 Leonhard von Meyenn
Leonhard von Meyenn Nicole Leonie Bertschi
Nicole Leonie Bertschi Christoph Schlapbach
Christoph Schlapbach
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 September 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02285
This article is part of the Research Topic Skin Autoimmunity View all 69 articles
A properly functioning T cell compartment is crucial to protect the host from infections, tumors, and environmental substances. In recent years, it has become increasingly clear that the processes underlying proper T cell activation, proliferation, and differentiation require well-tuned and dynamic changes in T cell metabolism. Thus, proper metabolic reprogramming in T cells is crucial to ensure proper immunity in the context of infection and anti-tumor immunity. Conversely, aberrant regulation of T cell metabolism can impair T cell function and thereby contribute to T cell-mediated disease. In this review, the relevance of recent insights into T cell metabolism for prototypical T cell-mediated skin diseases will be discussed and their therapeutic potential will be outlined. First, the major modules of T cell metabolism are summarized. Then, the importance of T cell metabolism for T cell-mediated skin diseases such as psoriasis and allergic contact dermatitis is discussed, based on the current state of our understanding thereof. Finally, novel therapeutic opportunities for inflammatory skin disease that might emerge from investigations in T cell metabolism are outlined.
T cells are key players of the adaptive immune system by acting as potent effectors and regulators of immunity. Adaptive T cell immunity results from clonal expansion of antigen-specific T cells that were activated by their cognate antigen presented by dendritic cells in secondary lymphoid organs (1). After priming of naive T cells, multiple subsets of effector and memory T cells with different phenotypic and functional properties are generated. Effector T cells (TEFF) migrate to peripheral tissues and inflamed sites to fight infection. After successful clearance of the invading antigen, the vast majority of effector cells die, leaving behind a small population of long-lived memory T cells (2). In case of re-exposure to previously encountered pathogens or antigens, memory T cells provide rapid and effective protective immunity.
Based on their migratory properties, memory T cells can be categorized in three major subsets: Central memory T (TCM) cells, effector memory T (TEM) cells, and the recently identified tissue-resident (TRM) cells. TCM cells express the chemokine receptor CCR7 and the vascular addressin L selectin (CD62L), enabling them to circulate between lymph nodes and blood. TEM cells express tissue-homing receptors that grant them access to peripheral tissues. Skin-homing TEM, for instance, express the E-selectin ligand Cutaneous Lymphocyte Antigen (CLA), which allows them to enter the skin, whereas the integrin α4β7 permits gut-homing TEM to enter the gut (3). TRM cells are non-recirculating memory T cells that persist long-term in epithelial barrier tissues and provide rapid front-line immune protection in peripheral tissues (4).
It is now increasingly understood that T cell identity and function are intertwined with metabolic pathways that are required for energy and substrate generation on the one hand, but also act to regulate effector functions, differentiation, and gene expression on the other hand (5). In this review, we will first summarize the current literature on T cell immunometabolism. Then, we will explore how the cutaneous environment in inflammatory skin disease might shape and influence T cell function via the modulation of T cell metabolism and look ahead on therapeutic opportunities that might arise therefrom.
After activation by cognate antigen, T cells rapidly increase their glucose uptake by upregulating the cell surface expression of glucose transporters such as GLUT1 (6). Once in the cytosol, glucose immediately enters the glycolysis pathway in which glucose is metabolized to pyruvate by a series of enzymatic reactions (Figure 1). At the same time, lactate excretion increases as a consequence of enhanced conversion of pyruvate to lactate, indicating that T cells engage in aerobic glycolysis (Figures 1, 2). Aerobic glycolysis describes the reduction of pyruvate to lactate in the presence of oxygen, instead of its oxidative phosphorylation (OXPHOS) in the mitochondria via the tricarboxylic acid (TCA) cycle. This process of aerobic glycolysis is also referred to as the “Warburg effect” in cancer cells (7). While aerobic glycolysis generates less ATP per molecule of glucose than OXPHOS, aerobic glycolysis yields metabolic intermediates important for cell growth and proliferation, and contributes to the cellular redox balance (NAD+/NADH) (8). During the 10 enzymatic steps of glycolysis in which glucose is converted to pyruvate, a variety of intermediates are generated that are used in different biosynthetic pathways. These include the pentose phosphate pathway, serine biosynthesis, de novo fatty acid synthesis, and hexosamine biosynthesis. The pentose phosphate pathway, for instance, donates important precursors for nucleotide synthesis and is thus of crucial importance for cell growth (9–11). As a consequence, glycolysis is not only an energy-generating pathway for T cells, but also a metabolic basis for anabolic biosynthesis and proliferation (10).
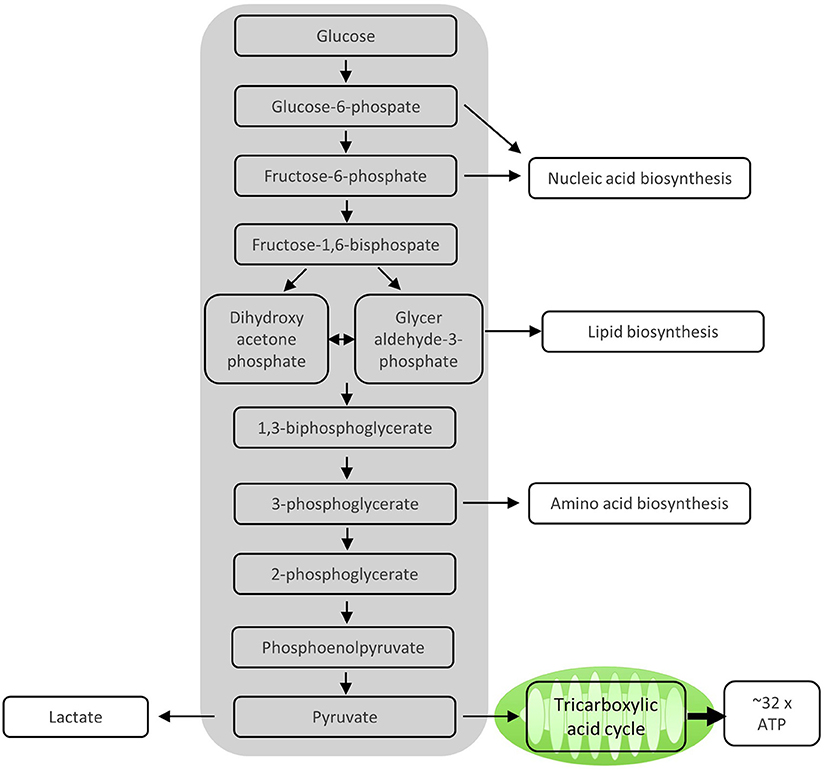
Figure 1. Enzymatic steps of glycolysis. During the 10 enzymatic steps of glycolysis in which glucose is converted to pyruvate, important intermediate molecules for nucleic acid synthesis, lipid biosynthesis, and amino acid biosynthesis, all of which are important for proliferating T cells, are generated. The sequential enzymatic reactions of glycolysis occurring in the cytosol generate ATP. The remaining pyruvate can be oxidized in the mitochondria via the tricarboxylic acid cycle and subsequent oxidative phosphorylation (OXPHOS) to generate ATP. Alternatively, pyruvate can be reduced to lactate and excreted, which mainly happens in proliferating T cells.
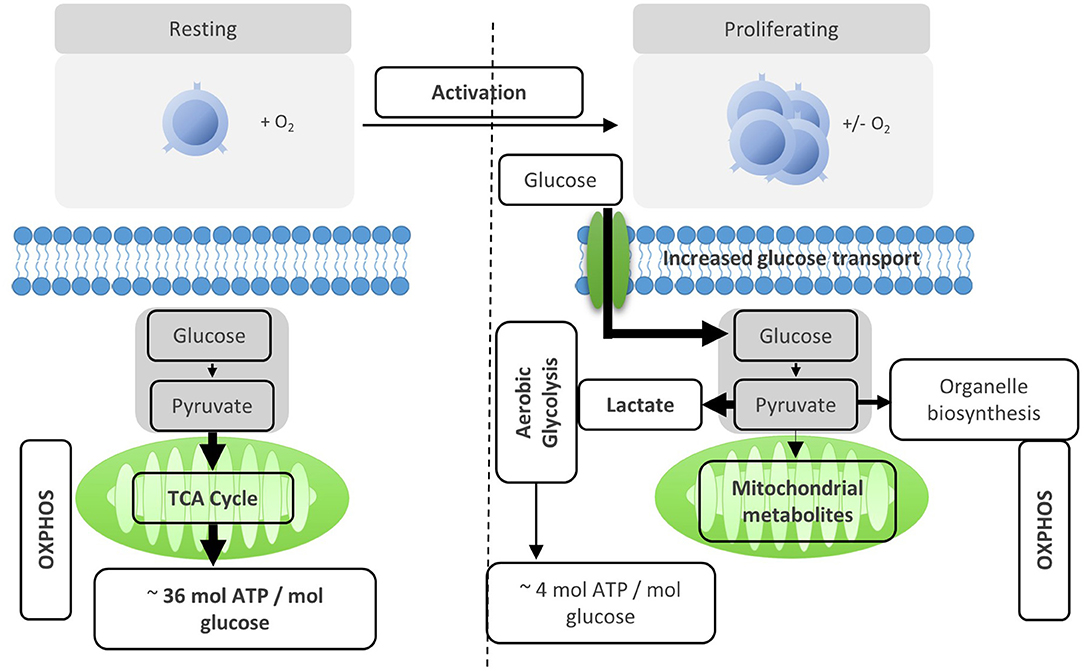
Figure 2. Activation reprograms T cells to increased usage of aerobic glycolysis. Resting T cells engage OXPHOS and glycolysis to generate energy for homeostasis and survival. Upon activation, T cells are reprogramed to increase aerobic glycolysis. The relative increase of anaerobic glycolysis after activation provides less energy, but generates molecules for biosynthesis.
