
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 12 June 2019
Sec. NK and Innate Lymphoid Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01208
This article is part of the Research Topic Fc-Mediated Antibody Functions and Fc-Receptor Polymorphism View all 19 articles
 Pascale Paul1,2*
Pascale Paul1,2* Pascal Pedini3
Pascal Pedini3 Luc Lyonnet1
Luc Lyonnet1 Julie Di Cristofaro4
Julie Di Cristofaro4 Anderson Loundou5Mathieu Pelardy3Agnes Basire3Françoise Dignat-George1,2Jacques Chiaroni3,4Pascal Thomas6Martine Reynaud-Gaubert7
Anderson Loundou5Mathieu Pelardy3Agnes Basire3Françoise Dignat-George1,2Jacques Chiaroni3,4Pascal Thomas6Martine Reynaud-Gaubert7 Christophe Picard3,4
Christophe Picard3,4Fc gamma receptors (FcγRs) play a major role in the regulation of humoral immune responses. Single-nucleotide polymorphisms (SNPs) of FCGR2A and FCGR3A can impact the expression level, IgG affinity and function of the CD32 and CD16 FcγRs in response to their engagement by the Fc fragment of IgG. The CD16 isoform encoded by FCGR3A [158V/V] controls the intensity of antibody-dependent cytotoxic alloimmune responses of natural killer cells (NK) and has been identified as a susceptibility marker predisposing patients to cardiac allograft vasculopathy after heart transplant. This study aimed to investigate whether FCGR2A and FCGR3A polymorphisms can also be associated with the clinical outcome of lung transplant recipients (LTRs). The SNPs of FCGR2A ([131R/H], rs1801274) and FCGR3A ([158V/F], rs396991) were identified in 158 LTRs and 184 Controls (CTL). The corresponding distribution of genotypic and allelic combinations was analyzed for potential links with the development of circulating donor-specific anti-HLA alloantibodies (DSA) detected at months 1 and 3 after lung transplant (LTx), the occurrence of acute rejection (AR) and chronic lung allograft dysfunction (CLAD), and the overall survival of LTRs. The FCGR3A [158V/V] genotype was identified as an independent susceptibility factor associated with higher rates of AR during the first trimester after LTx (HR 4.8, p < 0.0001, 95% CI 2.37–9.61), but it could not be associated with the level of CD16- mediated NK cell activation in response to the LTR's DSA, whatever the MFI intensity and C1q binding profiles of the DSA evaluated. The FCGR2A [131R/R] genotype was associated with lower CLAD-free survival of LTRs, independently of the presence of DSA at 3 months (HR 1.8, p = 0.024, 95% CI 1.08–3.03). Our data indicate that FCGR SNPs differentially affect the clinical outcome of LTRs and may be of use to stratify patients at higher risk of experiencing graft rejection. Furthermore, these data suggest that in the LTx setting, specific mechanisms of humoral alloreactivity, which cannot be solely explained by the complement and CD16-mediated pathogenic effects of DSA, may be involved in the development of acute and chronic lung allograft rejection.
Lung transplantationi (LTx) remains a challenging therapeutic option for patients with end-stage pulmonary disease. Significant improvement in immunosuppressive strategies has led to decreased lung allograft loss in the early post-transplant period. When compared to other solid organ transplantation settings, LTx remains associated with the lowest survival rates with a median survival of 6 years after transplant (1). Chronic lung allograft dysfunction (CLAD) is the main cause of chronic lung allograft rejection and is characterized by an irreversible loss of lung function associated with a high prevalence of complications such as bronchiolitis obliterans syndrome (BOS) and restrictive allograft syndrome (RAS) (2). Factors that relate to the HLA mismatch, to the graft procedure (ischemia, unilateral or bilateral surgery) and initial lung disease can lead to highly variable levels of recipient immune response to the lung allograft. Inflammatory biomarkers and antibodies, occurrence of acute cellular rejection (ACR), infections/colonization, auto-immunity, and air pollution have been analyzed for their potential predictive value in anticipating development of immunological responses associated with CLAD. Toll receptors, pro-inflammatory cytokines and non-classical HLA molecules, i.e., HLA-G and HLA-E polymorphisms or haplotypes, have been associated with CLAD (3–9), but the underlying mechanisms involved in this devastating outcome of LTx in a given recipient are still poorly understood. Antibody-mediated rejection (ABMR) has been associated with a higher incidence of chronic lung allograft dysfunction (CLAD) and mortality after LTx but the specific mechanisms and histological or immunological biomarkers that allow to define the clinical ABMR entity still await further comprehension (10). Detection of preexisting or development of de novo donor-specific human leukocyte antigen (HLA) alloantibodies (DSAs) have been extensively investigated for their potential value as biomarkers of humoral responses that may predict adverse outcome of LTx.
Various studies suggest that the pre-transplant detection of circulating DSA is not associated with an increased risk of developing CLAD or related death if prospective cross-match testing was negative (11, 12). The pretransplant detection of DSA and antibodies directed againd non-HLA antigens such as angiotensin type 1 receptor (AT1R) and endothelin type A receptor (ETAR) is reported to have a negative impact on lung transplant outcome (13).
Early detection of de novo DSA detected at 1 month after LTx has been significantly associated with a worse outcome (14). Multiplex solid phase single antigen bead assay (SAFB) detection of de novo DSAs, notably anti-HLA DQ DSA, have also been associated with acute cellular rejection (ACR) (15) CLAD (16–18). The presence of circulating DSA at the time of lung allograft biopsy has been identified as a risk factor for graft loss (19).
Although there is more and more direct and indirect evidence regarding the role of de novo DSA in CLAD occurrence, the mechanisms that sustain the variable toxicity of these anti-HLA antibodies to the lung allograft are still unclear. One of the well-known cytotoxic mechanisms of DSA occurs through the IgG-mediated activation of the complement cascade that results in C4d deposition within the graft. C4d staining in lung allograft biopsies is considered as a diagnosis criteria for ABMR (10, 20–28). However, although recent evidence suggests that detection of complement-binding DSA can be associated with lung allograft failure (29), the predictive value of complement-binding DSA on LTx clinical outcome remains to be firmly demonstrated.
IgG antibodies can also activate the immune system through ligation of functional Fc gamma receptors (FcyRs). Since these receptors are expressed by a variety of immune cells, including B cells, natural killer cells, platelets, dendritic cells and macrophages, FcR receptors constitute a major checkpoint that regulates the intensity of auto- and allo-immune responsiveness of the host in response to infectious and humoral threats (30, 31). As infections and the immune alloreactivity of the recipient toward the transplant remain a leading cause of graft rejection and death during the first year after LTx, there is a need for biomarkers that may improve the monitoring of early humoral responses in LTRs (32–34) and open therapeutic perspectives to dampen antibody-driven inflammation in immunized patients (35, 36).
Polymorphisms of FcγRIIA (FCGR2A [131R/H], rs1801274) FcγRIIIA (FCGR3A [158V/F], rs396991) genes, that respectively, encode for the CD32 and CD16 receptors for the Fc segment of IgG, have an impact on the level of expression and immune function of these activating FcγRs. Various reports have highlighted the clinical relevance of these SNPs in controlling the host immune response to monoclonal antibody therapy. The presence of a histidine (H) rather than an arginine (R) at position 131 results in higher affinity for IgG1 and IgG2. The FCGR2A [131H/H] genotype has been associated with higher efficacy of Rituximab-mediated B cell depletion strategies before transplant of ABO-incompatible organs. This FCGR2A [131H/H] SNP has also been identified as a susceptibility marker associated with the severity of community-acquired pneumonia (37). Genetic variation in the FCGR2A gene has also been shown to be associated with an increased prevalence of invasive pneumococcal diseases and respiratory infections after LTx (38, 39), regardless of the site of pneumococcal infection (40). Pro-inflammatory effects associated with the FCGR2A [131R/R] genotype have also been reported as a consequence of activating signals that result from the dynamic interactions between CD32 and its cognate C-reactive protein (CRP) and immunoglobulin ligands.
The clinical relevance of CD16 inflammatory pathways in antibody-mediated rejection of kidney and heart allografts has also been illuminated by studies that deciphered the molecular landscape of immune and endothelial cell transcripts that associate with ABMR lesions evaluated in allograft biopsies (41–43). Ligation of the Fc domain of IgG has also been identified as a mechanism that allows CD16-dependant clearance of viral pathogens by immune NK cells (44). Recent studies suggest that the Fc fragment of DSA can indeed exert its adverse cytotoxic and inflammatory effects though a complement-independent mechanism that relies on level of expression of the NK-cell surface CD16 receptor and its affinity for the Fc fragment of alloantibodies. The intensity of host antibody dependent cell cytotoxicity (ADCC) is known to be conditioned by the capacity of NK effector cells to form conjugates with antibody-coated allogeneic cells, which is in part influenced by the FCGR3A [158V/F] genotype (45–47). We and others have recently shown that polymorphic variation in the FCGR3A genes is also likely to affect the pathogenic effects of IgG alloantibodies by controlling the level of the CD16 dependent recipient's immunological cytotoxic responses to allogeneic donor cells exposed to chronic alloantibody threat (35, 48–51).
Genotypic variation in the FCGR3A receptor can thus impact the strength of FcR-dependent ADCC responses of NK cells and regulate their capacity to secrete inflammatory cytokines and release CD107a/Lamp1+ cytotoxic granules containing perforin and granzyme (50, 52, 53). The inter-individual variability in the functional CD16 receptor-dependent engagement by the Fc fragment of alloantibodies has been shown to condition the intensity of antibody-dependent cellular cytotoxicity (ADCC) of NK cells and the response to IgG immunotherapy.
The strength of FcR-mediated ADCC can also be affected by the isotype and glycosylation status of alloantibodies. The IgG1 and IgG3 isoforms have higher affinity for the CD16 FCGR3A [158V/V] variant when compared to FCGR3A [158F/F], thereby spurring increased effector cell activity in response to these IgG subclasses. We have shown that this high-affinity homozygous FCGR3A [158V/V] genotype is an independent predictor of cardiac allograft vasculopathy (48) and may be a clinically relevant underlying mechanism that sustains the level of DSA-mediated allograft injury (48–50). The recent evidence that the NK cell infiltration of the graft can predict kidney graft failure also sustains the clinical relevance of these NK cell mediated mechanisms of allograft injury (54).
Considering the central contribution of polymorphisms affecting FcγRIIA and FcγRIIIA genes and receptor function in the individual shaping of antibody-mediated inflammatory and cytotoxic alloimmune responses of the recipient, deciphering the mechanisms and FcγR susceptibility profiles that may be associated with LTx outcome is necessary to optimize risk stratification (8).
This study thus aimed to investigate whether FcγR polymorphisms may be linked to the pathogenic mechanisms of DSA toxicity and be associated with adverse clinical complications that impair patient and allograft outcome after LTx.
We conducted a retrospective single-center study enrolling 158 adult patients who underwent lung transplants (LTx) at the Marseille Lung Transplant Center between December 2006 and December 2013. All patients from the French cohort (COLT, Cohort in Lung Transplantation, l'Institut du Thorax, INSERM UMR1087/CNRS UMR 6291, CNIL 911142) were recruited in this study. All subjects gave written informed consent in accordance with the Declaration of Helsinki. A group of 184 healthy unrelated volunteer French bone marrow donors were also recruited to constitute a control cohort, allowing the analysis of the FcγR genotype. Blood donations were collected in the “Etablissement Francais du Sang,” in accordance with BSL-2 practices. A medical interview was carried out prior to blood donation to exclude donors with medical contraindications. This study was carried out in accordance with the French Public Health Code (art L1221-1), approved by an institutional ethics committee and conducted in compliance with the Good Clinical Practice Guidelines declaration of Helsinki and Istanbul.
