 Fei Qian
Fei Qian Liuqiang Zhang
Liuqiang Zhang Shaodong Lu2
Shaodong Lu2 Ping Liu
Ping Liu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 16 May 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01103
Background: Mast cells are considered an attractive therapeutic target for treating allergic diseases, and the Lyn–FcεRIβ interaction is essential for mast cell activation. This study investigated the antiallergic effect of scrodentoid A (SA) on mast cells and mast cell–mediated anaphylaxis.
Methods: For in vitro experiments, mast cells were treated with SA. Cell proliferation was tested using the XTT assay. The mRNA expression of various cytokines and chemokines was measured using qPCR. The levels of histamine, eicosanoids (PGD2, LTC4), and cytokines were measured using enzyme immunoassay kits. Signaling was investigated using Western blotting and immunoprecipitation. For in vivo experiments, the antiallergic activity of SA was evaluated using two mouse models of passive anaphylaxis as passive cutaneous and systemic anaphylaxis. The mechanism was investigated through immunohistochemistry and immunofluorescence.
Results: SA considerably inhibited immunoglobulin (Ig) E-mediated mast cell activation, including β-hexosaminidase release, mRNA and protein expression of various cytokines, and PGD2 and LTC4 release. Oral administration of SA effectively and dose-dependently suppressed mast cell–mediated passive cutaneous and systemic anaphylaxis. SA significantly attenuated the activation of Lyn, Syk, LAT, PLCγ, JNK, Erk1/2, and Ca2+ mobilization without Fyn, Akt, and P38 activation by blocking the Lyn–FcεRIβ interaction.
Conclusions: SA suppresses mast cell–mediated allergic response by blocking the Lyn–FcεRIβ interaction in vitro and in vivo. SA may be a promising therapeutic agent for allergic and other mast cell–related diseases.
Allergy prevalence has been increasing worldwide, which has contributed directly or indirectly to health and economic burdens (1, 2). Mast cells are central players in both the development and maintenance of allergic diseases. Mast cell activation releases various mediators, including preformed granule-associated chemical mediators, lipid mediators, and de novo synthesized cytokines, which are essential in allergies (3). Immunoglobulin (Ig) E and the high-affinity receptor FcεRI are vital in mast cell activation in an allergy context (4). FcεRI, present on mast cell surface, is a tetrameric complex comprising an IgE-binding α, signal-modulating β, and two signal-transducing γ subunits. The signaling cascades elicited by FcεRI aggregation start with Lyn phosphorylation, which transphosphylate the immunoreceptor tyrosine-based activation motif (ITAM) within FcεRIβ and FcεRIγ. Subsequently, another tyrosine kinase, Syk, is recruited and binds to phosphorylated ITAM, resulting in the phosphorylation of the adaptor proteins (LAT) and phospholipase Cγ (PLCγ). The principal axis pathway is then initated, activating the downstream signaling pathways, including the mitogen-activated protein kinase (MAPK), protein kinase C (PKC), and calcium flux pathways (5–7).
Binding of FcεRIα to the allergen–IgE complex initiates mast cell activation. FcεRIβ accelerates FcεRIα maturation in mast cells, thereby increasing FcεRI receptor expression on the cell membrane (8). FcεRIβ also amplifies the cell activation signal by enhancing the FcεRIγ signal by five to seven times, accelerating mast cell activation (9). Lyn is critical for ITAM phosphorylation on FcεRIβ (10), and a weak Lyn–FcεRIβ interaction is noted before FcεRI aggregation (11–13). Lyn next binds to the phosphorylated Y219 site of ITAM within FcεRIβ, and the interaction between Lyn and FcεRI increases considerably (14, 15). The Lyn–FcεRIβ interaction is essential for human mast cell activation (16). Thus, we hypothesized that blocking the Lyn–FcεRIβ interaction may be a new direction for allergic disease treatment.
In our previous studies, five 19(4 → 3)-abeo-abietane diterpenoids (scrodentoids A-E) were firstly isolated from the whole plant of Scrophularia dentata. Among them, Scrodentoid B (SB) was considered as a potential immunosuppressive agent (17), and the other biological activities of these compounds have not been reported. Recently, it was suggested that some diterpenoid compounds have antiallergic activity, particularly in mast cell–mediated allergies (18, 19). In our further anti-allergic screening, only Scrodentoid A (SA) could modify IgE/Ag-stimulated mast cell activation and mast cell mediated anaphylaxis in vitro. However, the content of SA in S. dentata is very low, and its mechanism of action as an anti-allergic compound remains unknown.
This is the first study to describe the chemical conversion from SB to SA and the inhibitory effects of SA on mast cell activation, evidenced by suppressed degranulation, lipid mediator and cytokine production, and reduced downstream signaling pathway activation. In agreement with this, SA considerably inhibits mast cell–mediated anaphylactic response by blocking the Lyn–FcεRIβ interaction. Therefore, SA might serve as a novel therapeutic target for allergic diseases.
Whole plants of Scrophularia dentata were collected from Tibet, China in October 2015. The plant was identified by Professor Zhili Zhao of School of Pharmacy, Shanghai University of Traditional Chinese Medicine. A voucher sample (No. CX2015) was deposited with the Herbarium of the Department of TCM Chemistry, School of Pharmacy of Shanghai University of Traditional Chinese Medicine (Shanghai, China).
ESIMS was obtained on a Bruker Daltonics Esquire 3000 plus (Bruker Daltonics, Bremen, Germany). The NMR spectra were recorded on a Bruker AM-400 spectrometer at 400 MHz for 1H and 100 MHz for 13C in CDCl3 Bruker AVANCE-III instrument operating at 600 MHz for 1H and 150 MHz for 13C. Silica gel (200 mesh to 300 mesh, Qingdao Haiyang Chemical Co., Ltd., Qingdao, China), C18 reversed-phase silica gel (150 to 200 mesh, Fuji Silysia Chemical, Ltd., Aichi, Japan) were used for column chromatography (CC). Semipreparative High-performance liquid chromatography (HPLC) was performed on an Angilent 1200 HPLC apparatus with an Eclipse XDB-C18 column (250 × 9.4 mm, 5 μm). All solvents used for CC were of analytical grade (Sinopharm Chemical Reagent Co., Ltd), and all solvents used for HPLC were of HPLC grade.
Mouse anti-dinitrophenyl (DNP) IgE, cell proliferation Kit II, glycine, 4-Nitrophenyl N-acetyl-β-D-glucosaminide (p-NAG), BSA, Evans Blue, Citric acid monohydrate, DMSO, o-phthaldialdehyde, and Ketotifen were purchased from Sigma-Aldrich (ST. Louis, MO, USA). The antibodies specific for phospho (p)-Syk (Tyr525/526), Syk, p-PLCγ (Tyr783), p-AMPK (Thr172), p-AMPK (Ser485/491), AMPK, p-P38 (Thr180/Tyr182), P38, p-JNK (Thr183/Tyr185), JNK, p-Erk1/2 (Thr202/Tyr204), Erk1/2, p-cPLA2 (Ser505), p-Akt (Ser473), Akt, Phospho-Tyrosine (P-Tyr-1000), Normal rabbit IgG were from Cell Signaling Technology (Danvers, MA, USA). The antibodies specific for p-Lyn (Tyr 507), Lyn, p-Fyn (Y530), Fyn, p-LAT (Y191), LAT were from Abcam China. The antibodies specific for PLCγ, FcεRIβ, FcεRIγ, GAPDH, Goat anti-rabbit-IgG HRP, Goat anti-mouse-IgG HRP were from Santa Cruz Biotechnology (Dallas, Texas, USA). Fluo-4 NW calcium assay kit, Fetal Bovine Serum (FBS), mouse IL-6 ELISA kit, mouse TNF alpha ELISA kit, Trizol Reagent, and the High-Capacity cDNA Reverse Transcription Kit were from Thermo Fisher. DNP-HSA was from Biosearch technology. Recombinatant Mouse IL-3 and recombinatant mouse SCF/c-kit Ligand were from RD systems China. LTC4, PGD2 ELISA Kit were from Cayman Chemicals (Ann Arbor, MI, USA). ECL Immobilon Western Cheniluminescent HRP Substrate was from Millipore (Millipore. Billerica, USA).
Extracts from the dried whole plant of S. dentata (8 kg) were obtained with 95% EtOH (3×, each 80 L), using a reflux apparatus for 1.5 h. The extract (1,080 g) was obtained after in vacuo removal of the solvent. The extract was suspended in water and sequentially extracted using CH2Cl2, The CH2Cl2 extract was evaporated to dryness in vacuo, and the resultant CH2Cl2 fraction (243.6 g) was subjected to silica gel column chromatography (CC), eluted with EtOAc in petroleumether (PE) (0–100%, stepwise) to yield 12 fractions (Fr. 1–Fr. 12). Fr. 5 was separated repeatedly using CC and was further separated through reversed-phase HPLC by using CH3CN-H2O (83:17) to yield SA (17) (1, 21 mg). Fr. 7 was separated repeatedly using CC and was further separated through ODS CC with an MeOH gradient (80–100%) in H2O to yield SB (17) (2, 367 mg).