The rapid changes in T cell metabolism after activation are regulated by a number of transcription factors and signaling pathways (8). Signals from cytokines such as IL-2 lead to upregulation of nutrient transporters. Ligation of the co-stimulatory molecule CD28 induces phosphatidylinositol-3-kinase (PI3K)-dependent phosphorylation of Akt (12, 13). Phosphorylated Akt induces expression and translocation of glucose transporters to the cell surface, enhances activity of glycolytic enzymes and activates key metabolic regulators such as mTOR (14). mTOR is a central orchestrator of T cell metabolism and is crucial for upregulation of glycolysis in activated T cells (15) (Figure 2). Further transcription factors involved in regulating activation-induced glycolysis are c-Myc, estrogen receptor α (ERRα) and hypoxia inducible factor-1α (HIF-1α) which coordinately drive the expression of genes involved in metabolism to fuel the bioenergetic needs of clonal expansion (8, 16–18).
Glycolytic activity in T cells is crucial for effector functions in T cells. This is best described for interferon-γ (INF-γ) production of CD8+ and CD4+ TEM cells, with multiple mechanisms linking glycolysis to INF-γ production. For instance, activation-induced glycolysis is accompanied by increased activity of GAPDH which promotes chromatin remodeling at the IFNG locus (15). In resting T cells, on the other hand, GAPDH is not engaged in glycolysis but is able to inhibit translation of IFNG mRNA by binding to its untranslated region (19). More broadly, glycolysis has been linked to pathogenic effector T cell responses in a variety of disease models, such as allogeneic transplantation (20), experimental autoimmune encephalitis (21), and systemic lupus erythematosus (SLE) (22). In these models, inhibiting glycolysis in T cells ameliorated the disease (21), suggesting that manipulation of glycolysis represents a promising target for T cell mediated disease. However, the role of T cell metabolism seems to be context and disease dependent, because reduced glycolysis has also been reported to play a pathogenic role in other disease settings, such as rheumatoid arthritis (23). In contrast to effector T cells, the importance of glycolysis for regulatory T cells (TREG) is less well-understood. Depending on the setting within which TREG cells have been investigated, glycolysis appears to be necessary or not for their suppressive function (24–26). Taken together, well-coordinated glycolytic activity is crucial for proper effector functions in T cells and represents an intriguing leverage point for therapeutic intervention even though the precise conditions for this remain to be elucidated.
Activation of T cells also leads to an increase in their mitochondrial function. Even in the presence of ongoing aerobic glycolysis, enough pyruvate is generated to enter the mitochondria via the mitochondrial pyruvate carrier and fuel the TCA cycle (14) (Figure 3). This process generates further molecules for biosynthesis and drives OXPHOS and, thereby, efficient ATP production. Once in the mitochondria, pyruvate is converted to acetyl-CoA by pyruvate dehydrogenase, and then enters the TCA cycle. In the TCA cycle, acetyl-CoA is oxidized to carbon dioxide and water, which generates GTP and reduces NAD and FADH, the electron carriers that later fuel OXPHOS (14). In addition, the TCA cycle generates a number of metabolic intermediates for anabolic processes such as amino acid and lipid biosynthesis (27). Other substrates can also be metabolized in the TCA cycle, such as glutamine via glutaminolysis or fatty acids via β oxidation (8). Thus, the TCA cycle integrates and mediates the metabolism of glucose, amino acids, and fatty acids (Figure 4).
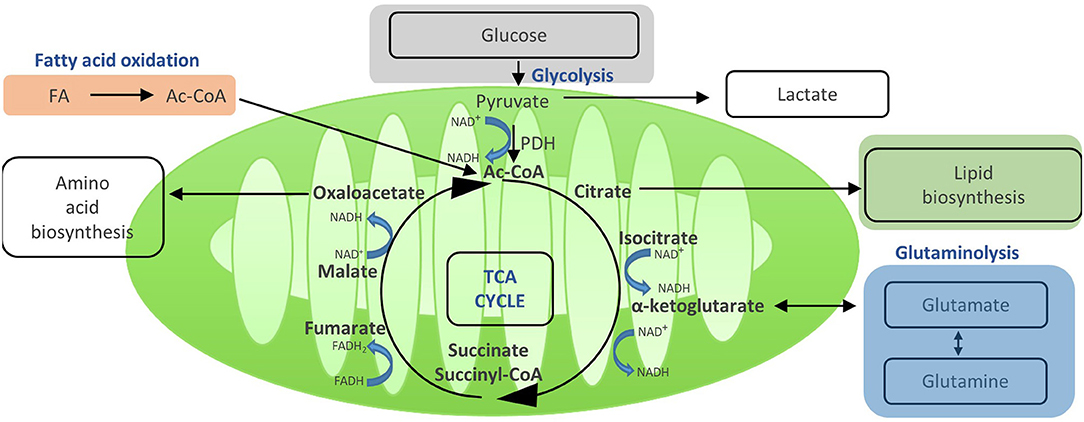
Figure 3. Acetyi-CoA enters the TCA and yields reduced electron carriers (NADH and FADH2) that drive OXPHOS. Once in the mitochondria, pyruvate is converted to acetyl-GoA by pyruvate dehydrogenase, and then enters the TGA cycle. In the TGA cycle, acetyl-GoA is oxidized to carbon dioxide and water, which generates GTP and reduces NAD+ and FADH, the electron carriers that later fuel OXPHOS. In addition, the TGA cycle generates a number of metabolic intermediates for anabolic processes such as amino acid and lipid biosynthesis. Other substrates can also be metabolized in the TGA cycle, such as glutamine via glutaminolysis or fatty acids via oxidation. Thus, the TGA cycle integrates and mediates the metabolism of glucose, amino acids, and fatty acids.
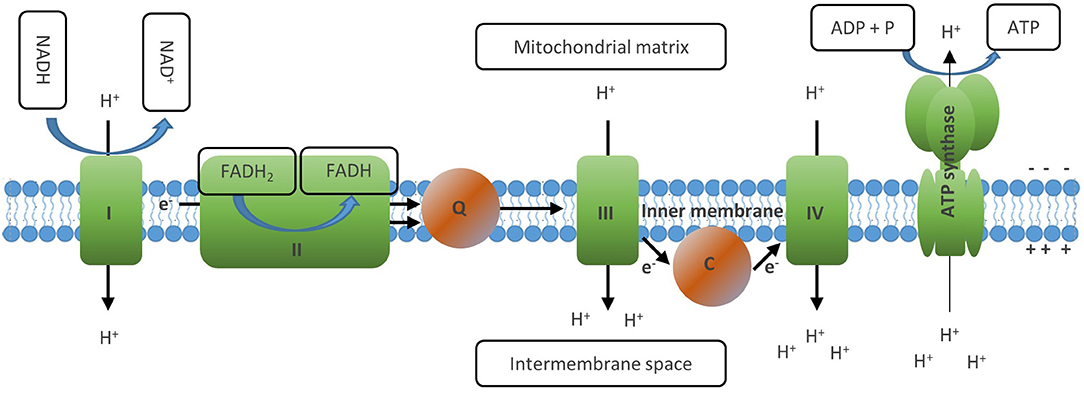
Figure 4. The electron transport chain generates ATP via oxidative phosphorylation. The ETC consists of a series of transmembrane proteins located on the inner mitochondrial membrane (IMM). Complex I and II of the ETC oxidize NADH and FADH2, respectively. Then complex II donates the electrons obtained to complex Ill via Coenzyme Q and later to complex IV via cytochrome C, where finally they are transferred to molecular oxygen which generates water. This process is linked to the transfer of protons into the intermembrane space of the mitochondria, which creates an electrochemical proton gradient between the opposite sides of the membrane. The last member of the ETC, the ATP synthase, finally uses this membrane potential to phosphorylate ADP to ATP, thus letting protons back to the mitochondrial matrix. Through this process, OXPHOS mediates maximal usage of the energy stored in nutrient molecules such as glucose or fatty acids by generating 30–36 ATP molecules from one molecule of glucose and 106 ATP from one molecule of palmitate. Full oxidation of NADH and FADH2 generated in the TCA, however, requires the presence of molecular oxygen to serve as acceptor of the electrons generated by the ETC reactions.
