 Tiffany Shih1
Tiffany Shih1 Saurav De
Saurav De Betsy J. Barnes
Betsy J. Barnes- 1Center for Autoimmune Musculoskeletal and Hematopoietic Disease, Northwell Health, The Feinstein Institute for Medical Research, Manhasset, NY, United States
- 2Graduate School of Biomedical Sciences Rutgers, The State University of New Jersey, Newark, NJ, United States
- 3Departments of Molecular Medicine and Pediatrics, Zucker School of Medicine at Hofstra/Northwell Health, Hempstead, NY, United States
Upon antigen recognition, naïve B cells undergo rapid proliferation followed by differentiation to specialized antibody secreting cells (ASCs), called plasma cells. Increased circulating plasma cells are reported in patients with B cell-associated malignancies, chronic graft-vs.-host disease, and autoimmune disorders. Our aim was to optimize an RNAi-based method that efficiently and reproducibly knocks-down genes of interest in human primary peripheral B cells for the targeted analysis of ASC differentiation. The unique contributions of transcriptional diversity in species-specific regulatory networks and the mechanisms of gene function need to be approached directly in human B cells with tools to hone our basic inferences from animal models to human biology. To date, methods for gene knockdown in human primary B cells, which tend to be more refractory to transfection than immortalized B cell lines, have been limited by losses in cell viability and ineffective penetrance. Our single-step siRNA nucleofector-based approach for human primary naïve B cells demonstrates reproducible knockdown efficiency (~40–60%). We focused on genes already known to play key roles in murine ASC differentiation, such as interferon regulatory factor 4 (IRF4) and AID. This study reports a validated non-viral method of siRNA delivery into human primary B cells that can be applied to study gene regulatory networks that control human ASC differentiation.
Introduction
B lymphocytes are critical members of the adaptive immune system as they are uniquely capable of secreting high titers of antigen-neutralizing antibody. B cells and their associated antibody-mediated response to antigen are important in the clearance of viral, bacterial, and fungal pathogens. Recognition of these foreign antigens by B cells triggers rapid proliferation and differentiation to specialized antibody secreting cells (ASCs) known as plasma cells. The process of ASC differentiation is a tightly regulated one that relies on synergistic signaling from multiple pathways (1). A large gene-regulatory network of transcription factors is required for regulating this multi-step process. One key player in the differentiation of naïve B cells to ASCs is the transcription factor interferon regulatory factor 4 (IRF4). Its role in ASC differentiation has been well-characterized in mice (2–4). Expression of IRF4 is high in murine ASCs and is critical for upregulating AID and BLIMP1 expression during ASC differentiation to plasma cells (5).
Very few of these murine-based B cell differentiation studies, however, have been replicated in human primary B cells. This delay in data replication is primarily due to difficulties in achieving gene knockdown in human primary naïve B cells, which tend to be more refractory to transfection than immortalized B cell lines, and have been limited by losses in cell viability and ineffective penetrance. While genetic approaches in mice provide invaluable physiological insights for identifying pathways which drive imbalance of B cell subsets, the exclusive use of inbred mice with limited diversity may mask pathways and gene functions that exist uniquely in humans (6, 7). Thus, methods for manipulating gene expression in human primary B cell subsets is essential for transferring findings in mice to humans. More importantly, an in vitro approach is necessary to understand how gene dysregulation may contribute to the development of human disease, including post-transplantation systemic persistence of alloimmune and autoimmune responses in chronic graft-vs.-host disease (8–14), as well as the severe consequences of B cell dysfunction in indolently incurable or aggressively fatal B cell-associated malignancies (15, 16), and autoimmunity (17).
In peripheral blood mononuclear cells (PBMCs) isolated from circulating blood, human naïve B cells constitute 0.7–4.9% of leukocytes (18). The variable frequency among individual donors and the refractory nature of primary naïve B cells to gene modification, by lentiviral vector or RNA transfection, have been limiting factors in the study of human ASC differentiation. Gene silencing by transfecting cells with small interfering RNA (siRNA) leads to the rapid degradation of corresponding mRNA and reduced target protein expression. Nucleofection is an electroporation technique that enables efficient introduction of siRNAs into cells and detectable silencing of target genes. Here, we describe an optimized non-viral method for transient knockdown by siRNA delivery into human primary naïve B cells for the study of key genes regulating ASC differentiation and effector function. We focused on genes already known to play a role in murine ASC differentiation, such as IRF4 and AID. This method has been optimized for efficient knockdown of four genes—IRF4, IRF5, AID, and GAPD—with minimal effects on cell viability and maximal effects on cell recovery and functional analysis after nucleofection.
Materials and Methods
Ethics Statement
This study was carried out in accordance with the Declaration of Helsinki. This study used blood from leukopaks of human healthy donors purchased from the New York Blood Center. These types of de-identified, publicly and commercially available specimens are exempt from ethics approval as they are fully anonymized.
Human PBMC Isolation and Primary B Cell Purification
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll [Corning, Manassas, VA, 25-072-CV] density centrifugation from buffy coats purchased from the NY Blood Center (Long Island City, NY). Naïve or total B cell purifications was performed by negative selection with magnetic separation according to manufacturer instructions (Stem Cell Technologies, Vancouver, Canada) using EasySep Human naïve B cell enrichment kit [19254] or naïve B cell isolation kit [17254]. Total B cell experiments were performed with cells purified using EasySep Human total B cell enrichment kit [19054] to achieve a >95% enriched population of naïve B cells (CD19+IgD+) or total B cells (CD19+). Isolated naïve B cells ranged from 5 × 106 to 26 × 106 from individual donor leukopaks containing 4 × 108 to 1 × 109 PBMCs.
Targeted siRNA Nucleofection
Isolated naïve B cells were centrifuged in antibiotic-free, serum-containing media as recommended by the Amaxa P3 Primary Cell 4D-Nucloefector X Kit L [Lonza, Cologne, Germany, V4XP-3024] at 300 × g for 10 min at room temperature. Cells were resuspended in room temperature Amaxa buffer as suggested by the manufacturer for primary cells. 2–3 × 106 cells/100 μL cuvette was the final concentration of cells used for nucleofection. For optimal results, siRNA was resuspended in 1X siRNA buffer composed of 5X buffer [GE Lifesciences, Lafayette, CO, B-002000-UB-100] diluted in nuclease-free water [Ambion, USA, AM9938] and used for nucleofection the same day. Reconstituted siRNA stored at −80°C for up to 2–4 weeks will generally retain knockdown efficiency, as determined by nucleofection and monitoring knockdown efficiency over time (data not shown). B cells were nucleofected with either mock (no siRNA), 1.5 μM of ON-TARGETplus Non-targeting Control Pool [Dharmacon, Lafayette, CO, D-001810-10-05] or SMARTpool ON-TARGETplus human IRF4 siRNA [Dharmacon, LU-019668-00-0005]. 1–1.5 μM ON-TARGETplus Targeted Control GAPD Pool [Dharmacon, D-001830-10-05], 1–1.5 μM of ON-TARGETplus AICDA siRNA [Dharmacon, LU-021409-00-0005], and 1.5 μM siGLO green transfection indicator siRNA [Dharmacon, D-001630-01-05] were also used. Cells were nucleofected using program EO-117 for primary human B cells of the Amaxa 4D Nucleofector system [Lonza] composed of the core unit and the X unit.
Immediately after nucleofection, 500 μL of pre-warmed (37°C) antibiotic-free media (10% fetal bovine serum (FBS) in Iscove's Modified Dulbecco's Media (IMDM) without antibiotics) was added to the cuvette by slowly releasing the media along the wall of the cuvette. The final suspension was then transferred into wells of a 24-well plate that each contained 1 mL of pre-warmed antibiotic-free media [Sigma, USA, F4135] per cuvette and cells allowed to rest in culture for 24 h at 37°C in 5% CO2. After resting, cells were transferred to a 14 mL Falcon tube to be pelleted, counted and then cultured with the appropriate cocktails for B cell activation or plasmablast differentiation.
Viability Post-nucleofection
Viability was determined by staining cells with trypan blue [Life Technologies, Carlsbad, CA, 15250-061] after resting nucleofected cells for 24 h and assessing by hemocytometer. Percent viable was calculated using the equation 100 × (total cells—blue cells)/total number.
