 Alison Wilson
Alison Wilson Margeaux Hodgson-Garms
Margeaux Hodgson-Garms Jessica E. Frith
Jessica E. Frith Paul Genever
Paul Genever
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 16 May 2019
Sec. Alloimmunity and Transplantation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01112
This article is part of the Research TopicSafety, Efficacy and Mechanisms of Action of Mesenchymal Stem Cell TherapiesView all 22 articles
Over the last decade, the acceleration in the clinical use of mesenchymal stromal cells (MSCs) has been nothing short of spectacular. Perhaps most surprising is how little we know about the “MSC product.” Although MSCs are being delivered to patients at an alarming rate, the regulatory requirements for MSC therapies (for example in terms of quality assurance and quality control) are nowhere near the expectations of traditional pharmaceuticals. That said, the standards that define a chemical compound or purified recombinant protein cannot be applied with the same stringency to a cell-based therapy. Biological processes are dynamic, adaptive and variable. Heterogeneity will always exist or emerge within even the most rigorously sorted clonal cell populations. With MSCs, perhaps more so than any other therapeutic cell, heterogeneity pervades at multiple levels, from the sample source to the single cell. The research and clinical communities collectively need to recognize and take steps to address this troublesome truth, to ensure that the promise of MSC-based therapies is fulfilled.
The term “MSCs” is used to describe a heterogeneous population of stromal cells, the exact nature and composition of which remains the subject of much debate. They are often characterized using criteria proposed by the International Society for Cell Therapy (ISCT) as plastic-adherent cells, expressing a distinct set of surface antigens and with the ability to differentiate in vitro into osteogenic, adipogenic, and chondrogenic lineages (1). This minimal definition, however, is far from definitive. MSCs exhibit unique immunomodulatory properties, support the hematopoietic niche and participate in tissue regeneration through diverse biological activities including engraftment-independent paracrine signaling. Though initially described and sourced from bone marrow we are now able to isolate MSC-like cells from a variety of tissues including adipose tissue, dental pulp, placenta, umbilical cord, and umbilical cord blood.
Although MSCs first appeared in the clinic in 1995 (2) and have since become one of the most clinically studied cell therapy platforms worldwide (3) many fundamental aspects of MSC biology remain undetermined; primarily a direct consequence of the pervasive heterogeneity that manifests itself between MSC donors, tissue sources, culture methods and individual cells within a clonal population. Furthermore, MSCs exhibit a remarkable level of plasticity over time and when presented with different microenvironments (4, 5). MSC multiplicity, and a lack of consensus in the scientific community, complicates MSC characterization and their translation into the clinic. This review will consider the multilevel origins of heterogeneity in MSCs (see Figure 1) and how we should be doing more to identify, track and quantify heterogeneity in MSCs to help determine its biological importance and impact in in vitro and in vivo contexts.
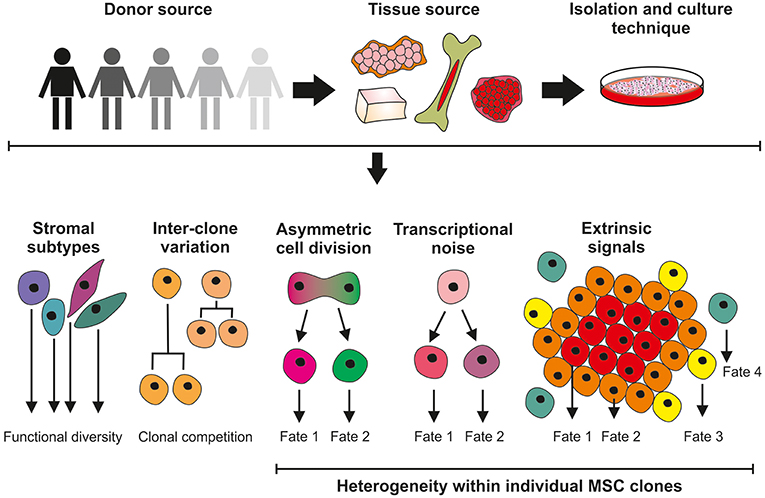
Figure 1. Sources of MSC heterogeneity; considerations for the clinical application of culture-expanded MSCs. Significant variation exists in MSC cultures isolated from different donors and different tissue sites. Unrefined and non-standardized isolation and culture techniques do not select for homogeneous cell populations and are likely to give rise to a mixture of stromal cell with different functions. Differences in the growth properties of MSC clones can result in cultures being dominated by the faster-growing lines. Further levels of heterogeneity can be introduced within MSC clones through asymmetric cell division and the effects of stochastic transcriptional noise, generating cells with modified phenotypes. MSC properties will also be determined by, for example, proximity to neighboring cells and extrinsic signaling factors.
MSC heterogeneity has certainly obscured our understanding of MSC biology and, correctly, prompted calls to re-evaluate the use of MSCs in therapy (6–10). However, the origins of heterogeneity are complex, fascinating and a constant theme in biology. It is clear from other work, particularly in microbial systems, that heterogeneity arising in genetically identical populations can have a positive impact on overall population fitness (11–14). Stochastic fluctuations in gene expression, or “noise,” can lead to phenotypic variability in clonal cell populations (11, 15) and “bet hedging” can confer survival advantages on individual cells within mixed communities when faced with environmental change (16, 17). It has been proposed that stochastic non-genetic variations (i.e., those not caused by genetic mutations) contribute to the evolution of tumors using bet hedging-like strategies (18–20) and the dynamic switching between subtly different phenotypes has been shown to influence cell fate in different adult and embryonic stem cell populations (21–23). Gene expression noise in MSCs is also likely to give rise to individual cells with different characteristics and therefore influence the aggregate function of the population. It is also clear that MSC heterogeneity is due at least in part to the existence of different subpopulations with distinct expression profiles and functional properties (24–26). It has not been determined if discrete stromal subpopulations evolve through stochastic or deterministic means, but many appear to possess properties that support general tissue maintenance [for example, immune control, vascular remodeling, hematopoiesis (25)] that are unrelated to stem cell function. Therefore, the umbrella “MSC” descriptor may actually cover a range of related but distinct cell types that are yet to be fully defined.
