
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 02 July 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01459
 Adrià Aterido1,2
Adrià Aterido1,2 Juan D. Cañete3Jesús Tornero4Francisco Blanco5Benjamín Fernández-Gutierrez6Carolina Pérez7Mercedes Alperi-López8Alex Olivè9Héctor Corominas10Víctor Martínez-Taboada11Isidoro González12Antonio Fernández-Nebro13Alba Erra14María López-Lasanta1Mireia López Corbeto1Núria Palau1Sara Marsal1
Juan D. Cañete3Jesús Tornero4Francisco Blanco5Benjamín Fernández-Gutierrez6Carolina Pérez7Mercedes Alperi-López8Alex Olivè9Héctor Corominas10Víctor Martínez-Taboada11Isidoro González12Antonio Fernández-Nebro13Alba Erra14María López-Lasanta1Mireia López Corbeto1Núria Palau1Sara Marsal1 Antonio Julià1*
Antonio Julià1*Background: Rheumatoid arthritis (RA) is the most frequent autoimmune disease involving the joints. Although anti-TNF therapies have proven effective in the management of RA, approximately one third of patients do not show a significant clinical response. The objective of this study was to identify new genetic variation associated with the clinical response to anti-TNF therapy in RA.
Methods: We performed a sequential multi-omic analysis integrating different sources of molecular information. First, we extracted the RNA from synovial biopsies of 11 RA patients starting anti-TNF therapy to identify gene coexpression modules (GCMs) in the RA synovium. Second, we analyzed the transcriptomic association between each GCM and the clinical response to anti-TNF therapy. The clinical response was determined at week 14 using the EULAR criteria. Third, we analyzed the association between the GCMs and anti-TNF response at the genetic level. For this objective, we used genome-wide data from a cohort of 348 anti-TNF treated patients from Spain. The GCMs that were significantly associated with the anti-TNF response were then tested for validation in an independent cohort of 2,706 anti-TNF treated patients. Finally, the functional implication of the validated GCMs was evaluated via pathway and cell type epigenetic enrichment analyses.
Results: A total of 149 GCMs were identified in the RA synovium. From these, 13 GCMs were found to be significantly associated with anti-TNF response (P < 0.05). At the genetic level, we detected two of the 13 GCMs to be significantly associated with the response to adalimumab (P = 0.0015) and infliximab (P = 0.021) in the Spain cohort. Using the independent cohort of RA patients, we replicated the association of the GCM associated with the response to adalimumab (P = 0.0019). The validated module was found to be significantly enriched for genes involved in the nucleotide metabolism (P = 2.41e-5) and epigenetic marks from immune cells, including CD4+ regulatory T cells (P = 0.041).
Conclusions: These findings show the existence of a drug-specific genetic basis for anti-TNF response, thereby supporting treatment stratification in the search for response biomarkers in RA.
Rheumatoid arthritis (RA) is the most common autoimmune-inflammatory arthritis affecting up to 1% of the worldwide population (1). RA is characterized by the chronic infiltration of immune cells in the synovial membrane, leading to progressive destruction of the joint cartilage and bone (2). The most notable success in the treatment of RA has been the introduction of Tumor Necrosis Factor (TNF) inhibitors. Anti-TNF agents have radically changed the prognosis of many RA patients (3), providing an important improvement of clinical signs and symptoms, quality of life and long-term protection of the synovial joint integrity. Despite this major accomplishment, there is a large fraction of anti-TNF treated patients (30–40%) that do not show a significant clinical improvement (4). To date, little is known on the biological mechanisms that underlie this differential response to anti-TNF agents. Understanding the basis of the lack of response could not only help to personalize patient therapy but also to gain insights into RA heterogeneity at the molecular level.
The two main classes of anti-TNF agents are monoclonal antibodies against TNF, like adalimumab and infliximab (5, 6), and the recombinant fusion protein containing the soluble TNF receptor p75 etanercept (7). Despite targeting the same molecule, the different anti-TNF drugs do not show the same level of efficacy in all patients. At the cellular and molecular level, there is evidence that the clinical response is partially mediated by the activation of drug-specific biological mechanisms (8–10). Supporting this, clinical observations have shown that patients who fail one anti-TNF treatment may still respond to a different anti-TNF drug (11). Therefore, there is a need to identify the genetic variability underlying the response to anti-TNF agents.
To date, a small number of genome-wide transcriptomic studies have been conducted on the RA synovium to investigate the biological processes associated with anti-TNF response (12–17). The gene expression signatures obtained in these studies, however, have shown a modest association with the treatment outcome. As a result, the overlap of differentially expressed genes between these studies is relatively low (18). This evidence suggests the existence of a high biological variability between RA patients. Identifying the source of this biological variation would be a major advance toward personalized medicine in RA (19).
Associating genetic variation to disease risk has provided a wealth of genes and biological pathways relevant for RA (20). The use of this approach to characterize the genetic basis of anti-TNF response in RA has, however, proven less productive. More than 40 candidate-gene association studies have been performed so far, but there has been very limited or non-existent replicability (21). Genome-wide association studies (GWAS) have proven to be a more successful approach for this objective. To date, eight GWAS on anti-TNF response in RA have been performed (22–29), identifying several loci associated at a genome-wide scale. From these, variation at MED15, GFRA1, PDE3A-SLCO1C1, and CD84 has been replicated in, at least, an independent cohort of patients (30). However, these few loci are insufficient to explain the heritability of anti-TNF response and, consequently, alternative analysis approaches must be devised (31, 32).
The integration of high-throughput transcriptomic and genomic data offers a new opportunity to characterize the biological basis underlying complex traits (33–37). In RA, the integrative analysis of gene expression levels and genetic variation has proven effective to identify novel causal genes as well as cell-type specific mechanisms associated with the disease pathogenesis (38–41). As an example, gene expression data from peripheral blood mononuclear cells has been successfully used to guide the selection of candidate genes for genetic association analysis (42). Accordingly, analyzing the expression profile associated with anti-TNF response at the target site of inflammation in RA -the synovial membrane- should be a powerful means to identify genetic variation associated with the therapeutic response. To date, this type of integrative analysis has not been performed in RA.
To gain a better understanding of the biological mechanisms of anti-TNF response in RA, we have performed a combined transcriptomic and genomic analysis. Using transcriptomic data from the RA synovium, we first identified the modules of co-expressed genes that are associated with anti-TNF response. We next tested the association of these modules at the genetic risk level using two independent GWAS cohorts of RA patients. Finally, we investigated the functional implication of the genetic modules associated with anti-TNF response at the two levels of molecular variation. These findings demonstrate the power of integrating multi-omic data to characterize the genetic basis of drug response in a complex disease like RA.
A total of 11 RA patients with clinically active RA from Spain were recruited by the outpatient's clinics of the rheumatology department of the Hospital Clinic de Barcelona. All patients fulfilled the 1987 American College of Rheumatology (ACR) classification criteria for RA (43). The main epidemiological and clinical variables of these patients are summarized in Table 1.
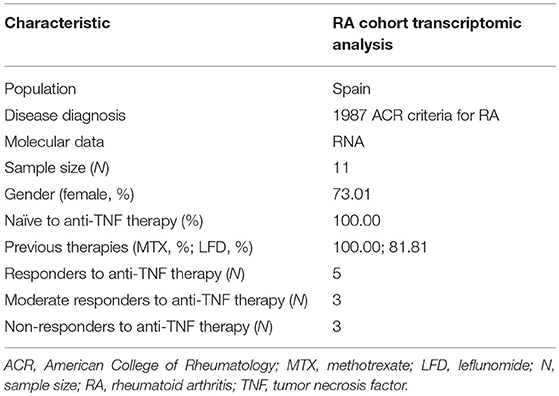
Table 1. Clinical and epidemiological characteristics of the patient cohort used for the genome-wide expression profiling.