In terms of T cell function, the TCA cycle appears to be critical for proper T cell activation, proliferation, and production of effector molecules (14). Fatty acid oxidation (FAO) has been shown to be particularly active and important in TREG (24, 28), memory T cells, and in skin-resident TRM (29). Glutaminolysis, the process of breaking down glutamine to α-ketoglutarate which then enters the TCA cycle, has equally been observed to be crucial for T cell function (8). After activation, T cells upregulate their expression of glutamine transporters, increase glutamine uptake, and promote expression of enzymes required for glutaminolysis (13, 16, 30, 31). While glycolysis-derived pyruvate is a critical substrate for oxidative phosphorylation, these “alternative” substrates described above seem to be particularly important to fuel the TCA and thereby T cell function when there is limited availability of glucose (32). The importance of the TCA cycle, FAO, or glutaminolysis for T cell function is exemplified by studies showing that interference with either FAO or glutamine metabolism has the potential to alleviate pathology in T cell-mediated diseases (20, 33, 34).
An important function of the TCA cycle is to generate the electron carriers NADH and FADH2 by reducing NAD+ and FADH, respectively. These molecules are then oxidized at the inner membrane of the mitochondrion, which drives the electron transport chain (ETC) and finally generates ATP (Figure 4) (14). The ETC consists of a series of transmembrane proteins located on the inner mitochondrial membrane (IMM). Complex I and II of the ETC oxidize NADH and FADH2, respectively, and then donate the electrons obtained to complex III and later to complex IV, where they are finally transferred to molecular oxygen which generates water (10). This process is linked to the transfer of protons into the intermembrane space of the mitochondria, which creates an electrical charge between the opposite sides of the membrane. The last member of the ETC, the ATP synthase, finally uses this gradient to phosphorylate ADP to ATP, thus transferring protons back to the mitochondrial matrix. Through this process, OXPHOS mediates maximal usage of the energy stored in nutrient molecules such as glucose or fatty acids by generating 30–36 ATP molecules from one molecule of glucose and 106 ATP from 1 molecule of palmitate (10, 35). Full oxidation of NADH and FADH2 generated in the TCA, however, requires the presence of molecular oxygen to serve as acceptor of the electrons generated by the ETC reactions.
A cell's mitochondrial mass determines the amount of OXPHOS a cell can engage in. In response to metabolic stress, cells can increase their mitochondrial mass in a process referred to as mitochondrial biogenesis (36). In line with their increased functional repertoire, memory T cells feature increased mitochondrial mass and function when compared to naive T cells (14, 37, 38). This is represented by a higher spare respiratory capacity (SRC) of memory T cells. SRC reflects a cell's ability to increase OXPHOS for energy generation in response to metabolic stress or increased demand as present under hypoxic conditions, for instance (39). Thus, memory T cells are able to sustain ATP levels and thereby cellular function even under hypoxia. It is thought that this represents a metabolic adaptation to the challenges of immune surveillance in peripheral tissues under inflammatory conditions where nutrients and oxygen are scarce (14). Increased respiratory capacity of memory T cells is at least in part related to increased FAO in the mitochondria (40). T cells with defects in FAO have impaired memory T cell formation, whereas enhanced FAO leads to increases in SRC and memory T-cell generation (37). In circulating memory T cells, the lipids that fuel FAO seem to derive from triacylglycerols that are synthesized in the endoplasmic reticulum from glucose-derived carbon, and not from fatty acid uptake from the environment (41). Conversely, skin TRM seem to be specialized to take up fatty acids from the cutaneous environment and use them for FAO, which is crucial for their function and survival (29). More broadly, memory T cells critically depend on highly functional mitochondria for the mounting of recall responses, for rapid proliferation, calcium signaling, and effector function (14). It is thus not surprising that impaired mitochondrial function leads to decreased T-cell functionality, whereas promotion of mitochondrial function promotes memory T-cell generation and function in a number of infectious models and in T cell mediated diseases (14, 22, 42–44).
In addition to glucose, amino acids are crucial nutrients for T cells as well (8). Adequate supply of amino acids is critical for T cell growth and function. Amino acids are essential building blocks for protein synthesis, provide an important backbone for de novo nucleotide synthesis, and act as metabolic fuel source that can be fed into multiple pathways (45, 46). Therefore, T cells upregulate amino acids transporters and increase their uptake of amino acids immediately after activation (30, 31) which is an important step in the metabolic reprograming of T cells. Influx of branched chain amino acids such as leucine lead to activation of the mTORC1 pathway which is a key event in metabolic programing of T cells.
In agreement with a central role of amino acid metabolism in T cell biology, modulation of amino acid levels either by downregulation of transporters or by limiting amino acid availability strongly affects T cell activation and function (31, 47). For instance, T helper cells deficient for LAT1 (Slc7a5), a transporter for phenylalanine, tyrosine, leucine, arginine and tryptophan, are unable to differentiate into TH1 or TH17 cells, whereas T reg cell differentiation is still functional (31). This impaired amino acid metabolism has been linked to reduction in TORC1 activity and lower expression of Myc, which in turn impaired the upregulation of the metabolic machinery required for differentiation in activated T cells. Accordingly, therapeutic manipulation of amino acid metabolism such as glutamine metabolism has been shown to inhibit unwanted T cell responses in animal models of allograft rejection (20). Yet, it appears that manipulating amino acid metabolism alone is not sufficient to prevent allograft rejection, since other metabolic pathways had to be inhibited in parallel to achieve adequate T cell inhibition. Interestingly, however, inhibiting amino acid metabolism had reciprocal effects on T regs as compared to effector T cells, as these two T cell subsets differentially depend on it. As a consequence, interference with amino acid metabolism has the potential to generate antigen-specific regulatory T cells while impairing pathological effector T cell responses. Furthermore, amino acids may also directly affect T cell metabolism and function. Elevating L-arginine levels has been shown to induce a shift from glycolysis to oxidative phosphorylation in activated T cells. This was associated with enhanced generation of central memory-like cells with higher survival and function (48).
Lipids and fatty acids make up another group of crucial T cells substrates. They are important building blocks for cell membranes, store high amounts of energy, and provide substrates for cell signaling and post-translational modifications (8). Not surprisingly, T cells therefore induce cellular lipid biosynthesis pathways as an integral part of their activation program (14). These processes are coordinated, at least in part, by c-Myc and mTOR and transcriptionally regulated by sterol regulatory element-binding protein (SREBF) transcription factors (16, 49, 50). SREBFs transcription factors induce gene expression of enzymes and regulators of fatty acid synthesis and of the mevalonate pathways, which are involved in synthesis of cholesterol. Consequently, impairment of SREBF function or other regulators of cholesterol homeostasis leads to failure of T cells to efficiently proliferate (50, 51). Furthermore, the mevalonate pathway may also affect T cell differentiation and function, either by providing cholesterol precursor or indirectly via provision of isoprenoids (52).
Fatty acid synthesis also has important effects on T cell function. Activated T cells can take up fatty acids from the extracellular space or synthesize them de novo. Interestingly, different T cell subsets appear to rely differentially on de novo synthesized fatty acids. In particular, inhibition of fatty acid synthesis limits TH17 cell differentiation while promoting generation of TREG cells and these effects translated into improved disease outcomes in experimental autoimmune encephalomyelitis (EAE) (8, 28). However, even when T cells have defective de novo fatty acid synthesis (because of inhibition/deletion of ACC1 for instance), they can compensate for the lack of fatty acid synthesis if there are abundant amounts of fatty acids in the extracellular space, likely through fatty acid uptake (53). Indeed, skin-resident CD8+ TRM have a highly effective lipid uptake machinery and are able to metabolize fatty acids from the cutaneous environment via β-oxidation to support their long-term survival and function in the skin (29, 54). In addition, non-TRM effector T cells have been shown to engage in fatty acid oxidation, although the amount of FAO that occurs in effector T cells seems to be highly context dependent (8, 32).
The metabolic modules described above have to be finely tuned in order to support the diverse metabolic needs of T cells during different states of activation and memory formation. The metabolic pathways preferably used by T cells based on their activation and differentiation status are summarized in Table 1. Failure to adequately meet the demand in energy, biomolecules and other metabolic mediators leads to T cell dysfunction and may contribute to disease (14). Although the regulatory mechanisms that orchestrate these complex metabolic processes are incompletely understood, two metabolic regulators have been found to be central regulators of T cell metabolism: mTOR and AMPK (8).

Table 1. Metabolic pathways of T cell subsets according to differentiation and activation status.
Mechanistic target of rapamycin (mTOR) has emerged as a central integrator of environmental signals and cellular metabolic state to regulate T cell development, activation, and differentiation. The mTOR protein is a serine/threonine kinase that can form two different complexes, mTORC1 and mTORC2, depending on the specific scaffolding proteins it associates with (10). The different mTOR complexes perform different functions and are regulated by different stimuli. mTORC1 senses the availability of factors that are necessary for cell growth and cell division and links this information with the appropriate metabolic processes. Signals capable of activating mTORC1 are amino acids, oxygen, energy availability, and growth factors (56–59). In situations that are unfavorable for cell growth or division (low ATP levels, hypoxia, etc.), mTORC1 is not active and cell growth is inhibited. mTORC2, on the other hand, is stimulated by growth factors and cytokines while being irresponsive to nutrient-availability (10, 60). mTORC2 primarily regulates metabolism in support of cell survival and the cytoskeleton (60).
