 Kelly A. Hogan
Kelly A. Hogan Claudia C. S. Chini
Claudia C. S. Chini Eduardo N. Chini
Eduardo N. Chini
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 31 May 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01187
This article is part of the Research Topic The Role of Ectonucleotidases in Acute and Chronic Inflammation View all 6 articles
CD38 (Cluster of Differentiation 38) is a multifunctional ecto-enzyme that metabolizes NAD+ and mediates nicotinamide dinucleotide (NAD+) and extracellular nucleotide homeostasis as well as intracellular calcium. CD38 is also an emerging therapeutic target under conditions in which metabolism is altered including infection, aging, and tumorigenesis. We describe multiple enzymatic activities of CD38, which may explain the breadth of biological roles observed for this enzyme. Of greatest significance is the role of CD38 as an ecto-enzyme capable of modulating extracellular NAD+ precursor availability: 1 to bacteria unable to perform de novo synthesis of NAD+; and 2 in aged parenchyma impacted by the accumulation of immune cells during the process of ‘inflammaging’. We also discuss the paradoxical role of CD38 as a modulator of intracellular NAD+, particularly in tumor immunity. Finally, we provide a summary of therapeutic approaches to CD38 inhibition and ‘NAD+ boosting’ for treatment of metabolic dysfunction observed during aging and in tumor immunity. The present review summarizes the role of CD38 in nicotinamide nucleotide homeostasis with special emphasis on the role of CD38 as an immunomodulator and druggable target.
Historically, CD38 was considered a surface marker used to identify T cells (1). Our discovery that CD38 is the main nicotinamide dinucleotide (NAD+) catabolic enzyme has shed light on the relevance of this enzyme in organismal NAD+ metabolism and has implications for several pathophysiological conditions including infection, aging, tumorigenesis (2–6). Furthermore, a more sophisticated understanding of the structure and function of CD38 has led to identification of therapeutic targets and potential treatments for conditions associated with metabolic dysfunction in infection, aging, and cancer (7–10). The role of CD38 in other organ systems, such as the central nervous system, has also been described. These studies focus on oxytocin signaling and social behavior in mice (11–14) and have been reviewed previously (15). Thus, the present review will provide an overview of CD38 enzymology; focus on recent advances in CD38-mediated age-related metabolic dysfunction and tumor immunometabolism; and summarize pharmacological approaches to CD38 inhibition.
The surface marker and multifunctional enzyme CD38 appears to provide a link between inflammation and age- and disease-related decline in tissue homeostasis and, therefore, represents a critical target for therapeutic intervention. CD38 is expressed predominately on immune cells in response to stimulation by cytokines, endotoxins, and interferon (16–18). Expression of the enzyme is regulated by a promoter region containing binding sites for NF-kB, RXR, LXR, and STAT suggesting that it plays a key role in the inflammatory response (18). CD38 expression causes a substantial decline in cellular NAD+ levels, thus altering the availability of substrates for enzymes regulating cellular homeostasis. Thus, infiltration of CD38-expressing immune cells during infection, aging, or tumorigenesis has the potential to: alter NAD+ homeostasis in parenchymal tissues or the tumor microenvironment; disrupt normal metabolic processes; and undermine tissue integrity.
The identification of CD38 as a key modulator of NAD+ metabolism in the context of cell signaling, aging, and tumor biology suggests that the enzyme is a target of promising therapeutic potential. CD38 is paradoxical in its mode of action, diverse in its locale, and functionally pleotropic, and thus presents numerous drug design challenges and opportunities. CD38 is positioned in the cellular membrane with its catalytic site facing toward the extracellular environment in a type II orientation (Figure 1) (19–21). Functionally, over 90% of CD38 acts as an ecto-NADase that catabolizes β-NAD+ (20). Given the abundance of intracellular NAD+, the presence of an extracellular catalytic site presents a topological paradox (19, 21). Our laboratory and others demonstrate that, in addition to NAD+, CD38 metabolizes extracellular NAD+ precursors (NMN and NR) prior to their transport into the cell for NAD+ biosynthesis (5). We have shown that the ecto-NMNase activity of CD38 plays a critical role in the regulation of nicotinamide nucleotides during the aging process (5). These findings suggest that the role of type II membrane-bound CD38 is to maintain NAD+ homeostasis by regulating precursors of NAD+ synthesis in the extracellular environment, thus calling into question the veracity of a topological paradox. Another outstanding question in the field is the role of intracellular forms of CD38. As discussed above, a small percent of CD38 can have its catalytic site facing the intracellular environment. Examples include the transmembrane Type III CD38 that has its c-terminal facing intracellularly; CD38 present in the nucleus and mitochondrial membrane; and a likely soluble form of CD38 present in the cytoplasm (Figure 1). In these configurations CD38 would have access to the intracellular pools of NAD+ and, without significant regulation, could lead to a severe decline in intracellular NAD+ and result in metabolic collapse (7, 19, 21, 22).
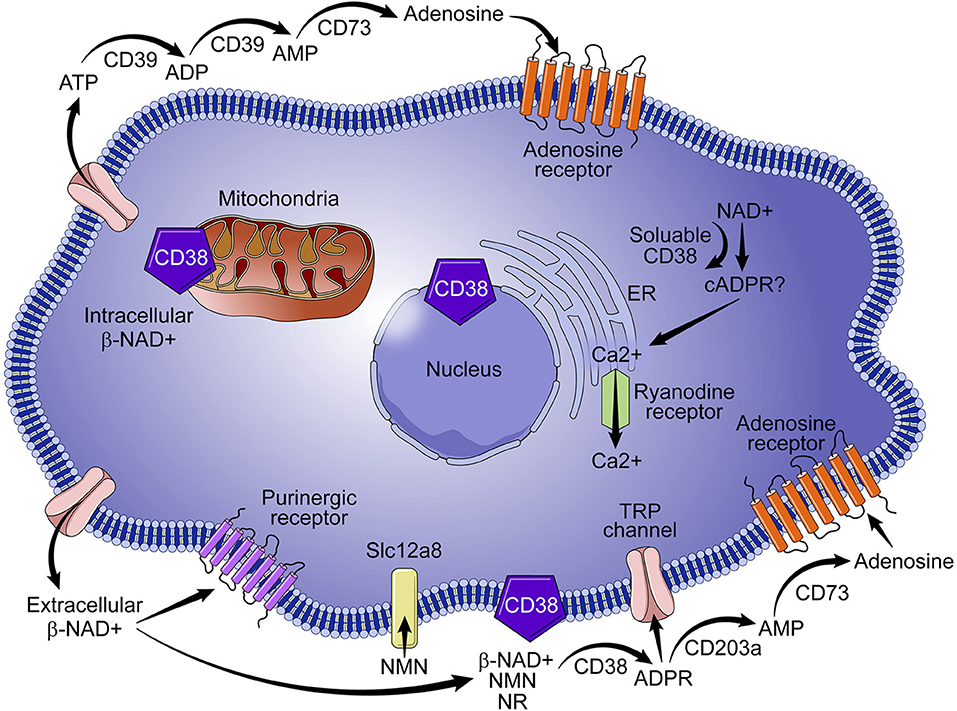
Figure 1. Role of CD38 in NAD+ metabolism. CD38 is predominantly expressed on immune cells and metabolizes nicotinamide nucleotides (NAD+ and NMN) to ADPR and cADPR, which results in the mobilization of calcium. Although intracellular CD38 is present in the cytoplasm and in the membranes of organelles, a vast majority of CD38 activity is extracellular, which results in degradation of NAD+ precursors (e. g., NMN) necessary for NAD+ synthesis. Extracellular activity of CD38 has wide ranging implications for NAD+ homeostasis in the context of infection, metabolic dysfunction, aging, and tumor biology.