Trifluoroacetic acid (0.3 mL) was added into a solution of SB (300 mg, 1.007 mmol) in CH2Cl2 (20 mL), then the mixture was stirred at 25°C for 12 h. The reaction mixture was poured into ice water, adjusted to pH = 7 with NaHCO3 (a.q.), then the residue was extracted with CH2Cl2 (40 mL×3), dried over Na2SO4, and concentrated to give the product of compound 3 (238 mg, yield: 80%). 1H NMR (CDCl3, 400 MHz) δ 2.28 (1H, dt, J = 13.1/3.1 Hz, H-1a), 1.75 (1H, td, J = 13.1/4.0 Hz, H-1b), 1.87 (1H, dq, J = 13.0/3.1 Hz, H-2a), 1.40 (1H, qd, J = 13.0/4.0 Hz, H-2b), 2.06 (1H, m, H-3), 2.71 (1H, m, H-5), 2.64 (1H, m, H-6a), 2.76 (1H,m, H-6b), 6.96 (1H, s, H-11), 7.93 (1H, s, H-14), 5.15 (1H, s, H-16a), 5.44 (1H, s, H-16b), 2.13 (3H, s, H-17), 4.93 (1H, s, H-18a), 4.70 (1H, s, H-18b), 1.12 (3H, d, J = 6.5 Hz, H-19), 1.07 (3H, s, H-20). ESIMS m/z 295 [M-H]−.
To a solution of compound 3 (200 mg) in ethyl acetate (40 mL), was added Pd/C (50 mg) and the mixture was stirred under H2 for 0.5 h. The precipitate was removed by filtration through Celite, and the filtrate was evaporated under reduced pressure. The residue was chromatographed on a silica gel (PE:EtOAc = 8:1) to give SA (200 mg) as a white amorphous powder in quantitative yield. 1H NMR (CDCl3, 400 MHz) δ 2.20 (1H, dt, J = 13.2/3.0 Hz, H-1a), 1.72 (1H, td, J = 13.2/4.0 Hz, H-1b), 1.84 (1H, dq, J = 13.0/3.0 Hz, H-2a), 1.37 (1H, qd, J = 13.0/4.0 Hz, H-2b), 2.05 (1H, m, H-3), 2.66 (1H, m, H-5), 2.60 (1H, m, H-6a), 2.71 (1H,m, H-6b), 6.77 (1H, s, H-11), 7.96 (1H, s, H-14), 3.15 (1H, seq, J = 6.8 Hz, H-15), 1.24 (3H, d, J = 6.8 Hz, H-16), 1.25 (3H, d, J = 6.8 Hz, H-17), 4.89 (1H, s, H-18a), 4.67 (1H, s, H-18b), 1.09 (3H, d, J = 6.5 Hz, H-19), 1.02 (3H, s, H-20). ESIMS m/z 297 [M-H]−.
BMMCs were derived from the bone marrow of Balb/C mice. Cells were maintained in RPMI-1640, 50 μmol/L 2-mercaptoethanol, 1 mmol/L sodium pyruvate, 100 U/mL penicillin−100 μg/mL streptomycin, 0.1 mmol/L non-essential amino acids, 25 mmol/L HEPES, 10% FBS, and 10 ng/mL mouse recombinant IL-3 and SCF. Cells were fed every 7 days. After 4 weeks, cell purity was determined by measuring CD117 and FcεRI expression through flow cytometry: 95% of cells were positive for both CD117 and FcεRI.
BMMCs were dispensed into 96-well plates (5 × 104 cells/well), and the cells were treated with SA (1, 10, 25, and 50 μmol/L) for 24 h. Cell viability was evaluated using a XTT-based cell proliferation assay kit, according to the manufacturer's protocol. Cell viability was expressed as the percentage ODtest/ODcontrol, where ODtest and ODcontrol indicate the optical densities of cells exposed to the test and control compounds, respectively.
BMMCs were first incubated at 5 × 105 cells/mL with 0.5 μg/mL anti-DNP-IgE in complete media overnight for sensitization. Cells were resuspended in HEPES buffer (10 mM HEPES, 137 mM NaCl, 2.7 mM KCl, 0.4 mM Na2HPO4·7H2O, 5.6 mM glucose, 1.8 mM CaCl2·2H2O, 1.3 mM MgSO4·7H2O; Sigma-Aldrich) and transferred to a round-bottom 96-well plate. Then, cells were treated with SA for 30 min. Finally, cells were treated with 0.1 μg/mL DNP-HSA for 30 min for stimulation. Mast cell degranulation was then assessed by measuring β-hexosaminidase (β-hex) release.
RBL-2H3 cells were dispensed into a 96-well plate and incubated for 5 h to allow cells to adhere. The cells were sensitized with 0.1 μg/mL anti-DNP-IgE for 20 h. The next day, the medium was discarded and the cells were washed two times with HEPES buffer. The cells were then treated with SA for 30 min, followed by stimulation with DNP-HSA for 30 min. Finally, mast cell degranulation was assessed by measuring β-hex release.
β-hex in the supernatants and pellet was quantified theough the hydrolysis of p-NAG in 0.1 M sodium citrate buffer (pH 4.5) for 90 min at 37°C. After adding 50 μL glycine (pH 10.7) to each fraction, the absorbance was measured at A405 and A570 in a microplate reader. The amount of total β-hex released was calculated using the following formula: % release = 100 × supernatant absorbance/(0.5 × supernatant absorbance + pellet absorbance).
IgE-sensitized BMMCs were washed, resuspended in HEPES at 2.5 × 106 cells/mL, and incubated with SA for 30 min at 37°C and 5% CO2. The BMMCs were then stimulated using 0.1 μg/mL DNP-HSA for 30 min at 37°C and 5% CO2. Samples were centrifuged at 300 × g for 5 min to obtain the supernatant. Histamine (working standards of 7.8–500 ng/mL) was freshly prepared. Histamine standards and supernatants were transferred to a 96-well flat-bottom plate and mixed with 12 μL of 1 M NaOH and 3 μL of 10 mg/mL o-phthalaldehyde. After 4 min at room temperature, 6 μL of 3 M HCl was added to stop the reaction. The fluorescence intensity was read using a 360-nm excitation filter and a 450-nm emission filter.
IgE-sensitized BMMCs or RBL-2H3 cells treatment with SA for 30 min stimulated with DNP-HSA for 1 h, total RNA was extracted using Trizol Reagent followed the manufacturer's instructions. Complementary DNA (cDNA) was then synthesized from 1 μg of total RNA using the High-Capacity cDNA Reverse Transcription Kit. Real-time PCR was performed using SYBR Green Supermix with sequence-specific primers in the 7500 Fast Real-Time PCR System. The GAPDH mRNA expression was used as an internal control. The following sequence-specific primers were used in this study: rat IL-4 (Forward 5′ to 3′: GAGGACCAGAACGAGACA; Reverse 5′ to 3′: GTGGAAGAGCATCAGGAG), rat CCL2 (Forward 5′ to 3′: GCAGAGACACAGACAGAGG; Reverse 5′ to 3′: CCAGAAGCGTGACAGAGA), rat TNF-α (Forward 5′ to 3′: CCCTGTTCTGCTTTCTCA; Reverse 5′ to 3′: GTTCTCCGTGGTGTTCCT), rat IL-13 (Forward 5′ to 3′: AGCAACATCACACAAGACC; Reverse 5′ to 3′: GGTTACAGAGGCCATTCA), rat GAPDH (Forward 5′ to 3′: GGCACAGTCAAGGCTGAGAATG; Reverse 5′ to 3′: ATGGTGGTGAAGACGCCAGTA), mouse IL-3 (Forward 5′ to 3′: AGGGTCCTTCATCATCAGTTTT; Reverse 5′ to 3′: GCTCTACCACCAGCATCCACA), mouse IL-5 (Forward 5′ to 3′: GGCTTCCTGCTCCTATCTAACTTCA; Reverse 5′ to 3′: ACAGTCATGGCACAGTCTGATTCAT), mouse IL-13 (Forward 5′ to 3′: CTCTTGCTTGCCTTGGTGGTC; Reverse 5′ to 3′: AGGGGAGTCTGGTCTTGTGTGAT), mouse IL-33 (Forward 5′ to 3′: CGGAGTAGTCCTTGTCGTTGG; Reverse 5′ to 3′: GGATGGGAAGAAGCTGATGGT), mouse IL-1β (Forward 5′ to 3′: TGTGTTTTCCTCCTTGCCTCTGAT; Reverse 5′ to 3′: TGCTGCCTAATGTCCCCTTGAAT), mouse IL-6 (Forward 5′ to 3′: TCACAGAAGGAGTGGCTAAGGACC; Reverse 5′ to 3′: ACGCACTAGGTTTGCCGAGTAGAT), mouse GM-SF (Forward 5′ to 3′: CACAAGTTACCACCTATGCGGA; Reverse 5′ to 3′: GAGTTCCTGGCTCATTACGCA), mouse MIP-1a (Forward 5′ to 3′: GCTCCCAGCCAGGTGTCATTTT; Reverse 5′ to 3′: AAGACTCTCAGGCATTCAGTTCCAG), mouse RANTES (Forward 5′ to 3′: GTGTGCCAACCCAGAGAAGAAGT; Reverse 5′ to 3′: AGCAAGCAATGACAGGGAAGCT), mouse GAPDH (Forward 5′ to 3′: AAGAAGGTGGTGAAGCAGG; Reverse 5′ to 3′: GAAGGTGGAAGAGTGGGAGT).
