
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 September 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.02099
 Sachin Kumar1*
Sachin Kumar1* Madhu Dikshit2*
Madhu Dikshit2*Neutrophils are the most abundant, short lived, and terminally differentiated leukocytes with distinct tiers of arsenals to counter pathogens. Neutrophils were traditionally considered transcriptionally inactive cells, but recent researches in the field led to a paradigm shift in neutrophil biology and revealed subpopulation heterogeneity, and functions pivotal to immunity and inflammation. Furthermore, recent unfolding of metabolic plasticity in neutrophils has challenged the long-standing concept of their sole dependence on glycolytic pathway. Metabolic adaptations and distinct regulations have been identified which are critical for neutrophil differentiation and functions. The metabolic reprogramming of neutrophils by inflammatory mediators or during pathologies such as sepsis, diabetes, glucose-6-phosphate dehydrogenase deficiency, glycogen storage diseases (GSDs), systemic lupus erythematosus (SLE), rheumatoid arthritis, and cancer are now being explored. In this review, we discuss recent developments in understanding of the metabolic regulation, that may provide clues for better management and newer therapeutic opportunities for neutrophil centric immuno-deficiencies and inflammatory disorders.
Neutrophils, the most abundant leukocytes in human, are commonly considered as terminally differentiated effector cells (1–3). These cells reach early at the site of insult to diminish intruders and further direct the adaptive immune responses (2, 4). Indeed neutropenic patients exhibit higher susceptibility to recurrent infections (2, 3). While, aberrant neutrophil responses also lead to tissue damage and are associated with pathological conditions like sepsis, asthma, ischemia-reperfusion injury, and rheumatoid arthritis (2, 3). Neutrophils are produced in the bone marrow through a tightly regulated process of granulopoiesis. Neutrophils perform diverse functions including phagocytosis, oxidative burst, neutrophil extracellular traps (NETs) to execute microbial killing (2, 5, 6). These functions often rely on cytoskeleton reorganization and energy, but for-long neutrophils were believed to depend mainly on glycolytic metabolism (7, 8). Successively, most of the neutrophil functions were studied only in the presence of glucose that delayed identification of other metabolic pathways in neutrophil biology.
Interestingly in the last decade, a mere dumb suicidal killer view about these cells was challenged due to the identification of a number of novel functions and their crosstalk with other immune cells (1–3, 9). Most vibrant and prominent change in the field of neutrophil biology has been the identification of distinct subset heterogeneity and cellular plasticity (10–12), that for a long time remained ambiguous possibly because of their short-lived nature and high turnover. Furthermore, though initially considered as transcriptionally inactive, recent research in the field has also suggested transcriptional regulations even in the mature neutrophils during their transit to blood, under inflammatory environment, activation, and NETosis (13–15). Furthermore, new findings have strengthened the concept that neutrophils are critical regulator of immunity.
Cellular metabolism plays a decisive role in the function and plasticity of diverse immune cells including T cells and macrophages (16, 17), though metabolic regulations in neutrophil biology are continuously unfolding. Recent studies seem to challenge the commitment of neutrophils only to glycolysis, as different metabolic routes (Figure 1) including tricarboxylic acid (TCA) cycle (also known as Krebs cycle), oxidative phosphorylation (OXPHOS), pentose phosphate pathway (PPP), fatty acid oxidation (FAO) are being recognized to fulfill the energetic, biosynthetic, and functional requirements of neutrophils (18–23). Importantly, these cells are exposed to a variety of metabolic fuels in different organs and also at the site of infection/inflammation. Neutrophils use carbohydrates, proteins, lipids, as well as amino-acids for energy productions and uptake them by diverse transporters, and also respond to nutrients by binding to the receptors (24). Neutrophils in respond to diverse stimuli by enhancing the uptake of glucose, suggesting functional role of glucose metabolism in modulating neutrophil functions. Neutrophils exhibit differences in the expression of GLUT1, GLUT3, and GLUT4 under resting and stimulated conditions (25). Neutrophils also express cholesterol and lipid receptors including LDL receptor (LDL-R), scavenger receptor and cholesterol efflux transporter like ATP-binding cassette transporters ABCA1 and ABCG1, that regulate neutrophil adhesion and activation (26, 27). Recent studies have identified role of free fatty acid signaling in neutrophil activation and functions. Various fatty acid receptors including free fatty acid receptor-1 (FFAR1/GPR40), free fatty acid receptor 2 (FFAR2/GPR43), and GPR84 that function against LCFA (>C12), SCFA (C2-C6), and MCFA (C7-C12) are expressed on neutrophils (28). Neutrophils also express Toll-like receptors to detect pathogenic stimuli and also lipoproteins (29). In addition, role of glutamine and arginine are investigated for their effect on neutrophil functions, while others have not received much attention. Still, information on these surface receptors and transporters in neutrophil metabolic adaptations remain limited. Hopefully technical advancements in the field in relation to metabolomics will pave way for the better understanding of neutrophil production, differentiation and pivotal functions viz a viz neutrophil metabolism. Recent developments in the field suggest that neutrophils adapt to different metabolic pathways in the presence of inflammatory signals or under diverse disease conditions (18, 21). This review focuses on key neutrophil functions and their dependence on different metabolic pathways. We further discuss that how does metabolic reprogramming impact neutrophil biology under different environments or disease conditions.
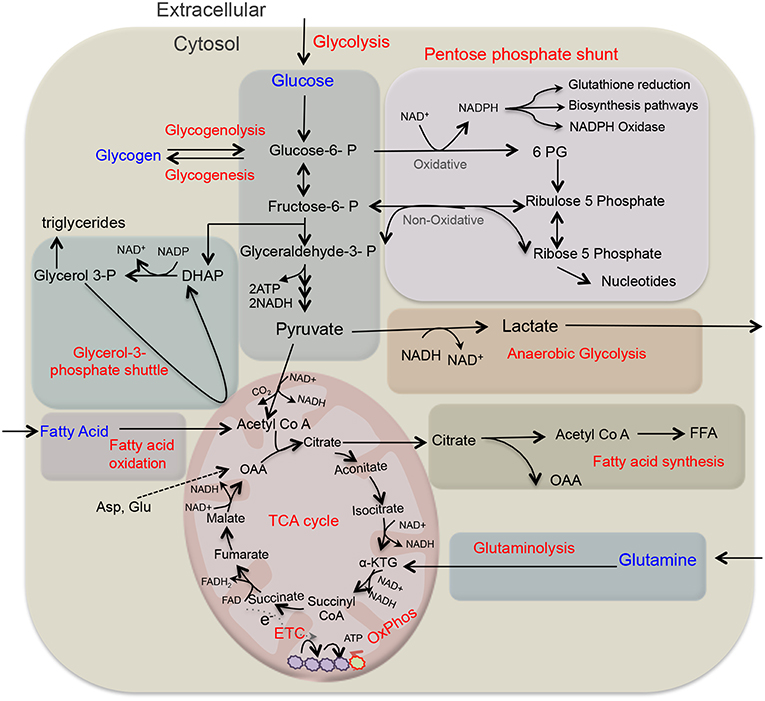
Figure 1. Overview of key metabolic pathways in neutrophils. Glycolysis is a major metabolic pathway in neutrophil cytosol, which converts glucose to pyruvate through a series of enzymes and reaction. In the absence of oxygen through anaerobic glycolysis, pyruvate gets reduce to lactate, and secreted out. While in the presence of oxygen, pyruvate participates in TCA cycle by its conversion to acetyl-coenzyme A (acetyl Co A) that provides reducing energy intermediates NADH and FADH2 which trough ETC generate ATPs. Neutrophils also utilize additional pentose-phosphate pathway (PPP) by using glucose-6-phosphate, an intermediate of the glycolytic pathway, as an entry point and using oxidative and non-oxidative phases produce NADPH and riboses that subsequently generate nucleotides. NADPH is critical as key NADPH oxidase dependent ROS generation in neutrophils and also regulates redox signaling. Depending on glucose abundance, glycogen stores get enriched in neutrophils that on-demand provides glucose based glycolytic intermediate supply. TCA cycle intermediate citrate through fatty acid synthesis (FAS) can lead to the generation of free fatty acid, while these endogenous FFA or transported from the extracellular environment through FAO pathway yields acetyl-CoA that fuels TCA cycle and produce significantly more energy in form of ATPs. Glutamine through glutaminolysis produces α-ketoglutarate and thus supports the TCA cycle. In addition, neutrophils also utilize glycerol-3- phosphate shuttle to generate NAD+ from NADH and helps in maintaining mitochondrial membrane potential. ATP, adenosine triphosphate; ETC, electron transport chain; FAO, fatty acid oxidation; NADPH, the reduced form of nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species.
Recent studies identified that neutrophils by utilizing various metabolic intermediates, formed by different metabolic pathways operative in the cytosol and mitochondria, meet their energy requirements (18, 20, 30–33). Though these metabolic pathways have been extensively reviewed earlier (16, 17), here we briefly summarized key metabolic routes important during neutrophil differentiation and functions (Table 1). During glycolysis, glucose is enzymatically converted to pyruvate in the cytosol, providing relatively low level of ATP and NADH. Under anaerobic condition, lactate is formed from pyruvate, and this conversion is catalyzed by the enzyme lactate dehydrogenase. While in the presence of oxygen, pyruvate forms acetyl coenzyme A, which enters TCA cycle in the mitochondria to efficiently generate energy (ATP) through electron transport chain (ETC) (16, 17). The PPP or hexose monophosphate (HMP) shunt utilizes glycolytic intermediate glucose-6-phosphate to generate ribose- 5-phosphate and NADPH in the cytosol. NADPH is a key substrate for NADPH oxidase (NOX2) to sustain ROS production by the activated neutrophils (40). Under fasting conditions or due to limited availability of glucose, cells adapt to fatty acid metabolism. Mitochondrial fatty acid β-oxidation (FAO) converts fatty acids to acyl-CoAs to yield acetyl-CoA that enters the TCA cycle. Glutamine is yet another metabolic substrate utilized by neutrophils particularly under glucose-limiting conditions (41–43). Glutaminolysis via glutamate dehydrogenase forms α-ketoglutarate that fuels TCA cycle (41). Metabolic plasticity during diverse stress thus helps to meet the energy requirement by efficiently diverting the intermediate metabolites being generated by the operative metabolic pathways (44).
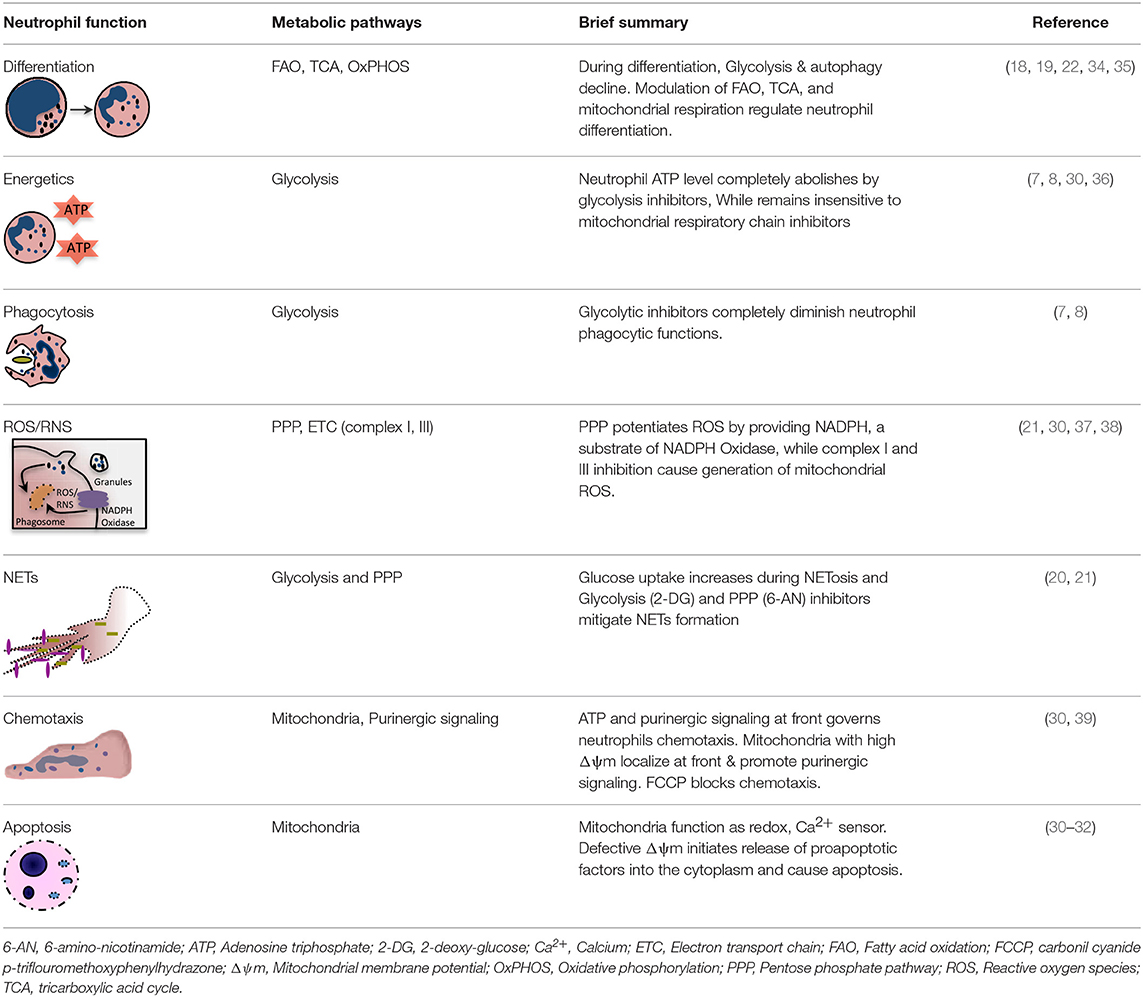
Table 1. Key metabolic pathways regulating distinct neutrophil functions.