A total of 348 RA patients that had received an anti-TNF treatment as their first biological treatment (adalimumab, infliximab, or etanercept) were included in the discovery stage of the genetic association study. This cohort of RA patients was collected from the outpatient's clinics of the rheumatology departments of 12 Spanish University Hospitals involved in the Immune-Mediated Inflammatory Disease Consortium (IMIDC) (44). For this study, all patients fulfilled the 1987 ACR classification criteria for RA and had more than 2 years of follow-up since diagnosis (43). All recruited individuals had an erosive disease defined as erosions in more than one joint group including hands and/or feet.
All RA patients included in this cohort were Caucasian European and born in Spain. Only those RA patients with all grandparents born in Spain were selected for the set-based genetic association analysis. The main clinical and epidemiological characteristics of this cohort of RA patients are summarized in Table S1.
Validation of the associated modules was performed using the RA anti-TNF therapy pharmacogenetic cohort described previously (22). This cohort consists of 2,706 RA patients of European ancestry compiled from 13 collections across five countries that had received an anti-TNF treatment (adalimumab, infliximab, or etanercept). All patients fulfilled the 1987 ACR criteria for RA or were diagnosed by a board-certified rheumatologist as previously described (22). The main clinical and epidemiological characteristics of this independent cohort are summarized in Table S1.
This study was carried out in accordance with the recommendations of the guidelines and regulations of the Hospital Universitari Vall d'Hebron Clinical Research Ethics Committee and local institutional review boards with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Hospital Universitari Vall d'Hebron Clinical Research Ethics Committee.
The clinical response to anti-TNF therapy was measured using the European League Against Rheumatism (EULAR) treatment response criteria (45). For all patients, the Disease Activity Score (DAS28) was measured at baseline and after 3–6 months of anti-TNF therapy (46). According to the DAS28 variation and the DAS28 at the endpoint, RA patients were categorized into good, moderate, and non-responders (Table S1).
Synovial biopsies were obtained by guided arthroscopy of the inflamed knee joint from 11 RA patients using a 2.7 mm arthroscope (Storz, Tuttlingen, Germany) under local anesthesia. In all patients, 6–8 biopsies were extracted from the suprapatellar pouch and medial and lateral gutters with a 3 mm grasping forceps. According to previous pharmacogenetic studies demonstrating that the clinical response to anti-TNF therapy in RA is associated with the pre-treatment transcriptomic profile (12), synovial biopsies were obtained before the treatment initiation. Each synovial sample was snap-frozen in liquid nitrogen and stored at −80°C for later RNA extraction. Total RNA was extracted from the synovial biopsies using the RNA Mini Kit (Qiagen, USA) and the integrity was assessed using BioAnalyzer microfluidic gel analysis (Agilent, USA). All samples were of high quality (RNA Integrity Number > 8). After RNA isolation, biotin-labeled cRNA (1.5 μg) was prepared using the Ambion Illumina RNA amplification kit (Ambion, USA) and Illumina TotalPrep RNA Amplification Kit (Ambion, US).
Whole genome transcript abundance from the RA inflamed synovium of the 11 RA patients was performed using the Illumina HumanWG-6 expression array system (Illumina, San Diego, CA, USA). This microarray platform measures the gene expression levels of more than 47,000 different transcripts. Data collection was performed using BeadStudio 3.1.1.0 software (Illumina, San Diego, USA). In order to use the most recent human genome annotation information, only microarray probes matching curated gene sequences from NCBI RefSeq database (release 51) (47) were selected. A final number of 21,189 curated probes representing the expression of 18,524 different human genes were finally selected for analysis. The gene expression intensities were then normalized on the log2-scale using the quantile normalization method (48). The presence of a potential bias between batches of microarray was minimized using the Bayesian ComBat procedure (49). The Raw and normalized data analyzed in the present study are publicly available in the NCBI's Gene Expression Omnibus database (accession number: GSE47726) (50).
There is compelling evidence that the expression of human genes is highly coordinated to develop biological functions (51). In order to identify modules of genes that might share transcriptional regulatory mechanisms in the RA inflamed synovium, we performed a genome-wide co-expression analysis using transcriptomic data from the synovial biopsies of 11 RA patients. For this objective, we used the weighted correlation network analysis (WGCNA) implemented in R software (52). In this method, the absolute pairwise gene expression correlation is raised to a soft thresholding power (β = 14) to compute a network adjacency matrix that emphasizes high correlations at the expense of low correlations. This matrix determines the gene coexpression network that is subsequently used to identify groups of genes with a high topological overlap (53). In this step, an unsupervised hierarchical clustering approach was implemented to detect the most representative modules of co-expressed genes in the RA inflamed synovium. Those modules composed by 10–300 genes were selected for follow-up analyses (54).
In order to describe how each GCM fits with genetic findings from previous studies on anti-TNF response in RA, we assessed the gene overlap of each GCM in genes that have been associated with anti-TNF response at the transcriptomic (N > 500 differentially expressed genes in the RA synovium) (12, 14–16, 18) and genetic level (N = 78 genes, P < 1e-05 in GWAS catalog) (55). In addition, we have investigated whether the GCMs include susceptibility genes for RA (N = 200 genes, P < 5e-08 in GWAS catalog) (55).
In order to analyze the association between each gene coexpression module (GCM) and the clinical response to anti-TNF therapy, we performed a principal component analysis (PCA). The first principal component, which captures the largest variability of the GCM (i.e., GCM eigengene), was used to test for association with anti-TNF response using a logistic regression model adjusted by gender. Based on the hypothesis that more extreme response phenotypes provide improved discrimination, we compared EULAR good responders (N = 5 patients) and EULAR non-responders (N = 3 patients) as previously described (22). Individuals showing an EULAR moderate response were excluded from the analysis (N = 3 patients). The complete list of association results for each GCM are shown in Table S2.
The GCMs that showed a significant association with the clinical response to anti-TNF were further characterized by computing the module significance and intramodular connectivity using the WGCNA software. The module significance is a measure of the biological significance of a given module for the phenotype tested for association. This measure is defined as the absolute value of the average biological significance of each gene and ranges from 0 to 1 (i.e., the higher the module significance, the more biologically significant is the module for the phenotype tested for association). The intramodular connectivity is an average measure of the gene connectivity within a given module.
The association between GCMs and anti-TNF response was also studied at the genetic level. For this objective, we used genotype data from the discovery cohort of RA patients to investigate the impact of genetic variation at these GCMs on the clinical response to anti-TNF therapy (25). The detailed procedure that was followed to perform genome-wide genotyping with the Illumina Quad610 BeadChip (Illumina, San Diego, California, USA) and the quality control analysis have been described previously (25). To evaluate the presence of population stratification in the anti-TNF treated patients, we conducted a PCA implemented in EIGENSOFT (v4.2) software (56). Using the first 10 principal components of variation over 10 iterations, none of the samples showed an outlier genetic background (Figure S1). To increase the number of genetic variants available for association testing, we performed genome-wide imputation. After pre-phasing the haplotypes using SHAPEIT V2 (Oxford, UK), imputation was conducted with IMPUTE V2 (Oxford, UK) (57). As a reference, we used the Caucasian European cohort (N = 379 samples) data generated by the 1,000 Genomes Project (phase 1, version 3) (58). Only those SNPs showing a MAF > 0.05 and having a high-quality imputation score (i.e., info quality metric > 0.8) were selected for the set-based genetic association analysis. After imputation, a total of 1,387,382 genetic variants were finally available to be tested for association with the clinical response to anti-TNF therapy.