BMMCs were dispensed into 24-well plates (2.5 × 105 cells per well) and sensitized with 0.5 μg/mL anti-DNP-IgE for 20 h. SA was dissolved in a medium containing DMSO and added to the cells to a final concentration of 5, 10, and 25 μmol/L for 30 min. Then cells were stimulated with DNP-HSA for 24 h for TNF, IL-6, and IL-13 or 6 h for LTC4 and PGD2.
The supernatant was collected, centrifuged at 300 × g for 5 min to remove cell debris, and stored at −80°C for ELISA. The supernatant was analyzed for TNF, IL-6, and IL-13 release using ELISA kits and for LTC4 and PGD2 with an ELISA kit, according to the manufacturer's protocol.
BMMC Ca2+ mobilization was measured using a Fluo-4 NW Calcium Assay kit. IgE-sensitized BMMCs were washed and resuspended in assay buffer at 2.5 × 106 cells/mL. A 50 μL suspension was transferred to a 96-well plate, loaded with dye for 30 min at 37°C, and treated with various concentrations of SA and PP2 for 30 min at room temperature. Fluorescence was read at 485-nm excitation and 516-nm emission filters. DNP-HSA (0.1 μg/mL) was added at 100 s, and calcium responses were recorded simultaneously.
IgE-sensitized BMMCs were washed, resuspended in HEPES buffer at 5 × 107 cells/mL, treated with SA (5, 10, and 25 μmol/L) or PP2 (10 μmol/L) for 30 min, and stimulated with 0.1 μg/mL DNP-HSA for 5 or 15 min at 37°C and 5% CO2. Or non-stimulated BMMCs (5 × 107 cells/mL) were treated with SA (5, 10, and 25 μmol/L) for 30 min. The reactions were terminated by adding a lysis buffer containing 1% Triton X-100, 1 × complete protease inhibitor cocktail, 50 μg/mL 3,4-dichloroisocoumarim (Roche Molecular Biochemicals, Indianapolis, Indiana, USA), 1 × protease inhibitor cocktail, 1 mM benzamidine, 1 mM sodium orthovanadate, 5.4 mM sodium pyrophosphate, and 50 mM sodium fluoride. Samples were boiled at 95°C for 5 min in 1 × NuPAGE sample buffer (Life Technologies). After measuring the protein concentration using the Bradford protein assay, samples were adjusted to the same concentration.
Cell protein samples were separated on a NuPAGE Novex 4–12% Bis-Tris gel under non-reduced condition. The proteins were then transferred to a nitrocellulose membrane at 20 V for 1 h. The membrane was blocked with 4% BSA-TBST for 1 h and probed with appropriate antibodies overnight at 4°C. The membrane was washed three times with TBST and incubated with HRP-conjugated goat anti-rabbit IgG or HRP-conjugated goat anti-mouse IgG diluted with 4% BSA-TBST for 1 h at room temperature. Finally, the membrane was developed with a chemiluminescent peroxidase substrate for 1 min and exposed to chemiluminescence film.
IgE-sensitized BMMCs were washed, resuspended in HEPES buffer at 5 × 107 cells/mL, treated with or without SA (25 μmol/L) for 30 min, and stimulated with DNP-HSA (0.1 μg/mL) for 10 min at 37°C and 5% CO2. Or non-stimulated BMMCs (5 × 107 cells/mL) were treated with SA (5, 10, and 25 μmol/L) for 30 min. The reactions were terminated by adding the lysis buffer. Samples were put on ice for 30 min, span at 12,000 rpm, 15 min to get the soluble lysates. The soluble lysates were immunoprecipitated with the appropriate antibodies for 2 h at 4°C. Protein A/G Sepharose beads (Santa Cruz Biotechnology) were then added, and the samples were incubated overnight at 4°C. After washing three times, the samples were boiled at 95°C for 5 min in 1 × NuPAGE sample buffer. Then, the samples were resolved on SDS-PAGE gels (10%) under non-reduced condition and blotted as described.
Six-week-old male ICR mice and Balb/c mice were obtained from the B&K Laboratory Animal Corp. Ltd. (Shanghai, China). All animal experiments were performed according to the Health Guidelines of the Shanghai University of Traditional Chinese Medicine, China, and protocols were approved by the Institutional Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine (SZY201710088).
Passive cutaneous anaphylaxis (PCA) was performed as described previously (9). In brief, 1 μg of anti-DNP-IgE was intradermally injected into the right ear of 7-week-old male mice. The next day, the mice received oral administration of 25, 50, or 100 mg/kg SA or 50 mg/kg ketotifen (Sigma-Aldrich). After 30 min, the mice were challenged for 30 min by intravenous injection of 100 μg DNP-HSA in 300 μL saline containing 0.5% Evans blue. Finally, Evans blue was extracted after 24 h at room temperature with 300 μL of formamide and measured by absorbance at 630 nm.
Seven-week-old male mice were sensitized by intravenous injection of 2 μg of anti-DNP-IgE for 24 h. The next day, the mice received oral administration of 25, 50 mg/kg SA or 50 mg/kg ketotifen. After 30 min, the mice were challenged by intravenous injection of 1 mg of DNP-HSA. After antigen challenge, rectal temperature was measured and recorded every 5 min for 50 min with a digital thermometer. Mice were sacrificed 1 h after antigen challenge. Blood serum was collected. Ear tissues were fixed with 4% paraformaldehyde and embedded in paraffin. Sections were stained with H&E, toluidine blue. For immunofluorescence (Lyn and FcεRIβ), mice were sacrificed 10 min after antigen challenge. Briefly, sections were incubated overnight with anti-Lyn and anti-FcεRIβ. Staining was detected using the appropriated secondary antibody. Nuclei were stained with DAPI. The stained sections were scanned using panoramic slide scanner (3D HISTECH, Hungary). The images were calculated using Image-Pro plus 6.0.
Experiments were conducted in triplicate, and data are presented as mean (n = 3) ± standard error of the mean (SEM). P values were determined using one-way ANOVA using GraphPad Prism 6.0. Statistical significance was set at P of ≤ 0.05, ≤ 0.01, or ≤ 0.001.
In order to enrich enough SA to meet the needs of pharmacological experiments, we obtained a total of 200 mg of SA through extraction, separation and chemical transformation (Figure 1A).
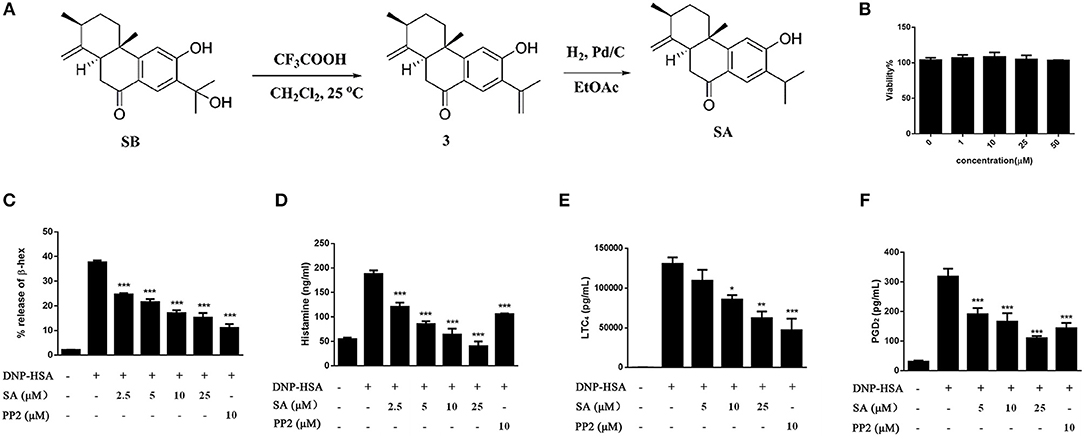
Figure 1. The preparation of SA and the effect of SA on viability and degranulation of mast cells. (A) A schematic diagram for the preparation of SA through chemical transformation. (B) BMMCs were incubated in the presence of 1, 10, 25, or 50 μmol/L of SA for 24 h. Cell viability was assayed using an XTT assay. (C–F) BMMCs were sensitized with anti-DNP IgE with or without 5, 10, or 25 μmol/L of SA for 30 min, challenged with DNP-HSA-induced release of β-hex (C) and histamine (D) at 30 min, and secretion of LTC4 (E) and PGD2 (F) at 6 h was assessed. The results are expressed as mean ± SEM from three individual experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the IgE/Ag-treated group.
