (2006) 396:23–30. doi: 10.1042/BJ20051568
60. Resta R, Yamashita Y, Thompson LF. Ecto-enzyme and signaling functions of lymphocyte CD73. Immunol Rev. (1998) 161:95–109. doi: 10.1111/j.1600-065X.1998.tb01574.x
61. Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J. Anti-CD73 therapy impairs tumor angiogenesis. Int J Cancer. (2014) 134:1466–73. doi: 10.1002/ijc.28456
62. Allard D, Allard B, Gaudreau PO, Chrobak P, Stagg J. CD73-adenosine: a next-generation target in immuno-oncology. Immunotherapy. (2016) 8:145–63. doi: 10.2217/imt.15.106
63. Chatterjee D, Tufa DM, Baehre H, Hass R, Schmidt RE, Jacobs R. Natural killer cells acquire CD73 expression upon exposure to mesenchymal stem cells. Blood. (2014) 123:594–5. doi: 10.1182/blood-2013-09-524827
64. Maliszewski CR, Delespesse GJ, Schoenborn MA, Armitage RJ, Fanslow WC, Nakajima T, et al. The CD39 lymphoid cell activation antigen. Molecular cloning and structural characterization. J Immunol. (1994) 153:3574–83.
65. Dwyer KM, Deaglio S, Gao W, Friedman D, Strom TB, Robson SC. CD39 and control of cellular immune responses. Purinergic Signal. (2007) 3:171–80. doi: 10.1007/s11302-006-9050-y
66. Saze Z, Schuler PJ, Hong CS, Cheng D, Jackson EK, Whiteside TL. Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood. (2013) 122:9–18. doi: 10.1182/blood-2013-02-482406
67. Figueiro F, Muller L, Funk S, Jackson EK, Battastini AM, Whiteside TL. Phenotypic and functional characteristics of CD39(high) human regulatory B cells (Breg). Oncoimmunology. (2016) 5:e1082703. doi: 10.1080/2162402X.2015.1082703
68. Schena F, Volpi S, Faliti CE, Penco F, Santi S, Proietti M, et al. Dependence of immunoglobulin class switch recombination in B cells on vesicular release of ATP and CD73 ectonucleotidase activity. Cell Rep. (2013) 3:1824–31. doi: 10.1016/j.celrep.2013.05.022
69. Bai A, Moss A, Rothweiler S, Longhi MS, Wu Y, Junger WG, et al. NADH oxidase-dependent CD39 expression by CD8(+) T cells modulates interferon gamma responses via generation of adenosine. Nat Commun. (2015) 6:8819. doi: 10.1038/ncomms9819
70. Fang F, Yu M, Cavanagh MM, Hutter Saunders J, Qi Q, Ye Z, et al. Expression of CD39 on activated T cells impairs their survival in older individuals. Cell Rep. (2016) 14:1218–31. doi: 10.1016/j.celrep.2016.01.002
71. Noble A, Mehta H, Lovell A, Papaioannou E, Fairbanks L. IL-12 and IL-4 activate a CD39-dependent intrinsic peripheral tolerance mechanism in CD8(+) T cells. Eur J Immunol. (2016) 46:1438–48. doi: 10.1002/eji.201545939
72. Doherty GA, Bai A, Hanidziar D, Longhi MS, Lawlor GO, Putheti P, et al. CD73 is a phenotypic marker of effector memory Th17 cells in inflammatory bowel disease. Eur J Immunol. (2012) 42:3062–72. doi: 10.1002/eji.201242623
73. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
74. Fletcher JM, Lonergan R, Costelloe L, Kinsella K, Moran B, O'Farrelly C, et al. CD39+Foxp3+ regulatory T Cells suppress pathogenic Th17 cells and are impaired in multiple sclerosis. J Immunol. (2009) 183:7602–10. doi: 10.4049/jimmunol.0901881