Furthermore, the role of mitochondria has been remained less understood in neutrophils and is discussed here. Initial investigations using electron microcopy have observed very few functional mitochondria in mature neutrophils (45, 46). Later other studies have implied the dispensable role of mitochondria for respiration, or energy production, as mitochondrial respiration inhibitors failed to alter ATP levels in neutrophils (8, 32, 36). As neutrophils utilize oxygen mainly for NOX-2 dependent superoxide generation, functional investigations on mitochondrial involvement have remained challenging. Recent advancements in mitochondrial probes, Seahorse assay for oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) analysis and metabolomics methods are identifying novel role of mitochondria in neutrophil biology. Consistently, neutrophils contain significantly low levels of oxidative phosphorylation complexes (47) and mitochondrial enzymes including glutamate dehydrogenase (GDH) and fumarase (32). In contrast, Fossati et al. have challenged the view of so-called few functional mitochondria in neutrophils, by using MitoTracker Red/JC-1 and demonstrated presence of a complex mitochondrial network in the neutrophils (30). This study has revived the thrust of research on mitochondria and identified specific and differential role of mitochondria in respiratory burst, chemotaxis, and apoptosis (30). Various studies have also demonstrated role of purinergic signaling (derived from purine nucleotide/nucleoside analogs such as adenosine and ATP) in the activation, migration, and other functions of neutrophils (48–50). Together, functional role of mitochondria as the redox regulator, rheostat for survival and migration have been identified in neutrophils (30). These functions and their metabolic regulation are being further discussed in the following sections.
Neutrophils, the highly dynamic cells, perform different functions in diverse microenvironments. Though the glycolytic view prevailed very long for neutrophils, recent researches have however, identified unique metabolic decisions of neutrophils during differentiation, survival, and apoptosis. Multiple approaches using diverse substrates, inhibitors, and/or knockout of genes affecting metabolism have identified utility of different metabolic pathways during neutrophil development and various functions (18–23). Key proteins and genes that are involved in the metabolic regulation during neutrophil development and performance of various functions are listed in Table 2. Specific modulations of TCA cycle, ETC, PPP, glutaminolysis, and fatty acid metabolic pathways seem to regulate neutrophils under patho-physiological settings. Following sections describe major metabolic requirements of neutrophils during development, functions, and survival.
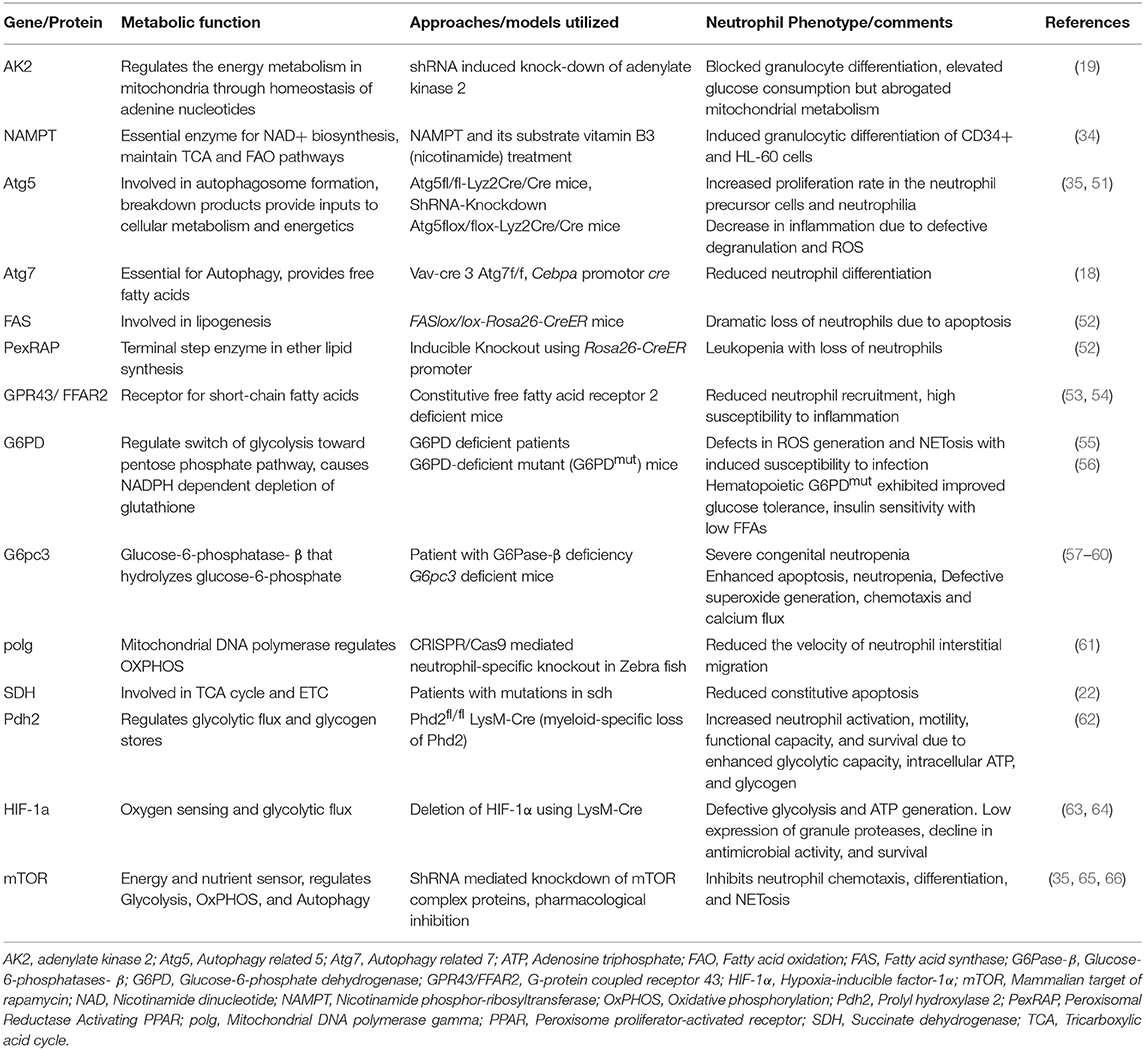
Table 2. Summary of distinct genes/proteins involved in metabolism and regulating neutrophil functions.
Neutrophils are normally produced in the bone marrow at a very high rate of 0.5–1.0 × 1011 cells per day through a highly controlled process of granulopoiesis (67) that includes generation of granulocyte-monocyte progenitors (GMPs) from hematopoietic stem cells (HSCs) and multipotent progenitors (MPPs). GMPs generate mature neutrophils through distinct developmental stages like myeloblasts (MBs), promyelocytes (PMs), myelocytes (MCs), metamyelocytes (MMs), band cells (BCs) (67). Interestingly, neutrophil differentiation can be drastically increased under stress or infection conditions, that is critical to maintain patho-physiological demands. Several lines of evidence suggested distinct metabolic regulation of neutrophil development with specific role of mitochondrial respiration, TCA cycle and oxidative phosphorylation (OXPHOS) in terminally differentiated neutrophils (19, 22, 34). In particular, deficiency of adenylate kinase 2 (AK2), that regulates adenine nucleotides homeostasis, exhibited defective mitochondrial activity (19). AK2 deficiency blocked granulocyte differentiation in promyelocytic HL60 cell line that was associated with elevated glucose consumption and significant accumulation of lactate and pyruvate suggesting a metabolic shift in the granulocyte progenitors leading to differentiation block (19). In another study, nicotinamide phosphor-ribosyltransferase (NAMPT), an essential enzyme for nicotinamide dinucleotide (NAD+) biosynthesis, maintained TCA and fatty acid oxidation (FAO) metabolic pathways, and triggered G-CSF induced granulopoiesis (34). Furthermore, NAMPT substrate vitamin B3 (a metabolic precursor of NAD) also induced neutrophilic differentiation, suggesting a decisive role of the NAD+ metabolic pathway in neutrophil development (34).
The role of FAO in neutrophil differentiation has also been observed through autophagy, a key process of auto-degradation that provides free fatty acids (FFA) (18, 35, 51). Interestingly, autophagic activity was observed to consistently reduce during neutrophil differentiation (18, 35). Deficiency of Atg5 (Autophagy related 5 protein) in neutrophilic lineage exhibited increased and more rapid neutrophil production leading to neutrophilia in vivo, without affecting functions like phagocytosis, ROS generation, and migration (35). In contrast, Bhattacharya et al. suggested reduced degranulation, ROS production and neutrophilic inflammation (51). These conflicting findings might be due to the utilization of different experimental approaches, emphasizing for better understanding of the role of autophagy in neutrophil differentiation. In a recent study, Riffelmacher et al. utilized Cebpa promotor cre to delete autophagy gene Atg7 at GMP-MB-stage in neutrophil precursors to evaluate early granulopoiesis and avoiding any HSC level defect. Atg7 deletion also led to neutrophilia with an accumulation of immature neutrophils, as deficiency of Atg7 or Atg5 caused differentiation defect in the earliest committed neutrophil precursors (MBs-MCs) (18). Further investigations focusing on metabolic regulation during neutrophil development and differentiation revealed a decline in most of the genes involved in glycolytic pathway from myeloblasts (MBs) to neutrophils, while mitochondrial content up-surged 2 fold during neutrophil differentiation (18), this was surprising and in contrast to the glycolytic view of mature neutrophils. Atg7 deficient neutrophil precursors exhibited increased glycolytic activity but impaired mitochondrial respiration, decreased ATP production, and accumulated lipid droplets, again suggesting autophagy dependent metabolism switch from glycolysis to FAO and mitochondrial respiration during neutrophil development (18). Consistently, conditional ablation of Atg7 also depleted FFA like palmitic, oleic, and eicosanoic acids that induced neutrophilia due to increase in immature neutrophils with inefficient phagocytosis and bacterial killing. Moreover, inhibition of FAO using etomoxir also led to the accumulation of lipid droplets and suppression of neutrophil maturation. Similarly, inhibition of autophagy-mediated lipid degradation using lipase inhibitors DEUP/orlistat caused a glycolytic shift in the metabolism and led to defective neutrophil differentiation. Importantly, administration of fatty acids or pyruvate that fuel the mitochondrial respiration rescued differentiation of autophagy-deficient neutrophil precursors (18). These findings thus identified importance of oxidative phosphorylation/mitochondrial respiration and fatty acid metabolism during neutrophil development and maturation.
A characteristic function of phagocytic cells, such as neutrophils and macrophages, is intricately regulated by various membrane receptors and intracellular signaling. During phagocytosis, receptor-mediated uptake of an opsonized pathogen into plasma membrane-derived vacuoles, further lead to the fusion of granules and lysosomes with phagosomal membrane to deliver anti-microbial peptides and proteases.
