 Madeleine A. Wemyss1,2†
Madeleine A. Wemyss1,2† Jaclyn S. Pearson1,2*†
Jaclyn S. Pearson1,2*†- 1Department of Molecular and Translational Research, Monash University, Clayton, VIC, Australia
- 2Department of Microbiology, Monash University, Clayton, VIC, Australia
Salmonella enterica subsp. enterica serovar Typhimurium (S. Typhimurium) is a Gram-negative bacterium with a broad host range that causes non-typhoidal salmonellosis in humans. S. Typhimurium infects epithelial cells and macrophages in the small intestine where it replicates in a specialized intracellular niche called the Salmonella-containing vacuole (SCV) and promotes inflammation of the mucosa to induce typically self-limiting gastroenteritis. Virulence and spread of the bacterium is determined in part by the host individual's ability to limit the infection through innate immune responses at the gastrointestinal mucosa, including programmed cell death. S. Typhimurium however, has evolved a myriad of mechanisms to counteract or exploit host responses through the use of Type III Secretion Systems (T3SS), which allow the translocation of virulence (effector) proteins into the host cell for the benefit of optimal bacterial replication and dissemination. T3SS effectors have been found to interact with apoptotic, necroptotic, and pyroptotic cell death cascades, interfering with both efficient clearance of the bacteria and the recruitment of neutrophils or dendritic cells to the area of infection. The interplay of host inflammation, programmed cell death responses, and bacterial defenses in the context of non-typhoidal Salmonella (NTS) infection is a continuing area of interest within the field, and as such has been reviewed here.
Pathogenicity and Virulence of Salmonella enterica Serovar Typhimurium
Infections caused by Salmonella enterica are a major challenge in both human and animal health. Salmonella enterica subsp. enterica serovars are categorized by their disease phenotypes into typhoidal (Typhi and Paratyphi) and non-typhoidal Salmonella (NTS) serovars (e.g., S. Typhimurium). Whereas, typhoidal serovars cause invasive disease and are human restricted, NTS serovars cause disease in a wide range of mammals and birds and typically cause self-limiting gastroenteritis (salmonellosis) in humans, with the bacteria restricted to the gastrointestinal mucosa (1, 2). S. Typhimurium is acquired via the fecal-oral route from consumption of raw or contaminated poultry products, and causes the majority of notified NTS infections in Australia (3, 4). In immunocompromised individuals, S. Typhimurium can cause invasive disease that requires antibiotic treatment or hospitalization. Murine infections with S. Typhimurium result in invasive disease and bacteremia, and thus are a more representative model of invasive salmonellosis but are nevertheless used to great effect to study the pathogenesis of S. Typhimurium in vivo.
The interactions between NTS and host cell processes during host invasion and the initial establishment of infection have been reviewed previously by LaRock et al. and as such are only briefly described here (5). Once ingested, S. Typhimurium enters the gastrointestinal tract and uses flagella to access the epithelial layer of the terminal ileum. Inflammatory responses in the epithelium release key nutrients required by the bacteria, also causing diarrheal symptoms that promote transmission (6, 7). Following contact with the epithelium, S. Typhimurium utilizes a Type III Secretion System (T3SS) encoded on Salmonella pathogenicity island-1 (SPI-1) to translocate effector proteins (such as SopE2, SipA, and SopB) into the epithelial cell cytosol, inducing actin rearrangement, membrane ruffling, and non-phagocytic cellular uptake of the bacteria into the host cell (Figure 1) (8–10). Inside the intracellular space, flagella are no longer required for motility of the bacteria, and are typically downregulated in order to avoid host immune responses (11–14). Internalization of S. Typhimurium causes formation of an endosome termed the early Salmonella-containing vacuole (SCV). Here, a second T3SS (encoded by the SPI-2 locus) is used to translocate virulence proteins such as SifA, SopD2, and SseJ, acidifying the vacuole and maturing the SCV into the ideal replicative niche for the bacteria (15). The late stage SCV enables efficient bacterial replication, while interconnected networks of Salmonella-induced filaments (SIFs) allow enclosed bacteria to acquire nutrients (15, 16). Other SPI-2 effectors prevent lysosomal fusion with the SCV, inhibiting recruitment of lysosomal enzymes and trafficking markers that would promote degradation of the vacuole (15, 17). In epithelial cells, subpopulations of S. Typhimurium have been observed in the cytosol, resulting in bacterial hyper-replication and host cell extrusion (18). Host guanylate-binding proteins (GBPs), expressed following Type I or II interferon (IFN) signaling, can also lyse the SCV, exposing S. Typhimurium to the cytosol (19–21).
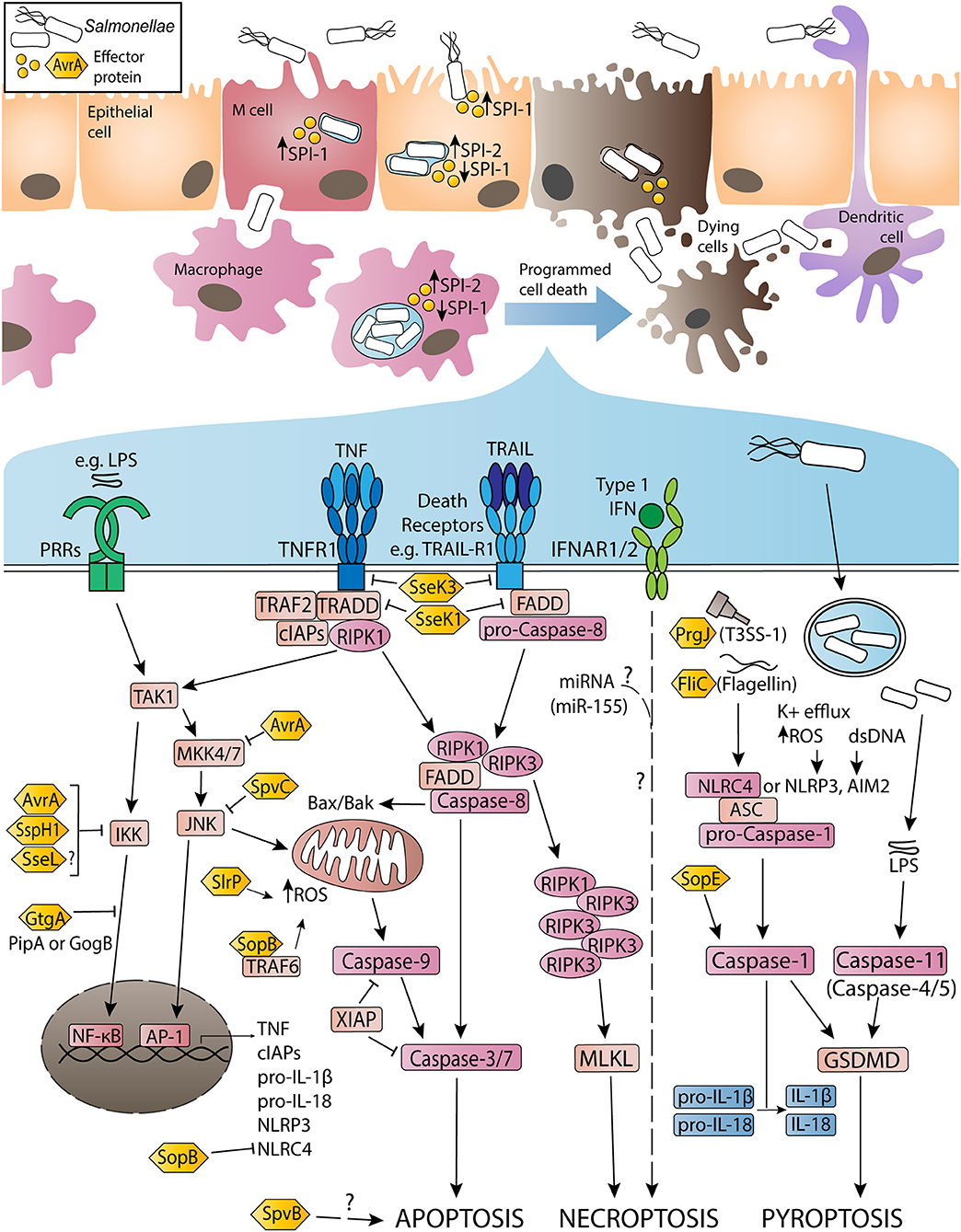
Figure 1. Activation and inhibition of apoptosis, necroptosis and pyroptosis by non-typhoidal Salmonella virulence (effector) proteins and other stimuli during Salmonella enterica serovar Typhimurium infection. Non-typhoidal Salmonella species invade intestinal epithelial cells through the use of SPI-1 effectors to induce membrane ruffling and actin rearrangement, resulting in non-phagocytic uptake of the bacteria. Alternatively, Salmonella uptake can occur due to M cell mediated transport across the epithelial barrier, or through sampling by phagocytic cells such as dendritic cells or macrophages. Once internalized, the SPI-1 T3SS and effectors are downregulated, while SPI-2 is upregulated to promote SCV formation and facilitate Salmonella replication. Throughout the infection, both SPI-1 and SPI-2 effector proteins interact with host innate immune pathways to either activate or inhibit inflammatory responses and programmed cell death. Signaling cascades have been simplified for clarity and are discussed in more detail in-text.
