 Isabella Lengfelder1
Isabella Lengfelder1 Irina G. Sava1Jonathan J. Hansen2
Irina G. Sava1Jonathan J. Hansen2 Karin Kleigrewe3Jeremy Herzog2
Karin Kleigrewe3Jeremy Herzog2 Klaus Neuhaus4,5
Klaus Neuhaus4,5 Thomas Hofmann3,4R. Balfour Sartor2Dirk Haller1,4*
Thomas Hofmann3,4R. Balfour Sartor2Dirk Haller1,4*- 1Chair of Nutrition and Immunology, Technische Universität München, Freising, Germany
- 2Division of Gastroenterology and Hepatology, University of North Carolina, Chapel Hill, NC, United States
- 3Bavarian Center for Biomolecular Mass Spectrometry, Technische Universität München, Freising, Germany
- 4ZIEL - Institute for Food & Health, Technische Universität München, Freising, Germany
- 5ZIEL Core Facility Microbiome, Technische Universität München, Freising, Germany
Inflammatory bowel diseases (IBD) are associated with compositional and functional changes of the intestinal microbiota, but specific contributions of individual bacteria to chronic intestinal inflammation remain unclear. Enterococcus faecalis is a resident member of the human intestinal core microbiota that has been linked to the pathogenesis of IBD and induces chronic colitis in susceptible monoassociated IL-10-deficient (IL-10−/−) mice. In this study, we characterized the colitogenic activity of E. faecalis as part of a simplified human microbial consortium based on seven enteric bacterial strains (SIHUMI). RNA sequencing analysis of E. faecalis isolated from monoassociated wild type and IL-10−/− mice identified 408 genes including 14 genes of the ethanolamine utilization (eut) locus that were significantly up-regulated in response to inflammation. Despite considerable up-regulation of eut genes, deletion of ethanolamine utilization (ΔeutVW) had no impact on E. faecalis colitogenic activity in monoassociated IL-10−/− mice. However, replacement of the E. faecalis wild type bacteria by a ΔeutVW mutant in SIHUMI-colonized IL-10−/− mice resulted in exacerbated colitis, suggesting protective functions of E. faecalis ethanolamine utilization in complex bacterial communities. To better understand E. faecalis gene response in the presence of other microbes, we purified wild type E. faecalis cells from the colon content of SIHUMI-colonized wild type and IL-10−/− mice using immuno-magnetic separation and performed RNA sequencing. Transcriptional profiling revealed that the bacterial environment reprograms E. faecalis gene expression in response to inflammation, with the majority of differentially expressed genes not being shared between monocolonized and SIHUMI conditions. While in E. faecalis monoassociation a general bacterial stress response could be observed, expression of E. faecalis genes in SIHUMI-colonized mice was characterized by up-regulation of genes involved in growth and replication. Interestingly, in mice colonized with SIHUMI lacking E. faecalis enhanced inflammation was observed in comparison to SIHUMI-colonized mice, supporting the hypothesis that E. faecalis ethanolamine metabolism protects against colitis in complex consortia. In conclusion, this study demonstrates that complex bacterial consortia interactions reprogram the gene expression profile and colitogenic activity of the opportunistic pathogen E. faecalis toward a protective function.
Introduction
Inflammatory bowel diseases (IBD) with the two dominant types Crohn's disease (CD) and ulcerative colitis (UC) are chronic relapsing inflammatory disorders affecting the distal intestine. Several factors appear to be involved in the pathogenesis of IBD, including genetic susceptibility (1, 2) together with diverse environmental triggers resulting in an inappropriate T-cell mediated activation of immunity toward components of the intestinal microbiota (3–5). IBD is associated with compositional (6–10) and functional (11, 12) changes of the intestinal microbiota referred to as dysbiosis. Dysbiotic changes associated with IBD are characterized by an overrepresentation of opportunistic pathogens and a loss of beneficial commensal organisms, indicating that the pathogenic potential of a dysbiotic community can be linked to certain organisms. Consequently, the specific contribution of individual bacteria to disease pathogenesis needs to be investigated in detail.
A relevant bacterial species in the context of IBD is Enterococcus faecalis. E. faecalis is a Gram-positive resident member of the human intestinal core microbiota harboring a number of pathogenic traits, which explains the association of this bacterium with inflammatory diseases and fatal nosocomial infections (13–15). Relative to healthy volunteers, the abundance of enterococci is increased in fecal samples of CD patients (16–18). This is in line with a high frequency of E. faecalis housekeeping and virulence genes in CD cohorts (19) and a high likelihood of the presence of virulence factors in E. faecalis isolates originating from inflamed IBD mucosa (20). UC patients have increased numbers of mucosa-associated E. faecalis correlating with disease activity (21) and a high E. faecalis-specific IgG sero-reactivity (22).
The dual characteristics of E. faecalis as core member of the human intestinal microbiota and as an opportunistic pathogen make this bacterium an ideal model to study microbe-host interactions relevant to the development of chronic colitis in a genetically susceptible host. In the IL10tm1Cgn mouse (IL-10−/−), which is a well-characterized model of human chronic colitis, monoassociation with E. faecalis induces severe intestinal inflammation, whereas wild type mice remain disease-free (23–26). In previous studies, we could show that certain bacterial structures contribute to the colitogenic activity of E. faecalis in monoassociated IL-10−/− mice (27, 28).
Using germ-free wild type and IL-10−/− mice monoassociated with E. faecalis or colonized with a colitogenic human enteric bacterial consortium (SIHUMI) (29), we aim to unravel the functional relevance of E. faecalis in colitis development with regard to gene expression and interactions with co-colonizing bacteria. Based on RNA sequencing (RNA seq) analysis, we characterize the role of ethanolamine (EA) utilization for E. faecalis survival and colitogenic activity in a susceptible host.
The intestine is a rich source of the metabolite EA, which is derived from phospholipid phosphatidylethanolamine of bacterial and eukaryotic cell membranes (30, 31). Bacteria capable of EA catabolism can utilize this compound as a source of carbon and/or nitrogen to promote growth as well as a signal influencing virulence during host colonization (32). EA utilization is a well-recognized property of diverse pathogens, for example Salmonella and enterohemorrhagic Escherichia coli (EHEC) (33–37). The only commensals known to carry EA utilization genes are E. faecalis and some commensal strains of E. coli (32, 38–41). E. faecalis catabolizes EA using enzymes encoded by the eut genes that are contained in a locus consisting of 19 genes. The eut gene expression is controlled by the EutVW two-component system, comprising the sensor histidine kinase EutW and the response regulator EutV (42, 43). Using an E. faecalis eutVW double deletion mutant (43), we are able to establish a novel correlation between E. faecalis EA utilization and an attenuation of the pro-inflammatory host response in the presence of a complex bacterial consortium.
Using immuno-magnetic separation, we are able to purify wild type E. faecalis cells from the colon content of SIHUMI-colonized wild type and IL-10−/− mice and analyze the gene expression in the presence of other bacteria. We show that E. faecalis adapts transcriptionally to the other co-colonizing bacteria. To unravel whether these transcriptional alterations have functional consequences, we colonized wild type and IL-10−/− mice with the SIHUMI consortium in the presence and absence of E. faecalis. We determine a protective activity of E. faecalis in SIHUMI colonized susceptible mice, demonstrating that complex bacterial consortia interactions can reprogram the colitogenic activity of the opportunistic pathogen E. faecalis toward a protective function.
Materials and Methods
Bacterial Strains and Cultivation
For this study we used the characterized human oral E. faecalis strain OG1RF and an isogenic eutVW double deletion mutant kindly provided by Danielle Garsin (Department of Microbiology and Molecular Genetics, The University of Texas Health Science Center at Houston, Houston, Texas, USA) (43). E. faecalis strains (Table 1) were cultivated in Brain Heart Infusion (BHI) broth or on BHI agar (BD Difco) at 37°C under aerobic conditions. The SIHUMI (29) consortium (Table 1) was kindly provided by R. Balfour Sartor (Division of Gastroenterology and Hepatology, University of North Carolina, Chapel Hill, NC, USA). All SIHUMI strains were cultivated under anaerobic conditions on Wilkins-Chalgren-Agar (WCA, Thermo Fisher Scientific) or in WCA broth supplemented with 0.05% (w/v) L-cystein (Carl Roth) and 0.002% (w/v) dithiothreitol (Sigma Aldrich). Of note, a different strain set for rats is also called SIHUMI and should not be mistaken (46). For colonization of germ-free mice with the SIHUMI consortium, the bacterial mixture was prepared as follows. Strains were grown individually in Hungate tubes for 24 h. A mixture of all strains with an equal cell density of 1 × 109 cells/ml was prepared in sterile glass tubes, mixed 1:1 with sterile glycerol in culture medium (40% v/v, gassed with N2) and sealed with a rubber septum. The bacterial mixture was stored at −80°C until use.
Table 1. Bacterial strains used in this study.
Animal Experiments—Animal Housing
Colonization of germ-free 129S6/SvEv wild type and IL-10−/− mice was performed in the National Gnotobiotic Rodent Research Center, University of North Carolina, USA and in the Core Facility Gnotobiology of the ZIEL—Institute for Food & Health, Technical University Munich, Germany. Mice were fed a standard chow diet (V1124-300, ssniff Spezialdiäten GmbH) ad libitum. The mice were kept in flexible film isolators ventilated via HEPA-filtered air at 22 ± 1°C with a 12-h light/dark cycle. Littermates were used when possible, or siblings from multiple breeders were combined and randomly assigned before assignment to experimental groups. The group size is a maximum of 5 mice per cage (floor area ~540 cm2). For enrichment the animals were provided with bedding, nesting material and an autoclavable cottage.
Animal Experiments—Experimental Design
For monoassociation, germ-free 129S6/SvEv wild type and IL-10−/− mice were colonized at the age of 10–12 weeks with either E. faecalis wild type OG1RF or the ΔeutVW mutant strain by oral and rectal swab. The mice were sacrificed after different periods of colonization (1, 4, 8, 12, 16, 20, and 24 weeks).
For colonization with the SIHUMI consortium, the bacterial mixture was prepared as described above. Germ-free 129S6/SvEv wild type and IL-10−/− mice were colonized at the age 10 weeks with 1 × 108 bacteria/strain by repetitive oral gavage (at week 0 and week 1). The composition of the bacterial consortia differed between the experimental groups as shown in Table 2. The mice were sacrificed after colonization periods of 4 or 16 weeks. A schematic overview of all experimental setups is included in the Supplementary Material.

Table 2. Composition of the bacterial consortium for the respective experimental groups.
Preparation of Bacterial Lysates
Bacterial lysates for stimulation of mesenteric lymph node (MLN) cell cultures were prepared as described previously (29). Details are provided in the Supplementary Material.
Bacterial DNA Extraction
Bacterial cells were lysed by mechanical lysis using a FastPrep-24 (MP Biomedicals). DNA was isolated using phenol/chloroform/iso-amyl alcohol (Carl Roth) and contaminating RNA was digested with 0.1 μg/μl RNase A (VWR) for 30 min at 37°C. Subsequently, the DNA was purified using the gDNA clean-up kit (Macherey-Nagel) according to the manufacturer's instructions.
Bacterial Community Analysis via Quantitative Real-Time Polymerase Chain Reaction (qPCR) Assay
For bacterial community analysis, oligonucleotide primers were used to target species-specific regions of the 16S ribosomal RNA (rRNA) gene. Quantification was performed using the LightCycler 480 Universal Probe Library System (UPL, Roche). Primers (Sigma Aldrich) and corresponding probes (Roche) are listed in Table 3. Positive and negative controls were included in every run. Standard curves were generated for each bacterial strain and used to enumerate the 16S rRNA gene copy numbers per species in intestinal content. 16S rRNA gene copy number per ng DNA was calculated via the molecular weight of the genome and 16S rRNA gene copy numbers/genome. The abundance of each bacterium was calculated as previously described (29) using the following formula: [(copies of 16S rRNA gene for a specific SIHUMI bacterium/cumulative copies of 16S rRNA gene of all bacteria) × 100].