Cells that currently meet this broad MSC descriptor have been identified in virtually all post-natal organs and tissues (27) and while bone marrow derived MSCs (BM-MSC) are still considered the gold standard, MSCs are now frequently also isolated from adipose tissue (AT-MSCs) and umbilical cord or cord blood (UC/UCB-MSCs) (28–33). There are well-documented disparities in proliferation, differentiation potential, surface markers, transcriptional, and proteomic profile of MSCs from different sources (34–36); an overarching consensus is hard to come by. For example, prevailing MSC characteristics such as tri-lineage differentiation potential present contradictory evidence in terms of lineage preference and full tri-lineage capacity (29, 30, 32, 37). Even when derived from the same tissue of origin, MSCs demonstrate prodigious donor-to-donor variation. This may be a factor of donor health influencing MSC availability and function (38, 39). Donor age can also affect self-renewal capacity and differentiation potential, which have been reported to decline in older donors (40–43). However, differences are also apparent in healthy donors of a similar age in proliferation rate, differentiation capacity, and ultimate clinical utility (44) leading to a further addition of complexity when directly comparing samples. It is tempting to speculate that MSC heterogeneity mirrors the diversity of environments from which they may be isolated, the reality is however that our understanding of MSCs in vivo is still in its infancy (8).
The multiplicity of MSCs and the absence of a meaningful consensus on definitions and characterization parameters makes comparing studies within the field difficult and translating them into clinical practice even more so. Because heterogeneity is seldom accounted for, and unique cell populations used in individual research projects are rarely fully defined, many studies are not only difficult to reproduce but difficult to evaluate for comparability and impact within the field. Incomplete knowledge of the characteristics of MSCs in vivo and how these will relate to clinical outcomes further exacerbate the problem when considering quality control requirements for MSCs as therapeutic agents. Changes in the source materials of clinical products, e.g., a different donor, prompt regulatory authorities to require re-characterization and evidence of “comparability.” In the event that comparability could not be demonstrated, product from the original and subsequent sources would be considered to be essentially different products. Thus, during clinical development, data on early product iterations could be invalidated, and post-authorization could, in the worst-case scenario, require re-authorization. In conjunction with the need for adequate cell numbers, this represents a major challenge to the acceptance of cell-based therapies as mainstream treatments; the options of extended culture or multiple donors each imply unavoidable heterogeneity. Consequently the manufacture of MSC products using processes that rely on a continuous supply of new tissue donations run the significant risk of supply constraint, interruption, and inconsistencies (10).
A typical bone marrow aspirate contains just 0.01–0.001% MSCs (45) and trials for the regeneration of bone and cartilage tissue commonly use in the order of 10 million cells. The need for high levels of culture expansion adds to the challenge of generating an MSC population that retains the ability to differentiate effectively or secrete the appropriate biomolecules to induce a beneficial paracrine response. Banfi et al. investigated the growth kinetics and differentiation potential of MSCs, using fresh isolates from different donors through to passage five, and showed a dramatic decrease in MSC functionality over time (46). MSCs from the same donor and same source (iliac crest marrow aspirate) isolated at different timepoints over a period of 6 months also show significant variation in growth rates (44). Other studies have confirmed this loss of MSC function, demonstrating reduced proliferation, colony-forming (CFU-f) efficiency, telomere length and differentiation capacity with increasing time in culture (4, 40, 47). With the mounting interest in the use of MSCs for their paracrine effect it is also noteworthy that the secreted output of MSCs has been shown to differ with number of passages (48). This reduction in therapeutic potency at the population level can mask changes within clonal MSCs. Schellenberg et al. assessed MSC clones following expansion and observed a continual decrease in CFU-f efficiency and differentiation capacity over time (49). Earlier analyses identified a complex hierarchy of MSC clones at varying stages of potency (50), so it may be that the diminishing clonal potential observed during MSC expansion is driven by subsets of cells reaching their proliferative limit or by entering the hierarchy of different stages through which cells pass during differentiation. Subsequent studies to track individual clones from MSC explant cultures showed that clonal complexity decreased markedly over 12 passages resulting in the clonal selection of a few dominant MSC clones (51).
Given the impact that culture expansion has on MSC fate, the in vitro environment and its influence on MSC properties is worth considering. In the majority of research laboratories, MSCs are expanded as a monolayer using standard tissue culture flasks with a plasma-treated polystyrene surface and medium containing fetal bovine serum. Surprisingly, given the detrimental effects on MSC proliferation, differentiation and paracrine activity of these basic methods, the industrial expansion of MSCs for clinical applications often still retains the same basic features (52). Scale-up can be achieved through the use of multilayered cell culture flasks (cell factories) or culture vessels specifically tailored for use with closed-box and automated systems. More advanced systems use roller bottles, hollow-fiber or stirred tank bioreactors [reviewed by (53)]. A major problem with this approach is that that these in vitro conditions are very different from the in vivo MSC microenvironment, lacking much of the complexity in terms of matrix composition, geometry, mechanical properties and interactions with other cell types. All of these microenvironmental factors are interpreted by the cell and have been shown to impact upon their behavior (54–59). At its worst, the non-physiological conditions of typical cell cultures can cause mutations or cellular defects (60) but even the best-case scenario results in cells whose behavior is markedly changed. Together, this results in loss of potential from the whole population, but MSC heterogeneity may also be driven by cells responding to local changes in the microenvironment, such as through poorly controlled substrate properties or local changes in oxygen and nutrient concentration driven by the static nature of the setup (61).
