 Xin Li
Xin Li Yanwei Xiang
Yanwei Xiang Fulun Li1
Fulun Li1 Chengqian Yin
Chengqian Yin Bin Li
Bin Li Xisong Ke
Xisong Ke
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 September 2019
Sec. Cancer Immunity and Immunotherapy
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02293
This article is part of the Research TopicWnt Signaling in Immune Cell Regulation During Microbial Infection and CancerView all 10 articles
Immunotherapy with checkpoint inhibitors has greatly prolonged the overall survival of cancer patients in melanoma and many other cancer types. However, only a subset of patients shows clinical responses from these interventions, which was predicated by the T cell-inflamed tumor microenvironment. T cell-inflamed phenotype is characterized by the infiltration of CD8+ T cells, CD8α/CD103-lineage dendritic cells (DCs), as well as high density of forkhead box P3 (FoxP3)+ regulatory T cells (Tregs) that are associated with the efficacy of immune checkpoint blockade. A number of regulators has been associated with T cell-inflammation in the tumor microenvironment, and WNT/β-catenin signaling is one of the best characterized. The tumor-intrinsic WNT/β-catenin signaling activation is frequently associated with poor spontaneous T cell infiltration across most human cancers. In this article, we review the essential roles of WNT/β-catenin signaling in the T cell-inflamed and non-T cell-inflamed tumor microenvironment, including the development and function of immune cells, activation of immune exclusion of tumor cells, and cancer immunosurveillance. We also discuss the impact of this pathway in driving the non-T cell-inflamed tumor microenvironment in other tumor types. To improve immunotherapy efficacy, we argue that targeting Wnt/β-catenin signaling should be a high priority for combinational cancer therapy to restore T cell infiltration.
Immunotherapy with immune checkpoint inhibitors (ICIs) is now common for melanoma and many other cancer types (1). An important property of the immune system is to distinguish “foreign” cells from normal cells in the body, which allows the body to attack foreign cells while retaining normal cells. Therefore, it uses “checkpoints”; specifically, molecules on certain immune cells need to be activated (or inactivated) to initiate an immune response (2). As a checkpoint protein on T cells, programmed death-1 (PD-1) normally acts as a type of “off switch” to ensure that T cells do not target other cells in the body (3). Upon binding to PD-L1, which is a ligand protein expressed by some normal (and cancer) cells, it signals T cells to abstain from attacking. Antibodies that target either PD-1 or PD-L1 block this binding and boost the immune response against cancer cells (2). The therapeutic targeting of this axis has shown impressive activity in various tumors including melanoma of the skin, non-small cell lung cancer, head and neck squamous cell carcinoma, renal cell carcinoma, urothelial carcinoma, Hodgkin lymphoma, Merkel cell carcinoma, and microsatellite unstable or mismatch repair-deficient tumors (2). CTLA-4 is another type of checkpoint inhibitor, and monoclonal antibodies that target this marker can also boost the body's immune response against cancer cells (4). For ICIs, and especially CTLA-4 antibodies, one concern is their side effects in some patients as they permit the immune system to attack some normal organs in the body (4).
Despite improvements in clinical outcomes, only the minority of patients responds to ICIs (5). In patients susceptible to immunotherapy, an active immune response is usually observed prior to treatment, which is characterized by infiltrating antigen-specific T cells (6). This phenotype has been described as a T cell-inflamed tumor microenvironment (TME) and can be used to predict responding and non-responding tumors (7). The TME is the environment surrounding the tumor and consists of peripheral blood vessels, immune cells, fibroblasts, signaling molecules, and extracellular matrix (8). Tumors are closely related to the surrounding microenvironment and interact with it continually. Specifically, by releasing extracellular signals, promoting tumor angiogenesis, and inducing peripheral immune tolerance, tumors can affect the microenvironment; in turn, the growth and evolution of cancer cells are also affected by immune cells in the microenvironment (9). In the TME, chemokines support the influx of CD8+ effector T cells, which subsequently are functionally inhibited by PD-L1, IDO, Treg cells, and anergy. The development of TME is promoted in part by type I interferon signaling and the CD8a+ dendritic cell (DC) lineage (10). In normal tissue microenvironments, chemokine expression is poor and T cell infiltration is lacking, and this is also associated with the minimal presence of defined immune inhibitory pathways (11). In non-T cell inflammatory tumors, T cell inflammatory gene expression is significantly lower than that in matched normal tissues, which is associated with the loss of the native immune phenotype (6). Notably, a recent TCGA database study found that most tumor types are inversely associated with a T-cell-inflamed gene expression signature, and that WNT/β-catenin pathway activation is one potential causal pathway (6).
A correlation between WNT/β-catenin signaling and the T-cell-inflamed TME has been established (6). Moreover, identification of molecular factors associated with non-T cell-inflamed TME is important for the development of combinational immunotherapy regimens. Mechanistic studies based on genetically-engineered mice have also shown that activation of tumor cell-intrinsic β-catenin prevents spontaneous T-cell priming and infiltration into the TME, rendering cells resistant to combinational ICI therapy (12, 13). Thus, pharmacologically targeting the WNT/β-catenin pathway could potentially improve the efficacy of immunotherapy.
Here, we review the mechanisms wherein WNT signaling interrupts immune functions in the T cell-inflamed and non-T cell-inflamed TME, including the development and function of immune cells, activation of immune exclusion in tumor cells, and cancer immunosurveillance. We also discuss the therapeutic potential of harnessing currently available WNT modulators to augment cancer immunotherapy.
The T cell-inflamed subset of tumors is dominated by T cell markers and chemokines, which might mediate effector T cell recruitment (14–16). The expression of chemokines C-C motif chemokine ligand (CCL) 2, CCL3, CCL4, CCL5, C-X-C motif chemokine ligand (CXCL) 9, and CXCL10 is associated with the infiltration of T cells, and each of these chemokines is sufficient to recruit CD8+ effector T cells in vitro (16). Previous studies have confirmed that the T cell-inflamed subset contains variable numbers of CD8+ T cells and CD8α/CD103-lineage DCs, but also possesses the highest density of FoxP3+ regulatory T cells (Tregs) (16). Additionally, many conventional T cells have a dysfunctional anergic phenotype. It has been found that CXCR3-binding chemokines (such as CXCL9 and CXCL10) are critical and essential for the recruitment of activated CD8+ T cells to tumor sites (17). As a major driver of Treg recruitment, CCL22 is partially produced by activated CD8+ T cells (18). Despite the presence of specific adaptive immunity in this subset of patients, the cause of tumor progression is likely secondary to immunosuppressive mechanisms that act to some extent in the TME (19). Furthermore, T cell dysfunction in the TME is antigen-specific and restricted to tumor reactive T cells (19).
In contrast, T cell markers and chemokines that mediate T cell recruitment in the non-T cell-inflamed TME are lacking. Macrophages, vascular endothelial cells, fibroblasts, extracellular matrices, and immature DCs in some cases are still present in these tumors (20–24). Moreover, both the priming and effector phases of the anti-tumor immune response are deficient in non-T cell inflammatory tumors (19). Effector T cell trafficking into the TME is complex and dependent on adhesion molecules and homing receptors on vascular endothelial cells, consistent with the fact that chemokines are produced by tumor cells and stromal cells within the TME (19). In most cases, this process is necessary for the clinical response of immunotherapy.