75. Longhi MS, Moss A, Bai A, Wu Y, Huang H, Cheifetz A, et al. Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS ONE. (2014) 9:e87956. doi: 10.1371/journal.pone.0087956
76. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. (2011) 71:2892–900. doi: 10.1158/0008-5472.CAN-10-4246
77. Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol. (2014) 177:531–43. doi: 10.1111/cei.12354
78. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol. (2012) 3:190. doi: 10.3389/fimmu.2012.00190
79. Boyiadzis M, Whiteside TL. The emerging roles of tumor-derived exosomes in hematological malignancies. Leukemia. (2017) 31:1259–68. doi: 10.1038/leu.2017.91
80. Whiteside TL. Exosomes carrying immunoinhibitory proteins and their role in cancer. Clin Exp Immunol. (2017) 189:259–67. doi: 10.1111/cei.12974
81. Wilkin F, Duhant X, Bruyns C, Suarez-Huerta N, Boeynaems JM, Robaye B. The P2Y11 receptor mediates the ATP-induced maturation of human monocyte-derived dendritic cells. J Immunol. (2001) 166:7172–7. doi: 10.4049/jimmunol.166.12.7172
82. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. (2008) 112:1822–31. doi: 10.1182/blood-2008-02-136325
83. Zanin RF, Braganhol E, Bergamin LS, Campesato LF, Filho AZ, Moreira JC, et al. Differential macrophage activation alters the expression profile of NTPDase and ecto-5′-nucleotidase. PLoS ONE. (2012) 7:e31205. doi: 10.1371/journal.pone.0031205
84. Zhu YP, Brown JR, Sag D, Zhang L, Suttles J. Adenosine 5′-monophosphate-activated protein kinase regulates IL-10-mediated anti-inflammatory signaling pathways in macrophages. J Immunol. (2015) 194:584–94. doi: 10.4049/jimmunol.1401024
85. Keyhani A, Huh YO, Jendiroba D, Pagliaro L, Cortez J, Pierce S, et al. Increased CD38 expression is associated with favorable prognosis in adult acute leukemia. Leuk Res. (2000) 24:153–9. doi: 10.1016/S0145-2126(99)00147-2
86. Marinov J, Koubek K, Stary J. Immunophenotypic significance of the “lymphoid” CD38 antigen in myeloid blood malignancies. Neoplasma. (1993) 40:355–8.
87. Naik J, Themeli M, de Jong-Korlaar R, Ruiter RWJ, Poddighe PJ, Yuan H, et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica. (2018) 104:e100–3. doi: 10.3324/haematol.2018.192757
88. Parry-Jones N, Matutes E, Morilla R, Brito-Babapulle V, Wotherspoon A, Swansbury GJ, et al. Cytogenetic abnormalities additional to t(11;14) correlate with clinical features in leukaemic presentation of mantle cell lymphoma, and may influence prognosis: a study of 60 cases by FISH. Br J Haematol. (2007) 137:117–24. doi: 10.1111/j.1365-2141.2007.06526.x
89. Damle RN, Temburni S, Calissano C, Yancopoulos S, Banapour T, Sison C, et al. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood. (2007) 110:3352–9. doi: 10.1182/blood-2007-04-083832
90. Malavasi F, Deaglio S, Damle R, Cutrona G, Ferrarini M, Chiorazzi N. CD38 and chronic lymphocytic leukemia: a decade later. Blood. (2011) 118:3470–8. doi: 10.1182/blood-2011-06-275610
91. Ocio EM, Richardson PG, Rajkumar SV, Palumbo A, Mateos MV, Orlowski R, et al. New drugs and novel mechanisms of action in multiple myeloma in 2013: a report from the International Myeloma Working Group (IMWG). Leukemia. (2014) 28:525–42. doi: 10.1038/leu.2013.350