It is clear that the requirement for extended in vitro expansion is a major contributor to the heterogeneity of MSC populations. A deeper understanding of the impacts of different environmental cues and the mechanisms by which they drive change, will be integral to the development of technologies for the large-scale production of quality MSC populations for clinical use.
MSC heterogeneity is multifactorial and functionally influential. Nonetheless the clinical application of MSCs does not appear to take this into account, with a selection of recent trial publications suggesting a comparatively limited assessment of cellular phenotype (Table 1). The criteria established for MSCs by the ISCT (1) are sometimes referenced in these studies but not necessarily met. It is of course possible that additional criteria were specified during manufacture but not published, however publication of more detail would increase our understanding of the MSC phenotypes in clinical use.
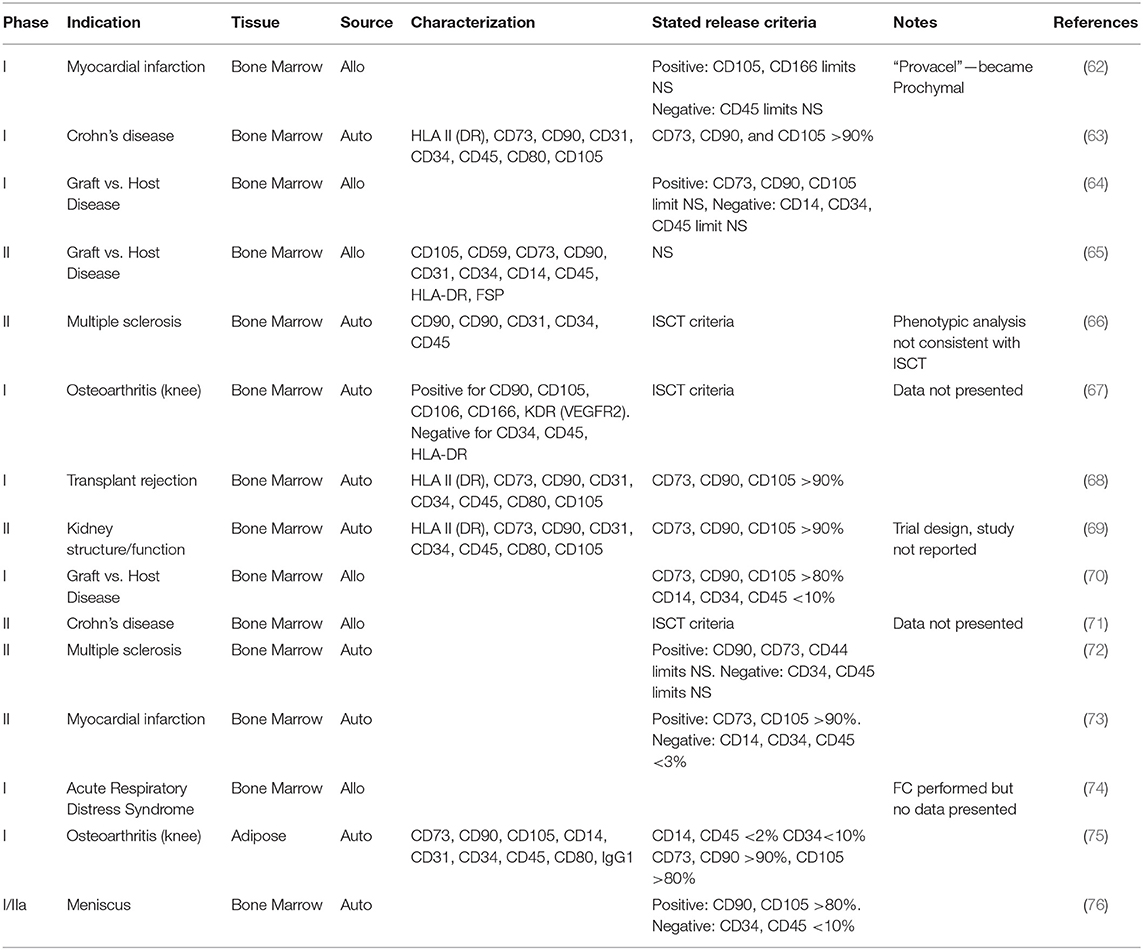
Table 1. Sample characterization and release criteria reported in clinical trials using MSCs.
Basic requirements for all biological medicines include the necessity to define the identity, the purity and the potency of the product. The developers of cell-based medicinal products must define the “active substance”; the cell type on which the therapeutic action of the product depends. Specification limits must be established for unique identification of the active substance within the product and for quantitation of its purity. Other phenotypes present, for example those arising from a tissue biopsy or culture contaminant, and non-viable cells, are generally regarded as impurities. These impurities should be reduced as far as possible and their content in the finished product limited and defined by specifications. Cellular impurities aside, major regulatory authorities do not always require cell-based medicinal products to consist of a pure population of cells. One of the first authorized cellular therapies was the immunotherapy Provenge (Dendreon Inc), approved by the US Food and Drug Administration (FDA) in 2010 for treatment of certain prostate cancers. Provenge contains autologous peripheral blood mononuclear cells (PBMC), which are cultured with PAP-GM-CSF, a fusion protein combining granulocyte-macrophage colony-stimulating factor (GM-CSF) with a prostate cancer antigen (prostatic acid phosphatase, PAP). Antigen-presenting cells within the PBMC fraction are activated by the fusion protein, providing a tumor-directed action. The exact composition of the Provenge dose varies depending on the cellular composition of each patient's leukapheresis sample, but may contain, amongst others, T and B lymphocytes and natural killer cells so the therapy is inherently heterogeneous (77, 78). In 2015 the European Union (EU) authorized its first stem cell-based product, Holoclar (Chiesi Farmaceutici SPA, Italy). Holoclar is a population of cultured autologous human corneal epithelial cells containing limbal stem cells (LSCs) intended for treatment of ocular burns. The active substance contains only approximately 3.5% of p63bright LSCs, in a mixed population with transient amplifying meroclones and paraclones and terminally differentiated corneal epithelial cells (79). The extensive heterogeneity of the overall product, which arises from the inherent cellular variation in the patient's biopsy, was justified by evidence of relevant supportive properties provided by the non-stem majority population; these were therefore not considered to be cellular impurities (80).