The T cell-inflamed phenotype is associated with the efficacy of immune checkpoint blockade, whereas non-T cell-inflamed tumors rarely benefit. Recently, a series of studies has linked alterations in WNT signaling to oncogenesis, disease progression, and resistance to treatment in the TME (25, 26). Furthermore, dysregulated WNT signaling supports malignant transformation and disease progression through a variety of mechanisms in the TME (27). The high expression of specific immune cell genes in the TME, known as the T-cell-inflamed phenotype, has been associated with response to multiple immunotherapies including therapeutic vaccines and checkpoint blocking antibodies (11, 15, 16, 28–31). In contrast, the non-T-cell-inflamed TME appears to be closely related to a lack of clinical benefit from immunotherapy, particularly in relation to anti-PD-1 antibodies (30, 31). Despite a variety of molecular mechanisms that could be theoretically detrimental to the T-cell-inflamed microenvironment, several studies have indicated that oncogenic molecular aberrations are sufficient to drive the immune exclusion phenotype in some cases (6). In a study using a genetically-engineered mouse model, tumor cell-intrinsic WNT/β-catenin signaling in melanoma was found to be the first somatic alteration associated with the non-T-cell-inflamed TME in patients (13). In addition, the transcriptional repression of key chemokine genes leads to a lack of basic leucine zipper ATF-like transcription factor 3 (Batf3)-lineage DC recruitment, and the subsequent failure to prime and recruit CD8+ T cells appears to be involved in this effect (12, 13). This effect is dominant in the TME and results in decreased pre-clinical efficacy for checkpoint blockade, tumor antigen vaccination, and adoptive T-cell transfer immunotherapy approaches (12, 13). In addition, blocking the β-catenin pathway enhances the influx of CD8+ T cells and increases IFNγ-related gene targets in syngeneic murine models of B16F10 melanoma, 4T1 mammary carcinoma, Neuro2A neuroblastoma, and Renca renal adenocarcinoma (32). Therefore, strategies to overcome barriers that restrict T cell migration into tumor sites might ultimately promote immunotherapy efficacy in non-T cell-inflamed tumors. The Wnt/β-catenin pathway could thus represent a high-priority target for combinational cancer immunotherapy.
The WNT signaling pathway is highly conserved between species and has been shown to play an important role in controlling multiple developmental processes including asymmetric cell division, stem cell pluripotency, and cell fate specification (33, 34). In addition to the importance of WNT signaling in stem cells and hematopoiesis, its role in the development of T lymphocytes in the thymus is indispensable (35).
T cell factor (TCF), the effector transcription factor of the WNT signaling pathway, was named for its indispensable role in T cell development and proliferation in the thymus (36). The TCF family consists of four members, specifically TCF-1 (encoded by the TCF-7 gene), TCF-3, TCF-4, and LEF-1 (37, 38). TCF/LEF transcription factors are the major end-point mediators of Wnt signaling throughout metazoans. Although there are other transcription factors that can bind β-catenin and activate transcription, TCFs are major nuclear effector molecules of this pathway (39).
CD8+ T cells are key effectors of the tumor-immune cycle. In the tumor immune system, CD8+ T cells are activated by DCs and co-stimulatory molecules, and then infiltrate into the tumor site to kill target cancer cells (40). By preventing the infiltration of CD8+ T cells during tumor progression, tumor cells evade immune elimination, excluding or inactivating CD8+ T cells (41). Owing to prolonged antigen exposure and the suppressive TME, tumor-infiltrating CD8+ T cells affect the progressive loss of effector functions (42). Wnt/β-catenin signaling is essential for T cell differentiation, effector functions, and migration (43). The activation of TCF-1/β-catenin signaling can result in stem cell-like phenotypes resulting in the formation of memory CD8+ T cells or differentiation into a Tfh cell-like gene expression profile in vivo (44). Naive CD8+ T cells differentiate into effector T cells and kill tumor cells in the tumor-immune cycle (45). Moreover, the activation and maintenance of memory T cells is indispensable to maintain anti-tumor immunity. It has been confirmed that Wnt/β-catenin signaling and TCF1 are highly activated and expressed in undifferentiated CD8+ T and memory CD8+ T cells, and that TCF1 is downregulated when naive CD8+ T cells differentiate into effector CD8+ T cells (46).
Collectively, the differentiation of naïve CD8+ T cells into CD8+ T effector cells is inhibited, the development of memory precursor and central memory CD8+ T cells is promoted, and expansion is stimulated by TCF-1 (Figure 1). However, the requirement for β-catenin in memory T cell functions, as a coactivator of TCF-1, remains controversial.
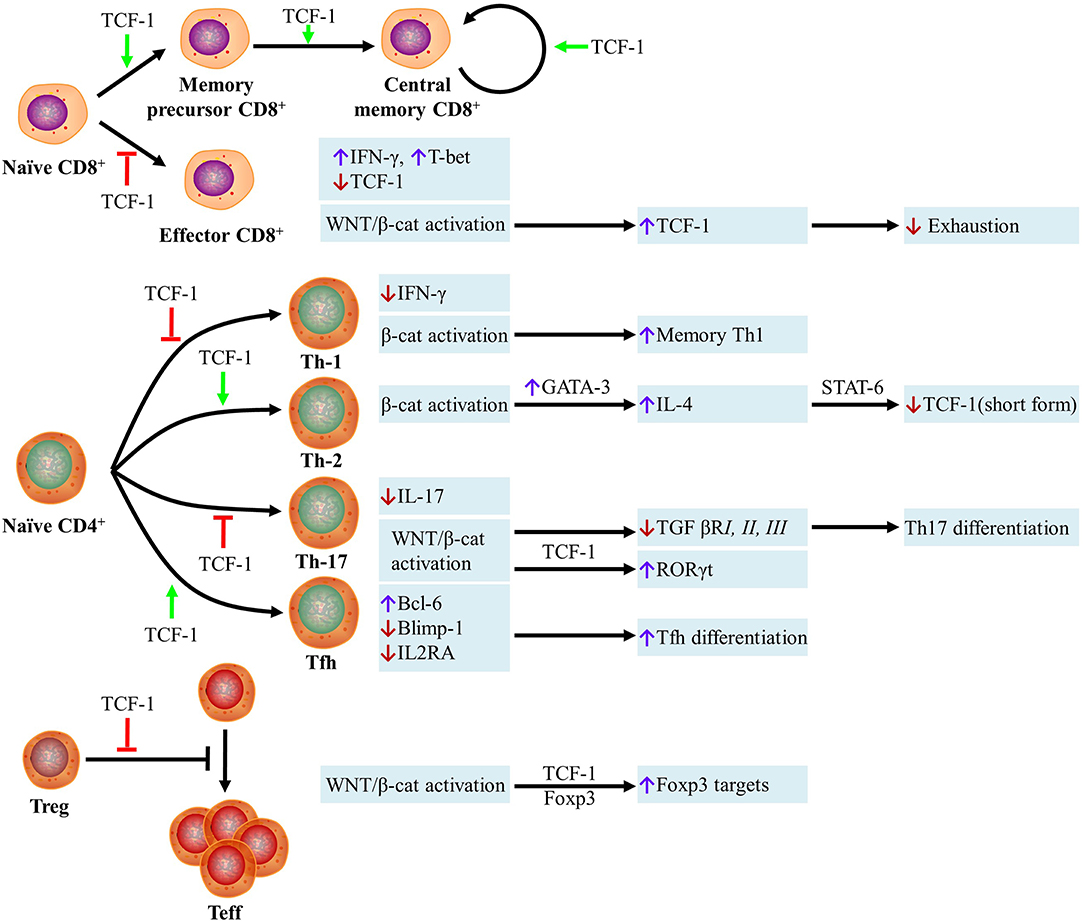
Figure 1. Role of Wnt Proteins in T cells. (i) The differentiation of naïve CD8+ T cells into CD8+ T effector cells is inhibited, the development of memory precursor and central memory CD8+ T cells is promoted, and the expansion is stimulated by TCF-1. Naïve CD8+ T cells are resistant to becoming CD8 effector T cells if they are TCF-1high cells, whereas TCF-1low cells become active IFN-γ+ T-bet+ effector T cells. WNT/β-catenin-mediated activation of effector CD8+ T cells can stimulate the transcription of TCF-1, which inhibits type I IFN-mediated cell exhaustion. (ii) The differentiation of naïve CD4+ T cells into Th1 and Th17 cells is inhibited and differentiation into Th2 and Tfh subsets is promoted by TCF-1. Unlike TCF-1, all types of T cell differentiation and function are enhanced by the expression of β-catenin. The transcription of TCF-1 is inhibited by T-bet, which is the Th1 hallmark transcription factor, thus preventing the inhibition of T-bet-mediated transcription of IFN-γ. Beta-catenin activation enhances memory Th1 cell formation. In Th2 cells, the transcription of GATA-binding protein-3 (GATA-3) can be induced by β-catenin. GATA-3 stimulates the expression of IL-4, which can bind IL-4R and suppress transcription of the short form of TCF-1 via STAT6. In Th17 cells, IL-17 transcription can be suppressed by TCF-1 directly. Wnt signaling activation inhibits the transcription of TGF-βRI, II, and III, which has an important effect on Th17 differentiation. Upon β-catenin activation, RORγt transcription can be increased by TCF-1 directly. TCF-1 promotes Tfh differentiation by increasing Bcl-6 expression and inhibiting the transcription of Blimp-1 and IL2RA. (iii) TCF-1 inhibits the Treg cell-mediated suppression of effector T cell (Teff) proliferation. In the absence of WNT, the transcriptional repressor Foxp3 inhibits many genes including IL-2. TCF-1 activity is prioritized over Foxp3 when WNT is present, which results in the expression of various transcriptional targets shared by Foxp3 and TCF-1. TCF-1, T-cell factor 1; IFN-γ, interferon gamma; GATA3, GATA binding protein 3; IL-4, interleukin 4; TGFβRI, transforming growth factor beta receptor I; TGFβRII, transforming growth factor beta receptor II; TGFβRIII, transforming growth factor beta receptor III; Blimp-1, B lymphocyte-induced maturation protein-1; IL2RA, interleukin 2 receptor alpha.