Cytosolic S. Typhimurium enable the detection of pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS) and flagellin, by pattern recognition receptors (PRRs) or Nod-like receptors (NLRs). PRRs act to recruit immune cells to infected tissues and limit bacterial virulence through the activation of pro-inflammatory signaling. The ability of Salmonella infection to induce tumor necrosis factor (TNF) production in epithelial cells and macrophages is well-documented (22–24). TNF signaling typically reinforces the production of pro-survival cytokines and anti-apoptotic factors via nuclear factor kappa B (NF-κB) or mitogen-activated protein kinase (MAPK) signaling cascades (25–27). However, effector proteins such as GtgA, SspH1, SptP, and potentially SseL can prevent the activation of these pathways, instead driving TNF signaling toward programmed cell death cascades (28–34). These include apoptosis, necroptosis, and pyroptosis, and are triggered by TNF and other death receptor ligands, or inflammasome activation (Figure 1). Death of the host cell allows escape of S. Typhimurium into the extracellular space, and uptake of the bacteria by professional phagocytes. Neutrophils play a key role in the overall clearance of S. Typhimurium, killing the phagocytosed bacteria through the activity of reactive oxygen species (ROS), while infected dendritic cells and macrophages can spread the bacteria to the mesenteric lymph nodes, spleen, and liver (12, 35–37).
Apoptotic Cell Death Pathways During Salmonella Infection
Apoptosis is a caspase-dependent form of programmed cell death, induced in damaged or stressed cells in response to intrinsic or extrinsic signaling cascades (38). The apoptotic process results in DNA fragmentation, phosphatidylserine exposure, formation of apoptotic bodies, and the display of “eat me” signals to prompt phagocytic clearance of the dying cell. Intrinsic apoptosis is triggered by DNA damage, accumulation of ROS or endoplasmic reticulum (ER) stress, resulting in mitochondrial outer membrane permeabilization and activation of caspase-9. Caspase-9 catalyzes the activation of caspase-3 and caspase-7, which execute the biochemical and morphological changes characteristic of apoptosis (38). In contrast, extrinsic apoptosis responds to ligand or cytokine binding to transmembrane death receptors on the surface of the cell. Death receptors possess an apoptosis-activating death domain, and include receptors such as Fas, TNFR1, and TRAIL-R1. Upon TNF stimulation, TNFR1 recruits adaptor proteins such as TNFR1-associated death domain protein (TRADD), TNFR-associated factor 2 (TRAF2) and receptor-interacting serine/threonine-protein kinase 1 (RIPK1) (Figure 1). RIPK1 is subject to ubiquitylation and phosphorylation events that direct TNFR1 signaling toward pro-survival NF-κB activation (39). In the absence of modifications, RIPK1 associates with pro-caspase-8, TRADD and Fas-associated protein with death domain (FADD) to form a cytosolic secondary signaling complex (40, 41). Cellular FLICE-like inhibitory protein (cFLIP) also regulates complex assembly by inhibiting caspase-8 activation (42). Secondary complex activation allows caspase-8 to activate caspase-3/-7 and subsequent apoptosis.
During S. Typhimurium infection, autocrine or paracrine TNF signaling triggers cell death responses by initiating extrinsic apoptosis. Salmonella effector proteins also induce apoptosis via these signaling pathways. SlrP is an E3 ubiquitin ligase translocated by both SPI-1 and SPI-2 that interacts with thioredoxin-1 (Trx1) and the ER chaperone protein, ERdj3 (43–46). Expression of SlrP increased cytotoxicity in infected HeLa cells, suggesting a role for SlrP in inducing intrinsic apoptosis in infected epithelial cells (43, 44). Additionally, translocation of SPI-2 effector SpvB (an ADP-ribosylase) promotes apoptosis in human monocyte-derived macrophages (HMDMs), potentially due to loss of polymerized F-actin (47–50). SpvB may have a similar effect in S. Dublin-infected HT-29 cells, although apoptosis was markedly delayed in these cells (28 h post-infection in vitro) (51). However, the mechanism by which SpvB promotes apoptosis remains unclear.
Alternatively, effectors such as SopB may have a role in preventing intrinsic apoptosis. SopB (also known as SigD) is a phosphoinositide phosphatase translocated by SPI-1 that has multiple reported virulence functions (8, 52, 53). Infection of mouse embryonic fibroblasts revealed that SopB is ubiquitylated by TRAF6, potentially as a mechanism of directing SopB activity within the host cell (54, 55). SopB-TRAF6 interactions prevent the recruitment of TRAF6 to the mitochondria, inhibiting accumulation of ROS in the organelle, thus preventing intrinsic apoptosis (56). SopB phosphatase activity in epithelial cells also mediates the recruitment of Rho and Ras family GTPases to the site of infection, promoting pro-survival Akt signaling and inhibiting apoptotic responses downstream (57–59). Another SPI-1 translocated effector that alters apoptotic pathways is AvrA, which displays deubiquitinase and acetyl-transferase activity. Studies of S. Typhimurium-infected HeLa or HCT116 cells demonstrated that AvrA deubiquitylates IκBα to suppress pro-survival NF-κB activation (60, 61). Interestingly, in vivo mouse infections, as well as transfection of AvrA into HEK293T cells, indicated that AvrA also prevents apoptotic responses by acetylating MAPK kinase 4 (MKK4) and inhibiting the c-Jun N-terminal-kinase (JNK) pathway (62–64). Similarly, SpvC (a phosphothreonine lyase) acts to both dampen inflammation and suppress apoptosis by inactivating members of the MAPK pathway (65, 66). While suppression of both pro-survival signaling and pro-apoptotic pathways may initially seem counterintuitive, it is likely that this duality allows Salmonella to prolong infection of epithelial cells, allowing greater opportunities for replication in this cell type.