92. Suzuki R, Suzumiya J, Nakamura S, Aoki S, Notoya A, Ozaki S, et al. Aggressive natural killer-cell leukemia revisited: large granular lymphocyte leukemia of cytotoxic NK cells. Leukemia. (2004) 18:763–70. doi: 10.1038/sj.leu.2403262
93. Wang L, Wang H, Li PF, Lu Y, Xia ZJ, Huang HQ, et al. CD38 expression predicts poor prognosis and might be a potential therapy target in extranodal NK/T cell lymphoma, nasal type. Ann Hematol. (2015) 94:1381–8. doi: 10.1007/s00277-015-2359-2
94. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. (1994) 367:645–8. doi: 10.1038/367645a0
95. Kong Y, Yoshida S, Saito Y, Doi T, Nagatoshi Y, Fukata M, et al. CD34+CD38+CD19+ as well as CD34+CD38-CD19+ cells are leukemia-initiating cells with self-renewal capacity in human B-precursor ALL. Leukemia. (2008) 22:1207–13. doi: 10.1038/leu.2008.83
96. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. (2012) 12:767–75. doi: 10.1038/nrc3368
97. Kong Y, Chang YJ, Liu YR, Wang YZ, Jiang Q, Jiang H, et al. CD34(+)CD38(-)CD58(-) cells are leukemia-propagating cells in Philadelphia chromosome-positive acute lymphoblastic leukemia. Leukemia. (2014) 28:2398–401. doi: 10.1038/leu.2014.228
98. van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, et al. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clin Cancer Res. (2005) 11:6520–7. doi: 10.1158/1078-0432.CCR-05-0468
99. Gerber JM, Smith BD, Ngwang B, Zhang H, Vala MS, Morsberger L, et al. A clinically relevant population of leukemic CD34(+)CD38(-) cells in acute myeloid leukemia. Blood. (2012) 119:3571–7. doi: 10.1182/blood-2011-06-364182
100. Zeijlemaker W, Grob T, Meijer R, Hanekamp D, Kelder A, Carbaat-Ham JC, et al. CD34(+)CD38(-) leukemic stem cell frequency to predict outcome in acute myeloid leukemia. Leukemia. (2018) 33:1102–12. doi: 10.1038/s41375-018-0326-3
101. Kong Y, Xu LP, Liu YR, Qin YZ, Sun YQ, Wang Y, et al. Presence of CD34(+)CD38(-)CD58(-) leukemia-propagating cells at diagnosis identifies patients at high risk of relapse with Ph chromosome-positive ALL after allo-hematopoietic SCT. Bone Marrow Transplant. (2015) 50:348–53. doi: 10.1038/bmt.2014.274
102. Long J, Liu S, Li K, Zhou X, Zhang P, Zou L. High proportion of CD34+/CD38-cells is positively correlated with poor prognosis in newly diagnosed childhood acute lymphoblastic leukemia. Leuk Lymphoma. (2014) 55:611–7. doi: 10.3109/10428194.2013.807924
103. Krupka C, Lichtenegger FS, Kohnke T, Bogeholz J, Bucklein V, Roiss M, et al. Targeting CD157 in AML using a novel, Fc-engineered antibody construct. Oncotarget. (2017) 8:35707–17. doi: 10.18632/oncotarget.16060
104. Mirkowska P, Hofmann A, Sedek L, Slamova L, Mejstrikova E, Szczepanski T, et al. Leukemia surfaceome analysis reveals new disease-associated features. Blood. (2013) 121:e149–159. doi: 10.1182/blood-2012-11-468702
105. Morandi F, Morandi B, Horenstein AL, Chillemi A, Quarona V, Zaccarello G, et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget. (2015) 6:25602–18. doi: 10.18632/oncotarget.4693
106. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. (1999) 94:1840–7.