In order to test for replication of the GCMs that were found to be associated with anti-TNF response in the discovery cohort, we used GWAS data from an independent cohort of 2,706 RA patients that had received anti-TNF treatment. These GWAS data were obtained from the Synapse public repository (syn3280809, doi: 10.7303/syn3280809)). The genotyping and imputation procedures have been previously described (22). A total of 2,539,607 genetic variants were available for replication testing of the GCMs associated in the discovery cohort.
The set-based genetic association analysis is a powerful approach to test the association between groups of biologically-related genes and complex traits (59, 60). Using this analytical strategy we have successfully identified new genetic pathways associated with psoriasis, psoriatic arthritis, and with clinically relevant phenotypes of systemic lupus erythematosus (61–63). In the present study, we have used the set-based test implemented in PLINK. In order to use this analytical methodology, SNPs need to be assigned to genes and, subsequently, to GCMs. For this objective, we conducted the SNP-gene mapping using proximity-based criteria, which is the pre-dominant approach in genetic association analyses at the set level (54, 59). According to the reference studies using this statistical approach (59, 64, 65), we used a SNP-gene distance window of 20 Kb and the NCBI RefSeq database for gene annotation (Release 63, 12th October 2017) (66). Those SNPs mapping to genes that are included in GCMs (Table S2) were subsequently assigned to their corresponding GCMs. A total of 249,220 SNPs mapping to the transcriptomically-associated GCMs were finally available for set-based analysis at the genetic level. In the set based-testing, an average association statistic is computed for each GCM and its empirical significance is calculated by a permutation-based approach. Briefly, SNPs are pruned for LD and then each one is individually tested for association with the phenotype, in this case, response to anti-TNF therapy. Association testing was here performed using a logistic regression model incorporating the principal components of variation to control for potential ancestry variation. In the same way of the transcriptomic analysis, genetic association of each GCM was performed using only patients with more distinct responses: that is, EULAR good responders and EULAR non-responders. The P-values of the associated GCMs in the discovery and validation cohorts were combined using the logit method implemented in R (67).
In order to investigate the biological relevance of the GCM associated with the clinical response to adalimumab, we conducted a functional enrichment analysis on common biological processes. For this objective, we used the reference databases for biological pathway annotation: (i) BioCarta (www.biocarta.com), (ii) Kyoto Encyclopedia of Genes and Genomes (KEGG) (68), and Reactome (69). To ensure that the enrichment results are representative of the biology underlying the adalimumab response, we used exclusively those genes that were associated with treatment response in both the discovery and replication cohorts (P < 0.05). The enrichment analysis was performed using the hypergeometric statistical test (70) and the False Discovery Rate (FDR) method was used to account for multiple testing (71).
Many of the pathological cell types responsible of the clinical heterogeneity of autoimmune diseases are still unknown (72). In the present study, we hypothesized that genetic variation from the associated modules is a valuable source of biological information to identify cell types influencing the anti-TNF response. Accordingly, we analyzed the enrichment of the module-associated variation on the cell-type-specific H3K4me3 chromatin mark using the EPIGWAS software (73). For this analysis we used the module-associated variation (P < 0.05 in either the discovery or replication cohorts), epigenetic data from H3K4me3 peaks of 34 cell types generated by the Roadmap project (74), and reference genotyping data from the Caucasian European population included in the 1,000 Genomes Project (58). The EPIGWAS software estimates the regulatory score of each variant (i.e., normalized ratio between the nearest H3K4me3 peak and distance to the summit of the peak). For each cell type, the loci scores are summed to assess the cell type regulatory score. Using a sampling-based approach (N = 10,000 matched sets of SNPs from the HapMap Project), the statistical significance of the enrichment analysis is defined as the proportion of matched sets exceeding the observed cell type specific regulatory score. In addition to this cell type epigenetic enrichment analysis, we have also conducted an epigenetic fine-mapping of the module-associated variation in enhancer histone marks previously described as cell-type specific (i.e., H3K27ac and H3K4me1) (75). This analysis has been performed using HaploR software (76, 77).
To determine the expression patterns that characterize the inflamed synovium in RA, we performed a genome-wide weighted coexpression analysis on transcriptomic data generated from synovial biopsies from patients starting anti-TNF therapy. Using this approach, we identified a total of 149 GCMs (Figure 1). The module content ranged from 14 to 251 genes (Table S2). From the total of 149 GCMs identified in the genome-wide coexpression analysis, we found that 74 GCMs (49.66%) include genes that are differentially expressed between responders and non-responders to anti-TNF therapy, 9 GCMs (6.05%) include genetic variation underlying anti-TNF response, and 37 GCMs (24.83%) include susceptibility genes for RA (Table S3).
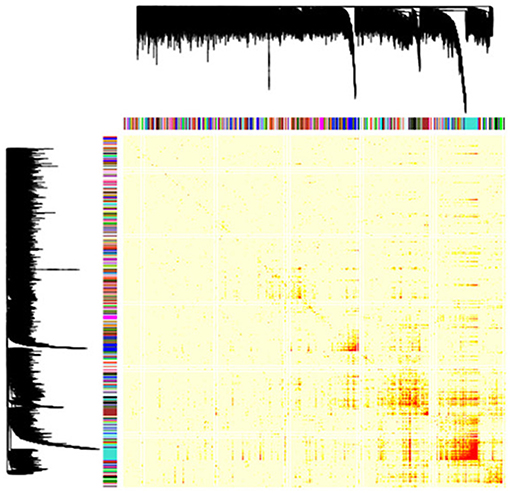
Figure 1. Gene coexpression modules identified in the RA inflamed synovium. Dendrogram showing the 149 gene coexpression modules identified with the unsupervised hierarchical clustering approach in the genome-wide coexpression analysis. Each dendrogram branch corresponds to a gene coexpression module, as shown in the color bar below. The height of the dendrogram represents the co-expression distance among genes. The heatmap represents the adjacency matrix that was built using the pairwise gene expression correlation raised to a soft thresholding power of β = 14. The heatmap is colored according to the absolute value of the pairwise gene expression correlation, ranging from yellow (i.e., weak correlation) to red (i.e., strong correlation). The x and y axis represent the total of 18,524 genes that were included in the genome-wide coexpression analysis.
We then analyzed the association between the 149 GCMs and the response to anti-TNF therapy at week 14. We found that 13 GCMs were significantly associated to anti-TNF response (P < 0.05, Table 2).
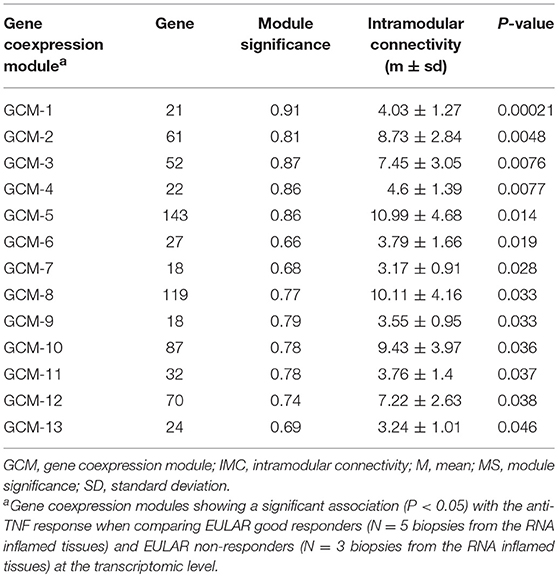
Table 2. Gene coexpression modules from the RA inflamed synovium that are associated with anti-TNF response at the transcriptomic level.
After identifying the genetic modules associated with anti-TNF response at the transcript level, we sought to test their association at the genetic level. Using a set-based analysis approach on GWAS data from 348 anti-TNF treated RA patients from Spain, we tested the association of the 13 GCMs with the response to anti-TNF therapy (Table S4). GCM-7 and GCM-10 were found to be significantly associated with the response to adalimumab and infliximab treatments, respectively (P < 0.05, Table 3). Using GWAS data from a large international cohort of 2,706 RA patients, we validated the association between GCM-7 and the response to adalimumab (P = 0.0019, Table 3).