AMP-activated protein kinase (AMPK) acts to counterbalance the effects of mTOR. AMPK is an enzyme that functions as a sensor of cellular energy levels. It is activated by high AMP/ATP ratios, which result from shortage of nutrients or other forms of cellular stress. Consequently, activated AMPK downregulates energy consuming pathways such as biomolecule synthesis while upregulating those that generate energy, such as oxidation of glucose and fatty acids (10, 61). Mechanistically, AMPK has been shown to inhibit activation of mTOR (62). Given its central role in inhibiting cellular anabolism, it is not surprising that activation of AMPK suppresses the differentiation of T cells, as shown in a number of studies (10, 63–65). It has been proposed that activation of AMPK might specifically impair differentiation of TH1 and TH17 cells and that this effect is beneficial in animal models of T cell mediated inflammatory disease (10). However, in most of these study, it has not been addressed to what extend these AMPK-mediated effects are primarily T cell intrinsic or whether they include T cell independent effects of AMPK modulation. Furthermore, inhibitory effects of AMPK on T cell differentiation appear to depend on the metabolic environment. In fact, AMPK has been shown to be dispensable for T cell differentiation under normal metabolic conditions, since T cells which lack a functional AMPK are still able to acquire effector functions but fail to differentiate under suboptimal environmental conditions (32, 66). Taken together, AMPK might thus primarily function to mediate T cell adaptation to situations of low nutrient availability as encountered in hypoxic and glucose-deprived microenvironments of peripheral tissues under inflammatory conditions.
In recent years, research on T cell metabolism has attracted great attention and generated substantial insights into the intricate links between T cell function and T cell metabolism. Although many aspects of the complex mechanisms that link these central aspects of T cell biology remain to be elucidated, it is unquestioned that the therapeutic manipulation of T cell metabolism holds great potential in immune mediated disease. It seems possible that 1 day, overactive T cells in inflammatory disease can be curbed and exhausted T cells in cancer can be activated through manipulation of their metabolism. Before such therapies can be developed, however, many obstacles remain to be overcome, one of which is the fact that metabolism is a central function of all cells. Developing means to specifically target T cells will thus be a key step on the way to metabolism based T cell manipulation in health and disease.
As outlined above, a T cell's ability to survive, proliferate, and exert effector functions is highly dependent on its metabolic state. In turn, a T cell's metabolic state is highly dependent on the microenvironment, since it is the source from which T cells take up the nutrients they require for their proper functioning (32, 67). Studies in the field of cancer immunology have established that limited nutrient availability in the microenvironment can profoundly affect T cell effector functions (68). Conversely, high concentrations of tumor derived metabolites such as lactate can hinder activation of tumor-infiltrating T cells (69, 70). Although there is a lack of studies actually quantifying the metabolic microenvironment in inflammatory skin disease, it is likely that these interactions between T cells and the cutaneous environment of inflamed skin are equally important for T cell function.
Secondary lymphoid tissues such as the lymph nodes provide a nutrient-rich environment optimal for survival, activation, and growth of T cells (71, 72). However, once T cells migrate into peripheral tissues like the skin, they are forced to metabolically adapt to the local conditions, which may differ substantially in terms of nutrient availability (55). It is assumed that the microenvironment of the skin is restricted in many nutrients, particularly glucose, but is rich in lipids. While this is a reasonable assumption to make, based on the anatomy and biochemical make-up of the skin, it is important to note that cutaneous interstitial glucose, amino acid, or fatty acid levels have never been quantified comprehensively, neither under homeostatic nor under inflammatory conditions (73, 74). Future studies are needed to address this in more detail. At any rate, it is likely that T cells encounter a metabolic environment in the skin that is very different from that in blood or in lymphoid tissues and therefore have to adapt metabolically.
An outstanding example of such a metabolic adaptation is the specialization of skin TRM for uptake and β-oxidation of exogenous free fatty acids to support their long-term survival and function (29). The skin is a primary interface between the body and the environment and thus represents a site of first line defense against microbial pathogens, physical damage, and chemical harm (54). This “immunological hotbed” is populated by large numbers of sessile, non-migratory skin-resident T cells (skin TRM) which function to provide rapid on-site immune protection against known pathogens (4). Besides the skin, TRM cells are also found in all other major epithelial barrier tissues, such as the lung and the gastro-intestinal tract (55, 75, 76). In a landmark study by Pan et al., the transcriptional program of CD8+ TRM in skin was determined before and after their generation by vaccination with vaccinia virus (skin scarification). Strikingly, CD8+ TRM upregulated a transcriptional program that was strongly enriched for genes associated with uptake and metabolism of exogenous free fatty acids as they differentiated into bona fide TRM 90 days post-vaccination. Amongst the top upregulated genes were fatty acid binding proteins 4 and 5 (Fabp4/5), Cd36, and lipoprotein lipase (Lpl), all of which are genes involved in lipid uptake and fatty acid metabolism. The lipoprotein lipase Lpl cleaves triglycerides into free fatty acids and diacylglycerol. Cd36 is a lipid scavenger receptor able to bind and internalize free fatty acids. Fatty acid binding proteins are intracellular chaperones able to shuttle FFAs from the cytoplasm to the mitochondria for β-oxidation. This transcriptional signature of TRM was further confirmed to have important functional relevance. TRM were shown to have a higher capacity to take up free fatty acids from the environment and a higher capacity to perform mitochondrial fatty acid β-oxidation when compared to TCM or TEM. This was crucial for the proper functioning of TRM, as inhibition of these metabolic pathways resulted in decreased generation, persistence and function of TRM in the skin. For instance, pre-treatment of CD8+ T cells with etomoxir, an irreversible inhibitor of carnitine palmitoyltransferase 1 (CPT1a) which catalyzes the rate limiting step for mitochondrial fatty acid β-oxidation, strongly impaired the generation and maintenance of skin TRM.
In the study by Pan et al., the expression of Fabp4/5 in TRM was shown to be regulated by peroxisome proliferator-activated receptor gamma (PPARγ) (29). Pharmacological inhibition or genetic deletion of PPARγ in T cells resulted in decreased generation and function of TRM post-skin scarification. Interestingly, PPARγ is also increasingly implicated in the regulation of “pathogenic” TH2 cells in both humans and mice (77). Pathogenic TH2 cells are a subset of TH2 cells that contain all allergen-specific TH cells in humans (78), provide superior protection in models of parasite infection (79) and mediate tissue pathology in models of allergic inflammation (80). In addition to their dependence on PPARγ, pathogenic TH2 cells have been shown to express high levels of IL-9 (78). Intriguingly, IL-9 is a cytokine that is preferentially expressed by skin-homing and skin-resident TH cells (81, 82). Furthermore, this population of IL-9+ TH cells was recently found to express high levels of PPARγ which in turn acts as positive regulator of IL-9 expression in these cells (83). These PPARγ+ IL-9+ TH cells were further found to infiltrate lesions of allergic contact dermatitis, a prototypical TRM mediated disease (84). Taken together, PPARγ emerges as an important regulator of both tissue resident and pathogenic T cells. The underlying reason for this link might be that PPARγ confers a metabolic advantage to T cells that enter the cutaneous microenvironment by mediating metabolic adaptation. Given that PPARγ is a well-established drug target, it appears possible that both CD8+ and CD4+ TRM will 1 day be targeted via this metabolically relevant transcription factor (85).
Tissue inflammation is intricately linked to hypoxia (86). Hypoxia is a strong inducer of metabolic reprogramming in T cells and activation of the hypoxia response has been shown to cause T cell dysfunction and contribute to disease (87). In particular, hypoxia drives glycolysis to maintain ATP production, thus compensating for reduced OXPHOS activity under conditions of limited oxygen pressure (87). This process is critically regulated by the transcription factor HIF-1α, whose activation induces the expression of glycolytic enzymes such as hexokinase and phosphofructokinase, to ramp up glycolysis (see also section Regulation of T Cell Metabolism) (5, 88). Overactive glycolysis, in turn, has been identified as a hallmark of pathogenic T cells in inflammatory and autoimmune disease (20–22, 67). Activation of HIF-1α may also directly affect the balance between pathogenic TH17 cells and regulatory TREG cells. HIF-1α directly binds FOXP3 and promotes its proteasomal degradation, thereby impeding TREG differentiation. Conversely, HIF-1α directly activates RORγt and thus promotes IL-17 expression and TH17 differentiation (89). Addressing the hypoxia response in inflammatory skin disease might thus represent a promising new leverage point for therapy. In fact, certain drugs currently used to treat inflammatory skin disease may act via the modulation of the hypoxia response in T cells. For instance, dimethyl fumarate (DMF), used in the treatment of psoriasis, is thought to impair the HIF-1α-induced response by promoting its degradation (90). Inhibition of glycolysis by 2-Desoxy-D-glucose (2-DG) or other inhibitors of glycolysis might further help to restore the balance between TH17 and TREG cells in skin diseases such as psoriasis.