The role of CD38 as a second messenger enzyme for the synthesis of cADPR is also proposed (Figure 1) (21–24). Interestingly, CD38 is a very inefficient cyclase and must degrade nearly 100 molecules of NAD+ to generate one molecule of cADPR (Figure 2) (7). It is possible that the generation of cADPR could be part of a byproduct of the NAD+ glycohydrolase activity of CD38. Furthermore, the structurally unrelated enzyme SARM1 has NAD+ glycohydrolase activity and generates a very small fraction of cADPR during its catalysis (25). This may indicate that the NADase activity of SARM also generates cADPR as a byproduct. Thus, the widely accepted notion that CD38 is mainly an ADP-ribosyl cyclase is misleading based on the fact that its primary role is to degrade nicotinamide nucleotides such as NAD+ and NMN. Therefore, we propose that the main enzymatic activity of CD38 is not an ADP-ribosyl cyclase, but rather an NAD+ glycohydrolase (Figure 2) and that both CD38 and SARM1 should be characterize based on their main enzymatic activity, namely the NAD/NMNase. In spite of these enzyme classifications, it is clear that cADPR is a second messenger that regulates intracellular calcium homeostasis in several cells (Figure 1) (24, 26, 27).
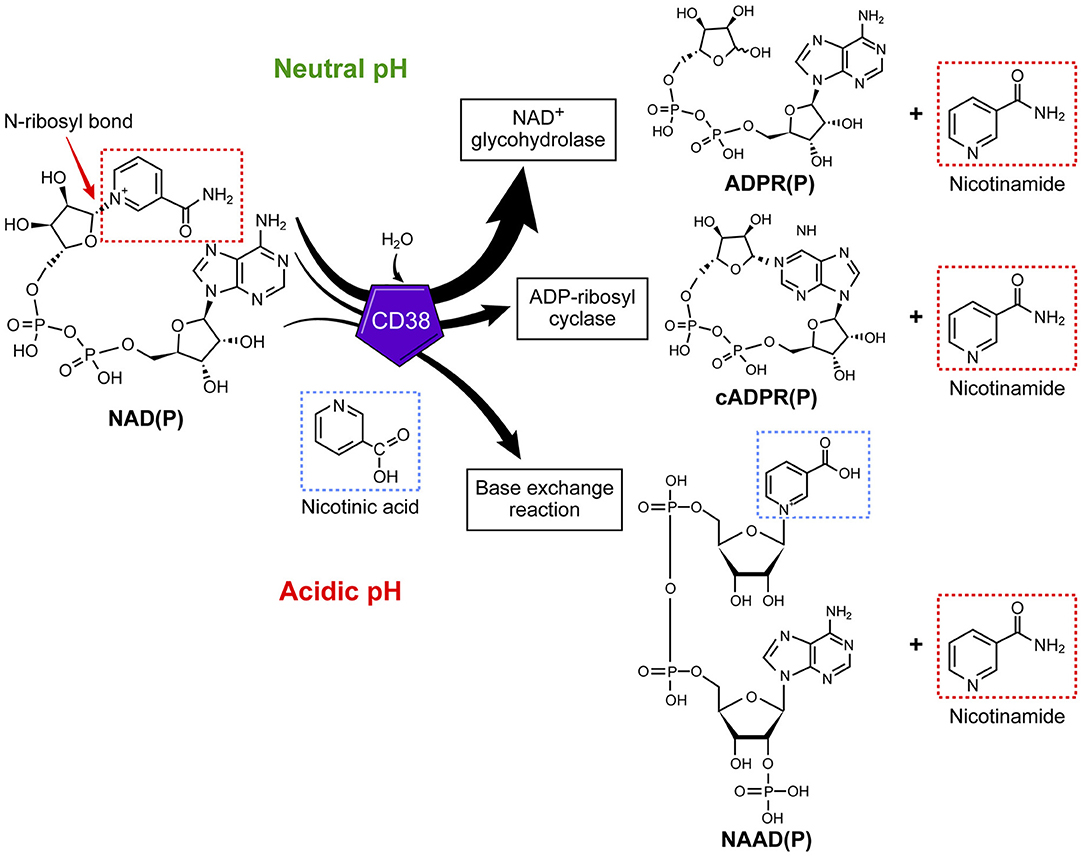
Figure 2. CD38 is primarily an NAD+ glycohydrolase, which degrades nicotinamide nucleotides. In addition to the NAD+ glycohydrolase activity, CD38 exhibits inefficient ADP-ribosyl cyclase activity and mediates a base-exchange of vitamin B3 bases in the presence of excess nicotinamide analogs.
Another role for CD38 is the regulation of extracellular adenosine, which requires consumption of NAD+. The synthesis of adenosine from NAD+ is a complementary mechanism similar to the CD39/CD73-mediated catabolism of ATP to adenosine (Figure 1) (28, 29). Adenosine is important in immune modulation because it has been implicated in immune suppression through purinergic receptor binding (Figure 1) and in the immunomodulation of multiple myeloma (28, 29) and lung cancer (30). Thus, involvement of CD38 in the regulation of NAD+ and adenosine homeostasis has led to some speculation that CD38 may function as an immune check point molecule (31, 32).
In addition to the NAD+ glycohydrolase and ecto-NMNase activity of CD38, the enzyme catalyzes the base-exchange reaction that leads to an exchange of vitamin B3 bases on a molecule (Figure 2). This reaction is optimal in the presence of excess nicotinamide analogs and at low pH; however, this reaction can also be catalyzed at physiological pH (33–35). One of the potential molecules that can be generated by this reaction is the nicotinic acid derivative of NADP (NAADP) (Figure 2) (36). Whether this reaction occurs in cells naturally expressing CD38 or in vivo is unknown; however, the synthesis of NAADP by CD38 by a base-exchange reaction in lysosomes would have implications for intracellular calcium homeostasis. Interestingly, NAADP levels in tissues and cells appear to be independent of the CD38 expression (33–36), indicating that CD38 independent NAADP synthesis exists in mammalian tissues. One of the potential candidates for the synthesis of NAADP is the newly discovered NADase SARM1.
In any case, it appears that the base-exchange reaction operates in vivo when excess nicotinamide analogs are available to tissues (Figure 2) (37). For example, we have demonstrated that CD38 is responsible for the synthesis of isoniazid derivatives of NAD+ and NADP+ in animals given toxic doses of this anti-tuberculosis medication (37). Isoniazid is a nicotinamide derivative that can serve as a substrate for the CD38-mediated base-exchange reaction (37). Thus, it is possible that the CD38 base-exchange reaction is partially responsible for the in vivo toxicity of isoniazid through the formation of toxic NAD+ intermediates during drug metabolism. It is important to highlight that, in addition to its enzymatic activity, CD38 could have enzymatic-independent roles in cell migration and homing through interaction with adhesion molecules such as CD31 (38).
An important and still outstanding question is why inflammatory cells express CD38. The inability of bacteria such as H. influenza to recycle or perform de novo synthesis of NAD+ may provide insight into the role of CD38 in response to infection (39, 40). These pathogens, including H. aegyptius, H. influenzae, H. haemolyticus, H. parainfluenzae, and H. parahaemolyticus, are responsible for a spectrum of infections, and lack the ability to synthesize NAD+ and rely instead on uptake of NAD+ and NAD+ precursor molecules (e. g., NMN, NR) to support metabolism and growth (40). In fact, NAD+ and its precursors are necessary for the growth of bacteria and have to be included in culture media as the V-factor (Figure 3) (39). Interestingly, these bacteria express nucleotide transporters to facilitate intracellular incorporation (40). The presence of CD38 ecto-nicotinamide nucleotidase in activated immune cells may reduce the availability of NAD+ to bacteria and other pathogens and could limit the development or progression of infections. This process may occur not only extracellularly but also in the intracellular space. Indeed, we have demonstrated previously that CD38+ macrophages limit the growth of some obligatory intracellular bacteria (18). We propose that CD38, as either an ecto-enzyme or as an intracellular enzyme, promotes metabolic collapse in pathogens by degrading NAD+ and its precursors.