Table 3. Gene coexpression modules showing treatment specific associations at the genetic level.
To characterize the functional role of the module associated to adalimumab response, we performed two complementary enrichment analyses. Using reference data from common biological pathways, we found that GCM-7 is significantly enriched in genes that participate in the nucleotide metabolism (P = 2.41e-05, Table 4; Table S5; Figure S2). Using cell-type specific H3K4me3 epigenetic data, we found that genetic variation at GCM-7 is significantly enriched in epigenetic marks from six different cell types (Figure 2), including CD4+ regulatory T cells (Tregs, P = 0.041) and CD34+ myeloid lineage precursors (P = 0.021). No significant differences between the number of module-associated variants mapping to the enhancer marks H3K27ac and H3K4me1 were detected in any cell type (P > 0.05). Enhancer histone marks from fibroblast primary cells were found to include the largest number of module-associated variants (Figure S3).
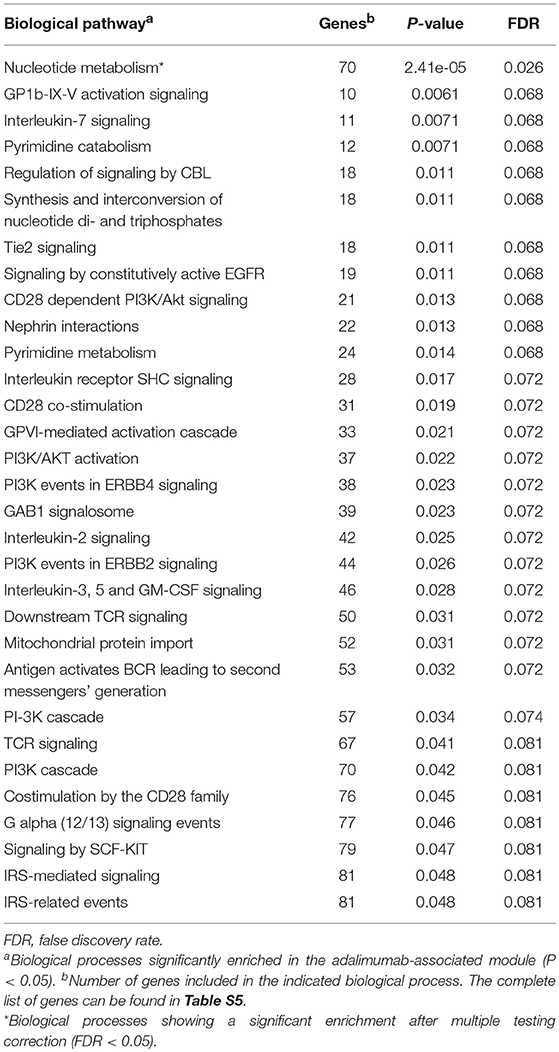
Table 4. Common biological processes enriched in genes from the adalimumab-associated module.
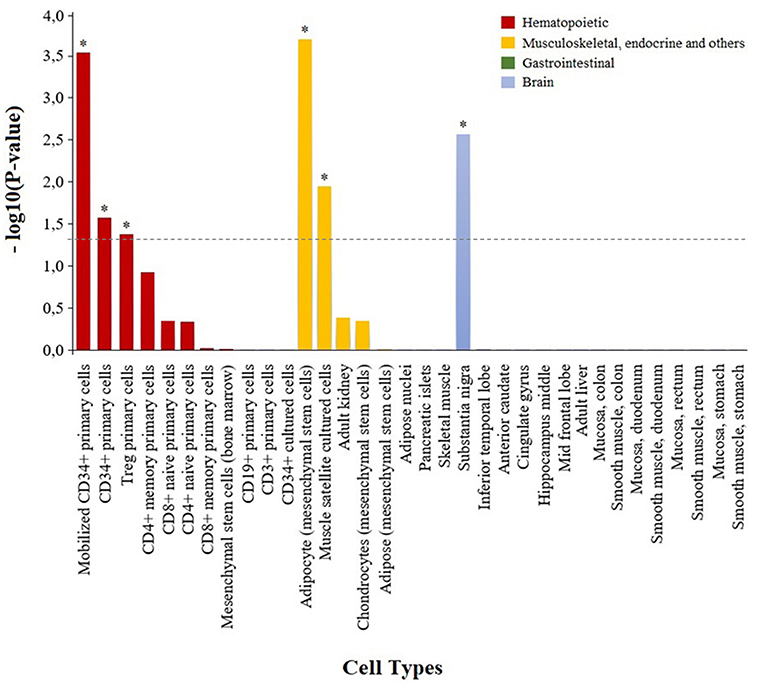
Figure 2. Enrichment of genetic variation from the adalimumab-associated module in H3K4me3 histone marks in specific cell types. Bar plot showing the statistical significance of the adalimumab-associated genetic module and H3K4me3 histone marks from the 34 cell types analyzed. Cell types with H3K4me3 histone marks significantly enriched in adalimumab-associated variants are represented with an asterisk (P < 0.05).
One of the major challenges in the treatment of RA is to understand the biological mechanisms influencing the clinical response to anti-TNF therapy. Genetic and transcriptomic analyses have been used separately to characterize the molecular basis of treatment efficacy, but with limited success. To investigate the genetic basis of anti-TNF response in RA, we have performed a combined transcriptomic and genomic analysis. Analyzing gene expression data from the RA inflamed synovium, we have first identified the GCMs that are associated with anti-TNF response. We have then tested the association of these GCMs at the genetic level using two independent patient cohorts. This combined genomic approach has enabled to identify a genetic module that is associated with the response to adalimumab. Functional analysis of this module suggests that nucleotide metabolism and Tregs could mediate this response.
In this study, we have found a genetic basis for the clinical response to adalimumab that is not shared with other anti-TNF drugs. These results are in line with previous genetic findings derived from both GWAS and candidate-gene analyses. Genome-wide significant loci MED15 and CD84 were found to be associated with the clinical response to etanercept, but not to adalimumab or infliximab (22, 25). At the candidate-gene level, we have previously found that variation at FCGR2A gene, which here mapped to a GCM composed by >300 genes that was excluded from the analysis, is associated with the clinical response to adalimumab but not to etanercept (78). There is also evidence of treatment-specific variation at the transcriptomic level. Treatment with adalimumab has been associated with a gene signature in the synovial membrane that is involved in cellular proliferation (16). This gene signature, however, has not been observed after infliximab treatment, thereby suggesting a different mode of action in apparently similar drugs (12). Taken together, our results provide additional support to the existence of specific biological mechanisms that mediate the response to adalimumab.
The genetic module associated with adalimumab response was found to be enriched in genes that participate in the nucleotide metabolism. In addition to the essential role that this biological process plays in DNA replication, nucleotide metabolism is responsible for the synthesis of adenosine, a purine nucleoside that exhibits a potent anti-inflammatory activity when bound to its cognate adenosine receptors (79). Binding of adenosine to the A2A receptor in M1 macrophages, the principal producers of TNF in the synovial joint in RA, induces the switch to the anti-inflammatory M2 phenotype and subsequent reduction of pro-inflammatory mediators (80–82). In turn, adenosine receptors are upregulated by cytokines that activate NFκB, like TNF, and their expression has been found to be high in RA patients (83). Despite this overexpression, adenosine receptors display a weaker affinity for adenosine in RA patients compared to controls, thereby dampening their anti-inflammatory effect. There is evidence that treatment with adalimumab and not methotrexate restores the binding parameters of adenosine receptors of RA patients to those of healthy individuals (84, 85). Our results are in line with this evidence and provide a functional link between the effectivity of an anti-TNF therapy and the local production of adenosine in the synovial joint.