In vitro B Cell Activation and Plasmablast Differentiation
After resting, nucleofected naïve B cells were cultured in 96-well U-bottom plates [Costar, USA, 3799] at a minimal density of 0.5 × 106 in 250 μL of IMDM supplemented with 10% FBS and 1X penicillin-streptomycin [Corning, 30-002-Cl] per well. B cell cultures of 3 days or less were treated with or without 10 μg/mL unconjugated goat anti-human IgM antibody [Southern Biotech, Birmingham, AL 2020-01] and 2.5 μg/mL CpG-B oligodeoxynucleotide (ODN) 2006 [Hycult Tech, Uden, The Netherlands, HC4309]. For plasmablast differentiation, purified naïve B cells were cultured for 7 days in the presence of 200 ng/mL sCD40L [Peprotech, Rocky Hill, NJ 310-02-10UG] alone or a “C4” cocktail, consisting of 200 ng/mL sCD40L, 100 ng/mL IL-21 [Peprotech, 200-21-2UG], 10 μg/mL unconjugated goat anti-human IgM antibody, and 2.5 μg/mL CpG-B ODN 2006 (17, 19–24). For activation prior to nucleofection, 2 × 106 cells/mL were stimulated in a 24-well flat bottom plate overnight with or without CpG-B plus anti-IgM in a final volume of 1 mL IMDM supplemented with 10% FBS and penicillin-streptomycin. After pre-activation, cells were washed twice with 0.5% BSA in 1 × PBS and counted for the nucleofection protocol described above.
Flow Cytometry
B cells were washed with 1X PBS and stained with Live/Dead Fixable Yellow Dead Cell Stain Kit viability discrimination dye [Life Technologies, L34959]. Cells were subsequently blocked in 2% bovine serum albumin (BSA) supplemented with human TruStain FcX Blocker [Biolegend, San Diego, CA, 422302] for 5 min and then stained with antibodies against B cell surface markers (Table 1). After staining, cells were washed in 1X PBS and then fixed in 2% PFA before analysis on a BD Fortessa flow cytometer [BD Biosciences]. For intracellular protein staining, after overnight fixation, cells were permeabilized in 0.1% Triton X-100 and rinsed in 1X PBS 2 times before blocking in 5% BSA solution. Intracellular AID was detected with unconjugated goat polyclonal primary antibody [Santa Cruz Biotech., CA, sc-14680], and subsequently stained with donkey anti-goat IgG-AF488 secondary [Invitrogen, USA, A-11055]. Intracellular GAPD was stained with unconjugated rabbit anti-human GAPDH antibody EPR16891 [Abcam, USA, ab181602] and subsequently with goat anti-rabbit AF488 [Life Technologies, A11034]. Intracellular IRF4 was detected with rat anti-human/mouse IRF4-phycoerythrin (PE) [Biolegend, 646404] and rat immunoglobulin (Ig)G1, k isotype control [Biolegend, 400408]. Cells staining positive for the live/dead stain were excluded from the flow cytometry analysis. Doublets were excluded from our analysis of FSC-A vs. FSC-H gating. Naïve B cells were defined by CD19+IgD+ surface expression, and plasmablasts were defined by CD19+CD20+IgD−CD27+CD38+ surface expression (Supplementary Figure 1). FCS files were analyzed using FlowJo v9.3.2 (Tree Star Inc., Ashland, OR).
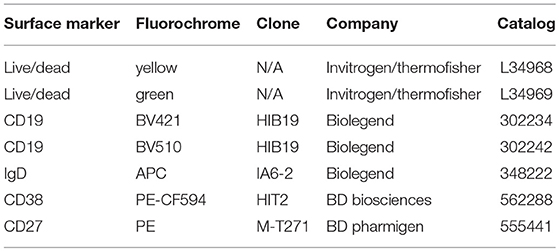
Table 1. Antibodies used for surface staining and flow cytometry.
Statistical Analysis
All statistical analyses were performed using Prism v6.2 (GraphPad Software, San Diego, CA). Student's t-test was used for comparisons between two samples with normal distribution. Prior to test, graph kurtosis was analyzed to ensure normal distribution. Data are reported as mean ± SEM. P-value < 0.05 was considered significant.
Results
Analysis of Cell Viability After Nucleofection and Optimization of Cell Number for ASC Differentiation
We previously attempted shRNA lentiviral transduction of human primary naïve B cells and were unsuccessful. We later developed a siRNA nucleofection protocol that required two rounds of nucleofection with low concentrations of IRF5 siRNAs over 48 h to obtain ~40–60% knockdown efficiency of IRF5 proteins in human primary naïve B cells (17). We have now optimized the protocol further for single siRNA nucleofection and knockdown of other genes involved in ASC differentiation (Figure 1A). We initially optimized the protocol for IRF4 knockdown, as it is known to play essential roles in murine ASC differentiation. In mice lacking Irf4, B and T cells were unable to proliferate in response to B cell receptor (BCR), T cell receptor (TCR), CD40, or LPS stimulation (25). Studies in mice revealed that Irf4 is necessary for AID upregulation, class-switch recombination (CSR), and generation of plasma cells in response to BCR signaling (5, 26, 27).
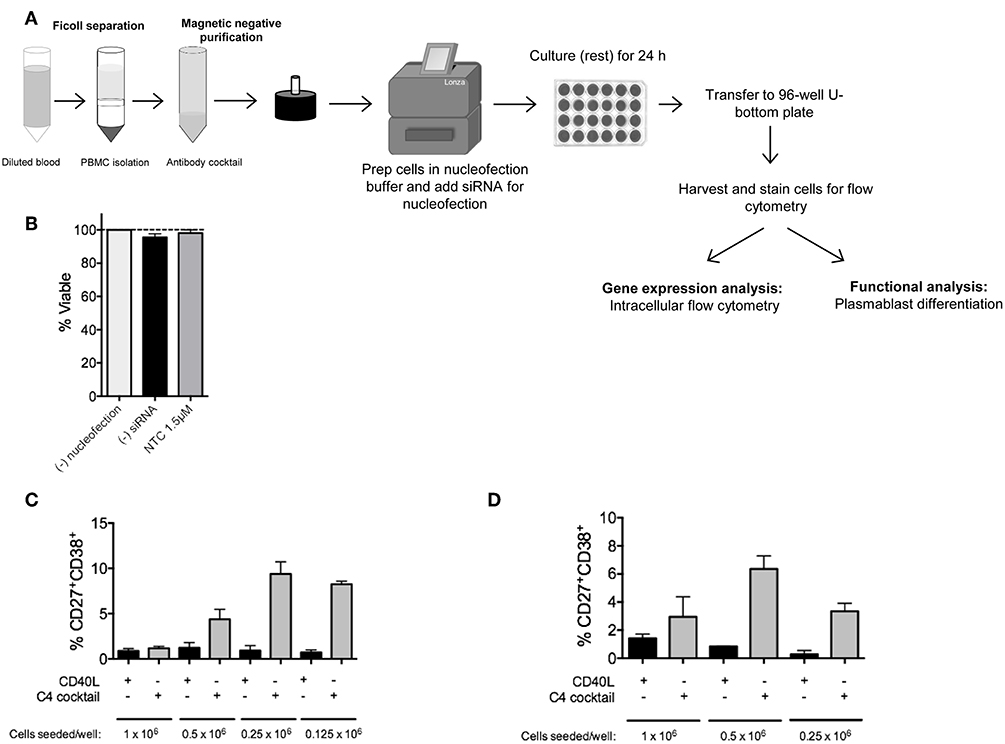
Figure 1. RNAi knockdown in primary human naïve B cells by Amaxa nucleofection protocol. (A) Overview of nucleofection protocol and experimental readouts. (B) Isolated human naïve B cells were nucleofected with or without non-targeting control (NTC) siRNA and rested for 24 h. Viability of cells after resting was determined by trypan blue staining (n = 4). (C) Optimal seeding density of non-nucleofected primary human naïve B cells to generate plasmablasts in a 96-well plate via 7 day in vitro culture with C4 cocktail (n = 4). (D) Same as (C) except optimal seeding density is shown for nucleofected cells (n = 4).
Since primary B cells are notoriously difficult-to-transfect, and we are interested in one of the more rare subsets, plasma cells, we first optimized naïve B cell numbers and viability after nucleofection for downstream analysis of plasma cells by flow cytometry (17). In this assay, cells were either left untouched, mock-nucleofected, or nucleofected with 1.5 μM ON-TARGETplus Non-targeting Control (NTC) Pool siRNA. By trypan blue staining, no significant difference in cell viability was detected 24 h post-nucleofection of primary naïve B cells (Figure 1B).