Before investigating the effect of SA (Figure 1A) on mast cell activation, we examined the cytotoxicity of SA on BMMCs by using XTT assay. BMMCs were treatmed with various concentrations (1, 10, 25, and 50 μmol/L) of SA for 24 h (Figure 1B); SA did not exhibit any cytotoxicity, even at 50 μmol/L. Therefore, we used SA at concentrations of up to 25 μmol/L for all in vitro experiments. To determine the effect of SA on BMMC degranulation, we first sensitized BMMCs with anti-DNP-IgE for 20 h and then incubated cells with various concentrations (2.5, 5, 10, and 25 μmol/L) of SA. After 30 min, BMMCs were stimulated with DNP-HSA for 30 min, and then mast cells released β-hex and histamine. SA treatment significantly decreased β-hex (Figure 1C) and histamine (Figure 1D) release in a dose-dependent manner. Furthermore, the inhibitory effect of SA on histamine was better than that obtained by PP2, a tyrosine kinase inhibitor.
FcεRI-mediated mast cells also release lipid mediators after activation—prostaglandin D2 (PGD2) and LTC4. To further clarify the effect of SA, we sensitized BMMCs with anti-DNP-IgE, followed by incubation with or without SA for 30 min. The cells were then activated with DNP-HSA for 6 h. When BMMCs were activated for 6 h, cells released a large amount of LTC4 (Figure 1E), but SA treatment significantly decreased LTC4 release in a dose-dependent manner. SA also considerably attenuated PGD2 generation after cells were stimulated with DNP-HSA for 6 h (Figure 1F).
Engagement of the FcεRI receptor leads to the production of multiple cytokines and chemokines essential for mast cell function and allergic response. To determine the effect of SA on FcεRI-mediated cytokines and chemokines, BMMCs were sensitized with anti-DNP-IgE overnight, and pretreatment with SA for 30 min, then stimulated with DNP-HSA for 1 h. We found that SA significantly inhibited the expression of cytokine mRNAs in FcεRI-mediated BMMCs. These include Th2-related cytokines and inflammation-related cytokines. However, the expression of MIP-1α which belongs to chemokines remained unaltered (Figure 2A). Subsequently, we used ELISA to confirm the effect of SA on FcεRI-mediated secretion of TNF-α, IL-6, and IL-13. As shown in Figure 2B, treatment with SA significantly reduced TNF-α, IL-6, and IL-13 secretion in a dose-dependent manner. In particular, SA at 25 μmol/L considerably inhibited IL-6 release, and the inhibition rate was up to 79.9%. These data indicate that SA plays a role in stabilizing mast cells, inhibiting their activation.
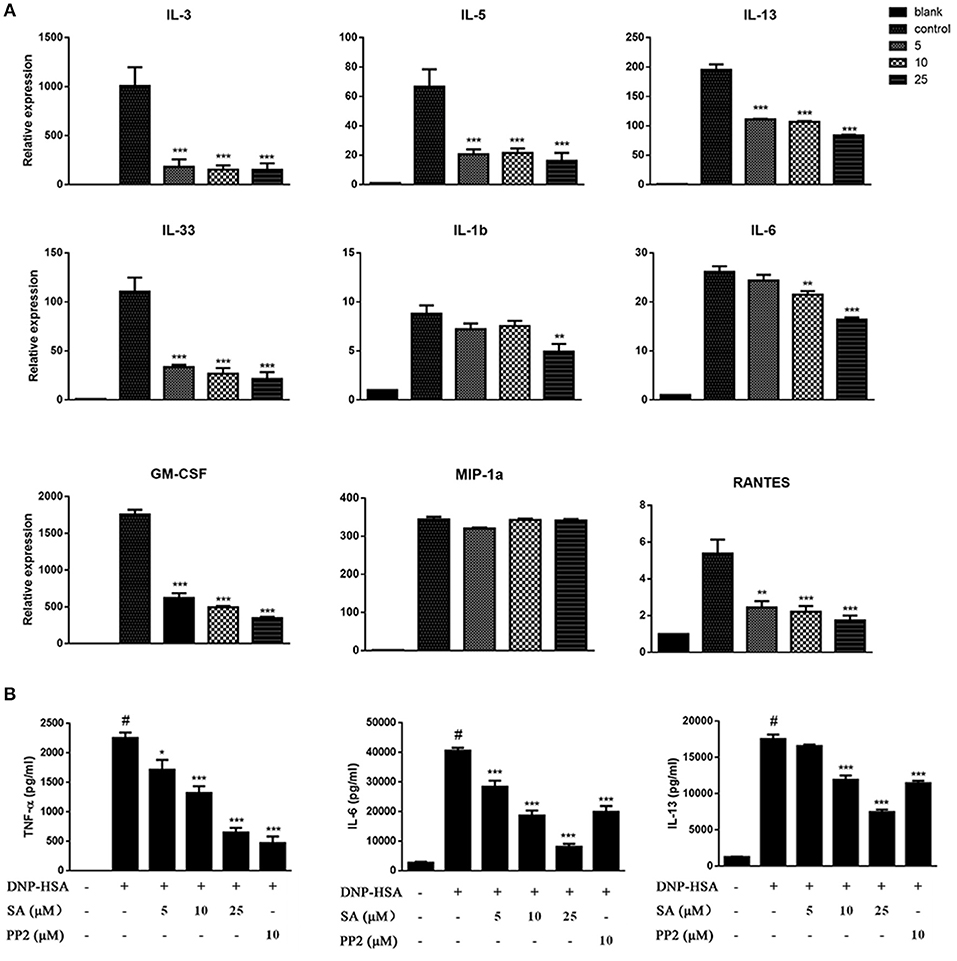
Figure 2. SA inhibits IgE/Ag-stimulated cytokine production. (A) BMMCs were sensitized with anti-DNP IgE with or without 5, 10, or 25 μmol/L of SA for 30 min and challenged with DNP-HSA for 1 h. Cytokine and chemokine mRNA expression was measured by RT-PCR. (B) TNF-α, IL-6, and IL-13 levels were measured by ELISA at 24 h. The results are expressed as mean ± SEM from three individual experiments. #P < 0.05 vs. the non-stimulated mast cell group. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the IgE/Ag- treated group.
We next evaluated the effect of SA on IgE-activated RBL-2H3 cells. IgE-sensitized RBL-2H3 cells were pretreatment with SA for 30 min, then stimulated with DNP-HSA for 30 min or 1 h. After 30 min of DNP-HSA stimulation, the supernatant was assayed for β-hex release (Figure 3A), SA significantly inhibited β-hex release in a concentration-dependent manner. And SA also attenuated the mRNA expressions of TNF-α (Figure 3B), CCL2 (Figure 3C), IL-13 (Figure 3D), and IL-4 (Figure 3E) after RBL-2H3 cells were stimulated with DNP-HSA for 1 h. This further demonstrated that SA suppressed the release of allergic mediators from mast cells.
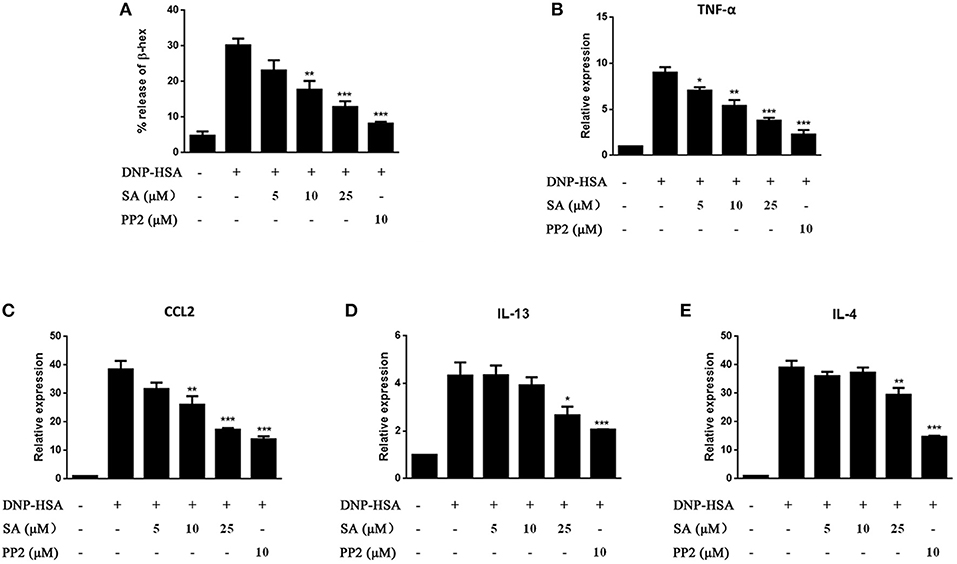
Figure 3. Effect of SA on IgE/Ag-stimulated RBL-2H3 cells. RBL-2H3 cells were sensitized with anti-DNP IgE with or without 5, 10, or 25 μmol/L of SA for 30 min, challenged with DNP-HSA induced release of β-hex (A) at 30 min, the mRNA expression of TNF-α (B), CCL2 (C), IL-13 (D), and IL-4 (E) was measured by RT-PCR at 1 h. The results are expressed as mean ± SEM from three individual experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the IgE/Ag-treated group.