Table 3. Species specific 16S rRNA gene-targeted primers and UPL probes used for bacterial community analysis.
Isolation of E. faecalis Cells From Intestinal Content by Immuno-Magnetic Separation
The isolation method was modified according to protocols of Nolle (48). Frozen colonic content from SIHUMI-colonized mice was disrupted by bead beating in 1 ml RNAlater (Sigma Aldrich) using glass beads (1 mm, Carl Roth). Subsequently, samples were diluted with wash buffer (PBS, 0.5% biotin-free BSA, 10% RNAlater) and filtered through three cell strainers with decreasing pore size (VWR; 100, 70, and 30 μm, respectively). The samples were kept on ice throughout and only pre-chilled buffer solutions were used. The bacterial cells were pelleted by centrifugation and resuspended in 1 ml wash buffer. The samples were mixed with 10 μl Enterococcus-specific antibody (biotinylated, Thermo Fisher Scientific PA1-73123) and incubated for 10 min at 4°C under gentle agitation. The samples were centrifuged for 2 min at 9600 × g. After a washing step, the cells were resuspended in 100 μl wash buffer. Ten μl Streptavidin MicroBeads (Miltenyi Biotec) were added per sample. After incubation for 15 min at 4°C under gentle agitation, the cells were washed with wash buffer and resuspended in 500 μl separation buffer (PBS, 0.5% biotin-free BSA, 2 mM EDTA, 10% RNAlater). Magnetically labeled E. faecalis cells were isolated using MACS filter columns (LS, Miltenyi Biotec) in a MidiMACS Separator (Miltenyi Biotec) according to the manufacturer's instructions and total bacterial RNA was isolated.
Bacterial RNA Isolation
Bacterial total RNA was isolated as described previously (49). Details are provided in the Supplementary Material.
Microbial Total RNA Sequencing (RNA Seq)
RNA Seq of E. faecalis Isolated From Monoassociated Mice
Bacterial RNA prepared from the colonic content of E. faecalis OG1RF monoassociated wild type and IL-10−/− mice (n = 8 mice/group; colonization period: 16 weeks) was sequenced as described previously (28). RNA isolation and rRNA depletion was done by Jonathan Hansen and Sandrine Tchaptchet from the Division of Gastroenterology and Hepatology of the University of North Carolina in Chapel Hill. Library preparation and sequencing was performed at the Washington University St. Louis Genome Technology Access Center generating in total 190 million unidirectional 50-bp reads using an Illumina HiSeq2000 instrument (Illumina).
RNA Seq of E. faecalis Isolated From SIHUMI Colonized Mice
E. faecalis cells in the colonic content of SIHUMI colonized wild type (n = 6 mice/group; colonization period: 16 weeks) and IL-10−/− mice (n = 13 mice/group; colonization period: 16 weeks) were isolated by immuno-magnetic separation. Total bacterial RNA was isolated as described above and purified using the RNeasy MinElute Cleanup kit (Qiagen). Contaminating genomic DNA was eliminated using TURBO DNase (Thermo Fisher Scientific) and RNA integrity was validated by agarose gel electrophoresis. Ribosomal RNA was depleted by using the RiboMinus™ Transcriptome Isolation Kit (Bacteria, Thermo Fisher Scientific) according to the manufacturer's instructions. Subsequently, the RNA was fragmented for 180 s using a Covaris E220 sonicator set at 10% duty cycle, intensity of 5 and 200 cycles/burst in 120 μl 1 mM Tris-EDTA pH 8. The fragmented RNA was precipitated together with 1 μl glycogen (Thermo Fisher Scientific), 1:10 sample volume 3 M sodium acetate (Ambion) and 2.5 sample volume of 100% EtOH. Next, the RNA was dephosphorylated using 10 U Antarctic phosphatase per 300 ng RNA at 37°C for 30 min (New England Biolabs). The RNA fragments were recovered using the miRNeasy Mini kit (Qiagen) followed by 5'-phosphorylation using 20 U T4 polynucleotide kinase at 37°C for 60 min (Thermo Fisher Scientific). Again, the miRNeasy Mini kit was used for recovery. RNA fragment size was examined using the BioAnalyzer on an RNA Pico Chip (Agilent Technologies). The cDNA library was prepared as described in the TruSeq Small RNA Library Prep Reference Guide (Illumina) with the following exceptions: After adapter ligation and reverse transcription, fragments were size-selected between 145 and 300 bp corresponding to an insert length between 18 and 173 bp. The sequencing was conducted at the Core Facility Microbiome, generating in total 300 million unidirectional 50 bp reads using an Illumina HiSeq2000 instrument.
Bioinformatics
Data analysis was performed using the Galaxy web platform (50) (server: usegalaxy.eu). Quality of the raw sequence data was assessed with the FastQC tool (version 0.71). After adapter clipping with the Clip adapter sequences tool (version 1.0.3), Illumina FASTQ files were mapped to the reference genome of E. faecalis OG1RF (GenBank: NC_017316.1) using Bowtie2 (version 2.3.4.2). Using htseq-count, the aligned reads overlapping with genomic features were counted. Differentially expressed genes between bacteria isolated from inflamed IL-10−/− vs. healthy wild type mice were determined using DEseq2 (version 2.11.40.2). KEGG gene set enrichment analysis was executed using the Bioconductor clusterProfiler package (version 3.8.1) implemented in R (51).
Histopathological Analysis
The intestinal tract of gnotobiotic wild type and IL-10−/− mice was removed immediately after sacrificing the animals and cleaned from stool. Cross-sections or “swiss-roles” (52) of parts from the intestinal tract were fixed in 4% formalin, dehydrated, and embedded in paraffin. For histopathological analysis, tissue samples were cut into 5 μm sections, deparaffinized and stained with hematoxylin and eosin. The histopathology was evaluated by an independent examiner in a blinded manner as described previously (53), resulting in scores from 0 (not inflamed) to 12 (highly inflamed).
RNA Isolation (Mouse Tissue)
Tissue sections of distal colon were stored in RNAlater (Sigma Aldrich) at −80°C. Tissue samples were transferred into 500 μl RA1 buffer (Macherey Nagel) supplemented with 10 mM DTT (Sigma Aldrich) and the tissue was homogenized using a Miccra D-1 homogenizer. RNA was isolated using the NucleoSpin RNA Kit (Macherey Nagel) according to the manufacturer's instructions.
Reverse Transcription and Gene Expression Analysis via qPCR (RNA From Mouse Tissue)
Complementary DNA (cDNA) was synthesized from 1 μg RNA using the Moloney murine leukemia virus (MMLV) Point Mutant Synthesis System (Promega) and random hexamers. Quantification was performed using the LightCycler 480 Universal Probe Library System (UPL, Roche). Primers (Sigma Aldrich) and corresponding UPL probes are listed in Table 4. Relative quantification of gene expression was calculated by means of 2−ΔΔCt values: . Relative gene expression values were normalized to GAPDH and HPRT expression.

Table 4. Primers and corresponding UPL probes used for mouse gene expression analysis.
MLN Cell Cultures
MLNs were aseptically harvested from gnotobiotic wild type and IL-10−/− mice associated with the SIHUMI consortium. Subsequently, tissue was homogenized and filtrated through 70 μm cell strainers (VWR) resulting in unfractionated single cell suspensions. MLN cells, isolated from individual mice (for IL-10−/− mice) or a pool of mice (wild type mice), were cultured (5 × 105 cells per well) in RPMI-1640 medium (Sigma Aldrich) containing 10% fetal calf serum (Biochrom) and 1% antibiotics/antimycotics (Sigma Aldrich) and stimulated with 15 μg/ml of bacterial lysate from the respective colonizing species. After incubation for 72 h, the supernatants were collected for cytokine analysis and stored at −80°C.
Cytokine Quantification
IL-12p40 and IFNγ concentration in cell culture supernatant was quantified using commercially available ELISA kits (eBioscience).
Ethanolamine Quantitation in Colonic Content by LC-MS/MS Analysis
Quantitation of ethanolamine in colonic content from wild type and IL-10−/− mice monoassociated with E. faecalis OG1RF or ΔeutVW was performed at the Bavarian Center for Biomolecular Mass Spectrometry (BayBioMS, Freising, Germany). Sample preparation and mass spectrometry quantitation were modified after (54). Details are provided in the Supplementary Material.
Statistical Analysis
Statistical analyses were performed using Prism version 6.05 (GraphPad). Unless otherwise stated, data are presented as mean ± standard deviation. Shapiro-Wilk or D'Agostino-Pearson omnibus test was used to assess the normality of distribution of investigated parameters. Statistical significant differences between two groups were determined using two-tailed unpaired parametric t-test. Statistical significant differences between three or more unpaired groups were determined using one-way ANOVA followed by pairwise testing (Tukey). Effects of genotype and colonization groups were compared using two-way ANOVA followed by pairwise testing (Tukey). Differences were considered significant for *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Analysis of RNA seq data was performed using Galaxy platform web platform (50) or R as described in detail above.
Results
E. faecalis Adapts to a Chronic Inflammatory Environment by Altering Its Gene Expression Profile
To analyse the kinetics of E. faecalis-driven inflammation, we monoassociated germ-free wild type and IL-10−/− mice at the age of 10 weeks with E. faecalis wild type OG1RF for different periods of time. After 4, 8, and 12 weeks of colonization, IL-10−/− mice showed mild signs of inflammation in the distal colon as indicated by the histological score. Between the 12th and the 20th week of colonization, IL-10−/− mice started to develop severe distal colonic inflammation reaching a plateau after week 20. A similar disease pattern was also observed in mid colon with an overall lower level of inflammation (Supplementary Figure 1A). In proximal colon, the inflammation persisted at a very low level throughout the analyzed colonization periods (Supplementary Figure 1B). All wild type mice remained disease-free (Figure 1A). The colonization density as detected by countable colony forming units (CFU) in luminal colon contents was not affected by the level of inflammation (Figure 1B). Based on the kinetic of distal colonic inflammation and on previous publications, 16 weeks of colonization were used in subsequent experiments. At this time, inflammation was still in the ascending phase and therefore advantageous for the analysis of factors modulating disease severity.