In 2016, the EU approved Strimvelis [Orchard Therapeutics (Netherlands) BV], a gene therapy for treatment of adenosine deaminase (ADA) severe combined immunodeficiency (ADA-SCID), in which autologous CD34+ hematopoietic stem cells (HSCs) were transduced with ADA cDNA to provide the missing gene sequence. The active substance of Strimvelis includes not only the transduced CD34+ cells, but also the non-genetically modified CD34+ fraction, based on the fact that HSC transplantation is itself a standard treatment for ADA-SCID (81) These examples provide illustrations of the general acceptability, where justified, of heterogeneous cell populations within authorized cellular therapies. In the latter two cases, the heterogeneity specifically contributes to the overall clinical effect of the product and is not merely a consequence of the manufacturing process. The complexity associated with using fundamentally variable starting materials which are then processed, inducing further heterogeneity, implies that the purity of most cell-based products will be challenging to define. The regulators' expectation of quantitation of the population being administered in terms of identity and purity (82, 83) will be difficult to achieve definitively; it is probably more reasonable to demonstrate a degree of reproducibility across product batches and to relate the composition of each batch to those used in clinical trials than to provide exact percentages of each minor cellular component (84). The identification of relevant mechanisms of action will be of crucial importance in determining the acceptability of a degree of heterogeneity, since MSC activity in a specific clinical application should help inform selection of an ideal MSC population, whether this may be a heterogeneous preparation or a specified subset.
The inevitability of MSC heterogeneity and the consequences of culture expansion for the production of cell therapies, discussed earlier, raise key questions for developers of regenerative medicines. Whilst, as illustrated above, there is no obligation to demonstrate that a product contains only the specific cell type of interest, the challenges of definition and identification are accentuated when considering MSCs. The apparent absence of major concerns around cellular heterogeneity in whole organ and HSC transplantation is sometimes highlighted as support for a less rigorous approach to the characterization and control of cell-based therapies. However, acceptance of heterogeneity in these situations may be due in part to the fact that organ and HSC transplants are procedures which are considered to fall within the practice of medicine rather than items externally regulated as medicinal products.
In order to advance the clinical utility of MSCs, it is essential that strategies to quantify heterogeneity are agreed. As a starting point, it is important to define the biological properties of the different stromal cell types within a mixed population. It is likely that stem-cell and non-stem-cell fractions are co-extracted using current protocols for MSC isolation. For regenerative therapies, it would seem logical that the stem-cell component is the essential active ingredient, however non-differentiating stromal cells could play important supporting roles, for example in immune control; precisely why we need a full biological understanding that relates to mechanism of action. This can be achieved by exploiting techniques suitable for phenotyping individual cells, including flow cytometry, electrophysiology, microscopy (in various forms), image/morphometric analysis, lineage tracing, and powerful new single cell-omic technologies. Effective strategies will be required to ensure data are integrated, interpreted correctly and shared. The key to clinical translation will be to develop the most appropriate non-destructive biomarker identification techniques that provide functional discrimination. Reliable subtype-specific biomarkers will also support the development of treatments to target MSCs in situ, potentially negating the need for culture expansion. Alongside these, improved methods for MSC expansion that retain, or even promote selection of the desired MSC properties will be essential for the production of MSC products with a more defined set of characteristics and high therapeutic efficacy. Such technologies will likely incorporate biophysical as well as biochemical cues and provide platforms for scale-up culture in bioreactors. With the role of the paracrine effect of MSCs coming to the fore (85), therapies based on the MSC secretome or MSC-derived extracellular vesicles (EVs) may emerge to complement the MSC therapeutic toolkit. However, different MSC populations (or cells within that population) are still likely to produce different secretomes and so many of the fundamental challenges relating to MSC heterogeneity will remain.
Given the challenges associated with providing consistency in an MSC product from multiple tissue isolates, the generation of MSCs from pluripotent stem cell populations has garnered interest (86–92). The expansion capability of pluripotent cells means that a single clonal population can potentially be manufactured and subsequently differentiated into a virtually limitless supply of MSCs. This type of platform relieves the need for continuous tissue donations, simplifies the subject of donor-donor variation and bypasses many of the sources of MSC heterogeneity that arise when working with ex vivo cells. Induced pluripotent stem cells (iPSC)-derived MSCs offer the potential for large-scale production of more homogenous, off-the-shelf products with limited batch-to-batch variation that could deliver more consistent clinical outcomes. The first phase I clinical trial using iPSC-derived MSCs was completed in 2018 with promising results from Cynata Therapeutics's lead Cymerus™CYP-001 product for the treatment of graft vs. host disease (93), although the full findings have not yet been published. While the clinical use of iPSC-MSCs holds promise, an effective comparison of pluripotent cell-derived MSCs to their adult tissue counterparts is required, with appropriate safety profiling. Clonal immortalized MSC lines (both iPSC-derived and genetically modified adult MSCs) may also be developed for bulk harvesting of secreted products, proteins, and EV cargoes, which could ultimately dispense with the need for the transplantation of MSCs as a whole-cell product, however the issue of stochastic heterogeneity arising in clonal cell populations will always persist.
MSCs can offer widespread therapeutic benefits but we must balance enthusiastic demands for clinical progress against the need for better mechanistic understanding. Unraveling MSC multiplicity is the essential first step in that process.
AW, MH-G, JF, and PG wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
This work was supported by the Tissue Engineering and Regenerative Therapies Centre Versus Arthritis (21156).