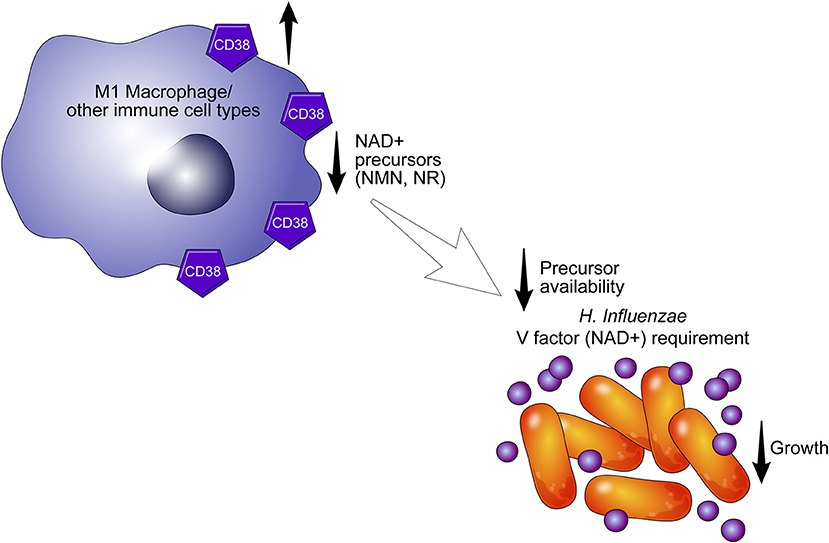
Figure 3. CD38 may alter the availability of NAD+ precursors. CD38+ cells may limit the growth of pathogens such as H. aegyptius, H. influenzae, H. haemolyticus, H. parainfluenzae, and H. parahaemolyticus by degrading extracellular NAD+ precursors required for NAD+ synthesis in bacteria. In the absence of NAD and its precursors (V factor), pathogens undergo metabolic collapse.
In addition to macrophages, which express CD38 in M1 polarization (18, 41, 42), others have reported a role for CD38 in neutrophil- and T cell-mediated immune response (43, 44). Neutrophils lacking CD38 demonstrate altered mobilization from the bone marrow and migration to sites of infection (43, 45, 46). CD38+ T cells play a myriad of roles in acute and chronic infections including the ability to be cytotoxic (47) as well as inhibit an immune response (48). What remains unknown is whether some effects are mediated by the enzymatic or non-enzymatic roles of CD38. Taken together, expression of CD38 on immune cells appears to play a role in the immune system, particularly in the context of infection.
Unlike an immune response to infection, inflammaging is a “sterile” inflammatory response which is cytokine-mediated and prompted by damage to DNA and proteins as well as a diminished capacity for cell repair in the aging organism (49, 50). In age-related decline, there is a reduction of NAD+, a master regulator of metabolism, which when reduced is a cofactor in electron transport during oxidation-reduction reactions. Furthermore, NAD+ is a critical molecule in cell signaling, intracellular calcium regulation, and chromatin remodeling (24, 26, 27, 36, 43, 51–53). NAD+ has emerged as a molecule that provides a link between signaling and metabolism. Decline of NAD+ levels is very likely a key player in the pathogenesis of several diseases including age-related conditions (Figure 4) (4–6, 9, 52, 54–61).
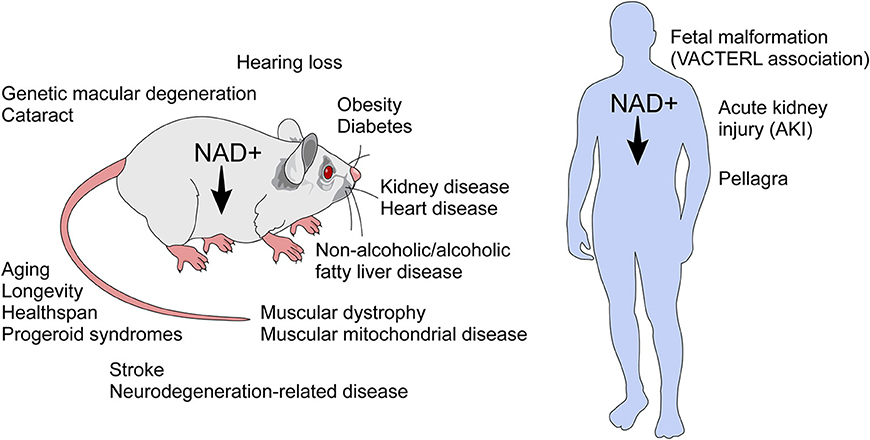
Figure 4. NAD+ decline has been implicated in several diseases and age-related conditions. NAD+ decline has been implicated in the biology of aging and in several conditions in rodents. In humans, NAD decline has been implicated in pellagra, acute kidney injury, and the fetal malformation syndrome (VACTERL association), which affects many organ systems during development.
Until recently, age-related NAD+ decline was thought to be a consequence of activation of NAD+-consuming DNA-repair enzymes such as Poly-ADP-ribosylating enzymes (PARPs) or decrease in NAD+ synthesis (9, 62). Unlike CD38, PARPs are found exclusively in the intracellular compartment, more specifically in the nuclei (9). PARPs are activated by DNA damage and catalyze the NAD-dependent polymerization of a chain of polymeric adenosine diphosphate ribose (poly (ADP-ribose) or PAR, which signals repair of DNA. PARPs lead to intranuclear and intracellular NAD+ consumption and NAD+ collapse (9). Additionally, levels of the rate-limiting NAD+ synthesis enzyme NAMPT decrease in some aging tissues (9, 63, 64) indicating a diminished capacity to generate intracellular NAD+ by salvage pathway synthesis and reliance on import of extracellular NAD+ precursors NMN and NR.
We have shown that genetic ablation of CD38 protects against age-related NAD+ decline and mitochondrial dysfunction. Furthermore, levels and activity of CD38 increase during aging (5). Recently, we proposed that NAD+ decline during aging in cells/tissues is related to exposure to factors related to the senescence associated secretory phenotype (SASP) which may increase CD38 expression in these cells/tissues. In fact, we demonstrated that SASP conditioned media from senescence cells can induce CD38 expression in both macrophages and endothelial cells (65). Thus, it is possible that the senescent phenotype drives the accumulation of CD38+ inflammatory cells, which modulate the availability of NAD+ precursors, and plays a crucial role in the metabolism of nicotinamide nucleotide (Figure 5) (65).
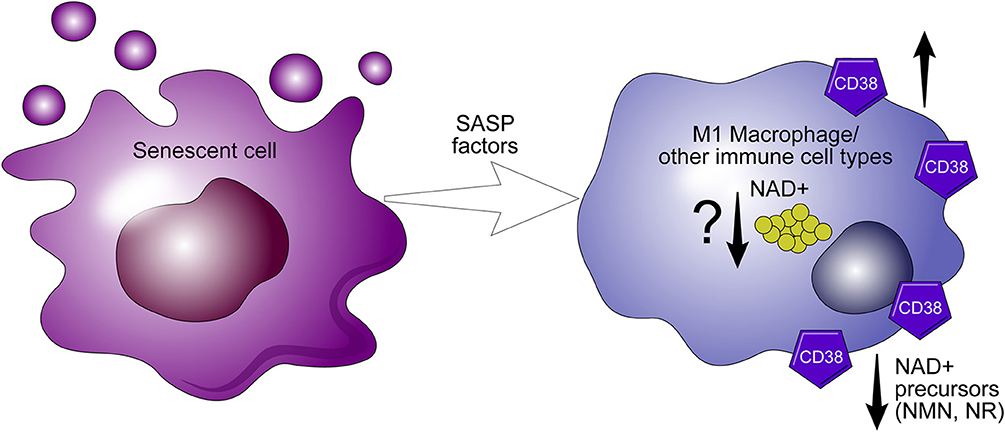
Figure 5. CD38 expression in macrophages mediate “inflammaging.” The senescence associated senescent phenotype (SASP) appears to drive the expression of CD38 in macrophages. During inflammaging, CD38+ macrophages accumulate in aging tissues and potentiate the decline of precursors necessary for NAD+ synthesis.
Age-related diseases such as cancer provide an interesting context for considering ways in which immune cells are modulated by the tumor microenvironment as well as targeted by immune-based therapies. Emerging data demonstrate that CD38 knockout mice under high metabolic pressure, such as high fat diets, are protected against the development of cancers and have increased longevity (62). Interestingly, the role of CD38 in the tumor cell provides somewhat conflicting data. For example, pancreatic and prostate cancer, which exhibit low CD38 expression and increased cellular NAD+ levels, exhibit increased tumor cell survival (66, 67). Increased CD38 activity in both adenocarcinomas, results in decreased intracellular NAD+, diminished cell growth, and increased apoptosis and cellular senescence (66–68).
On the other hand, metabolic reprogramming of NAD+ regulation via inhibition of CD38 has been proposed as a strategy for improving efficacy of immune-based therapies. CD38, in particular, appears to play a significant role in the regulation of metabolism and immunomodulation of the tumor microenvironment (1, 31, 38, 69–76).