All recipients received a similar standardized immunosuppressive regimen in accordance with our institutional protocols. Induction therapy consisted of intravenous administration (IV) of rabbit anti-thymocyte globulins (rATG, Pasteur Merieux, Lyon, France) given for the first 3 post-operative days [except when daily lymphocyte count was below 200/mm3, and when there were cytomegalovirus (CMV) and/or EBV mismatches (i.e., seronegative recipient and seropositive donor)] and high dose methylprednisolone (6 mg/kg/d Day 1, 2 mg/kg/d Day 2 and Day 3, and 1 mg/kg/d thereafter). The standard triple maintenance immunosuppressive regimen consisted of tacrolimus (adjusted to maintain whole blood levels varying between 12 and 14 ng/ml), mycophenolate mofetil in 5 patients (adjusted to a white blood cell count above 4,000 mm3), and steroids (prednisone) tapered to 0.25 mg/kg/d over the first 3 months and stopped around 12 months after surgery.
According to CMV recipient positive (R+) and donor positive/recipient negative (D+/R–) status, patients received CMV prophylaxis with IV ganciclovir switch to oral valganciclovir.
Postoperatively, recipients received prophylactic or preemptive anti-infection treatment (antibiotic, antiviral, and antifungal therapies) according to their preoperative and/or concomitant infectious status.
Recipients had regular visits to the transplant center for clinical radiological and functional evaluation. At our institution, surveillance transbronchial biopsies are routinely performed at the end of the first month, or earlier if clinically indicated. All transbronchial biopsies were graded for ACR (A grade) and lymphocytic bronchiolitis (B grade) (LB) by lung transplant pathologists. ACR and LB were defined, respectively, as perivascular or peribronchial mononuclear inflammation according to the International Society for Heart and Lung Transplantation (ISHLT) criteria. Histologic appraisal of ACR and LB was conducted in accordance with accepted ISHLT standards in terms of the minimum number of biopsy samples and exclusion of opportunistic infection (27, 28). Pulmonary function tests (PFTs) were routinely conducted at our center on a monthly basis for the first 12 post-operative months, at M2 intervals in the second year and at M3 intervals thereafter. The baseline FEV1 value was calculated as the average of the 2 best FEV1 values at least 2 measure gap. Baseline values of total lung capacity (TLC) and FEV1/FVC were defined as the average of the 2 measurements obtained at the same time as the best 2 FEV1 measurements. Chronic lung allograft dysfunction (CLAD) was defined according to the standardized international criteria (55). The phenotype BOS or RAS was specified according to ISHLT guidelines (56). The following histological patterns compatible with AMR were also analyzed: (i) neutrophil capillaritis and (ii) acute lung injury with or without diffuse alveolar damage and with or without organizing pneumonia.
Recipient serum samples from 2006 to 2013 were collected routinely prior to transplant, at the minimum when the patient was placed on the waiting list and before the transplant procedure (D0: Day 0) and serially after LTx (at month 1 and 3). All sera samples obtained from the 158 recipients were further assessed using Luminex single-antigen flow beads (SAFB) to determine antibody specificity using Single Antigen—One Lambda reagents (LABScreen® Single Antigen class I or LABScreen® Single Antigen class II, One Lambda, Thermo Fisher Scientific, Canoga Park, California, USA) according to the recommendations of the manufacturers. The mean fluorescence intensity (MFI) used the baseline formula proposed by the FusionTM v 3.2 software. All beads with a normalized MFI threshold >1,000 were considered positive. Since detected circulating alloantibodies were often directed against distinct HLA class I and/or class II antigens, the cumulative mean fluorescence intensity (cMFI) of DSA was calculated as the sum of the MFI for each of the individual DSAs detected. DSA were considered to be HLA antibodies directed against donor HLA antigen. Putative HLA-Cw and -DQA1 DSA were identified in accordance with the conventional linkage disequilibrium that is reported between HLA-B and HLA-C loci and between HLA-DQB1 and HLA-DQA1 loci, as respectively, described in the French population (57) and by supplementary extensive genotyping of HLA- genes in the recipient. Putative HLA-DR51, -DR52 and -DR53 DSA were determined in accordance with the linkage disequilibrium between DRB3, DRB4, DRB5 loci and DRB1 loci of recipient. The allelic specificities of HLA-Cw, DQA and -DR51, -DR52 and -DR53 DSA were assigned accordingly.
All patients with HLA antibodies were tested for the presence of C1q. The binding level was determined by the C1qScreen™ assay per manufacturer instructions (One Lambda, Thermo Fisher Scientific, Canoga Park, California, USA). Fluorescence intensity was measured using Luminex-based LABScan™ 100 flow analyzer. C1q specific antibody specificity and binding levels were analyzed and determined through the FusionTM v 3.2 software. C1q MFI > 1,000 was considered positive.
The detection of single nucleotide polymorphisms (SNPs) allowed for the genotyping of FCGR3A ([158V/F], rs396991) and FCGR2A ([131R/H], rs1801274) as described (5, 48) using the SNAPSHOT technique. Genomic DNA was extracted from a 200-μl whole blood sample using the QIAmp Blood DNA Mini kit (Qiagen, Courtaboeuf, France) according to manufacturer instructions. A previously described homemade primer extension method (58) was used to simultaneously analyze the SNPs of the FcγR genes. The forward and reverse primers sequences were, respectively: FCGR3A-F 5′ TCCTAATAGGTTTGGCAGTG 3′ and FCGR3A-R 5′ AAATGTTCAGAGATGCTGCT 3′, FCGR2A-F 5′ CCAGGAGGGAGAAACCATC 3′ and FCGR2A-R 5′ CTCTTCTCCCCTCCCTACAT 3′. The extension primers are, respectively: FCGR3A_176-F: 30T-CCTACTTCTGCAGGGGGCTT and FCGR2A_166F 57T-CCAAAAGCCACACTCAAAGA. Data were analyzed using GeneMapper 4.0 with specific detection parameters. Using an in-house computer program, output files (.txt) exported from GeneMapper 4.0 were automatically formatted into files readable by the “Phenotype” application of the Gene[Rate] computer tool package (http://geneva.unige.ch/generate). For each different allele obtained, PCR products were sequenced on both strands using the BigDye Terminator v1.1 Sequencing Kit (Applied Biosystems) and analyzed on an automated fluorescence-based ABI PRISM 3130 XL genetic analyzer according to manufacturer protocol. FcR polymorphisms at these 4 loci were analyzed for allele frequencies, or homozygous or heterozygous allelic combination of FcγR alleles defining the corresponding genotype.
Recipients and deceased donors were genotyped for low resolution HLA-A, -B, -DRB1, and -DQB1 loci by LABType® SSO (One Lambda, Thermo Fisher Scientific, Canoga Park, California, USA) according to the manufacturer's specifications and the retrieved output was analyzed for allele identification using HLA FusionTM v 1.2.1. Software (One Lambda, Thermo Fisher Scientific, Canoga Park, California, USA).
Evaluation of the CD16-dependent alloreactive potential of DSA was assessed using the previously described NK cell humoral activation test (NK-CHAT) (50). The level of serum-induced CD16 engagement and the degranulating potential of NK cells was assessed by flow cytometry analysis of the level of CD16 cell surface expression within the CD3−CD56+ NK cell compartment, gated within PBMC effector cells after exposure to target B cells that had been precoated in the presence of LTRs or control sera. Effector cells used in the standardized assay were prepared from a healthy donor displaying the FCGR3A [158V/V] encoding the high-affinity CD16 variant. The HLA typing of the two B cell lines used as targets were A2/A2, B44/B56, Cw1/Cw5, DR1/DR4, DR53, DQ5/DQ7 and A3/A3 B7/B35, Cw4/Cw5, DR10/DR15, DR51, DQ5/DQ6). Sera were selected for the presence of DSA recognizing a limited set of HLA antigens expressed by EBV target cell lines. Briefly, 500,000 target B cells (B-EBV cell lines expressing the cognate DSA target alloantigens detected in LTR) were incubated with control (CTL) unsensitized human male AB serum (Lonza) to block FcR, then rinsed and incubated for 15 min in the presence of 50% LTR serum [or CTL serum supplemented or not with 10 γg/ml of Rituximab IgG (positive control)] and then rinsed to remove unbound antibodies. Incubation of effector PBMC and pre-coated allogeneic B-EBV cell targets (1:1 ratio) was performed for 3 h at 37°C in presence of GolgiStop (Becton Dickinson 554724) and CD107-PC5 (Becton Dickinson 555802). Cells were then washed and labeled with CD3-ECD (Beckman Coulter A07748), CD16-PE (Beckman Coulter A07766), CD56-PC7 (Beckman Coulter A21692) for 15 min at room temperature protected from light. After 1 wash step, cells were resuspended in 500 μl PBS 2% SVF. Data acquisition and analysis was performed on a Beckman Coulter Navios cytometer. The mean fluorescence intensity of CD16 and percent of NK cells expressing Lamp1/CD107a expression was analyzed within the CD3-CD56+ NK cell subset. The NK-CHAT CD16 down regulation index (CD16DRI) was evaluated as a ratio between CD16 MFI measured on NK cells incubated with target cells in presence of effector cell autologous CTL serum/CD16 MFI of NK cells incubated with target cells and recipient serum to be tested. CD16DRI = (MFI CD16 control serum)/(MFI CD16 test serum). Alternatively, when serum at time of LTx was available for the assay, the baseline CD16 expression was evaluated in reference to the DSA negative serum collected at time of LTx. Sera containing HLA-DQ7 DSA obtained from kidney transplant recipients (KTR) with antibody-mediated rejection (ABMR) at time of diagnosis, previously described to induce high levels of CD16-dependent NK cell alloreactivity, were introduced as positive controls of the experiments, indexing the CD16 down regulation induced by LTx sera.
Variables used to perform univariate and multivariable analyses included preoperative donor variables (donor age, sex, CMV status, HLA typing), preoperative donor–recipient matching parameters (age, sex, CMV status, and HLA mismatch), operative variables such as ischemia time, type of procedure (single vs. bilateral LTx) and preoperative and post-operative recipient variables (initial lung disease, HLA typing, pre- and post-transplant immunization status, occurrence of AR BOS, RAS, CLAD, or infectious bacterial episodes occurring during the first year post-transplant), and notably during the early post-Tx period at months 1 (M1) and 3 (M3) after LTx (Table 1).
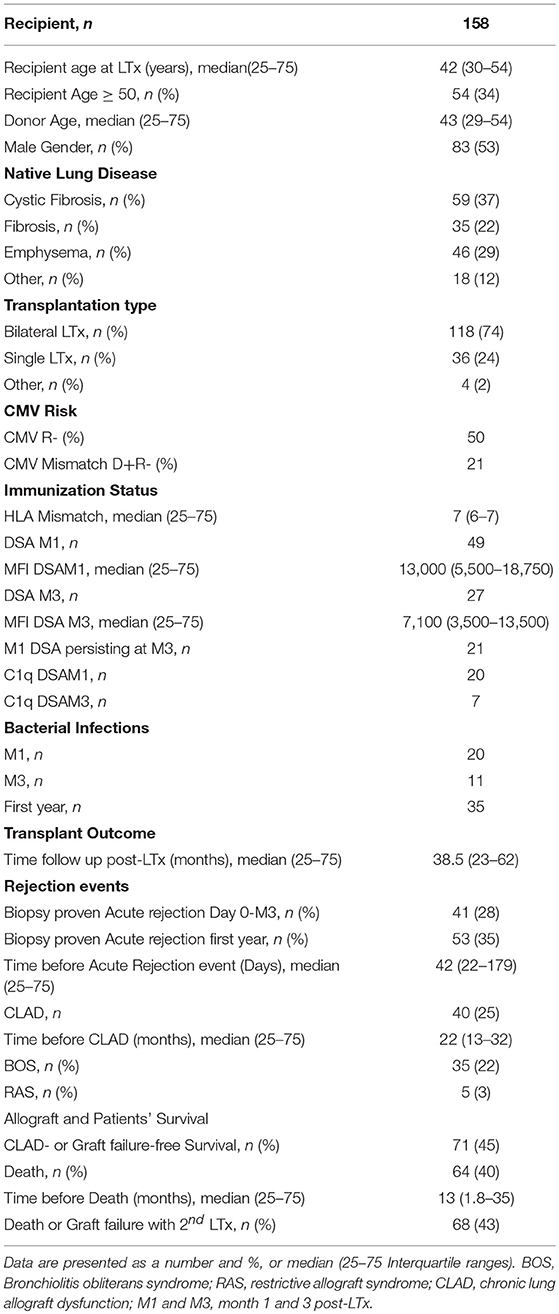
Table 1. Demographic and Clinical characteristics of LTRs.