Salmonella Infection and Necroptotic Cell Death
Necroptosis is a caspase-independent lytic form of programmed cell death that results in characteristic pore formation and the release of cellular contents and highly inflammatory damage-associated molecular patterns (DAMPs) into the extracellular space (67). Initially triggered by TNF binding to TNFR1, necroptosis occurs when caspase-8 is non-functional or inhibited. In the absence of active caspase-8, deubiquitylated RIPK1 is able to interact with RIPK3, subsequently forming an amyloid-like complex (called the necrosome), activating RIPK3 via autophosphorylation events (68). Active RIPK3 then mediates the phosphorylation of mixed lineage kinase domain-like protein (MLKL), enabling MLKL oligomerization and migration to the plasma membrane, triggering membrane permeabilization and lytic cell death (Figure 1) (68). Although RIPK3 and MLKL are critical for the induction of necroptosis, the precise mechanism by which active MLKL executes necroptosis remains unclear (38, 69, 70). Released DAMPs induce inflammatory responses in neighboring cells, promoting recruitment of innate immune cells and mediating tissue pathology in the immediate area (71).
Observations of necroptosis in response to S. Typhimurium infection have included studies comparing infected C57BL/6J wild type (WT) or type I IFN alpha/beta receptor 1 deficient (Ifnar1−/−) mice (72). Type I IFNs act through heterodimeric IFNAR1/IFNAR2 complexes to activate Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling cascades, resulting in the transcription of interferon-stimulated genes (ISGs) (73). Following intravenous S. Typhimurium infection, Ifnar1−/− mice experienced improved survival compared to WT mice, while infected Ifnar1−/− bone marrow derived macrophages (BMDMs) experienced reduced rates of cytotoxicity in vitro, with decreased activation of RIPK1 and RIPK3 (72). Immunoprecipitation of IFNAR1 in WT BMDMs indicated RIPK1 associates with IFNAR1 following Type I IFN stimulation, while in vivo infection of Ripk3−/− mice induced similar cytotoxicity to Ifnar1−/− mice (72). Robinson et al. thus proposed a role for Type I IFN signaling in inducing necroptosis in S. Typhimurium-infected macrophages (72). Later work found that signaling downstream of IFNAR1/RIPK1/RIPK3 interactions resulted in recruitment of phosphoglycerate mutase family member 5 (PGAM5) (74). PGAM5 recruitment by RIPK3 was suggested as a mechanism of promoting or executing necroptosis in S. Typhimurium-infected BMDMs via impaired production of antioxidants, resulting in ROS-mediated mitochondrial damage (74, 75). However, studies outside the S. Typhimurium infection context did not support PGAM5 as a mediator of necroptosis, instead proposing that PGAM5 counteracts necroptosis by promoting autophagic degradation of mitochondria (inhibiting ROS production) (70, 76, 77).
Other explorations of necroptosis in the context of S. Typhimurium infection involved the use of qRT-PCR techniques to assess the expression of micro RNAs (miRNAs) induced by infection in RAW264.7 cells (78). A highly upregulated miRNA, miR-155, mediated cytotoxicity levels similar to S. Typhimurium-infected cells when transfected into RAW264.7s (78). Further in vitro transfections indicated that miR-155 induced RIPK1 and RIPK3 phosphorylation (indicative of necroptosis) by 18 h post-treatment, as well as cleavage of poly (ADP-ribose) polymerase-1 (PARP-1) in a similar manner to S. Typhimurium infection (78). Treatment with RIPK1 inhibitor necrostatin-1s partially rescued cell viability in miR-155 transfected cells, supporting a role for necroptosis in contributing to cytotoxicity (78). The authors suggested that PARP-1 activation occurs downstream of RIPK1/RIPK3 activation, however existing work in TNF-stimulated L929 cells instead proposes that PARP-1 contributes to a separate programmed necrosis pathway (78, 79).
Virulence proteins may also play a role in mediating host necroptotic responses during S. Typhimurium infection. Salmonella secreted effector K1 (SseK1), SseK2, and SseK3 are a family of related virulence proteins with glycosyltransferase activity that share high sequence homology with the Arg-GlcNAc transferase, NleB, found in attaching and effacing (A/E) pathogens (80–82). SseK effectors reportedly inhibit TNF-induced NF-κB signaling and cell death in macrophages, through arginine glycosylation of FADD and TRADD by SseK1 and SseK3, respectively (83). In vitro infections of RAW264.7 cells with ΔsseK123 S. Typhimurium showed similar levels of caspase-3/-7 activation when compared to WT infection, but resulted in higher levels of MLKL phosphorylation, indicating that SseK1 and SseK3 may specifically inhibit necroptotic cell death (83). Reports of SseK binding targets remain inconclusive, with suggested glycosylation targets for SseK1 including GAPDH, FADD, and TRADD, while SseK2 may glycosylate FADD (83–85). Recently, mass spectrometry-based screens have identified TNFR1 and TRAIL-R as novel glycosylation targets of SseK3, and demonstrated that TRADD is the preferred binding target of SseK1 (85). Although the specific actions of SseK effectors have yet to be confirmed, collectively these results suggest that SseK1 and SseK3 modify TNFR superfamily members as well as TRADD or FADD, thus inhibiting both TNF-mediated NF-κB signaling and cell death via apoptosis or necroptosis.
Inflammasome Activation and Pyroptotic Cell Death During Salmonella Infection
Pyroptosis is a highly inflammatory, caspase-dependent form of lytic cell death characterized by pore formation and release of active IL-1β and IL-18 (86). Originally thought to be a caspase-1 dependent form of apoptosis or necrosis, pyroptosis is an important host defense mechanism against S. Typhimurium (87–89). Typically, pyroptosis in S. Typhimurium-infected cells is triggered by the sensing of flagellin (FliC and FljB) or PrgJ (a SPI-1 rod protein) by NLR family apoptosis inhibitory proteins (NAIPs), which then interact with NLR family caspase activation and recruitment domain (CARD)-containing protein 4 (NLRC4) to trigger assembly of a multiprotein complex called the NLRC4 inflammasome (90–92). NLRC4 recruits pro-caspase-1 via shared CARD domains, and can also recruit apoptosis-associated speck-like protein containing a CARD domain (ASC), to assemble the inflammasome and induce the proteolytic activation of caspase-1 (Figure 1) (92). Active caspase-1 mediates pyroptosis by cleaving gasdermin-D (GSDMD), producing an N-terminal segment that forms multimeric pores in the cell membrane and releases cellular contents into the extracellular space (93, 94). Caspase-1 also cleaves IL-1β and IL-18 into their active forms, allowing their release through the GSDMD-N pores, or following the process of necrosis or others [as reviewed by Eder et al. (95)] (93, 95–98).