Given that SA downregulated the release of allergic mediators in vitro, we used PCA in the ear and passive systemic anaphylaxis (PSA) to evaluate the action of SA in anaphylactic responses in vivo. PCA was elicited by subcutaneous injection of anti-DNP-IgE into the ear. After 24 h, mice treated with various concentrations (25, 50, and 100 mg/kg) of SA for 30 min. Then, mice received intravenous DNP-HSA solution containing 0.5% Evans blue dye. Mice challenged with DNP-HSA showed a clear PCA response in the ear. SA significantly reduced the PCA response on visual inspection (Figure 4A) and the Evans blue dye extracted from the reaction site of the ear (Figure 4B), but no difference was found between SA at 50 and 100 mg/kg. Therefore, we used SA at 25 and 50 mg/kg in the PSA assay.
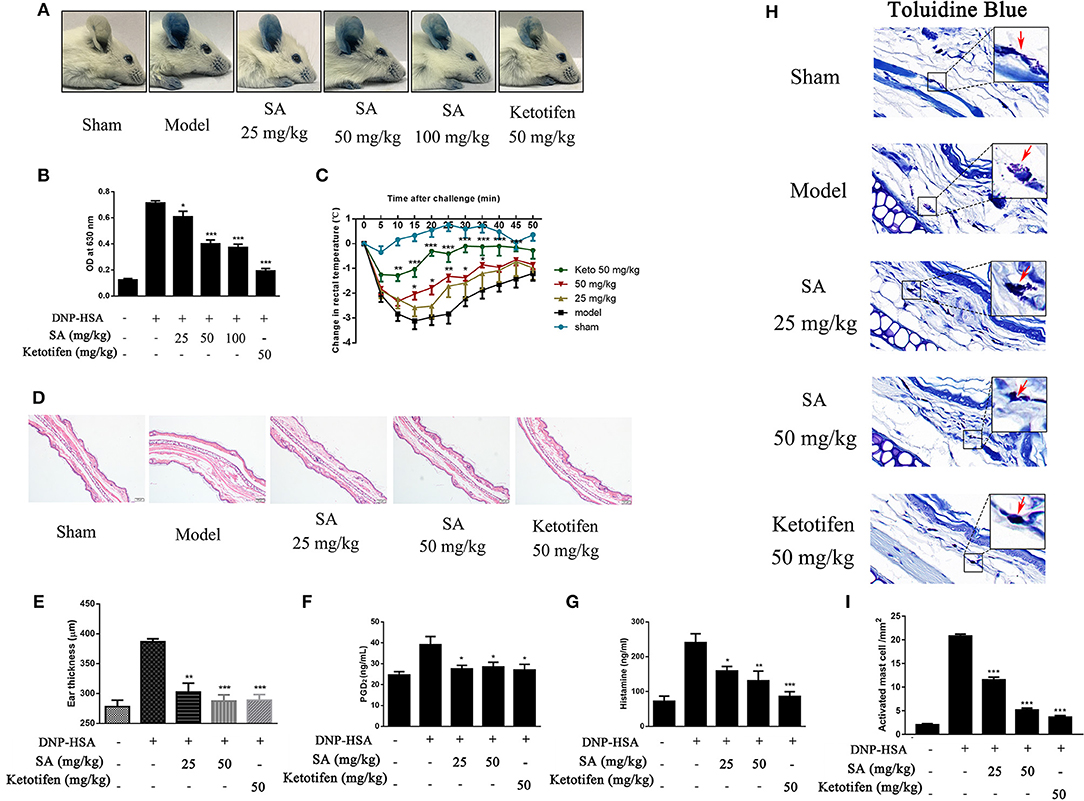
Figure 4. Effect of SA on IgE-mediated PCA and PSA reaction. (A,B) In the PCA test, ICR mice were sensitized with anti-DNP-IgE or saline for 24 h. Then, mice were treated with SA or ketotifen for 30 min, followed by intravenous injection with DNP-HSA containing 0.5% Evans blue. (A) Representative photos of ears showing dye extravasation. (B) Evans blue was extracted in formaldehyde and quantified as absorbance at 630 nm. (C–H) In the PSA test, mice were injected with anti-DNP-IgE (2 μg) or saline and 24 h later were orally administered SA or ketotifen; after 30 min, DNP-HSA (2 mg) was intravenously injected. Blood was collected 1 h after antigen challenge, and serum histamine and PGD2 were assessed. (C) Rectal temperatures were assessed every 5 min for 50 min. (D) Histological section of ears of mice, stained with HE. Bar, 100 μm. (E) Ear thickness was calculated based on histological section. (F) Serum levels of PGD2. (G) Serum levels of histamine. (H) Tissue mast cells in ear were assessed using toluidine blue staining. Arrowheads indicate individual mast cells. Bars, 20 μm. (I) Absolute numbers of activated mast cell per mm2 (n = 8 mice in each group. Data are represented as mean ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001 vs. the IgE/Ag-treated mice).
PSA is an IgE-mediated type I immediate hypersensitivity reaction. Mice were sensitized with anti-DNP-IgE overnight and treated with or without SA for 30 min; then, they were intravenously injected with DNP-HSA to elicit an anaphylactic response. Rectal temperature was recorded every 5 min to monitor the magnitude of the PSA response. Mice challenged with DNP-HSA showed an evident rectal temperature drop by 10–25 min after injection. By contrast, mice treated with SA or ketotifen showed significantly reduced rectal temperature drop (Figure 4C). Histological analysis of ear sections at 1 h after injection of DNP-HSA also confirmed the antiallergic effect of SA (Figures 4D,E). In addition, SA significantly inhibited serum levels of PGD2 (Figure 4F) and histamine (Figure 4G). In particular, at the same concentration, SA was comparable to ketotifen, a H1 blocker and mast cell stabilizer.
Because SA inhibited mast cell activation in vitro, we hypothesized that SA achieves the inhibition effect of anaphylactic responses by stabilizing mast cells. Thus, we examined whether mast cell degranulation occurs or not in PSA with SA. Toluidine blue staining of ear sections showed nearly activated mast cells after allergen exposure (red arrow), whereas SA significantly reduced mast cell activation, particularly at 50 mg/kg, most mast cells were at rest status (Figure 4H). In addition, as shown as Figure 4I, SA suppressed the absolute numbers of activated mast cell. These data indicate that SA inhibits allergic response in vivo by suppressing mast cell activation.
Intracellular calcium is an important second messenger in cells, and a transient increase in intracellular calcium is crucial for the activation of mast cells (6, 20). Therefore, we explored the effect of SA on Ca2+ influx in BMMCs. BMMCs had high levels of calcium flux after allergen exposure and reached a peak after BMMCs challenged DNP-HSA for 50 s (Figure 5A). As expected, SA dramatically decreased intracellular calcium levels (Figure 5A), and effectively inhibited the amount of calcium levels which was accumulated from BMMCs challenged DNP-HSA 0 s to 300 s (Figure 5B).
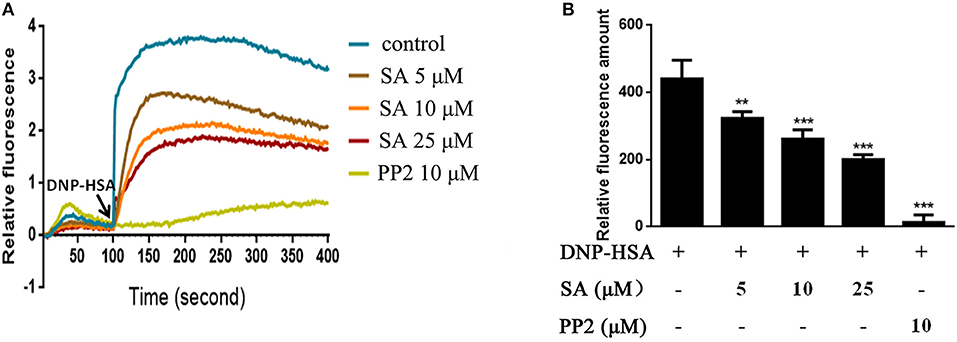
Figure 5. SA inhibits Ca2+ mobilization in IgE/Ag-stimulated BMMCs. (A) Calcium flux was measured by Fluo-4 NW Calcium Assay kit at the indicated times after stimulation. (B) The amount of calcium flux that was accumulated from BMMCs challenged DNP-HSA 0 s to 300 s. Data are represented as mean ± SEM from three experiments. **P < 0.01, ***P < 0.001 vs. the IgE/Ag-treated group.
The aforementioned data indicated that SA inhibits FcεRI-mediated mast cell activation both in vitro and in vivo. We next examined whether SA affects the activation of intracellular signaling pathways by FcεRI in BMMCs. IgE-sensitized BMMCs were pretreated with SA for 30 min, followed by DNP-HSA crosslinking for 5 min or 15 min. After 5 min of DNP-HSA stimulation, SA dose-dependently inhibited the phosphorylation of Lyn (Figures 6A,E), which is a FcεRI-proximal tyrosine kinase. We next detected the downstream signaling molecules of Lyn: FcεRIβ, FcεRIγ, Syk, LAT, and PLCγ1; as expected, SA significantly suppressed their phosphorylation (Figures 6B,C,F–J). However, Fyn, another FcεRI–proximal tyrosine kinase, was not significantly changed after treatment with SA (Figures 6A,D). Consistently, SA could not change the phosphorylation of Akt (Figures 7A,F), a downstream Fyn-dependent protein (21).