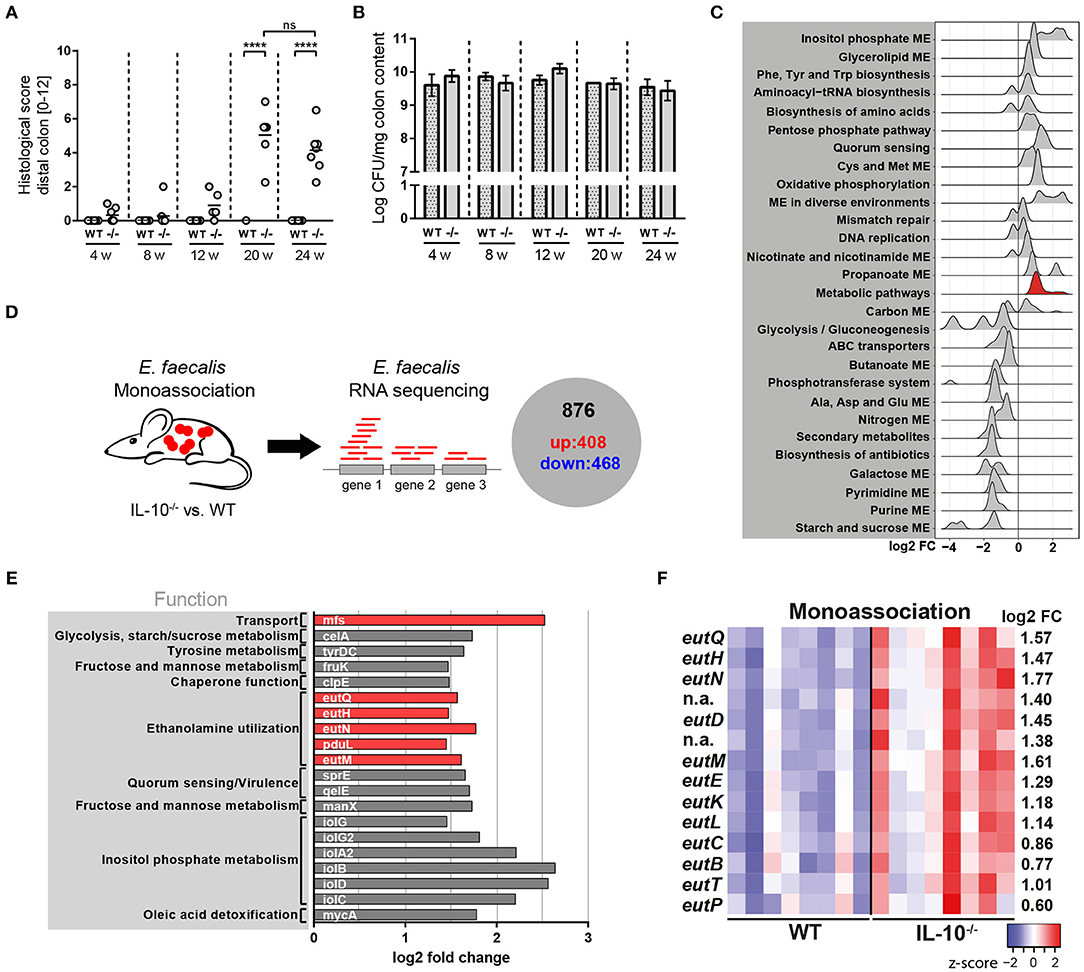
Figure 1. E. faecalis adapts to a chronic inflammatory environment by altering its gene expression profile. (A,B) Wild type (WT) and IL-10−/− (–/–) mice were monoassociated with E. faecalis for 4, 8, 12, and 24 weeks (w). (A) Histological inflammation score in the distal colon. (B) E. faecalis counts/mg in luminal contents from colon. (C) Experimental setup: Transcriptional profiling of luminal bacteria isolated from the colon of E. faecalis-monoassociated IL-10−/− (inflamed) and healthy wild type mice (n = 8/group) revealed 408 significantly up-regulated genes and 486 down-regulated genes in E. faecalis under inflammatory conditions. (D–F) Expression profiles of E. faecalis in an inflamed (IL-10−/−) vs. a non-inflamed (WT) environment in monoassociated mice. (D) Expression distribution (log2 lower or higher) of genes for GSEA-enriched categories (KEGG). The gene group containing eut-genes is highlighted red. (E) Top 20 up-regulated genes. Only annotated genes with known function are shown. Genes of the eut locus are highlighted in red. Gene functions were assigned according to KEGG database. (F) Differential expression of genes in relation to a chronically inflamed environment is shown for significantly regulated eut genes. The log2 ratio of mean abundance (IL-10−/− vs. WT) of normalized expression levels is shown (up regulation is indicated red, down regulation is indicated blue). Differences were considered significant for ****p ≤ 0.0001.
We hypothesized that E. faecalis quickly adapts to a chronic inflammatory environment by altering its gene expression profile. These adaptation mechanisms are likely responsible for the persistence and virulence of these bacteria in a susceptible host. To assess E. faecalis disease-associated transcriptome, we monoassociated mice with E. faecalis OG1RF for 16 weeks and performed RNA sequencing of bacteria isolated from luminal colon content of healthy wild type and inflamed IL-10−/− mice. Transcriptional profiling identified 408 significantly up-regulated and 468 significantly down-regulated genes under conditions of chronic inflammation (Figure 1C and Supplementary Figure 1C, Supplementary Table 1). The samples clearly clustered according to mouse genotype as detected by principal component analysis (PCA) indicating that inflammation modulates E. faecalis gene expression (Supplementary Figure 1D). E. faecalis response to inflammation was characterized by an up regulation of genes associated with a bacterial stress response toward unfavorable growth conditions. KEGG pathway enrichment analysis showed an enrichment of the biosynthesis of amino acids, quorum sensing, mismatch repair, and oxidative phosphorylation among up-regulated genes (Figure 1D). Induction of these processes has also been observed in previous studies analyzing E. faecalis and E. coli adaptation during periods of stress (55–57). Nucleotide synthesis genes were in general down regulated, as indicated by an enrichment of purine- and pyrimidine metabolism transcripts in wild type mice. Furthermore, a large number of genes involved in metabolism and nutrient acquisition were differentially expressed. Sugar metabolism and transport were repressed with an enrichment of starch and sucrose metabolism, ABC transport, PTS transport and glycolysis among down-regulated genes. On the other hand, genes necessary for the utilization of alternative carbon sources were up regulated with an enrichment of inositol phosphate metabolism and glycerolipid metabolism (Figure 1D). Interestingly, genes of the ethanolamine utilization (eut) locus were also identified among the top 20 up-regulated genes in response to an inflammatory environment (Figure 1E). In total, 14 E. faecalis eut genes were significantly up regulated in inflamed IL-10−/− mice indicating an important function of this metabolic pathway for the survival and virulence of E. faecalis in the inflamed intestine (Figure 1F).
E. faecalis Ethanolamine Utilization Has Protective Functions in Gnotobiotic Il-10−/− Mice Colonized With a Simplified Microbial Consortium
To determine whether the utilization of ethanolamine (EA) is relevant for E. faecalis colonization and colitogenic activity, we monoassociated germ-free wild type and IL-10−/− mice with E. faecalis wild type OG1RF or an ΔeutVW mutant strain (Δeut) for 1 and 16 weeks (Figure 2A). The Δeut strain does not express the eut genes and is not able to utilize EA (43, 58, 59). After 1 week of colonization, wild type and IL-10−/− mice showed no signs of inflammation. After 16 weeks of colonization, IL-10−/− mice developed severe inflammation in the distal colon, independent of the E. faecalis strain used for monoassociation (OG1RF histopathological score: 5.0 ± 0.9, Δeut histopathological score: 4.6 ± 0.5). All wild type mice remained disease-free (Figures 2B,C). E. faecalis OG1RF and Δeut mutant strain showed a similar colonization density as detected by CFU in luminal contents from colon (Figure 2D). The colon inflammation in IL-10−/− mice was accompanied by increased spleen weights (Supplementary Figure 2A) and increased expression of pro-inflammatory markers in the distal colon tissue. The expression levels of TNF, IL-12p40, and IL-17 were similar for both colonization groups, only IFNγ expression levels were increased in E. faecalis wild type OG1RF compared to Δeut mutant monoassociated IL-10−/− mice (Figure 2E). To determine whether E. faecalis catabolizes EA in vivo, we measured EA concentrations in intestinal fluid. Luminal EA concentrations were significantly higher in wild type mice monoassociated with Δeut mutant, indicating catabolism of EA by E. faecalis OG1RF. This trend was also observed in IL-10−/− mice after 4 weeks of colonization, but after 16 weeks of colonization intestinal EA concentrations were similar in both E. faecalis OG1RF and Δeut-mutant monoassociated mice (Figure 2F).
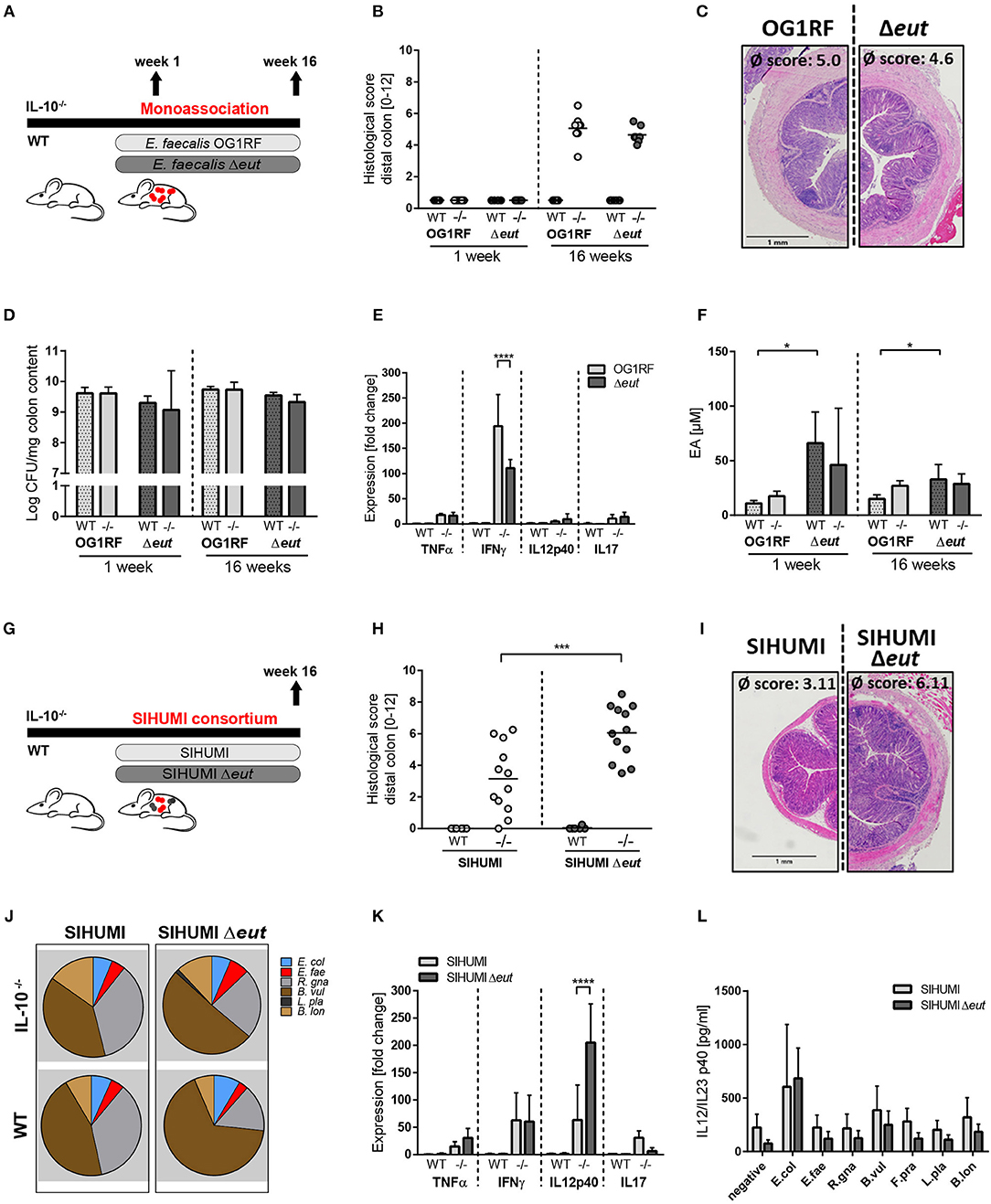
Figure 2. E. faecalis ethanolamine utilization has protective functions in gnotobiotic IL-10 −/− mice colonized with a simplified microbial consortium. (A–F) Germ-free wild type (WT) and IL-10−/− (–/–) mice were monoassociated with E. faecalis wild type (OG1RF) or the Δeut mutant for 1 and 16 weeks, respectively. (A) Experimental setup. (B) Histological inflammation in the distal colon. (C) Representative hematoxylin/eosin-stained sections of distal colon after 16 weeks of colonization. (D) E. faecalis presence in luminal contents from colon as log CFU counts/mg (E) Cytokine expression in colon tissue sections. Values are shown as fold-change normalized to cytokine expression levels in wild type mice colonized with E. faecalis wild type strain. (F) Quantitation of ethanolamine in luminal contents from colon. (G–L) Germ-free wild type (WT) and IL-10−/− (–/–) mice were colonized with SIHUMI consortium including E. faecalis wild type (SIHUMI) or the Δeut mutant (SIHUMI Δeut) for 16 weeks. (G) Experimental setup. (H) Histological inflammation in the distal colon. (I) Representative hematoxylin/eosin-stained sections of distal colon after 16 weeks of colonization. (J) Relative abundances of SIHUMI bacterial species in luminal colon content by 16S rRNA gene targeted qPCR. (K) Cytokine expression in colon tissue sections shown as fold-change normalized to cytokine expression levels in wild type mice colonized with the SIHUMI consortium. (L) IL-12p40 secretion of MLN cells isolated from SIHUMI or SIHUMI Δeut colonized mice that were re-activated with the respective bacterial lysate for 72 h. E. col, Escherichia coli; E. fae, Enterococcus faecalis; R. gna, Ruminococcus gnavus; B. vul, Bacteroides vulgatus; L. pla, Lactobacillus plantarum; B. lon, Bifidobacterium longum. Differences were considered significant for *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