107. Del Poeta G, Maurillo L, Venditti A, Buccisano F, Epiceno AM, Capelli G, et al. Clinical significance of CD38 expression in chronic lymphocytic leukemia. Blood. (2001) 98:2633–9. doi: 10.1182/blood.V98.9.2633
108. Ibrahim S, Keating M, Do KA, O'Brien S, Huh YO, Jilani I, et al. CD38 expression as an important prognostic factor in B-cell chronic lymphocytic leukemia. Blood. (2001) 98:181–6. doi: 10.1182/blood.V98.1.181
109. Jelinek DF, Tschumper RC, Geyer SM, Bone ND, Dewald GW, Hanson CA, et al. Analysis of clonal B-cell CD38 and immunoglobulin variable region sequence status in relation to clinical outcome for B-chronic lymphocytic leukaemia. Br J Haematol. (2001) 115:854–61. doi: 10.1046/j.1365-2141.2001.03149.x
110. Morabito F, Mangiola M, Oliva B, Stelitano C, Callea V, Deaglio S, et al. Peripheral blood CD38 expression predicts survival in B-cell chronic lymphocytic leukemia. Leuk Res. (2001) 25:927–32. doi: 10.1016/S0145-2126(01)00049-2
111. Durig J, Naschar M, Schmucker U, Renzing-Kohler K, Holter T, Huttmann A, et al. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia. (2002) 16:30–5. doi: 10.1038/sj.leu.2402339
112. Schroers R, Griesinger F, Trumper L, Haase D, Kulle B, Klein-Hitpass L, et al. Combined analysis of ZAP-70 and CD38 expression as a predictor of disease progression in B-cell chronic lymphocytic leukemia. Leukemia. (2005) 19:750–8. doi: 10.1038/sj.leu.2403707
113. Zucchetto A, Bomben R, Dal Bo M, Bulian P, Benedetti D, Nanni P, et al. CD49d in B-cell chronic lymphocytic leukemia: correlated expression with CD38 and prognostic relevance. Leukemia. (2006) 20:523–5; author reply: 528–9. doi: 10.1038/sj.leu.2404087
114. Krober A, Seiler T, Benner A, Bullinger L, Bruckle E, Lichter P, et al. V(H) mutation status, CD38 expression level, genomic aberrations, and survival in chronic lymphocytic leukemia. Blood. (2002) 100:1410–6.
115. Cramer P, Hallek M. Prognostic factors in chronic lymphocytic leukemia-what do we need to know? Nat Rev Clin Oncol. (2011) 8:38–47. doi: 10.1038/nrclinonc.2010.167
116. Martinez-Trillos A, Pinyol M, Delgado J, Aymerich M, Rozman M, Baumann T, et al. The mutational landscape of small lymphocytic lymphoma compared to non-early stage chronic lymphocytic leukemia. Leuk Lymphoma. (2018) 59:2318–26. doi: 10.1080/10428194.2017.1397660
117. Ouillette P, Collins R, Shakhan S, Li J, Peres E, Kujawski L, et al. Acquired genomic copy number aberrations and survival in chronic lymphocytic leukemia. Blood. (2011) 118:3051–61. doi: 10.1182/blood-2010-12-327858
118. Braggio E, Kay NE, VanWier S, Tschumper RC, Smoley S, Eckel-Passow JE, et al. Longitudinal genome-wide analysis of patients with chronic lymphocytic leukemia reveals complex evolution of clonal architecture at disease progression and at the time of relapse. Leukemia. (2012) 26:1698–701. doi: 10.1038/leu.2012.14
119. Medd PG, Clark N, Leyden K, Turner S, Strefford JA, Butler C, et al. A novel scoring system combining expression of CD23, CD20, and CD38 with platelet count predicts for the presence of the t(11;14) translocation of mantle cell lymphoma. Cytometry B Clin Cytom. (2011) 80:230–7. doi: 10.1002/cyto.b.20590
120. Camacho FI, Algara P, Rodriguez A, Ruiz-Ballesteros E, Mollejo M, Martinez N, et al. Molecular heterogeneity in MCL defined by the use of specific VH genes and the frequency of somatic mutations. Blood. (2003) 101:4042–6. doi: 10.1182/blood-2002-11-3456
121. Orchard J, Garand R, Davis Z, Babbage G, Sahota S, Matutes E, et al. A subset of t(11;14) lymphoma with mantle cell features displays mutated IgVH genes and includes patients with good prognosis, nonnodal disease. Blood. (2003) 101:4975–81. doi: 10.1182/blood-2002-06-1864
122. Hari P, Raj RV, Olteanu H. Targeting CD38 in refractory extranodal natural killer cell-T-cell lymphoma. N Engl J Med. (2016) 375:1501–2. doi: 10.1056/NEJMc1605684
123. de The H, Chen Z. Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat Rev Cancer. (2010) 10:775–83. doi: 10.1038/nrc2943
124. Kamimura T, Miyamoto T, Harada M, Akashi K. Advances in therapies for acute promyelocytic leukemia. Cancer Sci. (2011) 102:1929–37. doi: 10.1111/j.1349-7006.2011.02045.x
125. Drach J, McQueen T, Engel H, Andreeff M, Robertson KA, Collins SJ, et al. Retinoic acid-induced expression of CD38 antigen in myeloid cells is mediated through retinoic acid receptor-alpha. Cancer Res. (1994) 54:1746–52.