CD38 has emerged recently as a component of mitochondrial transfer/trafficking between cells (77, 78). For many years, it has been demonstrated that cells can transfer their mitochondria to other cells, particularly cancer cells (79–84). However, components of this intriguing biological process have not been identified. In particular, it has been shown that multiple myeloma cells may take or receive mitochondria from non-malignant bone marrow stromal cells. This transfer occurs via a CD38-dependnet tumor-derived tunneling nanotubes (78). The precise enzymatic, structural, and signaling roles for CD38 in this process have not been described.
Recently, metabolic reprogramming of the CD38-NAD+ axis in CD4+ T cells has shown to be a promising approach to adoptive T cell (ACT) therapy (75). ACT therapy involves the ex vivo alteration, expansion, and transfer of tumor epitope reactive autologous T cells to a tumor bearing host. What results is a population of T cells with enhanced anti-tumor potential. The dependence of CD4+ T cells on an array of metabolites deems this population a particularly robust target for metabolic reprogramming (85). Chatterjee et al. report a strategy in which Th1 and Th17 cells are merged into hybrid Th1/17 cells, which rely on glutamine-driven oxidative phosphorylation (glutaminolysis) (75). This shift toward glutaminolysis confers greater survival of Th1/17 cells, elevated secretion of IFN-γ and IL-17 production, and enhanced anti-tumor activity (Figure 6). Curiously, Th1/17 cells demonstrate robust activity of the transcription factor Foxo1, which controls T cell memory and is responsible for the anti-tumor phenotype of the Th1/17 hybrid cell type. Induction of Foxo1 occurs by deacetylation by NAD+-dependent Sirt1 and is essential to the immune response of T cells. Chatterjee et al. demonstrate that, in addition to increased Foxo1 activity, Th1/17 exhibit higher levels of NAD+ and reduced CD38 expression (75). The significance of this finding is that increased NAD+ levels improve the anti-tumor response of Th1/17 by providing a substrate for Sirt1. Chatterjee observed a similar phenotype in CD38-deficient natural killer (NK) cells, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs) (75). What remains unknown, however, is how glutaminolysis controls production of NAD+ and whether CD38 inhibition is a function of glutamine catabolism.
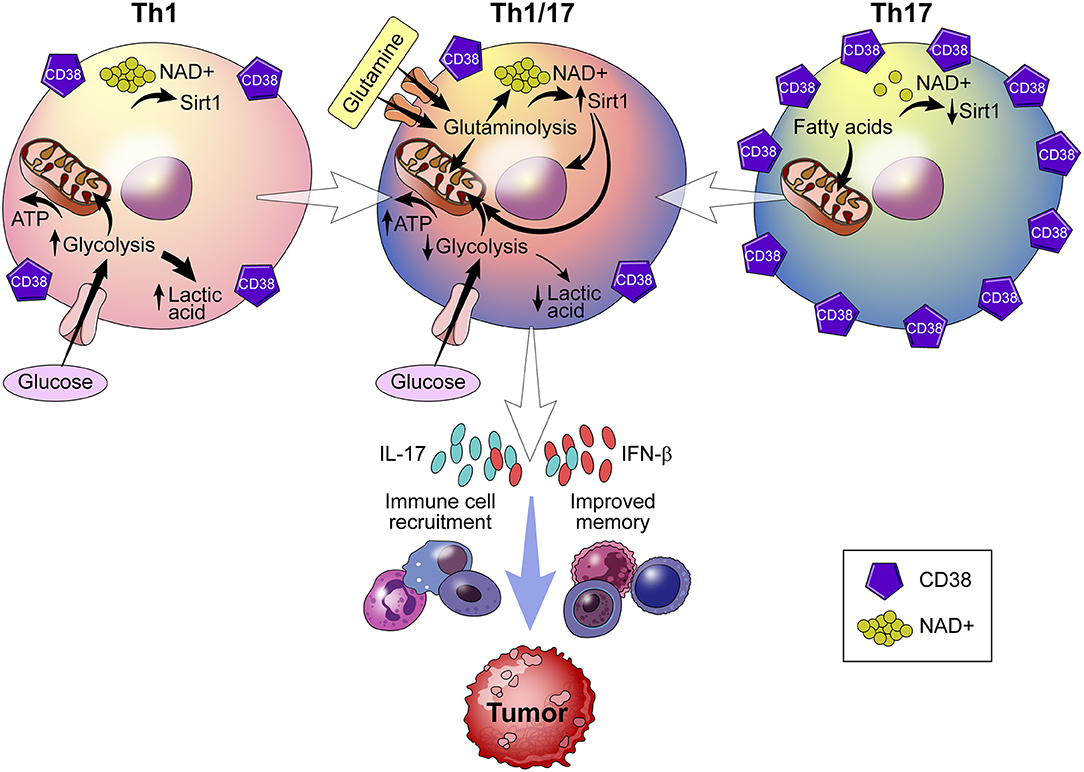
Figure 6. Role of CD38 in immunometabolism and tumor biology. Using a strategy that merges Th1 and Th17 cells into a hybrid Th1/Th17 cell, the CD38-NAD+ axis undergoes metabolic reprogramming. Reprogramming results in a shift toward glutamine-driven oxidative phosphorylation, which improves T cell survival, immune cell recruitment to the tumor, and T cell memory. Th1/Th17 hybrid cells also demonstrate reduced expression of CD38 and higher levels of NAD+, which serves as a substrate for NAD+ -dependent Sirt1 deacetylase activity that is essential to the T cell response.
Programmed death-ligand 1 (PD-L1, B7-H1, or CD274) is present on the surface of dendritic cells or macrophages, but is also frequently over-expressed in tumor cells. Tumor cells co-opt the PD-L1 in order to evade T cell surveillance (86). CD8+ cytotoxic T cells treated with checkpoint inhibitors such as PD-1/PD-L1 blocking antibodies demonstrate temporal upregulation of CD38 (87). Increased CD38 activity, in turn, results in CD8+ T cell suppression (73). This phenomenon may in part explain the high drug resistance rates observed in patients treated with PD-1/PD-L1 blockers (88). The mechanism by which CD38 mediated PD-1/PD-L1 blockade resistance likely includes perturbation of adenosine receptor signaling in tumor microenvironment leading to immune modulation (87). CD38 expression in tumors is a biomarker of poor response to checkpoint inhibitor therapy (88). One possible approach to ameliorate resistance to PD-1/PD-L1blockade is co-inhibition of CD38, which may re-establish the immune response of T cells to the tumor. Taken together, CD38 inhibition is emerging as a potential therapeutic strategy for shaping the immunometabolome of host T cells in order to enhance immune response to tumor cells.
Successful pregnancy requires the establishment of maternal immune tolerance to the fetus (89). For the uterine environment to be receptive to pregnancy, there must be activation, differentiation, and expansion of toleragenic dendritic cells (tDCs) and CD4+CD25+ Treg cells (90, 91) Evidence from a CBAxDBA/2 mouse model of pregnancy failure suggests that components of seminal fluid prime the immunological landscape of the uterine environment for implantation (92). Several years ago our group demonstrated that human seminal fluid contains high amounts of a soluble 49 kDa CD38 enzyme (93). Interestingly, the glycohydrolase activity of CD38 in the seminal fluid is significantly inhibited by the high zinc content of the seminal fluid (93). These data raise the possibility that seminal fluid CD38 could control the metabolic fate of NAD+ and adenosine and regulate sperm and fetomaternal tolerance. A study by Kim and colleagues demonstrated that in matings between CD38−/− male mice and wild type females, embryo reabsorption rates are higher, and fewer total and viable implantations sites are observed suggesting a role for CD38 in establishing pregnancy (94). Indeed, tDCs and Foxp3+ Tregs were induced by soluble seminal CD38 through a CD31- independent pathway (94). These data indicate that CD38 may play a similar role in fetomaternal tolerance in humans and further highlight the physiological role of CD38 in immunomodulation. What remains poorly understood, however, is the specific mechanism of CD38 enzymatic activity at the fetomaternal tolerance. For example, it is not known if CD38-mediated regulation of NAD+ metabolism or the accumulation of adenosine is involved in establishing tolerance between mother and fetus.
Based on the multi-faceted roles of CD38, there are extensive opportunities for design of molecules capable of inhibiting this enzyme. Below we summarize those molecules and discuss their potential as immunomodulatory therapeutics.