Other sensors capable of inducing pyroptosis via ASC-caspase-1 inflammasomes include NLRP3 (senses K+ efflux or increased ROS), AIM2 (detects cytosolic dsDNA) and pyrin (senses inhibition of RhoA GTPase activity) (99–103). Both NLRP3 and NLRC4 contribute to IL-1β and IL-18 maturation and pyroptosis in S. Typhimurium-infected macrophages (104). Activated NLRC4 amplifies caspase-1 activation in infected macrophages by recruiting NLRP3, forming a single inflammasome complex with ASC that mediates pyroptotic responses downstream (104–106). Alternatively, non-canonical inflammasome pathways can induce pyroptosis through the sensing of cytosolic LPS by murine caspase-11 (or human caspase-4/-5) which cleaves GSDMD independent of caspase-1 activation, however caspase-11 does not cleave IL-1β or IL-18, thus reducing pro-inflammatory cytokine release (Figure 1) (107–112). Both NLRC4 and non-canonical inflammasome activation play a role in epithelial cell responses to infection, and may help reduce bacterial dissemination throughout the intestinal mucosa (108, 113–116).
Crosstalk with caspase-8 and apoptotic pathways can also promote inflammasome activation in S. Typhimurium-infected cells. Studies of NLRP3 and NLRC4 interactions during S. Typhimurium infection detected IL-1β maturation mediated by ASC-caspase-8 specks, suggesting a role for caspase-8 as an inflammasome effector (105, 117). Other studies have proposed roles for caspase-8 in priming inflammasome activation, or coordinating cleavage of caspase-1 in the absence of NLRP3 or NLRC4 (118). Although not yet demonstrated, effectors such as SlrP, which induce downstream ROS accumulation, could contribute to inflammasome activation and pyroptotic responses due to NLRP3 detection of ROS. However, a study of IL-1β release in a murine S. Typhimurium in vivo infection context found that SlrP signaling inhibited IL-1β activation, contradicting this idea (119). Aside from SlrP, effectors such as SipB, SopE, or SopB may influence pyroptosis in S. Typhimurium-infected macrophage. Following secretion, SipB interacts with SipC to form a translocon pore, facilitating SPI-1 effector translocation into the host cell (120). SipB is reportedly sufficient to induce caspase-1-mediated “apoptosis” and IL-18 maturation in SipB transfected or S. Typhimurium-infected dendritic cells and peritoneal macrophages, potentially via direct interactions with caspase-1 (121–123). These results likely indicate pyroptosis, however the mechanisms by which SipB interact with caspase-1 or the inflammasome remain unclear.
S. Typhimurium SPI-1 effector SopE is a guanine nucleotide exchange factor that catalyzes the activation of host cell Rho GTPases such as Cdc42 and Rac1 (124). Activation of Rac1 by SopE has been reported to induce caspase-1 activation and IL-1β secretion during S. Typhimurium infection of HeLa or RAW264.7 cells, and in vivo infection of murine enterocytes (125). SopE-induced caspase-1 activation in macrophages was not due to NLRC4 sensing of flagellin, suggesting an alternative sensor mechanism (126). Other Gram-negative bacteria possess effectors that modify Rho GTPase activity, for instance Yersinia spp. effector YopT, which inhibits the activity of RhoA (102, 127). This RhoA inactivation allows assembly of the pyrin inflammasome, resulting in downstream caspase-1 activity and pyroptosis in infected cells (102). This suggests interesting avenues of research for SopE-induced caspase-1 activation; however, pyrin activation has not been observed in response to changes in Rac1 or Cdc42 activity (127). In contrast to SopE, SPI-1 effector SopB plays a role in dampening inflammasome activation. SopB has been associated with the downregulation of NLRC4 in S. Typhimurium-infected macrophages and B cells (128–130). NLRC4 depletion was associated with reductions in both IL-1β maturation and cytotoxicity in S. Typhimurium-infected B cells, and was determined to be the result of Akt/YAP pathway activation (128, 130). Loss of NLRC4 inhibits the dominant inflammasome involved in the pyroptotic response to S. Typhimurium infection, thus allowing the bacteria better opportunities for replication before escaping the host cell.
Lastly, although S. Typhimurium effectors both activate and inhibit inflammasome activation, current understandings of these effectors suggest that their translocation is under temporal and spatial control by the bacteria due to their translocating T3SS type. A recent study demonstrated that mutation of the SPI-1 T3SS resulted in decreased HMDM cytotoxicity and IL-1β release, while infection with ΔSPI-2 S. Typhimurium induced rapid cell death and IL-1β production in these cells (131). SPI-2 mutation also resulted in increased expression of SPI-1 effectors detectable by NLRC4 (FljB, PrgI, and PrgJ), suggesting that SPI-2 activity helps suppress the translocation of SPI-1 effectors later in infection (131).
Concluding Remarks
Investigating cell death in the context of S. Typhimurium infection has revealed highly complex interactions between host signaling cascades and bacterial virulence effectors. Tightly regulated control of T3SS effector translocation supports bacterial requirements at different infection stages, allowing S. Typhimurium to evade or promote cell death responses. Our understanding of Salmonella-host interactions is continually evolving, with virulence mechanisms and effector proteins still to be characterized and improved in vitro and in vivo models for testing hypotheses frequently emerging. While the current literature does not describe immediate applications for exploiting programmed cell death in treatment of salmonellosis, further exploration of NTS virulence factors could help characterize clinical isolates, leading to personalized therapies and improved patient outcomes. Additionally, the high specificity of Salmonella effector proteins could prove crucial to the development of novel genome or proteome editing tools (such as the recently described use of effectors from Shigella flexneri) (132). Overall, exploration of pathogen-mediated cell death provides crucial insights into how bacteria can mediate survival and dissemination between host cells and can further improve our general understanding of the importance of cell death in counteracting bacterial pathogenesis.
Author Contributions
MW wrote the initial manuscript and designed the figure. JP and MW edited and revised the manuscript. Both authors read and approved the final manuscript.
Funding
MW was supported by an Australian Government Research Training Program (RTP) Scholarship at Monash University, Melbourne, Australia. JP was supported by a National Health and Medical Research Council (NHMRC) Career Development Fellowship (APP1159230).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Dr. Deborah Williamson and Dr. Rebecca Ambrose for critical reading of the manuscript.
Abbreviations
A/E, attaching and effacing; AIM2, absent in melanoma 2; AP-1, activator protein 1; ASC, apoptosis-associated speck-like CARD-containing protein; AvrA, avirulence gene A; CARD, caspase activation and recruitment domain; cIAP, cellular inhibitor of apoptosis; EHEC, enterohemorrhagic Escherichia coli, EPEC, enteropathogenic E. coli; FADD, Fas-associated protein with death domain; GBP, guanylate-binding protein; GSDMD, gasdermin D; IFN, interferon; IFNAR1, interferon alpha/beta receptor alpha chain; IL-1β, interleukin 1β; IKK, inhibitor of kappa kinase complex; JNK, c-Jun N-terminal kinase; LUBAC, linear ubiquitin chain assembly complex; MAPK, mitogen activated protein kinase; MLKL, mixed lineage kinase domain-like protein; MKK4, MAPK kinase 4; NF-κB, nuclear factor kappa B; NLR, Nod-like receptor; NLRC4, NLR family CARD domain-containing 4; NLRP3, NLR family pyrin domain-containing protein 3; PAMP, pathogen-associated molecular pattern; PipA, pathogenicity island-encoded protein A; PRR, pattern recognition receptor; RIPK, receptor interacting serine/threonine protein kinase; TAK1, transforming growth factor beta-activated kinase 1; TLR, Toll-like receptor; TNF, tumor necrosis factor; TNFR1, TNF receptor 1; TRADD, TNFR1-associated death domain protein; TRAF, TNFR associated factor; TRAIL, TNF-related apoptosis-inducing ligand; TRAIL-R1, TRAIL receptor 1; SIF, Salmonella induced filaments; SlrP, Salmonella leucine-rich repeat protein; SopB, Salmonella outer protein B; SpvB, Salmonella plasmid virulence gene B; SseL, Salmonella secreted effector L.