Continuous variables were presented as median values and 25–75 interquartile ranges or mean ± sd according to their distribution evaluated using the Agostino & Pearson omnibus normality test performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, La Jolla, California, USA). Categorical variables were presented as percentages. Fisher's exact test and the chi-square test were used to compare categorical data and t-tests or Mann Whitney tests were used for the comparison of continuous variables.
The primary endpoints of this study were overall survival (OS) and disease/CLAD-free survival (DFS). OS was defined as the interval between the date of transplant and the last follow-up visit or death. DFS was defined as the time interval from transplant to the first event: either the graft failure or diagnosis of acute rejection or CLAD in living recipients, or the death of the patient. The Kaplan-Meier method was used to estimate overall survival and rejection-free survival. The log-rank test was used to assess the univariate effects on OS and DFS. For all analyses, a 2-sided p < 0.05 was considered statistically significant. Variables with a p < 0.2 were also considered to construct multivariate regression models. Multivariate analyses were performed using Fine and Gray's proportional hazards regression model. All analyses were performed using IBM SPSS 15.0 software (SPSS Inc., Chicago, IL) and the cmprsk package (developed by Gray, June 2001) on R2.3.0 software (http://www.R-project.org).
The demographic characteristics and clinical features of the 158 adult LTRs (median age: 42 years, 75 females and 83 males) are summarized in Table 1. Patients received a first LTx for cystic fibrosis (37%), emphysema (29%), pulmonary fibrosis (22%), or another diagnosis (12%). The median follow-up time was 38 months after LTx. Median survival time of the 94 patients who were alive at time of follow-up was 4.2 years. Sixty-four patients (40.5%) died during the study follow-up (median time before death 12.6 months, 25–75 interquartile range: 1.8–35.5 years) and 5 patients experienced CLAD-associated graft failure with indication of a second lung transplant or death. Among deceased patients, 32 patients died during the first year post-LTx, among which 12 patients died during the first month post-LTx and 9 between the first and third months post-LTx. Acute rejection diagnosis was confirmed in the lung transplant biopsy of 53 patients (36%), the first rejection episode occurring before the third month in 41 LTRs (occurring before the fourth month in 24 LTRs and during the third month and the twelfth month post-LTx in 11 LTRs). The occurrence of acute rejection could be associated with the presence of DSA in 49% of these patients (n = 20). During the study period, 40 LTRs (25%) developed CLAD, of which 35 (87%) had BOS and 5 (12%) had RAS. Seventeen of the patients with a CLAD diagnosis did not survive and 5 were considered for a second transplant procedure. CLAD-free survival of the graft was observed in 71 patients (45%) with a median disease-free survival time of 4.2 years (25–75 percentile: 3.1–5.8).
Genotyping of the FCGR2A and FCGR3A polymorphisms was performed in 158 LTRs and analyzed in reference to a control cohort of 184 healthy donors (CTLs) (Figure 1). The FCGR2A-H allele was detected in 75.3% of LTRs and 83.1% of CTLs (p = 0.0734) while the -R allele was detected in 67.7% of LTRs and in 76% of CTLs (p = 0.0851). Although the distribution of FCGR2A [131R/R] and FCGR2A [131H/H] was not significantly altered in LTRs, the frequency of FCGR2A [131H/R] was found to be significantly lower in LTRs analyzed in reference to CTLs (chi-square p = 0.0028). The distribution of FCGR3A alleles did not significantly differ between patients and controls; presence of the -F allele was detected in 81% of LTRs and in 83.7% of CTLs while the -V allele distribution was in 64.6% of LTRs and in 68.5% in CTLs. The genotype frequencies of the various genotype combinations resulting from analysis of these SNPs in the LTR and CTL cohorts are illustrated in Figure 1. The frequency of the FCGR3A [158F/F] genotype was positively correlated with the FCGR2A [131R/R] genotype in CTLs and in LTRs (p = 0.008 and p < 0.0001, respectively). Presence of the FCGR2A [131R/R] was thus significantly associated with lower rates of patients with the FCGR3A [158V/V] good responder genotypes in LTRs (p = 0.038), but this inverse correlation did not reach significance in the control cohort (Table 2). Analysis of the groups stratified according for FCGR SNPs did not reveal any significant difference regarding the age of the LTR and type of underlying initial lung disease. The FCGR2A [131H/R] SNP with was found to be associated with recipient gender (p = 0.029). The FCGR2A [131H/H] genotype was observed to be significantly more common in women LTRs (32 out of 51, p = 0.008).
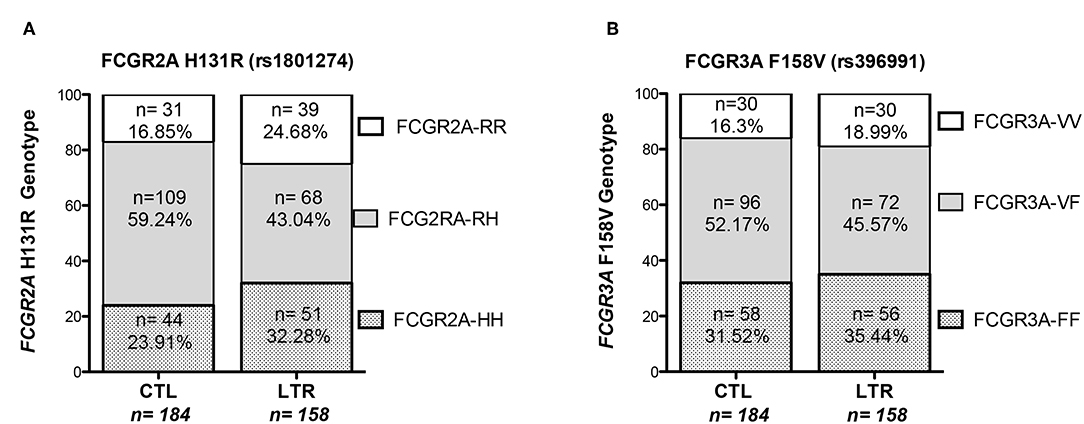
Figure 1. Distribution of FCGR3A and FCGR2A genotypes in LTRs and CTLs. The proportion of the FCGR2A (left Panel, A) and FCGR3A (right panel, B) genotypes resulting from the SNP allelic combination were analyzed in a cohort of 158 LTRs and 184 CTLs recruited in southern France.
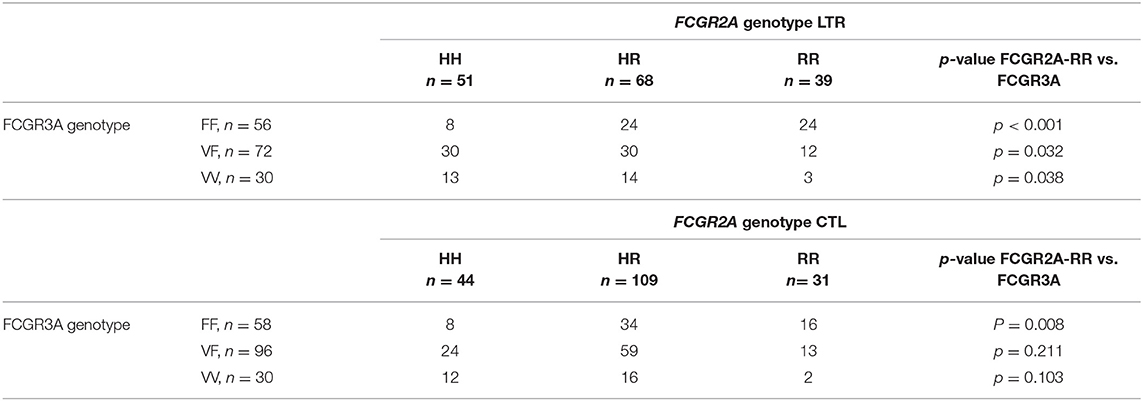
Table 2. Association between the FCGR2A [131R/R] and FCGR3A [158V/F] genotypes observed in LTRs and CTLs.
During the first 3 months post-transplant, 55 patients developed de novo circulating DSA. During the first month post-LTx (M1), DSA developed in 49 LTRs (12 HLA class I, 20 HLA class II, and 17 HLA class I and II). The median MFI of DSA detected at M1 was 7,000 for HLA Class I DSA and 12,100 for HLA Class II DSA and 12 patients (8%) exhibited high cMFI values of DSA (over the 18,750 threshold corresponding to the 75 percentile value observed for MFI analyzed at M1) (Table 1). Circulating DSA could be detected in 27 LTRs analyzed at month 3 (M3) post-LTx (11 HLA class I, 13 HLA class II and 3 HLA class I and II) (Figure 2). The development of de novo DSA was observed at M3 in 6 LTRs with no detectable DSA at M1 (4 HLA class I, 1 HLA class II and 1 HLA Class I and Class II). The median MFI values of DSA detected at M1 and at M3 were, respectively, 13,000 and 7,100 (Table 1, Figure 2). DSA detected at M1 persisted at M3 in 21 LTRs (44% of DSA-positive MTR at M1) and 4 of these M3 DSA were also detectable 1 year post-transplant. HLA-DQ DSA were the only HLA class II persisting at M3. The median cMFI level of DSA detected at M1 and persisting at M3 (14,600, 25–75 percentile, 7,000–23,250) could not be linked to their persistence at M3 and did not significantly differ from the cMFI of DSA observed at M1 but undetectable at M3 (median MFI: 12,200, 25–75 percentile 4,000–16,000). Circulating DSA directed against DQ specificities were detected in 33 LTRs at M1 (median DQ-DSA MFI: 12,000 25–75 percentile 7,600–14,500) and in 14 LTRs at M3 (median 12,000, 5,000–15,850, 11 HLA-DQ DSA detected at M1 persist at M3 while 3 of the 14 HLA-DQ DSA detected at M3 were de novo DSA). C1q Binding DSA at M1 were detected in 20 LTRs (2 HLA class I, 15 HLA class II, and 3 HLA class I and II) with a median cMFI of C1q-binding DSA detected at M1 of 23,000 (25–75 percentile 6,300–30,000). Seven of the 27 DSA detected at M3 were found to bind C1q (1 HLA class I, 5 class II HLA DSA and 1 HLA class I and class II), with a median cMFI of 17,150 (25–75 percentile 7,000–31,000). All C1q binding DSA detected at M3 were also detected at M1.