WNT/β-catenin signaling also regulates the differentiation of CD4+ helper T (TH) cells. TCF-1 and β-catenin support Th2 polarization through activation of the expression of the Th2 master transcription factor GATA binding protein 3 (GATA3) via a special AT-rich sequence binding protein-1 (SATB1), which is a chromatin organizer that plays pivotal roles in T cell development (47, 48). A subsequent study confirmed that the sustained activation of β-catenin in mouse CD4+ thymocytes results in the up-regulation of RAR related orphan receptor C (RARC) and consequent Th17 polarization, ultimately resulting in the production of pro-inflammatory cytokines that favor tumorigenesis (49). Another recent study suggested that Wnt10b regulates type 2 inflammation and the activation of Th2 cells, since Wnt10b deficiency was found to exacerbate house dust mite-induced asthma in mice (50). In addition, the selective ablation of TCF-1 or LEF-1 via two plausible mechanisms leads to Tfh deficiency in the LCMV acute infection model (51).
Overall, the differentiation of naïve CD4+ T cells into Th1 and Th17 cells is inhibited, whereas differentiation into Th2 and Tfh subsets is promoted, by TCF-1. Further, unlike the effects of TCF-1, all types of T cell differentiation and function are enhanced by the expression of β-catenin (Figure 1).
It is been shown that the infiltration of Treg cells into the TME results in inhibition of the anti-tumor immune response (41). Treg cells are recruited into the TME by chemokines (such as CCL-28) secreted by tumor cells and innate immune cells (52). Wnt/β-catenin signaling limits the immunosuppressive activity of those cells by modulating the TCF-1-dependent inhibition of FoxP3 transcriptional activity (53). Production of the negative immunomodulators Foxp3, TGFβ, and IL-10 can be reduced by blocking the Wnt/β-catenin signaling pathway in Treg cells (54). Further, if wild-type Treg cells are inactivated, the functional integrity of Tregs will be ensured by the direct activation of genes encoding PD-1 and GITR to increase β-catenin expression, whereas β-catenin levels are reduced and Treg-mediated immune suppression is impaired by liver kinase B1 (LKB1) deletion (55).
In summary, TCF-1 inhibits the Treg cell-mediated suppression of effector T cell proliferation. When WNT is present, TCF-1 activity is prioritized over Foxp3, which results in the expression of various transcriptional targets shared by Foxp3 and TCF-1 (Figure 1).
The TME regulates DCs through a variety of mechanisms, inhibiting their ability to induce anti-tumor responses. It has been confirmed that the Wnt/β-catenin signaling pathway plays a critical role in crosstalk between tumor cells and DCs within the TME (56). Upon conditional knockout of the Wnt co-receptors LRP5 and LRP6 on DCs, DC-mediated anti-tumor immunity is enhanced, ultimately resulting in delayed tumor growth (57). The recruitment of T cells and DCs into solid tumors is inhibited by β-catenin signaling in melanoma cells (13). In tumor-bearing mouse studies, the activation of β-catenin in DCs resulted in more tolerogenic phenotypes than those mediated by the DC vaccine-induced cross-priming inhibition of anti-tumor CD8+ T cells by IL-10 (58). Furthermore, it has been demonstrated that melanoma induces DC-mediated tolerance to inhibit the efficacy of immunotherapy via Wnt5a-mediated mechanisms in mice (59). Notably, DC-specific β-catenin ablation was found to improve anti-PD-1 immunotherapy efficacy in a syngeneic tumor model (59). In antitumor immunotherapy, DCs are crucial subsets that can modulate T cell responses, and studies have summarized the potential to therapeutically target the Wnt pathway in DCs.
The role of the Wnt-β-catenin pathway might be related to the generation of tolerogenic DCs. However, caution is needed before concluding that the Wnt pathway plays a major role in DC function.
Understanding the mechanisms of T cell exclusion in the TME is critical to improve cancer immunotherapy. The Wnt/β-catenin pathway has been identified as one of the most important oncogenic signaling pathways associated with immune evasion (58, 60). The pan-cancer association between WNT/β-catenin signaling and immune exclusion was previously confirmed by analyzing activated WNT/β-catenin signaling based on somatic mutations, copy number alterations, gene expression, and reverse-phase protein arrays (6). In addition to WNT/β-catenin signaling, other oncogenic events can also contribute to immune exclusion (19, 61). Accordingly, complex interactions among multiple oncogenic events are coupled to mediate more potent immune exclusion in some tumors, such as colorectal cancer, which exhibits the concurrent activation of WNT/β-catenin, MYC, and RAS (62–64).
Mutations in Wnt/β-catenin lead to a non-T cell inflammatory tumor phenotype and might be a biomarker to predict resistance to immunotherapy with ICIs in hepatocellular carcinoma (HCC) (65). Activation of β-catenin reduces T cell infiltrations, increases progression in immunocompetent hosts, and suppresses ICIs in a mouse model of hepatocarcinogenesis (66). The Wnt/β-catenin pathway is also involved in the regulation of innate immunity, such as DCs (67). One study detected an association between activation of Wnt/β-catenin signaling and the loss of T cell gene expression in human metastatic melanoma (13). Beta-catenin activation occurs in 48% of non-T cell inflammatory melanomas (6), and thus, other oncogenic pathways likely contribute to immune exclusion in the remainder of these tumors. Tumor-cell intrinsic activation of the WNT/β-catenin pathway is associated with a lack of T cells in the microenvironment of metastatic melanoma and other cancer types (68). Beta-catenin induces expression of the transcriptional repressor ATF3 and inhibits the transcription of CCL4 in mouse models (69). Furthermore, the defective production of CCL4 results in the impaired infiltration and activation of Batf3-lineage CD103+ DCs, reduced CD8+ T cell priming and infiltration, and a subsequent lack of responses to immune checkpoint blockade (69). In the absence of active β-catenin signaling, the normal production of CCL4 was found to be restored, resulting in CD103+ DC activation and the infiltration and proficient priming of CD8+ T cells (69). Wnt/β-catenin signaling within intestinal DCs regulates the balance between inflammatory and regulatory responses in the gut (69). In intestinal DCs, β-catenin was found to be associated with anti-inflammatory mediators, such as retinoic acid-metabolizing enzymes, IL-10, and transforming growth factor-β, as well as Treg-induced stimulation (70).
It has also been shown that Wnt/β-catenin signaling is involved in crosstalk between cancer cells and tumor-associated macrophages. One study demonstrated that interleukin-1β, secreted by tumor-associated macrophages (TAMs), might increase the availability of β-catenin via the phosphorylation of GSK3β in colon cancer cells, thereby disrupting the function of the β-catenin destruction complex (71). Colorectal cancer cells stimulate IL-β production in macrophages via Snail, which is a soluble factor and product of Wnt target genes (72).
In summary, the Wnt/beta-catenin pathway mediates immune exclusion via three mechanisms as follows: (i) inhibiting the production of CCL4 in Batf3-lineage CD103+ DCs through induction of the expression of the transcriptional repressor ATF3, which in turn reduces the initiation and infiltration of CD8+ T cells; (ii) increasing the interaction between Snail (a soluble factor and product of a Wnt-regulated gene) and TAMs, which in turn increases β-catenin activity via IL-1β; (iii) enhancing Treg survival (Figure 2).