References
1. Broz P, Ohlson MB, Monack DM. Innate immune response to Salmonella Typhimurium, a model enteric pathogen. Gut Microbes. (2012) 3:62–70. doi: 10.4161/gmic.19141
2. Gal-Mor O, Boyle EC, Grassl GA. Same species, different diseases: how and why typhoidal and non-typhoidal Salmonella enterica serovars differ. Front Microbiol. (2014) 5:391. doi: 10.3389/fmicb.2014.00391
3. Moffatt CR, Musto J, Pingault N, Miller M, Stafford R, Gregory J, et al. Salmonella Typhimurium and outbreaks of egg-associated disease in Australia, 2001 to 2011. Foodborne Pathog Dis. (2016) 13:379–85. doi: 10.1089/fpd.2015.2110
4. Wilson HL, Kennedy KJ, Moffatt CRM. Epidemiology of non-typhoid Salmonella infection in the Australian Capital Territory over a 10-year period. Intern Med J. (2018) 48:316–23. doi: 10.1111/imj.13625
5. LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol. (2015) 13:191–205. doi: 10.1038/nrmicro3420
6. Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. (2010) 467:426–9. doi: 10.1038/nature09415
7. Byndloss MX, Rivera-Chavez F, Tsolis RM, Baumler AJ. How bacterial pathogens use type III and type IV secretion systems to facilitate their transmission. Curr Opin Microbiol. (2017) 35:1–7. doi: 10.1016/j.mib.2016.08.007
8. Raffatellu M, Wilson RP, Chessa D, Andrews-Polymenis H, Tran QT, Lawhon S, et al. SipA, SopA, SopB, SopD, and SopE2 contribute to Salmonella enterica serotype Typhimurium invasion of epithelial cells. Infect Immun. (2005) 73:146–54. doi: 10.1128/IAI.73.1.146-154.2005
9. Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS, Wang D, et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe. (2012) 12:645–56. doi: 10.1016/j.chom.2012.10.009
10. Zhang K, Riba A, Nietschke M, Torow N, Repnik U, Putz A, et al. Minimal SPI1-T3SS effector requirement for Salmonella enterocyte invasion and intracellular proliferation in vivo. PLoS Pathog. (2018) 14:e1006925. doi: 10.1371/journal.ppat.1006925
11. Cummings LA, Wilkerson WD, Bergsbaken T, Cookson BT. In vivo, fliC expression by Salmonella enterica serovar Typhimurium is heterogeneous, regulated by ClpX, and anatomically restricted. Mol Microbiol. (2006) 61:795–809. doi: 10.1111/j.1365-2958.2006.05271.x
12. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. (2010) 11:1136–42. doi: 10.1038/ni.1960
13. Lai MA, Quarles EK, Lopez-Yglesias AH, Zhao X, Hajjar AM, Smith KD. Innate immune detection of flagellin positively and negatively regulates Salmonella infection. PLoS ONE. (2013) 8:e72047. doi: 10.1371/journal.pone.0072047
14. Sporing I, Felgner S, Preusse M, Eckweiler D, Rohde M, Haussler S, et al. Regulation of flagellum biosynthesis in response to cell envelope stress in Salmonella enterica serovar Typhimurium. MBio. (2018) 9:e00736-17. doi: 10.1128/mBio.00736-17
15. Knuff K, Finlay BB. What the SIF is happening-the role of intracellular Salmonella-induced filaments. Front Cell Infect Microbiol. (2017) 7:335. doi: 10.3389/fcimb.2017.00335
16. Liss V, Swart AL, Kehl A, Hermanns N, Zhang Y, Chikkaballi D, et al. Salmonella enterica remodels the host cell endosomal system for efficient intravacuolar nutrition. Cell Host Microbe. (2017) 21:390–402. doi: 10.1016/j.chom.2017.02.005
17. Jennings E, Thurston TLM, Holden DW. Salmonella SPI-2 type III secretion system effectors: molecular mechanisms and physiological consequences. Cell Host Microbe. (2017) 22:217–31. doi: 10.1016/j.chom.2017.07.009
18. Castanheira S, Garcia-Del Portillo F. Salmonella populations inside host cells. Front Cell Infect Microbiol. (2017) 7:432. doi: 10.3389/fcimb.2017.00432
19. Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature. (2014) 509:366–70. doi: 10.1038/nature13157
20. Ingram JP, Brodsky IE, Balachandran S. Interferon-gamma in Salmonella pathogenesis: new tricks for an old dog. Cytokine. (2017) 98:27–32. doi: 10.1016/j.cyto.2016.10.009
21. Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J Leukoc Biol. (2017) 101:143–50. doi: 10.1189/jlb.4MR0516-223R
22. Jung HC, Eckmann L, Yang SK, Panja A, Fierer J, Morzycka-Wroblewska E, et al. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest. (1995) 95:55–65. doi: 10.1172/JCI117676
23. Ciacci-Woolwine F, Blomfield IC, Richardson SH, Mizel SB. Salmonella flagellin induces tumor necrosis factor alpha in a human promonocytic cell line. Infect Immun. (1998) 66:1127–34.