The integration of cell type epigenetic data with our genetic association results suggested an important role of immune cells for mediating the response to adalimumab in RA. In this analysis, both Tregs and CD34+ cells are associated with anti-TNF response. Tregs are central anti-inflammatory and self-tolerance mediators, producing high levels of anti-inflammatory cytokines TGF-β and IL-10 to inhibit the overactivation of effector T cells (86–88). In RA, Tregs have been found to be functionally defective, and treatment with anti-TNF agents has shown to restore their T cell suppressor capacity (89, 90). There is evidence, however, that this beneficial effect on Tregs is reached via treatment-specific mechanisms (91). In particular, adalimumab, but not etanercept, has been shown to induce a particular Treg phenotype that restrains IL-17-mediated inflammation by downregulating the production of IL-6 by monocytes (92). In line with the specific effects that anti-TNF agents can have on different T cell subsets (93), our findings indicate that genetic variation could influence the activity of Tregs of RA patients treated with adalimumab.
In this study, we have identified a GCM that is enriched in epigenetic marks of CD34+ myeloid precursor cells and associated to adalimumab response. There is evidence that circulating bone marrow-derived stem cells like CD34+ cells migrate to the inflamed RA synovium (94), where they form de novo blood vessels during the acute phase of the disease (95, 96). Neoangiogenesis is an essential mechanism for the recruitment of immune cells into the RA synovium and perpetuation of the synovial inflammation (97). After recruitment, the high immune cell proliferation generates and hypoxic environment that stimulates the production of more neoangiogenic mediators (79), which have been found highly expressed in the RA inflamed synovium (98–100). From a pharmacological perspective, anti-TNF agents have been shown to ameliorate inflammation by effectively reducing RA synovium vascularity (101, 102). As expected, this reduction has been found to be stronger in responder patients (103). Based on this evidence, the modulation of the neoangiogenic activity of CD34+ cells could be one of the possible mechanisms by which anti-TNF agents could ameliorate synovial inflammation in RA. Supporting this hypothesis, recent studies have identified myeloid signatures and phenotypes in RA synovium that are associated to treatment response (17, 104). Consequently, genetic variation regulating the neoangiogenic function of the CD34+ cells infiltrating the RA synovium could also influence the clinical response to adalimumab.
The present study has limitations. The number of RA patients included in the transcriptomic analysis was relatively low. For this reason, patients could not be stratified by neither anti-TNF agent nor synovial phenotype. It is likely that, by analyzing a larger number of synovial biopsies, additional GCMs and biological mechanisms underlying anti-TNF response in the RA synovium could be identified. Although extracting synovial biopsies from larger cohorts of patients is clinically challenging, this will help to expand the present genetic association results. Another limitation is that the clinical response for the two GWAS cohorts was not identical. While both patient cohorts used the same clinical score (i.e., EULAR response criteria), the Spain sample recorded the response at 3 months of treatment, and the replication sample included clinical responses in a 3–6 months window. The clinical efficacy at these two time points tends to be correlated, however, this difference could have led to a loss of statistical power to validate the genetic module associated with the response to infliximab (GCM-10). Finally, one caveat of the set-based method used here is that the genetic association between GCMs and anti-TNF response was tested using SNPs within or proximal to the GCMs' genes. It is well-known that many regulatory SNPs lie in the non-coding genome and modulate gene expression through 3D interactions and eQTL mechanisms. Our strategy did not account for this. To our knowledge, there is yet no set-based method that has successfully integrated this regulatory information. One of the major problems for this approach is the context-dependent nature of many 3D interactions and eQTLs. In particular, there is evidence that both spatial DNA organization and eQTLs are cell-type and cell-state dependent (105–111). The integration of this regulatory information is therefore still a challenge for the set-based genetic analysis. With the increasing regulatory information that is currently being derived from eQTL analysis and Hi-C experiments on separate cell types, a more profound ascertainment of the impact of SNPs on gene expression will be obtained and, eventually, more comprehensive set-based methods will be developed. These new findings, however, will exponentiate the number of possible regulatory sites, and careful selection of disease-relevant cell types should be performed. To this regard, our study suggests that regulatory T cells and CD34+ progenitors are relevant cell types for the mediation of the response to anti-TNF agents. It is nonetheless important to note that the lack of publicly available epigenetic data from synovial fibroblasts has precluded to assess how genetic variation from the adalimumab-associated GCM impact on this relevant cell type and, consequently, on the adalimumab efficacy in RA. Moreover, recent studies using the single-cell RNA-seq technology have identified new cell types in the RA synovium (112). Future studies incorporating this technology are therefore warranted not only to corroborate our findings, but also to identify new and cell type specific GCMs influencing anti-TNF response in RA.
In conclusion, integrating transcriptomic and genetic data, we have identified a genetic module that is associated with the clinical response to adalimumab. These results provide new insights into the biological basis underlying the differential response to anti-TNF agents and suggest that drug diversity should be considered in the search for treatment response biomarkers.
This study was carried out in accordance with the recommendations of the guidelines and regulations of the Hospital Universitari Vall d'Hebron Clinical Research Ethics Committee and local institutional review boards with written informed consent from all subjects. All subjects gave written informed consent in accordance with the Declaration of Helsinki. The protocol was approved by the Hospital Universitari Vall d'Hebron Clinical Research Ethics Committee.
All the authors were involved in the design, analysis, and interpretation of data. All authors revised the manuscript and gave final approval for its submission.