Despite considerable up regulation of eut genes, the deletion of ethanolamine utilization revealed no impact on E. faecalis colitogenic activity in monoassociated mice. To test the hypothesis that E. faecalis EA utilization is only important for bacterial fitness in competition with other microbes, we characterized the colitogenic activity of E. faecalis Δeut mutant in the context of a simplified human microbial consortium (SIHUMI). We colonized germ-free wild type and IL-10−/− mice with SIHUMI consortium either containing E. faecalis wild type OG1RF (SIHUMI) or E. faecalis Δeut (SIHUMI Δeut) mutant strain for a period of 16 weeks (Figure 2G). When we introduced the bacterial consortium to germ-free mice, 6 out of 7 species successfully colonized the mouse intestine, as determined by 16S rRNA gene-based quantitative qPCR (Figure 2J). Faecalibacterium prausnitzii could not be detected by qPCR and droplet digital PCR at any time (Figure 2J, Supplementary Figures 2B,C) in intestinal content (taken at week 4 and 16 of colonization) and feces (taken at week 2, 4, 8, and 16 of colonization). While IL-10−/− mice colonized with the SIHUMI consortium gradually developed inflammation in the distal colon (SIHUMI histopathological score: 3.1 ± 2.3), colitis severity was significantly increased when E. faecalis OG1RF was replaced by Δeut mutant strain (SIHUMI Δeut histopathological score: 6.1 ± 1.8) (Figures 2H, I). These results surprisingly suggest protective functions of E. faecalis EA utilization in the context of chronic intestinal inflammation. Tissue pathology was accompanied by increased spleen weights in IL-10−/− mice colonized with the SIHUMI Δeut consortium (Supplementary Figure 2D). As expected, all colonized wild type mice remained disease-free. The replacement of E. faecalis wild type OG1RF strain by Δeut mutant strain resulted in a shift of the bacterial community with a significant increase of Bacteroides vulgatus relative abundance accompanied by a decrease in Ruminococcus gnavus abundance (Figure 2J, Supplementary Figure 5). Interestingly, the E. faecalis Δeut mutant strain showed an increased colonization density compared to the wild type OG1RF strain, as detected by CFU in luminal contents from colon (Supplementary Figure 2E). However, E. faecalis abundance did not correlate with the histopathology score of the distal colon (Supplementary Figure 2F). The only significant correlation between bacterial abundance and the level of colonic inflammation was detected for R. gnavus with a decreased abundance being associated with higher inflammatory scores (Supplementary Figures 2G, 5). Tissue sections from the distal colon of IL-10−/− mice showed high expression of pro-inflammatory cytokines in comparison to wild type mice. Tissue expression of TNF and IL-12p40 reflected the degree of histopathology with higher expression levels in SIHUMI Δeut colonized compared to SIHUMI colonized IL-10−/− mice. Expression levels of IFNγ were similar between SIHUMI and SIHUMI Δeut colonized IL-10−/− mice, while IL-17 expression was decreased in SIHUMI Δeut colonized IL-10−/− mice (Figure 2K). To address the bacterial antigen-specific immune responses in colonized mice, we isolated MLN cells from IL-10−/− mice colonized with SIHUMI or SIHUMI Δeut consortium and re-stimulated them with respective lysates of each of the colonizing species. Unfractionated MLN cells produced high levels of IL-12p40 when stimulated with E. coli lysate, while IL-12p40 secretion in response to all other lysates was similar to the negative control. The level of MLN activation was similar for all E. faecalis strains (Figure 2L). Since replacement of E. faecalis wild type by the Δeut mutant resulted in a more severe inflammatory phenotype, we investigated whether increased inflammation was associated with an enhanced humoral immune response against specific SIHUMI species. It has been demonstrated by others that certain commensal bacteria induce serum IgA responses that confer immunity against polymicrobial sepsis (60). When we probed lysates of SIHUMI species with sera from SIHUMI or SIHUMI Δeut colonized IL-10−/− mice, we observed the highest number of IgA immunoreactive bands for lysates of B. vulgatus and R. gnavus, the two most abundant species in colonized mice (Supplementary Figure 2H and Figure 2J). Similar results have been obtained for serum IgG antibodies (data not shown). The serum IgA response against SIHUMI species was not influenced by the level of inflammation, as the number and intensity of immunoreactive bands was similar for lysates probed with SIHUMI or SIHUMI Δeut sera (Supplementary Figure 2H).
Complex Bacterial Consortia Reprogram E. faecalis Gene Expression
Since we observed a protective effect of E. faecalis EA utilization only in a complex bacterial community and not under conditions of E. faecalis monoassociation, we aimed to gain a better understanding of E. faecalis gene expression in the presence of other microbes. We purified E. faecalis cells from the colon content of SIHUMI colonized wild type and IL-10−/− mice using immuno-magnetic separation and performed RNA sequencing (Figure 3A). Transcriptional profiling identified 180 significantly up- and 148 significantly down-regulated genes under inflammatory conditions (Supplementary Figure 3A). The samples clustered according to mouse genotype, as shown by PCA analysis (Supplementary Figure 3B). Among the top 20 up-regulated genes, several genes associated with arginine biosynthesis and glycerol metabolism were identified (Figure 3B). Arginine biosynthesis genes have been shown to be up-regulated in lactic acid bacteria in response to acid stress (55) and genes involved in glycerol metabolism have been shown to be induced during E. faecalis-mediated mouse peritonitis as well as intestinal colonization (61, 62). Additionally, an E. faecalis mutant affected in glycerol metabolism showed attenuated virulence in a mouse peritonitis model (62).
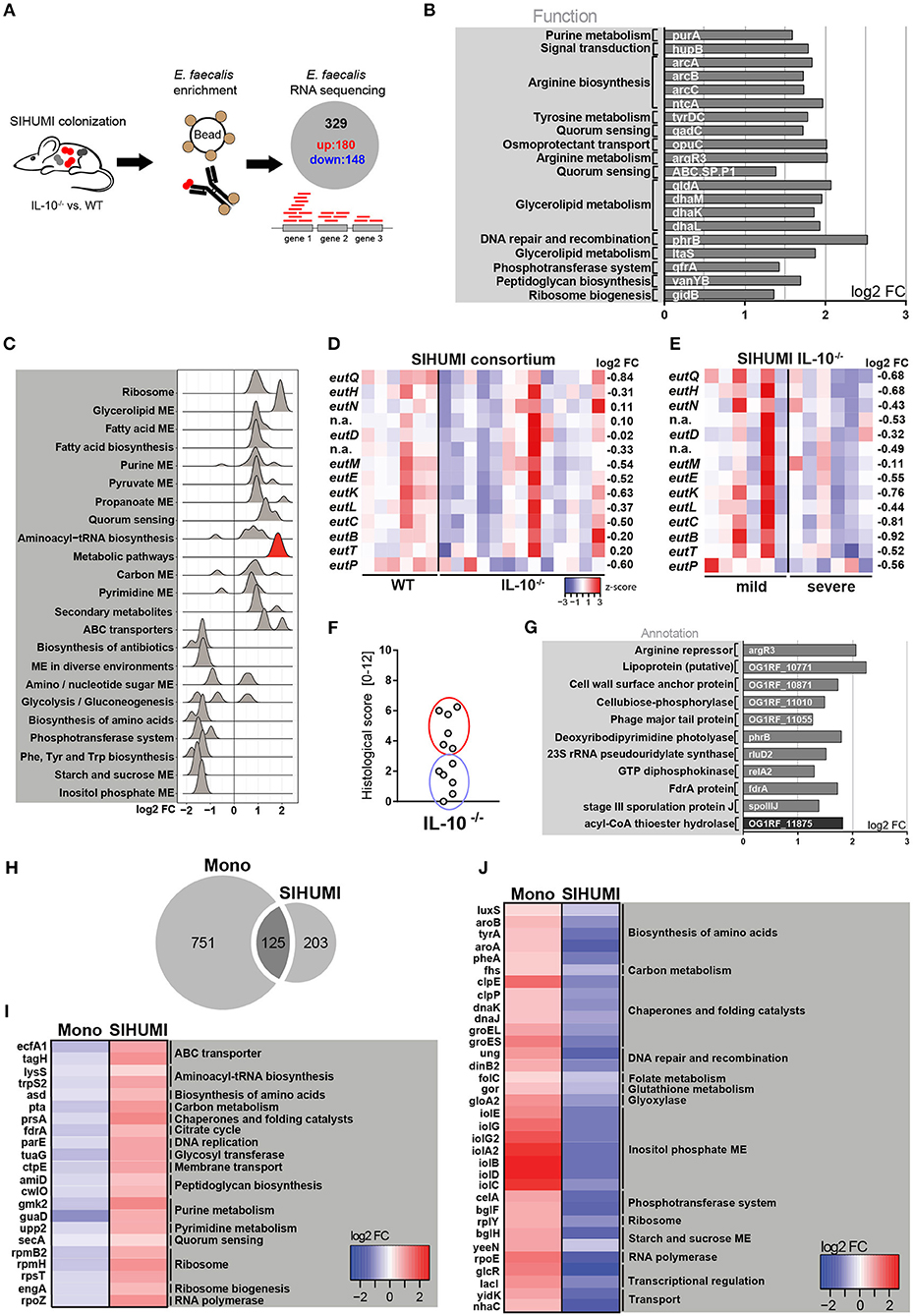
Figure 3. Complex bacterial consortia reprogram E. faecalis gene expression in response to inflammation. Luminal E. faecalis cells were isolated from the colon of inflamed IL-10−/− (n = 13) and healthy wild type (WT) (n = 6) mice, colonized with SIHUMI consortium (16 weeks), using immuno-magnetic separation and subjected to RNA sequencing analysis. (A) Experimental setup: Transcriptional profiling revealed 180 significantly up-regulated and 148 significantly down-regulated genes in E. faecalis isolated from IL-10−/− mice compared to E. faecalis isolated from wild type mice colonized with the SIHUMI consortium. (B) Top 20 up-regulated genes in E. faecalis isolated from SIHUMI colonized mice (IL-10−/− vs. WT). Only annotated genes with known function are shown. Gene functions were assigned according to KEGG database. (C) Expression distribution of enriched genes for GSEA enriched categories (KEGG) in E. faecalis isolated from SIHUMI colonized mice (IL-10−/− vs. WT). The gene group highlighted red contains the eut-genes. (D) Differential expression of eut genes in E. faecalis of an inflamed vs. non-inflamed environment. The log2 ratios of mean abundance (IL-10−/− vs. WT) of normalized expression levels are shown (up regulation is indicated red, down regulation is indicated blue). (E) Differential expression of eut genes in E. faecalis of a severely inflamed vs. mildly inflamed environment. The log2 ratios of mean abundances of normalized expression levels are shown (up regulation is indicated red, down regulation is indicated blue). (F) Histological inflammation scores in the distal colon of SIHUMI colonized IL-10−/− mice comparing severely inflamed mice (red) and mildly inflamed mice (blue). (G) Significantly differentially regulated genes of E. faecalis isolated either from severely inflamed or mildly inflamed SIHUMI colonized IL-10−/− mice (up regulation is indicated gray, down regulation is indicated black). (H–J) Differentially regulated genes in response to inflammation of E. faecalis isolated from monoassociated (IL-10−/− vs. WT) compared to SIHUMI-colonized mice (IL-10−/− vs. WT). (H) Venn diagram showing the differentially regulated genes shared. (I) Genes down regulated in monoassociated and up regulated in SIHUMI-associated mice, respectively. The log2 ratios of mean abundances of normalized expression levels are shown (up regulation is indicated red, down regulation is indicated blue). Gene functions were assigned according to the KEGG database; only annotated genes with known function are shown. (J) As before, but genes up regulated in monoassociated and down regulated in SIHUMI associated mice, respectively.