JF and MH-G acknowledge funding from Cynata Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. (2006) 8:315–17. doi: 10.1080/14653240600855905
2. Lazarus HM, Haynesworth SE, Gerson SL, Rosenthal NS, Caplan AI. Ex vivo expansion and subsequent infusion of human bone marrow-derived stromal progenitor cells (mesenchymal progenitor cells): implications for therapeutic use. Bone Marrow Transplant. (1995) 16:557–64.
3. Fung M, Yuan Y, Atkins H, Shi Q, Bubela T. Responsible translation of stem cell research: an assessment of clinical trial registration and publications. Stem Cell Rep. (2017) 8:1190–201. doi: 10.1016/j.stemcr.2017.03.013
4. Baxter MA, Wynn RF, Jowitt SN, Wraith JE, Fairbairn LJ, Bellantuono I. Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells. (2004) 22:675–82. doi: 10.1634/stemcells.22-5-675
5. Digirolamo CM, Stokes D, Colter D, Phinney DG, Class R, Prockop DJ. Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br J Haematol. (1999) 107:275–81. doi: 10.1046/j.1365-2141.1999.01715.x
6. Bianco P, Cao X, Frenette PS, Mao JJ, Robey PG, Simmons PJ, et al. The meaning, the sense and the significance: translating the science of mesenchymal stem cells into medicine. Nat Med. (2013) 19:35–42. doi: 10.1038/nm.3028
7. Phinney DG. Functional heterogeneity of mesenchymal stem cells: implications for cell therapy. J Cell Biochem. (2012) 113:2806–12. doi: 10.1002/jcb.24166
8. McLeod CM, Mauck RL. On the origin and impact of mesenchymal stem cell heterogeneity: new insights and emerging tools for single cell analysis. Eur Cell Mater. (2017) 34:217–31. doi: 10.22203/eCM.v034a14
9. Pevsner-Fischer M, Levin S, Zipori D. The origins of mesenchymal stromal cell heterogeneity. Stem Cell Rev. (2011) 7:560–8. doi: 10.1007/s12015-011-9229-7
10. Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. (2018) 22:824–33. doi: 10.1016/j.stem.2018.05.004
11. Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. (2002) 297:1183–6. doi: 10.1126/science.1070919
12. Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial persistence as a phenotypic switch. Science. (2004) 305:1622–5. doi: 10.1126/science.1099390
13. Levy SF. Cellular heterogeneity: benefits besides bet-hedging. Curr Biol. (2016) 26:R355–7. doi: 10.1016/j.cub.2016.03.034
14. Altschuler SJ, Wu LF. Cellular heterogeneity: do differences make a difference? Cell. (2010) 141:559–63. doi: 10.1016/j.cell.2010.04.033
15. Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. (2005) 307:1965–9. doi: 10.1126/science.1109090
16. Veening J-W, Smits WK, Kuipers OP. Bistability, epigenetics, and bet-hedging in bacteria. Annu Rev Microbiol. (2008) 62:193–210. doi: 10.1146/annurev.micro.62.081307.163002
17. de Jong IG, Haccou P, Kuipers OP. Bet hedging or not? A guide to proper classification of microbial survival strategies. Bioessays. (2011) 33:215–23. doi: 10.1002/bies.201000127
18. Brock A, Chang H, Huang S. Non-genetic heterogeneity–a mutation-independent driving force for the somatic evolution of tumours. Nat Rev Genet. (2009) 10:336–42. doi: 10.1038/nrg2556
19. Gravenmier CA, Siddique M, Gatenby RA. Adaptation to stochastic temporal variations in intratumoral blood flow: the Warburg effect as a bet hedging strategy. Bull Math Biol. (2018) 80:954–70. doi: 10.1007/s11538-017-0261-x
20. Jolly MK, Kulkarni P, Weninger K, Orban J, Levine H. Phenotypic plasticity, bet-hedging, and androgen independence in prostate cancer: role of non-genetic heterogeneity. Front Oncol. (2018) 8:50. doi: 10.3389/fonc.2018.00050
21. Chang HH, Hemberg M, Barahona M, Ingber DE, Huang S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature. (2008) 453:544–7. doi: 10.1038/nature06965
22. Enver T, Pera M, Peterson C, Andrews PW. Stem cell states, fates, and the rules of attraction. Cell Stem Cell. (2009) 4:387–97. doi: 10.1016/j.stem.2009.04.011
23. Krieger T, Simons BD. Dynamic stem cell heterogeneity. Development. (2015) 142:1396–406. doi: 10.1242/dev.101063
24. Phinney DG, Hill K, Michelson C, DuTreil M, Hughes C, Humphries S, et al. Biological activities encoded by the murine mesenchymal stem cell transcriptome provide a basis for their developmental potential and broad therapeutic efficacy. Stem Cells. (2006) 24:186–98. doi: 10.1634/stemcells.2004-0236
25. Phinney DG. Biochemical heterogeneity of mesenchymal stem cell populations: clues to their therapeutic efficacy. Cell Cycle. (2007) 6:2884–9. doi: 10.4161/cc.6.23.5095
26. James S, Fox J, Afsari F, Lee J, Clough S, Knight C, et al. Multiparameter analysis of human bone marrow stromal cells identifies distinct immunomodulatory and differentiation-competent subtypes. Stem Cell Rep. (2015) 4:1004–15. doi: 10.1016/j.stemcr.2015.05.005
27. Da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. (2006) 119:2204–13. doi: 10.1242/jcs.02932
28. Bieback K, Kern S, Klüter H, Eichler H. Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells. (2004) 22:625–34. doi: 10.1634/stemcells.22-4-625
29. Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. (2000) 109:235–42. doi: 10.1046/j.1365-2141.2000.01986.x
30. Goodwin HS, Bicknese AR, Chien SN, Bogucki BD, Quinn CO, Wall DA. Multilineage differentiation activity by cells isolated from umbilical cord blood: expression of bone, fat, and neural markers. Biol Blood Marrow Transplant. (2001) 7:581–8. doi: 10.1053/bbmt.2001.v7.pm11760145
31. Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA. (2000) 97:13625–30. doi: 10.1073/pnas.240309797
32. Kögler G, Sensken S, Airey JA, Trapp T, Müschen M, Feldhahn N, et al. A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med. (2004) 200:123–35. doi: 10.1084/jem.20040440