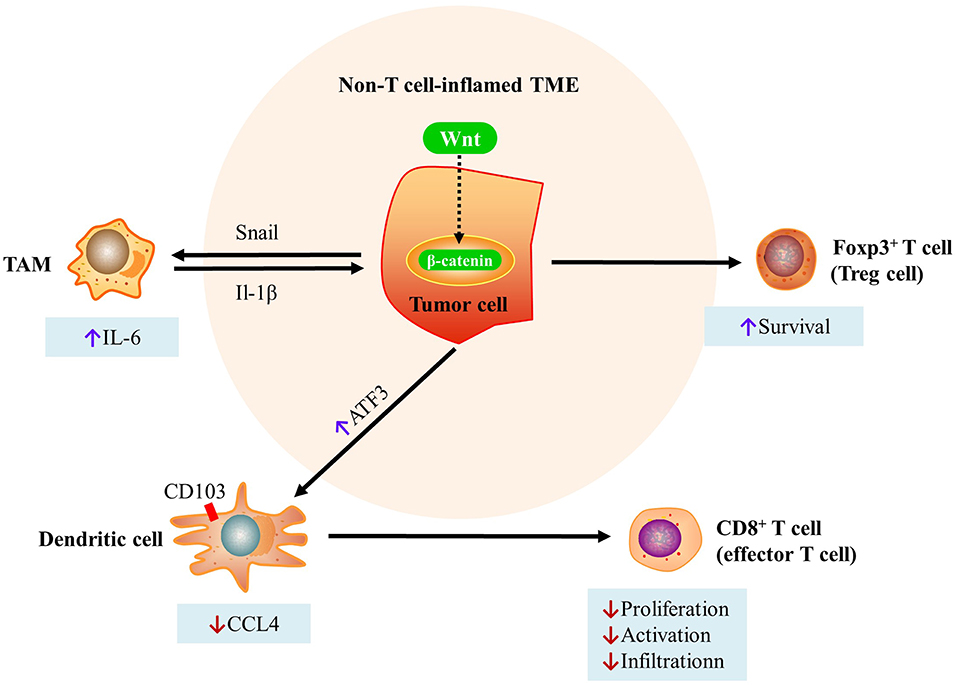
Figure 2. Mechanisms of immune exclusion through the Wnt/beta-catenin pathway. (i) Inhibition of the production of CCL4 in Batf3-lineage CD103+ DCs through induction of the expression of the transcriptional repressor ATF3. This in turn reduces the initiation and infiltration of CD8+ T cells. (ii) Increases in the interaction between Snail (a soluble factor and product of a Wnt-regulated gene) and TAMs, which in turn increases β-catenin activity via IL-1β. (iii) Enhanced Treg survival [modified from Pai et al. (73)]. DC, dendritic cell; TAMs, tumor-associated macrophages; CCL4, C-C motif chemokine ligand 4; ATF3, activating transcription factor 3; TME, tumor microenvironment.
It has been widely accepted that WNT/β-catenin signaling affects cancer immunosurveillance across cancer types (27). By modulating various aspects of tumor-immune cell interactions including the immunogenicity of cancer cells and the ability of immune cells, such as DCs, natural killer (NK) cells, Treg cells, myeloid-derived suppressor cells (MDSCs), and cytotoxic T lymphocytes (CTLs) to elicit effective tumor-targeting immune responses, it has been discovered that WNT signaling positively or negatively affects anticancer immunosurveillance (Figure 3).
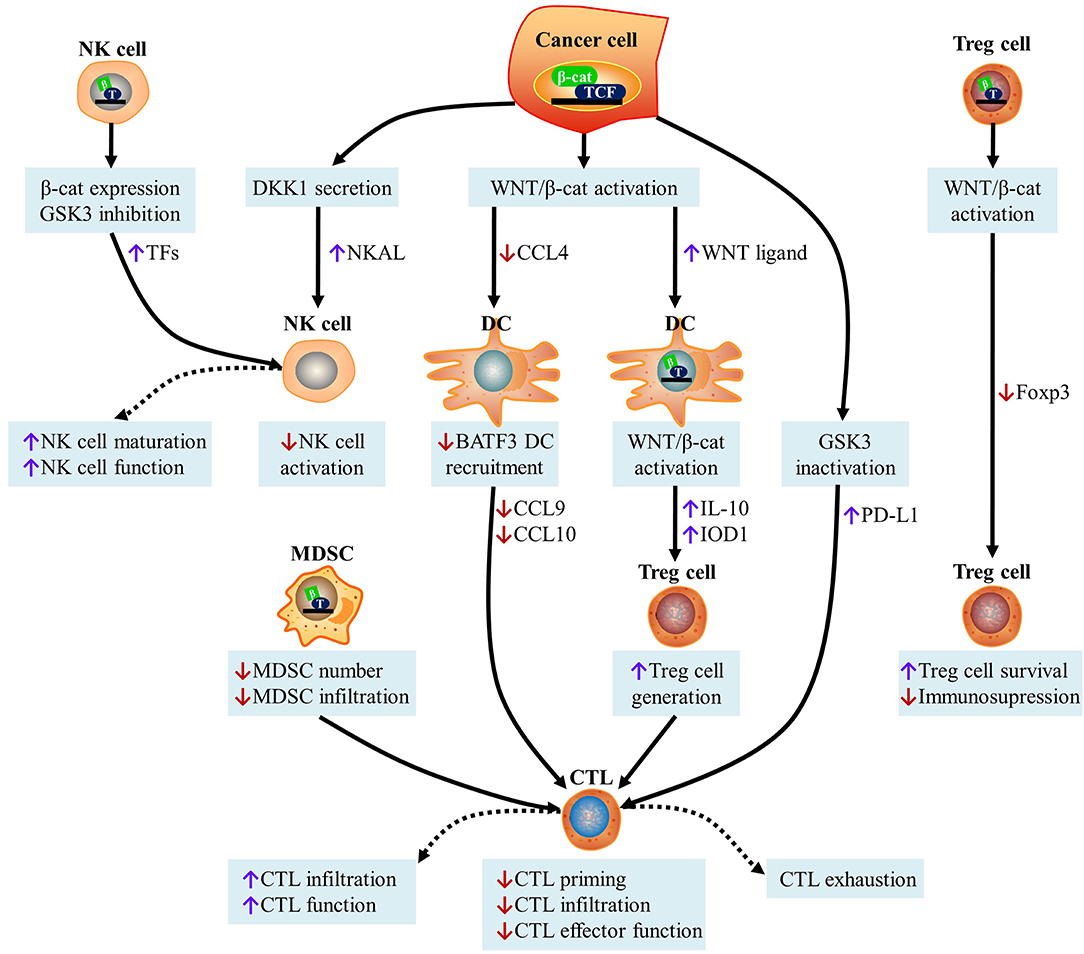
Figure 3. Effect of WNT signaling on cancer immunosurveillance. (i) Activation of the WNT/β-catenin pathway in cancer cells inhibits the secretion of CCL4 and recruits BATF3-dependent DCs to the TME. This causes a decrease in DC-derived CXCL10 levels and limits CD8+ CTL infiltration, leading to defective cross-priming. WNT ligands released by cancer cells can induce the canonical WNT signaling pathway in DCs, leading to the increased secretion of IL-10, restricted production of IL-12, and up-regulation of indoleamine IDO1. This contributes to the production of Treg cells and the subsequent inhibition of CTL activity. The inhibition of GSK3 in cancer cells promotes β-catenin activation and subsequently stabilizes PD-L1, which drives CTL exhaustion upon interactions with PD-1. Canonical WNT/β-catenin in MDSCs inhibits their ability to accumulate and infiltrate into malignant lesions, leading to the increased recruitment of CTLs. (ii) DKK1 secreted by latent metastatic cells prevents NK cell-dependent cancer cell lysis, which is caused by the low expression of NKALs, which engage NKARs. GSK3 inhibition and β-catenin overexpression in NK cells lead to the up-regulation of transcription factors responsible for NK cell maturation, thereby stimulating the function of NK cells as effectors. (iii) Although the expression of β-catenin is beneficial for the survival of Treg cells in vitro, the activation of TCF-1, downstream of canonical WNT signaling, blocks Foxp3 transcriptional functions and impairs the immunosuppressive activity of Treg cells. CTL, cytotoxic T cell; DKK1, Dickkopf WNT signaling pathway inhibitor 1; NKALs, NK cell activating ligands; NKARs, NK-cell-activating receptors; MDSCs, myeloid-derived suppressor cells; DC, dendritic cell; CCL4, C-C motif chemokine ligand 4; TCF, T cell factor; BATF3, basic leucine zipper ATF-Like transcription factor 3; GSK3, glycogen synthase kinase 3; PD-L1, programmed cell death ligand 1; NK, natural killer; IL-10, interleukin 10; IDO1, indoleamine 2,3-dioxygenase 1; TFs, transcription factors; Foxp3, forkhead box p3.