The present study was funded by the Spanish Ministry of Economy and Competitiveness (Grant Nos. PSE-010000-2006-6 and IPT-010000-2010-36) and by the Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR, FI-DGR 2016, Grant No. 00587), which is supported by the Secretaria d'Universitats i Recerca (Economy and Knowledge Department, Generalitat de Catalunya), and co-funded by the European Social Fund. The study sponsor had no role in the collection, analysis, or interpretation of the data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and clinical specialists collaborating in the IMID Consortium for participation. We thank the data contributors that provided genotype data from anti-TNF treated patients through the Synapse public repository.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01459/full#supplementary-material
1. Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. (2003) 423:356–61. doi: 10.1038/nature01661
2. Klareskog L, Catrina AI, Paget S. Rheumatoid arthritis. Lancet. (2009) 373:659–72. doi: 10.1016/S0140-6736(09)60008-8
3. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
4. Kievit W, Adang EM, Fransen J, Kuper HH, van de Laar MA, Jansen TL, et al. The effectiveness and medication costs of three anti-tumour necrosis factor alpha agents in the treatment of rheumatoid arthritis from prospective clinical practice data. Ann Rheum Dis. (2008) 67:1229–34. doi: 10.1136/ard.2007.083675
5. Kempeni J. Preliminary results of early clinical trials with the fully human anti-TNFalpha monoclonal antibody D2E7. Ann Rheum Dis. (1999) 58(Suppl 1):I70–2. doi: 10.1136/ard.58.2008.i70
6. Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, et al. Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet. (1994) 344:1105–10. doi: 10.1016/S0140-6736(94)90628-9
7. Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, et al. Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med. (1997) 337:141–7. doi: 10.1056/NEJM199707173370301
8. Agnholt J, Dahlerup JF, Kaltoft K. The effect of etanercept and infliximab on the production of tumour necrosis factor alpha, interferon-gamma and GM-CSF in in vivo activated intestinal T lymphocyte cultures. Cytokine. (2003) 23:76–85. doi: 10.1016/S1043-4666(03)00201-1
9. Aeberli D, Seitz M, Juni P, Villiger PM. Increase of peripheral CXCR3 positive T lymphocytes upon treatment of RA patients with TNF-alpha inhibitors. Rheumatology. (2005) 44:172–5. doi: 10.1093/rheumatology/keh437
10. Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y, et al. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. (2005) 52:61–72. doi: 10.1002/art.20764
11. Kekow J, Mueller-Ladner U, Schulze-Koops H. Rituximab is more effective than second anti-TNF therapy in rheumatoid arthritis patients and previous TNFalpha blocker failure. Biologics. (2012) 6:191–9. doi: 10.2147/BTT.S32244
12. van der Pouw Kraan TC, Wijbrandts CA, van Baarsen LG, Rustenburg F, Baggen JM, Verweij CL, et al. Responsiveness to anti-tumour necrosis factor alpha therapy is related to pre-treatment tissue inflammation levels in rheumatoid arthritis patients. Ann Rheum Dis. (2008) 67:563–6. doi: 10.1136/ard.2007.081950
13. van der Pouw Kraan TC, van Gaalen FA, Huizinga TW, Pieterman E, Breedveld FC, Verweij CL. Discovery of distinctive gene expression profiles in rheumatoid synovium using cDNA microarray technology: evidence for the existence of multiple pathways of tissue destruction and repair. Genes Immun. (2003) 4:187–96. doi: 10.1038/sj.gene.6363975
14. Lindberg J, Wijbrandts CA, van Baarsen LG, Nader G, Klareskog L, Catrina A, et al. The gene expression profile in the synovium as a predictor of the clinical response to infliximab treatment in rheumatoid arthritis. PLoS ONE. (2010) 5:e11310. doi: 10.1371/journal.pone.0011310
15. Lindberg J, af Klint E, Catrina AI, Nilsson P, Klareskog L, Ulfgren AK, et al. Effect of infliximab on mRNA expression profiles in synovial tissue of rheumatoid arthritis patients. Arthritis Res Ther. (2006) 8:R179. doi: 10.1186/ar2090
16. Badot V, Galant C, Nzeusseu Toukap A, Theate I, Maudoux AL, Van den Eynde BJ, et al. Gene expression profiling in the synovium identifies a predictive signature of absence of response to adalimumab therapy in rheumatoid arthritis. Arthritis Res Ther. (2009) 11:R57. doi: 10.1186/ar2678
17. Dennis G Jr, Holweg CT, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. (2014) 16:R90. doi: 10.1186/ar4555
18. Toonen EJ, Gilissen C, Franke B, Kievit W, Eijsbouts AM, den Broeder AA, et al. Validation study of existing gene expression signatures for anti-TNF treatment in patients with rheumatoid arthritis. PLoS ONE. (2012) 7:e33199. doi: 10.1371/journal.pone.0033199
19. Folkersen L, Brynedal B, Diaz-Gallo LM, Ramskold D, Shchetynsky K, Westerlind H, et al. Integration of known DNA, RNA and protein biomarkers provides prediction of anti-TNF response in rheumatoid arthritis: results from the COMBINE study. Mol Med. (2016) 22:322–8. doi: 10.2119/molmed.2016.00078
20. Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. (2014) 506:376–81. doi: 10.1038/nature12873
21. Bek S, Bojesen AB, Nielsen JV, Sode J, Bank S, Vogel U, et al. Systematic review and meta-analysis: pharmacogenetics of anti-TNF treatment response in rheumatoid arthritis. Pharmacogenomics J. (2017) 17:403–11. doi: 10.1038/tpj.2017.26
22. Cui J, Stahl EA, Saevarsdottir S, Miceli C, Diogo D, Trynka G, et al. Genome-wide association study and gene expression analysis identifies CD84 as a predictor of response to etanercept therapy in rheumatoid arthritis. PLoS Genet. (2013) 9:e1003394. doi: 10.1371/journal.pgen.1003394
23. Umicevic Mirkov M, Cui J, Vermeulen SH, Stahl EA, Toonen EJ, Makkinje RR, et al. Genome-wide association analysis of anti-TNF drug response in patients with rheumatoid arthritis. Ann Rheum Dis. (2013) 72:1375–81. doi: 10.1136/annrheumdis-2012-202405
24. Honne K, Hallgrimsdottir I, Wu C, Sebro R, Jewell NP, Sakurai T, et al. A longitudinal genome-wide association study of anti-tumor necrosis factor response among Japanese patients with rheumatoid arthritis. Arthritis Res Ther. (2016) 18:12. doi: 10.1186/s13075-016-0920-6
25. Julia A, Fernandez-Nebro A, Blanco F, Ortiz A, Canete JD, Maymo J, et al. A genome-wide association study identifies a new locus associated with the response to anti-TNF therapy in rheumatoid arthritis. Pharmacogenomics J. (2016) 16:147–50. doi: 10.1038/tpj.2015.31
26. Krintel SB, Palermo G, Johansen JS, Germer S, Essioux L, Benayed R, et al. Investigation of single nucleotide polymorphisms and biological pathways associated with response to TNFalpha inhibitors in patients with rheumatoid arthritis. Pharmacogenet Genomics. (2012) 22:577–89. doi: 10.1097/FPC.0b013e3283544043
27. Plant D, Bowes J, Potter C, Hyrich KL, Morgan AW, Wilson AG, et al. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. (2011) 63:645–53. doi: 10.1002/art.30130
28. Liu C, Batliwalla F, Li W, Lee A, Roubenoff R, Beckman E, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol Med. (2008) 14:575–81. doi: 10.2119/2008-00056.Liu
29. Massey J, Plant D, Hyrich K, Morgan AW, Wilson AG, Spiliopoulou A, et al. Genome-wide association study of response to tumour necrosis factor inhibitor therapy in rheumatoid arthritis. Pharmacogenomics J. (2018) 18:657–64. doi: 10.1038/s41397-018-0040-6
30. Acosta-Colman I, Palau N, Tornero J, Fernandez-Nebro A, Blanco F, Gonzalez-Alvaro I, et al. GWAS replication study confirms the association of PDE3A-SLCO1C1 with anti-TNF therapy response in rheumatoid arthritis. Pharmacogenomics. (2013) 14:727–34. doi: 10.2217/pgs.13.60
31. Umicevic Mirkov M, Janss L, Vermeulen SH, van de Laar MA, van Riel PL, Guchelaar HJ, et al. Estimation of heritability of different outcomes for genetic studies of TNFi response in patients with rheumatoid arthritis. Ann Rheum Dis. (2015) 74:2183–7. doi: 10.1136/annrheumdis-2014-205541