33. Zuk PA, Zhu M, Mizuno H, Huang J, Futrell JW, Katz AJ, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. (2001) 7:211–28. doi: 10.1089/107632701300062859
34. Kern S, Eichler H, Stoeve J, Klüter H, Bieback K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells. (2006) 24:1294–301. doi: 10.1634/stemcells.2005-0342
35. Mattar P, Bieback K. Comparing the immunomodulatory properties of bone marrow, adipose tissue, and birth-associated tissue mesenchymal stromal cells. Front Immunol. (2015) 6:560. doi: 10.3389/fimmu.2015.00560
36. Strioga M, Viswanathan S, Darinskas A, Slaby O, Michalek J. Same or not the same? Comparison of adipose tissue-derived versus bone marrow-derived mesenchymal stem and stromal cells. Stem Cells Dev. (2012) 21:2724–52. doi: 10.1089/scd.2011.0722
37. Lee OK, Kuo TK, Chen W-M, Lee K-D, Hsieh S-L, Chen T-H. Isolation of multipotent mesenchymal stem cells from umbilical cord blood. Blood. (2004) 103:1669–75. doi: 10.1182/blood-2003-05-1670
38. Kuznetsov SA, Mankani MH, Bianco P, Robey PG. Enumeration of the colony-forming units-fibroblast from mouse and human bone marrow in normal and pathological conditions. Stem Cell Res. (2009) 2:83–94. doi: 10.1016/j.scr.2008.07.007
39. Wang J, Liao L, Wang S, Tan J. Cell therapy with autologous mesenchymal stem cells-how the disease process impacts clinical considerations. Cytotherapy. (2013) 15:893–904. doi: 10.1016/j.jcyt.2013.01.218
40. Yang Y-HK, Ogando CR, Wang See C, Chang T-Y, Barabino GA. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res Ther. (2018) 9:131. doi: 10.1186/s13287-018-0876-3
41. D'Ippolito G, Schiller PC, Ricordi C, Roos BA, Howard GA. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res. (1999) 14:1115–22. doi: 10.1359/jbmr.1999.14.7.1115
42. Katsara O, Mahaira LG, Iliopoulou EG, Moustaki A, Antsaklis A, Loutradis D, et al. Effects of donor age, gender, and in vitro cellular aging on the phenotypic, functional, and molecular characteristics of mouse bone marrow-derived mesenchymal stem cells. Stem Cells Dev. (2011) 20:1549–61. doi: 10.1089/scd.2010.0280
43. Stenderup K, Justesen J, Clausen C, Kassem M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone. (2003) 33:919–26. doi: 10.1016/j.bone.2003.07.005
44. Phinney DG, Kopen G, Righter W, Webster S, Tremain N, Prockop DJ. Donor variation in the growth properties and osteogenic potential of human marrow stromal cells. J Cell Biochem. (1999) 75:424–36.
45. Roobrouck VD, Ulloa-Montoya F, Verfaillie CM. Self-renewal and differentiation capacity of young and aged stem cells. Exp Cell Res. (2008) 314:1937–44. doi: 10.1016/j.yexcr.2008.03.006
46. Banfi A, Muraglia A, Dozin B, Mastrogiacomo M, Cancedda R, Quarto R. Proliferation kinetics and differentiation potential of ex vivo expanded human bone marrow stromal cells: implications for their use in cell therapy. Exp Hematol. (2000) 28:707–15. doi: 10.1016/S0301-472X(00)00160-0
47. Reiser J, Zhang X-Y, Hemenway CS, Mondal D, Pradhan L, La Russa VF. Potential of mesenchymal stem cells in gene therapy approaches for inherited and acquired diseases. Expert Opin Biol Ther. (2005) 5:1571–84. doi: 10.1517/14712598.5.12.1571
48. Russell AL, Lefavor R, Durand N, Glover L, Zubair AC. Modifiers of mesenchymal stem cell quantity and quality. Transfusion. (2018) 58:1434–40. doi: 10.1111/trf.14597
49. Schellenberg A, Stiehl T, Horn P, Joussen S, Pallua N, Ho AD, et al. Population dynamics of mesenchymal stromal cells during culture expansion. Cytotherapy. (2012) 14:401–11. doi: 10.3109/14653249.2011.640669
50. Russell KC, Phinney DG, Lacey MR, Barrilleaux BL, Meyertholen KE, O'Connor KC. In vitro high-capacity assay to quantify the clonal heterogeneity in trilineage potential of mesenchymal stem cells reveals a complex hierarchy of lineage commitment. Stem Cells. (2010) 28:788–98. doi: 10.1002/stem.312
51. Selich A, Daudert J, Hass R, Philipp F, von Kaisenberg C, Paul G, et al. Massive clonal selection and transiently contributing clones during expansion of mesenchymal stem cell cultures revealed by lentiviral RGB-barcode technology. Stem Cells Transl Med. (2016) 5:591–601. doi: 10.5966/sctm.2015-0176
52. Lechanteur C, Briquet A, Giet O, Delloye O, Baudoux E, Beguin Y. Clinical-scale expansion of mesenchymal stromal cells: a large banking experience. J Transl Med. (2016) 14:145. doi: 10.1186/s12967-016-0892-y
53. Mizukami A, Swiech K. Mesenchymal stromal cells: from discovery to manufacturing and commercialization. Stem Cells Int. (2018) 2018:4083921. doi: 10.1155/2018/4083921
54. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. (2006) 126:677–89. doi: 10.1016/j.cell.2006.06.044
55. Frith JE, Thomson B, Genever PG. Dynamic three-dimensional culture methods enhance mesenchymal stem cell properties and increase therapeutic potential. Tissue Eng Part C Methods. (2010) 16:735–49. doi: 10.1089/ten.tec.2009.0432
56. Frith JE, Mills RJ, Cooper-White JJ. Lateral spacing of adhesion peptides influences human mesenchymal stem cell behaviour. J Cell Sci. (2012) 125:317–27. doi: 10.1242/jcs.087916
57. Kusuma GD, Brennecke SP, O'Connor AJ, Kalionis B, Heath DE. Decellularized extracellular matrices produced from immortal cell lines derived from different parts of the placenta support primary mesenchymal stem cell expansion. PLoS ONE. (2017) 12:e0171488. doi: 10.1371/journal.pone.0171488
58. Etheridge L, Mason RA, Saleh F, Genever P. Cell-cell signaling pathways that regulate mesenchymal stromal cell differentiation. In: Atkinson K, editor. The Biology and Therapeutic Application of Mesenchymal Cells. Hoboken, NJ: John Wiley & Sons, Inc. (2016) p. 91–103.