24. Pietilä TE, Veckman V, Kyllönen P, Lähteenmäki K, Korhonen TK, Julkunen I. Activation, cytokine production, and intracellular survival of bacteria in Salmonella-infected human monocyte-derived macrophages and dendritic cells. J Leukoc Biol. (2005) 78:909–20. doi: 10.1189/jlb.1204721
25. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. (2013) 13:679–92. doi: 10.1038/nri3495
26. Liu T, Zhang L, Joo D, Sun SC. NF-κB signaling in inflammation. Signal Transduct Target Ther. (2017) 2:17023. doi: 10.1038/sigtrans.2017.23
27. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. (2017) 17:545–58. doi: 10.1038/nri.2017.52
28. Lin SL, Le TX, Cowen DS. SptP, a Salmonella typhimurium type III-secreted protein, inhibits the mitogen-activated protein kinase pathway by inhibiting Raf activation. Cell Microbiol. (2003) 5:267–75. doi: 10.1046/j.1462-5822.2003.t01-1-00274.x
29. Haraga A, Miller SI. A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cell Microbiol. (2006) 8:837–46. doi: 10.1111/j.1462-5822.2005.00670.x
30. Rytkonen A, Poh J, Garmendia J, Boyle C, Thompson A, Liu M, et al. SseL, a Salmonella deubiquitinase required for macrophage killing and virulence. Proc Natl Acad Sci USA. (2007) 104:3502–7. doi: 10.1073/pnas.0610095104
31. Le Negrate G, Faustin B, Welsh K, Loeffler M, Krajewska M, Hasegawa P, et al. Salmonella secreted factor L deubiquitinase of Salmonella Typhimurium inhibits NF-κB, suppresses IκBα ubiquitination and modulates innate immune responses. J Immunol. (2008) 180:5045–56. doi: 10.4049/jimmunol.180.7.5045
32. Mesquita FS, Holden DW, Rolhion N. Lack of effect of the Salmonella deubiquitinase SseL on the NF-kappaB pathway. PLoS ONE. (2013) 8:e53064. doi: 10.1371/journal.pone.0053064
33. Sun H, Kamanova J, Lara-Tejero M, Galan JE. A family of Salmonella type III secretion effector proteins selectively targets the NF-kappaB signaling pathway to preserve host homeostasis. PLoS Pathog. (2016) 12:e1005484. doi: 10.1371/journal.ppat.1005484
34. Wu M, El Qaidi S, Hardwidge PR. SseL deubiquitinates RPS3 to inhibit its nuclear translocation. Pathogens. (2018) 7:86. doi: 10.3390/pathogens7040086
35. Pietro M. Immunity to systemic Salmonella infections. Curr Mol Med. (2002) 2:393–406. doi: 10.2174/1566524023362492
36. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. (2011) 21:103–15. doi: 10.1038/cr.2010.178
37. Voedisch S, Koenecke C, David S, Herbrand H, Forster R, Rhen M, et al. Mesenteric lymph nodes confine dendritic cell-mediated dissemination of Salmonella enterica serovar Typhimurium and limit systemic disease in mice. Infect Immun. (2009) 77:3170–80. doi: 10.1128/IAI.00272-09
38. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
39. Silke J, Meier P. Inhibitor of apoptosis (IAP) proteins-modulators of cell death and inflammation. Cold Spring Harb Perspect Biol. (2013) 5:a008730. doi: 10.1101/cshperspect.a008730
40. Moquin DM, McQuade T, Chan FK. CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE. (2013) 8:e76841. doi: 10.1371/journal.pone.0076841
41. Annibaldi A, Meier P. Checkpoints in TNF-induced cell death: implications in inflammation and cancer. Trends Mol Med. (2018) 24:49–65. doi: 10.1016/j.molmed.2017.11.002
42. Tsuchiya Y, Nakabayashi O, Nakano H. FLIP the switch: regulation of apoptosis and necroptosis by cFLIP. Int J Mol Sci. (2015) 16:30321–41. doi: 10.3390/ijms161226232
43. Bernal-Bayard J, Ramos-Morales F. Salmonella type III secretion effector SlrP is an E3 ubiquitin ligase for mammalian thioredoxin. J Biol Chem. (2009) 284:27587–95. doi: 10.1074/jbc.M109.010363
44. Bernal-Bayard J, Cardenal-Munoz E, Ramos-Morales F. The Salmonella type III secretion effector, Salmonella leucine-rich repeat protein (SlrP), targets the human chaperone ERdj3. J Biol Chem. (2010) 285:16360–8. doi: 10.1074/jbc.M110.100669
45. Zouhir S, Bernal-Bayard J, Cordero-Alba M, Cardenal-Munoz E, Guimaraes B, Lazar N, et al. The structure of the Slrp-Trx1 complex sheds light on the autoinhibition mechanism of the type III secretion system effectors of the NEL family. Biochem J. (2014) 464:135–44. doi: 10.1042/BJ20140587
46. Cordero-Alba M, Garcia-Gomez JJ, Aguilera-Herce J, Ramos-Morales F. Proteomic insight into the effects of the Salmonella ubiquitin ligase SlrP on host cells. Biochem Biophys Res Commun. (2016) 472:539–44. doi: 10.1016/j.bbrc.2016.03.014
47. Libby SJ, Lesnick M, Hasegawa P, Weidenhammer E, Guiney DG. The Salmonella virulence plasmid spv genes are required for cytopathology in human monocyte-derived macrophages. Cell Microbiol. (2000) 2:49–58. doi: 10.1046/j.1462-5822.2000.00030.x
48. Lesnick ML, Reiner NE, Fierer J, Guiney DG. The Salmonella spvB virulence gene encodes an enzyme that ADP-ribosylates actin and destabilizes the cytoskeleton of eukaryotic cells. Mol Microbiol. (2001) 39:1464–70. doi: 10.1046/j.1365-2958.2001.02360.x
49. Browne SH, Lesnick ML, Guiney DG. Genetic requirements for Salmonella-induced cytopathology in human monocyte-derived macrophages. Infect Immun. (2002) 70:7126. doi: 10.1128/IAI.70.12.7126-7135.2002
50. Mesa-Pereira B, Medina C, Camacho EM, Flores A, Santero E. Novel tools to analyze the function of Salmonella effectors show that SvpB ectopic expression induces cell cycle arrest in tumor cells. PLoS ONE. (2013) 8:e78458. doi: 10.1371/journal.pone.0078458
51. Paesold G, Guiney DG, Eckmann L, Kagnoff MF. Genes in the Salmonella pathogenicity island 2 and the Salmonella virulence plasmid are essential for Salmonella-induced apoptosis in intestinal epithelial cells. Cell Microbiol. (2002) 4:771–81. doi: 10.1046/j.1462-5822.2002.00233.x
52. Bakowski MA, Cirulis JT, Brown NF, Finlay BB, Brumell JH. SopD acts cooperatively with SopB during Salmonella enterica serovar Typhimurium invasion. Cell Microbiol. (2007) 9:2839–55. doi: 10.1111/j.1462-5822.2007.01000.x
53. Bakowski MA, Braun V, Brumell JH. Salmonella-containing vacuoles: directing traffic and nesting to grow. Traffic. (2008) 9:2022–31. doi: 10.1111/j.1600-0854.2008.00827.x
54. Ruan HH, Li Y, Zhang XX, Liu Q, Ren H, Zhang KS, et al. Identification of TRAF6 as a ubiquitin ligase engaged in the ubiquitination of SopB, a virulence effector protein secreted by Salmonella Typhimurium. Biochem Biophys Res Commun. (2014) 447:172–7. doi: 10.1016/j.bbrc.2014.03.126
55. Ruan HH, Zhang Z, Wang SY, Nickels LM, Tian L, Qiao JJ, et al. Tumor necrosis factor receptor-associated factor 6 (TRAF6) mediates ubiquitination-dependent STAT3 activation upon Salmonella enterica serovar Typhimurium infection. Infect Immun. (2017) 85:e00081-17. doi: 10.1128/IAI.00081-17
56. Ruan H, Zhang Z, Tian L, Wang S, Hu S, Qiao JJ. The Salmonella effector SopB prevents ROS-induced apoptosis of epithelial cells by retarding TRAF6 recruitment to mitochondria. Biochem Biophys Res Commun. (2016) 478:618–23. doi: 10.1016/j.bbrc.2016.07.116
57. Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia. (2003) 17:590–603. doi: 10.1038/sj.leu.2402824
58. Knodler LA, Finlay BB, Steele-Mortimer O. The Salmonella effector protein SopB protects epithelial cells from apoptosis by sustained activation of Akt. J Biol Chem. (2005) 280:9058–64. doi: 10.1074/jbc.M412588200
59. Truong D, Boddy KC, Canadien V, Brabant D, Fairn GD, D'Costa VM, et al. Salmonella exploits host Rho GTPase signalling pathways through the phosphatase activity of SopB. Cell Microbiol. (2018) 20:e12938. doi: 10.1111/cmi.12938
60. Collier-Hyams LS, Zeng H, Sun J, Tomlinson AD, Bao ZQ, Chen H, et al. Cutting edge: Salmonella AvrA effector inhibits the key proinflammatory, anti-apoptotic NF- B pathway. J Immunol. (2002) 169:2846–50. doi: 10.4049/jimmunol.169.6.2846
61. Ye Z, Petrof EO, Boone D, Claud EC, Sun J. Salmonella effector AvrA regulation of colonic epithelial cell inflammation by deubiquitination. Am J Pathol. (2007) 171:882–92. doi: 10.2353/ajpath.2007.070220
62. Jones RM, Wu H, Wentworth C, Luo L, Collier-Hyams L, Neish AS. Salmonella AvrA coordinates suppression of host immune and apoptotic defenses via JNK pathway blockade. Cell Host Microbe. (2008) 3:233–44. doi: 10.1016/j.chom.2008.02.016
63. Du F, Galan JE. Selective inhibition of type III secretion activated signaling by the Salmonella effector AvrA. PLoS Pathog. (2009) 5:e1000595. doi: 10.1371/journal.ppat.1000595
64. Wu H, Jones RM, Neish AS. The Salmonella effector AvrA mediates bacterial intracellular survival during infection in vivo. Cell Microbiol. (2012) 14:28–39. doi: 10.1111/j.1462-5822.2011.01694.x
65. Mazurkiewicz P, Thomas J, Thompson JA, Liu M, Arbibe L, Sansonetti P, et al. SpvC is a Salmonella effector with phosphothreonine lyase activity on host mitogen-activated protein kinases. Mol Microbiol. (2008) 67:1371–83. doi: 10.1111/j.1365-2958.2008.06134.x
66. Haneda T, Ishii Y, Shimizu H, Ohshima K, Iida N, Danbara H, et al. Salmonella type III effector SpvC, a phosphothreonine lyase, contributes to reduction in inflammatory response during intestinal phase of infection. Cell Microbiol. (2012) 14:485–99. doi: 10.1111/j.1462-5822.2011.01733.x
67. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
68. He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol. (2018) 19:912–22. doi: 10.1038/s41590-018-0188-x
69. Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. (2013) 39:443–53. doi: 10.1016/j.immuni.2013.06.018
70. Remijsen Q, Goossens V, Grootjans S, Van den Haute C, Vanlangenakker N, Dondelinger Y, et al. Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. (2014) 5:e1004. doi: 10.1038/cddis.2013.531
71. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. (2013) 38:209–23. doi: 10.1016/j.immuni.2013.02.003
72. Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat Immunol. (2012) 13:954–62. doi: 10.1038/ni.2397
73. de Weerd NA, Nguyen T. The interferons and their receptors–distribution and regulation. Immunol Cell Biol. (2012) 90:483–91. doi: 10.1038/icb.2012.9
74. Hos NJ, Ganesan R, Gutierrez S, Hos D, Klimek J, Abdullah Z, et al. Type I interferon enhances necroptosis of Salmonella Typhimurium-infected macrophages by impairing antioxidative stress responses. J Cell Biol. (2017) 216:4107–21. doi: 10.1083/jcb.201701107
75. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. (2012) 148:228–43. doi: 10.1016/j.cell.2011.11.030
76. Lu W, Sun J, Yoon JS, Zhang Y, Zheng L, Murphy E, et al. Mitochondrial protein PGAM5 regulates mitophagic protection against cell necroptosis. PLoS ONE. (2016) 11:e0147792. doi: 10.1371/journal.pone.0147792
77. Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O'Donnell CL, Gough PJ, et al. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol. (2016) 196:407–15. doi: 10.4049/jimmunol.1501662
78. Ro YT, Jo GH, Jung SA, Lee EH, Shin J, Lee JH. Salmonella induced miR155 enhances necroptotic death in macrophage cells via targeting RIP1/3. Mol Med Rep. (2018) 18:5133–40. doi: 10.3892/mmr.2018.9525
79. Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P, et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci. (2014) 71:331–48. doi: 10.1007/s00018-013-1381-6
80. Kujat Choy SL, Boyle EC, Gal-Mor O, Goode DL, Valdez Y, Vallance BA, et al. SseK1 and SseK2 are novel translocated proteins of Salmonella enterica serovar Typhimurium. Infect Immun. (2004) 72:5115–25. doi: 10.1128/IAI.72.9.5115-5125.2004
81. Yang Z, Soderholm A, Lung TW, Giogha C, Hill MM, Brown NF, et al. SseK3 is a Salmonella effector that binds TRIM32 and modulates the host's NF-kappaB signalling activity. PLoS ONE. (2015) 10:e0138529. doi: 10.1371/journal.pone.0138529
82. Pearson JS, Giogha C, Ong SY, Kennedy CL, Kelly M, Robinson KS, et al. A type III effector antagonizes death receptor signalling during bacterial gut infection. Nature. (2013) 501:247–51. doi: 10.1038/nature12524
83. Gunster RA, Matthews SA, Holden DW, Thurston TL. SseK1 and SseK3 type III secretion system effectors inhibit NF-kappaB signaling and necroptotic cell death in Salmonella-infected macrophages. Infect Immun. (2017) 85:e00010–17. doi: 10.1128/IAI.00242-17
84. El Qaidi S, Chen K, Halim A, Siukstaite L, Rueter C, Hurtado-Guerrero R, et al. NleB/SseK effectors from Citrobacter rodentium, Escherichia coli, and Salmonella enterica display distinct differences in host substrate specificity. J Biol Chem. (2017) 292:11423–30. doi: 10.1074/jbc.M117.790675
85. Newson JP, Scott NE, Yeuk Wah Chung I, Wong Fok Lung T, Giogha C, Gan J, et al. Salmonella effectors SseK1 and SseK3 target death domain proteins in the TNF and TRAIL signaling pathways. Mol Cell Proteomics. (2019) 18:1138–56. doi: 10.1101/359117
86. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
87. Brennan MA, Cookson BT. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol. (2000) 38:31–40. doi: 10.1046/j.1365-2958.2000.02103.x
88. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. (2001) 9:113–4. doi: 10.1016/S0966-842X(00)01936-3
89. Monack DM, Navarre WW, Falkow S. Salmonella-induced macrophage death: the role of caspase-1 in death and inflammation. Microbes Infect. (2001) 3:1201–12. doi: 10.1016/S1286-4579(01)01480-0
90. Rayamajhi M, Zak DE, Chavarria-Smith J, Vance RE, Miao EA. Cutting edge: mouse NAIP1 detects the type III secretion system needle protein. J Immunol. (2013) 191:3986–9. doi: 10.4049/jimmunol.1301549
91. Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA. (2013) 110:14408–13. doi: 10.1073/pnas.1306376110
92. Vance RE. The NAIP/NLRC4 inflammasomes. Curr Opin Immunol. (2015) 32:84–9. doi: 10.1016/j.coi.2015.01.010
93. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. (2015) 526:660–5. doi: 10.1038/nature15514
94. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