A single leukopak of blood (~35–38 mL) yields between 5 and 26 × 106 naïve B cells. To determine the appropriate number of purified primary naïve B cells that will lead to sufficient plasma cell numbers for functional analysis, we examined differentiation of naïve B cells, at different seeding densities, to plasmablasts by 7 day in vitro culture with C4 cocktail (anti-IgM, CpG-B, IL21, and CD40L) (17, 19–24, 28). Similar to previously published work, in non-nucleofected naïve B cells, a seeding density of 0.25 × 106 cells per well of a 96-well plate was required for the optimal generation of CD19+IgD−CD27+CD38+ plasmablasts (Figure 1C) (29, 30). Representative gating strategy for plasmablasts is shown in Supplementary Figure 1. As recommended by the manufacturer, all Amaxa nucleofection reagents were kept at room temperature prior to use. For primary naïve B cells, we found that an optimal cell concentration for nucleofection with maximal siRNA entry and cell viability post-nucleofection was 2–2.5 × 106 cells per cuvette (in 100 μL volume); below 1.5 × 106 or above 3 × 106 million cells reduced viability and nucleofection efficiency. After nucleofection, 500 μL of 37°C pre-warmed IMDM supplemented with 10% FBS and no antibiotics was added to each cuvette and gently aspirated with pipettes provided in the kit for gentle transfer of cells into wells of a 24-well plate that had been pre-equilibrated to 37°C at 5% CO2 with 1.0 mL of IMDM/well. Cells were then rested for 24 h at 37°C in 5% CO2. Cell loss can occur at this point when transferring from the 24-well plate to the tube for washing. To address this, wells were thoroughly washed with 1 mL sterile 1X PBS or media using a 1,000 μL pipette to physically detach cells along the bottom surface and the circumference of the well. This step can be repeated as needed. Distinct from non-nucleofected naïve B cells, we found that 0.5 × 106 cells per well of a 96-well plate were required for optimal plasmablast generation after mock nucleofection (Figure 1D) (17). Thus, a critical step in the process is to re-count your cells after nucleofection and 24 h resting before transferring naïve B cells to a 96-well plate for 7 day in vitro culture to plasmablasts using the C4 cocktail. A good rule of thumb for calculating cell number for downstream functional analysis is to begin with nearly twice the number of cells that you want to end with for functional analysis post-nucleofection.
Analysis of Knockdown Efficiency and ASC Differentiation
IRF4 expression is high in plasma cells and low in naïve B cells but expression increases within 48 h after stimulation with anti-IgM for BCR cross-linking or CD40L (31, 32). siRNA-mediated knockdown of IRF4 has been previously described in total CD19+ B cells using a final concentration of 1.5 μM siRNA (33). We thus used IRF4 siRNAs in the range of 1–1.5 μM for knockdown in human primary naïve B cells. Unlike IRF5 that is expressed at sufficient basal levels in naïve B cells to detect knockdown without stimulation (17), both IRF4 and AID are expressed at very low levels and thus require B cell activation to detect knockdown. We found that IRF4 expression peaked at 48 h in CD19+IgD+ B cells after stimulation of PBMC with anti-IgM and the TLR9 agonist CpG-B; AID expression peaked at 72–96 h (Supplementary Figures 2A,B). We thus used these time points for analysis of IRF4 and AID protein expression after knockdown. An a priori understanding of mRNA and protein expression patterns of the particular gene of interest is required to determine appropriate time points for knockdown analysis.
Although we previously found that efficient knockdown of IRF5 in human primary naïve B cells required a dual nucleofection protocol with low siRNA concentrations (17), for IRF4, we were able to optimize knockdown by single nucleofection of 1.5 μM siRNAs (Figure 2A). Using the single nucleofection protocol, we examined IRF4 knockdown efficiency after single nucleofection with mock, SMARTpool ON-TARGETplus human IRF4 siRNA or NTC siRNA. Results revealed a range of 30–50% knockdown of IRF4 proteins by IRF4 siRNA, and not NTC siRNA, at 72 h post-nucleofection (24 h rest plus 48 h stimulation) (Figures 2B–D and Supplementary Figures 2C–F) with >95% post-nucleofection viability (Supplementary Figure 3A). Given that IRF4 knockdown with IRF4 siRNAs gave a Gaussian distribution of knockdown levels (Figure 2B), these data suggest that all cells, rather than a small subset of cells, were nucleofected with siRNA.
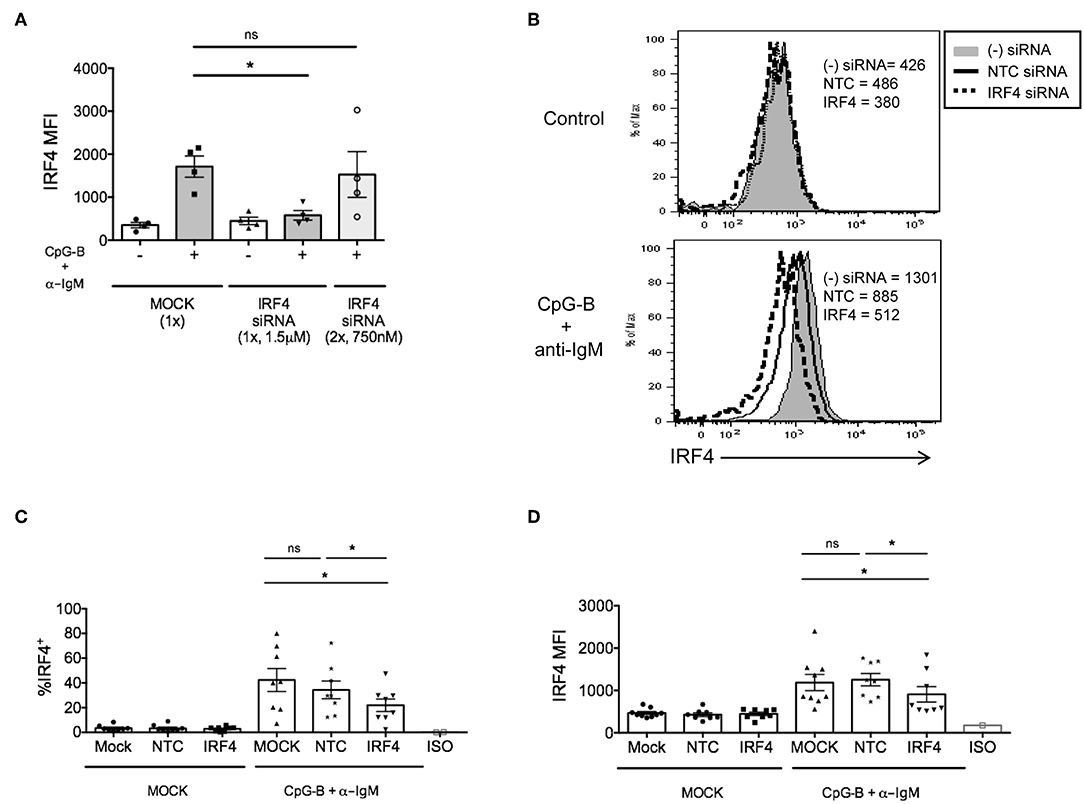
Figure 2. IRF4 knockdown in primary human naïve B cells by Amaxa nucleofection protocol. Isolated human naive B cells were nucleofected with or without IRF4 targeting siRNA and stimulated with or without anti-IgM + CpG-B for 48 h. (A) Comparison of single vs. dual nucleofection protocols (n = 4). (B) Isolated human naive B cells were nucleofected with or without 1.5 μM IRF4 targeting siRNA or non-targeting control (NTC) siRNA, rested for 24 h, and stimulated with or without anti-IgM + CpG-B for 48 h. Representative histogram overlays show IRF4 expression after stimulation. (C,D) Similar to B except data are summarized from multiple independent experiments showing % IRF4+ CD19+IgD+ B cells (C) and MFI of IRF4 in CD19+IgD+B cells (D) from n = 8 independent donors. Paired t-test for significance was performed (*p < 0.05).
In addition to negative control siRNAs, positive control siRNAs are recommended to confirm knockdown efficiency and specificity of siRNA function (34). We utilized a positive control siRNA targeting GAPD (ON-TARGETplus GAPD Control Pool siRNA). All targeted and non-targeted siRNAs were used at the same concentration for direct comparison of effects (either 1 or 1.5 μM) (Supplementary Figure 3B). Nucleofection with GAPD siRNAs resulted in a significant reduction in GAPD protein at 72 h post-nucleofection (24 h rest plus 48 h stimulation) (Figures 3A,B), with little significant impact on IRF4 expression (Figure 3C) or post-nucleofection viability (Supplementary Figure 3A). Notably, GAPD siRNA also provided a Gaussian distribution of knockdown levels, suggesting that all cells are getting nucleofected with siRNAs equally. ASC differentiation was then examined after mock-nucleofection, nucleofection with IRF4 siRNA, NTC siRNA, or GAPD Targeted siRNA. Similar to findings in Irf4−/− mice, knockdown of IRF4 in human primary naïve B cells correlated with a significant reduction (~30–40%) in ASC differentiation (Figures 3D,E). While NTC siRNA had no significant effect on plasmablast generation, we were surprised to detect a strong trend in plasmablast reduction after GAPD knockdown suggesting a potential role for GAPD in plasma cell differentiation (Figures 3D,F). Indeed, GAPD is an important regulator of cell growth, proliferation and survival due, in part, to its role in regulating the generation of glycolytic ATP (35–38). A subpopulation of naïve B cells, in response to B cell activation, will survive and undergo clonal expansion, CSR and differentiation to ASCs, in which GAPD has been implicated in Migliaccio et al. (39). Thus, while positive control siRNAs can serve as important tools for determining specificity of function, they can also complicate the system, depending on the downstream assays used to determine functional consequence(s) of siRNA knockdown. Nonetheless, data clearly show that the single nucleofection protocol can be used to knockdown IRF4 efficiently, leading to a significant reduction in human ASC differentiation.