Surprisingly, we observed no overlap with the top up-regulated genes in E. faecalis isolated from monoassociated mice (Figures 3B, 1E). Additionally, the functional categories enriched among all differentially regulated genes were mainly different between E. faecalis monoassociation or in the context of the SIHUMI community (Figures 3C, 1D). In the SIHUMI consortium, E. faecalis response to inflammation was characterized by an induction of cellular pathways required for growth and replication, including ribosome, aminoacyl-tRNA biosynthesis, nucleotide synthesis, pyruvate and propanoate metabolism, ABC transport and fatty acid biosynthesis and metabolism. In contrast to E. faecalis monoassociation, inositol phosphate metabolism and the biosynthesis of amino acids were repressed. Quorum sensing and glycerolipid metabolism were induced, whereas phosphotransferase systems and starch and sucrose metabolism were repressed in both mono- and complex association of IL-10−/− mice (Figures 3C, 1D). Interestingly, E. faecalis isolated from SIHUMI colonized mice showed higher expression of eut genes in healthy (Figure 3D) and mildly inflamed (Figure 3E) compared to inflamed animals.
In IL-10−/− mice colonized with the SIHUMI consortium, we observed a gradient of colitis severity. Six out of 12 mice had a colitis score <3 and were defined as mildly inflamed animals, the remaining 6 mice with a colitis score >3 were defined as severely inflamed animals (Figure 3F). When comparing E. faecalis gene expression between severely and mildly inflamed IL-10−/− mice, only 11 genes showed statistical significant regulation (Supplementary Figures 3C,D). In particular, the lipoprotein SpoIIIJ (OG1RF_12576), which has been shown to be important for bile resistance of E. faecalis (63) and a cell wall surface anchor family protein (OG1RF_10871), which is associated with E. faecalis biofilm formation (64), appeared to be interesting candidates for E. faecalis virulence (Figure 3G).
When comparing E. faecalis transcriptional response to inflammation between bacteria isolated from mono- and complex-associated mice on the gene level, the majority of differentially regulated genes were not shared. This suggests that the bacterial environment reprograms E. faecalis gene expression. Only 125 genes were shared between both colonization environments, whereas 751 genes were exclusively regulated in monoassociation and 203 genes were exclusively regulated in the SIHUMI community (Figure 3H). In addition, the majority of shared genes showed opposite patterns of regulation. Several genes associated with fundamental cellular pathways were down regulated in E. faecalis monoassociated mice, whereas these genes were strongly up regulated in E. faecalis of the SIHUMI community (Figure 3I). This includes genes associated with the translation apparatus (lysS, trpS2, rpmB2, rpmH, rpsT, engA), RNA synthesis (rpoZ), DNA replication (parE), citrate cycle (fdrA), peptidoglycan biosynthesis (amiD, cwlO), nucleotide metabolism (gmk2, guaD, upp2), and transport (ecfA1, tagH, ctpE, secA) (Figure 3I). Vice versa, we observed an up regulation of several stress-response genes in E. faecalis of monoassociated mice, including genes that encode for proteases and chaperones (clpE, clpP, dnaK, dnaJ, groEL, groES), a ribosomal stress protein (rplY), a detoxifying enzyme (gloA2) and proteins associated with DNA repair (ung, dinB2) and oxidative stress resistance (gor) (55, 65). In addition, genes associated with the biosynthesis of amino acids (luxS, aroA, aroB, tyrA, pheA), transport (yidK, nhaC), transcriptional regulation (glcR, lacI) and a protein homologous to RNA polymerase subunit RpoE, were up-regulated in E. faecalis of monoassociated mice, but down-regulated in E. faecalis SIHUMI colonized mice (Figure 3J).
Complex Microbial Consortia Interactions Reprogram E. faecalis Colitogenic Activity
We next addressed the question of whether the transcriptional differences of E. faecalis in mono- or complex-associated mice have functional consequences by applying the SIHUMI consortium with or without E. faecalis (SIHUMI-E. fae). Wild type and IL-10−/− mice were colonized at the age of 10 weeks for a period of 4 or 16 weeks (Figure 4A). Surprisingly, the colonization of IL-10−/− mice with SIHUMI in the absence of E. faecalis induced an aggravated inflammatory host response compared to the control. After 16 weeks of colonization, IL-10−/− mice colonized with SIHUMI-E. fae consortium developed severe inflammation in the distal colon (score: 4.8 ± 2.4), while the degree of colitis was significantly reduced in IL-10−/− mice colonized with SIHUMI in the presence of E. faecalis (score: 2.8 ± 2.5) (Figures 4B,C). After 4 weeks of colonization, IL-10−/− mice showed mild pancolitis independent of the colonizing bacteria. All colonized wild type mice remained disease-free (Figures 4B,C). The histological inflammation was accompanied by increased spleen weights (Supplementary Figure 4A).
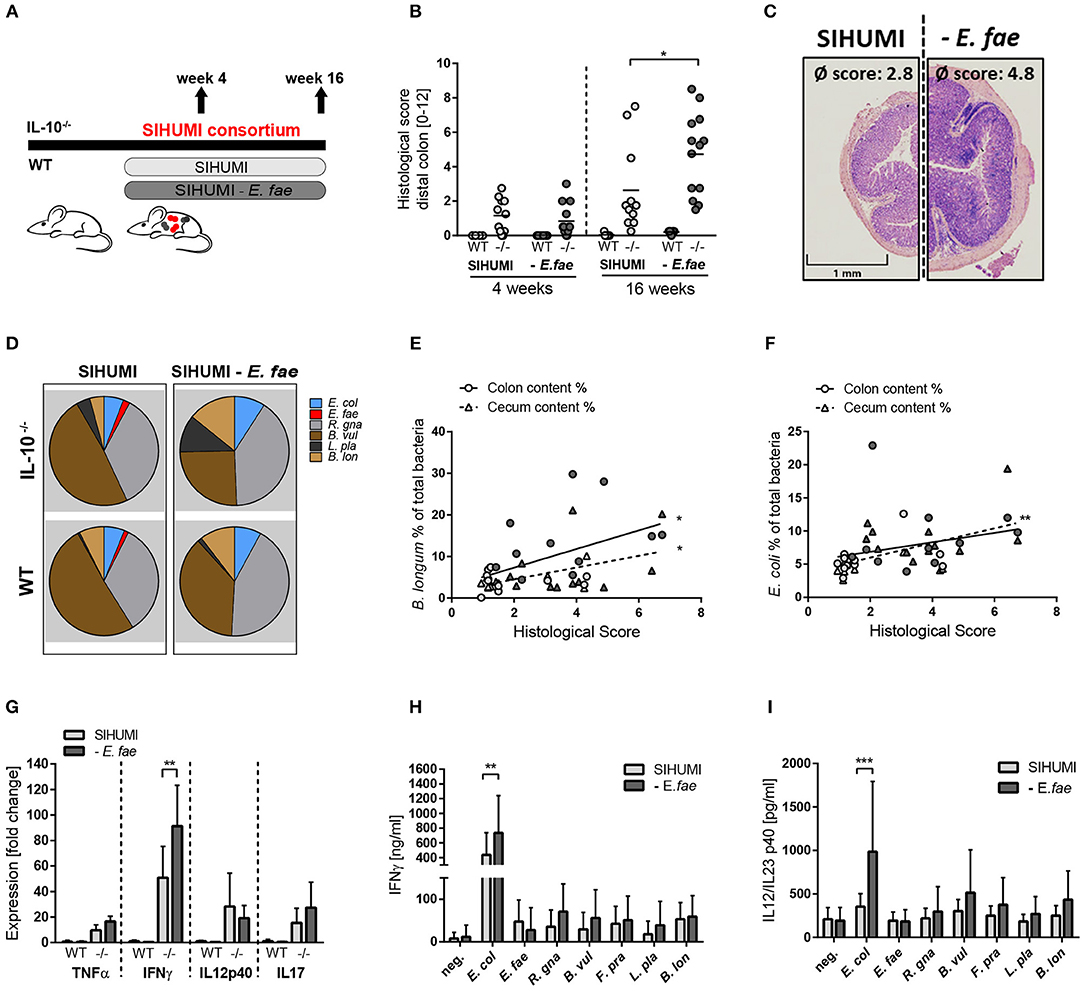
Figure 4. Complex microbial consortia interactions reprogram colitogenic activity of E. faecalis in gnotobiotic IL-10−/− mice. (A) Experimental setup: Germ-free IL-10−/− (–/–) and wild type (WT) mice were colonized with SIHUMI consortium or SIHUMI consortium without E. faecalis (- E. fae) for a period of 4 and 16 weeks. (B) Histological inflammation in the distal colon. (C) Representative hematoxylin/eosin-stained sections of distal colon after 16 weeks of colonization. (D) Relative abundances of SIHUMI bacterial species in luminal colon content by 16S rRNA gene targeted qPCR. (E) Correlation between B. longum abundance (colon and cecum luminal content) and mean histological scores of cecum tip and distal colon. (F) As before, correlation of E. coli abundance. (G) Cytokine expression in colon tissue sections shown as fold change, normalized to cytokine expression levels in wild type mice colonized with the SIHUMI consortium. (H) IFNγ secretion of MLN cells isolated from SIHUMI or SIHUMI without E. faecalis colonized mice (colonization period: 16 weeks) that were re-activated with the respective bacterial lysate for 72 h. (I) As before, IL-12p40 secretion. E. col, Escherichia coli; E. fae, Enterococcus faecalis; R. gna, Ruminococcus gnavus; B. vul, Bacteroides vulgatus; L. pla, Lactobacillus plantarum; B. lon, Bifidobacterium longum. Differences were considered significant for *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
The absence of E. faecalis resulted in changes of the relative abundances of specific SIHUMI species in the colonic content. Compared to SIHUMI colonized mice, the relative concentrations of E. coli and B. longum were slightly higher, while B. vulgatus concentrations were lower in SIHUMI-E. fae colonized mice (Figure 4D, Supplementary Figure 6). In addition, the relative abundance of E. coli and B. longum correlated positively with the development of histopathology. E. faecalis abundance was not correlated with colitis activity (Figures 4E,F, Supplementary Figure 4D). The expression of the pro-inflammatory cytokine IFNγ was significantly increased in IL-10−/− mice colonized with SIHUMI-E. fae, supporting the observation at the level of tissue pathology. TNF and IL-17 expression levels followed the same pattern, whereas IL-12p40 expression was not influenced by the composition of the colonizing consortium (Figure 4G). To address the bacterial antigen-specific immune responses of colonized mice, we isolated MLN cells from IL-10−/− mice colonized with SIHUMI in the presence and absence of E. faecalis and re-stimulated them with respective lysates of each of the colonizing SIHUMI species. After 4 weeks of colonization, the secretion of IFNγ and IL-12p40 in response to E. coli lysate stimulation was slightly increased in MLN cells isolated from mice colonized with SIHUMI. All other lysates failed to activate the MLN cell cultures (Supplementary Figures 4B,C). After 16 weeks of colonization, MLN cells produced maximal levels of IFNγ and IL-12p40 upon stimulation with E. coli lysate, although the relative luminal concentrations of this bacterium were quite low (Figures 4D,H,I). The MLN response toward E. coli lysate was even more pronounced in MLNs isolated from mice colonized with SIHUMI—E. fae consortium (Figures 4H,I), suggesting compensatory mechanisms in the absence of E. faecalis. The level of serum IgA antibodies against SIHUMI species was only slightly influenced by the composition of the bacterial consortium. When we probed lysates of SIHUMI species with sera from SIHUMI or SIHUMI-E. fae colonized IL-10−/− mice, we observed the highest number of immunoreactive bands for lysates of B. vulgatus, R. gnavus, and E. coli. The number and intensity of immunoreactive bands against E. coli and B. vulgatus lysates were slightly increased when probed with SIHUMI-E. fae sera, indicating increased levels of IgA antibodies against these species in mice colonized with SIHUMI in the absence of E. faecalis (Supplementary Figure 4E).