Early studies have suggested that neutrophil phagocytic functions predominantly depend on glycolytic pathway, based on their sensitivity to glycolysis inhibitors, sodium iodoacetate (SIA) and sodium fluoride and insensitivity to mitochondrial respiratory chain inhibitors, potassium cyanide (KCN), and antimycin A (7, 8). Furthermore, neutrophils depend on glucose for the energy requirement, as both sodium iodoacetate and 2-deoxyglucose (2-DG) diminished ATP levels. 2-DG competitively blocked the production of glucose-6-phosphate (G6P) thus both glycolysis and PPP pathways were inhibited, while SIA irreversibly inhibited glyceraldehyde-3-phosphate dehydrogenase (GAPDH) enzyme thus only glycolytic pathway was stopped. Importantly antimycin A, sodium azide, or cyanide had a negligible effect on ATP generation and oxygen consumption in phagocytizing neutrophils (7, 8), suggesting a dispensable role of mitochondrial metabolism in phagocytosis. Consistently, Fossati et al. have also observed that phagocytosis remains un-affected in the presence of FCCP or oligomycin, that disrupt mitochondrial membrane potential and Fo-ATPase, respectively (30). Interestingly, inflammatory environment that is commonly associated with low oxygen tension seems to sustain neutrophils on glycolysis (8). Phagocytosis also depends on stored glycogen in the neutrophils (7) so as to provide glucose through glycogenolysis for phagocytic functions (7, 8, 30, 68). Molecular regulations of glycogenolysis, glucose uptake, and glucose utilization in phagocytosing neutrophils warrant further investigations for better understanding.
Respiratory burst or superoxide () generation by the neutrophils is catalyzed by NOX-2, a multi-subunit enzymatic complex by utilizing oxygen and NADPH (5, 69, 70). Importantly, patients with the chronic granulomatous disease (CGD) that exhibit functional mutations in NOX2 subunit, counter life-threatening infections (70). NOX-2 activation involves translocation of cytoplasmic subunits including p47phox to the membrane. Rac 2, a protein activated only in GTP bound state initiates the NOX-2 activation and is an energy dependent process. NADPH, the NOX-2 substrate is produced by PPP utilizing G-6-P, an intermediate generated by glycolysis or glutaminolysis. A few studies have implied the additional role of mitochondria in ROS production, as complex I inhibition with rotenone or metformin and mitigation of complex III with myxothiazol or antimycin A, lead to an increase in superoxide and hydrogen peroxide generation (37, 38).
Neutrophils primarily depend on a rapid consumption of oxygen by NOX2 system for superoxide and ROS generation, thus ROS production can be intriguingly mimicked by OCR in neutrophils (71). Diphenyleneiodonium (DPI), a NOX-2 inhibitor and 2DG, inhibitor of glycolysis completely suppress PMA dependent increase in OCR (71), thus confirming role of glycolysis in NOX-2 activation/ROS generation. Rice et al. recently dissected involvement of diverse metabolic pathways in ROS generation (72). 2DG mediated block of glycolysis only reduced the initial-earlier phase OCR, in contrast the later phase of OCR profile remained unaffected. Interestingly, mitochondrial inhibition using rotenone/antimycin A, FCCP, and fatty acid metabolism inhibition by etomoxir, led to dramatic reductions in OCR during 2DG treatment (72). This suggests requirement of glucose metabolism for the early phase of high and intense ROS production, while both fatty acid metabolism and mitochondrial function facilitated prolonged H2O2 production during the late phase.
Neutrophils from glycogen storage disease (GSDs) patients having glucose-6-phosphate transporter (G6PT) deficiency also exhibited impaired ROS generation, suggesting the importance of glucose homeostasis in NOX-2 activation (57). Interestingly, neutrophils meet their requirement of energy by utilizing different metabolic intermediates/nutrients during phagocytosis and chemotaxis (68, 73). Various soluble chemotaxins accelerate the transmembrane glucose uptake without affecting stored endogenous glycogen (73). In a recent study, glycolytic enzyme 6-phosphofructo-2-kinase (PFK-2) was found to localize with NOX-2 following neutrophil activation. While inhibition/depletion of PFK-2 reduced the rate of glycolysis and also NOX2 activity (74). Surprisingly, NOX2 inhibition also reduced the rate of PFK-2 catalyzed glycolysis, this revealed an unexpected function of NOX2 in the activation dependent increase in glycolytic metabolism through PFK-2 (74). Interestingly, addition of α-ketoglutarate to the neutrophil suspension significantly increased superoxide and H2O2 generation as well as intracellular pyruvate, asparagine, glutamine, aspartate, and glutamate (75). Similar results were also observed following treatment with pyruvate on ROS production (76), suggesting metabolic adaptation/plasticity of neutrophils to utilize distinct metabolic intermediates for catabolic and anabolic processes. Moreover, role of short chain fatty acids (SCFA), in particular acetate via GPR43 receptor has also been suggested in enhanced ROS generation and phagocytosis (77).
Neutrophils release chromatin traps along with antimicrobial granular contents through NETosis to kill and prevent the dissemination of pathogens (6). NETosis is also energy driven catabolic process that requires inter-mixing of cytoplasmic granules with decondensed nuclear chromatin, and expulsion of filamentous traps (6, 21). Mediators promoting NETosis have been categorized on the basis of their dependence/independence on ROS. Recent studies are focusing on finding the metabolic requirement of NETosis (20, 21), which suggest its dependent on glucose but not on glutamine (20). Consistently uptake of glucose was increased during phorbol myristate acetate (PMA) induced NETosis (20). Moreover, ROS, a known driver of NETosis was less dependent on glucose during the initial phase of chromatin decondensation (20). Rodriguez-Espinosa et al. thus proposed an essential role of glucose and glycolysis for the second phase of NETosis i.e., NETs release (20). Indeed, 2-DG that diminished NETosis (20), simultaneously inhibited glycolysis as well as PPP. Subsequent study conducted by Azevedo et al. revealed a metabolic shift toward PPP during NETosis (21), which was coupled with increased activity of glucose-6-phosphate dehydrogenase (G6PD) that diverts glycolytic intermediate G6P to PPP. Consistently, G6PD inhibitor, 6-aminonicotinamide (6-AN) has also reduced ROS production and DNA release in PMA and amyloid fibrils stimulated cells, suggesting G6P as a fuel for NOX2 activation (21). Though incomplete inhibition of ROS as well as NETs with G6PD inhibitor advocates the possibility of additional NADPH source than PPP or concurrently active glycolytic pathway, warranting further investigations. It is also important to mention that due to short life of neutrophils most of metabolic studies are conducted using pharmacological agents, which also have non-specific effects.
Until recently, it has been well-accepted that under anaerobic conditions glucose produces lactate as metabolic waste. Recently, two independent breakthrough studies using 13C-lactate tracing identified that lactate can be converted to pyruvate and fuel metabolism through TCA cycle (78, 79). Lactate can be converted back to glucose in the liver through Cori cycle (78, 79). Neutrophils are commonly present in hypoxic and inflammatory sites that are often enriched with lactate, but role of lactate in neutrophil function is relatively less explored. Rodriguez-Espinosa et al. have linked PMA induced lactate release and NETs formation (20). In light of the recent paradigm shift in role of lactate in metabolism, lactate might play a unique role in NETosis. Consist to this, Alarcon et al. demonstrated that d(–) lactic acid, which can be formed by bacteria, induced NETosis in bovine neutrophils (80). Significant changes in the lactate level during diverse metabolic and inflammatory diseases, like diabetes, sepsis etc, might modulate neutrophil responses and NETosis. Recent study conducted in our lab has demonstrated that lactate was formed during PMA and A23187 induced NETosis, and oxamate, a LDH inhibitor reduced NETs release by these mediators (81). Moreover, previous study from our lab has also demonstrated that oxidized LDL though TLR pathway induced NETosis in human PMNs (82), suggesting a possible role of FAO in NETosis and neutrophil-mediated tissue damage. NETosis has been linked with both physiological and pathological conditions (3), thus necessitating a proper understanding of the metabolic requirements for improved understanding and for developing future therapeutic interventions.
Participation of neutrophils in immunity is highly dependent on chemotaxis, a process of cell migration toward intruder/injury site (83). Importantly, chemotaxis is a coordination of cytoskeleton reorganizing events including adhesion, polarization following actin-enriched pseudopod formation in the front and myosin-dependent contraction toward uropod (83, 84). Together these cytoskeleton reorganization events are energy dependent and well-supported by small GTPases during neutrophil migration (84, 85).
Though in neutrophils primarily dispensable for bioenergetics, mitochondria and TCA cycle enzymes has been observed to regulate chemotaxis (33, 39, 61). FCCP mediated significant inhibition of neutrophil migration suggested the role of mitochondrial signaling in chemotaxis of neutrophils (30). Further study revealed that neutrophils release ATP at the front that amplifies purinergic signaling through P2Y2 nucleotide receptors (39). Indeed, in response to chemotactic cues mitochondria sense and release ATP to further stimulate P2Y2 receptors and promote mTOR signaling to fuel the mitochondrial activity at the leading edge (33). Activated mitochondria displaying high calcium uptake and Δψm localize to the front and deliver the ATP to promote purinergic signaling (33). Furthermore, inhibition of mitochondrial ATP production by cyanide, rotenone, and CCCP, an uncoupler completely blocked chemotaxis by mitigating gradient sensing and speed through alternative purinergic signaling (33). A recent study identified direct evidence of mitochondrial role in neutrophil migration, as neutrophil-specific disruption of mitochondrial DNA polymerase, polg led to reduced motility of neutrophils in Zebra fish (61). In yet another study, prolonged treatment with oligomycin, inhibited chemotaxis (30), suggesting an essential role of Fo-ATPase to uphold chemotaxis.
Iso-citrate dehydrogenase (IDH) is an important enzyme in cellular metabolism that decarboxylates isocitrate to α-ketoglutarate in TCA cycle. IDH-1 mutant neutrophils exhibited impaired chemotaxis, suggesting the importance of TCA metabolism in neutrophil function (86). Short-chain fatty acids (SCFAs) including acetate, propionate, and butyrate via L-selectin stimulation induced neutrophil migration in vivo (87). SCFAs by engaging with G-protein coupled receptor 43 (GPR43, also known as FFAR2) induced neutrophil chemotaxis (53), additionally GPR43 deficient neutrophils failed to migrate to propionate or GPR43 agonist phenylacetamide-1 (53, 54). SCFAs including acetate catabolize to acetyl-Co-A that generate a higher amount of ATP molecules through TCA and OXPHOS. Thus, these studies reveal role of mitochondria and TCA cycle in neutrophil chemotaxis. Moreover, neutrophils migrate to all the organs/tissues in the body, it is well-known that organs exhibit distinct preference for the metabolic fuels. How does diverse intermediates of distinct metabolic pathways in various organs impact neutrophil populations and their functions remain to be defined.
Neutrophil homeostasis is governed by the clearance of exhausted cells through the apoptotic process, while uncontrolled apoptosis can lead to neutropenia. Apoptosis is an energy-dependent process (88), similarly prolonged survival of neutrophils might be a result of the change in energy homeostasis and metabolism (89). Intriguingly, G-CSF exerts an anti-apoptotic effect through mitochondria-dependent mechanisms (31). In spite of minimal involvement in energetics, mitochondria in neutrophils maintain the membrane potential (Δψm), which depends on proton gradient derived from respiratory chain complexes and OXPHOS (47). Complex III can receive electrons from glycolysis via the glycerol-3-phosphate shuttle that helps to maintain Δψm using mitochondrial glycerol phosphate dehydrogenase (mGPD) enzyme in neutrophils (47). While a loss of Δψm in neutrophils precedes the phosphatidylserine exposure and initiates the release of cytochrome C and other pro-apoptotic factors into the cytoplasm to prime apoptosis (30–32).
A study using patients with mutations in succinate dehydrogenase (SDH) encoding gene indicates its role in neutrophil apoptosis (22). SDH oxidizes succinate to fumarate in the TCA cycle and also functions as ubiquinone oxidoreductase in ETC complex II (22). Neutrophils from these patients exhibited higher succinate levels, and reduced constitutive as well as hypoxia induced apoptosis, in an HIF-1α independent manner (22). Consistently, treatment with 3-nitropropionic acid, an irreversible SDH inhibitor led to reduced constitutive apoptosis in neutrophils from healthy individuals (22), implying role of TCA cycle intermediates in neutrophil apoptosis.