Tumor-intrinsic WNT/beta-catenin signaling affects the immunogenicity of carcinoma cells. Several components of the WNT signal transduction cascade that are overexpressed in carcinoma cells as tumor-associated antigens can be recognized by the immune system (74). Moreover, it has been confirmed that WNT signaling is a major regulator of tumor immune tolerance. Active forms of β-catenin promote immune evasion and resistance to immunotherapy with anti-PD-1, which was found to involve the deficient recruitment of DCs and impaired T cell activity in a novel mouse model of HCC (75). A previous study showed that tumor-induced β-catenin signaling plays a role in DCs, resulting in inhibition of the DC-dependent cross-sensitization of anti-tumor CTLs and infiltrating immune effector cells into a tolerant state (76).
The accumulation of β-catenin in human melanoma cells or DCs leads to the expression of IL-10, which diminishes the capacity of DCs to cross-prime CD8+ CTLs (77). In melanomas, canonical or non-canonical WNT signaling also limits antitumor immunity in DCs, which is associated with metabolic immunosuppression by IDO1 (78). This indicates that the activation of this pathway in DCs is important to maintain clonal amplified antigen-specific CTLs, although most studies indicate that classical WNT signaling inhibits antitumor CTL cross-priming. These studies indicate that the role of WNT signaling in the regulation of anticancer responses during CTL activation is stage-dependent (27).
Excluding CTL infiltration into the TME is the prominent mechanism of WNT-mediated immunoevasion in multiple types of cancer. Therefore, the conditional expression of oncogenic Braf plus Pten deletion and the engineered expression of β-catenin in mouse melanomas results in cells that are unable to express CCL4, leading to DC recruitment defects (13). Due to the absence of CD103+ DC-derived chemokines, such as CXCL9 and CXCL10, tumor infiltration by CTLs is prevented and antitumor immune responses are impaired (13). It is generally accepted that effector cell deficiency in the TME is associated with active classical WNT signaling, which is also the main cause of resistance to cancer immunotherapy (79). Actually, tumor-intrinsic, active β-catenin signaling in human melanoma causes resistance to monoclonal antibodies including anti-PD-L1and anti-CTLA4 via T cell exclusion (13). In the absence of WNT ligands, GSK3β mediates the phosphorylation-dependent proteasomal degradation of PD-L1 by β-TrCP, and tumor infiltration is ultimately increased by interferon-producing CTLs (80).
WNT signaling exerts significant anticancer effects. For example, limiting β-catenin signaling prevents carcinogenesis and the accumulation of CD11b+Gr-1+ MDSCs (81). Beta-catenin inhibits the downstream deletion of Plcg2 (82), Cul4b (83), or Mucl (84), as well as the increased availability of DKK1 in the microenvironment (85), leading to MDSC amplification and recruitment into the TME, attenuating specific immune responses in different types of tumors. Quiescence in lung metastatic cancer cells is caused by autocrine DKK1, which leads to the downregulation of certain cell surface markers recognized by NK cells, allowing cancer cells to evade innate immunity (86).
In summary, cancer immunosurveillance is influenced by WNT signaling in a complex and context-dependent manner.
The WNT/β-catenin pathway should be a high-priority molecular target for new drug development, in an effort to restore T cell infiltration and potentially expand the efficacy of immunotherapy. Several specific inhibitors have been developed and have entered preclinical trials (Table 1); however, the activity of monotherapy has not been sufficient to warrant further advances to registered trials (87). Beta-catenin signaling is widely utilized by many cell types, and thus on-target, off-tumor effects limit the potential of therapeutics that target this protein. The immune evasive mechanisms of β-catenin activation, including the inhibition of chemokines and cytokine gene expression by tumor cells (13), provide the opportunity to develop agents with more restricted activities that predominantly augment immunity while maintaining other essential cellular functions.
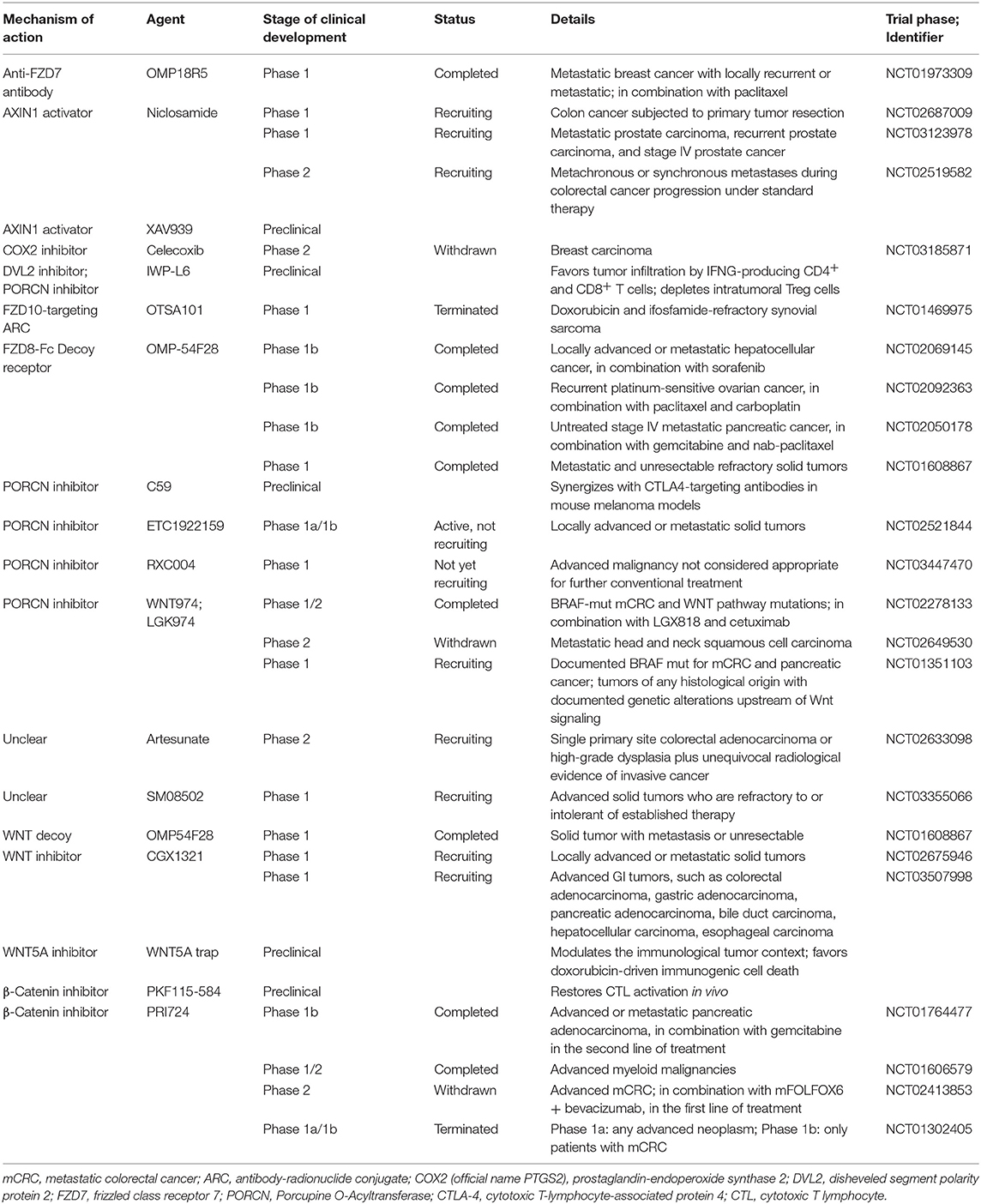
Table 1. WNT inhibitors for cancer immunotherapy currently in clinical development.