Discussion
In this study, we demonstrate that E. faecalis functionally adapts to chronic inflammation, supporting the hypothesis that host-related mechanisms contribute to the colitogenic activity of opportunistic pathogens. E. faecalis is a disease-relevant pathobiont and colonization of IBD-related mouse models, such as IL-10−/− mice, supports the concept that commensal bacteria harboring pathogenic traits drive chronic inflammation in genetically susceptible but not wild type hosts (66). In addition to previous identified genes modulating the colitogenic effect of E. faecalis (28), we performed transcriptional profiling of E. faecalis under inflammatory conditions and identified 876 genes to be differentially regulated compared to normal conditions. Despite considerable up regulation of 14 genes of the eut locus in inflamed IL-10−/− mice, the deletion of E. faecalis ethanolamine utilization (ΔeutVW) showed no effect on the colonization density and colitogenic activity of E. faecalis in monoassociated mice. Interestingly, E. coli responds to inflammation in monoassociated IL-10−/− mice by the up regulation of stress response genes, but targeted deletion of these genes aggravated the inflammatory phenotype (11, 12). This supports our observation that bacterial adaptation processes correlating with intestinal inflammation are not necessarily harmful for the host. In line with this, we have shown in a previous study that genes that produce two colitis-relevant structures in E. faecalis are not transcriptionally regulated in inflamed monoassociated IL-10−/− mice (28). The measurement of luminal intestinal EA allowed us to demonstrate that E. faecalis utilizes this substrate in-vivo in wild type mice and not-inflamed IL-10−/− mice. However, EA concentrations in inflamed IL-10−/− mice were similar for both E. faecalis wild type OG1RF and Δeut mutant colonization. This is surprising as E. faecalis eut genes were massively up regulated in inflamed IL-10−/− mice relative to wild type controls. This paradox could be explained by limitations of our model as we only analyzed gene expression and not eut protein levels. Additional studies to understand E. faecalis' eut-gene expression and in-vivo EA metabolism are needed.
We provide for the first time evidence for a protective effect of bacterial EA utilization in the context of chronic intestinal inflammation. Using a simplified microbial consortium based on human strains (SIHUMI), we demonstrate that E. faecalis EA utilization attenuates colitis in mice colonized with a complex bacterial community. The replacement of E. faecalis wild type strain by a Δeut mutant resulted in exacerbated colitis in SIHUMI colonized IL-10−/− mice, as indicated by increased histological inflammation, increased tissue expression of TNF and IL-12p40 and increased spleen weights. The colonization density of Δeut mutant was slightly increased compared to wild type E. faecalis. However, the relative abundance of E. faecalis did not correlate with the histological inflammation, suggesting that increased E. faecalis Δeut numbers do not explain the aggravated inflammatory phenotype. MLN cells secreted high levels of IL-12p40 in response to E. coli lysate, but the highest IgA immunoreactivity was observed for B. vulgatus and R. gnavus, the two most abundant species. The bacterial community changed when E. faecalis wild type OG1RF was replaced by Δeut mutant strain, with a significant increase in the relative abundance of B. vulgatus accompanied by a decrease in the abundance of R. gnavus. B. vulgatus induces colitis in HLA/B27-β2m rats, but does not induce inflammation in other colitis models (e.g., IL-10−/− or IL-2−/− mice) (67), which makes it difficult to assess whether this bacterium has colitogenic activity in a complex community in IL-10−/− mice. Taken together, our data suggest that E. faecalis is not the driver of inflammation in the SIHUMI community, but modulates disease by interaction with co-colonizing bacteria. The catabolism of EA by E. faecalis may prevent the utilization of this metabolite by the more colitogenic species E. coli. It has been shown that EA can promote E. coli growth and virulence (35, 54). However, the expression of selected E. coli virulence (fliC, fimH, ompA, ompC) and ethanolamine utilization genes (eutR, eutC, eutQ) was not affected in our model (data not shown) and also the relative abundance of E. coli was not increased in the presence of E. faecalis Δeut mutant.
Our data provide new insights into the role of EA utilization in microbiota-host interactions. EA plays a well-recognized role in host adaption and virulence of enteric pathogens including EHEC, Salmonella and Listeria monocytogenes (35, 36, 54, 68, 69). In contrast, we show that E. faecalis EA utilization has a protective function in IL-10−/− mice in the presence of other resident bacterial species. These discrepancies might arise from differences in bacterial physiology and pathogenicity. The pathogen Salmonella requires alternative electron acceptors released from inflamed host tissue for EA utilization (34, 70), whereas E. faecalis can grow anaerobically on EA in the absence of alternative electron acceptors (71). In addition, others have demonstrated a protective effect of EA utilization by opportunistic pathogens as well. The metabolism of EA resulted in reduced virulence together with a delayed onset of disease in a hamster model of Clostridium difficile infection (72). Furthermore, EA utilization by commensal E. coli isolates allowed them to outcompete the pathogen EHEC (41). Consistent with our data, the colonization efficiency of E. faecalis ΔeutVW in the murine gastrointestinal tract was increased relative to the wild type strain (73). This stands in contrast to enteric pathogens, where the loss of ethanolamine utilization resulted in a competitive disadvantage (34, 36, 54). In Caenorhabditis elegans, an E. faecalis eut mutant was attenuated in virulence, but in this host some E. faecalis strains are regarded as a pathogenic organism resulting in the death of infected nematodes (38). In contrast, probiotic E. faecalis protect C. elegans against EHEC, underlining the importance of strain-level based experiments (74). In any case, the interesting correlation between bacterial pathogenicity and the role of EA utilization for bacterial survival and virulence awaits further investigation.
To the best of our knowledge, this is the first published study to analyze the influence of co-colonization with a bacterial consortium on gene expression and function of an opportunistic pathogen. Most importantly, we demonstrate that complex bacterial consortia interactions reprogram the gene expression profile and the colitogenic activity of E. faecalis toward a protective function. Transcriptional interactions between bacteria have already been illustrated for two members of the dominant gut bacterial phyla. Eubacterium rectale and Bacteroides thetaiotaomicron adapt their profile of utilized substrates in response to the co-colonizing species in gnotobiotic mice (75). In addition, Plichta et al. (76) demonstrated transcriptional species-specific interactions in a complex bacterial community with central metabolisms being strongly affected by coexistence with other microbes. Using immuno-magnetic separation, we were able to isolate E. faecalis cells from SIHUMI colonized mice. This allowed us to study E. faecalis transcriptional response to host inflammation depending on the microbial environment. Interestingly, the majority of regulated genes were not shared between E. faecalis colonizing in monoassociation or in the SIHUMI community. This shows that E. faecalis adapts transcriptionally to the co-colonizing microbes.
In monoassociated IL-10−/− mice, E. faecalis gene expression was characterized by an enrichment of functions important for bacterial stress adaptation. General stress-response genes, amino acid biosynthesis genes and genes involved in the utilization of alternative carbon sources were up regulated. A similar regulation pattern has been observed by others analyzing E. faecalis transcriptome during mammalian infection. The intraperitoneal transcriptome of E. faecalis in gnotobiotic mice is characterized by the up regulation of stress-response genes and a metabolic adaptation to an inflamed environment (62). Frank et al. (77) demonstrated that E. faecalis cells undergo transcriptional adaptation and exist in a stringent response state in a rabbit subdermal abscess model. The stringent response, a bacterial stress adaptation mechanism, is characterized by the repression of fundamental cellular pathways, whereas amino acid biosynthesis genes and stress-survival genes are strongly induced. In addition, metabolic rearrangements including the utilization of alternative carbon sources are a hallmark of the stringent response (65, 78). Since we observe similar pathways regulated in response to inflammation and a link between E. faecalis stringent response and virulence has been demonstrated in a study by Gaca et al. (65), one could speculate that the gene expression of E. faecalis explains the colitogenic character of this bacterium in monoassociated IL-10−/− mice. The detailed investigation of E. faecalis genes and their biological role in the pathophysiology of chronic inflammation awaits further investigation.
In co-colonization with other microbes, however, E. faecalis gene expression in inflamed mice is not primarily characterized by the induction of stress-response pathways, but rather by an enrichment of pathways important for bacterial growth and replication. Although some genes important for bacterial adaptation to environmental stress were up regulated (e.g., arginine deaminase pathway genes, opuC, phrB) (38, 55), the transcriptional profile does not indicate a stringent response state as observed in monoassociated E. faecalis. Fundamental cellular pathways like replication and translation were induced and the expression of general stress-response genes like clpE, clpP, dnaK, dnaJ, groEL, and groES were repressed (62, 79).
Previous studies analyzing E. faecalis' function in chronic inflammation were conducted in monoassociated IL-10−/− mice and revealed a colitogenic behavior (23, 24, 27, 28). By characterizing E. faecalis as part of a microbial consortium, we are adding new knowledge to the complex interdependence of opportunistic pathogens, the genetically predisposed host and the luminal microbial environment. Colonization of IL-10−/− mice with SIHUMI under omission of E. faecalis or in the presence of E. faecalis with defective EA utilization resulted in a more severe phenotype as indicated by the histological inflammation, spleen weights and the pro-inflammatory cytokine expression in colon tissue. This supports the hypothesis that E. faecalis provides protective mechanisms in complex consortia by the utilization of EA. A massive pro-inflammatory response of reactivated MLN cells to E. coli lysate stimulation and a positive correlation between histological score and E. coli abundance point to this bacterium and not E. faecalis as a main driver of SIHUMI mediated colitis. One could speculate that E. faecalis outcompetes E. coli or other SIHUMI strains for EA, but when E. faecalis is missing, or E. faecalis EA utilization is defective, the virulence of E. coli or other EA-responsive strains is enhanced leading to an increased immune response and inflammation.