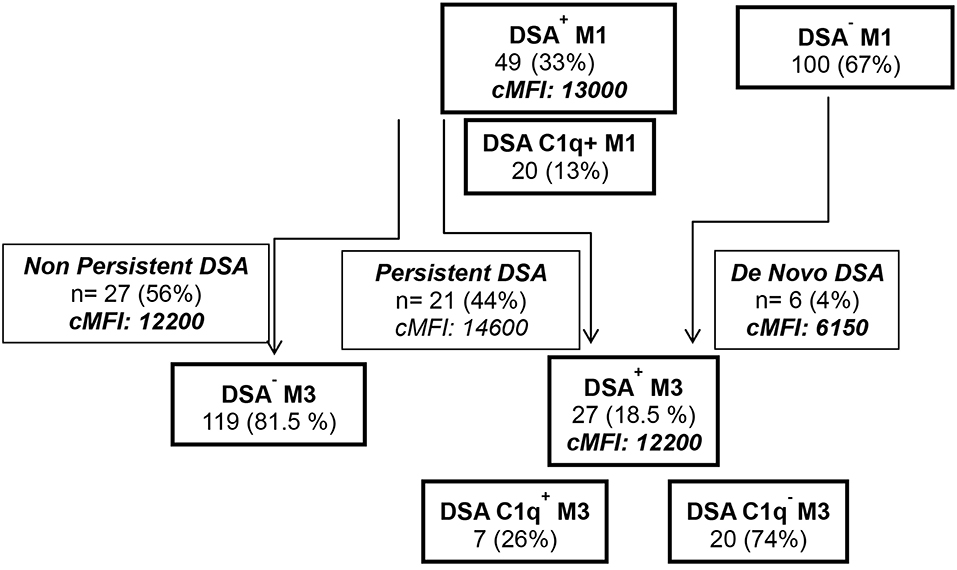
Figure 2. Study flow chart analysis of the detection by single antigen flow bead Luminex Assay of de novo DSA and C1q binding activity of DSA at M1 and at M3 post-LTx.
The FCGR2A [131R/R] genotype was also to found to correlate with the detection of circulating DSA at M3 (p = 0.011) and notably to the development of de novo DSA between the first and second months post-transplant (4 out of the 6 de novo DSA detected at M3 were genotyped as FCGR2A [131R/R], p = 0.017). FCGR3A [158V/V] tends to be inversely associated with the persistence of DSA at M3 (p = 0.07, Table 3), as only 1 out of the 21 patients with persistent DSA at M3 was genotyped as FCGR3A [158V/V]. The FCGR3A-F allele was significantly associated with the detection of circulating DSA at M3 (26 out of 27 LTRs with detectable circulating DSA at M3 were positive for the FCGR3A-F allele, p = 0.024) (Table 3).
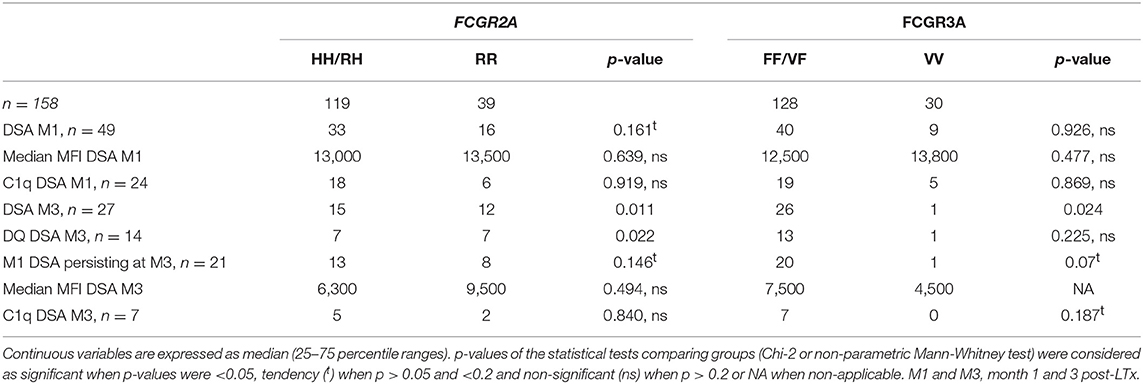
Table 3. Patients immunization characteristics according to the FCGR2A and FCGR3A genotypes.
The FCGR3A [158V/V] was also identified as a susceptibility genotype associated with a higher risk of acute rejection (log rank test for equality of survivor functions p = 0.0009, Figure 3A). Multivariate Cox regression analysis also revealed that the susceptibility conferred by the FCGR3A [158V/V] genotype (OR 4.8, p < 0.0001, 95% CI 2.375–9.607) was independent from the development of DSA at M3 and their C1q binding activity. FCGR2A [131R/H] was not observed to affect the rate of acute rejection. The FCGR2A [131R/R] genotype was instead associated with the detection of DSA occurring in the absence of acute rejection (p = 0.029).
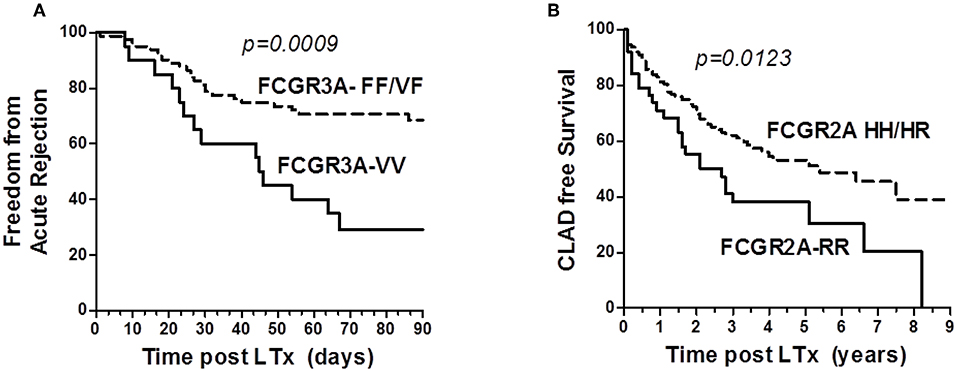
Figure 3. Analysis of the influence of Fc-gamma Receptor polymorphisms on Rejection Free survival. (A) Impact of FCGR3A genotype on freedom from acute lung rejection. Kaplan–Meir Survival analysis links the FCGR3A [158V/V] (solid line, n = 20) to lower acute-rejection-free survival after 3 months post-LTx in LTRs, when compared with the FCGR3A [158V/F] and FCGR3A [158F/F] group (dashed line, n = 81). LTRs with the FCGR3A [158V/V] show an increased rate of acute rejection events occurring during the first 3 months post-LTx. (B) Impact of FCGR2A genotype on CLAD free survival. Kaplan–Meir Survival analysis links the FCGR2A [131R/R] (solid line, n = 39) to a lower CLAD-free survival rate in LTRs over time or study follow-up (years, x axis), when compared with the FCGR2A [131H/H] and FCGR2A [131R/H] group (dashed line, n = 119).
The DSA immunization status of the patient evaluated during the first year post- LTx could not be associated with CLAD. Although the number of observations was small, 4 of the 5 LTRs who developed RAS-associated CLAD were homozygous for the FCGR3A–FF allele (p = 0.034). While the FCGR3A [158V/F] could not be significantly associated with the risk of CLAD, FCGR2A [131R/R] was identified as a susceptibility marker associated with lower CLAD-free survival of LTRs (n = 87, logrank test for equality of survivor functions p = 0.0123, Figure 3B). Multivariate Cox regression models adjusting co-variables identified using univariate analysis further identified FCGR2A [131R/R] as an independent susceptibility marker associated with CLAD (OR 2.2; p = 0.022, 95CI 1.126–4.435) (Table 4). Multivariate Cox analysis further showed that the FCGR2A [131R/R] genotype is significantly associated with the enhanced risk of developing a composite LTx adverse outcomes (CLAD, graft failure or death), independently of other factors such as detection of DSA at M3 or native emphysema lung disease (Table 4).
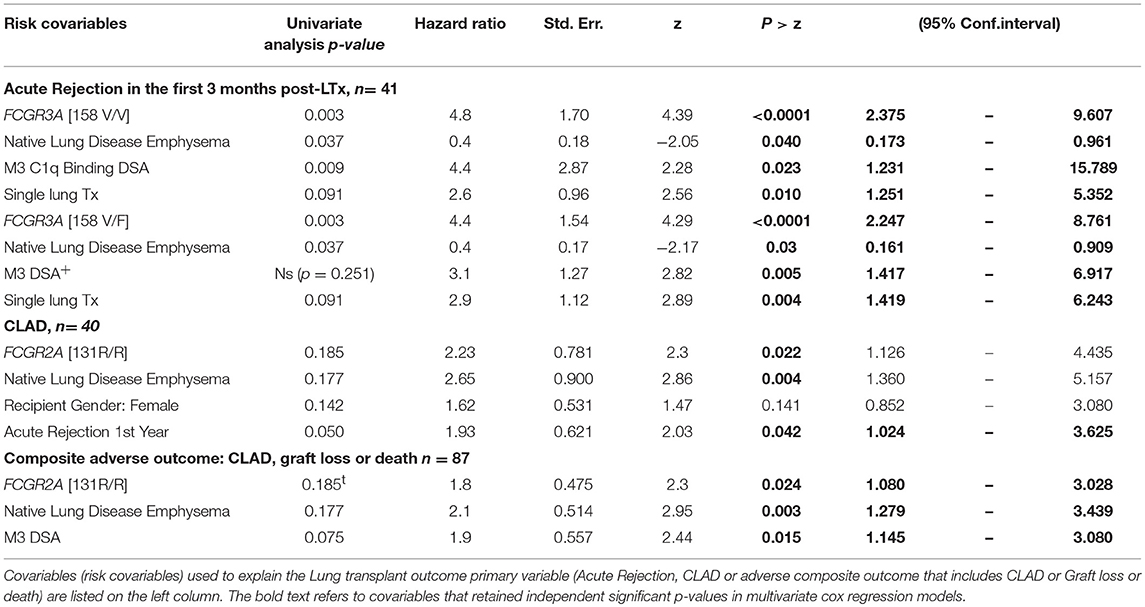
Table 4. Univariate analysis and multivariate Cox regression analysis of risk factors associated with acute or chronic rejection events.
Considering our previous finding that identifies the potential value of FCGR3A [158V/F] in predicting cardiac allograft vasculopathy (48), we further investigated whether the presence of association of acute rejection events with high affinity FCGR3A [158V/V] genotype could be associated with the DSA-mediated pathogenic effects that promote acute rejection mechanisms of lung allograft. Using the NK-cellular humoral activation test (NK-CHAT), previously designed to index the level of DSA-mediated engagement of CD16 and unravel potential ADCC-driven pathogenic effects of circulating DSA (41), we therefore aimed to investigate whether circulating DSA detected at M1 or M3 in LTRs have the potential to stimulate NK cell alloreactivity. Evaluation of the DSA-induced down regulation of NK cell CD16 expression (CD16 Down Regulation Index or CD16DRI) was performed in a standardized NK-CHAT assay analyzing alloreactivity of NK cells toward serum-coated CD20+ B lymphocytes expressing the cognate HLA target alloantigen, using 23 serum samples obtained from 20 distinct LTRs (Table 5). Thirteen sera were collected at M1 and ten at M3. Four sera had detectable DSA that persisted at M3. Seven DSA at M1 and Four DSA at M3 were C1q positive.