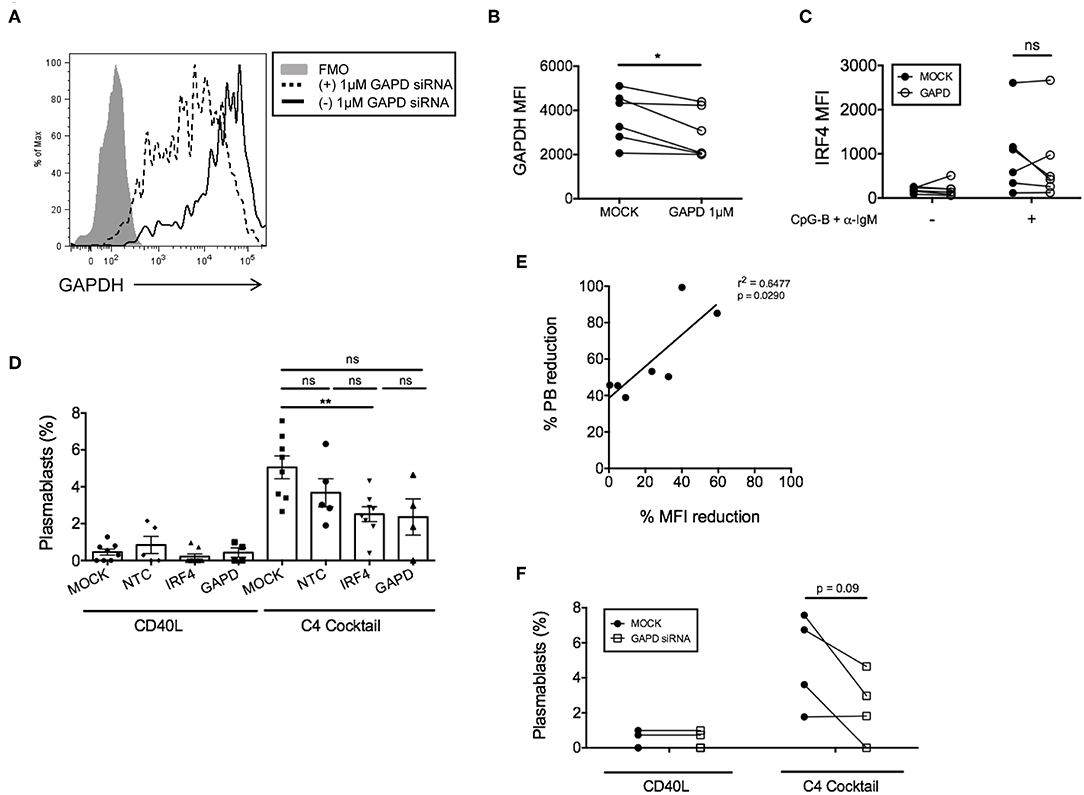
Figure 3. GAPD housekeeping gene as a positive control for nucleofection. (A) Representative figure showing histogram overlay of B cells nucleofected with 1 μM GAPD siRNA and then cultured for 48 h post-nucleofection. (B) Paired dots show the efficiency of GAPDH knockdown in CD19+IgD− B cells from matched independent donors after 48 h culture (n = 6). (C) Same as (B) except IRF4 expression was determined after nucleofection with 1 μM GAPD siRNA (n = 6). (D) Naïve B cells were cultured for 7 days with the C4 cocktail (anti-IgM + CpG-B + IL-21+ CD40L) to induce plasmablast differentiation post-nucleofection with 1.5 μM GAPD siRNA, 1.5 μM IRF4 siRNA or 1.5 μM NTC siRNA. Plasmablast differentiation was determined by flow cytometry analysis of CD19+IgD−CD27+CD38+ cells (n = 9). Bars represent mean ± SEM. (E) Correlation between % plasmablast reduction and % IRF4 MFI reduction. (F) Effect of GAPD siRNA on % plasmablasts from (D) are shown as paired dots to indicate matched donor effects. Paired t-test for significance was performed (*p < 0.05).
Applying the Single Nucleofection Knockdown Protocol to Other Genes Involved in ASC Differentiation
To determine whether this method is effective for silencing other genes that are induced after B cell activation, we examined knockdown of AID, a factor that is critical for CSR during ASC differentiation (40, 41). After titration, we identified the optimal concentration of 1–1.5μM AICDA siRNA that provided a significant, albeit, small reduction in the percentage of CD19+IgD+AID+ B cells (~10–20%) after anti-IgM + CpG-B stimulation for 48 h (Figure 4A). Somewhat surprising, this reduction did not translate into a significant reduction in AID MFI (Figure 4B). Based on AID expression kinetics (Supplementary Figure 2B), we extended the activation time point to 72 h and were still unable to detect significant effects on AID expression (Figures 4C,D).
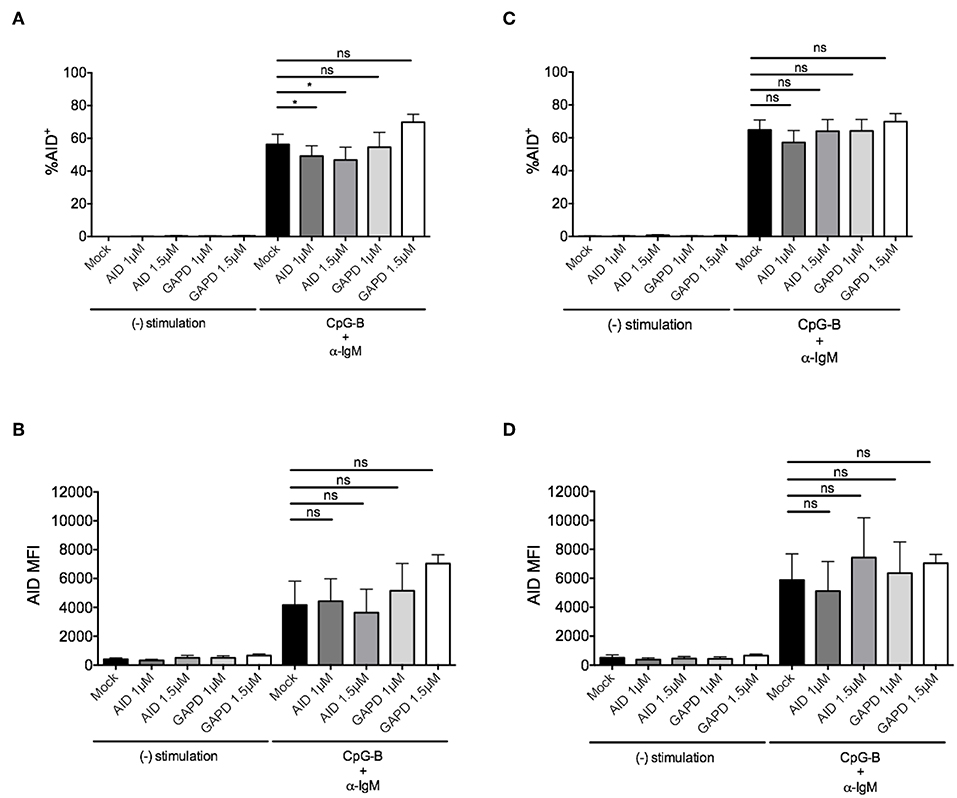
Figure 4. Efficiency of AID knockdown in primary naïve human B cells is marginal at 48 and 72 h post-nucleofection. Isolated naïve B cells were stimulated with anti-IgM + CpG-B after single nucleofection protocol with AID or GAPD siRNA (1 or 1.5 μM). (A) % AID+CD19+IgD+ B cells is shown at 48 h post-stimulation. (B) AID MFI is shown at same time point as (A). (C) Same as (A) except % AID+CD19+IgD+ B cells is shown 72 h post-stimulation. (D) AID MFI is shown at same time point as (C). Bars represent mean ± SEM (n = 4). Paired t-test for significance was performed (*p < 0.05).
Due to the low level of AID knockdown observed, we examined an alternate approach to achieve more robust knockdown of AID expression for functional analysis. We hypothesized that the inherent characteristics of the gene might require activation prior to nucleofection. Thus, purified naïve B cells were activated first with anti-IgM and CpG-B overnight prior to the standard nucleofection protocol and re-stimulation. Indeed, we detected a stronger knockdown effect after pre-activation (Figure 5A), revealing that 1.5 μM AICDA siRNA provides a significant reduction in AID MFI (ranging from ~30–50% knockdown) (Figures 5B,C). This level of AID knockdown resulted in ~90% loss of plasmablast differentiation (Figures 5D,E), with no significant effect on cell viability (Supplementary Figure 3A). Altogether, these data indicate that the single nucleofection protocol for knockdown in human primary naïve B cells can be applied to multiple genes.