Over the past few years, a variety of agents that target main components or modulators of WNT signaling have been developed and used as tumor therapies. Different clinical trials listed in the National Library of Medicine database are testing the effectiveness and safety of these drugs, which have not been approved by the regulatory agencies for clinical use. At first, drug development focused on disrupting WNT-driven tumor growth, including monoclonal antibodies to frizzled class receptor (FZD) receptors (e.g., OMP-54F28, OMP-18R5) (88), porcupine O-acyltransferase (PORCN) inhibitors (e.g., WNT974, RXC004, ETC1922159) (89), AXIN1 activators (e.g., Niclosamide, XAV939) (90), and β-catenin inhibitor (e.g., PRI724, PKF115-584) (91). Even though the immunomodulatory effects of these drugs have been neglected for a long time, there is increasing evidence suggesting that WNT inhibitors can also help to reestablish anticancer immunity. For example, the β-catenin inhibitor PKF115-584 effectively stimulates DC suppression, leading to a robust therapeutic response (77). Furthermore, both IWP-L6 and XAV939 deplete Treg cells from the TME, thus achieving a therapeutically-relevant immune response in an animal model of melanoma and lymphoma (57, 92). Various mechanisms associated with the cancer immunomodulatory effects of WNT inhibitors are listed in Table 1.
The immunological context of the TME has an important effect on the clinical efficacy of cancer immunotherapy, including its composition, activation status, and localization (93). Actually, the inhibition of local or systemic immunosuppression is the target of immunotherapy that is used to restore CTL depletion and reinstate the immunological control of tumor growth (94). ICIs limit the capability of co-inhibitory receptors including PD-1 and CTLA4, which are used to establish CTL depletion. However, when CTLs are absent or unable to infiltrate malignant cell niches, the efficacy of immunotherapy with ICIs is severely reduced (95). Since this phenotype is common in tumors that present with WNT activation, inhibitors of canonical WNT signaling can be combined with CTLA4-blocking mAbs to suppress the progression of melanoma in mice (78). Notably, molecular profiling of HCC has identified that patients with ICIs and activating alteration in WNT/β-catenin signaling have lower disease control rates, shorter median progression-free survival, and shorter median overall survival (96). One recent study developed potent, selective inhibitors targeting the interaction of β-catenin/Bcl9, which overcome resistance to ICIs by modulating Treg cells (97).
The activation of anticancer immune responses largely determines the efficacy of several conventional therapeutic strategies for the treatment of cancer, including a variety of chemotherapeutics, radiation therapy, and some targeted anticancer agents (98). Different anticancer molecules, such as oxaliplatin and doxorubicin can result in a noticeable immunogenic variant of cell death to establish adaptive anticancer immunity (99). Further, it has been confirmed that WNT signaling limits the efficacy of multiple anticancer therapies in this manner (10). Accordingly, it has been found that efficient local Wnt5a trapping results in significant remodeling of the immunosuppressive TME and promotes immunogenic cell-death-mediated immunotherapy (10).
The activity of chemotherapy and immunotherapy with ICIs can be boosted by WNT inhibitors. Studies have shown that CD8+ T cells and CD4+ TH17 cells can differentiate into stem-like cells with advantageous anticancer functions upon the intervention with GSK3 inhibitors (46, 100); they also stimulate NK cell-dependent anticancer immunity (86). The restoration of β-catenin expression in MDSCs and the use of an anti-DKK1 mAb also limit tumor growth by establishing a TME that is beneficial for immunocompetency (85). Therefore, based on the existing preclinical evidence, WNT activation can constitute a therapeutic target.
This review highlights the role of WNT signaling in tumor initiation, malignant tumor progression, and resistance to therapeutics. In the context of combinatorial treatment regimens, a promising candidate WNT modulator, to improve the efficacy of various immunotherapeutic agents, is currently being validated in a variety of preclinical tumor models.
Many preclinical studies have demonstrated that the efficacy of ICIs is affected by WNT/β-catenin signaling, which reduces the infiltration of CTLs into the TME. In contrast, the insensitivity of melanoma patients to immunotherapy with ICIs affects the expression levels of certain components of the WNT signaling cascade (3). In terms of the future direction, it is important to identify biomarkers associated with the non-T cell inflammatory tumor microenvironment. Although WNT modulators are in development for use in combination immunotherapy regimens for cancer, further preclinical and clinical studies are needed to confirm their utility.
XK and XL conceived and wrote this manuscript. XL, YX, FL, CY, BL, and XK discussed the literature and contributed to the revision of the manuscript. CY contributed to the revision of the manuscript. All authors have read and approved the final manuscript.
This project was supported by a grant from the National Key Research and Development Program of China (no. 2018YFC1705301). It was also supported by the NSFC of China (no. 81874470, 81874319), Shanghai Development Office of TCM [nos. ZY(2018–2020)-FWTX-1008, ZY(2018–2020)-CCCX-2004-08, ZY(2018–2020)-FWTX-4010], the Development Fund for Shanghai Talents (no. 2017047), Young Talent Supporting Program of the China Association of Traditional Chinese Medicine [no. CACM-2017-QNRC2-(B05)], and the Science and Technology Commission of Shanghai Municipality (18401933500), Shanghai Municipal Education Commission (2019-01-07-00-10-E00072).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Wang B, Tian T, Kalland KH, Ke X, Qu Y. Targeting Wnt/beta-catenin signaling for cancer immunotherapy. Trends Pharmacol Sci. (2018) 39:648–58. doi: 10.1016/j.tips.2018.03.008
2. Ramos RN, Piaggio E, Romano E. Mechanisms of resistance to immune checkpoint antibodies. Handb Exp Pharmacol. (2018) 249:109–28. doi: 10.1007/164_2017_11
3. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell. (2016) 165:35–44. doi: 10.1016/j.cell.2016.02.065
4. Kobold S, Duewell P, Schnurr M, Subklewe M, Rothenfusser S, Endres S. Immunotherapy in tumors. Dtsch Arztebl Int. (2015) 112:809–15. doi: 10.3238/arztebl.2015.0809
5. Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer Cell. (2016) 29:285–96. doi: 10.1016/j.ccell.2016.02.004
6. Luke JJ, Bao R, Sweis RF, Spranger S, Gajewski TF. WNT/beta-catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res. (2019) 25:3074–83. doi: 10.1158/1078-0432.ccr-18-1942
7. Olson DJ, Luke JJ. The T-cell-inflamed tumor microenvironment as a paradigm for immunotherapy drug development. Immunotherapy. (2019) 11:155–9. doi: 10.2217/imt-2018-0171
8. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. (2015) 348:74–80. doi: 10.1126/science.aaa6204
9. Korneev KV, Atretkhany KN, Drutskaya MS, Grivennikov SI, Kuprash DV, Nedospasov SA. TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis. Cytokine. (2017) 89:127–35. doi: 10.1016/j.cyto.2016.01.021
10. Liu Q, Zhu H, Tiruthani K, Shen L, Chen F, Gao K, et al. Nanoparticle-mediated trapping of Wnt family member 5A in tumor microenvironments enhances immunotherapy for B-Raf proto-oncogene mutant melanoma. ACS Nano. (2018) 12:1250–61. doi: 10.1021/acsnano.7b07384
11. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
12. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–23.e4. doi: 10.1016/j.ccell.2017.04.003
13. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
14. Ulloa-Montoya F, Louahed J, Dizier B, Gruselle O, Spiessens B, Lehmann FF, et al. Predictive gene signature in MAGE-A3 antigen-specific cancer immunotherapy. J Clin Oncol. (2013) 31:2388–95. doi: 10.1200/jco.2012.44.3762
15. Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. (2010) 16:399–403. doi: 10.1097/PPO.0b013e3181eacbd8
16. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.can-08-2281
17. Mikucki ME, Fisher DT, Matsuzaki J, Skitzki JJ, Gaulin NB, Muhitch JB, et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat Commun. (2015) 6:7458. doi: 10.1038/ncomms8458