59. Saleh FA, Genever PG. Turning round: multipotent stromal cells, a three-dimensional revolution? Cytotherapy. (2011) 13:903–12. doi: 10.3109/14653249.2011.586998
60. Wright WE, Shay JW. Historical claims and current interpretations of replicative aging. Nat Biotechnol. (2002) 20:682–8. doi: 10.1038/nbt0702-682
61. Rodrigues CAV, Fernandes TG, Diogo MM, da Silva CL, Cabral JMS. Stem cell cultivation in bioreactors. Biotechnol Adv. (2011) 29:815–29. doi: 10.1016/j.biotechadv.2011.06.009
62. Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. (2009) 54:22777–86. doi: 10.1016/j.jacc.2009.06.055
63. Duijvestein M, Vos ACW, Roelofs H, Wildenberg ME, Wendrich BB, Verspaget HW, et al. Autologous bone marrow-derived mesenchymal stromal cell treatment for refractory luminal Crohn's disease: results of a phase I study. Gut. (2010) 59:16622–69. doi: 10.1136/gut.2010.215152
64. Herrmann R, Sturm M, Shaw K, Purtill D, Cooney J, Wright M, et al. Mesenchymal stromal cell therapy for steroid-refractory acute and chronic graft versus host disease: a phase 1 study. Int J Hematol. (2012) 95:182–8. doi: 10.1007/s1s2185-011-09899-2
65. Kuzmina LA, Petinati NA, Parovichnikova EN, Lubimova LS, Gribanova EO, Gaponova TV, et al. Multipotent mesenchymal stromal cells for the prophylaxis of acute graft-versus-host disease-a phase II study. Stem Cells Int. (2012) 2012:968213. doi: 10.1155/2012/968213
66. Bonab MM, Sahraian MA, Aghsaie A, Karvigh SA, Hosseinian SM, Nikbin B, et al. Autologous mesenchymal stem cell therapy in progressive multiple sclerosis: an open label study. Curr Stem Cell Res Ther. (2012) 7:407–14. doi: 10.2174/157488812804484648
67. Orozco L, Munar A, Soler R, Alberca M, Soler F, Huguet M, et al. Treatment of knee osteoarthritis with autologous mesenchymal stem cells: a pilot study. Transplantation. (2013) 95:15355–41. doi: 10.1097/TP.0b00b0b13e33e3e18291a11a2ad22da
68. Reinders MEJ, de Fijter JW, Roelofs H, Bajema IM, de Vries DK, Schaapherder AF, et al. Autologous bone marrow-derived mesenchymal stromal cells for the treatment of allograft rejection after renal transplantation: results of a phase I study. Stem Cells Transl Med. (2013) 2:107–11. doi: 10.5966/sctm.20122-0114
69. Reinders MEJ, Bank JR, Dreyer GJ, Roelofs H, Heidt S, Roelen DL, et al. Autologous bone marrow derived mesenchymal stromal cell therapy in combination with everolimus to preserve renal structure and function in renal transplant recipients. J Transl Med. (2014) 12:331. doi: 10.1186/s1s2967-014-0331-x
70. Introna M, Lucchini G, Dander E, Galimberti S, Rovelli A, Balduzzi A, et al. Treatment of graft versus host disease with mesenchymal stromal cells: a phase I study on 40 adult and pediatric patients. Biol Blood Marrow Transplant. (2014) 20:375–81. doi: 10.1016/j.bbmt.2013.11.033
71. Forbes GM, Sturm MJ, Leong RW, Sparrow MP, Segarajasingam D, Cummins AG, et al. A phase 2 study of allogeneic mesenchymal stromal cells for luminal Crohn's disease refractory to biologic therapy. Clin Gastroenterol Hepatol. (2014) 12:64–71. doi: 10.1016/j.cgh.2013.06.021
72. Llufriu S, Sepúlveda M, Blanco Y, Marín P, Moreno B, Berenguer J, et al. Randomized placebo-controlled phase II trial of autologous mesenchymal stem cells in multiple sclerosis. PLoS ONE. (2014) 9:e1e13936. doi: 10.1371/journal.pone.0113936
73. Lee J-W, Lee S-H, Youn Y-J, Ahn M-S, Kim J-Y, Yoo B-S, et al. A randomized, open-label, multicenter trial for the safety and efficacy of adult mesenchymal stem cells after acute myocardial infarction. J Korean Med Sci. (2014) 29:23–31. doi: 10.3346/jkms.2014.29.1.23
74. Wilson JG, Liu KD, Zhuo H, Caballero L, McMillan M, Fang X, et al. Mesenchymal stem (stromal) cells for treatment of ARDS: a phase 1 clinical trial. Lancet Respir Med. (2015) 3:24–32. doi: 10.1016/S2S213-2600(14)702911-7
75. Pers Y-M, Rackwitz L, Ferreira R, Pullig O, Delfour C, Barry F, et al. Adipose mesenchymal stromal cell-based therapy for severe osteoarthritis of the knee: a phase I dose-escalation trial. Stem Cells Transl Med. (2016) 5:847–56. doi: 10.5966/sctm.20155-0245
76. Whitehouse MR, Howells NR, Parry MC, Austin E, Kafienah W, Brady K, et al. Repair of torn avascular meniscal cartilage using undifferentiated autologous mesenchymal stem cells: from in vitro optimization to a first-in-human study. Stem Cells Transl Med. (2017) 6:12377–48. doi: 10.1002/sctm.16-0199
77. Highlights of Prescribing Information. Provenge. Available online at: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/provenge-sipuleucel-t (accessed December 12, 2018).