95. Eder C. Mechanisms of interleukin-1beta release. Immunobiology. (2009) 214:543–53. doi: 10.1016/j.imbio.2008.11.007
96. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. (2015) 25:1285–98. doi: 10.1038/cr.2015.139
97. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
98. Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse activators of the NLRP3 inflammasome promote IL-1beta secretion by triggering necrosis. Cell Rep. (2015) 11:1535–48. doi: 10.1016/j.celrep.2015.05.003
99. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. (2013) 20:1149–60. doi: 10.1038/cdd.2013.37
100. Gross CJ, Mishra R, Schneider KS, Medard G, Wettmarshausen J, Dittlein DC, et al. K(+) Efflux-independent NLRP3 inflammasome activation by small molecules targeting mitochondria. Immunity. (2016) 45:761–73. doi: 10.1016/j.immuni.2016.08.010
101. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. (2016) 530:354–7. doi: 10.1038/nature16959
102. Heilig R, Broz P. Function and mechanism of the pyrin inflammasome. Eur J Immunol. (2018) 48:230–8. doi: 10.1002/eji.201746947
103. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
104. Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. (2010) 207:1745–55. doi: 10.1084/jem.20100257
105. Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci USA. (2014) 111:7403–8. doi: 10.1073/pnas.1402911111
106. Qu Y, Misaghi S, Newton K, Maltzman A, Izrael-Tomasevic A, Arnott D, et al. NLRP3 recruitment by NLRC4 during Salmonella infection. J Exp Med. (2016) 213:877–85. doi: 10.1084/jem.20132234
107. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. (2013) 341:1246–9. doi: 10.1126/science.1240248
108. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe. (2014) 16:249–56. doi: 10.1016/j.chom.2014.07.002
109. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
110. Yang J, Zhao Y, Shao F. Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol. (2015) 32:78–83. doi: 10.1016/j.coi.2015.01.007
111. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. (2012) 490:288–91. doi: 10.1038/nature11419
112. Man SM, Karki R, Briard B, Burton A, Gingras S, Pelletier S, et al. Differential roles of caspase-1 and caspase-11 in infection and inflammation. Sci Rep. (2017) 7:45126. doi: 10.1038/srep45126
113. Knodler LA, Vallance BA, Celli J, Winfree S, Hansen B, Montero M, et al. Dissemination of invasive Salmonella via bacterial-induced extrusion of mucosal epithelia. Proc Natl Acad Sci USA. (2010) 107:17733–8. doi: 10.1073/pnas.1006098107
114. Sellin ME, Muller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. (2014) 16:237–48. doi: 10.1016/j.chom.2014.07.001
115. Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of caspase-1 and−8. Immunity. (2017) 46:649–59. doi: 10.1016/j.immuni.2017.03.016
116. Lei-Leston AC, Murphy AG, Maloy KJ. Epithelial cell inflammasomes in intestinal immunity and inflammation. Front Immunol. (2017) 8:1168. doi: 10.3389/fimmu.2017.01168
117. Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol. (2013) 191:5239–46. doi: 10.4049/jimmunol.1301581
118. Feltham R, Vince JE, Lawlor KE. Caspase-8: not so silently deadly. Clin Transl Immunology. (2017) 6:e124. doi: 10.1038/cti.2016.83
119. Rao S, Schieber AMP, O'Connor CP, Leblanc M, Michel D, Ayres JS. Pathogen-mediated inhibition of anorexia promotes host survival and transmission. Cell. (2017) 168:503–16.e12. doi: 10.1016/j.cell.2017.01.006
120. Ly KT, Casanova JE. Mechanisms of Salmonella entry into host cells. Cell Microbiol. (2007) 9:2103–11. doi: 10.1111/j.1462-5822.2007.00992.x
121. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci USA. (1999) 96:2396. doi: 10.1073/pnas.96.5.2396
122. Dreher D, Kok M, Obregon C, Kiama SG, Gehr P, Nicod LP. Salmonella virulence factor SipB induces activation and release of IL-18 in human dendritic cells. J Leukoc Biol. (2002) 72:743–51. Available online at: https://jlb.onlinelibrary.wiley.com/doi/full/10.1189/jlb.72.4.743
123. van der Velden AWM, Velasquez M, Starnbach MN. Salmonella rapidly kill dendritic cells via a caspase-1- dependent mechanism. J Immunol. (2003) 171:6742–9. doi: 10.4049/jimmunol.171.12.6742
124. Friebel A, Ilchmann H, Aepfelbacher M, Ehrbar K, Machleidt W, Hardt WD. SopE and SopE2 from Salmonella Typhimurium activate different sets of RhoGTPases of the host cell. J Biol Chem. (2001) 276:34035–40. doi: 10.1074/jbc.M100609200
125. Muller AJ, Hoffmann C, Galle M, Van Den Broeke A, Heikenwalder M, Falter L, et al. The S. Typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe. (2009) 6:125–36. doi: 10.1016/j.chom.2009.07.007
126. Hoffmann C, Galle M, Dilling S, Kappeli R, Muller AJ, Songhet P, et al. In macrophages, caspase-1 activation by SopE and the type III secretion system-1 of S. Typhimurium can proceed in the absence of flagellin. PLoS ONE. (2010) 5:e12477. doi: 10.1371/journal.pone.0012477
127. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. (2014) 513:237–41. doi: 10.1038/nature13449
128. Perez-Lopez A, Rosales-Reyes R, Alpuche-Aranda CM, Ortiz-Navarrete V. Salmonella downregulates Nod-like receptor family CARD domain containing protein 4 expression to promote its survival in B cells by preventing inflammasome activation and cell death. J Immunol. (2013) 190:1201–9. doi: 10.4049/jimmunol.1200415
129. Hu GQ, Song PX, Chen W, Qi S, Yu SX, Du CT, et al. Critical role for Salmonella effector SopB in regulating inflammasome activation. Mol Immunol. (2017) 90:280–6. doi: 10.1016/j.molimm.2017.07.011
130. Garcia-Gil A, Galan-Enriquez CS, Perez-Lopez A, Nava P, Alpuche-Aranda C, Ortiz-Navarrete V. SopB activates the Akt-YAP pathway to promote Salmonella survival within B cells. Virulence. (2018) 9:1390–402. doi: 10.1080/21505594.2018.1509664
131. Bierschenk D, Monteleone M, Moghaddas F, Baker PJ, Masters SL, Boucher D, et al. The Salmonella pathogenicity island-2 subverts human NLRP3 and NLRC4 inflammasome responses. J Leukoc Biol. (2019) 105:401–10. doi: 10.1002/JLB.MA0318-112RR
Keywords: non-typhoidal Salmonella, programmed cell death, innate immunity, T3SS effector protein, immune evasion, host-pathogen interaction
Citation: Wemyss MA and Pearson JS (2019) Host Cell Death Responses to Non-typhoidal Salmonella Infection. Front. Immunol. 10:1758. doi: 10.3389/fimmu.2019.01758
Received: 02 May 2019; Accepted: 11 July 2019;
Published: 26 July 2019.
Edited by:
Barbara Bottazzi, Milan University, ItalyReviewed by:
Silvia Guglietta, Medical University of South Carolina, United StatesElsa Anes, University of Lisbon, Portugal
Copyright © 2019 Wemyss and Pearson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaclyn S. Pearson, amFjbHluLnBlYXJzb25AaHVkc29uLm9yZy5hdQ==
†Present address: Madeleine A. Wemyss and Jaclyn S. Pearson, Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, Clayton, VIC, Australia