126. Mehta K, Cheema S. Retinoid-mediated signaling pathways in CD38 antigen expression in myeloid leukemia cells. Leuk Lymphoma. (1999) 32:441–9. doi: 10.3109/10428199909058401
127. MacDonald RJ, Shrimp JH, Jiang H, Zhang L, Lin H, Yen A. Probing the requirement for CD38 in retinoic acid-induced HL-60 cell differentiation with a small molecule dimerizer and genetic knockout. Sci Rep. (2017) 7:17406. doi: 10.1038/s41598-017-17720-4
128. Gao Y, Camacho LH, Mehta K. Retinoic acid-induced CD38 antigen promotes leukemia cells attachment and interferon-gamma/interleukin-1beta-dependent apoptosis of endothelial cells: implications in the etiology of retinoic acid syndrome. Leuk Res. (2007) 31:455–63. doi: 10.1016/j.leukres.2006.07.004
129. Zupo S, Isnardi L, Megna M, Massara R, Malavasi F, Dono M, et al. CD38 expression distinguishes two groups of B-cell chronic lymphocytic leukemias with different responses to anti-IgM antibodies and propensity to apoptosis. Blood. (1996) 88:1365–74.
130. Lanham S, Hamblin T, Oscier D, Ibbotson R, Stevenson F, Packham G. Differential signaling via surface IgM is associated with VH gene mutational status and CD38 expression in chronic lymphocytic leukemia. Blood. (2003) 101:1087–93. doi: 10.1182/blood-2002-06-1822
131. Morabito F, Cutrona G, Gentile M, Fabbi M, Matis S, Colombo M, et al. Prognostic relevance of in vitro response to cell stimulation via surface IgD in binet stage a CLL. Br J Haematol. (2010) 149:160–3. doi: 10.1111/j.1365-2141.2009.08032.x
132. Deaglio S, Capobianco A, Bergui L, Durig J, Morabito F, Duhrsen U, et al. CD38 is a signaling molecule in B-cell chronic lymphocytic leukemia cells. Blood. (2003) 102:2146–55. doi: 10.1182/blood-2003-03-0989
133. Deaglio S, Vaisitti T, Bergui L, Bonello L, Horenstein AL, Tamagnone L, et al. CD38 and CD100 lead a network of surface receptors relaying positive signals for B-CLL growth and survival. Blood. (2005) 105:3042–50. doi: 10.1182/blood-2004-10-3873
134. Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D'Arena G, et al. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol Med. (2010) 16:87–91. doi: 10.2119/molmed.2009.00146
135. Burger JA, Tsukada N, Burger M, Zvaifler NJ, Dell'Aquila M, Kipps TJ. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood. (2000) 96:2655–63.