78. Thara E, Dorff TB, Pinski JK, Quinn DI. Vaccine therapy with sipuleucel-T (Provenge) for prostate cancer. Maturitas. (2011) 69:296–303. doi: 10.1016/j.maturitas.2011.04.012
79. Holoclar European Public Assessment Report. Ex vivo Expanded Autologous Human Corneal Epithelial Cells Containing Stem Cells. Available online at: https://www.ema.europa.eu/documents/product-information/holoclar-epar-product-information_en.pdf (accessed December 12, 2018).
80. Flory E, Gasparini P, Jekerle V, Palomäki T, Salmikangas P. Regulatory viewpoints on the development of advanced stem cell-based medicinal products in light of the first EU-approved stem cell product. Cell Gene Ther Insights. (2015) 1:109–27. doi: 10.18609/cgti.2015.010
81. Strimvelis. Common Name - Autologous CD34+ Enriched Cell Fraction That Contains CD34+ Cells Transduced With Retroviral Vector That Encodes for the Human ADA cDNA Sequence. Available online at: https://www.ema.europa.eu/documents/assessment-report/strimvelis-epar-public-assessment-report_en.pdf (accessed December 14, 2018).
82. Guidance for FDA Reviewers and Sponsors: Content and Review of Chemistry Manufacturing and Control (CMC) Information for Human Somatic Cell Therapy Investigational New Drug Applications (INDs). Available online at: https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/xenotransplantation/ucm092705.pdf (accessed December 14, 2018).
83. European Medicines Agency. Guideline on Human Cell-Based Medicinal Products (EMEA/CHMP/410869/2006) (2008).
84. Carmen J, Burger SR, McCaman M, Rowley JA. Developing assays to address identity, potency, purity and safety: cell characterization in cell therapy process development. Regen Med. (2012) 7:85–100. doi: 10.2217/rme.11.105
85. Kusuma GD, Carthew J, Lim R, Frith JE. Effect of the microenvironment on mesenchymal stem cell paracrine signaling: opportunities to engineer the therapeutic effect. Stem Cells Dev. (2017) 26:617–31. doi: 10.1089/scd.2016.0349
86. Arpornmaeklong P, Brown SE, Wang Z, Krebsbach PH. Phenotypic characterization, osteoblastic differentiation, and bone regeneration capacity of human embryonic stem cell-derived mesenchymal stem cells. Stem Cells Dev. (2009) 18:955–68. doi: 10.1089/scd.2008.0310
87. Boyd NL, Robbins KR, Dhara SK, West FD, Stice SL. Human embryonic stem cell-derived mesoderm-like epithelium transitions to mesenchymal progenitor cells. Tissue Eng Part A. (2009) 15:1897–907. doi: 10.1089/ten.tea.2008.0351
88. Brown PT, Squire MW, Li W-J. Characterization and evaluation of mesenchymal stem cells derived from human embryonic stem cells and bone marrow. Cell Tissue Res. (2014) 358:149–64. doi: 10.1007/s00441-014-1926-5
89. De Peppo GM, Marcos-Campos I, Kahler DJ, Alsalman D, Shang L, Vunjak-Novakovic G, et al. Engineering bone tissue substitutes from human induced pluripotent stem cells. Proc Natl Acad Sci USA. (2013) 110:8680–5. doi: 10.1073/pnas.1301190110
90. Hynes K, Menicanin D, Mrozik K, Gronthos S, Bartold PM. Generation of functional mesenchymal stem cells from different induced pluripotent stem cell lines. Stem Cells Dev. (2014) 23:1084–96. doi: 10.1089/scd.2013.0111
91. Lian Q, Zhang Y, Zhang J, Zhang HK, Wu X, Zhang Y, et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. (2010) 121:1113–23. doi: 10.1161/CIRCULATIONAHA.109.898312
92. Vodyanik MA, Yu J, Zhang X, Tian S, Stewart R, Thomson JA, et al. A mesoderm-derived precursor for mesenchymal stem and endothelial cells. Cell Stem Cell. (2010) 7:718–29. doi: 10.1016/j.stem.2010.11.011
Keywords: mesenchymal stromal cell, heterogeneity, cell subpopulations, cell-based therapy, single cell technologies
Citation: Wilson A, Hodgson-Garms M, Frith JE and Genever P (2019) Multiplicity of Mesenchymal Stromal Cells: Finding the Right Route to Therapy. Front. Immunol. 10:1112. doi: 10.3389/fimmu.2019.01112
Received: 26 February 2019; Accepted: 01 May 2019;
Published: 16 May 2019.
Edited by:
Guido Moll, Charité Medical University of Berlin, GermanyReviewed by:
Evren Alici, Karolinska Institute (KI), SwedenCopyright © 2019 Wilson, Hodgson-Garms, Frith and Genever. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jessica E. Frith, amVzc2ljYS5mcml0aEBtb25hc2guZWR1
Paul Genever, cGF1bC5nZW5ldmVyQHlvcmsuYWMudWs=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.