Anti-CD38 monoclonal antibodies are shown to be highly efficacious in the treatment of multiple myeloma (MM) and pre-clinical studies highlight the potential use in other tumors such as CLL, lung cancer, prostate cancer and melanoma (29, 70, 72–74, 76, 95–99) and in a preclinical model of melanoma (87). Currently, there are four monoclonal antibodies in clinical trials for the treatment of CD38+ malignancies (76, 95, 96). These include Daratumumab (Janssen Biotech) (31, 32, 66, 72, 74, 76, 95–97), Isatuximab (Sanofi-Aventis) (73, 76, 98), MOR202 (Morphosys) (76), and TAK079 (Takeda) (100) (Table 1), which produce antibody-dependent cell-mediated toxicity (ADCC) and have comparable binding affinities and similar safety profiles. These antibody-based therapies, however, differ in their ability to induce direct apoptosis, mount complement-mediated cytotoxicity (CDC), inhibit CD38 directly, and induce antibody-dependent cell-mediated phagocytosis (ADCP) (Table 1) (76, 96). The prevailing mechanism of action of anti-CD38 antibodies currently under investigation is hypothesized to be ADCC; however the therapeutic benefit may also involve inhibition of CD38 NADase activity and subsequent NAD+ boosting. Recently, Chatterjee et al. raised the possibility that T cell function in the tumor microenvironment can be enhanced by inhibition of CD38 and increased availability of NAD+ (75). This finding supports the tandem use of anti-CD38 antibodies and PD-1/PD-L1 checkpoint blockers to enhance the immune response to cancer, thereby providing a two-pronged approach to immunomodulation of the tumor microenvironment.
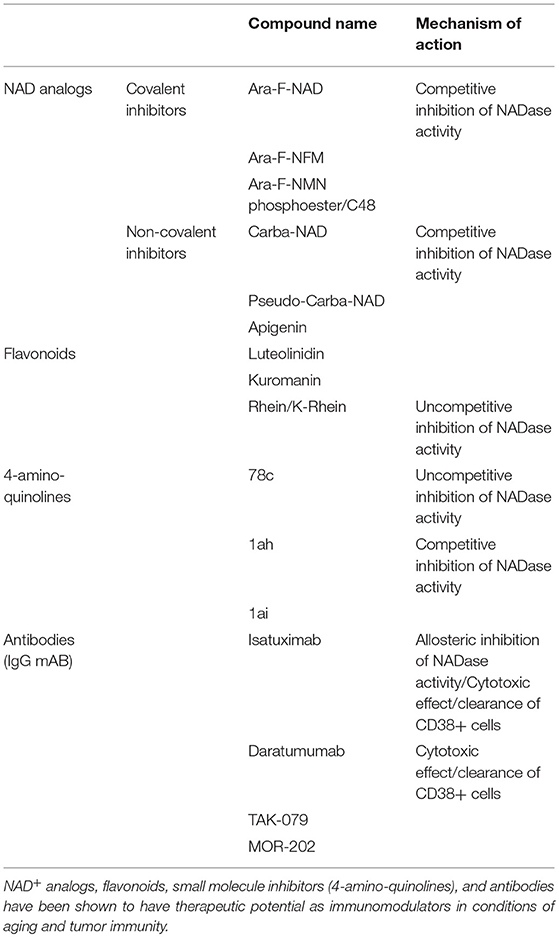
Table 1. Therapeutic approaches to CD38 inhibition and immunomodulation.
There are over 200 compounds capable of inhibiting CD38 (Table 1). These compounds are classified as NAD analogs (101), flavonoids (102), and heterocyclic compounds (103, 104) and can form either covalent or non-covalent bonds to amino acids located in the active site of CD38 (105). Elucidation of CD38 catalysis and active site crystallography has resulted in design of high affinity CD38 small molecule inhibitors (26, 103, 106). Small molecule derivatives of 4-amino-quinoline, in particular, are non-covalent and demonstrate an IC50 values in the nanomolar range (103, 104) and include three lead compounds: 78c (IC50 7.3 nM), 1ai (IC50 46 nM), and Iah (IC50 115 nM). Mutagenesis studies implicate binding site residues that are important for NADase activity (105). Changes to these residues result in loss of glycohydrolase activity and increased cyclase activity. Small molecule blockade of CD38, therefore, compromises interactions between endogenous substrates (NAD+, NMN) and the active site. CD38 small molecule inhibitors show promise as NAD+ boosting drugs in pre-clinical studies (103, 104, 106) and are likely to be important tools for understanding disease-specific roles for the multi-functional enzyme.
Several naturally occurring compounds are reported to inhibit the catalytic activity of CD38 including flavonoid compounds apigenin, quercetin, and leteolinidin (8, 102, 107) (Table 1). Most flavonoids, with the exception of 4,5-dihydroxyanthrquinone-2-carboxylic acid (RHein), are competitive antagonists of CD38, which likely lead to an increase in cellular NAD+ levels and activation of NAD-dependent enzymes such as SIRTUINs (8). Flavonoid CD38 inhibitors demonstrate a lack of toxicity in humans (8, 102) and beneficial effects in animal models of obesity, heart ischemia, kidney injury, viral infection, and cancer (8, 42, 71, 102, 107, 108). However, these compounds lack specificity and demonstrate off-target effects and a poor oral pharmacokinetic profile (8, 102).
Based on the mechanisms of CD38 catalysis and crystal structure, NAD+ analog inhibitors were developed by modifying nicotinamide ribose group within NAD+ and NMN (101, 109–111). These inhibitors include the low affinity, non-covalent inhibitor carba-NAD and ara-NAD (Table 1). Although these drugs may have potential as immunomodulators capable of increasing tumor suppressive effects (75), their utility may be limited by their inhibitory effects on NAD+ consuming enzymes.
CD38 has emerged as a major NAD+ consuming enzyme and as a regulator of NAD+ homeostasis in several conditions. The primary role of CD38 in immunity has not been completely understood. However, it is possible that CD38 may have a key role in the immune response to bacterial infections. It has also been described that the CD38-NAD+ axis controls metabolic reprogramming of T cells in the tumor microenvironment and contributes to drug resistance plaguing PD-1/PD-L1checkpoint inhibitors. As a central regulator of metabolism, maintenance of NAD+ homeostasis is taking center stage as a modifier of health and disease. These findings highlight the potential drugability of CD38 as a target in metabolic dysfunction typically observed in the contexts of aging and tumor immunity. In addition, CD38 cytotoxic antibodies has been used to treat CD38 positive tumors such as multiple myeloma and potentially CLL. Thus, CD38 has both physiological and pathological roles as well as great potential as a therapeutic target in various human diseases.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
The work in EC laboratory is supported in part by grants from the Ted Nash Long Life Foundation, the Glenn Foundation for Medical Research via the Paul F. Glenn Laboratories for Senescence at the Mayo Clinic, National Institutes of Health (NIH) grants from the National Institute of Aging (NIA, grants AG-26094, AG-58812), and the National Cancer Institute (NCI) via the Mayo Clinic pancreatic cancer SPORE program (CA 102701-14P2).
Funding from Calico Life Sciences. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
EC has a patent on CD38 inhibitors and is a consultant for Teneobio.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
NAD+, Nicotinamide adenine dinucleotide; NMN, Nicotinamide mononucleotide; NR, Nicotinamide riboside; NAADP, Nicotinic acid adenine dinucleotide phosphate; ADP, Adenosine diphosphate; ADPR, ADP-ribose; cADPR, cyclic ADP-ribose; NADP+, Nicotinamide adenine dinucleotide phosphate; SARM1, Sterile alpha and TIR Motif Containing 1; VACTERL, Vertebral defects, trachea-esophageal fistula, renal anomalies, and limb abnormalities; RXR, Retinoid X receptors; LXR, Liver X receptor; STAT, Signal transducer and activator of transcription; PARP, Poly ADP ribose polymerase; PD-1, Programmed cell death protein 1; PD-L1, Programmed death ligand 1.