32. Sieberts SK, Zhu F, Garcia-Garcia J, Stahl E, Pratap A, Pandey G, et al. Crowdsourced assessment of common genetic contribution to predicting anti-TNF treatment response in rheumatoid arthritis. Nat Commun. (2016) 7:12460. doi: 10.1038/ncomms12460
33. Mehta D, Heim K, Herder C, Carstensen M, Eckstein G, Schurmann C, et al. Impact of common regulatory single-nucleotide variants on gene expression profiles in whole blood. Eur J Hum Genet. (2013) 21:48–54. doi: 10.1038/ejhg.2012.106
34. Goring HH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, Cole SA, et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet. (2007) 39:1208–16. doi: 10.1038/ng2119
35. Montgomery SB, Dermitzakis ET. From expression QTLs to personalized transcriptomics. Nat Rev Genet. (2011) 12:277–82. doi: 10.1038/nrg2969
36. Lappalainen T, Sammeth M, Friedlander MR, t Hoen PA, Monlong J, Rivas MA, et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature. (2013) 501:506–11. doi: 10.1038/nature12531
37. Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. (2007) 315:848–53. doi: 10.1126/science.1136678
38. Okada Y, Suzuki A, Ikari K, Terao C, Kochi Y, Ohmura K, et al. Contribution of a non-classical HLA gene, HLA-DOA, to the risk of rheumatoid arthritis. Am J Hum Genet. (2016) 99:366–74. doi: 10.1016/j.ajhg.2016.06.019
39. Ishigaki K, Kochi Y, Suzuki A, Tsuchida Y, Tsuchiya H, Sumitomo S, et al. Polygenic burdens on cell-specific pathways underlie the risk of rheumatoid arthritis. Nat Genet. (2017) 49:1120–5. doi: 10.1038/ng.3885
40. Thalayasingam N, Nair N, Skelton AJ, Massey J, Anderson AE, Clark AD, et al. CD4+ and B lymphocyte expression quantitative traits at rheumatoid arthritis risk loci in untreated early arthritis: implications for causal gene identification. Arthritis Rheum. (2017) 70:361–70. doi: 10.1136/annrheumdis-2017-eular.4028
41. Aterido A, Palacio C, Marsal S, Avila G, Julia A. Novel insights into the regulatory architecture of CD4+ T cells in rheumatoid arthritis. PLoS ONE. (2014) 9:e100690. doi: 10.1371/journal.pone.0100690
42. Mathews RJ, Robinson JI, Battellino M, Wong C, Taylor JC, Eyre S, et al. Evidence of NLRP3-inflammasome activation in rheumatoid arthritis (RA); genetic variants within the NLRP3-inflammasome complex in relation to susceptibility to RA and response to anti-TNF treatment. Ann Rheum Dis. (2014) 73:1202–10. doi: 10.1136/annrheumdis-2013-203276
43. Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. (1988) 31:315–24. doi: 10.1002/art.1780310302
44. Julia A, Tortosa R, Hernanz JM, Canete JD, Fonseca E, Ferrandiz C, et al. Risk variants for psoriasis vulgaris in a large case-control collection and association with clinical subphenotypes. Hum Mol Genet. (2012) 21:4549–57. doi: 10.1093/hmg/dds295
45. van Gestel AM, Haagsma CJ, van Riel PL. Validation of rheumatoid arthritis improvement criteria that include simplified joint counts. Arthritis Rheum. (1998) 41:1845–50. doi: 10.1002/1529-0131(199810)41:10<1845::AID-ART17>3.3.CO;2-B
46. Prevoo ML, van 't Hof MA, Kuper HH, van Leeuwen MA, van de Putte LB, van Riel PL. Modified disease activity scores that include twenty-eight-joint counts. Development and validation in a prospective longitudinal study of patients with rheumatoid arthritis. Arthritis Rheum. (1995) 38:44–8. doi: 10.1002/art.1780380107
47. Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence project: update and current status. Nucleic Acids Res. (2003) 31:34–7. doi: 10.1093/nar/gkg111
48. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. (2003) 19:185–93. doi: 10.1093/bioinformatics/19.2.185
49. Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. (2007) 8:118–27. doi: 10.1093/biostatistics/kxj037
50. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. (2002) 30:207–10. doi: 10.1093/nar/30.1.207
51. Komili S, Silver PA. Coupling and coordination in gene expression processes: a systems biology view. Nat Rev Genet. (2008) 9:38–48. doi: 10.1038/nrg2223
52. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. (2008) 9:559. doi: 10.1186/1471-2105-9-559
53. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. (2005) 4. doi: 10.2202/1544-6115.1128
54. Ramanan VK, Shen L, Moore JH, Saykin AJ. Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet. (2012) 28:323–32. doi: 10.1016/j.tig.2012.03.004
55. Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. (2019) 47:D1005–12. doi: 10.1093/nar/gky1120
56. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. (2006) 38:904–9. doi: 10.1038/ng1847
57. Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda, Md). (2011) 1:457–70. doi: 10.1534/g3.111.001198
58. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE. An integrated map of genetic variation from 1,092 human genomes. Nature. (2012) 491:56–65. doi: 10.1038/nature11632
59. Wang K, Li M, Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat Rev Genet. (2010) 11:843–54. doi: 10.1038/nrg2884
60. de Leeuw CA, Neale BM, Heskes T, Posthuma D. The statistical properties of gene-set analysis. Nat Rev Genet. (2016) 17:353–64. doi: 10.1038/nrg.2016.29
61. Aterido A, Julia A, Ferrandiz C, Puig L, Fonseca E, Fernandez-Lopez E, et al. Genome-wide pathway analysis identifies genetic pathways associated with psoriasis. J Invest Dermatol. (2016) 136:593–602. doi: 10.1016/j.jid.2015.11.026
62. Aterido A, Julia A, Carreira P, Blanco R, Lopez-Longo JJ, Venegas JJP, et al. Genome-wide pathway analysis identifies VEGF pathway association with oral ulceration in systemic lupus erythematosus. Arthritis Res Ther. (2017) 19:138. doi: 10.1186/s13075-017-1345-6
63. Aterido A, Canete JD, Tornero J, Ferrandiz C, Pinto JA, Gratacos J, et al. Genetic variation at the glycosaminoglycan metabolism pathway contributes to the risk of psoriatic arthritis but not psoriasis. Ann Rheum Dis. (2019) 78. doi: 10.1136/annrheumdis-2018-214158
64. Wu MC, Kraft P, Epstein MP, Taylor DM, Chanock SJ, Hunter DJ, et al. Powerful SNP-set analysis for case-control genome-wide association studies. Am J Hum Genet. (2010) 86:929–42. doi: 10.1016/j.ajhg.2010.05.002
65. Edwards YJ, Beecham GW, Scott WK, Khuri S, Bademci G, Tekin D, et al. Identifying consensus disease pathways in Parkinson's disease using an integrative systems biology approach. PLoS ONE. (2011) 6:e16917. doi: 10.1371/journal.pone.0016917
66. Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. (2012) 40:D130–5. doi: 10.1093/nar/gkr1079
67. Schroder MS, Culhane AC, Quackenbush J, Haibe-Kains B. survcomp: an R/Bioconductor package for performance assessment and comparison of survival models. Bioinformatics. (2011) 27:3206–8. doi: 10.1093/bioinformatics/btr511
68. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. (2000) 28:27–30. doi: 10.1093/nar/28.1.27
69. Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. (2014) 42:D472–7. doi: 10.1093/nar/gkv1351
70. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. (2011) 27:1739–40. doi: 10.1093/bioinformatics/btr260
71. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. (1990) 9:811–8. doi: 10.1002/sim.4780090710
72. Glinos DA, Soskic B, Trynka G. Immunogenomic approaches to understand the function of immune disease variants. Immunology. (2017) 152:527–35. doi: 10.1111/imm.12796
73. Trynka G, Sandor C, Han B, Xu H, Stranger BE, Liu XS, et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat Genet. (2013) 45:124–30. doi: 10.1038/ng.2504
74. Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, et al. The NIH roadmap epigenomics mapping consortium. Nat Biotechnol. (2010) 28:1045–8. doi: 10.1038/nbt1010-1045
75. Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. (2015) 518:337–43. doi: 10.1038/nature13835
76. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. (2012) 40:D930–4. doi: 10.1093/nar/gkr917