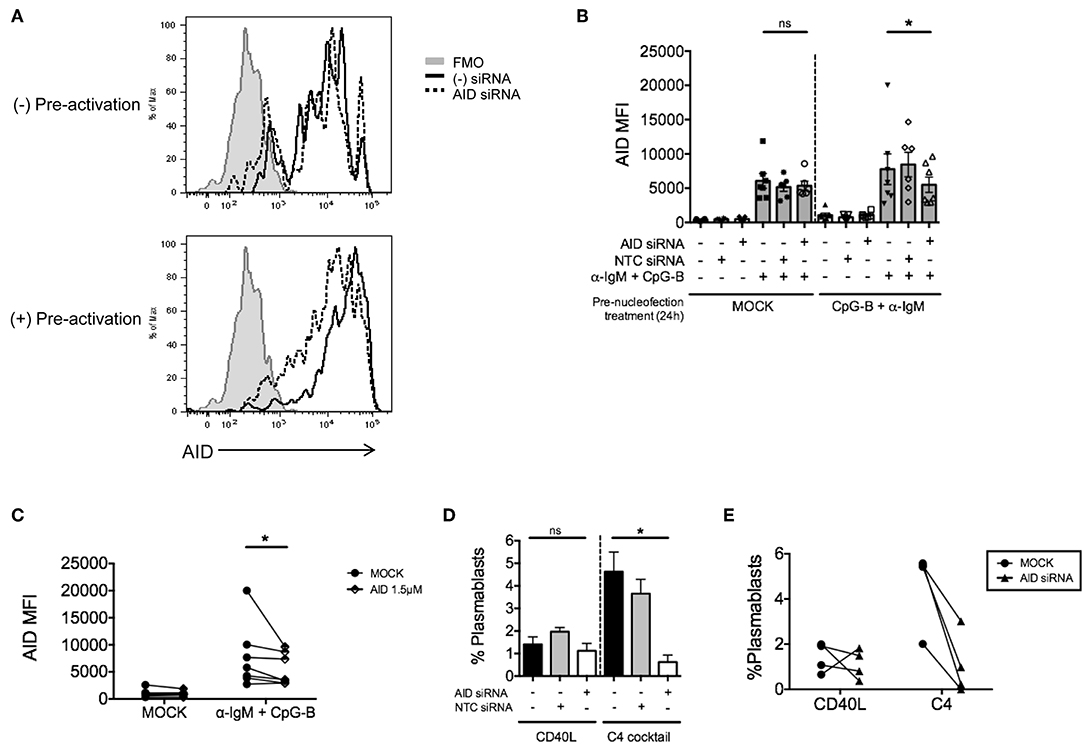
Figure 5. Pre-activation of primary naïve human B cells enhances the effect of AID knockdown. Isolated naïve B cells were cultured for 24 h (+Pre-activation) or without (-Pre-activation) anti-IgM + CpG-B then nucleofected one time with AID or NTC siRNA (1.5 μM). After 24 h resting, cells were re-stimulated with or without anti-IgM + CpG-B for 72 h. (A) Representative histogram overlay of AID expression after knockdown in the presence or absence of pre-activation. (B) AID MFI in CD19+IgD+ B cells was determined at 72 h post-stimulation (n = 7). (C) Paired dots show the efficiency of AID knockdown in CD19+IgD− B cells from matched independent donors in (B). (D) Following pre-activation and nucleofection with AID or NTC siRNA, cells were in vitro cultured for 7 days with C4 cocktail to induce plasmablast differentiation (CD19+IgD−CD27+CD38+) (n = 4). (E) Effect of AID siRNA on % plasmablasts from (D) are shown as paired dots to indicate matched donor effects. Bars represent mean ± SEM. P-values were determined by paired t-test for significance (*p < 0.05).
Sorting of Knockdown Cells by Co-nucleofection With siGLO
It was previously reported that the abundance of target gene expression is a critical factor that determines the efficiency of siRNA-mediated gene silencing (42). IRF4 and GAPD siRNAs showed a Gaussian distribution of knockdown levels suggesting that most cells were nucleofected equally with siRNAs (Figures 2B, 3A), while AID siRNAs showed a more disparate level of knockdown distribution. Given that gene expression can vary based on cellular context resulting in unequal knockdown effects, we attempted to optimize a method of co-nucleofection for sorting and functional analysis of nucleofected cells with knockdown.
We previously explored co-nucleofection of IRF5 siRNA with GFP mRNA (Trinity Biotech: L601) or pmaxGFPTM vector (Lonza) as a method to sort for cells with knockdown (17). Unfortunately, we were unsuccessful as co-nucleofection with GFP mRNA resulted in ~20% GFP+ naïve B cells with no correlation between GFP and IRF5 knockdown; both GFP+ and GFP− cells showed equivalent IRF5 knockdown levels (17). Similarly, the pmaxGFPTM resulted in a low yield of GFP+ cells with only ~2–4% of naïve B cells expressing GFP (17). At the time, we hypothesized that the failed attempts might be due to size restrictions on B cell uptake; green fluorescent protein (GFP) is larger than most siRNAs. Here, we attempted a new strategy for knockdown and selection using Dharmacon's siGLO green reagent that is used to examine siRNA transfection efficiency. Primary naïve B cells were co-nucleofected with equal parts of IRF4 siRNA and siGLO green. After 24 and 48 h post-nucleofection, nucleofecion efficiency and IRF4 knockdown were examined by flow cytometry. We detected a range in siGLO nucleofection efficiency with 10 to 45% of cells being siGLO+; similar levels were seen in co-nucleofected cells (Figures 6A–C). Importantly, IRF4 knockdown levels were similar between co-nucleofected and IRF4 siRNA nucleofected samples (Figure 6D) suggesting that siGLO does not compete with IRF4 for entry into cells during nucleofection. Unfortunately though, we found that the siGLO signal dramatically decreases, independent of concentration, 24 h post-nucleofection (Figure 6A), which is before we are able to detect significant IRF4 knockdown (Figure 2). Thus, to further assess the use of siGLO for tracking nucleofection with knockdown, we co-nucleofected cells with siGLO and NTC siRNA or GAPD siRNA and performed the similar analysis at the earlier time point of 24 h post-nucleofection. We detected similar siGLO nucleofection efficiency as seen before (~10–30%, Supplementary Figure 4A) and knockdown of GAPD was retained independent of siGLO (Figure 6E). Importantly, while the overall nucleofection efficiency was low with siGLO (Supplementary Figure 4B), data in Figure 6F suggest that knockdown of genes with abundant baseline expression (Supplementary Figure 4C) may be tracked with siGLO since we detected ~40% reduction of GAPD protein levels in siGLO+ cells at 24 h post-nucleofection.
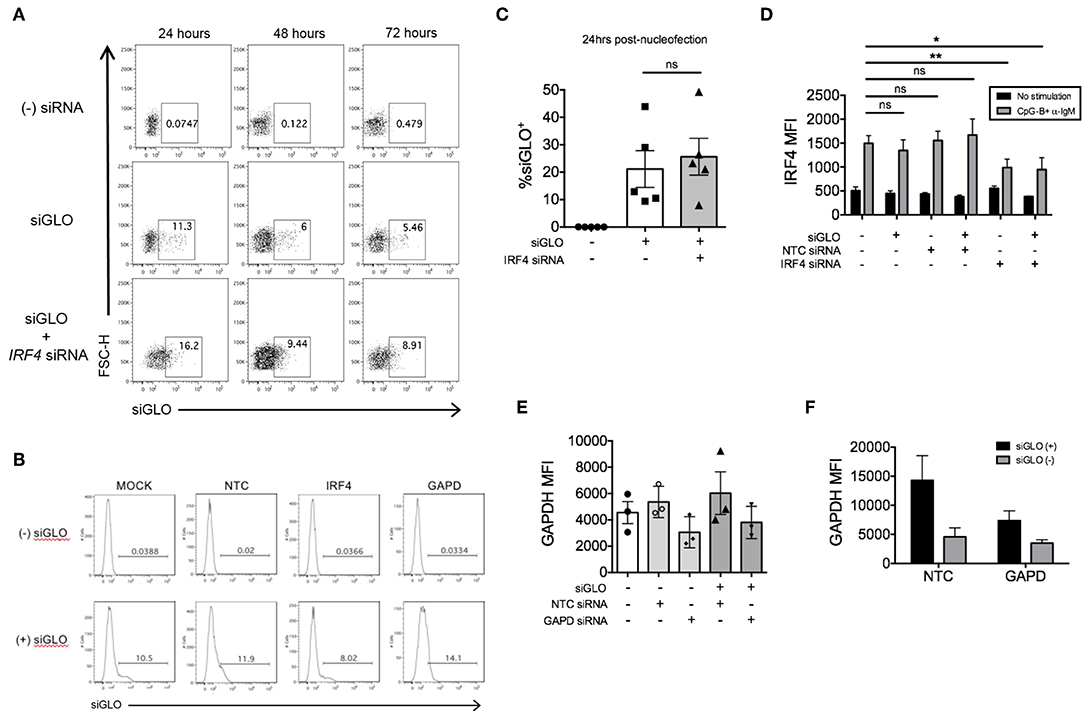
Figure 6. siGLO green nucleofection of purified primary naïve B is transient and lost by 48 h post-nucleofection. (A) Representative dot plots showing purified naïve B cells nucleofected with 1.5 μM siGLO or 1.5 μM siGLO + 1.5 μM IRF4 siRNA cultured for 24, 48, and 72 h after nucleofection and analyzed for siGLO+ cells. (B) Similar to (A) except representative histograms show the different siGLO co-nucleofection efficiencies in naïve B cells at 24 h post-nucleofection. (C) % siGLO+ B cells are shown from multiple independent experiments of (B) at 24 h post-nucleofection (n = 5). (D) IRF4 knockdown effect is retained in naïve B cells co-nucleofected with siGLO and IRF4 siRNA after stimulation with anti-IgM + CpG-B for 48 h (n = 4). (E) Same as (D) except GAPD MFI was examined 24 h post-nucleofection (n = 3). (F) Comparison of GAPD knockdown from (E) in siGLO+ vs. siGLO− cells. Bars represent mean ± SEM. P-values were determined by paired t-test for significance (*p < 0.05, **p < 0.01).