Just as T cells themselves, other cells of the skin organ also critically depend on nutrients for their functioning. Innate immune cells (91, 92), keratinocytes (93), endothelial cells (94, 95), stromal cells (96), amongst others, thus all engage in a competition for metabolites with T cells and thereby establish a bidirectional metabolic relationship (55). This competition will be especially active under inflammatory conditions, where cells proliferate and undergo metabolic reprograming. For instance, keratinocytes critically depend on increased glucose uptake via the glucose transporter Glut1 for injury- or inflammation-associated keratinocyte proliferation (93). Consequently, inactivation of Glut1 in keratinocytes decreased psoriasis-like inflammation in mouse models and in human psoriatic skin organoids. Interestingly, when Glut1 was uniquely deleted in keratinocytes by means of conditional knock-out, the immune response in the skin in animal models of psoriasis was not abrogated. Conversely, topical inhibition of glucose transport using a chemical GLUT1 inhibitor inhibited not only keratinocyte proliferation but also prevented infiltration and activation of immune cells. This discrepancy between genetic deletion of GLUT1 in keratinocytes and cell type-independent pharmacological inhibition of Glut1 suggests that this glucose transporter plays a crucial role in the facilitation of the immune response in psoriasis. Thus, inhibiting GLUT1 on T cells (and other immune cells) may be a highly effective therapeutic approach in psoriasis, given that GLUT1 is critical for T cell effector function (6). In fact, there is evidence that regulating glycolysis in T cells might be used to modulate autoimmunity. Overexpression of Glut1 in murine T cells boosts activation-induced production of IL-2 and IFN-γ. In mice carrying such T cells, accumulation of highly activated memory T cells was observed in conjunction with high levels of circulating IgG and deposit of IgG in the kidney. This suggests that Glut1 overexpression in T cells alone may contribute to autoimmunity (12). Conversely, knock-out of Glut1 in TH cells is sufficient to reduce their glycolytic activity, to impair generation of effector T cells, and to reduce inflammatory disease in vivo (6, 97).
Despite considerable recent advances in the field, many insights into the metabolic pathways critical for T cell function derive from in vitro T cell assays. However, these are reductionist representations of the complex metabolic situations in the microenvironment in vivo (68). Therefore, it remains to be discovered in more detail how exactly T cell immunometabolism is affected in inflammatory skin disease. Nevertheless, by observing how modulating the major metabolic modules affects experimental pathology, some interesting perspectives can be identified and are discussed below.
The mechanisms by which modulation of glycolysis in T cells affects inflammatory pathology has been investigated in some detail. Effector TH cells and CD8+ T cells show increased glycolytic activity as compared to naive T cells (16, 67), but not all TH cell lineages have equal glucose metabolism signatures. For instance, TH cells show reduced differentiation into TH1 and TH17 cells in vitro when glycolysis is inhibited, while inducible TREG differentiation is enhanced in low glucose conditions (24, 98). This affects TEFF/TREG balance which in turn has been related to reduced pathology in experimental autoimmune encephalomyelitis (98). To what extent modulation of glycolysis can be leveraged to specifically boost or inhibit distinct TH cell subsets is an active area of current research (67). In the context of skin inflammation, it will be crucial to consider the complex interplay between glucose availability in the tissue, glucose consumption by different skin-resident immune, stromal, and epithelial cell populations, and their differential glucose dependence for inflammatory reactions. A summary categorizing key metabolic properties of different T cell subsets in the context of inflammatory skin disease is shown in Table 2.
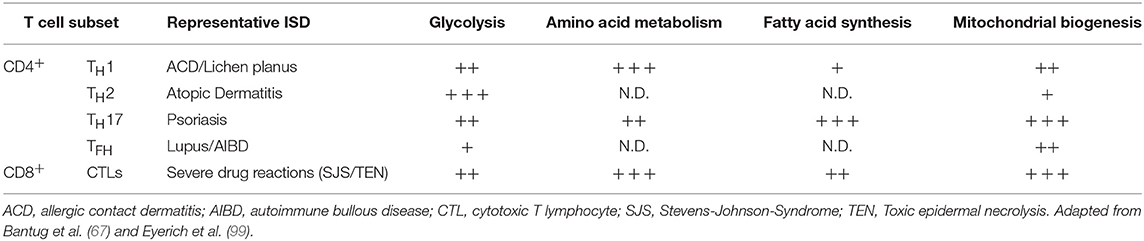
Table 2. T cell subsets in inflammatory skin disease (ISD) and their engagement of key metabolic pathways.
As outlined in more detail above, amino acids are another important substrate for T cell function. Availability of amino acids is likely to change significantly in inflamed skin, which in turn will shape T cell function. T cells require amino acids for protein and nucleic acid synthesis (67) and restriction of amino acids can differentially affect T cell activation via regulation of mTORC1 activation and other pathways (100). In the context of psoriasis, a TH17-driven skin disease, activating the amino acid starvation response through halofuginone or restricting amino acid flux into T cells might be of therapeutic interest, since it decreases TH17 differentiation, while leaving TH1 nor TH2 cell polarization unaffected (101, 102). Furthermore, glutamine availability for T cells also impacts differentiation of TH cells. If glutamine is restricted, TH cells polarized under TH1 conditions are skewed into a TREG phenotype, whilst again leaving TH2 polarization unaffected. Similar findings were made when uptake of glutamine into T cells is inhibited by genetic deletion of the glutamine transporter Slc1a5 (47). Taken together, interference with amino acid metabolism of T cells might be used to improve inflammatory skin disease, particularly psoriasis, as it has been shown to enable therapeutic intervention in multiple TH17-driven disease models (47, 67, 101). A number of molecules to inhibit cellular glutamine metabolism are in fact available, some of which are currently being tested clinically in oncological trials (see Table 3) (106, 108).
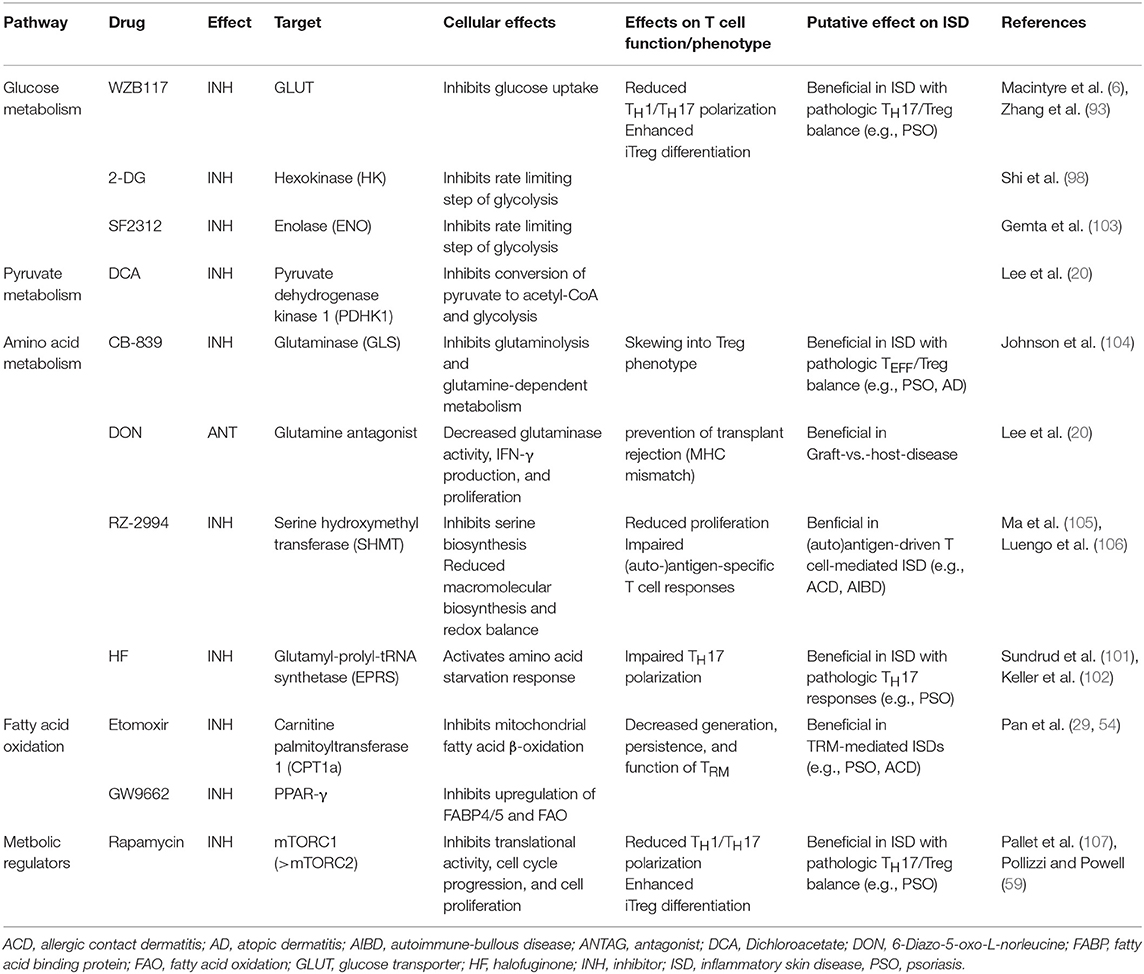
Table 3. Pharmacological modulators of T cell metabolism and their putative therapeutic application in inflammatory skin disease.