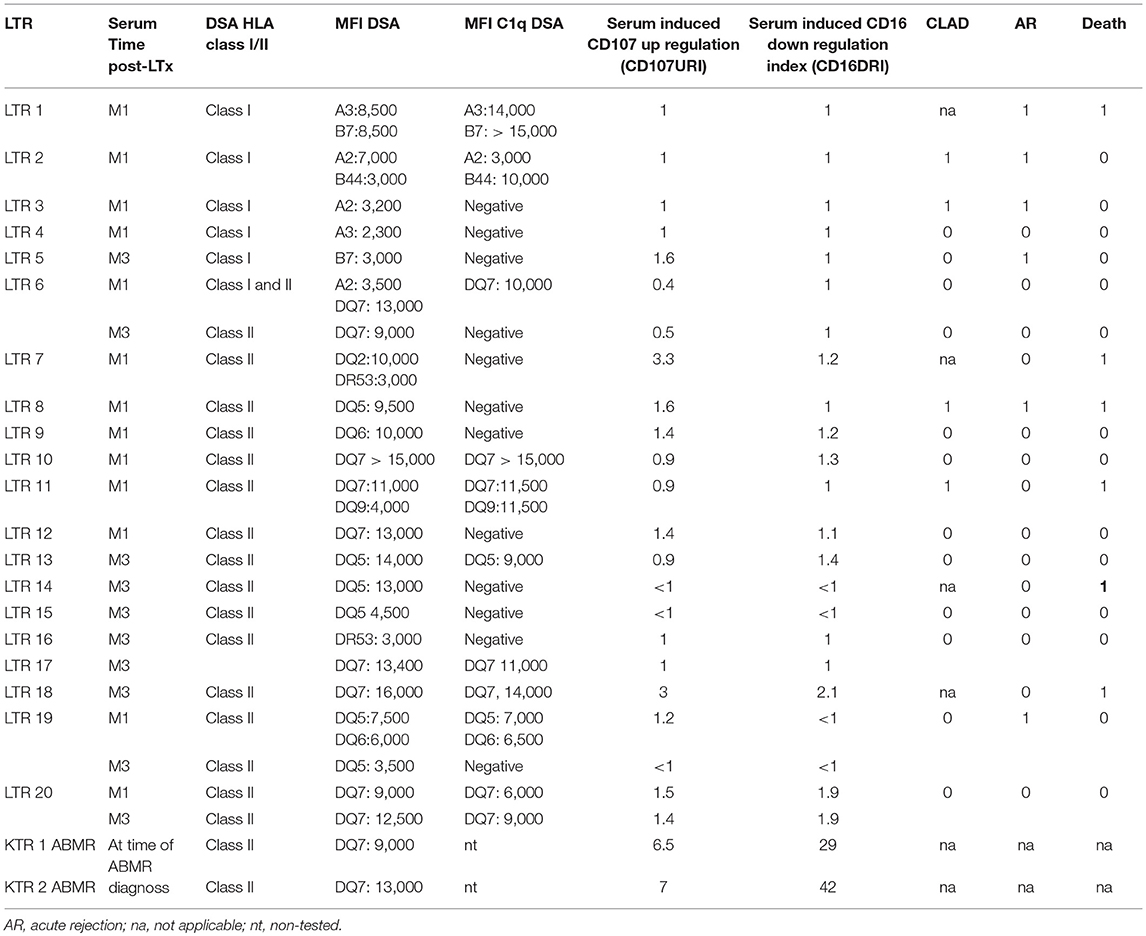
Table 5. NK-CHAT: Evaluation of DSA reactivity toward B cell targets expressed in sera collected from LTR and KTR, used as positive controls for NK-CHAT.
Surprisingly, the CD16DRI values induced by the DSA+ LTx sera were very low when compared to those obtained in response to Rituximab. These CD16 DRI values could not be associated with the intensity of DSA MFI and C1q binding activity of DSA. Furthermore, the CD16DRI of LTRs (LTR 17, 18, and 20) with HLA DQ7 DSA were lower than those obtained of ABMR kidney transplant recipients (KTR) with HLA DQ7 DSA and with similar MFI (Figure 4).
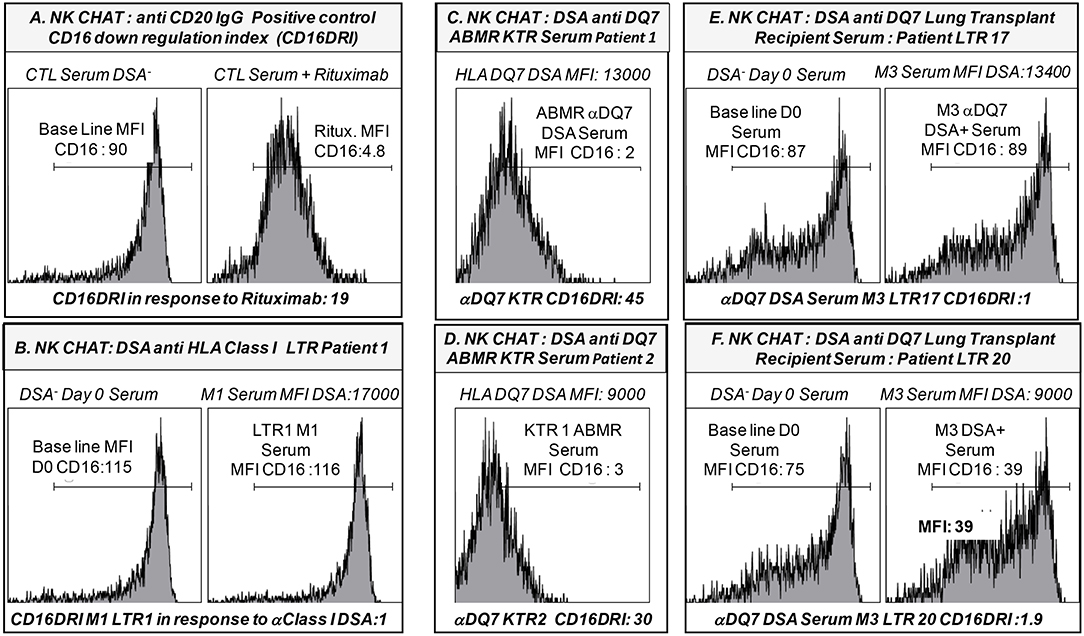
Figure 4. Representative illustration of the NK-Cellular Humoral test (NK-CHAT) used to evaluate serum- and DSA- induced NK cell activation toward B cell lines expressing cognate HLA class I or HLA-DQ7 donor-specific allo-antigens. (A) PBMC effectors obtained from FCGR3A [158 V/V] CTL were exposed to B-EBV cell lines previously coated with DSA-negative serum. This allowed for evaluation of the baseline expression level of CD16 expression (CD16MFI) within the CD3-CD56+CD16+ NK cell compartment when PBMC were exposed to B cell targets. As a positive control indexing the level of IgG-induced CD16 down regulation (CD16DRI), the same B-EBV cell lines were coated in the presence of 10γ/ml Rituximab (anti CD20 IgG) prior to exposure to the effector PBMC. This allowed for calculation of the CD16 down regulation index (CD16DRI: baseline CD16MFI NK cells exposed to B cells coated with CTL DSA-negative serum/CD16 MFI of NK cells exposed to the same B targets pre-coated in presence of Rituximab). In this context, CD16DRI in response to Rituximab = 19, i.e., 90/4.8. (B) The CD16 DRI of NK cells was evaluated in response to the serum collected at M1 in one LTR (LTR 1, Table 5) with detectable levels of DSA recognizing the HLA-A3 and -B7 expressed on the B cell targets. Non-immunized serum (DSA-negative) collected from LTR 1 at time of LTx (D0) was used as the reference baseline CD16 MFI value to calculate the CD16DRI. Despite a cumulative MFI of DSA >17,000, DSA detected at M1 in LTR1 failed to induce CD16-dependent NK cell alloreactivity (CD16DRI = 1). (C) The CD16 DRI of NK cells was evaluated in response to the serum collected at time of ABMR diagnosis in one kidney transplant recipients (KTR 1, Table 5) with detectable levels of DSA recognizing the HLA-DQ7 (MFI: 13,000) expressed on the B cell targets. The CD16DRI of KTR1 (CD16DRI: 45) was greater than that observed in response to the Rituximab (CD16DRI:19, A). (D) The CD16 DRI of NK cells was evaluated in response to the serum collected at time of ABMR diagnosis in a second KTR (KTR 2, Table 5) with detectable levels of DSA recognizing the HLA-DQ7 (MFI: 9,000) expressed on the B cell targets. The CD16DRI of KTR1 (CD16DRI: 30) was greater than that observed in response to the Rituximab (CD16DRI:19, A). (E) The CD16 DRI of NK cells was evaluated in response to the serum collected at M3 (time of acute rejection) in a second LTR (LTR 17, Table 5) with detectable levels of DSA recognizing the HLA- DQ7 (MFI: 13,400) expressed on the B cell targets. Non-immunized serum (DSA-negative) collected from LTR 17 at D0 was used as the reference baseline CD16 MFI value to calculate the CD16DRI (CD16DRI = 1). The MFI DSA of LTR 17 (MFI: 13,400) was similar to the MFI DSA of ABMR KTR 1 serum as illustrated in (C). (F) The CD16 DRI of NK cells was evaluated in response to the serum collected at M3 (time of acute rejection) in a third LTR (LTR 20, Table 5) with detectable levels of DSA recognizing the HLA- DQ7 (MFI DSA: 9,000) expressed on the B cell targets. The DSA-negative serum of LTR 20 collected at D0 was used as a baseline CD16 MFI value to calculate the CD16DRI (CD16DRI = 1.9). The MFI DSA of LTR 20 (MFI: 9,000) was similar to the MFI DSA of ABMR KTR 2 serum evaluated in (D).
In contrast to the results previously observed in the kidney and heart transplant setting, the association of the FCGR3A [158V/V] genotype with acute rejection appears to be independent of the presence of circulating DSA and the DSA MFI levels. In contrast to the KTR serum, NK-CHAT evaluation of the serum of immunized LTRs did not reveal significant alloreactive DSA toxicity and did not allow for determination of their pathogenic potential in eliciting CD16 and NK-cell cytotoxic activation.
Occurrence of acute and chronic events or infectious episodes was not shown to have a major impact on overall survival in the analyzed cohort.
Early development of DSA that persist at M3 (n = 21) were associated with lower graft or patient survival rates (p = 0.002) and were associated with the presence of the low-affinity F allelic variant of CD16. Persistence of DSA at M3 was thus lower in FCGR3A [158V/V] LTRs (p = 0.029). Other parameters such as high MFI DSA detected at M1 (>18,750 MFI, 75 percentile value of DSA MFI at M1 post-LTx, p = 0.109) or FCRGR2A [131R/R] (p = 0.052) tended to be associated with the risk of death. While the FCGR3A genotype could not be associated to overall survival, the FCRGR2A [131R/R] genotype was identified as a risk factor associated with the composite adverse outcome combining graft failure or patient death (n = 68, log rank test for equality of survivor functions p = 0.0417, Figure 5). Multivariate analysis of variables associated with death from all causes after LTx (n = 64, 40.5% of the LTR cohort) retained initial diseases other than cystic fibrosis (p = 0.048) DSA with MFI values > to the 75 percentile 18,750 MFI value for DSA detected at M1 as significant risk factors (Table 6). The FCRGR2A [131R/R] genotype was shown to be an independent factor associated with lower overall survival of LTRs using multivariate logistic Cox regression models (HR: 1.8, p = 0.047 95% CI: 1.008–3.121, Table 6).
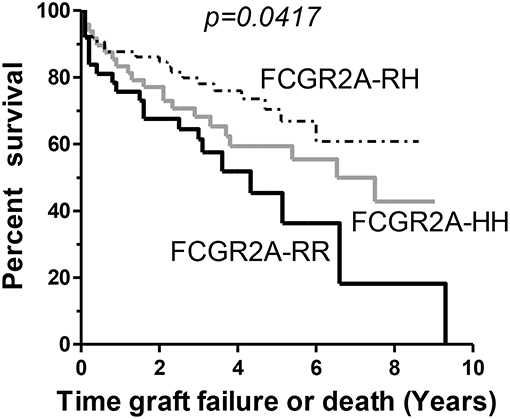
Figure 5. Kaplan-Meir survival analysis of lung allograft survival stratified according to FCGR2A [131R/H] genotypes. The FCGR2A [131R/R] homozygous genotype (solid black line, n = 39) is associated with lower survival rates in LTRs when compared to FCGR2A [131H/R] (gray line, n = 68) or the FCGR2A [131H/H] (dashed line, n = 51).