We next attempted co-nucleofection of total CD19+ B cells with siGLO and IRF4 siRNA to examine nucleofection efficiency and knockdown in multiple B cell subsets at one time. We detected similar but low levels of siGLO in both naïve B cells and plasmablasts 24 h post-nucleofection (Figures 7A–C). Analysis of IRF4 knockdown in naïve B cells revealed a similar knockdown level as that seen by nucleofection of purified naïve B cells (Figures 2, 7D) suggesting that this may be an alternate method for knockdown in naïve B cells. However, depending on the experimental outcome, sorting naïve B cells from nucleofected total B cells is unlikely to yield sufficient cell number for downstream functional analysis. Last, significant knockdown of IRF4 in plasmablasts was detected also by this method.
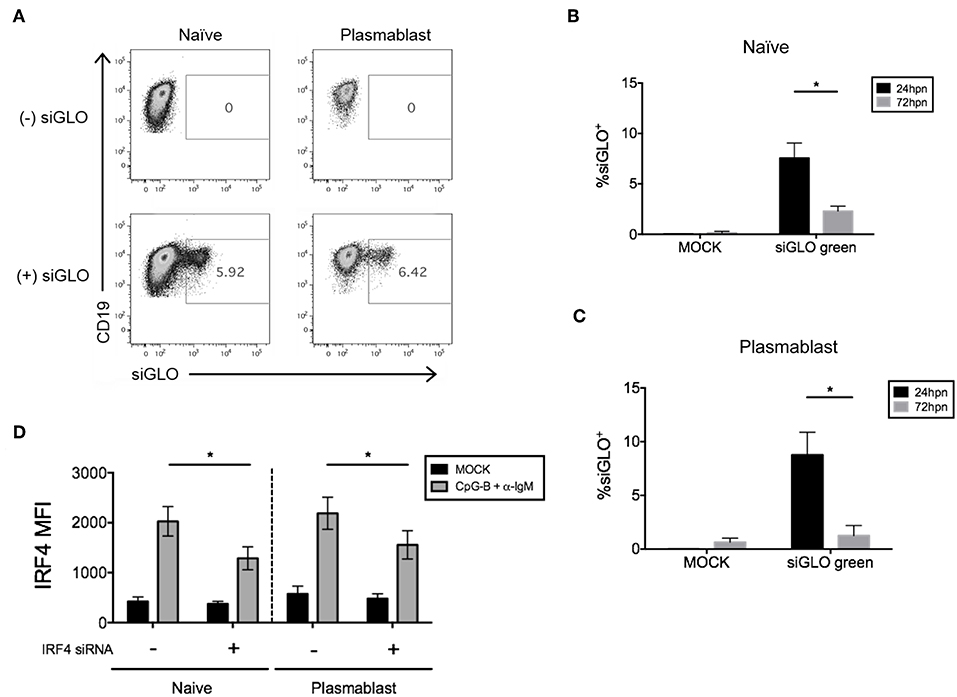
Figure 7. Efficiency of siGLO co-nucleofection with IRF4 siRNA in purified total CD19+ human B cells. (A) Representative dot plots of gated naïve B cells (CD19+IgD+) and plasmablasts (CD19+IgD−CD27+CD38+) that are siGLO+ at 24 h post-nucleofection with 1.5 μM siGLO. (B,C) Similar to (A) except data are summarized from multiple independent experiments showing siGLO nucleofection efficiency at 24 and 72 h post-nucleofection in naïve B cells (B) and plasmablasts (C) (n = 4). (D) IRF4 knockdown efficiency is retained in naïve B cells and plasmablasts after co-nucleofection of total B cells with siGLO and stimulation with anti-IgM + CpG-B for 48 h (n = 4). Bars represent mean ± SEM. P-values were determined by paired t-test for significance (*p < 0.05).
Discussion
In this report, we describe an optimized method for RNAi nucleofection of human primary naïve B cells to study the role of genes, such as IRF4 and AID that contribute to human ASC differentiation. The knockdown efficiency of both IRF4 and AID was sufficient to observe downstream functional effects on plasmablast differentiation. The central issue in optimizing gene knockdown, however, is to understand basal expression and expression induced after stimulation of your target gene. For transcription factors such as IRF5 that are sensitive to activation by nucleic acid-sensing innate immune sensors (43–47), we suggest optimizing with the low dose dual nucleofection protocol described by De et al. (17). Low concentrations of siRNA (500 nM), in two sequential nucleofections, provided an IRF5 knockdown with 40–60% efficiency (17). The lower siRNA concentrations likely minimize activation of RNA sensors and genes regulating the inflammatory response, which ultimately lead to IRF5 upregulation (48–50). In the case of IRF4, we were unable to detect knockdown after dual nucleofection with low concentrations of siRNA (Figure 2A). Dual nucleofection also results in more cell loss due to two transfer steps from cuvette to plate (data not shown). Further, genes such as IRF4 and AID that are expressed at low levels in naïve B cells require stimulation with a B cell activating trigger in order to detect knockdown. While optimizing our protocol for AID, we found that pre-activation was necessary to enhance the effect of knockdown. Similarly, other examples exist for genes with distinct patterns of expression in B cell differentiation, such as BACH2 (51), that requires alternate nucleofection protocols. Thus, the variation in knockdown efficiency between genes and amongst methods to determine knockdown efficiency (MFI vs. percent positivity) may be attributed to the inherent variation in gene expression (42).
Despite the limited and variable quantity of starting material, combined with the limited recovery of cells post-nucleofection, the bulk study of transient gene knockdown in human primary naïve B cells by flow cytometry after nucleofection can be achieved with the methods described herein (Figure 8). The optimal cell density of nucleofected naïve B cells when cultured long-term for plasmablast differentiation (7 days) was 5 × 105 after nucleofection. This number is likely necessary due to cell-cell contact required by B cells. Thus, key features of our method take into account cell density at the point of nucleofection and after nucleofection, as well as the kinetics of the target protein expression.
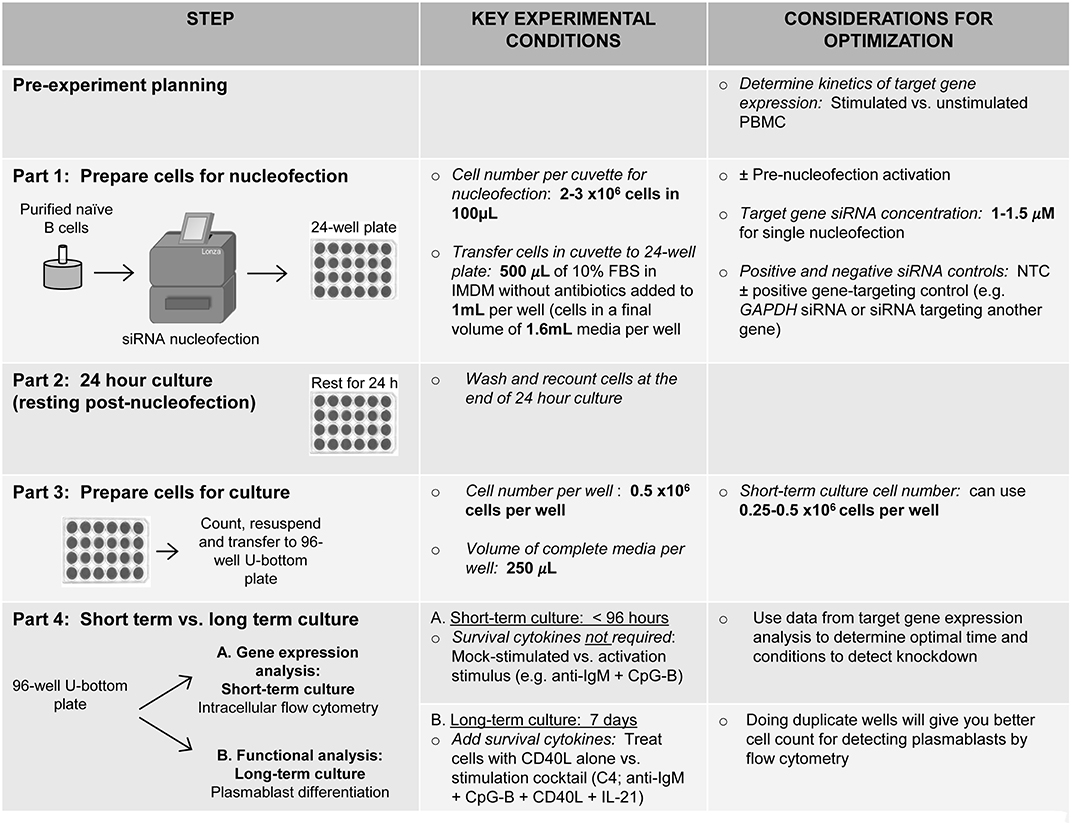
Figure 8. Schematic summarizing an optimized method for RNAi knockdown in primary human naïve B cells.