Modulation of T cell lipid metabolism might also be leveraged to improve inflammatory skin disease. Typically, skin diseases such as psoriasis and atopic dermatitis are characterized by exacerbated activation and proliferation of T cells in the skin (109). Proliferating cells depend on de novo fatty acid and cholesterol synthesis as building blocks for the plasma membrane and membranes of organelles. In this regard, there are intriguing differences in the dependence on de novo lipogenesis between TH17 cells and TREG cells. In particular, if fatty acid synthesis is inhibited by an inhibitor of acetyl-CoA Carboxylase 1 (ACC1, also known as ACACA), TH17 cell differentiation and proliferation is reduced while TREG cells are induced (28). ACC1 is an enzyme which catalyzes the carboxylation of acetyl-CoA to malonyl-CoA, the rate-limiting step in fatty acid synthesis. T cells that lack ACC1 show reduced de novo synthesis of palmitate and defective TH17 cell differentiation. This phenotype can be rescued by exogenous palmitate. Mice harboring ACC1-deficient T cells are less susceptible to EAE which underscores the crucial role of lipid metabolism in pathogenic TH17 cell responses and points to a novel therapeutic opportunity in psoriasis (Table 3).
Finally, the particular dependence of skin TRM on β-oxidation of exogenous free fatty acids from the cutaneous environment to fuel their metabolism might represent a weak spot that could be exploited therapeutically (29, 54). In fact, it is increasingly appreciated that chronic and/or aberrant activation of pathogenic TRM represents a key pathogenetic step in the onset and maintenance of many inflammatory skin diseases (3). Psoriasis (110), atopic dermatitis (111), vitiligo (112), cutaneous adverse drug eruption (113), and many other skin diseases have all been shown to be—at least in part—mediated by TRM. This provides an elegant explanation for the chronicity of T cell mediated skin diseases and their general tendency to relapse once immunosuppressive treatment is withdrawn. Thus, therapies that not only suppress but actually dislodge pathogenic TRM from their tissue niches in the skin may tackle these diseases at their very root and induce persistent remission. In mouse models, in vivo inhibition of mitochondrial β-oxidation with trimetazidine or etomoxir decreased the survival and maintenance of cutaneous TRM and hence show the fundamental feasibility of such therapeutic strategies (29). To what extend such approaches are transferrable to humans will have to be addressed by future studies.
Both severe psoriasis and severe atopic dermatitis are associated with diseases of the metabolic syndrome (114, 115). Although the reasons for this are incompletely understood, it is known that inflammation can modulate local and systemic metabolism and–vice versa local–that systemic metabolism can profoundly influence immune function (116, 117). Therefore, metabolism and immune function might interact in specific ways to drive aberrant inflammation in inflammatory skin disease. For instance, statins have been proposed to ameliorate psoriasis and their beneficial effects have been attributed to their effects on lipid metabolism in skin, as well as their proclaimed anti-inflammatory and immunomodulatory properties (118). In mice, it has indeed been shown that inhibition of HMG-CoA reductase has an inhibitory effect on TH17 cell differentiation (119). Future studies will have to address whether inhibition of HMG-CoA reductase with statins is truly beneficial in inflammatory skin diseases such as psoriasis and whether this potentially beneficial effect is mediated by interference with TH17 cell function. In fact, statins may also influence T cell function and inflammation via their inhibitory effect on mitochondrial function. Statins have been shown to inhibit mitochondrial complex III of the respiratory chain (120), leading to reduced generation of mitochondrial reactive oxygen species (mROS). Reduced levels of mROS, in turn, reduce activation of nuclear factor of activated T cells (NFAT) and IL-2 production in T cells (121), thus providing an additional putative mechanism by which statins may alleviate inflammatory skin disease.
In addition, dietary fatty acids from nutrition may also affect systemic immune responses and autoimmunity. In mice, dietary long-chain fatty acids enhance differentiation and proliferation of TH1 and TH17 cells whereas dietary short-chain FAs expanded gut TREG cells. These nutrition-dependent effects translated into exacerbated EAE via the expansion of pathogenic TH1 and/or TH17 cell populations (122). Furthermore, a diet high in saturated fatty acids is sufficient to exacerbate psoriatic skin inflammation independent of obesity (123) and this effect seems to be linked to an augmented TH17 response in the skin (124). Given that weight loss through diet adaptation can improve pre-existing psoriasis and prevent the onset of psoriasis in obese individuals it can be speculated that the beneficial effects thereof are at least in part mediated through an altered cross-talk between systemic metabolism and local T cell function (125). In addition to fatty acid metabolism, systemic amino acid levels may also affect T cell function in the skin. This has best been studied for the amino acid arginine which is an important modulator of T cell function (48, 126, 127). In animal models, increased dietary arginine enhanced T cell maturation and led to increased contact allergy reactions (128). It is thus possible that systemic and local cutaneous arginine levels may shape the immune response in inflammatory skin diseases.
At any rate, our understanding of the diverse processes that integrate with lymphocyte signaling, gene regulation, and function to shape T cell metabolism is still nascent. A better understanding of the metabolic regulation in T cells and the influence of nutrients in the microenvironment on T cell function will lead to novel insights into T cell function in the context of skin disease. This knowledge may open up new therapeutic approaches in the future.
LM has participated in writing the manuscript, researching the literature, and generating the tables and figures. CS has conceived the concept of the review and written the main part of the manuscript. NB has participated in revising the manuscript. This included adding of new text to the manuscript, revising the figures, and editing the existing parts of the manuscript.
This work was supported by the Peter Hans Hofschneider Professorship for Molecular Medicine and the Promedica Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MW and handling editor declared their shared affiliation.
1. Sallusto F, Lanzavecchia A. Heterogeneity of CD4+ memory T cells: functional modules for tailored immunity. Euro J Immunol. (2009) 39:2076–82. doi: 10.1002/eji.200939722
2. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who's who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol. (2013) 43:2797–809. doi: 10.1002/eji.201343751
3. Park CO, Kupper TS. The emerging role of resident memory T cells in protective immunity and inflammatory disease. Nat Med. (2015) 21:688–97. doi: 10.1038/nm.3883
4. Clark RA. Resident memory T cells in human health and disease. Sci Transl Med. (2015) 7:269rv261. doi: 10.1126/scitranslmed.3010641
5. Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic instruction of immunity. Cell. (2017) 169:570–86. doi: 10.1016/j.cell.2017.04.004
6. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
7. Warburg O. On the origin of cancer cells. Science. (1956) 123:309–14. doi: 10.1126/science.123.3191.309
8. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
9. Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. (2013) 342:1242454. doi: 10.1126/science.1242454
10. Almeida L, Lochner M, Berod L, Sparwasser T. Metabolic pathways in T cell activation and lineage differentiation. Sem. Immunol. (2016) 28:514–24. doi: 10.1016/j.smim.2016.10.009
11. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Ann Rev Cell Dev Biol. (2011) 27:441–64. doi: 10.1146/annurev-cellbio-092910-154237
12. Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
13. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. (2002) 16:769–77. doi: 10.1016/S1074-7613(02)00323-0
14. Dimeloe S, Burgener AV, Grahlert J, Hess C. T-cell metabolism governing activation, proliferation and differentiation; a modular view. Immunology. (2017) 150:35–44. doi: 10.1111/imm.12655
15. Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, et al. Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol. (2013) 14:1064–72. doi: 10.1038/ni.2687
16. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. (2011) 35:871–82. doi: 10.1016/j.immuni.2011.09.021
17. Michalek RD, Gerriets VA, Nichols AG, Inoue M, Kazmin D, Chang CY, et al. Estrogen-related receptor-alpha is a metabolic regulator of effector T-cell activation and differentiation. Proc Natl Acad Sci USA. (2011) 108:18348–53. doi: 10.1073/pnas.1108856108
18. Doedens AL, Phan AT, Stradner MH, Fujimoto JK, Nguyen JV, Yang E, et al. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat Immunol. (2013) 14:1173–82. doi: 10.1038/ni.2714