Lodhi et al. identified an important role of fatty acid synthase (FAS)-dependent de novo lipogenesis in neutrophil apoptosis (52). FAS catalyzes palmitate formation from malonyl-CoA. Conditional global FAS knockout mice die from neutropenic sepsis due to disrupted membrane phospholipid composition in FAS KO neutrophils. Loss of FAS impaired lipogenesis but enhanced programmed cell death without affecting granulocytic differentiation. Furthermore, peroxisomal reductase activating PPAR (PexRAP) knockout mice also exhibited loss of neutrophils, without drastically affecting other hematopoietic cells (52). Aoyama et al. have shown induction of caspase-dependent neutrophil apoptosis in the presence of SCFAs propionate and butyrate (90). In contrast, acetate induced neutrophil apoptosis was GPR43 dependent (77). Together these studies demonstrate role of FA metabolism in neutrophil apoptosis.
Enhanced neutrophil apoptosis during sepsis is associated with a decrease in glutamine concentration (91). Expectedly, glutamine supplementation protected mice from sepsis and organ failure (92–94), possibly by reducing neutrophil apoptosis (95–98). In yet another study, L-alanyl-L-glutamine led to a significant increase in alpha-ketoglutarate, pyruvate and ROS generation in neutrophils (99), while 6-diazo-5-oxo-L-norleucine (DON), a glutamine-analog that inhibits glutamine utilization reversed L-alanyl-L-glutamine effects (100). These studies, however overlooked ligand/stimuli induced functional responses. Thus, glutamine and its metabolic intermediates support functions of mitochondria and survival under diverse conditions in the neutrophils. Together these findings imply the importance of mitochondrial pathways, TCA cycle and fatty acid metabolism in neutrophil development, functions and survival.
Neutrophils constantly infiltrate almost all the organs and are therefore exposed to a variety of microenvironments in diverse organs/tissues. Furthermore, inflammatory sites are often associated with a low level of oxygen and glucose (101). Recent research identified key regulators of metabolic adaptability of neutrophils under these different conditions and is being discussed here (Figure 2). Hypoxia-inducible factor-1α (HIF-1α) and the mammalian target of rapamycin (mTOR) have been recognized as major regulators of metabolism in myeloid cells (63, 102). Importantly, neutrophils possess HIF-1α and factor inhibiting HIF (FIH) hydroxylase to sense hypoxia (101). Interestingly, in hypoxic environment neutrophil apoptosis is prevented by NF-κB activation (101). On the contrary, based on elevated oxygen consumption during respiratory burst, activated neutrophils at the sites of inflammation are advocated to induce oxygen depletion to promote inflammatory hypoxia (103). HIF-1α-null neutrophils exhibited low expression of granule proteases, including elastase and cathepsin G with reduced anti-microbial activity (64). In contrast, vHL-null cells with stabilized HIF-1α were observed to possess a high amount of proteases and bactericidal capacity (64). While, neutrophil development has remained unaffected by specific deletion of HIF-1α in the myeloid progenitors (63). however HIF-1α null macrophages have shown defects in hypoxia-induced Glut-1 expression (63), suggesting toward the role of HIF-1α in glucose utilization. In addition, neutrophils also express HIF-2α having overlapping functions with HIF-1α, and its expression has been up-regulated during inflammatory conditions. The specific role of HIF-2α in neutrophil apoptosis was observed using gain-of-function mutations that exhibited diminished neutrophil apoptosis (104). In contrast, HIF-2α-deficient neutrophils displayed enhanced apoptosis, leading to a reduction in neutrophil-mediated inflammation (104). HIFs are under tight regulation of oxygen tension and prolyl hydroxylases (PHDs) that cause ubiquitylation and proteasomal degradation of HIFs. Sadiku et al. demonstrated that the myeloid-specific deletion of PHD2 led to lung injury following enhanced neutrophil survival (62). Mechanistically, PHD2-deficient neutrophils displayed enhanced glycolytic capacity, generation of PPP intermediates, glycogen stores, and ATP levels. Further, glycolysis inhibition has protected these mice against enhanced neutrophilic responses, suggesting a direct connection of metabolic modulation during infection as well as inflammation (62). A recent study further confirmed that during anoxic condition, glucose and dimethyloxalylglycine (DMOG) maintained neutrophil viability and functionality for a longer period (89). Furthermore, ferroprotein sensing has been suggested to play a role in hypoxia-mediated inhibition of neutrophil apoptosis (105). Iron-chelating agents, desferrioxamine (DFO) and hydroxypyridines (CP-94) inhibited apoptosis under normoxic condition, while no effect was observed under hypoxia (105). Lactoferrin, an iron-binding protein released from activated neutrophils also enhanced neutrophil survival in the synovial fluid of rheumatoid arthritis patients (106).
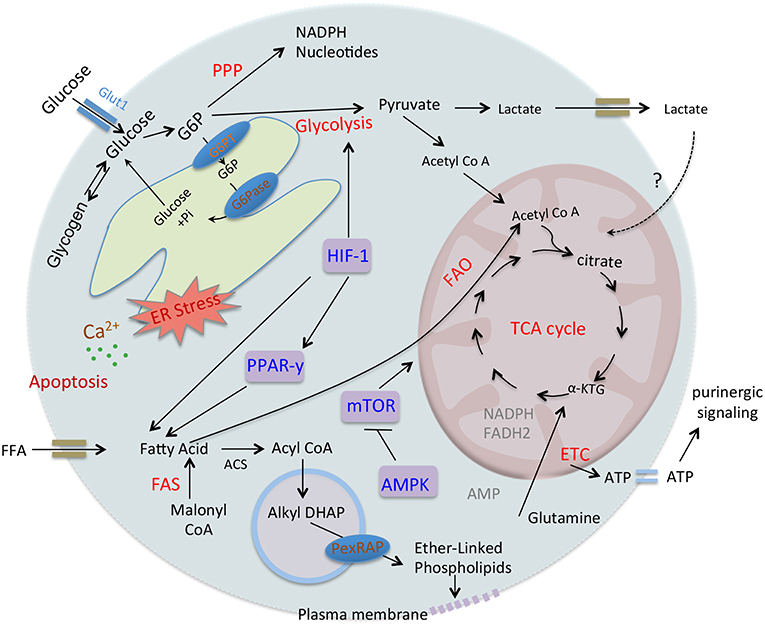
Figure 2. Regulation of neutrophil metabolism. Neutrophil utilizes distinct metabolic pathways including glucose oxidation, glycogen breakdown and β-oxidation of fatty acids. After transport of glucose in the cytosol, hexokinase converts it to G6P that participate in different pathways like glycolysis, PPP, glycogen synthesis, or ER cycling. Glycolysis converts glucose to pyruvate that can further utilize in TCA cycle or forms lactate. G6PT and G6Pase regulate recycling of G6P in ER and disruption of glucose/G6P balance and ER stress due to G6PT or G6Pase-β deficiencies result in impaired energy homeostasis, functions, and survivals of neutrophils. While FFA gets transport to the cytosol and autophagy also release FFA that undergoes β-oxidation and provides a high level of ATPs through mitochondrial OxPHOS pathway and regulates neutrophil differentiation. On the other hand, in neutrophils FAS and PexRAP produce peroxisomal lipids that get incorporated into the plasma membrane and thus maintaining membrane integrity and viability. Mitochondrial OxPHOS provides ATPs that also drives purinergic signaling in neutrophils. Transcriptional regulator HIF-1α regulates glucose transport as well as enzymes involved in the glycolysis. HIF-1α regulates PPARy, that also increases glucose uptake and regulates fatty acid metabolism. Interestingly, over-activation of PPARy in diverse diseases leads to defective energetics and functions of neutrophils. mTOR functions as a master regulator of mitochondrial metabolism and also regulates purinergic signaling. AMPK that senses AMP-ATP levels also controls neutrophil metabolism and functions.
mTOR, a known metabolic regulator (102), modulated chemotaxis, via mTORC2 mediated pseudopod protrusion, and cAMP accumulation at the neutrophil front (65). Furthermore, mTOR inhibition by rapamycin or PP242 reduced neutrophil chemotaxis via inhibition of chemokine-mediated ATP release. While at the uropod, A2a receptors mitigated mTORC2 signaling and blocked mitochondrial activation causing uropod retraction (33). Furthermore, rapamycin, an inducer of autophagy, mediated inhibition of mTORC1 also delayed neutrophil differentiation process (35). Consistently, pharmacological inhibition of mTOR or HIF-1α blocked NETs-dependent bacterial killing (66). Furthermore, expression of nuclear receptor peroxisome proliferator-activated receptors (PPARs) that drive fatty acid signaling, is enhanced in the neutrophils after activation (107). Moreover, sepsis patients exhibited high PPAR-y activity and reduced neutrophil chemotactic activity, which was prevented by PPAR-y antagonist (107). In addition, AMP-activated protein kinase (AMPK), a cellular energy sensor, maintains energy homeostasis via stimulation of glucose uptake and FAO in the cells. Interestingly, AMPK activation induced neutrophil chemotaxis, phagocytosis, and bactericidal activity (108). Together these studies demonstrate important metabolic regulators of neutrophil responses.
Neutrophil survival is enhanced during inflammatory conditions by modulating their apoptotic program possibly through metabolic reprogramming due to hypoxia and the nutrients availability at the inflammatory sites (101, 109, 110). Further, during distinct chronic, inflammatory and metabolic diseases, neutrophils also exhibit altered functions that are being discussed in the following section and are summarized in Table 3.
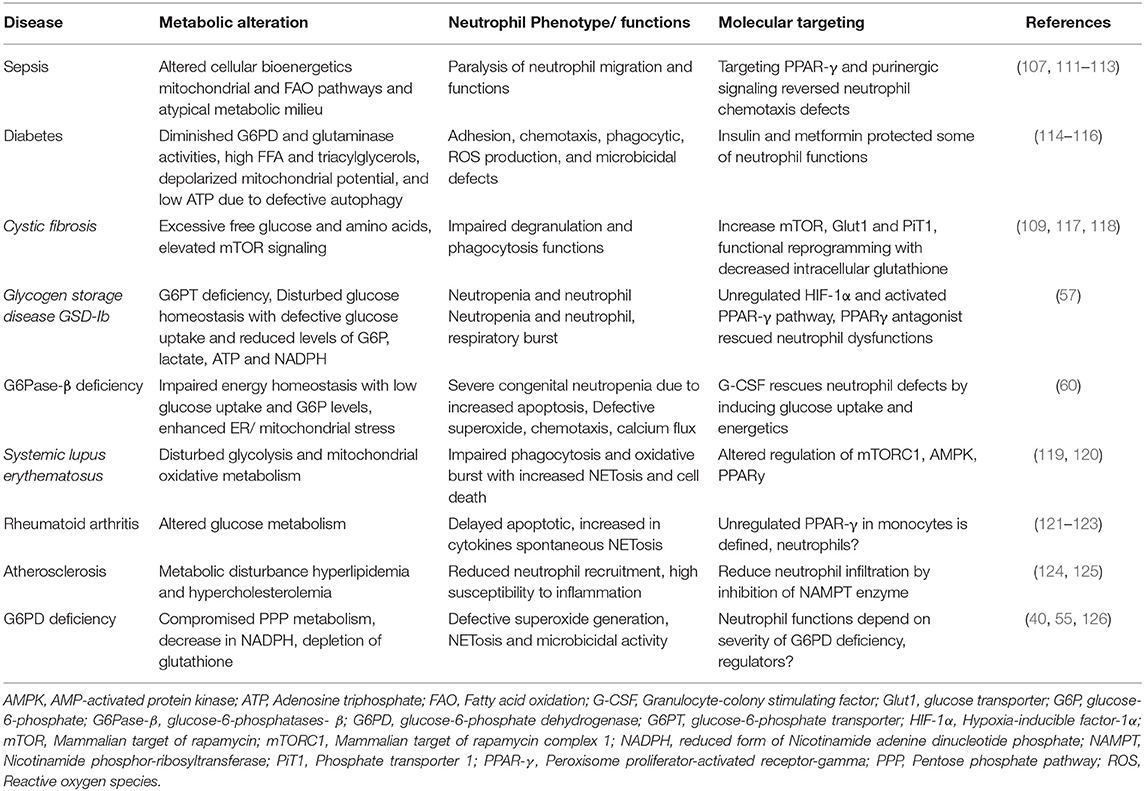
Table 3. Metabolic alteration responsible for neutrophil dysfunctions in disease conditions.