A good starting point for knocking down any gene of interest is to begin with 1 μM of siRNA; however, this may require further optimization depending on the kinetics of gene expression. Data presented herein suggests that knockdown in other B cell subsets can be obtained by nucleofection of total B cells, followed by sorting subsets of interest (Figure 7). However, some subsets, such as memory B cells and plasma cells that are low in the circulation will require alternative methods (52) for knockdown. For human primary GC plasma cells, Maarof et al. (53) describe a method for isolation and nucleofection to study IL-24 cytokine expression. Additional methods have been developed to study pre-B cells (54). Whether the general approach we describe can be applied to studies in patient samples is dependent on the specific considerations regarding amount of starting material available and relative frequency of the cell type being studied (55–58). Thus, depending on the cell subset of interest and functional read-out, methods may need to be further optimized to take into account reduced cell numbers.
Unfortunately, 100% transfection efficiency was not achieved in our study or by others (17, 51–54), possibly due to stochastic kinetics of siRNA entry into primary B cells (59). As death of primary human B cells following nucleofection correlates with size and structure of nucleic acids being transfected (60), siRNA knockdown efficiencies may vary between siRNAs and gene targets (61). Further, nucleofection of human B cells has been reported to be relatively inefficient when compared to human T cells (52). And, few, if any, studies have reported siRNA knockdown in human primary naïve B cells (17, 51). Applications of new technologies for genome editing, such as CRISPR-CAS9, in primary total B cells have reported comparable on-target efficacies as siRNA knockdown (62–64). We thus propose that the variables described in our optimized method using RNAi transfection technologies will provide a complementary approach for autologous therapeutic genome editing where stable modification to the host genome may entail long-term safety risks to the recipient. Further, these methods can be used more rapidly than CRISPR-CAS9 technology to strengthen the translation of findings from non-human models to human disease models.
Data Availability
All relevant data generated and analyzed for this study are included in the manuscript and Supplementary Files. For additional protocol details and/or original data, please contact Betsy J. Barnes (YmJhcm5lczFAbm9ydGh3ZWxsLmVkdQ==).
Ethics Statement
This study was carried out in accordance with the Institutional Review Board of Rutgers Biomedical and Health Sciences and the Feinstein Institute for Medical Research with written consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki.
Author Contributions
SD, TS, and BB were involved in the conception and design of the methodology and wrote the manuscript. TS performed all experiments. TS and BB analyzed the data. All authors have read and approved the final manuscript.
Funding
This work was supported by grants from the Lupus Research Alliance (BB), National Institutes of Health NIAMS AR065959-01 (BB), DOD Lupus Research Program W81XWH-18-1-0674 (BB), and the New Jersey Commission on Cancer Research (SD).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Flow Cytometry Facilities the Feinstein Institute for Medical Research for technical support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01652/full#supplementary-material
References
1. Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. (2015) 15:160–71. doi: 10.1038/nri3795
2. Ma S, Turetsky A, Trinh L, Lu R. IFN regulatory factor 4 and 8 promote Ig light chain kappa locus activation in pre-B cell development. J Immunol. (2006) 177:7898–904. doi: 10.4049/jimmunol.177.11.7898
3. Ma S, Pathak S, Trinh L, Lu R. Interferon regulatory factors 4 and 8 induce the expression of Ikaros and Aiolos to down-regulate pre-B-cell receptor and promote cell-cycle withdrawal in pre-B-cell development. Blood. (2008) 111:1396–403. doi: 10.1182/blood-2007-08-110106
4. Ochiai K, Maienschein-Cline M, Mandal M, Triggs JR, Bertolino E, Sciammas R, et al. A self-reinforcing regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation. Nat Immunol. (2012) 13:300–7. doi: 10.1038/ni.2210
5. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
6. Schroeder MA, DiPersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis Model Mech. (2011) 4:318–33. doi: 10.1242/dmm.006668
7. Richard ML, Gilkeson G. Mouse models of lupus: what they tell us and what they don't. Lupus Sci Med. (2018) 5:e000199. doi: 10.1136/lupus-2016-000199
8. Miklos DB, Kim HT, Miller KH, Guo L, Zorn E, Lee SJ, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. (2005) 105:2973–8. doi: 10.1182/blood-2004-09-3660
9. Sarantopoulos S, Stevenson KE, Kim HT, Cutler CS, Bhuiya NS, Schowalter M, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. (2009) 113:3865–74. doi: 10.1182/blood-2008-09-177840
10. Kharfan-Dabaja MA, Cutler CS. Rituximab for prevention and treatment of graft-versus-host disease. Int J Hematol. (2011) 93:578–85. doi: 10.1007/s12185-011-0855-2
11. Sarantopoulos S, Stevenson KE, Kim HT, Washel WS, Bhuiya NS, Cutler CS, et al. Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood. (2011) 117:2275–83. doi: 10.1182/blood-2010-10-307819
12. Allen JL, Tata PV, Fore MS, Wooten J, Rudra S, Deal AM, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood. (2014) 123:2108–15. doi: 10.1182/blood-2013-10-533562
13. Jacobson CA, Sun L, Kim HT, McDonough SM, Reynolds CG, Schowalter M, et al. Post-transplantation B cell activating factor and B cell recovery before onset of chronic graft-versus-host disease. Biol Blood Marrow Transplant. (2014) 20:668–75. doi: 10.1016/j.bbmt.2014.01.021
14. Sarantopoulos S, Blazar BR, Cutler C, Ritz J. B cells in chronic graft-versus-host disease. Biol Blood Marrow Transplant. (2015) 21:16–23. doi: 10.1016/j.bbmt.2014.10.029
15. Somasundaram R, Prasad MA, Ungerback J, Sigvardsson M. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood. (2015) 126:144–52. doi: 10.1182/blood-2014-12-575688
16. Koff JL, Flowers CR. B cells gone rogue: the intersection of diffuse large B cell lymphoma and autoimmune disease. Expert Rev Hematol. (2016) 9:553–61. doi: 10.1080/17474086.2016.1180972
17. De S, Zhang B, Shih T, Singh S, Winkler A, Donnelly R, et al. B cell-intrinsic role for IRF5 in TLR9/BCR-induced human B cell activation, proliferation, and plasmablast differentiation. Front Immunol. (2018) 8:1938. doi: 10.3389/fimmu.2017.01938
18. Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. (2010) 162:271–9. doi: 10.1111/j.1365-2249.2010.04206.x
19. Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, et al. IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol. (2005) 175:7867–79. doi: 10.4049/jimmunol.175.12.7867
20. Huggins J, Pellegrin T, Felgar RE, Wei C, Brown M, Zheng B, et al. CpG DNA activation and plasma-cell differentiation of CD27- naive human B cells. Blood. (2007) 109:1611–9. doi: 10.1182/blood-2006-03-008441
21. Genestier L, Taillardet M, Mondiere P, Gheit H, Bella C, Defrance T. TLR agonists selectively promote terminal plasma cell differentiation of B cell subsets specialized in thymus-independent responses. J Immunol. (2007) 178:7779–86. doi: 10.4049/jimmunol.178.12.7779
22. Avery DT, Deenick EK, Ma CS, Suryani S, Simpson N, Chew GY, et al. B cell-intrinsic signaling through IL-21 receptor and STAT3 is required for establishing long-lived antibody responses in humans. J Exp Med. (2010) 207:155–71. doi: 10.1084/jem.20091706
23. Berglund LJ, Avery DT, Ma CS, Moens L, Deenick EK, Bustamante J, et al. IL-21 signalling via STAT3 primes human naive B cells to respond to IL-2 to enhance their differentiation into plasmablasts. Blood. (2013) 122:3940–50. doi: 10.1182/blood-2013-06-506865
24. Agrawal S, Gupta S. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J Clin Immunol. (2011) 31:89–98. doi: 10.1007/s10875-010-9456-8
25. Mittrucker HW, Matsuyama T, Grossman A, Kundig TM, Potter J, Shahinian A, et al. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science. (1997) 275:540–3.