Our data emphasize the dualistic character of E. faecalis and show that the colitogenic function of a bacterial strain is not only defined its gene repertoire, but also by co-colonizing microbes. Simultaneous colonization of IL-10−/− mice with E. coli and E. faecalis resulted in more aggressive colitis as compared to disease induced by each species individually (80). Furthermore, Helicobacter hepaticus and Lactobacillus reuteri do not induce disease in monoassociated IL-10−/− mice, but when both strains are used in a dual-association setup they induce severe colitis (81). The infection of Rag2-deficient and wild type mice with Klebsiella pneumoniae, Proteus mirabilis, or a combination of both strains triggered inflammation in the presence of a complex specific-pathogen free (SPF) microbiota, but K. pneumoniae and P. mirabilis dual-association of germ-free mice did not result in the development of colitis (82). Similarly, in the severe combined immunodeficiency mouse model, segmented filamentous bacteria were only effective in triggering intestinal inflammation in combination with a complex SPF microbiota (83). Vice versa, E. coli induces colitis in gnotobiotic IL-2-deficient mice, but co-association with B. vulgatus prevents colitis development (84).
In summary, we demonstrate that E. faecalis gene expression and colitogenic activity is influenced by co-colonizing microbes. A protective effect of E. faecalis EA utilization was only apparent in SIHUMI colonized mice, but not under monocolonized conditions. Additionally, we report a shift of phenotype from a colitogenic to a protective activity of E. faecalis in the IL-10−/− mouse model depending on the bacterial environment. This model study with manageable bacterial complexity shows that monoassociation studies do not necessarily allow conclusions to be drawn about bacterial colitogenicity in IBD. This underlines that IBD is a complex disease in which both different host genes and bacterial genes play a role. The gradual development of a human-oriented complexity in carefully selected microbial consortia could be a viable way to unravel the complex microbe-host and microbe-microbe interactions in disease etiology. This may contribute to a basic understanding of the pathogenesis of human IBD and have therapeutic options.
Data Availability
The RNA seq datasets generated for this study can be found in the SRA archive of NCBI under the accession No. PRJNA527872 and PRJNA528098.
Ethics Statement
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of North Carolina, Chapel Hill, NC, USA or the Committee on Animal Health and Care of the local government (Regierung von Oberbayern, AZ 55.2-1-54-2532-109-2015) in Germany.
Author Contributions
IL, IS, RS, and DH conceived and designed the experiments. IL, IS, and JH performed mouse experiments and tissue analyses. KN and JJH supported RNA sequencing work. KK and TH supported LC-MS/MS analysis. JJH, KK, KN, TH, RS, and DH contributed reagents, materials, or analysis tools. IL and DH wrote the manuscript, with edits by RS, IS, JJH, and KN.
Funding
Funding was provided to DH from the Deutsche Forschungsgemeinschaft (DFG) including RTG 1482 and SPP 1656. Funding to RS was provided by NIH grants P40OD010995, P30 DK034987 and the Crohn's and Colitis Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Danielle Garsin for providing the E. faecalis Δeut VW strain. We thank Josef Sperl for the support with droplet digital PCR. We thank Maureen Bower for her support with gnotobiotic mice. We thank Lisa Holt and Nico Gebhard for their help with the samples. We thank Kurt Gedr for his support with biostatistics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2019.01420/full#supplementary-material
References
1. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
2. Turpin W, Goethel A, Bedrani L, Croitoru Mdcm K. Determinants of IBD heritability: genes, bugs, and more. Inflamm Bowel Dis. (2018) 24:1133–48. doi: 10.1093/ibd/izy085
3. Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. (2007) 448:427–34. doi: 10.1038/nature06005
4. Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. (2008) 134:577–94. doi: 10.1053/j.gastro.2007.11.059
5. Sartor RB, Wu GD. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology. (2017) 152:327–39 e324. doi: 10.1053/j.gastro.2016.10.012
6. Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, et al. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe. (2014) 15:382–92. doi: 10.1016/j.chom.2014.02.005
7. Berry D, Kuzyk O, Rauch I, Heider S, Schwab C, Hainzl E, et al. Intestinal microbiota signatures associated with inflammation history in mice experiencing recurring colitis. Front Microbiol. (2015) 6:1408. doi: 10.3389/fmicb.2015.01408
8. Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. (2017) 8:1784. doi: 10.1038/s41467-017-01973-8
9. Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, et al. A microbial signature for Crohn's disease. Gut. (2017) 66:813–22. doi: 10.1136/gutjnl-2016-313235
10. Schirmer M, Denson L, Vlamakis H, Franzosa EA, Thomas S, Gotman NM, et al. Compositional and temporal changes in the gut microbiome of pediatric Ulcerative Colitis patients are linked to disease course. Cell Host Microbe. (2018) 24, 600–10 e604. doi: 10.1016/j.chom.2018.09.009
11. Patwa LG, Fan TJ, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, et al. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology. (2011) 141:1842–51 e1841-10. doi: 10.1053/j.gastro.2011.06.064
12. Tchaptchet S, Fan TJ, Goeser L, Schoenborn A, Gulati AS, Sartor RB, et al. Inflammation-induced acid tolerance genes gadAB in luminal commensal Escherichia coli attenuate experimental colitis. Infect Immun. (2013) 81:3662–71. doi: 10.1128/IAI.00355-13
13. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. (2010) 464:59–65. doi: 10.1038/nature08821
14. Sava IG, Heikens E, Huebner J. Pathogenesis and immunity in enterococcal infections. Clin Microbiol Infect. (2010) 16:533–40. doi: 10.1111/j.1469-0691.2010.03213.x
15. Lebreton F, Willems RJL, Gilmore MS. Enterococcus diversity, origins in nature, and gut colonization. In: Gilmore MS, Clewell DB, Ike Y, Shankar N, editors. Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Boston, MA: Massachusetts Eye and Ear Infirmary (2014). p. 5–63.
16. Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, et al. Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis. (2010) 16:2034–42. doi: 10.1002/ibd.21319
17. Shiga H, Kajiura T, Shinozaki J, Takagi S, Kinouchi Y, Takahashi S, et al. Changes of faecal microbiota in patients with Crohn's disease treated with an elemental diet and total parenteral nutrition. Dig Liver Dis. (2012) 44:736–42. doi: 10.1016/j.dld.2012.04.014
18. Zhou Y, Chen H, He H, Du Y, Hu J, Li Y, et al. Increased Enterococcus faecalis infection is associated with clinically active Crohn disease. Medicine. (2016) 95:e5019. doi: 10.1097/MD.0000000000005019
19. Roche-Lima A, Carrasquillo-Carrion K, Gomez-Moreno R, Cruz JM, Velazquez-Morales DM, Rogozin IB, et al. The presence of genotoxic and/or pro-inflammatory bacterial genes in gut metagenomic databases and their possible link with inflammatory bowel diseases. Front Genet. (2018) 9:116. doi: 10.3389/fgene.2018.00116
20. Golinska E, Tomusiak A, Gosiewski T, Wiecek G, Machul A, Mikolajczyk D, et al. Virulence factors of Enterococcus strains isolated from patients with inflammatory bowel disease. World J Gastroenterol. (2013) 19:3562–72. doi: 10.3748/wjg.v19.i23.3562
21. Fite A, Macfarlane S, Furrie E, Bahrami B, Cummings JH, Steinke DT, et al. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol. (2013) 51:849–56. doi: 10.1128/JCM.02574-12
22. Furrie E, Macfarlane S, Cummings JH, Macfarlane GT. Systemic antibodies towards mucosal bacteria in ulcerative colitis and Crohn's disease differentially activate the innate immune response. Gut. (2004) 53:91–8. doi: 10.1136/gut.53.1.91
23. Balish E, Warner T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol. (2002) 160:2253–7. doi: 10.1016/S0002-9440(10)61172-8
24. Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, et al. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. (2005) 128:891–906. doi: 10.1053/j.gastro.2005.02.009
25. Ruiz PA, Shkoda A, Kim SC, Sartor RB, Haller D. IL-10 gene-deficient mice lack TGF-β/Smad signaling and fail to inhibit proinflammatory gene expression in intestinal epithelial cells after the colonization with colitogenic Enterococcus faecalis. J Immunol. (2005) 174:2990–9. doi: 10.4049/jimmunol.174.5.2990
26. Barnett MP, McNabb WC, Cookson AL, Zhu S, Davy M, Knoch B, et al. Changes in colon gene expression associated with increased colon inflammation in interleukin-10 gene-deficient mice inoculated with Enterococcus species. BMC Immunol. (2010) 11:39. doi: 10.1186/1471-2172-11-39
27. Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, Tonkonogy SL, et al. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology. (2011) 141:959–71. doi: 10.1053/j.gastro.2011.05.035
28. Ocvirk S, Sava IG, Lengfelder I, Lagkouvardos I, Steck N, Roh JH, et al. Surface-associated lipoproteins link Enterococcus faecalis virulence to colitogenic activity in IL-10-deficient mice independent of their expression levels. PLoS Pathog. (2015) 11:e1004911. doi: 10.1371/journal.ppat.1004911
29. Eun CS, Mishima Y, Wohlgemuth S, Liu B, Bower M, Carroll IM, et al. Induction of bacterial antigen-specific colitis by a simplified human microbiota consortium in gnotobiotic interleukin-10−/− mice. Infect Immun. (2014) 82:2239–46. doi: 10.1128/IAI.01513-13
30. Randle CL, Albro PW, Dittmer JC. The phosphoglyceride composition of Gram-negative bacteria and the changes in composition during growth. Biochim Biophys Acta. (1969) 187:214–20.
31. Patel D, Witt SN. Ethanolamine and phosphatidylethanolamine: partners in health and disease. Oxid Med Cell Longev. (2017) 2017:4829180. doi: 10.1155/2017/4829180
32. Kaval KG, Garsin DA. Ethanolamine utilization in bacteria. MBio. (2018) 9:e00066-18. doi: 10.1128/mBio.00066-18
33. Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. (2010) 467:426–9. doi: 10.1038/nature09415
34. Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, et al. Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci USA. (2011) 108:17480–5. doi: 10.1073/pnas.1107857108
35. Kendall MM, Gruber CC, Parker CT, Sperandio V. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic Escherichia coli O157:H7. MBio. (2012) 3:e00050-12. doi: 10.1128/mBio.00050-12
36. Anderson CJ, Clark DE, Adli M, Kendall MM. Ethanolamine signaling promotes Salmonella niche recognition and adaptation during infection. PLoS Pathog. (2015) 11:e1005278. doi: 10.1371/journal.ppat.1005278
37. Anderson CJ, Satkovich J, Koseoglu VK, Agaisse H, Kendall MM. The ethanolamine permease EutH promotes vacuole adaptation of Salmonella enterica and Listeria monocytogenes during macrophage infection. Infect Immun. (2018) 86:e00172-18. doi: 10.1128/IAI.00172-18
38. Maadani A, Fox KA, Mylonakis E, Garsin DA. Enterococcus faecalis mutations affecting virulence in the Caenorhabditis elegans model host. Infect Immun. (2007) 75:2634–7. doi: 10.1128/IAI.01372-06
39. Tsoy O, Ravcheev D, Mushegian A. Comparative genomics of ethanolamine utilization. J Bacteriol. (2009) 191:7157–64. doi: 10.1128/JB.00838-09
40. Winter SE, Winter MG, Xavier MN, Thiennimitr P, Poon V, Keestra AM, et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science. (2013) 339:708–11. doi: 10.1126/science.1232467
41. Rowley CA, Anderson CJ, Kendall MM. Ethanolamine influences human commensal Escherichia coli growth, gene expression, and competition with enterohemorrhagic E. coli O157:H7. MBio. (2018) 9:e01429-18. doi: 10.1128/mBio.01429-18
42. Del MF, Perego M. Ethanolamine activates a sensor histidine kinase regulating its utilization in Enterococcus faecalis. J Bacteriol. (2008) 190:7147–56. doi: 10.1128/JB.00952-08
43. Fox KA, Ramesh A, Stearns JE, Bourgogne A, Reyes-Jara A, Winkler WC, et al. Multiple posttranscriptional regulatory mechanisms partner to control ethanolamine utilization in Enterococcus faecalis. Proc Natl Acad Sci USA. (2009) 106:4435–40. doi: 10.1073/pnas.0812194106
44. Murray BE, Singh KV, Ross RP, Heath JD, Dunny GM, Weinstock GM. Generation of restriction map of Enterococcus faecalis OG1 and investigation of growth requirements and regions encoding biosynthetic function. J Bacteriol. (1993) 175:5216–23.
45. Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology. (2004) 127:412–21. doi: 10.1053/j.gastro.2004.04.061
46. Becker N, Kunath J, Loh G, Blaut M. Human intestinal microbiota: characterization of a simplified and stable gnotobiotic rat model. Gut Microbes. (2011) 2:25–33. doi: 10.4161/gmic.2.1.14651
47. Zhu L, Shen DX, Zhou Q, Liu CJ, Li Z, Fang X, et al. Universal Probe Library based real-time PCR for rapid detection of bacterial pathogens from positive blood culture bottles. World J Microbiol Biotechnol. (2014) 30:967–75. doi: 10.1007/s11274-013-1515-x
48. Nolle N. Salmonella Typhimurium-Infektion: Ernährungsabhängiges Transkriptom und Charakterisierung eines Galaktitol-spezifischen Aufnahmesystems. Dissertation, Technische Universität München (2017).
49. Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. (2013) 341:1241214. doi: 10.1126/science.1241214
50. Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Cech M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. (2018) 46:W537–44. doi: 10.1093/nar/gky379
51. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. (2012) 16:284–7. doi: 10.1089/omi.2011.0118
52. Moolenbeek C, Ruitenberg EJ. The “Swiss roll”: a simple technique for histological studies of the rodent intestine. Lab Anim. (1981) 15:57–9.
53. Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. (2005) 115:695–702. doi: 10.1172/JCI22996
54. Bertin Y, Girardeau JP, Chaucheyras-Durand F, Lyan B, Pujos-Guillot E, Harel J, et al. Enterohaemorrhagic Escherichia coli gains a competitive advantage by using ethanolamine as a nitrogen source in the bovine intestinal content. Environ Microbiol. (2011) 13:365–77. doi: 10.1111/j.1462-2920.2010.02334.x
55. van de Guchte M, Serror P, Chervaux C, Smokvina T, Ehrlich SD, Maguin E. Stress responses in lactic acid bacteria. Antonie Van Leeuwenhoek. (2002) 82, 187–216. doi: 10.1023/A:1020631532202
56. Durfee T, Hansen AM, Zhi H, Blattner FR, Jin DJ. Transcription profiling of the stringent response in Escherichia coli. J Bacteriol. (2008) 190:1084–96. doi: 10.1128/JB.01092-07
57. Ran S, Liu B, Jiang W, Sun Z, Liang J. Transcriptome analysis of Enterococcus faecalis in response to alkaline stress. Front Microbiol. (2015) 6:795. doi: 10.3389/fmicb.2015.00795
58. Debroy S, van der Hoeven R, Singh KV, Gao P, Harvey BR, Murray BE, et al. Development of a genomic site for gene integration and expression in Enterococcus faecalis. J Microbiol Methods. (2012) 90:1–8. doi: 10.1016/j.mimet.2012.04.011
59. Ramesh A, DebRoy S, Goodson JR, Fox KA, Faz H, Garsin DA, et al. The mechanism for RNA recognition by ANTAR regulators of gene expression. PLoS Genet. (2012) 8:e1002666. doi: 10.1371/journal.pgen.1002666
60. Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, et al. Commensal microbes induce serum IgA responses that protect against polymicrobial sepsis. Cell Host Microbe. (2018) 23, 302–11 e303. doi: 10.1016/j.chom.2018.01.005
61. Lindenstrauss AG, Ehrmann MA, Behr J, Landstorfer R, Haller D, Sartor RB, et al. Transcriptome analysis of Enterococcus faecalis toward its adaption to surviving in the mouse intestinal tract. Arch Microbiol. (2014) 196:423–33. doi: 10.1007/s00203-014-0982-2
62. Muller C, Cacaci M, Sauvageot N, Sanguinetti M, Rattei T, Eder T, et al. The intraperitoneal transcriptome of the opportunistic pathogen Enterococcus faecalis in mice. PLoS ONE. (2015) 10:e0126143. doi: 10.1371/journal.pone.0126143
63. Dale JL, Beckman KB, Willett JLE, Nilson JL, Palani NP, Baller JA, et al. Comprehensive functional analysis of the Enterococcus faecalis core genome using an ordered, sequence-defined collection of insertional mutations in strain OG1RF. mSystems. (2018) 3:e00062-18. doi: 10.1128/mSystems.00062-18
64. Nallapareddy SR, Singh KV, Sillanpaa J, Garsin DA, Hook M, Erlandsen SL, et al. Endocarditis and biofilm-associated pili of Enterococcus faecalis. J Clin Invest. (2006) 116:2799–807. doi: 10.1172/JCI29021
65. Gaca AO, Abranches J, Kajfasz JK, Lemos JA. Global transcriptional analysis of the stringent response in Enterococcus faecalis. Microbiology. (2012) 158(Pt 8):1994–2004. doi: 10.1099/mic.0.060236-0
66. Butto LF, Schaubeck M, Haller D. Mechanisms of microbe-host interaction in Crohn's Disease: dysbiosis vs. pathobiont selection. Front Immunol. (2015) 6:555. doi: 10.3389/fimmu.2015.00555
67. Hörmannsperger G, Schaubeck M, Haller D. Intestinal microbiota in animal models of inflammatory diseases. ILAR J. (2015) 56:179–91. doi: 10.1093/ilar/ilv019
68. Joseph B, Przybilla K, Stuhler C, Schauer K, Slaghuis J, Fuchs TM, et al. Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J Bacteriol. (2006) 188:556–68. doi: 10.1128/JB.188.2.556-568.2006
69. Luzader DH, Clark DE, Gonyar LA, Kendall MM. EutR is a direct regulator of genes that contribute to metabolism and virulence in enterohemorrhagic Escherichia coli O157:H7. J Bacteriol. (2013) 195:4947–53. doi: 10.1128/JB.00937-13
70. Price-Carter M, Tingey J, Bobik TA, Roth JR. The alternative electron acceptor tetrathionate supports B12-dependent anaerobic growth of Salmonella enterica serovar Typhimurium on ethanolamine or 1,2-propanediol. J Bacteriol. (2001) 183:2463–75. doi: 10.1128/JB.183.8.2463-2475.2001
71. Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol. (2010) 8:290–5. doi: 10.1038/nrmicro2334
72. Nawrocki KL, Wetzel D, Jones JB, Woods EC, McBride SM. Ethanolamine is a valuable nutrient source that impacts Clostridium difficile pathogenesis. Environ Microbiol. (2018) 20:1419–35. doi: 10.1111/1462-2920.14048
73. Kaval KG, Singh KV, Cruz MR, DebRoy S, Winkler WC, Murray BE, et al. Loss of ethanolamine utilization in Enterococcus faecalis increases gastrointestinal tract colonization. MBio. (2018) 9:e00790-18. doi: 10.1128/mBio.00790-18
74. Neuhaus K, Lamparter MC, Zolch B, Landstorfer R, Simon S, Spanier B, et al. Probiotic Enterococcus faecalis Symbioflor® down regulates virulence genes of EHEC in vitro and decrease pathogenicity in a Caenorhabditis elegans model. Arch Microbiol. (2017) 199:203–13. doi: 10.1007/s00203-016-1291-8
75. Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci USA. (2009) 106:5859–64. doi: 10.1073/pnas.0901529106
76. Plichta DR, Juncker AS, Bertalan M, Rettedal E, Gautier L, Varela E, et al. Transcriptional interactions suggest niche segregation among microorganisms in the human gut. Nat Microbiol. (2016) 1:16152. doi: 10.1038/nmicrobiol.2016.152
77. Frank KL, Colomer-Winter C, Grindle SM, Lemos JA, Schlievert PM, Dunny GM. Transcriptome analysis of Enterococcus faecalis during mammalian infection shows cells undergo adaptation and exist in a stringent response state. PLoS ONE. (2014) 9:e115839. doi: 10.1371/journal.pone.0115839
78. Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, et al. The global, ppGpp-mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol. (2008) 68:1128–48. doi: 10.1111/j.1365-2958.2008.06229.x
79. Giard JC, Laplace JM, Rince A, Pichereau V, Benachour A, Leboeuf C, et al. The stress proteome of Enterococcus faecalis. Electrophoresis. (2001) 22:2947–54. doi: 10.1002/1522-2683(200108)22:14<2947::AID-ELPS2947>3.0.CO;2-K
80. Kim SC, Tonkonogy SL, Karrasch T, Jobin C, Sartor RB. Dual-association of gnotobiotic IL-10-/- mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. (2007) 13:1457–66. doi: 10.1002/ibd.20246
81. Whary MT, Taylor NS, Feng Y, Ge Z, Muthupalani S, Versalovic J, et al. Lactobacillus reuteri promotes Helicobacter hepaticus-associated typhlocolitis in gnotobiotic B6.129P2-IL-10tm1Cgn (IL-10−/−) mice. Immunology. (2011) 133:165–78. doi: 10.1111/j.1365-2567.2011.03423.x
82. Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. (2010) 8:292–300. doi: 10.1016/j.chom.2010.08.004
83. Stepankova R, Powrie F, Kofronova O, Kozakova H, Hudcovic T, Hrncir T, et al. Segmented filamentous bacteria in a defined bacterial cocktail induce intestinal inflammation in SCID mice reconstituted with CD45RBhigh CD4+ T cells. Inflamm Bowel Dis. (2007) 13:1202–11. doi: 10.1002/ibd.20221
Keywords: Enterococcus faecalis, ethanolamine utilization, gnotobiotic mouse models, microbial consortium, SIHUMI, IBD, IL-10 deficient mouse, RNA sequencing
Citation: Lengfelder I, Sava IG, Hansen JJ, Kleigrewe K, Herzog J, Neuhaus K, Hofmann T, Sartor RB and Haller D (2019) Complex Bacterial Consortia Reprogram the Colitogenic Activity of Enterococcus faecalis in a Gnotobiotic Mouse Model of Chronic, Immune-Mediated Colitis. Front. Immunol. 10:1420. doi: 10.3389/fimmu.2019.01420
Received: 26 March 2019; Accepted: 05 June 2019;
Published: 20 June 2019.
Edited by:
Helena Tlaskalova-Hogenova, Institute of Microbiology (ASCR), CzechiaReviewed by:
Axel Kornerup Hansen, University of Copenhagen, DenmarkElena Verdu, McMaster University, Canada
Copyright © 2019 Lengfelder, Sava, Hansen, Kleigrewe, Herzog, Neuhaus, Hofmann, Sartor and Haller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dirk Haller, ZGlyay5oYWxsZXJAdHVtLmRl