Severe sepsis, a systemic inflammatory syndrome, is associated with impaired neutrophil migration and functions (127–129). Higher expression of chemokines and anti-apoptotic protein Bcl-xL and Mcl-1 during sepsis has been observed to delayed neutrophil apoptosis (127, 128). Defective neutrophil chemotaxis in sepsis has also been associated with reduced expression of chemokine receptor CXCR2, GPCR receptor kinase (GRK), and defective actin assembly (127, 129). Sepsis, due to persisting catabolism, alters cellular bioenergetics and metabolic milieu (111, 112). Role of mitochondrial activity, especially complex V (ATP synthase) and ETC complex III and IV, was associated with LPS-mediated dysfunction of neutrophil chemotaxis (108, 130), which was prevented by AMPK activator and GSK3β inhibitor (108, 130). Inhibition of chemotaxis via upregulation of PPAR-γ under septic conditions was reversed by PPAR-γ inhibition (107).
In yet another study, patients who survived sepsis were presented with increased autophagy that primed NETs formation (131). While dysregulated autophagy and reduced NETosis was evident in sepsis patients who did not survive (131). Consistently, augmentation of autophagy in a mouse model of sepsis also improved survival via a NETs-dependent mechanism (131). Together, functional rejuvenation of neutrophils during sepsis by autophagy provides diverse breakdown products and building blocks to modulate neutrophil metabolism. Importantly, Sumi et al. using a mouse model of cecal ligation and puncture have described the requirement of plasma ATP in neutrophil activation during sepsis (132). Conversely, systemic ATP in sepsis reduced neutrophil activation and chemotaxis by disrupting endogenous purinergic signaling mechanisms (113). Furthermore, suramin, P2-receptor antagonist blocked endogenous ATP signaling and impaired neutrophil activation and chemotaxis leading to increase in bacterial growth and mortality, while removal of systemic ATP by apyrase, ATP-diphosphatase improved bacterial clearance and survival in sepsis, advocating diverse approaches targeting the purinergic signaling for sepsis and other systemic inflammatory disorders (113).
Diabetes, a metabolic disorder lead to increase the systemic glucose levels, is associated with enhanced susceptibility for infections and defective neutrophil functions. Neutrophil adhesion, chemotaxis, phagocytosis, ROS production, and microbicidal activity defects have been observed in diabetic patients and animal models (114, 115). Hyperglycemia leads to diminished enzymatic activities of glucose-6-phosphate dehydrogenase, and glutaminase, while levels of phosphofructokinase (PFK) are augmented (114). Reduction in G6PD activity adversely affects pentose-phosphate pathway progression and also neutrophil functions (114, 133). Moreover, insulin improves neutrophil phagocytosis and ROS production ability even in the absence of normalized glycemic index, suggesting toward direct effect of insulin on neutrophils (114). Interestingly, under resting state neutrophils express glucose transporters GLUT1 and GLUT3 on surface that remain insensitive to insulin for glucose uptake (25). While in response to phorbol myristate acetate activation, GLUT4 mobilized to the cell surface in an insulin-sensitive manner (25). Defective glucose and glutamine metabolism during hyperglycemia seem to activate a putative compensatory FAO utilization by neutrophils (114). Moreover, enhanced levels of circulating FFA and triacylglycerols cause insulin resistance and also neutrophilic inflammation (53, 54). Neutrophils from streptozotocin-induced diabetic rats exhibit defective autophagy, depolarized mitochondrial potential, and low ATP levels (116). Homocysteine, a sulfur-containing amino acid, levels increased significantly during diabetes and also induced NETosis (134). Furthermore, homocysteine has been observed to enhance the rate of both oxidative phosphorylation and glycolysis along with accumulation of the PPP intermediates in the B cells through pyruvate Kinase M2 (PKM2) upregulation (135). Further investigations are required to understand the metabolic regulations in neutrophils during initiation, development, and progression of diabetes/insulin resistance.
Cystic fibrosis is a genetic disorder presented with recurrent respiratory tract infections. CF disease is also associated with large numbers of functionally dysregulated neutrophils (117, 136, 137). In CF, neutrophils fail to clear infections, due to functional reprogramming, as well as a reduction in CD16 and CD14 receptors (109). Moreover, increased lipid raft assembly, mobilization of granules and CD11b, CD66 expression were also observed, while elevated mTOR signaling suggested toward anabolic programming (109). Interestingly, CF airway neutrophils adapt to excessive free glucose and amino acids by substantially enhancing the expression of Glut1 and PiT1 (inorganic phosphate transporters) (118). However, it is not clear that how these adaptations affect neutrophil functionality in CF.
G6PD is an essential enzyme that switches glucose metabolism toward PPP (40), by catalyzing the conversion of glucose-6-phosphate to 6-phospho-D-glucono-1,5-lactone and NADPH, that fuels NOX2. G6PD deficiency is a common inherited disorder associated with a decrease in NADPH production leading to the depletion of glutathione, and hemolytic anemia. World Health Organization (WHO) has classified G6PD deficiency into different categories according to the residual G6PD activity and severity of hemolytic anemia (138). Interestingly reduced NOX2 activity strongly correlates with the levels of G6PD activity. G6PD deficiency mimics mild chronic granulomatous disease condition due to defects in the microbicidal and metabolic activity of neutrophils (126). A study using 37 G6PD patients clearly demonstrated a link between G6PD levels and superoxide generation in the neutrophils (139). On the contrary, Ardati et al. did not observe defects in ROS levels as detected by NBT assay in neutrophils having only 23% of normal G6PD activity (140). Conversely, almost 90% loss in G6PD activity led to reduction in ROS generation, NETosis and enhanced susceptibility to infections (55). Consistent to this, mice with hematopoietic G6PD deficiency exhibited improved glucose tolerance, insulin sensitivity, reduction in circulating FFAs, is most likely due to ameliorated inflammation (56). Together, these studies indicate impact of PPP on neutrophil NOX-2 activity and susceptibility to infections in G6PD deficiencies.
Glycogen is stored in the neutrophil cytosol (7, 141) and glycogenesis (glucose to glycogen conversion) is signaled by the abundance of glucose and ATP. While low glucose conditions, produce glucose-6-phosphate (G6P) through glycogenolysis. G6P is further hydrolyzed to glucose by glucose-6-phosphatases (G6Pase) by gluconeogenesis (58). Glycogen- glucose homeostasis is thus regulated by multi-enzyme glucose-6-phosphatases (G6Pase) complex that includes hydrolase and glucose-6-phosphate transporter (G6PT) subunits. G6PT localizes in the endoplasmic reticulum membrane and translocates G6P from the cytoplasm into the ER lumen, where hydrolase generates glucose. In neutrophils, cycling of G6P/glucose between cytoplasm and ER is utilized for glycolysis and PPP (58). Disturbance in its homeostasis causes GSD, particularly by mutations of G6Pase-α in GSD type Ia (GSD-Ia). GSD-Ia mice as well as patient exhibited elevated number of myeloid progenitor cells, and high G-CSF leading to neutrophilia, however neutrophil function remained unaltered (142). While GSD-Ib is due to the deficiency of G6PT, and it is associated with neutropenia, and neutrophil dysfunction (57, 143). Neutrophils from GSD-Ib patients exhibit defective glucose uptake and reduced levels of G6P, lactate, ATP, and NADPH, even though Glut-1, HIF1-α, and hexokinase (HK-3) expression were augmented (57). Moreover, treatment with PPARγ antagonist, GW9662, rescued neutrophil dysfunction in GSD-Ib patients (57). Similarly, mutations of G6Pase-β (encoded by G6PC3) also cause severe congenital neutropenia syndrome due to enhanced apoptosis (59). Furthermore, G-CSF mitigated neutropenia in G6Pase-β (G6PC3) deficiency by inhibiting apoptosis and inducing glucose uptake and modulating energetics (60). G-CSF also rescued defective superoxide generation, chemotaxis and calcium flux in G6pc3−/− neutrophils (60). These studies highlight importance of glycogenolysis mediated metabolic adaptation during neutrophil differentiation and functions through HIF-1α and PPAR-γ. Interestingly, these transcription factors also regulate FAO, TCA, and OXPHOS pathways.
Systemic lupus erythematosus (SLE) is an autoimmune disease with a high degree of immuno-inflammation and self-destruction. Interestingly, neutrophils in SLE disease condition display dysregulated aggregation, phagocytosis, oxidative burst, and enhanced cell death, NETosis (119). In addition, neutrophils modulate response of other immune cells by releasing self-DNA-peptide complexes that contain immunogenic complexes composed of neutrophil-derived antimicrobial peptides and chromatin (144, 145). Furthermore, SLE is also associated with defective clearance of NETs, however role of NETosis remain to be investigated in inducing autoimmunity (146). Importantly, low-density granulocytes (LDGs), a distinct subset of neutrophils with high levels of pro-inflammatory cytokines were seen elevated in SLE patients (147). Metabolic alteration in T and B cells in SLE has also been described (148). Increase in glycolysis and mitochondrial oxidative metabolism was found in CD4+ T cells in lupus (149). Treatment with 2-DG, metformin or rapamycin rescued lupus symptoms in mice model (149). Ribonucleoprotein immune complexes (RNP-ICs) are commonly found in SLE, which induce NETosis through mitochondrial ROS formation (120). Indeed, RNP-ICs, induced hypopolarization of mitochondria and their translocation to the cell surface. It is important to mention that RNP-ICs also induce NETosis in CGD patients with the consistent development of autoimmunity (120). Metabolic status of neutrophils in SLE has not been explored, even though there is enhanced NETosis, release of self-DNA–peptide complexes and pro-inflammatory phenotype with mitochondrial and mTOR alteration.
Another chronic autoimmune disease, rheumatoid arthritis (RA) is presented with extensive activation of the immune system causing synovitis and joint destruction. Neutrophils infiltration in the joint synovial fluid play an essential role in RA particularly during the early stages of the disease by releasing cytotoxic proteases and generating ROS (150). Neutrophils in the synovial fluid also exhibit distinct characteristics like the expression of cytokines and MHC molecules, that are in general expressed on macrophages and dendritic cells and their apoptosis is delayed (121, 122). Furthermore, the survival of SF neutrophils could be due to the hypoxic conditions prevailing in joints afflicted with RA (151). In addition, similar to SLE disease, in RA neutrophils also exhibit increased expression of inferron genes (152) and spontaneous NETosis (123). Furthermore, RA patients with altered glucose metabolism usually have a high risk of diabetes due to insulin resistance and impaired beta cell function (153). Neutrophil metabolic regulations in rheumatoid arthritis also remain less investigated.
Atherosclerosis is characterized by the plaques accumulated with low-density lipoproteins (LDLs) and cholesterol in the blood vessels in association with hyperlipidemia and inflammation. Researches in the last decade have established presence as well as causative role of neutrophils in atherosclerosis (124, 154). Metabolic disturbance including hyperlipidemia and hypercholesterolemia induce neutrophilia to trigger atherosclerosis (124). Moreover, oxidized-LDL, as well as cholesterol crystals also induce NETosis (82, 155) and inhibition of NAMPT, the enzyme responsible for NAD+ biosynthesis reduces neutrophil infiltration in the atherosclerotic plaque by decreasing CXCL1 levels in ApoE−/− mice (125). Together, metabolic changes in the atherosclerotic environment seem to modulate neutrophil functions, warranting more investigations for establishing the role of neutrophils in initiation/sustenance of atherosclerosis and plaque stability.