1. Golden-Mason L, Curry MP, Nolan N, Traynor O, McEntee G, Kelly J, et al. Differential expression of lymphoid and myeloid markers on differentiating hematopoietic stem cells in normal and tumor-bearing adult human liver. Hepatology. (2000) 31:1251–6. doi: 10.1053/jhep.2000.7713
2. Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, et al. Regulation of SIRT 1 mediated NAD dependent deacetylation: a novel role for the multifunctional enzyme CD38. Biochem Biophys Res Commun. (2006) 349:353–9. doi: 10.1016/j.bbrc.2006.08.066
3. Aksoy P, White TA, Thompson M, Chini EN. Regulation of intracellular levels of NAD: a novel role for CD38. Biochem Biophys Res Commun. (2006) 345:1386–92. doi: 10.1016/j.bbrc.2006.05.042
4. Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD (+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. (2013) 155:1624–38. doi: 10.1016/j.cell.2013.11.037
5. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM, et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. (2016) 23:1127–39. doi: 10.1016/j.cmet.2016.05.006
6. Schultz MB, Sinclair DA. Why NAD (+) declines during aging: it's destroyed. Cell Metab. (2016) 23:965–6. doi: 10.1016/j.cmet.2016.05.022
7. Chini EN. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr Pharm Des. (2009) 15:57–63.
8. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT, et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. (2013) 62:1084–93. doi: 10.2337/db12-1139
9. Imai S, Guarente L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. (2014) 24:464–71. doi: 10.1016/j.tcb.2014.04.002
10. Verdin E. NAD (+) in aging, metabolism, and neurodegeneration. Science. (2015) 350:1208–13. doi: 10.1126/science.aac4854
11. Higashida H, Lopatina O, Yoshihara T, Pichugina YA, Soumarokov AA, Munesue T, et al. Oxytocin signal and social behaviour: comparison among adult and infant oxytocin, oxytocin receptor and CD38 gene knockout mice. J Neuroendocrinol. (2010) 22:373–9. doi: 10.1111/j.1365-2826.2010.01976.x
12. Lopatina O, Liu HX, Amina S, Hashii M, Higashida H. Oxytocin-induced elevation of ADP-ribosyl cyclase activity, cyclic ADP-ribose or Ca (2+) concentrations is involved in autoregulation of oxytocin secretion in the hypothalamus and posterior pituitary in male mice. Neuropharmacology. (2010) 58:50–5. doi: 10.1016/j.neuropharm.2009.06.012
13. Higashida H, Yokoyama S, Munesue T, Kikuchi M, Minabe Y, Lopatina O. CD38 gene knockout juvenile mice: a model of oxytocin signal defects in autism. Biol Pharm Bull. (2011). 34:1369–72.
14. Higashida H, Yokoyama S, Huang JJ, Liu L, Ma WJ, Akther S, et al. Social memory, amnesia, and autism: brain oxytocin secretion is regulated by NAD+ metabolites and single nucleotide polymorphisms of CD38. Neurochem Int. (2012) 61:828–38. doi: 10.1016/j.neuint.2012.01.030
15. Lopatina O, Inzhutova A, Salmina AB, Higashida H. The roles of oxytocin and CD38 in social or parental behaviors. Front Neurosci. (2012) 6:182. doi: 10.3389/fnins.2012.00182
16. Musso T, Deaglio S, Franco L, Calosso L, Badolato R, Garbarino G, et al. CD38 expression and functional activities are up-regulated by IFN-gamma on human monocytes and monocytic cell lines. J Leukoc Biol. (2001) 69:605–12.
17. Kang BN, Tirumurugaan KG, Deshpande DA, Amrani Y, Panettieri RA, Walseth TF, et al. Transcriptional regulation of CD38 expression by tumor necrosis factor-alpha in human airway smooth muscle cells: role of NF-kappaB and sensitivity to glucocorticoids. Faseb J. (2006) 20:1000–2. doi: 10.1096/fj.05-4585fje
18. Matalonga J, Glaria E, Bresque M, Escande C, Carbo JM, Kiefer K, et al. The nuclear receptor LXR limits bacterial infection of host macrophages through a mechanism that impacts cellular NAD Metabolism. Cell Rep. (2017) 18:1241–55. doi: 10.1016/j.celrep.2017.01.007
19. Zhao YJ, Lam CM, Lee HC. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci Signal. (2012) 5:ra67. doi: 10.1126/scisignal.2002700
20. Shrimp JH, Hu J, Dong M, Wang BS, MacDonald R, Jiang H, et al. Revealing CD38 cellular localization using a cell permeable, mechanism-based fluorescent small-molecule probe. J Am Chem Soc. (2014) 136:5656–63. doi: 10.1021/ja411046j
21. Liu J, Zhao YJ, Li WH, Hou YN, Li T, Zhao ZY, et al. Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc Natl Acad Sci USA. (2017) 114, 8283–8. doi: 10.1073/pnas.1703718114
22. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. (2008) 88:841–86. doi: 10.1152/physrev.00035.2007
23. Liu Q, Graeff R, Kriksunov IA, Jiang H, Zhang B, Oppenheimer N, et al. Structural basis for enzymatic evolution from a dedicated ADP-ribosyl cyclase to a multifunctional NAD hydrolase. J Biol Chem. (2009) 284:27637–45. doi: 10.1074/jbc.M109.031005
24. Lin WK, Bolton EL, Cortopassi WA, Wang Y, O'Brien F, Maciejewska M, et al. Synthesis of the Ca (2+)-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in beta-adrenoceptor signaling. J Biol Chem. (2017) 292:13243–57. doi: 10.1074/jbc.M117.789347
25. Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD (+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron. (2017) 93:1334–1343.e1335. doi: 10.1016/j.neuron.2017.02.022
26. Kwong AK, Chen Z, Zhang H, Leung FP, Lam CM, Ting KY, et al. Catalysis-based inhibitors of the calcium signaling function of CD38. Biochemistry. (2012) 51:555–64. doi: 10.1021/bi201509f
27. Gul R, Park DR, Shawl AI, Im SY, Nam TS, Lee SH, et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) and Cyclic ADP-Ribose (cADPR) Mediate Ca2+ signaling in cardiac hypertrophy induced by beta-adrenergic stimulation. PLoS ONE. (2016) 11:e0149125. doi: 10.1371/journal.pone.0149125
28. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology. (2013) 2:e26246. doi: 10.4161/onci.26246
29. Horenstein AL, Quarona V, Toscani D, Costa F, Chillemi A, Pistoia V, et al. Adenosine generated in the bone marrow niche through a CD38-mediated pathway correlates with progression of human myeloma. Mol Med. (2016) 22:694–704. doi: 10.2119/molmed.2016.00198
30. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget. (2017) 8:8738–51. doi: 10.18632/oncotarget.14434
31. Shallis RM, Terry CM, Lim SH. The multi-faceted potential of CD38 antibody targeting in multiple myeloma. Cancer Immunol Immunother. (2017) 66:697–703. doi: 10.1007/s00262-017-1990-2
32. Assi R, Kantarjian H, Ravandi F, Daver N. Immune therapies in acute myeloid leukemia: a focus on monoclonal antibodies and immune checkpoint inhibitors. Curr Opin Hematol. (2018) 25:136–45. doi: 10.1097/moh.0000000000000401
33. Aarhus R, Graeff RM, Dickey DM, Walseth TF, Lee HC. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem. (1995) 270:30327–33.
34. Chini EN, Beers KW, Dousa TP. Nicotinate adenine dinucleotide phosphate (NAADP) triggers a specific calcium release system in sea urchin eggs. J Biol Chem. (1995) 270:3216–23.
35. Chini EN, Dousa TP. Enzymatic synthesis and degradation of nicotinate adenine dinucleotide phosphate (NAADP), a Ca (2+)-releasing agonist, in rat tissues. Biochem Biophys Res Commun. (1995) 209:167–74. doi: 10.1006/bbrc.1995.1485
36. Soares S, Thompson M, White T, Isbell A, Yamasaki M, Prakash Y, et al. NAADP as a second messenger: neither CD38 nor base-exchange reaction are necessary for in vivo generation of NAADP in myometrial cells. Am J Physiol Cell Physiol. (2007) 292:C227–39. doi: 10.1152/ajpcell.00638.2005
37. Li F, Wang P, Liu K, Tarrago MG, Lu J, Chini EN, et al. A high dose of isoniazid disturbs endobiotic homeostasis in mouse liver. Drug Metab Dispos. (2016) 44:1742–51. doi: 10.1124/dmd.116.070920
38. Deaglio S, Aydin S, Grand MM, Vaisitti T, Bergui L, D'Arena G, et al. CD38/CD31 interactions activate genetic pathways leading to proliferation and migration in chronic lymphocytic leukemia cells. Mol Med. (2010) 16:87–91. doi: 10.2119/molmed.2009.00146
39. Cynamon MH, Sorg TB, Patapow A. Utilization and metabolism of NAD by Haemophilus parainfluenzae. J Gen Microbiol. (1988) 134:2789–99. doi: 10.1099/00221287-134-10-2789
40. Herbert M, Sauer E, Smethurst G, Kraiss A, Hilpert AK, Reidl J. Nicotinamide ribosyl uptake mutants in Haemophilus influenzae. Infect Immun. (2003) 71:5398–401.
41. Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, et al. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. (2015) 10:e0145342. doi: 10.1371/journal.pone.0145342
42. Shu B, Feng Y, Gui Y, Lu Q, Wei W, Xue X, et al. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kappaB signaling suppression. Cell Signal. (2018) 42:249–58. doi: 10.1016/j.cellsig.2017.10.014
43. Partida-Sanchez S, Cockayne DA, Monard S, Jacobson EL, Oppenheimer N, Garvy B, et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat Med. (2001) 7:1209–16. doi: 10.1038/nm1101-1209
44. Naik J, Themeli M, de Jong-Korlaar R, Ruiter RWJ, Poddighe PJ, Yuan H, et al. CD38 as a therapeutic target for adult acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Haematologica. (2019) 104:e100–3. doi: 10.3324/haematol.2018.192757
45. Estrada-Figueroa LA, Ramirez-Jimenez Y, Osorio-Trujillo C, Shibayama M, Navarro-Garcia F, Garcia-Tovar C, et al. Absence of CD38 delays arrival of neutrophils to the liver and innate immune response development during hepatic amoebiasis by Entamoeba histolytica. Parasite Immunol. (2011) 33:661–8. doi: 10.1111/j.1365-3024.2011.01333.x
46. Lischke T, Heesch K, Schumacher V, Schneider M, Haag F, Koch-Nolte F, et al. CD38 controls the innate immune response against Listeria monocytogenes. Infect Immun. (2013) 81:4091–9. doi: 10.1128/iai.00340-13
47. Burel JG, Apte SH, Groves PL, Klein K, McCarthy JS, Doolan DL. Reduced plasmodium parasite burden associates with CD38+ CD4+ T Cells Displaying Cytolytic Potential and Impaired IFN-gamma production. PLoS Pathog. (2016) 12:e1005839. doi: 10.1371/journal.ppat.1005839
48. Bahri R, Bollinger A, Bollinger T, Orinska Z, Bulfone-Paus S. Ectonucleotidase CD38 demarcates regulatory, memory-like CD8+ T cells with IFN-gamma-mediated suppressor activities. PLoS ONE. (2012) 7:e45234. doi: 10.1371/journal.pone.0045234
49. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. (2019) 18:e12950. doi: 10.1111/acel.12950
50. van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. (2019) 40:113–27. doi: 10.1016/j.it.2018.12.007
51. Chini EN, Chini CC, Kato I, Takasawa S, Okamoto H. CD38 is the major enzyme responsible for synthesis of nicotinic acid-adenine dinucleotide phosphate in mammalian tissues. Biochem J. (2002) 362 (Pt 1):125–30.
52. Mouchiroud L, Houtkooper RH, Moullan N, Katsyuba E, Ryu D, Canto C, et al. The NAD (+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell. (2013) 154:430–41. doi: 10.1016/j.cell.2013.06.016
53. Aguilar-Arnal L, Katada S, Orozco-Solis R, Sassone-Corsi P. NAD (+)-SIRT1 control of H3K4 trimethylation through circadian deacetylation of MLL1. Nat Struct Mol Biol. (2015) 22:312–8. doi: 10.1038/nsmb.2990
54. Raghuramulu N, Srikantia SG, Rao BS, Gopalan C. Nicotinamide nucleotides in the erythrocytes of patients suffering from pellagra. Biochem J. (1965) 96:837–9.
55. Williams AC, Hill LJ, Ramsden DB. Nicotinamide, NAD (P) (H), and Methyl-Group homeostasis evolved and became a determinant of ageing diseases: hypotheses and lessons from pellagra. Curr Gerontol Geriatr Res. (2012) 2012:302875. doi: 10.1155/2012/302875
56. North BJ, Rosenberg MA, Jeganathan KB, Hafner AV, Michan S, Dai J, et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. Embo J. (2014) 33:1438–53. doi: 10.15252/embj.201386907
57. Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, et al. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. (2016) 24:795–806. doi: 10.1016/j.cmet.2016.09.013
58. Chini CCS, Tarrago MG, Chini EN. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. (2017) 455:62–74. doi: 10.1016/j.mce.2016.11.003
59. Guan Y, Wang SR, Huang XZ, Xie QH, Xu YY, Shang D, et al. Nicotinamide Mononucleotide, an NAD (+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J Am Soc Nephrol. (2017) 28:2337–52. doi: 10.1681/asn.2016040385
60. Shi H, Enriquez A, Rapadas M, Martin E, Wang R, Moreau J, et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N Engl J Med. (2017) 377:544–52. doi: 10.1056/NEJMoa1616361
61. Jacobson MK, Jacobson EL. Vitamin B3 in health and disease: toward the second century of discovery. Methods Mol Biol. (2018) 1813:3–8. doi: 10.1007/978-1-4939-8588-3_1
62. Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, et al. A high-fat diet and NAD (+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. (2014) 20:840–55. doi: 10.1016/j.cmet.2014.10.005
63. Garten A, Schuster S, Penke M, Gorski T, de Giorgis T, Kiess W. Physiological and pathophysiological roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. (2015) 11:535–46. doi: 10.1038/nrendo.2015.117
64. Imai SI. The NAD World 2.0: the importance of the inter-tissue communication mediated by NAMPT/NAD (+)/SIRT1 in mammalian aging and longevity control. NPJ Syst Biol Appl. (2016) 2:16018. doi: 10.1038/npjsba.2016.18
65. Chini C, Hogan KA, Warner GM, Tarrago MG, Peclat TR, Tchkonia T, et al. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD (+) decline. Biochem Biophys Res Commun. (2019) 513:486–93. doi: 10.1016/j.bbrc.2019.03.199
66. Bu X, Kato J, Hong JA, Merino MJ, Schrump DS, Lund FE, et al. CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis. (2018) 39:242–51. doi: 10.1093/carcin/bgx137
67. Mottahedeh J, Haffner MC, Grogan TR, Hashimoto T, Crowell PD, Beltran H, et al. CD38 is methylated in prostate cancer and regulates extracellular NAD (). Cancer Metab. (2018) 6:13. doi: 10.1186/s40170-018-0186-3
68. Chini CC, Guerrico AM, Nin V, Camacho-Pereira J, Escande C, Barbosa MT, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. (2014) 20:120–30. doi: 10.1158/1078-0432.ccr-13-0150
69. Grozio A, Sociali G, Sturla L, Caffa I, Soncini D, Salis A, et al. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J Biol Chem. (2013) 288:25938–49. doi: 10.1074/jbc.M113.470435
70. Deckert J, Wetzel MC, Bartle LM, Skaletskaya A, Goldmacher VS, Vallee F, et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin Cancer Res. (2014) 20:4574–83. doi: 10.1158/1078-0432.ccr-14-0695
71. Blacher E, Ben Baruch B, Levy A, Geva N, Green KD, Garneau-Tsodikova S, et al. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int J Cancer. (2015) 136:1422–33. doi: 10.1002/ijc.29095
72. Horenstein AL, Chillemi A, Quarona V, Zito A, Roato I, Morandi F, et al. NAD (+)-metabolizing ectoenzymes in remodeling tumor-host interactions: the human myeloma model. Cells. (2015) 4:520–37. doi: 10.3390/cells4030520
73. Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. (2017) 23:4290–300. doi: 10.1158/1078-0432.ccr-16-3192
74. Krejcik J, Frerichs KA, Nijhof IS, van Kessel B, van Velzen JF, Bloem AC, et al. Monocytes and Granulocytes Reduce CD38 expression levels on myeloma cells in patients treated with daratumumab. Clin Cancer Res. (2017) 23:7498–511. doi: 10.1158/1078-0432.ccr-17-2027
75. Chatterjee S, Daenthanasanmak A, Chakraborty P, Wyatt MW, Dhar P, Selvam SP, et al. CD38-NAD (+)Axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. (2018) 27:85–100.e108. doi: 10.1016/j.cmet.2017.10.006