77. Zhbannikov IY, Arbeev K, Ukraintseva S, Yashin AI. haploR: an R package for querying web-based annotation tools. F1000Res. (2017) 6:97. doi: 10.12688/f1000research.10742.2
78. Avila-Pedretti G, Tornero J, Fernandez-Nebro A, Blanco F, Gonzalez-Alvaro I, Canete JD, et al. Variation at FCGR2A and functionally related genes is associated with the response to anti-TNF therapy in rheumatoid arthritis. PLoS ONE. (2015) 10:e0122088. doi: 10.1371/journal.pone.0122088
79. Cronstein BN, Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol. (2017) 13:41–51. doi: 10.1038/nrrheum.2016.178
80. Bours MJ, Swennen EL, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5'-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. (2006) 112:358–404. doi: 10.1016/j.pharmthera.2005.04.013
81. Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types. Oncogene. (2003) 22:1206–18. doi: 10.1038/sj.onc.1206184
82. Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti-TNF-alpha therapies: the next generation. Nat Rev Drug Discov. (2003) 2:736–46. doi: 10.1038/nrd1175
83. Driessler F, Venstrom K, Sabat R, Asadullah K, Schottelius AJ. Molecular mechanisms of interleukin-10-mediated inhibition of NF-kappaB activity: a role for p50. Clin Exp Immunol. (2004) 135:64–73. doi: 10.1111/j.1365-2249.2004.02342.x
84. Varani K, Massara A, Vincenzi F, Tosi A, Padovan M, Trotta F, et al. Normalization of A2A and A3 adenosine receptor up-regulation in rheumatoid arthritis patients by treatment with anti-tumor necrosis factor alpha but not methotrexate. Arthritis Rheum. (2009) 60:2880–91. doi: 10.1002/art.24794
85. Vincenzi F, Padovan M, Targa M, Corciulo C, Giacuzzo S, Merighi S, et al. A(2A) adenosine receptors are differentially modulated by pharmacological treatments in rheumatoid arthritis patients and their stimulation ameliorates adjuvant-induced arthritis in rats. PLoS ONE. (2013) 8:e54195. doi: 10.1371/journal.pone.0054195
86. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. (2010) 11:7–13. doi: 10.1038/ni.1818
87. Lundy SK, Sarkar S, Tesmer LA, Fox DA. Cells of the synovium in rheumatoid arthritis. T lymphocytes. Arthritis Res Ther. (2007) 9:202. doi: 10.1186/ar2107
88. Bluestone JA, Tang Q. How do CD4+CD25+ regulatory T cells control autoimmunity? Curr Opin Immunol. (2005) 17:638–642. doi: 10.1016/j.coi.2005.09.002
89. Huang Z, Yang B, Shi Y, Cai B, Li Y, Feng W, et al. Anti-TNF-alpha therapy improves Treg and suppresses Teff in patients with rheumatoid arthritis. Cell Immunol. (2012) 279:25–9. doi: 10.1016/j.cellimm.2012.09.001
90. Ceeraz S, Hall C, Choy EH, Spencer J, Corrigall VM. Defective CD8+CD28+ regulatory T cell suppressor function in rheumatoid arthritis is restored by tumour necrosis factor inhibitor therapy. Clin Exp Immunol. (2013) 174:18–26. doi: 10.1111/cei.12161
91. Byng-Maddick R, Ehrenstein MR. The impact of biological therapy on regulatory T cells in rheumatoid arthritis. Rheumatology. (2015) 54:768–75. doi: 10.1093/rheumatology/keu487
92. McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. (2012) 64:3129–38. doi: 10.1002/art.34565
93. Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, et al. TNFalpha in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine. (2018) 101:4–13. doi: 10.1016/j.cyto.2016.09.001
94. Ruger B, Giurea A, Wanivenhaus AH, Zehetgruber H, Hollemann D, Yanagida G, et al. Endothelial precursor cells in the synovial tissue of patients with rheumatoid arthritis and osteoarthritis. Arthritis Rheum. (2004) 50:2157–66. doi: 10.1002/art.20506
95. Dai J, Agelan A, Yang A, Zuluaga V, Sexton D, Colman RW, et al. Role of plasma kallikrein-kinin system activation in synovial recruitment of endothelial progenitor cells in experimental arthritis. Arthritis Rheum. (2012) 64:3574–82. doi: 10.1002/art.34607
96. van der Strate BW, Popa ER, Schipper M, Brouwer LA, Hendriks M, Harmsen MC, et al. Circulating human CD34+ progenitor cells modulate neovascularization and inflammation in a nude mouse model. J Mol Cell Cardiol. (2007) 42:1086–97. doi: 10.1016/j.yjmcc.2007.03.907
97. Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. J Clin Invest. (1999) 103:3–4. doi: 10.1172/JCI5929
98. Muz B, Khan MN, Kiriakidis S, Paleolog EM. Hypoxia. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res Ther. (2009) 11:201. doi: 10.1186/ar2568
99. Hollander AP, Corke KP, Freemont AJ, Lewis CE. Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum. (2001) 44:1540–4. doi: 10.1002/1529-0131(200107)44:7<1540::AID-ART277>3.0.CO;2-7
100. Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. (2002) 46:2587–97. doi: 10.1002/art.10520
101. Criscione LG, St. Clair EW. Tumor necrosis factor-alpha antagonists for the treatment of rheumatic diseases. Curr Opin Rheumatol. (2002) 14:204–11. doi: 10.1097/00002281-200205000-00002
102. Feldmann M, Maini RN. Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. (2001) 19:163–96. doi: 10.1146/annurev.immunol.19.1.163
103. Izquierdo E, Canete JD, Celis R, Santiago B, Usategui A, Sanmarti R, et al. Immature blood vessels in rheumatoid synovium are selectively depleted in response to anti-TNF therapy. PLoS ONE. (2009) 4:e8131. doi: 10.1371/journal.pone.0008131
104. Humby F, Lewis M, Ramamoorthi N, Hackney JA, Barnes MR, Bombardieri M, et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann Rheum Dis. (2019) 78:761–72. doi: 10.1136/annrheumdis-2018-214539
105. Francesconi M, Lehner B. The effects of genetic variation on gene expression dynamics during development. Nature. (2014) 505:208–11. doi: 10.1038/nature12772
106. van der Wijst MGP, Brugge H, de Vries DH, Deelen P, Swertz MA, Franke L. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat Genet. (2018) 50:493–7. doi: 10.1038/s41588-018-0089-9
107. Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science. (2014) 343:1246949. doi: 10.1126/science.1246949
108. Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. (2013) 502:59–64. doi: 10.1038/nature12593
109. Heidari N, Phanstiel DH, He C, Grubert F, Jahanbani F, Kasowski M, et al. Genome-wide map of regulatory interactions in the human genome. Genome Res. (2014) 24:1905–17. doi: 10.1101/gr.176586.114
110. Smith EM, Lajoie BR, Jain G, Dekker J. Invariant TAD boundaries constrain cell-type-specific looping interactions between promoters and distal elements around the CFTR locus. Am J Hum Genet. (2016) 98:185–201. doi: 10.1016/j.ajhg.2015.12.002
111. Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, et al. Static and dynamic DNA loops form AP-1-bound activation hubs during macrophage development. Mol Cell. (2017) 67:1037–48. doi: 10.1016/j.molcel.2017.08.006
Keywords: rheumatoid arthritis, genomics, transcriptomics, multi-omics association analysis, anti-TNF therapy
Citation: Aterido A, Cañete JD, Tornero J, Blanco F, Fernández-Gutierrez B, Pérez C, Alperi-López M, Olivè A, Corominas H, Martínez-Taboada V, González I, Fernández-Nebro A, Erra A, López-Lasanta M, López Corbeto M, Palau N, Marsal S and Julià A (2019) A Combined Transcriptomic and Genomic Analysis Identifies a Gene Signature Associated With the Response to Anti-TNF Therapy in Rheumatoid Arthritis. Front. Immunol. 10:1459. doi: 10.3389/fimmu.2019.01459
Received: 31 January 2019; Accepted: 10 June 2019;
Published: 02 July 2019.
Edited by:
Laurence Morel, University of Florida, United StatesReviewed by:
Alessandra Nerviani, Queen Mary University of London, United KingdomCopyright © 2019 Aterido, Cañete, Tornero, Blanco, Fernández-Gutierrez, Pérez, Alperi-López, Olivè, Corominas, Martínez-Taboada, González, Fernández-Nebro, Erra, López-Lasanta, López Corbeto, Palau, Marsal and Julià. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Antonio Julià, dG9uaS5qdWxpYUB2aGlyLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.