136. Vaisitti T, Aydin S, Rossi D, Cottino F, Bergui L, D'Arena G, et al. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia. (2010) 24:958–69. doi: 10.1038/leu.2010.36
137. Pearce L, Morgan L, Lin TT, Hewamana S, Matthews RJ, Deaglio S, et al. Genetic modification of primary chronic lymphocytic leukemia cells with a lentivirus expressing CD38. Haematologica. (2010) 95:514–7. doi: 10.3324/haematol.2009.014381
138. Zucchetto A, Vaisitti T, Benedetti D, Tissino E, Bertagnolo V, Rossi D, et al. The CD49d/CD29 complex is physically and functionally associated with CD38 in B-cell chronic lymphocytic leukemia cells. Leukemia. (2012) 26:1301–12. doi: 10.1038/leu.2011.369
139. Vaisitti T, Serra S, Pepper C, Rossi D, Laurenti L, Gaidano G, et al. CD38 signals upregulate expression and functions of matrix metalloproteinase-9 in chronic lymphocytic leukemia cells. Leukemia. (2013) 27:1177–81. doi: 10.1038/leu.2012.260
140. Vaisitti T, Audrito V, Serra S, Buonincontri R, Sociali G, Mannino E, et al. The enzymatic activities of CD38 enhance CLL growth and trafficking: implications for therapeutic targeting. Leukemia. (2015) 29:356–68. doi: 10.1038/leu.2014.207
141. Kawano Y, Moschetta M, Manier S, Glavey S, Gorgun GT, Roccaro AM, et al. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. (2015) 263:160–72. doi: 10.1111/imr.12233
142. Morandi F, Marimpietri D, Horenstein AL, Bolzoni M, Toscani D, Costa F, et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD(). Oncoimmunology. (2018) 7:e1458809. doi: 10.1080/2162402X.2018.1458809
143. Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, et al. CD38-Driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. (2019) 79:2285–97. doi: 10.1158/0008-5472.CAN-18-0773
144. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. (2017) 23:4290–300. doi: 10.1158/1078-0432.CCR-16-3192
145. Coustan-Smith E, Song G, Clark C, Key L, Liu P, Mehrpooya M, et al. New markers for minimal residual disease detection in acute lymphoblastic leukemia. Blood. (2011) 117:6267–76. doi: 10.1182/blood-2010-12-324004
146. Wang W, Gao L, Li Y, Li ZL, Gong M, Huang FZ, et al. The application of CD73 in minimal residual disease monitoring using flow cytometry in B-cell acute lymphoblastic leukemia. Leuk Lymphoma. (2016) 57:1174–81. doi: 10.3109/10428194.2015.1070153
147. Sedek L, Theunissen P, Sobral da Costa E, van der Sluijs-Gelling A, Mejstrikova E, Gaipa G, et al. Differential expression of CD73, CD86 and CD304 in normal vs. leukemic B-cell precursors and their utility as stable minimal residual disease markers in childhood B-cell precursor acute lymphoblastic leukemia. J Immunol Methods. (2018). doi: 10.1016/j.jim.2018.03.005. [Epub ahead of print].
148. Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, et al. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol. (2001) 167:6021–30. doi: 10.4049/jimmunol.167.10.6021
149. Dulphy N, Henry G, Hemon P, Khaznadar Z, Dombret H, Boissel N, et al. Contribution of CD39 to the immunosuppressive microenvironment of acute myeloid leukaemia at diagnosis. Br J Haematol. (2014) 165:722–5. doi: 10.1111/bjh.12774
150. Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. ATP release from chemotherapy-treated dying leukemia cells elicits an immune suppressive effect by increasing regulatory T cells and tolerogenic dendritic cells. Front Immunol. (2017) 8:1918. doi: 10.3389/fimmu.2017.01918
151. Kong Y, Jia B, Zhao C, Claxton DF, Sharma A, Annageldiyev C, et al. Downregulation of CD73 associates with T cell exhaustion in AML patients. J Hematol Oncol. (2019) 12:40. doi: 10.1186/s13045-019-0728-3
152. Jakobsen JS, Laursen LG, Schuster MB, Pundhir S, Schoof E, Ge Y, et al. Mutant CEBPA directly drives the expression of the targetable tumor-promoting factor CD73 in AML. Sci Adv. (2019). 5:eaaw4304. doi: 10.1126/sciadv.aaw4304
153. Damle RN, Ghiotto F, Valetto A, Albesiano E, Fais F, Yan XJ, et al. B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood. (2002) 99:4087–93. doi: 10.1182/blood.V99.11.4087