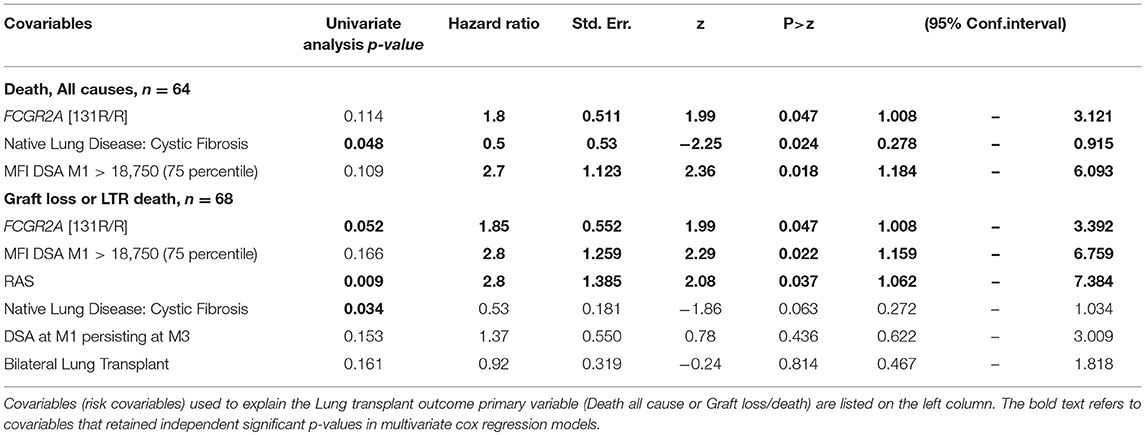
Table 6. Univariate analysis and multivariate Cox regression analysis of variables associated with graft loss and patient death post-LTx.
FcγR constitute a major marker of immune activation in response to infections, antibodies and CRP inflammatory ligands. Recent studies suggest that the engagement of FcR by the Fc fragment of alloantibodies can influence the cytotoxic activation level of NK cells toward the heart or kidney allograft. This study aimed to investigate whether the combined evaluation of FCGR3A [158F/V] and FCGR2A [131H/R] SNPs and biomarkers that index the level of CD16-dependent ADCC of NK cells might also be relevant to stratify patients at higher risk of lung allograft failure.
Our results show evidence that the FCGR3A [158V/V] genotype can be identified as a genetic marker that predisposes LTRs to acute rejection, independently of their DSA immunization status. Fifteen out of thirty patients with the FCGR3A [158V/V] genotype indeed developed acute rejection during the first trimester. Since this profile has been associated with higher NK cell responsiveness, we further investigated how it may relate to the NK cell mediated cytotoxic effects of DSA toward the lung allograft. Various studies have shown that the detection of circulating DSA with an ability to ligate the C1q component and activate the complement cascade can be associated with a greater risk of acute rejection and allograft loss after heart or kidney transplant. These complement-dependent mechanisms of DSA-mediated cytotoxic effects have also been recently documented in the lung transplant setting (19, 20). As previously reported (29), all C1q DSA detected at M3 were directed toward donor HLA-DQ alloantigens and C1q positive were shown to be significantly associated with acute rejection in the study cohort. A limitation of our study is the lack of elements allowing the distinction of cellular acute rejection (ACR) from ABMR. Endothelial deposition of C4d and microvascular inflammation were shown to be reliable markers of ABMR in renal and cardiac allografts, but the clinical relevance of C4d staining within lung allograft biopsies still remains controversial for lungs. These diagnostic criteria for ABMR were not always available on a routine basis at the time of evaluation until the Banff nomenclature released more accurate histologic features that characterize lung allograft grafts biopsies of DSA-positive LTRs.
However, the association between the FCGR3A [158V/V] genotype and presence of circulating DSA at M1 or at M3 did not appear to confer a higher risk of acute rejection, whatever their MFI and their C1q binding activity status, thus suggesting that the graft damage associated with the FCGR3A [158V/V] genotype is independent from DSA immunization status. This finding was unexpected, as we have previously shown that the intensity of antibody-dependent CD16 activation of NK cells, indexed by the non-invasive NK-CHAT, is significantly enhanced in recipients that bear the FCGR3A [158V/V] genotype and constitute a relevant cytotoxic mechanism sustaining toxic effects of DSA and allograft vasculopathy (48, 50). In contrast, NK-CHAT evaluation of DSA from LTRs actually shows evidence that the engagement of CD16 by LTx DSA is low when analyzed in reference to anti-HLA class II DSA that are found in the serum of kidney transplant recipients at time of ABMR diagnosis and that exhibit comparable HLA-DQ7 specificities and MFI values. This low potential of LTx DSA to induce CD16-mediated NK cell activation appears to be independent of their C1q binding activity and could not be associated with adverse outcome of LTx in the present study. As CD16 exhibits higher affinity for IgG1 and IgG3 alloantibodies, this failure of DSA to engage CD16 mediated cytotoxic functions may be due to the IgG2 and/or IgG4 isotypes of DSA, which have been shown to exhibit lower affinity for CD16. This finding may also indicate that the glycosylation pattern of IgG1 DSA interferes with the FcγRs-mediated recognition of the Fc fragment of IgG by immune cells or complement factors. These hypotheses are supported by the observation that only 30% of circulating DSA detected at M1 and at M3 were found to bind C1q in this study cohort and by previous findings that report high IgG4 levels in patients with cystic fibrosis lung disease (59). These data suggest that, in contrast to previous findings, the FCGR3A [158V/V] susceptibility genotype does not appear as a major mechanism of DSA-mediated lung allograft injury. CD16-dependent activation of NK cells in FCGR3A [158V/V] individuals that develop acute rejection may nevertheless be mediated by allo- or auto-antibodies that target non-HLA antigens and may not be revealed in the standardized NK-CHAT assay revealing CD16 cellular activation toward B lymphocyte cell targets. Several reports have indeed described ABMR lesions of the graft that occur in the absence of detectable levels of circulating DSA in the serum of kidney or heart transplant recipients. Antibodies directed against endothelial antigens or stress-induced antigens, such as vimentin, collagen V, Kα1 tubulin, AT1R, and MICA have also been reported in transplant recipients but the role of these non-HLA antibodies in the destruction and accelerated dysfunction of lung allograft remains poorly addressed (60). These reports have nevertheless raised interest in chronic injury resulting from humoral responses targeting non-HLA antigens, as these may be underestimated by the standard monitoring of patients' immunization status which is mainly restricted to the detection of anti-HLA alloantibodies (52). While the rate of acute rejection during the first year was previously identified as a risk factor for CLAD occurrence, occurrence of acute rejection in the first trimester could not be associated with an enhanced risk of developing chronic rejection nor with patient or graft survival in the present cohort. The F allelic variant of CD16 with low affinity for the Fc fragment of IgG was associated with the early development during the first month post-LTx of DSA that persist at M3. Persistence of DSA at M3 was thus less frequent in FCGR3A [158V/V] LTRs and was shown to be associated with lower survival times in LTR.
Our observations also identify a link between the presence of circulating DSA at M3 and the FCGR2A [131R/R] genotype, thus suggesting that FCGR2 polymorphisms may actually be associated with the persistence of harmful DSA at M3 rather than in the development of anti-HLA antibodies per se. The FCGR2A [131R/R] genotype was reported to be associated with shorter allograft survival in immunized kidney transplant recipients (KTR) (61, 62). We find that, independently of the risk associated with the FCGR2A [131R/R] genotype, detection and persistence of DSA at M3 constitute independent predictors of the adverse clinical composite outcome comprising CLAD or the patient's death. The persistence of DSA has been reported as a risk factor linked to BOS and to LTR death (16). A recent report showed that a majority of patients who were positive for de novo DSA during the first year after LTx developed BOS and were at higher risk of graft failure or death (63). In line with these reports, our study suggests that, independently of the presence of the susceptible FCGR2A [131R/R] genotype, the presence at M1 of DSA with high affinity for donor antigens (MFI DSA M1 > 18,750) can be identified as a risk factor associated with lower survival of LTRs.
The role of the complement-dependent pathogenicity of DSA is less well-documented in the lung transplant setting (19, 20). In a murine model of BOS, complement activation by antibodies to HLA class I was not required for the development of obliterative airway disease (OAD) that is similar to BOS in human LTx. Interestingly, in this study, at 90 days after LTx, only one out of 5 BOS patients had C1q DSA detected by SAFB whereas 3 out of 11 stable patients had C1q DSA. However, in our study, C1q DSA detected at M1 and M3 were mostly directed against HLA class II antigenic targets (90%) and do not appear as major contributors associated with the development of CLAD.
Although this is a limitation of the study, we cannot exclude the idea that auto-antibodies that target non-HLA antigens which were associated with the development of DSA, such as autoantibodies against K-α1 tubulin (K-α1T) and collagen V (ColV) (64), can participate in the chronic FcR-dependent reaction of recipient cells toward the lung allograft. This is supported by studies that report that development of col (V)-specific TH-17 cells may contribute to the pathogenesis of BOS (25, 59). Indeed, our standardized NK-CHAT evaluation of the DSA induced NK-cell activation was conducted toward B cell lines that express cognate HLA alloantigens. As B cells may not be relevant to evaluate the deleterious impact of non-HLA alloantibodies toward the lung allograft, this is a limit that prompts further studies that uses serum-coated lung epithelial or endothelial cells targets to evaluate NK cell ADCC.
FCGR2A [131R/H] is considered to be a heritable risk factor for a variety of infectious and inflammatory autoimmune diseases, including systemic lupus erythematosus, rheumatoid arthritis, malaria, multiple sclerosis, and anti-neutrophil cytoplasmic auto-antibody positive systemic vasculitis (65). The CD32 membrane receptor is expressed by a variety of immune cells that orchestrate the humoral immune response to pathogens, including B-lymphocytes, natural killer cells, macrophages, mast cells, and neutrophils. Its capacity to recognize IgGs bound to pathogens or infected cells has a protective effect against infections. While the FCGR2A allelic isoforms exhibit similar affinity for IgG1, the FCGR2A [131H/H] is the only FcγR variant that recognizes IgG2 subclasses, thus suggesting that the capacity to sense IgG2 antibodies may lead to impairment of pathogen surveillance in patients that lack the H allele. This SNP has been shown to play a role in the susceptibility to bacterial infections as FCGR2A [131H/H] individuals have greater potential to mediate IgG2-dependent bacterial phagocytosis than patients genotyped as FCGR2A [131R/R]. A distinct SNP (rs12746613) within the FCGR2A gene was previously associated with a higher risk of respiratory infections and mortality after LTx, but this variant was not associated with the risk of developing CLAD (34). Unlike other studies, our analysis of the present LTR cohort did not reveal any significant association between FCGR2A [131R/H] and occurrence of respiratory infections or the number of infection-related deaths. This discrepancy may relate to preventive and curative treatment of bacterial infections that differ between the transplantation centers. It may also reflect a complex interaction of these FCGR genotypes that encourages the overall survival of LTR for patients that have better capacities to thwart infections and overcome early acute rejection events. The observed finding of a inverse link between the FCGR2A [131R/R] susceptibility genotype and presence of the FCGR3A-V allele encoding the CD16 receptor variant with higher affinity for the IgG Fc fragment in LTRs, may in part explain a lack of association of the FcCR3A-VV genotype with DSA- mediated chronic lung allograft dysfunction. In this study, the presence of the FCGR2A [131H/H] was observed to be strongly associated with the presence of the “high IgG1 responder” FCGR3A [158V/V] genotype, and such linkage disequilibrium may in part explain how this intricate distribution of susceptible and protective FCR SNPs may participate in the complex tuning of the host immune response to early infectious and humoral challenges and may be associated with enhanced survival and lower rates of CLAD in patients who have the protective FCGR2A [131H/R or H/H] genotype, notably in female LTRs.