19. Chang CH, Curtis JD, Maggi LB, Faubert B, Villarino AV, O'Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. (2013) 153:1239–51. doi: 10.1016/j.cell.2013.05.016
20. Lee CF, Lo YC, Cheng CH, Furtmüller GJ, Oh B, Andrade-Oliveira V, et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep. (2015) 13:760–70. doi: 10.1016/j.celrep.2015.09.036
21. Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. (2015) 125:194–207. doi: 10.1172/JCI76012
22. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Trans Med. (2015) 7:274ra218. doi: 10.1126/scitranslmed.aaa0835
23. Yang Z, Fujii H, Mohan SV, Goronzy JJ, Weyand CM. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J Exp Med. (2013) 210:2119–34. doi: 10.1084/jem.20130252
24. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. (2011) 186:3299–303. doi: 10.4049/jimmunol.1003613
25. De Rosa V, Galgani M, Porcellini A, Colamatteo A, Santopaolo M, Zuchegna C, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. (2015) 16:1174–84. doi: 10.1038/ni.3269
26. Procaccini C, Carbone F, Di Silvestre D, Brambilla F, De Rosa V, Galgani M, et al. The proteomic landscape of human ex vivo regulatory and conventional T cells reveals specific metabolic requirements. Immunity. (2016) 44:712. doi: 10.1016/j.immuni.2016.02.022
27. Wettersten HI, Aboud OA, Lara PN Jr, Weiss RH. Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol. (2017) 13:410–9. doi: 10.1038/nrneph.2017.59
28. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med. (2014) 20:1327–33. doi: 10.1038/nm.3704
29. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. (2017) 543:252–6. doi: 10.1038/nature21379
30. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. (2010) 185:1037–44. doi: 10.4049/jimmunol.0903586
31. Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. (2013) 14:500–8. doi: 10.1038/ni.2556
32. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia-Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. (2015) 42:41–54. doi: 10.1016/j.immuni.2014.12.030
33. Byersdorfer CA, Tkachev V, Opipari AW, Goodell S, Swanson J, Sandquist S, et al. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood. (2013) 122:3230–7. doi: 10.1182/blood-2013-04-495515
34. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. (2016) 196:80–90. doi: 10.4049/jimmunol.1501537
35. Geltink RIK, Kyle RL, Pearce EL. Unraveling the complex interplay between T cell metabolism and function. Annu Rev Immunol. (2018) 36:461–88. doi: 10.1146/annurev-immunol-042617-053019
36. Sanchis-Gomar F, Garcia-Gimenez JL, Gomez-Cabrera MC, Pallardo FV. Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches. Curr Pharm Design. (2014) 20:5619–33. doi: 10.2174/1381612820666140306095106
37. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
38. Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U, et al. The immune-metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J Immunol. (2016) 196:106–14. doi: 10.4049/jimmunol.1501766
39. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. (2011) 435:297–312. doi: 10.1042/BJ20110162
40. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. (2009) 460:103–7. doi: 10.1038/nature08097
41. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8(+) T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. (2014) 41:75–88. doi: 10.1016/j.immuni.2014.06.005
42. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. (2016) 166:63–76. doi: 10.1016/j.cell.2016.05.035
43. Champagne DP, Hatle KM, Fortner KA, D'Alessandro A, Thornton TM, Yang R, et al. Fine-tuning of CD8(+) T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity. (2016) 44:1299–311. doi: 10.1016/j.immuni.2016.02.018
44. Gatza E, Wahl DR, Opipari AW, Sundberg TB, Reddy P, Liu C, et al. Manipulating the bioenergetics of alloreactive T cells causes their selective apoptosis and arrests graft-versus-host disease. Sci Trans Med. (2011) 3:67ra68. doi: 10.1126/scitranslmed.3001975
45. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) 16:619–34. doi: 10.1038/nrc.2016.71
46. Patel CH, Powell JD. Targeting T cell metabolism to regulate T cell activation, differentiation and function in disease. Curr Opin Immunol. (2017) 46:82–8. doi: 10.1016/j.coi.2017.04.006
47. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. (2014) 40:692–705. doi: 10.1016/j.immuni.2014.04.007
48. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. (2016) 167:829–42.e813. doi: 10.1016/j.cell.2016.09.031
49. Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. (2013) 39:1043–56. doi: 10.1016/j.immuni.2013.09.015
50. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP, et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. (2013) 14:489–99. doi: 10.1038/ni.2570
51. Bensinger SJ, Bradley MN, Joseph SB, Zelcer N, Janssen EM, Hausner MA, et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell. (2008) 134:97–111. doi: 10.1016/j.cell.2008.04.052
52. Youssef S, Stüve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. (2002) 420:78–84. doi: 10.1038/nature01158
53. Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol. (2014) 192:3190–3199. doi: 10.4049/jimmunol.1302985
54. Pan Y, Kupper TS. Metabolic reprogramming and longevity of tissue-resident memory T cells. Front Immunol. (2018) 9:1347. doi: 10.3389/fimmu.2018.01347
55. Veldhoen M, Blankenhaus B, Konjar S, Ferreira C. Metabolic wiring of murine T cell and intraepithelial lymphocyte maintenance and activation. Euro J Immunol. (2018) 48:1430–40. doi: 10.1002/eji.201646745
56. Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. (2016) 165:153–64. doi: 10.1016/j.cell.2016.02.035
57. Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, et al. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest. (2010) 90:762–73. doi: 10.1038/labinvest.2010.36
58. Waickman AT, Powell JD. Mammalian target of rapamycin integrates diverse inputs to guide the outcome of antigen recognition in T cells. J Immunol. (2012) 188:4721–9. doi: 10.4049/jimmunol.1103143
59. Pollizzi KN, Powell JD. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. (2015) 36:13–20. doi: 10.1016/j.it.2014.11.005
60. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. (2012) 149:274–93. doi: 10.1016/j.cell.2012.03.017
61. Steinberg GR, Carling D. AMP-activated protein kinase: the current landscape for drug development. Nat Rev Drug Disc. (2019) 18:1. doi: 10.1038/s41573-019-0019-2
62. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
63. Kang KY, Kim YK, Yi H, Kim J, Jung HR, Kim IJ, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol. (2013) 16:85–92. doi: 10.1016/j.intimp.2013.03.020
64. Bai A, Yong M, Ma AG, Ma Y, Weiss CR, Guan Q, et al. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J Pharmacol Exp Ther. (2010) 333:717–25. doi: 10.1124/jpet.109.164954
65. Lee SY, Lee SH, Yang EJ, Kim EK, Kim JK, Shin DY, et al. Metformin ameliorates inflammatory bowel disease by suppression of the STAT3 signaling pathway and regulation of the between Th17/treg balance. PLoS ONE. (2015) 10:e0135858. doi: 10.1371/journal.pone.0135858
66. Mayer A, Denanglaire S, Viollet B, Leo O, Andris F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Euro J Immunol. (2008) 38:948–56. doi: 10.1002/eji.200738045
67. Bantug GR, Galluzzi L, Kroemer G, Hess C. The spectrum of T cell metabolism in health and disease. Nat Rev Immunol. (2018) 18:19–34. doi: 10.1038/nri.2017.99
68. Ecker C, Riley JL. Translating in vitro T cell metabolic findings to in vivo tumor models of nutrient competition. Cell Metab. (2018) 28:190–5. doi: 10.1016/j.cmet.2018.07.009
69. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
70. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. (2007) 109:3812–9. doi: 10.1182/blood-2006-07-035972
71. Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. (2008) 29:848–62. doi: 10.1016/j.immuni.2008.11.002
72. Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol. (2010) 184:4062–8. doi: 10.4049/jimmunol.0903002
73. Zhang Y, Li Q, Rao E, Sun Y, Grossmann ME, Morris RJ, et al. Epidermal fatty acid binding protein promotes skin inflammation induced by high-fat diet. Immunity. (2015) 42:953–64. doi: 10.1016/j.immuni.2015.04.016
74. Khnykin D, Miner JH, Jahnsen F. Role of fatty acid transporters in epidermis: Implications for health and disease. Dermato Endocrinol. (2011) 3:53–61. doi: 10.4161/derm.3.2.14816
75. Konjar Š, Frising UC, Ferreira C, Hinterleitner R, Mayassi T, Zhang Q, et al. Mitochondria maintain controlled activation state of epithelial-resident T lymphocytes. Sci Immunol. (2018) 3:eaan2543. doi: 10.1126/sciimmunol.aan2543
76. Wang HC, Zhou Q, Dragoo J, Klein JR. Most murine CD8+ intestinal intraepithelial lymphocytes are partially but not fully activated T cells. J Immunol. (2002) 169:4717–22. doi: 10.4049/jimmunol.169.9.4717
77. Nobs SP, Kopf M. PPAR-gamma in innate and adaptive lung immunity. J Leukoc Biol. (2018) 104:737–41. doi: 10.1002/JLB.3MR0118-034R
78. Wambre E, Bajzik V, DeLong JH, O'Brien K, Nguyen QA, Speake C, et al. A phenotypically and functionally distinct human TH2 cell subpopulation is associated with allergic disorders. Sci Trans Med. (2017) 9:eaam9171. doi: 10.1126/scitranslmed.aam9171
79. Chen T, Tibbitt CA, Feng X, Stark JM, Rohrbeck L, Rausch L, et al. PPAR-gamma promotes type 2 immune responses in allergy and nematode infection. Sci Immunol. (2017) 2:aal5196. doi: 10.1126/sciimmunol.aal5196