154. Bennett F, Rawstron A, Plummer M, de Tute R, Moreton P, Jack A, et al. B-cell chronic lymphocytic leukaemia cells show specific changes in membrane protein expression during different stages of cell cycle. Br J Haematol. (2007) 139:600–4. doi: 10.1111/j.1365-2141.2007.06790.x
155. Serra S, Horenstein AL, Vaisitti T, Brusa D, Rossi D, Laurenti L, et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood. (2011) 118:6141–52. doi: 10.1182/blood-2011-08-374728
156. Jak M, Mous R, Remmerswaal EB, Spijker R, Jaspers A, Yague A, et al. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk Lymphoma. (2009) 50:788–801. doi: 10.1080/10428190902803677
157. Serra S, Vaisitti T, Audrito V, Bologna C, Buonincontri R, Chen SS, et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. (2016) 1:47–61. doi: 10.1182/bloodadvances.2016000984
158. Abousamra NK, Salah El-Din M, Hamza Elzahaf E, Esmael ME. Ectonucleoside triphosphate diphosphohydrolase-1 (E-NTPDase1/CD39) as a new prognostic marker in chronic lymphocytic leukemia. Leuk Lymphoma. (2015) 56:113–9. doi: 10.3109/10428194.2014.907893
159. Mackey JR, Galmarini CM, Graham KA, Joy AA, Delmer A, Dabbagh L, et al. Quantitative analysis of nucleoside transporter and metabolism gene expression in chronic lymphocytic leukemia (CLL): identification of fludarabine-sensitive and -insensitive populations. Blood. (2005) 105:767–74. doi: 10.1182/blood-2004-03-1046
160. Di Virgilio F, Adinolfi E. Extracellular purines, purinergic receptors and tumor growth. Oncogene. (2017) 36:293–303. doi: 10.1038/onc.2016.206
161. Wang X, Zhang T, Song Z, Li L, Zhang X, Liu J, et al. Tumor CD73/A2aR adenosine immunosuppressive axis and tumor-infiltrating lymphocytes in diffuse large B-cell lymphoma: correlations with clinicopathological characteristics and clinical outcome. Int J Cancer. (2019) 145:1414–22. doi: 10.1002/ijc.32144
162. Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, et al. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol. (2009) 183:6157–66. doi: 10.4049/jimmunol.0900475
163. Puente XS, Jares P, Campo E. Chronic lymphocytic leukemia and mantle cell lymphoma: crossroads of genetic and microenvironment interactions. Blood. (2018) 131:2283–96. doi: 10.1182/blood-2017-10-764373
164. Arruga F, Deaglio S. Mechanisms of resistance to targeted therapies in chronic lymphocytic leukemia. Handb Exp Pharmacol. (2018) 249:203–29. doi: 10.1007/164_2017_12
165. Shachar I, Cohen S, Marom A, Becker-Herman S. Regulation of CLL survival by hypoxia-inducible factor and its target genes. FEBS Lett. (2012) 586:2906–10. doi: 10.1016/j.febslet.2012.07.016
166. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5'-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. (2002) 110:993–1002. doi: 10.1172/JCI0215337
167. Vaupel P, Multhoff G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front Immunol. (2017) 8:1887. doi: 10.3389/fimmu.2017.01887
168. Bowser JL, Phan LH, Eltzschig HK. The hypoxia-adenosine link during intestinal inflammation. J Immunol. (2018) 200:897–907. doi: 10.4049/jimmunol.1701414
169. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. (2012) 122:787–95. doi: 10.1172/JCI59643
170. Koczula KM, Ludwig C, Hayden R, Cronin L, Pratt G, Parry H, et al. Metabolic plasticity in CLL: adaptation to the hypoxic niche. Leukemia. (2016) 30:65–73. doi: 10.1038/leu.2015.187
171. DiLillo DJ, Weinberg JB, Yoshizaki A, Horikawa M, Bryant JM, Iwata Y, et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia. (2013) 27:170–82. doi: 10.1038/leu.2012.165
172. Khagi Y, Mark TM. Potential role of daratumumab in the treatment of multiple myeloma. Onco Targets Ther. (2014) 7:1095–100. doi: 10.2147/OTT.S49480
173. Buteyn NJ, Fatehchand K, Santhanam R, Fang H, Dettorre GM, Gautam S, et al. Anti-leukemic effects of all-trans retinoic acid in combination with Daratumumab in acute myeloid leukemia. Int Immunol. (2018) 30:375–83. doi: 10.1093/intimm/dxy040