18. Martinenaite E, Munir Ahmad S, Hansen M, Met Ö, Westergaard MW, Larsen SK, et al. CCL22-specific T cells: modulating the immunosuppressive tumor microenvironment. Oncoimmunology. (2016) 5:e1238541. doi: 10.1080/2162402X.2016.1238541
19. Gajewski TF, Corrales L, Williams J, Horton B, Sivan A, Spranger S. Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv Exp Med Biol. (2017) 1036:19–31. doi: 10.1007/978-3-319-67577-0_2
20. Demoulin S, Herfs M, Delvenne P, Hubert P. Tumor microenvironment converts plasmacytoid dendritic cells into immunosuppressive/tolerogenic cells: insight into the molecular mechanisms. J Leuk Biol. (2013) 93:343–52. doi: 10.1189/jlb.0812397
21. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. (2012) 122:899–910. doi: 10.1172/jci45817
22. Watkins SK, Zhu Z, Riboldi E, Shafer-Weaver KA, Stagliano KE, Sklavos MM, et al. FOXO3 programs tumor-associated DCs to become tolerogenic in human and murine prostate cancer. J Clin Invest. (2011) 121:1361–72. doi: 10.1172/jci44325
23. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. (2010) 330:827–30. doi: 10.1126/science.1195300
24. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. (2008) 14:28–36. doi: 10.1038/nm1699
25. Murillo-Garzon V, Kypta R. WNT signalling in prostate cancer. Nat Rev Urol. (2017) 14:683–96. doi: 10.1038/nrurol.2017.144
26. Gagnaire A, Nadel B, Raoult D, Neefjes J, Gorvel JP. Collateral damage: insights into bacterial mechanisms that predispose host cells to cancer. Nat Rev Microbiol. (2017) 15:109–28. doi: 10.1038/nrmicro.2016.171
27. Galluzzi L, Spranger S, Fuchs E, Lopez-Soto A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. (2019) 29:44–65. doi: 10.1016/j.tcb.2018.08.005
28. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
29. Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. (2013) 25:268–76. doi: 10.1016/j.coi.2013.02.009
30. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. (2017) 127:2930–40. doi: 10.1172/jci91190
31. Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. (2012) 61:1019–31. doi: 10.1007/s00262-011-1172-6
32. Ganesh S, Shui X, Craig KP, Park J, Wang W, Brown BD, et al. RNAi-mediated beta-catenin inhibition promotes T cell infiltration and antitumor activity in combination with immune checkpoint blockade. Mol Ther. (2018) 26:2567–79. doi: 10.1016/j.ymthe.2018.09.005
33. Kim W, Kim M, Jho EH. Wnt/beta-catenin signalling: from plasma membrane to nucleus. Biochem J. (2013) 450:9–21. doi: 10.1042/bj20121284
34. Staal FJ, Luis TC, Tiemessen MM. WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol. (2008) 8:581–93. doi: 10.1038/nri2360
35. Zhu P, Zhu X, Wu J, He L, Lu T, Wang Y, et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat Immunol. (2019) 20:183–94. doi: 10.1038/s41590-018-0297-6
36. Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, et al. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature. (1995) 374:70–4. doi: 10.1038/374070a0
37. van Beest M, Dooijes D, van De Wetering M, Kjaerulff S, Bonvin A, Nielsen O, et al. Sequence-specific high mobility group box factors recognize 10-12-base pair minor groove motifs. J Biol Chem. (2000) 275:27266–73. doi: 10.1074/jbc.M004102200
38. van de Wetering M, Cavallo R, Dooijes D, van Beest M, van Es J, Loureiro J, et al. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. (1997) 88:789–99.
39. Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol. (2012) 4:a007906. doi: 10.1101/cshperspect.a007906
40. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. (2011) 208:1989–2003. doi: 10.1084/jem.20101158
41. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. (2017) 168:707–23. doi: 10.1016/j.cell.2017.01.017
42. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8(+) T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–56.e5. doi: 10.1016/j.cmet.2019.04.002
43. Gattinoni L, Ji Y, Restifo NP. Wnt/beta-catenin signaling in T-cell immunity and cancer immunotherapy. Clin Cancer Res. (2010) 16:4695–701. doi: 10.1158/1078-0432.CCR-10-0356
44. Ma J, Wang R, Fang X, Sun Z. β-catenin/TCF-1 pathway in T cell development and differentiation. J Neuroimmune Pharmacol. (2012) 7:750–62. doi: 10.1007/s11481-012-9367-y
45. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
46. Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. (2009) 15:808–13. doi: 10.1038/nm.1982
47. Notani D, Gottimukkala KP, Jayani RS, Limaye AS, Damle MV, Mehta S, et al. Global regulator SATB1 recruits beta-catenin and regulates T(H)2 differentiation in Wnt-dependent manner. PLoS Biol. (2010) 8:e1000296. doi: 10.1371/journal.pbio.1000296
48. Yu Q, Sharma A, Oh SY, Moon HG, Hossain MZ, Salay TM, et al. T cell factor 1 initiates the T helper type 2 fate by inducing the transcription factor GATA-3 and repressing interferon-gamma. Nat Immunol. (2009) 10:992–9. doi: 10.1038/ni.1762
49. Keerthivasan S, Aghajani K, Dose M, Molinero L, Khan MW, Venkateswaran V, et al. Beta-catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci Transl Med. (2014) 6:225ra28. doi: 10.1126/scitranslmed.3007607
50. Trischler J, Shiomi T, Turner DL, Sklepkiewicz PL, Goldklang MP, Tanaka KF, et al. Immune Modulation of the T Cell Response in Asthma through Wnt10b. Am J Respir Cell Mol Biol. (2016) 54:584–93. doi: 10.1165/rcmb.2014-0425OC
51. Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, et al. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nat Immunol. (2015) 16:980–90. doi: 10.1038/ni.3226
52. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines. (2016) 4:E28. doi: 10.3390/vaccines4030028
53. van Loosdregt J, Fleskens V, Tiemessen MM, Mokry M, van Boxtel R, Meerding J, et al. Canonical Wnt signaling negatively modulates regulatory T cell function. Immunity. (2013) 39:298–310. doi: 10.1016/j.immuni.2013.07.019
54. Dai W, Liu F, Li C, Lu Y, Lu X, Du S, et al. Blockade of Wnt/beta-catenin pathway aggravated silica-induced lung inflammation through Tregs regulation on Th immune responses. Mediat Inflamm. (2016) 2016:6235614. doi: 10.1155/2016/6235614
55. Yang K, Blanco DB, Neale G, Vogel P, Avila J, Clish CB, et al. Homeostatic control of metabolic and functional fitness of Treg cells by LKB1 signalling. Nature. (2017) 548:602–6. doi: 10.1038/nature23665
56. Hansen M, Andersen MH. The role of dendritic cells in cancer. Semin Immunopathol. (2017) 39:307–16. doi: 10.1007/s00281-016-0592-y
57. Hong Y, Manoharan I, Suryawanshi A, Majumdar T, Angus-Hill ML, Koni PA, et al. beta-catenin promotes regulatory T-cell responses in tumors by inducing vitamin A metabolism in dendritic cells. Cancer Res. (2015) 75:656–65. doi: 10.1158/0008-5472.can-14-2377
58. Fu C, Liang X, Cui W, Ober-Blobaum JL, Vazzana J, Shrikant PA, et al. Beta-catenin in dendritic cells exerts opposite functions in cross-priming and maintenance of CD8+ T cells through regulation of IL-10. Proc Natl Acad Sci USA. (2015) 112:2823–8. doi: 10.1073/pnas.1414167112
59. Zhao F, Xiao C, Evans KS, Theivanthiran T, DeVito N, Holtzhausen A, et al. Paracrine Wnt5a-beta-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. (2018) 48:147–60.e7. doi: 10.1016/j.immuni.2017.12.004
60. Spranger S, Gajewski TF. A new paradigm for tumor immune escape: β-catenin-driven immune exclusion. J Immunother Cancer. (2015) 3:43. doi: 10.1186/s40425-015-0089-6
61. Spranger S, Gajewski TF. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat Rev Cancer. (2018) 18:139–47. doi: 10.1038/nrc.2017.117
62. Lal N, White BS, Goussous G, Pickles O, Mason MJ, Beggs AD, et al. KRAS mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin Cancer Res. (2018) 24:224–33. doi: 10.1158/1078-0432.ccr-17-1090
63. Grasso CS, Giannakis M, Wells DK, Hamada T, Mu XJ, Quist M, et al. Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov. (2018) 8:730–49. doi: 10.1158/2159-8290.cd-17-1327
64. Guinney J, Dienstmann R, Wang X, de Reynies A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med. (2015) 21:1350–6. doi: 10.1038/nm.3967
65. Pinyol R, Sia D, Llovet JM. Immune exclusion-Wnt/CTNNB1 class predicts resistance to immunotherapies in HCC. Clin Cancer Res. (2019) 25:2021–3. doi: 10.1158/1078-0432.ccr-18-3778
66. Berraondo P, Ochoa MC, Olivera I, Melero I. Immune desertic landscapes in hepatocellular carcinoma shaped by beta-catenin activation. Cancer Discov. (2019) 9:1003–5. doi: 10.1158/2159-8290.cd-19-0696
67. Swafford D, Manicassamy S. Wnt signaling in dendritic cells: its role in regulation of immunity and tolerance. Discov Med. (2015) 19:303–10.