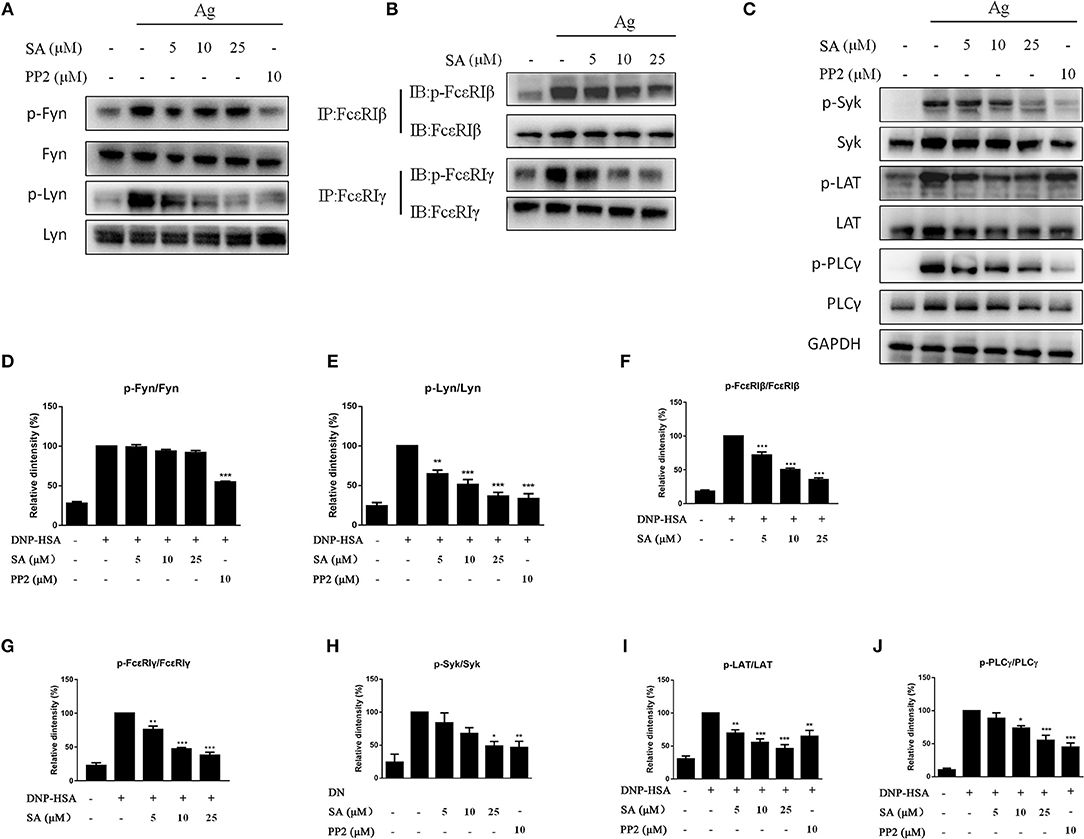
Figure 6. Effect of SA on FcεRI-mediated cell signaling in IgE/Ag-induced BMMCs. (A–C) IgE-sensitized BMMCs were treated with or without SA for 30 min and stimulated with DNP-HSA for 5 min. Then, protein lysates were analyzed for the phosphorylated and non-phosphorylated forms of Fyn, Lyn, FcεRIβ, FcεRIγ, Syk, LAT, PLCγ, using Western blotting or immunoprecipitation. (D–J) The related ratios of p-Fyn/Fyn (D), p-Lyn/Lyn (E), p-FcεRIβ/FcεRIβ (F), p-FcεRIγ/FcεRIγ (G), p-Syk/Syk (H), p-LAT/LAT (I), p-PLCγ/ PLCγ (J) protein levels were determined using densitometry statistical analysis. Data are represented as mean ± SEM from three experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the IgE/Ag-treated group.
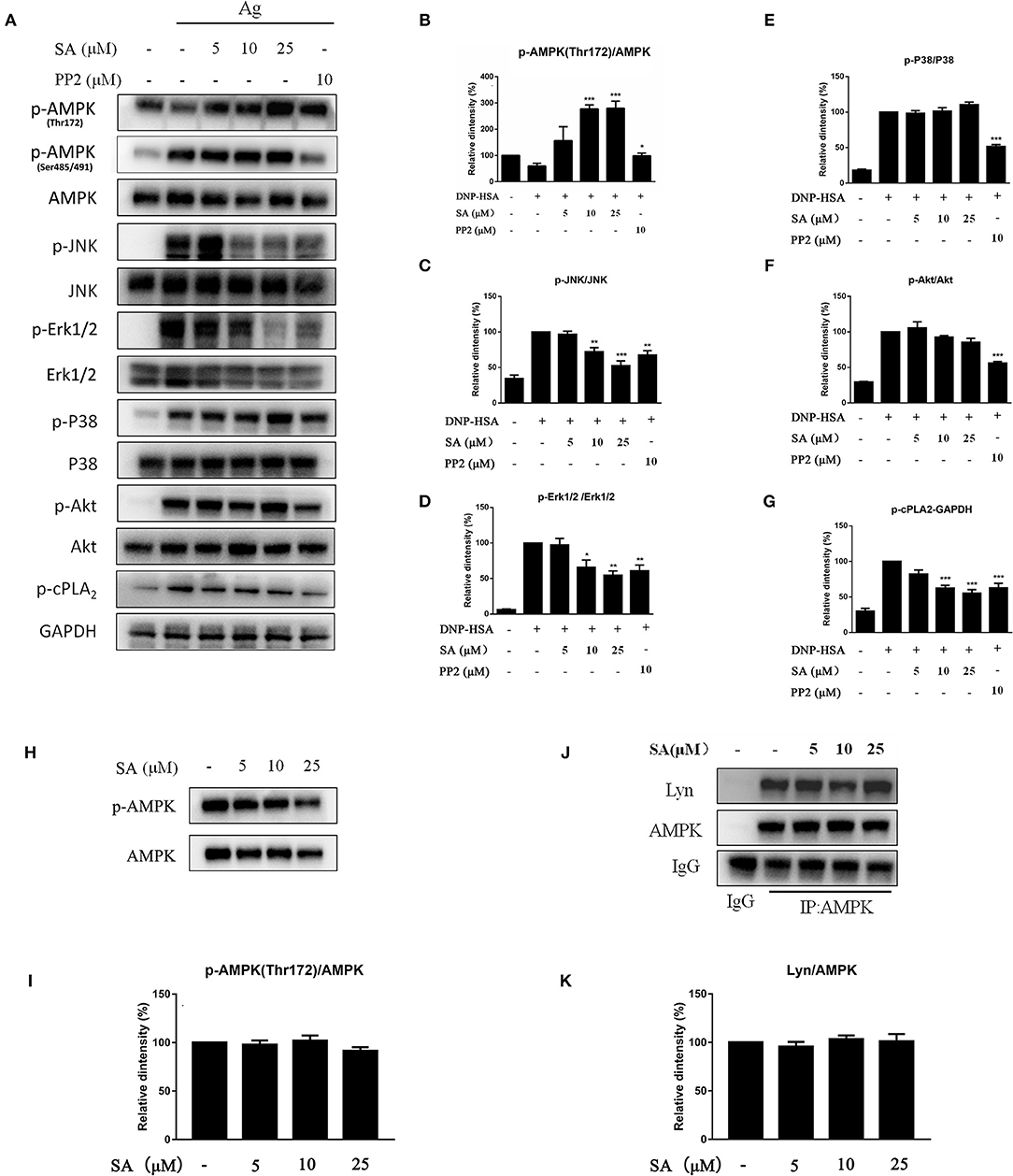
Figure 7. Effect of SA on AMPK and MAPK pathway. (A) IgE-sensitized BMMCs were treated with or without SA for 30 min and stimulated with DNP-HSA for 15 min. Then, protein lysates were analyzed for the phosphorylated and non-phosphorylated forms of AMPK, JNK, Erk1/2, P38, Akt, cPLA2, and GAPDH using Western blotting. (B–G) The related ratios of p-AMPK (Thr172)/AMPK (B), p-JNK/JNK (C), p-Erk1/2/Erk1/2 (D), p-P38/P38 (E), p-Akt/Akt (F), and p-cPLA2/GAPDH (G) protein levels were determined using densitometry statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001 vs. the IgE/Ag-treated group. (H) BMMCs were treated with or without SA for 30 min, and then protein lysates were analyzed for p-AMPK (Thr172) and AMPK. (I) The related ratios of p-AMPK (Thr172)/AMPK were determined using densitometry statistical analysis. (J) BMMCs were treated with or without SA for 30 min. Cell lysates were collected and subjected to immunoprecipitation with antibodies specific for AMPK, followed by Western blotting using the indicated antibodies. (K) The related ratios of Lyn/AMPK protein levels were determined by densitometry statistical analysis. Data are represented as mean ± SEM from three experiments.