In addition to this protective role against pathogens, the SNP dependent affinity of CD32 for the Fc fragment of IgG and/or CRP ligands was also identified as promoting inflammation. CD32-dependent triggering of immune cells is in part conditioned by the polymorphism and the expression profile of this functional receptor at the surface of immune cells. As is expressed by most leukocytes/macrophages that infiltrate the lung graft, FCGR2A [131R/H] could also influence the acquisition of an inflammatory-activated profile that favors tissue recruitment of activated lymphocytes to the lung (66). Interestingly, the FCGR2A [131R/R] susceptible genotype identified in this study has been associated with higher CRP binding avidity for the CD32 receptor expressed at the surface of monocytes and neutrophils.
Considering the growing evidence of the key role of CRP as an inflammatory mediator involved in the development of atherosclerosis and endothelial dysfunction, it is expected that CRP may be more powerful in triggering the pro-inflammatory function of CD32-expressing cell subsets such as platelets, endothelial cells, monocytes, and leukocytes in FCGR2A [131R/R] LTRs.
In conclusion, these data highlight that FCGR2A and FCGR3A polymorphisms constitute predisposing factors that are associated with the outcome of lung allografts. This study suggests that the combined assessment of the FCGR genotype and CRP or IgG ligands is thus an intriguing prospect to further decipher the complex mechanisms that shape the alloimmune and inflammatory responses in response to infectious and humoral threats. As shown in other organ transplant settings, our study indicates FCGR genotyping may favor early stratification of patients at risk and may create new perspectives to adapt personalized preventive and therapeutic approaches to prevent adverse outcomes of lung transplants.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
All patients from the French cohort (COLT, Cohort in Lung Transplantation, l'Institut du Thorax, INSERM UMR1087/CNRS UMR 6291, CNIL 911142) were recruited in this study and gave their written informed consent to participate to the study in accordance with the Declaration of Helsinki. A group of 184 healthy unrelated of volunteer French bone marrow donors were also recruited to constitute a control cohort allowing analysis of FcγR genotype. Blood donations were collected in the Etablissement Francais du Sang, in accordance with BSL-2 practices. A medical interview was carried out prior to blood donation to exclude donors with medical contraindications. This study was carried out in accordance with the French Public Health Code (art L1221-1), approved by institutional ethics committee and conducted in compliance with the Good Clinical Practice Guidelines, declaration of Helsinki and Istanbul.
CP and PPa designed and coordinated the study, analyzed the data, and wrote the paper. PPe, LL, and JD performed experiments. AL contributed to the methodological and statistical analysis. MP, AB, FD-G, and JC contributed to the research design. PT and MR-G contributed to the collection of patient material and to the clinical aspects of the study.
This work was supported in part by Vaincre la mucoviscidose through TP1008 funding and the Gregory Lemarchal association.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
HLA, Human Leukocyte Antigen; LTx, Lung Transplant; LTRx, Lung Transplant Recipient; Abs, Antibodies; Ag, Antigen; DSA, Donor-Specific Antibodies; BOS, Bronchiolitis Obliterans Syndrome; CLAD, Chronic Lung Allograft Dysfunction; ADCC, Antibody dependent cell cytotoxicity; CT, Computed Tomography; RAS, Restrictive Allograft Syndrome; ABMR, Antibody Mediated Rejection; ACR, Acute Cellular Rejection; SAFB, Single-Antigen Flow Beads; D, Days; M, Month; CMV, Cytomegalovirus; MFI, Mean Fluorescence Intensity; OS, Overall Survival; DFS, Disease Free Survival; CDC, Complement-Dependent Cytotoxicity; CF, Cystic Fibrosis.
1. Chambers DC, Yusen RD, Cherikh WS, Goldfarb SB, Kucheryavaya AY, Khusch K, et al. The registry of the international society for heart and lung transplantation: thirty-fourth adult lung and heart-lung transplantation report-2017; focus theme: allograft ischemic time. J Heart Lung Transplant. (2017) 36:1047–59. doi: 10.1016/j.healun.2017.07.016
2. Thabut G, Mal H. Outcomes after lung transplantation. J Thorac Dis. (2017) 9:2684–91. doi: 10.21037/jtd.2017.07.85
3. Brugiere O, Thabut G, Krawice-Radanne I, Rizzo R, Dauriat G, Danel C, et al. Role of HLA-G as a predictive marker of low risk of chronic rejection in lung transplant recipients: a clinical prospective study. Am J Transplant. (2015) 15:461–71. doi: 10.1111/ajt.12977
4. Verleden SE, Vos R, Vanaudenaerde BM, Verleden GM. Chronic lung allograft dysfunction phenotypes and treatment. J Thorac Dis. (2017) 9:2650–9. doi: 10.21037/jtd.2017.07.81
5. Di Cristofaro J, Pelardy M, Loundou A, Basire A, Gomez C, Chiaroni J, et al. HLA-E()01:03 allele in lung transplant recipients correlates with higher chronic lung allograft dysfunction occurrence. J Immunol Res. (2016) 2016:1910852. doi: 10.1155/2016/1910852
6. Di Cristofaro J, Reynaud-Gaubert M, Carlini F, Roubertoux P, Loundou A, Basire A, et al. HLA-G*01:04 approximately UTR3 recipient correlates with lower survival and higher frequency of chronic rejection after lung transplantation. Am J Transplant. (2015) 15:2413–20. doi: 10.1111/ajt.13305
7. Gregson AL. Infectious triggers of chronic lung allograft dysfunction. Curr Infect Dis Rep. (2016) 18:21. doi: 10.1007/s11908-016-0529-6
8. Ruttens D, Vandermeulen E, Verleden SE, Bellon H, Vos R, Van Raemdonck DE, et al. Role of genetics in lung transplant complications. Ann Med. (2015) 47:106–15. doi: 10.3109/07853890.2015.1004359
9. Agbor-Enoh S, Jackson AM, Tunc I, Berry GJ, Cochrane A, Grimm D, et al. Late manifestation of alloantibody-associated injury and clinical pulmonary antibody-mediated rejection: evidence from cell-free DNA analysis. J Heart Lung Transplant. (2018) 37:925–32. doi: 10.1016/j.healun.2018.01.1305
10. Levine DJ, Glanville AR, Aboyoun C, Belperio J, Benden C, Berry GJ, et al. Antibody-mediated rejection of the lung: a consensus report of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. (2016) 35:397–406. doi: 10.1016/j.healun.2016.01.1223
11. Chin N, Paraskeva M, Paul E, Cantwell L, Levvey B, Williams T, et al. Comparative analysis of how immune sensitization is defined prior to lung transplantation. Hum Immunol. (2015) 76:711–6. doi: 10.1016/j.humimm.2015.09.025
12. Zazueta OE, Preston SE, Moniodis A, Fried S, Kim M, Townsend K, et al. The presence of pretransplant HLA antibodies does not impact the development of chronic lung allograft dysfunction or CLAD-related death. Transplantation. (2017) 101:2207–12. doi: 10.1097/TP.0000000000001494
13. Reinsmoen NL, Mirocha J, Ensor CR, Marrari M, Chaux G, Levine DJ, et al. A 3-center study reveals new insights into the impact of non-HLA antibodies on lung transplantation outcome. Transplantation. (2017) 101:1215–21. doi: 10.1097/TP.0000000000001389
14. Le Pavec J, Suberbielle C, Lamrani L, Feuillet S, Savale L, Dorfmuller P, et al. De-novo donor-specific anti-HLA antibodies 30 days after lung transplantation are associated with a worse outcome. J Heart Lung Transplant. (2016) 35:1067–77. doi: 10.1016/j.healun.2016.05.020
15. Lobo LJ, Aris RM, Schmitz J, Neuringer IP. Donor-specific antibodies are associated with antibody-mediated rejection, acute cellular rejection, bronchiolitis obliterans syndrome, and cystic fibrosis after lung transplantation. J Heart Lung Transplant. (2013) 32:70–7. doi: 10.1016/j.healun.2012.10.007
16. Roux A, Bendib Le Lan I, Holifanjaniaina S, Thomas KA, Picard C, Grenet D, et al. Characteristics of donor-specific antibodies associated with antibody-mediated rejection in lung transplantation. Front Med. (2017) 4:155. doi: 10.3389/fmed.2017.00155
17. Verleden SE, Vanaudenaerde BM, Emonds MP, Van Raemdonck DE, Neyrinck AP, Verleden GM, et al. Donor-specific and -nonspecific HLA antibodies and outcome post lung transplantation. Eur Respir J. (2017) 50:1701248. doi: 10.1183/13993003.01248-2017
18. Tikkanen JM, Singer LG, Kim SJ, Li Y, Binnie M, Chaparro C, et al. De novo DQ donor-specific antibodies are associated with chronic lung allograft dysfunction after lung transplantation. Am J Respir Crit Care Med. (2016) 194:596–606. doi: 10.1164/rccm.201509-1857OC
19. Visentin J, Chartier A, Massara L, Linares G, Guidicelli G, Blanchard E, et al. Lung intragraft donor-specific antibodies as a risk factor for graft loss. J Heart Lung Transplant. (2016) 35:1418–26. doi: 10.1016/j.healun.2016.06.010
20. Vandermeulen E, Lammertyn E, Verleden SE, Ruttens D, Bellon H, Ricciardi M, et al. Immunological diversity in phenotypes of chronic lung allograft dysfunction: a comprehensive immunohistochemical analysis. Transpl Int. (2017) 30:134–43. doi: 10.1111/tri.12882
21. Kulkarni HS, Bemiss BC, Hachem RR. Antibody-mediated rejection in lung transplantation. Curr Transplant Rep. (2015) 2:316–23. doi: 10.1007/s40472-015-0074-5
22. Roden AC, Maleszewski JJ, Yi ES, Jenkins SM, Gandhi MJ, Scott JP, et al. Reproducibility of Complement 4d deposition by immunofluorescence and immunohistochemistry in lung allograft biopsies. J Heart Lung Transplant. (2014) 33:1223–32. doi: 10.1016/j.healun.2014.06.006
23. Hachem RR, Kamoun M, Budev MM, Askar M, Ahya VN, Lee JC, et al. Human leukocyte antigens antibodies after lung transplantation: primary results of the HALT study. Am J Transplant. (2018) 18:2285–94. doi: 10.1111/ajt.14893
24. Kozlowski T, Weimer ET, Andreoni K, Schmitz J. C1q test for identification of sensitized liver recipients at risk of early acute antibody-mediated rejection. Ann Transplant. (2017) 22:518–23. doi: 10.12659/AOT.904867
25. Kauke T, Oberhauser C, Lin V, Coenen M, Fischereder M, Dick A, et al. De novo donor-specific anti-HLA antibodies after kidney transplantation are associated with impaired graft outcome independently of their C1q-binding ability. Transpl Int. (2017) 30:360–70. doi: 10.1111/tri.12887
26. Wallace WD, Li N, Andersen CB, Arrossi AV, Askar M, Berry GJ, et al. Banff study of pathologic changes in lung allograft biopsy specimens with donor-specific antibodies. J Heart Lung Transplant. (2016) 35:40–8. doi: 10.1016/j.healun.2015.08.021