Recent research has highlighted heterogeneity and plasticity in neutrophil populations on the basis of their state of maturation, altered life span, release of cytokines like IL-10, IL-17A, antigen presentation, diverse surface proteins, distinct antibacterial responses, pro-inflammatory, pro-angiogenic, or immunosuppressive nature (9–12, 147, 156–159). Importantly, microenvironment seems to play a defining role in the neutrophil heterogeneity. For example, tumor-associated neutrophils (TANs) exhibit two distinct phenotypes, N1 cells with anti-neoplastic activity and N2 cells with immunosuppressive characteristics. Transforming growth factor (TGF)-β in the neoplasm microenvironment induces N2 population, which promote tumor development (157). In yet another study, a unique population of neutrophil was identified, expressing transcription factor RORγt and IL-17A after treating with IL-6 and IL-23 (156). Cellular and functional diversity, as well as neutrophil heterogeneity has been majorly found during inflammatory conditions and infectious disorders like sepsis, diabetes, rheumatoid arthritis, and SLE. Interestingly, during infectious and SLE conditions there is an increase in the number of neutrophils expressing programmed death-ligand 1 (PD-L1) (158, 159) and frequency of PD-L1 expressing neutrophils correlates with SLE disease severity (159). These neutrophils also have immunosuppressive characteristics and possibly exert lesser tissue damage. PD-L1+ neutrophils on the other hand suppress T-cell immunity and enhance tumor progression (160). A recent study revealed enhanced migration of neutrophils during early-stage of cancer (161) and these neutrophils exhibit high rates of oxidative phosphorylation and glycolysis and produce more ATP (161). Furthermore, tumor microenvironment induces metabolic adaptation in neutrophils with an increase in mitochondrial fitness and immune- suppression (72). Neutrophils in tumor-elicited environment, due to limited glucose supply adapt to mitochondrial FAO to support NOX-2 -dependent ROS production (72). Incidentally, neutrophils depend on glycolysis during early phase of intense ROS generation, while during late phase ROS production is supported by mitochondrial FAO (72). Taken together, recent developments unravel metabolic decisions in cellular plasticity and differential pro-inflammatory or immunosuppressive responses. Future studies to provide better understanding of neutrophil heterogeneity and their metabolic adaptations to perform microenvironment specific function are now needed.
Neutrophils pave the way for immunoregulation at the site of infection/inflammation. Recent research in the area of neutrophil biology has challenged the hypothesis of neutrophils dependence on glycolysis for their varied functional responses, differentiation, and survival. Neutrophils however utilize diverse metabolic pathways including glycolysis, glutaminolysis, PPP, FAO, TCA cycle to perform distinct functions. Thus, targeting these metabolic adaptations by neutrophils might be beneficial for combating infections and inflammation mediated tissue damage. Furthermore, metabolic switch(es), if any, remains least defined so far to impact neutrophil functionality and heterogeneity. Cutting-edge single cell metabolomics analyses in coordination with high-throughput approaches might be of immense importance to investigate heterogeneity in neutrophil population (162). Detailed untargeted metabolomics studies might help in identifying metabolic switch(es) during the apoptotic program and/or enhanced survival. It would also be interesting to decipher how neutrophil adapt to metabolism during various pathological conditions and hypoxia due to availability of distinct nutrients, metabolic intermediates, and a mixture of various mediators (see Outstanding Questions). Finally, improved understanding of the metabolic switch(es) in neutrophil populations migrating to inflammatory/infectious sites and survive at these sites could be helpful in unraveling the mechanistic understanding of various disorders.
• Is there any specific role of metabolic adaptation(s) if any during pro-inflammatory and immunosuppressive conditions in the various neutrophil subsets?
• Can distinct metabolic pathways be efficiently targeted/suppressed to modulate specific neutrophil functions and survival?
• Can neutrophils subsets be targeted to tackle infectious and inflammatory conditions?
• Can single cell metabolomics be helpful in understanding and defining the neutrophil plasticity?
• Metabolic activities in the subcellular organelle and phagosomes of the resting and activated neutrophils remain undefined so far.
• Comparison of metabolic pathways in neutrophils with other phagocytic cells is of fundamental importance to delineate the role of metabolism if any in the short-lived fate of neutrophils.
SK conceptualized the idea, collects relevant information, prepared figures, table, wrote the manuscript. MD provided critical suggestion and helped in writing the manuscript.
SK was supported by DBT-India Ramalingaswami Fellowship BT/RLF/Re-entry/28/2014 and DST-SERB early career grant ECR/2017/001274. MD was supported by JC Bose National fellowship from DST-SERB (SB/SE/JCB-017/2015).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Amit Lahiri for initial comments on the manuscript. This manuscript is CDRI communication no. 9883.
1. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
2. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
3. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
4. Swamydas M, Gao JL, Break TJ, Johnson MD, Jaeger M, Rodriguez CA, et al. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci Transl Med. (2016) 8:322ra10. doi: 10.1126/scitranslmed.aac7718
5. Segal AW. How neutrophils kill microbes. Annu Rev Immunol. (2005) 23:197–223. doi: 10.1146/annurev.immunol.23.021704.115653
6. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
7. Sbarra AJ, Karnovsky ML. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J Biol Chem. (1959) 234:1355–62.
8. Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Invest. (1982) 70:550–7. doi: 10.1172/JCI110647
9. Mocsai A. Diverse novel functions of neutrophils in immunity, inflammation, and beyond. J Exp Med. (2013) 210:1283–99. doi: 10.1084/jem.20122220
10. Silvestre-Roig C, Hidalgo A, Soehnlein O. Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood. (2016) 127:2173–81. doi: 10.1182/blood-2016-01-688887
11. Takashima A, Yao Y. Neutrophil plasticity: acquisition of phenotype and functionality of antigen-presenting cell. J Leukoc Biol. (2015) 98:489–96. doi: 10.1189/jlb.1MR1014-502R
12. Scapini P, Marini O, Tecchio C, Cassatella MA. Human neutrophils in the saga of cellular heterogeneity: insights and open questions. Immunol Rev. (2016) 273:48–60. doi: 10.1111/imr.12448
13. Khan MA, Palaniyar N. Transcriptional firing helps to drive NETosis. Sci Rep. (2017) 7:41749. doi: 10.1038/srep41749
14. Lakschevitz FS, Visser MB, Sun C, Glogauer M. Neutrophil transcriptional profile changes during transit from bone marrow to sites of inflammation. Cell Mol Immunol. (2015) 12:53–65. doi: 10.1038/cmi.2014.37
15. Hu Z, Jiang K, Frank MB, Chen Y, Jarvis JN. Complexity and specificity of the neutrophil transcriptomes in juvenile idiopathic arthritis. Sci Rep. (2016) 6:27453. doi: 10.1038/srep27453
16. O'Neill LA, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. (2016) 16:553–65. doi: 10.1038/nri.2016.70
17. Gaber T, Strehl C, Buttgereit F. Metabolic regulation of inflammation. Nat Rev Rheumatol. (2017) 13:267–79. doi: 10.1038/nrrheum.2017.37
18. Riffelmacher T, Clarke A, Richter FC, Stranks A, Pandey S, Danielli S, et al. Autophagy-Dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity. (2017) 47:466–80.e5. doi: 10.1016/j.immuni.2017.08.005
19. Six E, Lagresle-Peyrou C, Susini S, De Chappedelaine C, Sigrist N, Sadek H, et al. AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis. (2015) 6:e1856. doi: 10.1038/cddis.2015.211
20. Rodriguez-Espinosa O, Rojas-Espinosa O, Moreno-Altamirano MM, Lopez-Villegas EO, Sanchez-Garcia FJ. Metabolic requirements for neutrophil extracellular traps formation. Immunology. (2015) 145:213–24. doi: 10.1111/imm.12437
21. Azevedo EP, Rochael NC, Guimarães-Costa AB, de Souza-Vieira TS, Ganilho J, Saraiva EM, et al. A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J Biol Chem. (2015) 290:22174–83. doi: 10.1074/jbc.M115.640094
22. Jones R, McDonald KE, Willson JA, Ghesquière B, Sammut D, Daniel E, et al. Mutations in succinate dehydrogenase B (SDHB) enhance neutrophil survival independent of HIF-1α expression. Blood. (2016) 127:2641–4. doi: 10.1182/blood-2016-02-696922
23. Richer BC, Salei N, Laskay T, Seeger K. Changes in neutrophil metabolism upon activation and aging. Inflammation. (2018) 41:710–721. doi: 10.1007/s10753-017-0725-z
24. Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol. (2013) 17:638–50. doi: 10.1016/j.intimp.2013.06.034
25. Maratou E, Dimitriadis G, Kollias A, Boutati E, Lambadiari V, Mitrou P, et al. Glucose transporter expression on the plasma membrane of resting and activated white blood cells. Eur J Clin Invest. (2007) 37:282–90. doi: 10.1111/j.1365-2362.2007.01786.x
26. Lara LL, Rivera H, Perez PC, Blanca I, Bianco NE, et al. Low density lipoprotein receptor expression and function in human polymorphonuclear leucocytes. Clin Exp Immunol. (1997) 107:205–12. doi: 10.1046/j.1365-2249.1997.d01-888.x
27. Oh H, Mohler ER 3rd, Tian A, Baumgart T, Diamond SL. Membrane cholesterol is a biomechanical regulator of neutrophil adhesion. Arterioscler Thromb Vasc Biol. (2009) 29:1290–7. doi: 10.1161/ATVBAHA.109.189571
28. Alvarez-Curto E, Milligan G. Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol. (2016) 114:3–13. doi: 10.1016/j.bcp.2016.03.017
29. Hayashi F, Means TK, Luster AD. Toll-like receptors stimulate human neutrophil function. Blood. (2003) 102:2660–9. doi: 10.1182/blood-2003-04-1078
30. Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW. The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol. (2003) 170:1964–72. doi: 10.4049/jimmunol.170.4.1964
31. Maianski NA, Mul FP, van Buul JD, Roos D, Kuijpers TW. Granulocyte colony-stimulating factor inhibits the mitochondria-dependent activation of caspase-3 in neutrophils. Blood. (2002) 99:672–9. doi: 10.1182/blood.V99.2.672
32. Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Different. (2004) 11:143–53. doi: 10.1038/sj.cdd.4401320
33. Bao Y, Ledderose C, Graf AF, Brix B, Birsak T, Lee A, et al. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J Cell Biol. (2015) 210:1153–64. doi: 10.1083/jcb.201503066
34. Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, et al. NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. (2009) 15:151–8. doi: 10.1038/nm.1913
35. RoŽman S, Yousefi S, Oberson K, Kaufmann T, Benarafa C, Simon HU. The generation of neutrophils in the bone marrow is controlled by autophagy. Cell Death Different. (2015) 22:445–56. doi: 10.1038/cdd.2014.169
36. Reiss M, Roos D. Differences in oxygen metabolism of phagocytosing monocytes and neutrophils. J Clin Invest. (1978) 61:480–8. doi: 10.1172/JCI108959
37. Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G, et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respirat Crit Care Med. (2008) 178:168–79. doi: 10.1164/rccm.200710-1602OC
38. Zmijewski JW, Lorne E, Banerjee S, Abraham E. Participation of mitochondrial respiratory complex III in neutrophil activation and lung injury. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L624–34. doi: 10.1152/ajplung.90522.2008
39. Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, et al. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. (2006) 314:1792–5. doi: 10.1126/science.1132559
40. Stanton RC. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life. (2012) 64:362–9. doi: 10.1002/iub.1017
41. Newsholme P, Curi R, Pithon Curi TC, Murphy CJ, Garcia C, Pires de Melo M. Glutamine metabolism by lymphocytes, macrophages, and neutrophils: its importance in health and disease. J Nutr Biochem. (1999) 10:316–24. doi: 10.1016/S0955-2863(99)00022-4
42. Curi R, Newsholme P, Pithon-Curi TC, Pires-de-Melo M, Garcia C, Homem-de-Bittencourt Júnior PI, et al. Metabolic fate of glutamine in lymphocytes, macrophages and neutrophils. Braz J Med Biol Res. (1999) 32:15–21. doi: 10.1590/S0100-879X1999000100002
43. Curi TC, De Melo MP, De Azevedo RB, Zorn TM, Curi R. Glutamine utilization by rat neutrophils: presence of phosphate-dependent glutaminase. Am J Physiol. (1997) 273:C1124–9. doi: 10.1152/ajpcell.1997.273.4.C1124
44. Stincone A, Prigione A, Cramer T, Wamelink MM, Campbell K, Cheung E, et al. The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev Camb Philos Soc. (2015) 90:927–63. doi: 10.1111/brv.12140
45. Bainton DF, Ullyot JL, Farquhar MG. The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med. (1971) 134:907–34. doi: 10.1084/jem.134.4.907
46. Zucker-Franklin D. Electron microscopic studies of human granulocytes: structural variations related to function. Semin Hematol. (1968) 5:109–33.