MAPK signaling is distal signaling during mast cell activation and is vital for the FcεRI-mediated production of cytokines and chemokines in mast cells (6, 22, 23). As shown in Figures 7A,C–E, after 15 min of DNP-HSA stimulation, SA considerably inhibited the phosphorylation of Erk1/2 and JNK, but p-P38 remained unaltered; in addition, SA significantly inhibited p-cPLA2 (Figures 7A,G), which is consistent with the inhibition of LTC4 release (24). Moreover, SA significantly promoted AMP-activated protein kinase (AMPK) phosphorylation at Thr172 after 15 min of DNP-HSA stimulation (Figures 7A,B), while it did not alter p-AMPK (Ser485/491). In addition, SA pretreatment alone did not affect AMPK phosphorylation in non-stimulated mast cells (Figures 7H,I).
Previous report showed that Lyn-specific siRNA increase human mast cell activation (25). To further clarify the mechanism of SA in allergic response, we assessed the effects of SA on Lyn activity by using an in vitro enzyme system. However, SA did not inhibit Lyn kinase activation (data not shown). We then examined the effect of SA on AMPK/Lyn interaction before any FcεRI aggregation. As shown as Figures 7J,K, the interaction between AMPK and Lyn was not altered with SA. In the Lyn-related pathway, the Lyn–FcεRIβ interaction is indispensable for FcεRI-mediated mast cells (16). We examined whether SA influenced the Lyn–FcεRIβ interaction. As shown as Figures 8A,B, there was a weak binding between Lyn and FcεRIβ when mast cells were at rest status, and SA did not affect the binding between Lyn and FcεRIβ. However, allergen exposure for 10 min considerably enhanced the binding between Lyn and FcεRIβ, and SA blocked this binding, reduced it to that at mast cell rest status. On investigating whether SA blocked the binding between Lyn and FcεRIβ in vivo in the PSA model, we found that SA significantly suppressed the binding between Lyn and FcεRIβ after exposure to the allergen for 10 min, whereas ketotifen did not (Figures 8C,D).
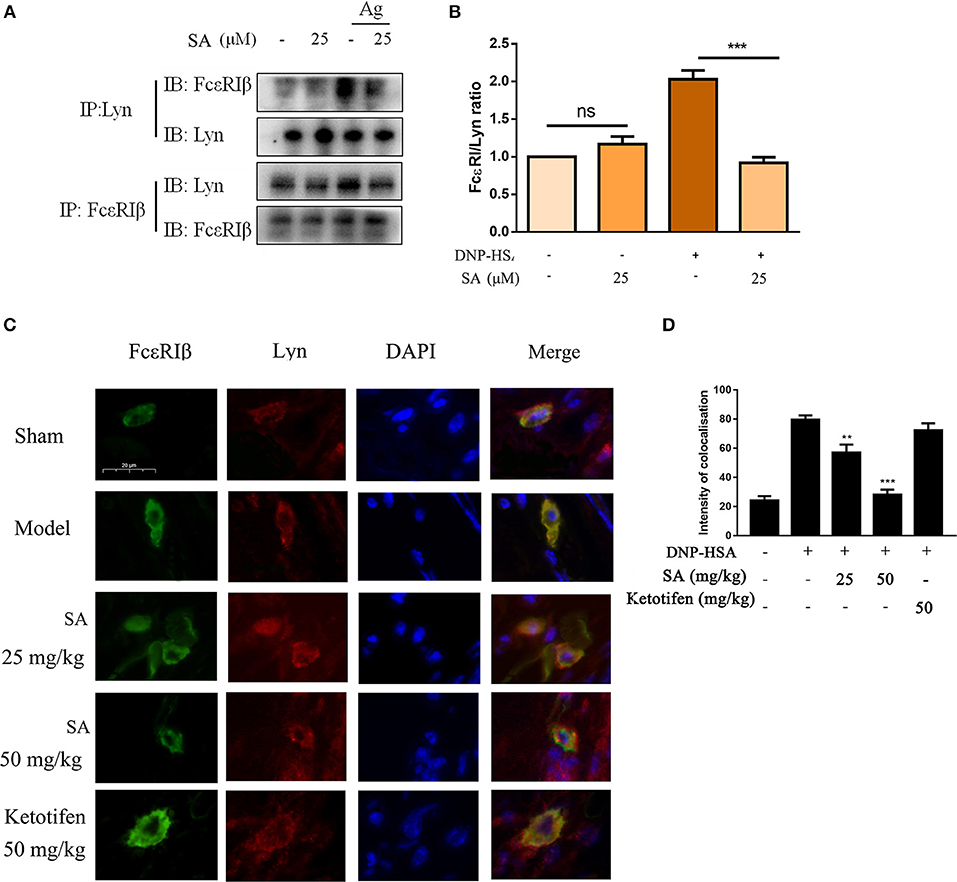
Figure 8. Effect of SA on the interaction between Lyn and FcεRI. (A) IgE-sensitized BMMCs were treated with or without SA for 30 min and then stimulated with DNP-HSA for 10 min. Cell lysates were collected and subjected to immunoprecipitation with antibodies specific for Lyn or FcεRIβ, followed by Western blotting using the indicated antibodies. (B) The related ratios of FcεRIβ/Lyn protein levels from anti-Lyn immunoprecipitation were determined by densitometry statistical analysis. Data are represented as mean ± SEM from three experiments. (C) Immunofluorescence images showing the interaction between Lyn (red) and FcεRIβ (green) in ear form PSA mice. Bars, 20 μm. (D) The intensity of colocalisation between Lyn and FcεRIβ, and quantified by Image-Pro Plus 6.0.
Abietane-type diterpenes are abundant in nature and have various biological activities and pharmacological properties, such as antitumor (26), antiviral (27), antibacterial (28, 29), and anti-inflammatory (30). However, their antiallergic effect has not been reported. SA is an abietane-type diterpene from S. dentata Royle ex Benth. In this study, we first report that SA inhibits mast cell activation and mast cell–mediated allergic reactions.
Mast cells play a key role in allergic response in conditions such as asthma, rhinitis, and atopic dermatitis. IgE/Ag-activated mast cells activate a complex intracellular signaling pathway. Lyn and Fyn are essential for initiating FcεRI-mediated mast cell activation: Lyn mediates a pivotal signaling pathway following aggregation of FcεRIs, whereas Fyn is key in a complementary pathway for mast cell activation (31). Several studies have reported compounds that suppress IgE-mediated allergic response through the inhibition of Lyn and Fyn. For example, AZD7762 suppressed IgE-mediated mast cells and allergic responses by inhibiting the activity of Lyn and Fyn kinases (32). Elaeocarpus, which was isolated from Elaeocarpus sylvestris L., inhibited Fyn and Lyn phosphorylation, thereby preventing mast cell degranulation and expression of proinflammatory cytokines (33).
Unlike the aforementioned compounds, SA inhibited the phosphorylation of only Lyn and not Fyn (Figure 6A), and had no inhibitory effect on Akt (Figure 7A), a protein that belongs to Fyn-regulated compensatory signaling pathways. In experiments with RBL-2H3 cells, where Fyn gene and protein expressions are hardly detected (34), SA inhibited the degranulation and expression of proinflammatory cytokines in a dose-dependent manner (Figure 3). Therefore, speculating that SA only inhibits the Lyn-dependent pathway in IgE/Ag-induced mast cells, we investigated the effect of SA on phosphorylation of FcεRIβ, FcεRIγ, Syk, LAT, PLCγ, and MAPKs. SA significantly suppressed the phosphorylation of Syk, LAT, PLCγ, Erk1/2, and JNK, but did not affect the phosphorylation of P38.
AMPK, a regulator of energy metabolism, negatively regulates mast cell activation and anaphylaxis. AMPK activation suppressed IgE/Ag-induced mast cell activation through the inhibition of Erk and JNK phosphorylation, but did not change P38 phosphorylation (35). Therefore, we investigated the involvement of SA in IgE/Ag-induced AMPK activation. As expected, SA significantly increased AMPK phosphorylation at Thr172 after 15 min of DNP-HSA stimulation (Figure 7A), while it did not alter p-AMPK (Ser485/491). However, the AMPK phosphorylation at Thr 172 is not affected with Lyn knockdown, whereas Fyn counter-regulates the AMPK signaling pathway in mast cells (35). Thus, we suggest that the target of SA in mast cells might be simultaneously related to Lyn and AMPK.
FcεRIβ amplifies the FcεRI-mediated activation signal. The Lyn–FcεRIβ interaction is indispensable for FcεRIβ-mediated human mast cell activation (16). Lyn can pre-associate with the FcεRIβ chain before receptor crosslinking, and receptor stimulation can further increase their binding (11). In addition, AMPK binds to Lyn when mast cells are at rest, and after mast cell activation, AMPK binds to FcεRIβ (36). We hypothesized that the AMPK–FcεRIβ binding might be caused by the binding of Lyn to FcεRIβ, and that the Lyn–FcεRIβ interaction might simultaneously affect Lyn and AMPK phosphorylation. Although previous study showed that AMPK activation blocked the formation of Lyn-FcεRIβ complexes, their results are based on the AMPK phosphorylation at Ser485/491 (36). In our study, we did not observe significant effect of SA on p-AMPK (Ser 485/491) after FcεRI activation; instead, we observed that SA significantly increased AMPK phosphorylation at Thr172 after 15 min of DNP-HSA stimulation (Figure 7A). In addition, we observed that SA did not alter the interaction between AMPK and Lyn in non-stimulated mast cell (Figure 7J). And SA blocked the binding Lyn to FcεRIβ in IgE/Ag-induced mast cell both in vitro (Figures 8A,B) and in vivo (Figures 8C,D). Above all, we suggest that SA suppressed mast cell–mediated allergic response by blocking the Lyn–FcεRIβ interaction in vitro and in vivo, which is a key step in mast cell activation. Thus, SA may be a promising therapeutic agent for allergic diseases.