27. Berry GJ, Burke MM, Andersen C, Bruneval P, Fedrigo M, Fishbein MC, et al. The 2013 International Society for Heart and Lung Transplantation Working Formulation for the standardization of nomenclature in the pathologic diagnosis of antibody-mediated rejection in heart transplantation. J Heart Lung Transplant. (2013) 32:1147–62. doi: 10.1016/j.healun.2013.08.011
28. Stewart S, Fishbein MC, Snell GI, Berry GJ, Boehler A, Burke MM, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. (2007) 26:1229–42. doi: 10.1016/j.healun.2007.10.017
29. Brugiere O, Roux A, Le Pavec J, Sroussi D, Parquin F, Pradere P, et al. Role of C1q-binding anti-HLA antibodies as a predictor of lung allograft outcome. Eur Respir J. (2018) 52:1701898. doi: 10.1183/13993003.01898-2017
30. Das LK, Ide K, Tanaka A, Morimoto H, Shimizu S, Tanimine N, et al. Fc-gamma receptor 3A polymorphism predicts the incidence of urinary tract infection in kidney-transplant recipients. Hum Immunol. (2017) 78:357–62. doi: 10.1016/j.humimm.2017.03.006
31. Shimizu S, Tanaka Y, Tazawa H, Verma S, Onoe T, Ishiyama K, et al. Fc-gamma receptor polymorphisms predispose patients to infectious complications after liver transplantation. Am J Transplant. (2016) 16:625–33. doi: 10.1111/ajt.13492
32. Alsaeed M, Husain S. Infections in heart and lung transplant recipients. Crit Care Clin. (2019) 35:75–93. doi: 10.1016/j.ccc.2018.08.010
33. Nosotti M, Tarsia P, Morlacchi LC. Infections after lung transplantation. J Thorac Dis. (2018) 10:3849–68. doi: 10.21037/jtd.2018.05.204
34. Sarmiento E, Cifrian J, Calahorra L, Bravo C, Lopez S, Laporta R, et al. Monitoring of early humoral immunity to identify lung recipients at risk for development of serious infections: a multicenter prospective study. J Heart Lung Transplant. (2018) 37:1001–12. doi: 10.1016/j.healun.2018.04.001
35. Castro-Dopico T, Clatworthy MR. Fcgamma receptors in solid organ transplantation. Curr Transplant Rep. (2016) 3:284–93. doi: 10.1007/s40472-016-0116-7
36. Ivan E, Colovai AI. Human Fc receptors: critical targets in the treatment of autoimmune diseases and transplant rejections. Hum Immunol. (2006) 67:479–91. doi: 10.1016/j.humimm.2005.12.001
37. Meletiadis J, Walsh TJ, Choi EH, Pappas PG, Ennis D, Douglas J, et al. Study of common functional genetic polymorphisms of FCGR2A, 3A and 3B genes and the risk for cryptococcosis in HIV-uninfected patients. Med Mycol. (2007) 45:513–8. doi: 10.1080/13693780701390140
38. Ruttens D, Verleden SE, Goeminne PC, Vandermeulen E, Wauters E, Cox B, et al. Genetic variation in immunoglobulin G receptor affects survival after lung transplantation. Am J Transplant. (2014) 14:1672–7. doi: 10.1111/ajt.12745
39. De Rose V, Arduino C, Cappello N, Piana R, Salmin P, Bardessono M, et al. Fcgamma receptor IIA genotype and susceptibility to P. aeruginosa infection in patients with cystic fibrosis. Eur J Hum Genet. (2005) 13:96–101. doi: 10.1038/sj.ejhg.5201285
40. Bougle A, Max A, Mongardon N, Grimaldi D, Pene F, Rousseau C, et al. Protective effects of FCGR2A polymorphism in invasive pneumococcal diseases. Chest. (2012) 142:1474–81. doi: 10.1378/chest.11-2516
41. Halloran PF, Venner JM, Madill-Thomsen KS, Einecke G, Parkes MD, Hidalgo LG, et al. Review: the transcripts associated with organ allograft rejection. Am J Transplant. (2018) 18:785–95. doi: 10.1111/ajt.14600
42. Loupy AJ, Duong Van Huyen P, Hidalgo L, Reeve J, Racape M, Aubert O, et al. Gene expression profiling for the identification and classification of antibody-mediated heart rejection. Circulation. (2017) 135:917–35. doi: 10.1161/CIRCULATIONAHA.116.022907
43. Roux A, Levine DJ, Zeevi A, Hachem R, Halloran K, Halloran PF, et al. Banff lung report: current knowledge and future research perspectives for diagnosis and treatment of pulmonary antibody-mediated rejection (AMR). Am J Transplant. (2018) 19:21–31. doi: 10.1111/ajt.14990
44. Dai HS, Griffin N, Bolyard C, Mao HC, Zhang J, Cripe TP, et al. The Fc domain of immunoglobulin is sufficient to bridge NK cells with virally infected cells. Immunity. (2017) 47, 159–170.e10. doi: 10.1016/j.immuni.2017.06.019
45. Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the Fc{gamma}RIIIa-158 V/V and V/F polymorphism. Blood. (2007) 110:2561–4. doi: 10.1182/blood-2007-01-070656
46. Hussain K, Hargreaves CE, Rowley TF, Sopp JM, Latham KV, Bhatta P, et al. Impact of human FcgammaR gene polymorphisms on IgG-triggered cytokine release: Critical importance of cell assay. Format Front Immunol. (2019) 10:390. doi: 10.3389/fimmu.2019.00390
47. Perez-Romero CA, Sanchez IP, Naranjo-Piedrahita L, Orrego-Arango JC, Muskus-Lopez CE, Rojas-Montoya W, et al. Frequency analysis of the g.7081T>G/A and g.10872T>G polymorphisms in the FCGR3A gene (CD16A) using nested PCR and their functional specific effects. Genes Immun. (2019) 20:39–45. doi: 10.1038/s41435-017-0001-0
48. Paul P, Picard C, Sampol E, Lyonnet L, Di Cristofaro J, Paul-Delvaux L, et al. Genetic and functional profiling of CD16-dependent natural killer activation identifies patients at higher risk of cardiac allograft vasculopathy. Circulation. (2018) 137:1049–59. doi: 10.1161/CIRCULATIONAHA.117.030435
49. Arnold ML, Kainz A, Hidalgo LG, Eskandary F, Kozakowski N, Wahrmann M, et al. Functional Fc gamma receptor gene polymorphisms and donor-specific antibody-triggered microcirculation inflammation. Am J Transplant. (2018) 18:2261–73. doi: 10.1111/ajt.14710
50. Legris T, Picard C, Todorova D, Lyonnet L, Laporte C, Dumoulin C, et al. Antibody-dependent NK cell activation is associated with late kidney allograft dysfunction and the complement-independent alloreactive potential of donor-specific antibodies. Front Immunol. (2016) 7:288. doi: 10.3389/fimmu.2016.00288
51. Sablik KA, Litjens NHR, Klepper M, Betjes MGH. Increased CD16 expression on NK cells is indicative of antibody-dependent cell-mediated cytotoxicity in chronic-active antibody-mediated rejection. Transpl Immunol. (2019) 54:52–8. doi: 10.1016/j.trim.2019.02.005
52. Delville M, Charreau B, Rabant M, Legendre C, Anglicheau D. Pathogenesis of non-HLA antibodies in solid organ transplantation: where do we stand? Hum Immunol. (2016) 77:1055–62. doi: 10.1016/j.humimm.2016.05.021
53. Venner JM, Hidalgo LG, Famulski KS, Chang J, Halloran PF. The molecular landscape of antibody-mediated kidney transplant rejection: evidence for NK involvement through CD16a Fc receptors. Am J Transplant. (2015) 15:1336–48. doi: 10.1111/ajt.13115
54. Yazdani S, Callemeyn J, Gazut S, Lerut E, de Loor H, Wevers M, et al. Natural killer cell infiltration is discriminative for antibody-mediated rejection and predicts outcome after kidney transplantation. Kidney Int. (2018) 95:188–98. doi: 10.1016/j.kint.2018.08.027
55. Verleden GM, Raghu G, Meyer KC, Glanville AR, Corris P. A new classification system for chronic lung allograft dysfunction. J Heart Lung Transplant. (2014) 33:127–33. doi: 10.1016/j.healun.2013.10.022
56. Kapila A, Baz MA, Valentine VG, Bhorade SM. Reliability of diagnostic criteria for bronchiolitis obliterans syndrome after lung transplantation: a survey. J Heart Lung Transplant. (2015) 34:65–74. doi: 10.1016/j.healun.2014.09.029
57. Pedron BV, Guerin-El Khourouj V, Dalle JH, Ouachee-Chardin M, Yakouben K, Corroyez F, et al. Contribution of HLA-A/B/C/DRB1/DQB1 common haplotypes to donor search outcome in unrelated hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. (2011) 17:1612–8. doi: 10.1016/j.bbmt.2011.03.009
58. Di Cristofaro J, Buhler S, Frassati C, Basire A, Galicher V, Baier C, et al. Linkage disequilibrium between HLA-G*0104 and HLA-E*0103 alleles in Tswa Pygmies. Tissue Antigens. (2011) 77:193–200. doi: 10.1111/j.1399-0039.2010.01599.x
59. Clerc A, Reynaud Q, Durupt S, Chapuis-Cellier C, Nove-Josserand R, Durieu I, et al. Elevated IgG4 serum levels in patients with cystic fibrosis. PLoS ONE. (2017) 12:e0181888. doi: 10.1371/journal.pone.0181888
60. Yousem SA, Zeevi A. The histopathology of lung allograft dysfunction associated with the development of donor-specific HLA alloantibodies. Am J Surg Pathol. (2012) 36:987–92. doi: 10.1097/PAS.0b013e31825197ae
61. Yuan FF, Watson N, Sullivan JS, Biffin S, Moses J, Geczy AF, et al. Association of Fc gamma receptor IIA polymorphisms with acute renal-allograft rejection. Transplantation. (2004) 78:766–9. doi: 10.1097/01.TP.0000132560.77496.CB
62. Arnold ML, Fuernrohr BG, Weiss KM, Harre U, Wiesener MS, Spriewald BM. Association of a coding polymorphism in Fc gamma receptor 2A and graft survival in re-transplant candidates. Hum Immunol. (2015) 76:759–64. doi: 10.1016/j.humimm.2015.09.034
63. Kauke T, Kneidinger N, Martin B, Dick A, Schneider C, Schramm R, et al. Bronchiolitis obliterans syndrome due to donor-specific HLA-antibodies. Tissue Antigens. (2015) 86:178–85. doi: 10.1111/tan.12626
64. Saini D, Weber J, Ramachandran S, Phelan D, Tiriveedhi V, Liu M, et al. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant. (2011) 30:624–31. doi: 10.1016/j.healun.2011.01.708
65. Zhang C, Wang W, Zhang H, Wei L, Guo S. Association of FCGR2A rs1801274 polymorphism with susceptibility to autoimmune diseases: a meta-analysis. Oncotarget. (2016) 7:39436–43. doi: 10.18632/oncotarget.9831
Keywords: Fc-gamma receptors, natural killer cells, lung transplantation, chronic lung allograft dysfunction, HLA antibodies, allograft rejection
Citation: Paul P, Pedini P, Lyonnet L, Di Cristofaro J, Loundou A, Pelardy M, Basire A, Dignat-George F, Chiaroni J, Thomas P, Reynaud-Gaubert M and Picard C (2019) FCGR3A and FCGR2A Genotypes Differentially Impact Allograft Rejection and Patients' Survival After Lung Transplant. Front. Immunol. 10:1208. doi: 10.3389/fimmu.2019.01208
Received: 19 February 2019; Accepted: 13 May 2019;
Published: 12 June 2019.
Edited by:
Guido Ferrari, Duke University, United StatesReviewed by:
Rajalingam Raja, University of California, San Francisco, United StatesCopyright © 2019 Paul, Pedini, Lyonnet, Di Cristofaro, Loundou, Pelardy, Basire, Dignat-George, Chiaroni, Thomas, Reynaud-Gaubert and Picard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pascale Paul, cGFzY2FsZS5wYXVsQHVuaXYtYW11LmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.