26. Klein U, Casola S, Cattoretti G, Shen Q, Lia M, Mo T, et al. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol. (2006) 7:773–82. doi: 10.1038/ni1357
27. Biswas PS, Gupta S, Stirzaker RA, Kumar V, Jessberger R, Lu TT, et al. Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J Exp Med. (2012) 209:581–96. doi: 10.1084/jem.20111195
28. Deenick EK, Avery DT, Chan A, Berglund LJ, Ives ML, Moens L, et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J Exp Med. (2013) 210:2739–53. doi: 10.1084/jem.20130323
29. Tangye SG, Avery DT, Deenick EK, Hodgkin PD. Intrinsic differences in the proliferation of naive and memory human B cells as a mechanism for enhanced secondary immune responses. J Immunol. (2003) 170:686–94. doi: 10.4049/jimmunol.170.2.686
30. Moens L, Tangye SG. Cytokine-mediated regulation of plasma cell generation: IL-21 takes center stage. Front Immunol. (2014) 5:65. doi: 10.3389/fimmu.2014.00065
31. Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, et al. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet. (1997) 17:226–30. doi: 10.1038/ng1097-226
32. Willis SN, Good-Jacobson KL, Curtis J, Light A, Tellier J, Shi W, et al. Transcription factor IRF4 regulates germinal center cell formation through a B cell-intrinsic mechanism. J Immunol. (2014) 192:3200–6. doi: 10.4049/jimmunol.1303216
33. Indrevaer RL, Moskaug JO, Paur I, Bohn SK, Jorgensen SF, Blomhoff R, et al. IRF4 is a critical gene in retinoic acid-mediated plasma cell formation and is deregulated in common variable immunodeficiency-derived B cells. J Immunol. (2015) 195:2601–11. doi: 10.4049/jimmunol.1500250
34. Mocellin S, Provenzano M. RNA interference: learning gene knock-down from cell physiology. J Transl Med. (2004) 2:39. doi: 10.1186/1479-5876-2-39
35. Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S, et al. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Biol Chem. (2008) 283:30632–41. doi: 10.1074/jbc.M801401200
36. Phadke MS, Krynetskaia NF, Mishra AK, Krynetskiy E. Glyceraldehyde 3-phosphate dehydrogenase depletion induces cell cycle arrest and resistance to antimetabolites in human carcinoma cell lines. J Pharmacol Exp Ther. (2009) 331:77–86. doi: 10.1124/jpet.109.155671
37. Zhang JY, Zhang F, Hong CQ, Giuliano AE, Cui XJ, Zhou GJ, et al. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med. (2015) 12:10–22. doi: 10.7497/j.issn.2095-3941.2014.0019
38. Chiche J, Reverso-Meinietti J, Mouchotte A, Rubio-Patino C, Mhaidly R, Villa E, et al. GAPDH expression predicts the response to R-CHOP, the tumor metabolic status, and the response of DLBCL patients to metabolic inhibitors. Cell Metab. (2019) 29:1243–57 e1210. doi: 10.1016/j.cmet.2019.02.002
39. Migliaccio N, Palmieri C, Ruggiero I, Fiume G, Martucci NM, Scala I, et al. B-cell receptor-guided delivery of peptide-siRNA complex for B-cell lymphoma therapy. Cancer Cell Int. (2015) 15:50. doi: 10.1186/s12935-015-0202-4
40. Ramiro A, Reina San-Martin B, McBride K, Jankovic M, Barreto V, Nussenzweig A, et al. The role of activation-induced deaminase in antibody diversification and chromosome translocations. Adv Immunol. (2007) 94:75–107. doi: 10.1016/S0065-2776(06)94003-6
41. Delker RK, Fugmann SD, Papavasiliou FN. A coming-of-age story: activation-induced cytidine deaminase turns 10. Nat Immunol. (2009) 10:1147–53. doi: 10.1038/ni.1799
42. Hong SW, Jiang Y, Kim S, Li CJ, Lee DK. Target gene abundance contributes to the efficiency of siRNA-mediated gene silencing. Nucleic Acid Ther. (2014) 24:192–8. doi: 10.1089/nat.2013.0466
43. Kim DH, Longo M, Han Y, Lundberg P, Cantin E, Rossi JJ. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat Biotechnol. (2004) 22:321–5. doi: 10.1038/nbt940
44. Hornung V, Guenthner-Biller M, Bourquin C, Ablasser A, Schlee M, Uematsu S, et al. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat Med. (2005) 11:263–70. doi: 10.1038/nm1191
45. Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, et al. 5'-Triphosphate RNA is the ligand for RIG-I. Science. (2006) 314:994–7. doi: 10.1126/science.1132505
46. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5'-phosphates. Science. (2006) 314:997–1001. doi: 10.1126/science.1132998
47. Kim S, Koo T, Jee HG, Cho HY, Lee G, Lim DG, et al. CRISPR RNAs trigger innate immune responses in human cells. Genome Res. (2018) 28:367–73. doi: 10.1101/gr.231936.117
48. Marques JT, Williams BR. Activation of the mammalian immune system by siRNAs. Nat Biotechnol. (2005) 23:1399–405. doi: 10.1038/nbt1161
49. Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. (2010) 9:57–67. doi: 10.1038/nrd3010
50. Meng Z, Lu M. RNA interference-induced innate immunity, off-target effect, or immune adjuvant? Front Immunol. (2017) 8:331. doi: 10.3389/fimmu.2017.00331
51. Hipp N, Symington H, Pastoret C, Caron G, Monvoisin C, Tarte K, et al. IL-2 imprints human naive B cell fate towards plasma cell through ERK/ELK1-mediated BACH2 repression. Nat Commun. (2017) 8:1443. doi: 10.1038/s41467-017-01475-7
52. Kardava L, Moir S, Wang W, Ho J, Buckner CM, Posada JG, et al. Attenuation of HIV-associated human B cell exhaustion by siRNA downregulation of inhibitory receptors. J Clin Invest. (2011) 121:2614–24. doi: 10.1172/JCI45685
53. Maarof G, Bouchet-Delbos L, Gary-Gouy H, Durand-Gasselin I, Krzysiek R, Dalloul A. Interleukin-24 inhibits the plasma cell differentiation program in human germinal center B cells. Blood. (2010) 115:1718–26. doi: 10.1182/blood-2009-05-220251
54. Kurosawa A, Saito S, Mori M, Adachi N. Nucleofection-based gene targeting in human pre-B cells. Gene. (2012) 492:305–8. doi: 10.1016/j.gene.2011.11.003
56. Tyner JW, Deininger MW, Loriaux MM, Chang BH, Gotlib JR, Willis SG, et al. RNAi screen for rapid therapeutic target identification in leukemia patients. Proc Natl Acad Sci USA. (2009) 106:8695–700. doi: 10.1073/pnas.0903233106
57. Maxson JE, Gotlib J, Pollyea DA, Fleischman AG, Agarwal A, Eide CA, et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia and atypical CML. N Engl J Med. (2013) 368:1781–90. doi: 10.1056/NEJMoa1214514
58. Agarwal A, Tyner JW. RNAi screening of leukemia cells using electroporation. Methods Mol Biol. (2016) 1470:85–94. doi: 10.1007/978-1-4939-6337-9_7
59. Bartlett DW, Davis ME. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic Acids Res. (2006) 34:322–33. doi: 10.1093/nar/gkj439
60. Seiffert M, Stilgenbauer S, Dohner H, Lichter P. Efficient nucleofection of primary human B cells and B-CLL cells induces apoptosis, which depends on the microenvironment and on the structure of transfected nucleic acids. Leukemia. (2007) 21:1977–83. doi: 10.1038/sj.leu.2404863
61. Aleman LM, Doench J, Sharp PA. Comparison of siRNA-induced off-target RNA and protein effects. RNA. (2007) 13:385–95. doi: 10.1261/rna.352507
62. Smith I, Greenside PG, Natoli T, Lahr DL, Wadden D, Tirosh I, et al. Evaluation of RNAi and CRISPR technologies by large-scale gene expression profiling in the Connectivity Map. PLoS Biol. (2017) 15:e2003213. doi: 10.1371/journal.pbio.2003213
63. Johnson MJ, Laoharawee K, Lahr WS, Webber BR, Moriarity BS. Engineering of primary human B cells with CRISPR/Cas9 targeted nuclease. Sci Rep. (2018) 8:12144. doi: 10.1038/s41598-018-30358-0
Keywords: IRF4, AID, siRNA knockdown, plasmablast, antibody secreting cells, B cell
Citation: Shih T, De S and Barnes BJ (2019) RNAi Transfection Optimized in Primary Naïve B Cells for the Targeted Analysis of Human Plasma Cell Differentiation. Front. Immunol. 10:1652. doi: 10.3389/fimmu.2019.01652
Received: 21 March 2019; Accepted: 03 July 2019;
Published: 23 July 2019.
Edited by:
Ana María Hernández, University of Havana, CubaReviewed by:
Michael Reth, University of Freiburg, GermanyChristopher Sundling, Karolinska Institute (KI), Sweden
Copyright © 2019 Shih, De and Barnes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Betsy J. Barnes, YmJhcm5lczFAbm9ydGh3ZWxsLmVkdQ==