68. Trujillo JA, Sweis RA-O, Bao R, Luke JJ. T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. (2018) 6:990–1000. doi: 10.1158/2326-6066.CIR-18-0277
69. Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. (2008) 14:162–9. doi: 10.1038/nm1707
70. Manicassamy S, Reizis B, Ravindran R, Nakaya H, Salazar-Gonzalez RM, Wang YC, et al. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science. (2010) 329:849–53. doi: 10.1126/science.1188510
71. Kaler P, Augenlicht L, Klampfer L. Macrophage-derived IL-1beta stimulates Wnt signaling and growth of colon cancer cells: a crosstalk interrupted by vitamin D3. Oncogene. (2009) 28:3892–902. doi: 10.1038/onc.2009.247
72. Kaler P, Augenlicht L, Klampfer L. Activating mutations in beta-catenin in colon cancer cells alter their interaction with macrophages; the role of snail. PLoS ONE. (2012) 7:e45462. doi: 10.1371/journal.pone.0045462
73. Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol. (2017) 10:101. doi: 10.1186/s13045-017-0471-6
74. Robbins PF, El-Gamil M, Li YF, Kawakami Y, Loftus D, Appella E, et al. A mutated beta-catenin gene encodes a melanoma-specific antigen recognized by tumor infiltrating lymphocytes. J Exp Med. (1996) 183:1185–92. doi: 10.1084/jem.183.3.1185
75. Ruiz de Galarreta M, Bresnahan E, Molina-Sanchez P, Lindblad KE, Maier B, Sia D, et al. Beta-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. (2019) 9:1124–41. doi: 10.1158/2159-8290.cd-19-0074
76. Baur AS, Lutz MB, Schierer S, Beltrame L, Theiner G, Zinser E, et al. Denileukin diftitox (ONTAK) induces a tolerogenic phenotype in dendritic cells and stimulates survival of resting Treg. Blood. (2013) 122:2185–94. doi: 10.1182/blood-2012-09-456988
77. Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N, et al. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J Immunol. (2012) 189:2110–7. doi: 10.4049/jimmunol.1102282
78. Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma-derived Wnt5a promotes local dendritic-cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res. (2015) 3:1082–95. doi: 10.1158/2326-6066.cir-14-0167
79. Galluzzi L, Yamazaki T, Kroemer G. Linking cellular stress responses to systemic homeostasis. Nat Rev Mol Cell Biol. (2018) 19:731–45. doi: 10.1038/s41580-018-0068-0
80. Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. (2016) 7:12632. doi: 10.1038/ncomms12632
81. Di Piazza M, Nowell CS, Koch U, Durham AD, Radtke F. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell. (2012) 22:479–93. doi: 10.1016/j.ccr.2012.08.016
82. Capietto AH, Kim S, Sanford DE, Linehan DC, Hikida M, Kumosaki T, et al. Down-regulation of PLCgamma2-beta-catenin pathway promotes activation and expansion of myeloid-derived suppressor cells in cancer. J Exp Med. (2013) 210:2257–71. doi: 10.1084/jem.20130281
83. Qian Y, Yuan J, Hu H, Yang Q, Li J, Zhang S, et al. The CUL4B/AKT/beta-catenin axis restricts the accumulation of myeloid-derived suppressor cells to prohibit the establishment of a tumor-permissive microenvironment. Cancer Res. (2015) 75:5070–83. doi: 10.1158/0008-5472.can-15-0898
84. Poh TW, Bradley JM, Mukherjee P, Gendler SJ. Lack of Muc1-regulated beta-catenin stability results in aberrant expansion of CD11b+Gr1+ myeloid-derived suppressor cells from the bone marrow. Cancer Res. (2009) 69:3554–62. doi: 10.1158/0008-5472.can-08-3806
85. D'Amico L, Mahajan S, Capietto AH, Yang Z, Zamani A, Ricci B, et al. Dickkopf-related protein 1 (Dkk1) regulates the accumulation and function of myeloid derived suppressor cells in cancer. J Exp Med. (2016) 213:827–40. doi: 10.1084/jem.20150950
86. Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. (2016) 165:45–60. doi: 10.1016/j.cell.2016.02.025
87. Voronkov A, Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharma Des. (2013) 19:634–64. doi: 10.2174/138161213804581837
88. Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, et al. Wnt pathway inhibition via the targeting of frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci USA. (2012) 109:11717–22. doi: 10.1073/pnas.1120068109
89. You L, Zhang C, Yarravarapu N, Morlock L, Wang X, Zhang L, et al. Development of a triazole class of highly potent Porcn inhibitors. Bioorg Med Chem Lett. (2016) 26:5891–5. doi: 10.1016/j.bmcl.2016.11.012
90. Li C, Zheng X, Han Y, Lv Y, Lan F, Zhao J. XAV939 inhibits the proliferation and migration of lung adenocarcinoma A549 cells through the WNT pathway. Oncol Lett. (2018) 15:8973–82. doi: 10.3892/ol.2018.8491
91. Landman EBM, Miclea RL, van Blitterswijk CA, Karperien M. Small molecule inhibitors of WNT/β-catenin signaling block IL-1β- and TNFα-induced cartilage degradation. Arthritis Res Ther. (2013) 15:R93. doi: 10.1186/ar4273
92. Hong Y, Manoharan I, Suryawanshi A, Shanmugam A, Swafford D, Ahmad S, et al. Deletion of LRP5 and LRP6 in dendritic cells enhances antitumor immunity. Oncoimmunology. (2016) 5:e1115941. doi: 10.1080/2162402x.2015.1115941
93. Galluzzi L, Chan TA, Kroemer G, Wolchok JD, Lopez-Soto A. The hallmarks of successful anticancer immunotherapy. Sci Transl Med. (2018) 10:eaat7807. doi: 10.1126/scitranslmed.aat7807
94. Galluzzi L, Vacchelli E, Bravo-San Pedro JM, Buque A, Senovilla L, Baracco EE, et al. Classification of current anticancer immunotherapies. Oncotarget. (2014) 5:12472–508. doi: 10.18632/oncotarget.2998
95. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. (2017) 541:321–30. doi: 10.1038/nature21349
96. Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res. (2019) 25:2116–26. doi: 10.1158/1078-0432.ccr-18-2293
97. Feng M, Jin JQ, Xia L, Xiao T, Mei S, Wang X, et al. Pharmacological inhibition of beta-catenin/BCL9 interaction overcomes resistance to immune checkpoint blockades by modulating Treg cells. Sci Adv. (2019) 5:eaau5240. doi: 10.1126/sciadv.aau5240
98. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell. (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
99. Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. (2017) 17:97–111. doi: 10.1038/nri.2016.107
Keywords: WNT/β-catenin signaling pathway, T cell-inflammation, tumor microenvironment, CD8+ T cells, immune exclusion, immunotherapy
Citation: Li X, Xiang Y, Li F, Yin C, Li B and Ke X (2019) WNT/β-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 10:2293. doi: 10.3389/fimmu.2019.02293
Received: 30 May 2019; Accepted: 11 September 2019;
Published: 26 September 2019.
Edited by:
Antje Blumenthal, University of Queensland, AustraliaReviewed by:
Fotini Gounari, University of Chicago, United StatesCopyright © 2019 Li, Xiang, Li, Yin, Li and Ke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xisong Ke, eGlzb25na2VAc2h1dGNtLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.