76. van de Donk N, Richardson PG, Malavasi F. CD38 antibodies in multiple myeloma: back to the future. Blood. (2018) 131:13–29. doi: 10.1182/blood-2017-06-740944
77. Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, et al. Transfer of mitochondria from astrocytes to neurons after stroke. Nature. (2016) 535:551–5. doi: 10.1038/nature18928
78. Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH, et al. CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res. (2019) 79:2285–97. doi: 10.1158/0008-5472.can-18-0773
79. Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH. Nanotubular highways for intercellular organelle transport. Science. (2004) 303:1007–10. doi: 10.1126/science.1093133
80. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA. (2006) 103:1283–8. doi: 10.1073/pnas.0510511103
81. Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med. (2013) 11:94. doi: 10.1186/1479-5876-11-94
82. Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. (2015) 21:81–94. doi: 10.1016/j.cmet.2014.12.003
83. Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B, et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife. (2017) 6:e22187. doi: 10.7554/eLife.22187
84. Lu J, Zheng X, Li F, Yu Y, Chen Z, Liu Z, et al. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget. (2017) 8:15539–52. doi: 10.18632/oncotarget.14695
85. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. (2015) 212:1345–60. doi: 10.1084/jem.20151159
86. Wang Y, Wang H, Yao H, Li C, Fang JY, Xu J. Regulation of PD-L1: emerging routes for targeting tumor immune evasion. Front Pharmacol. (2018) 9:536. doi: 10.3389/fphar.2018.00536
87. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. (2018) 8:1156–75. doi: 10.1158/2159-8290.cd-17-1033
88. Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. (2018) 17:129. doi: 10.1186/s12943-018-0864-3
89. Erlebacher A. Immunology of the maternal-fetal interface. Annu Rev Immunol. (2013) 31:387–411. doi: 10.1146/annurev-immunol-032712-100003
90. Aluvihare VR, Betz AG. The role of regulatory T cells in alloantigen tolerance. Immunol Rev. (2006) 212:330–43. doi: 10.1111/j.0105-2896.2006.00408.x
91. Robertson SA, Guerin LR, Moldenhauer LM, Hayball JD. Activating T regulatory cells for tolerance in early pregnancy - the contribution of seminal fluid. J Reprod Immunol. (2009) 83:109–16. doi: 10.1016/j.jri.2009.08.003
92. Clark DA, Rahmati M, Gohner C, Bensussan A, Markert UR, Chaouat G. Seminal plasma peptides may determine maternal immune response that alters success or failure of pregnancy in the abortion-prone CBAxDBA/2 model. J Reprod Immunol. (2013) 99:46–53. doi: 10.1016/j.jri.2013.03.006
93. Zielinska W, Barata H, Chini EN. Metabolism of cyclic ADP-ribose: Zinc is an endogenous modulator of the cyclase/NAD glycohydrolase ratio of a CD38-like enzyme from human seminal fluid. Life Sci. (2004) 74:1781–90.
94. Kim BJ, Choi YM, Rah SY, Park DR, Park SA, Chung YJ, et al. Seminal CD38 is a pivotal regulator for fetomaternal tolerance. Proc Natl Acad Sci USA. (2015) 112:1559–64. doi: 10.1073/pnas.1413493112
95. de Weers M, Tai YT, van der Veer MS, Bakker JM, Vink T, Jacobs DC, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. (2011) 186:1840–8. doi: 10.4049/jimmunol.1003032
96. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. (2015) 373:1207–19. doi: 10.1056/NEJMoa1506348
97. Nijhof IS, Casneuf T, van Velzen J, van Kessel B, Axel AE, Syed K, et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood. (2016) 128:959–70. doi: 10.1182/blood-2016-03-703439
98. Martin T, Baz R, Benson DM, Lendvai N, Wolf J, Munster P, et al. A phase 1b study of isatuximab plus lenalidomide and dexamethasone for relapsed/refractory multiple myeloma. Blood. (2017) 129:3294–303. doi: 10.1182/blood-2016-09-740787
99. Manna A, Aulakh S, Jani P, Ahmed S, Akhtar S, Coignet M, et al. Targeting CD38 enhances the antileukemic activity of ibrutinib in chronic lymphocytic leukemia (CLL). Clin Cancer Res. (2019). doi: 10.1158/1078-0432.ccr-18-3412. [Epub ahead of print].
100. Roepcke S, Plock N, Yuan J, Fedyk ER, Lahu G, Zhao L, et al. Pharmacokinetics and pharmacodynamics of the cytolytic anti-CD38 human monoclonal antibody TAK-079 in monkey - model assisted preparation for the first in human trial. Pharmacol Res Perspect. (2018) 6:e00402. doi: 10.1002/prp2.402
101. Slama JT, Simmons AM. Carbanicotinamide adenine dinucleotide: synthesis and enzymological properties of a carbocyclic analogue of oxidized nicotinamide adenine dinucleotide. Biochemistry. (1988) 27:183–93.
102. Kellenberger E, Kuhn I, Schuber F, Muller-Steffner H. Flavonoids as inhibitors of human CD38. Bioorg Med Chem Lett. (2011) 21:3939–42. doi: 10.1016/j.bmcl.2011.05.022
103. Becherer JD, Boros EE, Carpenter TY, Cowan DJ, Deaton DN, Haffner CD, et al. Discovery of 4-Amino-8-quinoline carboxamides as novel, submicromolar inhibitors of NAD-Hydrolyzing Enzyme CD38. J Med Chem. (2015) 58:7021–56. doi: 10.1021/acs.jmedchem.5b00992
104. Haffner CD, Becherer JD, Boros EE, Cadilla R, Carpenter T, Cowan D, et al. Discovery, synthesis, and biological evaluation of thiazoloquin (az)olin (on)es as potent CD38 inhibitors. J Med Chem. (2015) 58:3548–71. doi: 10.1021/jm502009h
105. Zhang S, Xue X, Zhang L, Zhang L, Liu Z. Comparative analysis of pharmacophore features and quantitative structure-activity relationships for CD38 covalent and non-covalent inhibitors. Chem Biol Drug Des. (2015) 86:1411–24. doi: 10.1111/cbdd.12606
106. Tarrago MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC, et al. A potent and specific CD38 inhibitor ameliorates age-related metabolic dysfunction by reversing tissue NAD (+) decline. Cell Metab. (2018) 27:1081–1095.e1010. doi: 10.1016/j.cmet.2018.03.016
107. Boslett J, Hemann C, Zhao YJ, Lee HC, Zweier JL. Luteolinidin protects the postischemic heart through CD38 inhibition with preservation of NAD (P) (H). J Pharmacol Exp Ther. (2017) 361:99–108. doi: 10.1124/jpet.116.239459
108. Schiavoni I, Scagnolari C, Horenstein AL, Leone P, Pierangeli A, Malavasi F, et al. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology. (2018) 154:122–31. doi: 10.1111/imm.12873
109. Muller-Steffner HM, Malver O, Hosie L, Oppenheimer NJ, Schuber F. Slow-binding inhibition of NAD+ glycohydrolase by arabino analogues of beta-NAD. J Biol Chem. (1992) 267:9606–11.
110. Sauve AA, Munshi C, Lee HC, Schramm VL. The reaction mechanism for CD38. A single intermediate is responsible for cyclization, hydrolysis, and base-exchange chemistries. Biochemistry. (1998) 37:13239–49. doi: 10.1021/bi981248s
Keywords: CD38, NADase, NAD+, aging, cancer, metabolism, senescence, macrophages
Citation: Hogan KA, Chini CCS and Chini EN (2019) The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 10:1187. doi: 10.3389/fimmu.2019.01187
Received: 01 April 2019; Accepted: 10 May 2019;
Published: 31 May 2019.
Edited by:
Silvia Deaglio, University of Turin, ItalyReviewed by:
Antje Garten, Leipzig University, GermanyCopyright © 2019 Hogan, Chini and Chini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eduardo N. Chini, Y2hpbmkuZWR1YXJkb0BtYXlvLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.