80. Nobs SP, Natali S, Pohlmeier L, Okreglicka K, Schneider C, Kurrer M, et al. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J Exp Med. (2017) 214:3015–35. doi: 10.1084/jem.20162069
81. Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, et al. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Sci Trans Med. (2014) 6:219ra218. doi: 10.1126/scitranslmed.3007828
82. Clark RA, Schlapbach C. TH9 cells in skin disorders. Sem Immunopathol. (2017) 39:47–54. doi: 10.1007/s00281-016-0607-8
83. Micossé C, von Meyenn L, Steck O, Kipfer E, Adam C, Simillion C, et al. Human “TH9” cells are a subpopulation of PPAR-gamma(+) TH2 cells. Sci Immunol. (2019) 4:aat5943. doi: 10.1126/sciimmunol.aat5943
84. Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med. (2015) 21:647–53. doi: 10.1038/nm.3860
85. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66. doi: 10.1038/nm.3159
86. Corcoran SE, O'Neill LA. HIF1alpha and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
87. Fearon U, Canavan M, Biniecka M, Veale DJ. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat Rev Rheumatol. (2016) 12:385–97. doi: 10.1038/nrrheum.2016.69
88. Obach M, Navarro-Sabaté A, Caro J, Kong X, Duran J, Gómez M, et al. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem. (2004) 279:53562–70. doi: 10.1074/jbc.M406096200
89. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
90. Zhao G, Liu Y, Fang J, Chen Y, Li H, Gao K. Dimethyl fumarate inhibits the expression and function of hypoxia-inducible factor-1alpha (HIF-1alpha). Biochem Biophys Res Commun. (2014) 448:303–7. doi: 10.1016/j.bbrc.2014.02.062
91. Russell DG, Huang L, VanderVen BC. Immunometabolism at the interface between macrophages and pathogens. Nat Rev Immunol. (2019) 19:291–304. doi: 10.1038/s41577-019-0124-9
92. Caslin HL, Taruselli MT, Haque T, Pondicherry N, Baldwin EA, Barnstein BO, et al. Inhibiting glycolysis and ATP production attenuates IL-33-mediated mast cell function and peritonitis. Front Immunol. (2018) 9:3026. doi: 10.3389/fimmu.2018.03026
93. Zhang Z, Zi Z, Lee EE, Zhao J, Contreras DC, South AP, et al. Differential glucose requirement in skin homeostasis and injury identifies a therapeutic target for psoriasis. Nat Med. (2018) 24:617–27. doi: 10.1038/s41591-018-0003-0
94. Bierhansl L, Conradi LC, Treps L, Dewerchin M, Carmeliet P. Central role of metabolism in endothelial cell function and vascular disease. Physiology. (2017) 32:126–40. doi: 10.1152/physiol.00031.2016
95. Zecchin A, Kalucka J, Dubois C, Carmeliet P. How endothelial cells adapt their metabolism to form vessels in tumors. Front Immunol. (2017) 8:1750. doi: 10.3389/fimmu.2017.01750
96. Zhao X, Psarianos P, Ghoraie LS, Yip K, Goldstein D, Gilbert R, et al. Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat Metab. (2019) 1:147–57. doi: 10.1038/s42255-018-0008-5
97. Piotrowski JT, Gomez TS, Schoon RA, Mangalam AK, Billadeau DD. WASH knockout T cells demonstrate defective receptor trafficking, proliferation, and effector function. Mol Cell Biol. (2013) 33:958–73. doi: 10.1128/MCB.01288-12
98. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
99. Eyerich K, Eyerich S. Immune response patterns in non-communicable inflammatory skin diseases. J Eur Acad Dermatol Venereol. (2018) 32:692–703. doi: 10.1111/jdv.14673
100. Kolev M, Dimeloe S, Le Friec G, Navarini A, Arbore G, Povoleri GA, et al. Complement regulates nutrient influx and metabolic reprogramming during Th1 cell responses. Immunity. (2015) 42:1033–47. doi: 10.1016/j.immuni.2015.05.024
101. Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. (2009) 324:1334–8. doi: 10.1126/science.1172638
102. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol. (2012) 8:311–7. doi: 10.1038/nchembio.790
103. Gemta LF, Siska PJ, Nelson ME, Gao X, Liu X, Locasale JW, et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci Immunol. (2019) 4:aap9520. doi: 10.1126/sciimmunol.aap9520
104. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175:1780–95.e1719. doi: 10.1016/j.cell.2018.10.001
105. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. (2017) 25:345–57. doi: 10.1016/j.cmet.2016.12.011
106. Luengo A, Gui DY, Vander Heiden MG. Targeting metabolism for cancer therapy. Cell Chem Biol. (2017) 24:1161–80. doi: 10.1016/j.chembiol.2017.08.028
107. Pallet N, Fernandez-Ramos AA, Loriot MA. Impact of immunosuppressive drugs on the metabolism of T cells. Int Rev Cell Mol Biol. (2018) 341:169–200. doi: 10.1016/bs.ircmb.2018.05.009
108. Choi YK, Park KG. Targeting glutamine metabolism for cancer treatment. Biomol Therap. (2018) 26:19–28. doi: 10.4062/biomolther.2017.178
109. Ho AW, Kupper TS. T cells and the skin: from protective immunity to inflammatory skin disorders. Nat Rev Immunol. (2019).
110. Matos TR, O'Malley JT, Lowry EL, Hamm D, Kirsch IR, Robins HS, et al. Clinically resolved psoriatic lesions contain psoriasis-specific IL-17-producing alphabeta T cell clones. J Clin Invest. (2017) 127:4031–41. doi: 10.1172/JCI93396
111. Brunner PM, Emerson RO, Tipton C, Garcet S, Khattri S, Coats I, et al. Nonlesional atopic dermatitis skin shares similar T-cell clones with lesional tissues. Allergy. (2017) 72:2017–25. doi: 10.1111/all.13223
112. Boniface K, Seneschal J. Vitiligo as a skin memory disease: the need for early intervention with immunomodulating agents and a maintenance therapy to target resident memory T cells. Exp Dermatol. (2019) 28:656–61. doi: 10.1111/exd.13879
113. Hoetzenecker W, Nägeli M, Mehra ET, Jensen AN, Saulite I, Schmid-Grendelmeier P, et al. Adverse cutaneous drug eruptions: current understanding. Semin Immunopathol. (2016) 38:75–86. doi: 10.1007/s00281-015-0540-2
114. Shalom G, Dreiher J, Kridin K, Horev A, Khoury R, Battat E, et al. Atopic dermatitis and the metabolic syndrome: a cross-sectional study of 116 816 patients. J Eur Acad Dermatol Venereol. (2019) 33:1762–7. doi: 10.1111/jdv.15642
115. Boehncke WH, Schon MP. Psoriasis. Lancet. (2015) 386:983–94. doi: 10.1016/S0140-6736(14)61909-7
116. Zhou H, Liu F. Regulation, communication, and functional roles of adipose tissue-resident CD4(+) T cells in the control of metabolic homeostasis. Front Immunol. (2018) 9:1961. doi: 10.3389/fimmu.2018.01961
117. Ganguly D. Do Type I interferons link systemic autoimmunities and metabolic syndrome in a pathogenetic continuum? Trends Immunol. (2018) 39:28–43. doi: 10.1016/j.it.2017.07.001
118. Mosiewicz J, Pietrzak A, Chodorowska G, Trojnar M, Szepietowski J, Reich K, et al. Rational for statin use in psoriatic patients. Arch Dermatol Res. (2013) 305:467–72. doi: 10.1007/s00403-013-1374-1
119. Zhang X, Tao Y, Troiani L, Markovic-Plese S. Simvastatin inhibits IFN regulatory factor 4 expression and Th17 cell differentiation in CD4+ T cells derived from patients with multiple sclerosis. J Immunol. (2011) 187:3431–7. doi: 10.4049/jimmunol.1100580
120. Schirris TJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BG, et al. Statin-induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab. (2015) 22:399–407. doi: 10.1016/j.cmet.2015.08.002
121. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
122. Haghikia A, Jörg S, Duscha A, Berg J, Manzel A, Waschbisch A, et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. (2015) 43:817–29. doi: 10.1016/j.immuni.2015.09.007
123. Herbert D, Franz S, Popkova Y, Anderegg U, Schiller J, Schwede K, et al. High-fat diet exacerbates early psoriatic skin inflammation independent of obesity: saturated fatty acids as key players. J Invest Dermatol. (2018) 138:1999–2009. doi: 10.1016/j.jid.2018.03.1522
124. Higashi Y, Yamakuchi M, Fukushige T, Ibusuki A, Hashiguchi T, Kanekura T. High-fat diet exacerbates imiquimod-induced psoriasis-like dermatitis in mice. Exp. Dermatol. (2018) 27:178–84. doi: 10.1111/exd.13484
125. Mahil SK, McSweeney SM, Kloczko E, McGowan B, Barker JN, Smith CH. Does weight loss reduce the severity and incidence of psoriasis or psoriatic arthritis? A critically appraised topic. Br J Dermatol. (2019). doi: 10.1111/bjd.17741. [Epub ahead of print].
126. Suwanpradid J, Shih M, Pontius L, Yang B, Birukova A, Guttman-Yassky E, et al. Arginase1 deficiency in monocytes/macrophages upregulates inducible nitric oxide synthase to promote cutaneous contact hypersensitivity. J Immunol. (2017) 199:1827–34. doi: 10.4049/jimmunol.1700739
127. Cao LY, Chung JS, Teshima T, Feigenbaum L, Cruz PD, Jacobe HT, et al. Myeloid-derived suppressor cells in psoriasis are an expanded population exhibiting diverse T-cell-suppressor mechanisms. J Invest Dermatol. (2016) 136:1801–10. doi: 10.1016/j.jid.2016.02.816
Keywords: T cell metabolism, inflammatory skin disease, glycolysis, OXPHOS, lipid metabolism
Citation: von Meyenn L, Bertschi NL and Schlapbach C (2019) Targeting T Cell Metabolism in Inflammatory Skin Disease. Front. Immunol. 10:2285. doi: 10.3389/fimmu.2019.02285
Received: 31 May 2019; Accepted: 10 September 2019;
Published: 24 September 2019.
Edited by:
Christian David Sadik, Universität zu Lübeck, GermanyReviewed by:
Amanda S. MacLeod, Duke University, United StatesCopyright © 2019 von Meyenn, Bertschi and Schlapbach. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christoph Schlapbach, Y2hyaXN0b3BoLnNjaGxhcGJhY2hAaW5zZWwuY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.