174. van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. (2018) 131:13–29. doi: 10.1182/blood-2017-06-740944
175. Varga C, Maglio M, Ghobrial IM, Richardson PG. Current use of monoclonal antibodies in the treatment of multiple myeloma. Br J Haematol. (2018) 181:447–59. doi: 10.1111/bjh.15121
176. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
177. Overdijk MB, Verploegen S, Bogels M, van Egmond M, Lammerts van Bueren JJ, Mutis T, et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. MAbs. (2015) 7:311–21. doi: 10.1080/19420862.2015.1007813
178. Overdijk MB, Jansen JH, Nederend M, Lammerts van Bueren JJ, Groen RW, Parren PW, et al. The therapeutic CD38 monoclonal antibody daratumumab induces programmed cell death via fcgamma receptor-mediated cross-linking. J Immunol. (2016) 197:807–13. doi: 10.4049/jimmunol.1501351
179. Moreno L, Perez C, Zabaleta A, Manrique I, Alignani D, Ajona D, et al. The mechanism of action of the anti-CD38 monoclonal antibody isatuximab in multiple myelmoa. Clin Cancer Res. (2019) 25:3176–87. doi: 10.1158/1078-0432.CCR-18-1597
180. van de Don kN, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. (2018) 9:2134. doi: 10.3389/fimmu.2018.02134
181. Kellenberger E, Kuhn I, Schuber F, Muller-Steffner H. Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett. (2011) 21:3939–42. doi: 10.1016/j.bmcl.2011.05.022
182. Cuesta-Mateos C, Alcaraz-Serna A, Somovilla-Crespo B, Munoz-Calleja C. Monoclonal antibody therapies for hematological malignancies: not just lineage-specific targets. Front Immunol. (2017) 8:1936. doi: 10.3389/fimmu.2017.01936
183. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. (2011) 334:1573–7. doi: 10.1126/science.1208347
184. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. (2010) 139:1030–40. doi: 10.1053/j.gastro.2010.05.007
185. Bastid J, Cottalorda-Regairaz A, Alberici G, Bonnefoy N, Eliaou JF, Bensussan A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene. (2013) 32:1743–51. doi: 10.1038/onc.2012.269
186. Hausler SF, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Honig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res. (2014) 6:129–39.
187. Hayes GM, Cairns B, Levashova Z, Chinn L, Perez M, Theunissen JW, et al. CD39 is a promising therapeutic antibody target for the treatment of soft tissue sarcoma. Am J Transl Res. (2015) 7:1181–8.
188. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
189. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
190. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. (2010) 70:2245–55. doi: 10.1158/0008-5472.CAN-09-3109
191. Bhattarai S, Freundlieb M, Pippel J, Meyer A, Abdelrahman A, Fiene A, et al. α,β-methylene-ADP (AOPCP) derivatives and analogues: development of potent and selective ecto-5'-nucleotidase (CD73) inhibitors. J Med Chem. (2015) 58:6248–63. doi: 10.1021/acs.jmedchem.5b00802
192. Menzel S, Schwarz N, Haag F, Koch-Nolte F. Nanobody-based biologics for modulating purinergic signaling in inflammation and immunity. Front Pharmacol. (2018) 9:266. doi: 10.3389/fphar.2018.00266
193. Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. (2012) 61:917–26. doi: 10.1007/s00262-011-1155-7
194. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA. (2013) 110:14711–6. doi: 10.1073/pnas.1308209110
Keywords: CD38, CD39, CD73, leukemias, myeloma, lymphoma, immunosuppression, tumor microenvironment
Citation: Vaisitti T, Arruga F, Guerra G and Deaglio S (2019) Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets. Front. Immunol. 10:2301. doi: 10.3389/fimmu.2019.02301
Received: 10 April 2019; Accepted: 11 September 2019;
Published: 04 October 2019.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceReviewed by:
Cemil Korcan Ayata, University Hospital of Basel, SwitzerlandCopyright © 2019 Vaisitti, Arruga, Guerra and Deaglio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Silvia Deaglio, c2lsdmlhLmRlYWdsaW9AdW5pdG8uaXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.