47. van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, Kuijpers TW. Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS ONE. (2008) 3:e2013. doi: 10.1371/journal.pone.0002013
48. Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol. (2012) 32:856–64. doi: 10.1161/ATVBAHA.111.226845
49. Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. (2004) 104:3986–92. doi: 10.1182/blood-2004-06-2066
50. Junger WG. Immune cell regulation by autocrine purinergic signalling. Nat Rev Immunol. (2011) 11:201–12. doi: 10.1038/nri2938
51. Bhattacharya A, Wei Q, Shin JN, Abdel Fattah E, Bonilla DL, Xiang Q, et al. Autophagy is required for neutrophil-mediated inflammation. Cell Rep. (2015) 12:1731–9. doi: 10.1016/j.celrep.2015.08.019
52. Lodhi IJ, Wei X, Yin L, Feng C, Adak S, Abou-Ezzi G, et al. Peroxisomal lipid synthesis regulates inflammation by sustaining neutrophil membrane phospholipid composition and viability. Cell Metab. (2015) 21:51–64. doi: 10.1016/j.cmet.2014.12.002
53. Vinolo MA, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly-Y M, et al. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS ONE. (2011) 6:e21205. doi: 10.1371/journal.pone.0021205
54. Sina C, Gavrilova O, Förster M, Till A, Derer S, Hildebrand F, et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J Immunol. (2009) 183:7514–22. doi: 10.4049/jimmunol.0900063
55. Siler U, Romao S, Tejera E, Pastukhov O, Kuzmenko E, Valencia RG, et al. Severe glucose-6-phosphate dehydrogenase deficiency leads to susceptibility to infection and absent NETosis. J Allergy Clin Immunol. (2017) 139:212–9.e3. doi: 10.1016/j.jaci.2016.04.041
56. Ham M, Choe SS, Shin KC, Choi G, Kim JW, Noh JR, et al. Glucose-6-Phosphate dehydrogenase deficiency improves insulin resistance with reduced adipose tissue inflammation in obesity. Diabetes. (2016) 65:2624–38. doi: 10.2337/db16-0060
57. Jun HS, Weinstein DA, Lee YM, Mansfield BC, Chou JY. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood. (2014) 123:2843–53. doi: 10.1182/blood-2013-05-502435
58. Jun HS, Lee YM, Cheung YY, McDermott DH, Murphy PM, De Ravin SS, et al. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood. (2010) 116:2783–92. doi: 10.1182/blood-2009-12-258491
59. Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, et al. Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J Clin Invest. (2007) 117:784–93. doi: 10.1172/JCI30443
60. Jun HS, Lee YM, Song KD, Mansfield BC, Chou JY. G-CSF improves murine G6PC3-deficient neutrophil function by modulating apoptosis and energy homeostasis. Blood. (2011) 117:3881–92. doi: 10.1182/blood-2010-08-302059
61. Zhou W, Cao L, Jeffries J, Zhu X, Staiger CJ, Deng Q. Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis Model Mech. (2018) 11:dmm033027. doi: 10.1242/dmm.033027
62. Sadiku P, Willson JA, Dickinson RS, Murphy F, Harris AJ, Lewis A, et al. Prolyl hydroxylase 2 inactivation enhances glycogen storage and promotes excessive neutrophilic responses. J Clin Invest. (2017) 127:3407–20. doi: 10.1172/JCI90848
63. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/S0092-8674(03)00154-5
64. Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. (2005) 115:1806–15. doi: 10.1172/JCI23865
65. Liu L, Das S, Losert W, Parent CA. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev Cell. (2010) 19:845–57. doi: 10.1016/j.devcel.2010.11.004
66. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1α. Blood. (2012) 120:3118–25. doi: 10.1182/blood-2012-01-405993
67. Borregaard N. Neutrophils, from marrow to microbes. Immunity. (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
68. Weisdorf DJ, Craddock PR, Jacob HS. Granulocytes utilize different energy sources for movement and phagocytosis. Inflammation. (1982) 6:245–56. doi: 10.1007/BF00916406
69. Bladridge CW, Gerard RW. The extra respiration of phagocytes. Am J Physiol. (1932) 103:235–6. doi: 10.1152/ajplegacy.1932.103.1.235
70. Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. (2002) 397:342–4. doi: 10.1006/abbi.2001.2642
71. Chacko BK, Kramer PA, Ravi S, Johnson MS, Hardy RW, Ballinger SW, et al. Methods for defining distinct bioenergetic profiles in platelets, lymphocytes, monocytes, and neutrophils, and the oxidative burst from human blood. Lab Invest. (2013) 93:690–700. doi: 10.1038/labinvest.2013.53
72. Rice CM, Davies LC, Subleski JJ, Maio N, Gonzalez-Cotto M, Andrews C, et al. Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat Commun. (2018) 9:5099. doi: 10.1038/s41467-018-07505-2
73. Weisdorf DJ, Craddock PR, Jacob HS. Glycogenolysis versus glucose transport in human granulocytes: differential activation in phagocytosis and chemotaxis. Blood. (1982) 60:888–93.
74. Baillet A, Hograindleur MA, El Benna J, Grichine A, Berthier S, Morel F, et al. Unexpected function of the phagocyte NADPH oxidase in supporting hyperglycolysis in stimulated neutrophils: key role of 6-phosphofructo-2-kinase. FASEB J. (2017) 31:663–73. doi: 10.1096/fj.201600720R
75. Mühling J, Tussing F, Nickolaus KA, Matejec R, Henrich M, Harbach H, et al. Effects of alpha-ketoglutarate on neutrophil intracellular amino and alpha-keto acid profiles and ROS production. Amino Acids. (2010) 38:167–77. doi: 10.1007/s00726-008-0224-5
76. Mathioudakis D, Engel J, Welters ID, Dehne MG, Matejec R, Harbach H, et al. Pyruvate: immunonutritional effects on neutrophil intracellular amino or alpha-keto acid profiles and reactive oxygen species production. Amino Acids. (2011) 40:1077–90. doi: 10.1007/s00726-010-0731-z
77. Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. (2009) 461:1282–6. doi: 10.1038/nature08530
78. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, et al. Lactate metabolism in human lung tumors. Cell. (2017) 171:358–71.e9. doi: 10.1016/j.cell.2017.09.019
79. Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, et al. Glucose feeds the TCA cycle via circulating lactate. Nature. (2017) 551:115–8. doi: 10.1038/nature24057
80. Alarcón P, Manosalva C, Conejeros I, Carretta MD, Muñoz-Caro T, Silva LMR, et al. d(-) Lactic acid-induced adhesion of bovine neutrophils onto endothelial cells is dependent on neutrophils extracellular traps formation and CD11b expression. Front Immunol. (2017) 8:975. doi: 10.3389/fimmu.2017.00975
81. Awasthi D, Nagarkoti S, Sadaf S, Chandra T, Kumar S, Dikshit M. Glycolysis dependent lactate formation in neutrophils: a metabolic link between NOX-dependent and independent NETosis. Biochim Biophys Acta. (2019). doi: 10.1016/j.bbadis.2019.165542
82. Awasthi D, Nagarkoti S, Kumar A, Dubey M, Singh AK, Pathak P, et al. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Rad Biol Med. (2016) 93:190–203. doi: 10.1016/j.freeradbiomed.2016.01.004
83. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
84. Kumar S, Xu J, Perkins C, Guo F, Snapper S, Finkelman FD, et al. Cdc42 regulates neutrophil migration via crosstalk between WASp, CD11b, and microtubules. Blood. (2012) 120:3563–74. doi: 10.1182/blood-2012-04-426981
85. Kumar S, Xu J, Kumar RS, Lakshmikanthan S, Kapur R, Kofron M, et al. The small GTPase Rap1b negatively regulates neutrophil chemotaxis and transcellular diapedesis by inhibiting Akt activation. J Exp Med. (2014) 211:1741–58. doi: 10.1084/jem.20131706
86. Amankulor NM, Kim Y, Arora S, Kargl J, Szulzewsky F, Hanke M, et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. (2017) 31:774–86. doi: 10.1101/gad.294991.116
87. Vinolo MA, Rodrigues HG, Hatanaka E, Hebeda CB, Farsky SH, Curi R. Short-chain fatty acids stimulate the migration of neutrophils to inflammatory sites. Clin Sci. (2009) 117:331–8. doi: 10.1042/CS20080642
88. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
89. Monceaux V, Chiche-Lapierre C, Chaput C, Witko-Sarsat V, Prevost MC, Taylor CT, et al. Anoxia and glucose supplementation preserve neutrophil viability and function. Blood. (2016) 128:993–1002. doi: 10.1182/blood-2015-11-680918
90. Aoyama M, Kotani J, Usami M. Butyrate and propionate induced activated or non-activated neutrophil apoptosis via HDAC inhibitor activity but without activating GPR-41/GPR-43 pathways. Nutrition. (2010) 26:653–61. doi: 10.1016/j.nut.2009.07.006
91. Karinch AM, Pan M, Lin CM, Strange R, Souba WW. Glutamine metabolism in sepsis and infection. J Nutr. (2001) 131:2535S−8S. discussion: 2550S−1S. doi: 10.1093/jn/131.9.2535S
92. Singleton KD, Wischmeyer PE. Glutamine's protection against sepsis and lung injury is dependent on heat shock protein 70 expression. Am J Physiol Regul Integr Comp Physiol. (2007) 292:R1839–45. doi: 10.1152/ajpregu.00755.2006
93. Singleton KD, Serkova N, Beckey VE, Wischmeyer PE. Glutamine attenuates lung injury and improves survival after sepsis: role of enhanced heat shock protein expression. Crit Care Med. (2005) 33:1206–13. doi: 10.1097/01.CCM.0000166357.10996.8A
94. de Oliveira GP, Silva JD, de Araújo CC, Prota LF, Abreu SC, Madeira C, et al. Intravenous glutamine administration reduces lung and distal organ injury in malnourished rats with sepsis. Shock. (2014) 41:222–32. doi: 10.1097/SHK.0000000000000102
95. Castell L, Vance C, Abbott R, Marquez J, Eggleton P. Granule localization of glutaminase in human neutrophils and the consequence of glutamine utilization for neutrophil activity. J Biol Chem. (2004) 279:13305–10. doi: 10.1074/jbc.M309520200
96. Furukawa S, Saito H, Inoue T, Matsuda T, Fukatsu K, Han I, et al. Supplemental glutamine augments phagocytosis and reactive oxygen intermediate production by neutrophils and monocytes from postoperative patients in vitro. Nutrition. (2000) 16:323–9. doi: 10.1016/S0899-9007(00)00228-8
97. Pithon-Curi TC, Schumacher RI, Freitas JJ, Lagranha C, Newsholme P, Palanch AC, et al. Glutamine delays spontaneous apoptosis in neutrophils. American journal of physiology. Cell Physiol. (2003) 284:C1355–61. doi: 10.1152/ajpcell.00224.2002
98. Sasaki E, Umeda T, Takahashi I, Arata K, Yamamoto Y, Tanabe M, et al. Effect of glutamine supplementation on neutrophil function in male judoists. Luminescence. (2013) 28:442–9. doi: 10.1002/bio.2474
99. Muhling J, Nickolaus KA, Halabi M, Fuchs M, Krull M, et al. Alterations in neutrophil (PMN) free intracellular alpha-keto acid profiles and immune functions induced by L-alanyl-L-glutamine, arginine or taurine. Amino Acids. (2005) 29:289–300. doi: 10.1007/s00726-005-0223-8
100. Muhling J, Burchert D, Langefeld TW, Matejec R, Harbach H, et al. Pathways involved in alanyl-glutamine-induced changes in neutrophil amino- and alpha-keto acid homeostasis or immunocompetence. Amino Acids. (2007) 33:511–24. doi: 10.1007/s00726-006-0395-x
101. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
102. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
103. Egners A, Erdem M, Cramer T. The response of macrophages and neutrophils to hypoxia in the context of cancer and other inflammatory diseases. Mediat Inflamm. (2016) 2016:2053646. doi: 10.1155/2016/2053646
104. Thompson AA, Elks PM, Marriott HM, Eamsamarng S, Higgins KR, Lewis A, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood. (2014) 123:366–76. doi: 10.1182/blood-2013-05-500207
105. Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, et al. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. (2002) 100:3008–16. doi: 10.1182/blood-2002-02-0454
106. Wong SH, Francis N, Chahal H, Raza K, Salmon M, Scheel-Toellner D, et al. Lactoferrin is a survival factor for neutrophils in rheumatoid synovial fluid. Rheumatology. (2009) 48:39–44. doi: 10.1093/rheumatology/ken412
107. Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, Standiford TJ. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-{gamma}. Blood. (2008) 112:4250–8. doi: 10.1182/blood-2007-12-128967
108. Park DW, Jiang S, Tadie JM, Stigler WS, Gao Y, Deshane J, et al. Activation of AMPK enhances neutrophil chemotaxis and bacterial killing. Mol Med. (2013) 19:387–98. doi: 10.2119/molmed.2013.00065
109. Tirouvanziam R, Gernez Y, Conrad CK, Moss RB, Schrijver I, Dunn CE, et al. Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc Natl Acad Sci USA. (2008) 105:4335–9. doi: 10.1073/pnas.0712386105
110. Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol. (2010) 184:4062–8. doi: 10.4049/jimmunol.0903002
111. Lewis AJ, Billiar TR, Rosengart MR. Biology and metabolism of sepsis: innate immunity, bioenergetics, and autophagy. Surg Infect. (2016) 17:286–93. doi: 10.1089/sur.2015.262
112. Chiolero R, Revelly JP, Tappy L. Energy metabolism in sepsis and injury. Nutrition. (1997) 13:45S−51S. doi: 10.1016/S0899-9007(97)00205-0
113. Li X, Kondo Y, Bao Y, Staudenmaier L, Lee A, Zhang J, et al. Systemic adenosine triphosphate impairs neutrophil chemotaxis and host defense in sepsis. Crit Care Med. (2017) 45:e97–104. doi: 10.1097/CCM.0000000000002052
114. Alba-Loureiro TC, Hirabara SM, Mendonca JR, Curi R, Pithon-Curi TC. Diabetes causes marked changes in function and metabolism of rat neutrophils. J Endocrinol. (2006) 188:295–303. doi: 10.1677/joe.1.06438
115. Alba-Loureiro TC, Munhoz CD, Martins JO, Cerchiaro GA, Scavone C, Curi R, et al. Neutrophil function and metabolism in individuals with diabetes mellitus. Braz J Med Biol Res. (2007) 40:1037–44. doi: 10.1590/S0100-879X2006005000143
116. Kuwabara WM, Curi R, Alba-Loureiro TC. Autophagy is impaired in neutrophils from streptozotocin-induced diabetic rats. Front Immunol. (2017) 8:24. doi: 10.3389/fimmu.2017.00024
117. Ng HP, Zhou Y, Song K, Hodges CA, Drumm ML, Wang G. Neutrophil-mediated phagocytic host defense defect in myeloid Cftr-inactivated mice. PLoS ONE. (2014) 9:e106813. doi: 10.1371/journal.pone.0106813
118. Laval J, Touhami J, Herzenberg LA, Conrad C, Taylor N, Battini JL, et al. Metabolic adaptation of neutrophils in cystic fibrosis airways involves distinct shifts in nutrient transporter expression. J Immunol. (2013) 190:6043–50. doi: 10.4049/jimmunol.1201755
119. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. (2011) 7:691–9. doi: 10.1038/nrrheum.2011.132
120. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
121. Wright HL, Chikura B, Bucknall RC, Moots RJ, Edwards SW. Changes in expression of membrane TNF, NF-{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann Rheum Dis. (2011) 70:537–43. doi: 10.1136/ard.2010.138065
122. Cross A, Bucknall RC, Cassatella MA, Edwards SW, Moots RJ. Synovial fluid neutrophils transcribe and express class II major histocompatibility complex molecules in rheumatoid arthritis. Arthritis Rheum. (2003) 48:2796–806. doi: 10.1002/art.11253
123. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. (2014) 16:R122. doi: 10.1186/ar4579
124. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. (2010) 122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
125. Nencioni A, da Silva RF, Fraga-Silva RA, Steffens S, Fabre M, Bauer I, et al. Nicotinamide phosphoribosyltransferase inhibition reduces intraplaque CXCL1 production and associated neutrophil infiltration in atherosclerotic mice. Thrombosis Haemostas. (2014) 111:308–22. doi: 10.1160/TH13-07-0531
126. Gray GR, Stamatoyannopoulos G, Naiman SC, Kliman MR, Klebanoff SJ, Austin T, et al. Neutrophil dysfunction, chronic granulomatous disease, and non-spherocytic haemolytic anaemia caused by complete deficiency of glucose-6-phosphate dehydrogenase. Lancet. (1973) 2:530–4. doi: 10.1016/S0140-6736(73)92350-7
127. Shen XF, Cao K, Jiang JP, Guan WX, Du JF. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med. (2017) 21:1687–97. doi: 10.1111/jcmm.13112
128. Guo RF, Sun L, Gao H, Shi KX, Rittirsch D, Sarma VJ, et al. In vivo regulation of neutrophil apoptosis by C5a during sepsis. J Leukoc Biol. (2006) 80:1575–83. doi: 10.1189/jlb.0106065
129. Arraes SM, Freitas MS, da Silva SV, de Paula Neto HA, Alves-Filho JC, Auxiliadora Martins M, et al. Impaired neutrophil chemotaxis in sepsis associates with GRK expression and inhibition of actin assembly and tyrosine phosphorylation. Blood. (2006) 108:2906–13. doi: 10.1182/blood-2006-05-024638
130. Liu Z, Bone N, Jiang S, Park DW, Tadie JM, Deshane J, et al. AMP-activated protein kinase and glycogen synthase kinase 3β modulate the severity of sepsis-induced lung injury. Mol Med. (2015) 21:937–50. doi: 10.2119/molmed.2015.00198
131. Park SY, Shrestha S, Youn YJ, Kim JK, Kim SY, Kim HJ, et al. Autophagy primes neutrophils for neutrophil extracellular trap formation during sepsis. Am J Respirat Crit Care Med. (2017) 196:577–89. doi: 10.1164/rccm.201603-0596OC
132. Sumi Y, Woehrle T, Chen Y, Bao Y, Li X, Yao Y, et al. Plasma ATP is required for neutrophil activation in a mouse sepsis model. Shock. (2014) 42:142–7. doi: 10.1097/SHK.0000000000000180
133. Perner A, Nielsen SE, Rask-Madsen J. High glucose impairs superoxide production from isolated blood neutrophils. Intensive Care Med. (2003) 29:642–5. doi: 10.1007/s00134-002-1628-4
134. Joshi MB, Baipadithaya G, Balakrishnan A, Hegde M, Vohra M, Ahamed R, et al. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci Rep. (2016) 6:36362. doi: 10.1038/srep36362
135. Deng J, Lü S, Liu H, Liu B, Jiang C, Xu Q, et al. Homocysteine activates B cells via regulating PKM2-dependent metabolic reprogramming. J Immunol. (2017) 198:170–83. doi: 10.4049/jimmunol.1600613
136. Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, et al. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J Leukoc Biol. (2008) 83:1345–53. doi: 10.1189/jlb.0907658
137. Laval J, Ralhan A, Hartl D. Neutrophils in cystic fibrosis. Biol Chem. (2016) 397:485–96. doi: 10.1515/hsz-2015-0271
138. Group WW. Glucose-6-phosphate dehydrogenase deficiency. Bull World Health Organ. (1989) 67:601–11.
139. Wolach B, Ashkenazi M, Grossmann R, Gavrieli R, Friedman Z, Bashan N, et al. Diurnal fluctuation of leukocyte G6PD activity. A possible explanation for the normal neutrophil bactericidal activity and the low incidence of pyogenic infections in patients with severe G6PD deficiency in Israel. Pediatr Res. (2004) 55:807–13. doi: 10.1203/01.PDR.0000120680.47846.47
140. Ardati KO, Bajakian KM, Tabbara KS. Effect of glucose-6-phosphate dehydrogenase deficiency on neutrophil function. Acta Haematol. (1997) 97:211–5. doi: 10.1159/000203685
141. Robinson JM, Karnovsky ML, Karnovsky MJ. Glycogen accumulation in polymorphonuclear leukocytes, and other intracellular alterations that occur during inflammation. J Cell Biol. (1982) 95:933–42. doi: 10.1083/jcb.95.3.933
142. Kim SY, Chen LY, Yiu WH, Weinstein DA, Chou JY. Neutrophilia and elevated serum cytokines are implicated in glycogen storage disease type Ia. FEBS Lett. (2007) 581:3833–8. doi: 10.1016/j.febslet.2007.07.013
143. Chou JY, Jun HS, Mansfield BC. Glycogen storage disease type I and G6Pase-β deficiency: etiology and therapy. Nat Rev Endocrinol. (2010) 6:676–88. doi: 10.1038/nrendo.2010.189
144. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra19. doi: 10.1126/scitranslmed.3001180
145. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, et al. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. (2011) 3:73ra20. doi: 10.1126/scitranslmed.3001201
146. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
147. Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. (2010) 184:3284–97. doi: 10.4049/jimmunol.0902199
148. Li W, Sivakumar R, Titov AA, Choi SC, Morel L. Metabolic factors that contribute to lupus pathogenesis. Crit Rev Immunol. (2016) 36:75–98. doi: 10.1615/CritRevImmunol.2016017164
149. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Transl Med. (2015) 7:274ra18. doi: 10.1126/scitranslmed.aaa0835
150. Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. (2001) 167:1601–8. doi: 10.4049/jimmunol.167.3.1601
151. Cross A, Barnes T, Bucknall RC, Edwards SW, Moots RJ. Neutrophil apoptosis in rheumatoid arthritis is regulated by local oxygen tensions within joints. J Leukoc Biol. (2006) 80:521–8. doi: 10.1189/jlb.0306178
152. Wright HL, Thomas HB, Moots RJ, Edwards SW. Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology. (2015) 54:188–93. doi: 10.1093/rheumatology/keu299
153. Pi H, Zhou H, Jin H, Ning Y, Wang Y. Abnormal glucose metabolism in rheumatoid arthritis. Biomed Res Int. (2017) 2017:9670434. doi: 10.1155/2017/9670434
154. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. (2012) 110:875–88. doi: 10.1161/CIRCRESAHA.111.257535
155. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
156. Taylor PR, Roy S, Leal SM Jr, Sun Y, Howell SJ, et al. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin-2. Nat Immunol. (2014) 15:143–51. doi: 10.1038/ni.2797
157. Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. (2009) 16:183–94. doi: 10.1016/j.ccr.2009.06.017
158. McNab FW, Berry MP, Graham CM, Bloch SA, Oni T, Wilkinson KA, et al. Programmed death ligand 1 is over-expressed by neutrophils in the blood of patients with active tuberculosis. Eur J Immunol. (2011) 41:1941–7. doi: 10.1002/eji.201141421
159. Luo Q, Huang Z, Ye J, Deng Y, Fang L, Li X, et al. PD-L1-expressing neutrophils as a novel indicator to assess disease activity and severity of systemic lupus erythematosus. Arthrit Res Ther. (2016) 18:47. doi: 10.1186/s13075-016-0942-0
160. Wang TT, Zhao YL, Peng LS, Chen N, Chen W, Lv YP, et al. Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut. (2017) 66:1900–11. doi: 10.1136/gutjnl-2016-313075
161. Patel S, Fu S, Mastio J, Dominguez GA, Purohit A, Kossenkov A, et al. Unique pattern of neutrophil migration and function during tumor progression. Nat Immunol. (2018) 19:1236–47. doi: 10.1038/s41590-018-0229-5
Keywords: neutrophil functions, glycolysis, TCA cycle, pentose phosphate pathway, glutaminolysis, fatty acid oxidation, metabolic adaptation, diseases
Citation: Kumar S and Dikshit M (2019) Metabolic Insight of Neutrophils in Health and Disease. Front. Immunol. 10:2099. doi: 10.3389/fimmu.2019.02099
Received: 22 February 2019; Accepted: 20 August 2019;
Published: 20 September 2019.
Edited by:
Ivan Zanoni, Harvard Medical School, United StatesReviewed by:
Marco Di Gioia, Boston Children's Hospital and Harvard Medical School, United StatesCopyright © 2019 Kumar and Dikshit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sachin Kumar, U2FjaGluLmt1QGNkcmkucmVzLmlu; Madhu Dikshit, bWFkaHVkaWtzaGl0QHRoc3RpLnJlcy5pbg==; ZHJtYWRodWRpa3NoaXRAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.