All animal experiments were performed according to the Health Guidelines of the Shanghai University of Traditional Chinese Medicine, China, and protocols were approved by the Institutional Animal Ethics Committee of Shanghai University of Traditional Chinese Medicine (No. SZY201710088).
YL and JX designed the study. FQ, LZ, SL, and GM preformed the experiments. FQ and YL analyzed the data and wrote the manuscript. FG and PL provided scientific discussion.
This work was supported by the National Natural Science Foundation of China (No. 81673570), the Excellent Academic Leaders Program of Shanghai (16XD1403500), Shanghai Sail Program, the China Postdoctoral Science Foundation (2018M642067), and the programs of High level University Innovation Team and Shanghai E-Research Institute of Bioactive Constituents in Traditional Chinese Medicine.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Marianna Kulka (University of Alberta) for technical assistance.
BSA, Bovine Serum Albumin; DMSO, Dimethyl sulfoxide; DNP-HSA, 2,4-dinitrophenyl -Human serum albumin; Erk1/2, extracellular regulated protein kinases; FcεRI, the high-affinity receptor for IgE; IgE, Immunoglobulin E; ITAM, immunoreceptor tyrosine-based activatory motif; JNK, c-Jun N-terminal kinase; LAT, Linker for activation of T cell; MAPK, mitogen-activated protein kinase; P38, P38 mitogen activated protein; cPLA2, cytosolic phospholipase A2; PGD2, prostaglandin D2; PLCγ, phospholiase C-gamma; XTT, 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)−5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide; β-Hex, β-hexosaminidase; AMPK, AMP-activated protein kinase.
1. Cruz AA, Cooper PJ, Figueiredo CA, Alcantara-Neves NM, Rodrigues LC, Barreto ML. Global issues in allergy and immunology: parasitic infections and allergy. J Allergy Clin Immunol. (2017) 140:1217–28. doi: 10.1016/j.jaci.2017.09.005
2. Pawankar R, Canonica GW, Holgate ST, Lockey R. Allergic diseases and asthma:, a major global health concern. Curr Opin Allergy Clin Immunol. (2012) 12:39–41 doi: 10.1097/ACI.0b013e32834ec13b
3. Galli S J, Tsai M. IgE and mast cells in allergic disease. Nat Med. (2012) 18:693–704. doi: 10.1038/nm.2755
4. Balbino B, Conde E, Marichal T, Starkl P, Reber LL. Approaches to target IgE antibodies in allergic diseases. Pharmacol Therap. (2018) 191:50–64. doi: 10.1016/j.pharmthera.2018.05.015
5. Getahun A, Cambier JC. Of ITIMs, ITAMs and ITAMis, revisiting Immunoglobulin Fc receptor signaling. Immunol Rev. (2015) 268:66–73. doi: 10.1111/imr.12336
6. Gilfillan AM, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol. (2006) 6:218–30. doi: 10.1038/nri1782
7. Al-Azzam N, Kondeti V, Duah E, Gombedza F, Thodeti CK. Modulation of mast cell proliferative and inflammatory responses by leukotriene d4 and stem cell factor signaling interactions. J Cell Physiol. (2015) 230:595–602. doi: 10.1002/jcp.24777
8. Donnadieu E, Jouvin MH, Kinet JP. A second amplifier function for the allergy-associated FceRI-β subunit. Immunity. (2000) 12:515–23. doi: 10.1016/S1074–7613(00)80203–4
9. Lin S, Cicala C, Scharenberg AM, Kinet JP. The FcεRIβ subunit functions as an Amplifier of FcεRIγ-mediated cell activation signals. Cell. (1996) 85:985–95. doi: 10.1016/S0092–8674(00)81300–8
10. Pribluda VS, Pribluda C, Metzger H. Transphosphorylation as the mechanism by which the highaffinity receptor for IgE is phosphorylated upon aggregation. Proc Natl Acad Sci USA. (1994) 91:11246–50. doi: 10.1073/pnas.91.23.11246
11. Vonakis BM, Haleem-Smith H, Benjamin P, Metzger H. Interaction between the unphosphorylated receptor with high affinity for IgE and Lyn kinase. J Biol Chem. (2001) 276:1041–50. doi: 10.1074/jbc.M003397200
12. Kovárová M, Tolar P, Arudchandran R, Dráberová L, Rivera J, Dráber P. Structure-function analysis of Lyn kinase association with lipid rafts and initiation of early signaling events after Fcepsilon receptor I aggregation. Mol Cell Biol. (2001) 21:8318–28. doi: 10.1128/MCB.21.24.8318–8328.2001
13. Kovarova M, Wassif CA, Odom S, Liao K, Porter F, Rivera J. Cholesterol-deficiency in a murine model of Smith-Lemli-Opitz Syndrome reveals increased mast cell responsiveness. J Exp Med. (2006) 203:1161–71. doi: 10.1084/jem.20051701
14. On M, Billingsley JM, Jouvin MH, Kinet JP. Molecular dissection of the FcεRIβ signaling amplifier. J Biol Chem. (2004) 279:45782–90. doi: 10.1074/jbc.M404890200
15. Furumoto Y, Nunomura S, Terada T, Rivera J, Ra C. The FcεRIβ immunoreceptor tyrosine-based activation motif exerts inhibitory control on MAPK and IκB kinase phosphorylation and mast cell cytokine production. J Biol Chem. (2004) 279:49177–87. doi: 10.1074/jbc.M404730200
16. Okayama Y, Kashiwakura J I, Matsuda A, Sasaki-Sakamoto T, Ra C. The interaction between Lyn and FcεRIβ is indispensable for FcεRI-mediated human mast cell activation. Allergy. (2012) 67:1241–9. doi: 10.1111/j.1398–9995.2012.02879.x
17. Zhang L, Zhang D, Jia Q, Wang R, Dorje G, Zhao ZL, et al. 19(4 → 3)-abeo-abietane diterpenoids from Scrophularia dentata Royle ex Benth. Fitoterapia. (2015) 106:72–7. doi: 10.1016/j.fitote.2015.08.005
18. Song EH, Chung KS, Kang YM, Lee JH, Lee M, An HJ. Eupatilin suppresses the allergic inflammatory response in vitro and in vivo. Phytomedicine. (2018) 42:1–8. doi: 10.1016/j.phymed.2017.08.027
19. Rossi A, Caiazzo E, Bilancia R, Riemma M, Pagano E, Cicala C, et al. Salvinorin A inhibits airway hyperreactivity induced by Ovalbumin Sensitization. Front Pharmacol. (2016) 7:525. doi: 10.3389/fphar.2016.00525
20. Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. (2008) 9:81–8. doi: 10.1038/ni1546
21. Sibilano R, Frossi B, Suzuki R, MSc FD, Gri G, Piconese S, et al. Modulation of FcεRI-dependent mast cell response by OX40L via Fyn, PI3K, and RhoA. J Allergy Clin Immunol. (2012) 130:751–60. doi: 10.1016/j.jaci.2012.03.032
22. Lu Y, Yang JH, Li X, Hwangbo K, Hwang SL, Taketomi Y. Emodin, a naturally occurring anthraquinone derivative, suppresses IgE-mediated anaphylactic reaction and mast cell activation. Biochem Pharmacol. (2011) 82:1700–8. doi: 10.1016/j.bcp.2011.08.022
23. Saitoh S, Odom S, Gomez G, Sommers CL, Young HA, Rivera J. The four distal tyrosines are required for LAT-dependent signaling in FcepsilonRI-mediated mast cell activation. J Exp Med. (2003) 198:831–4. doi: 10.1084/jem.20030574
24. Li X, Kwon O, Kim DY, Taketomi Y, Murakami M, Chang HW. NecroX-5 suppresses IgE/Ag-stimulated anaphylaxis and mast cell activation by regulating the SHP-1-Syk signaling module. Allergy. (2016) 71:198–209. doi: 10.1111/all.12786
25. Yamashita Y, Charles N, Furumoto Y, Odonm S, Yamashita T, Gilfillan AM, et al. Cutting edge: genetic variation influences Fc epsilonRI-induced mast cell activation and allergic responses. J Immunol. (2007) 179:740. doi: 10.4049/jimmunol.179.2.740
26. Jian B, Zhang H, Han C, Liu J. Anti-cancer activities of diterpenoids derived from Euphorbia fischeriana Steud. Molecules. (2018) 23:387. doi: 10.3390/molecules23020387
27. González M A, Zaragozá R J. Semisynthesis of the antiviral abietane diterpenoid jiadifenoic acid C from callitrisic acid (4-epidehydroabietic acid) isolated from sandarac resin. J Nat Prod. (2014) 77:2114–7. doi: 10.1021/np500569y
28. Abdissa N, Frese M, Sewald N. Antimicrobial abietane-type diterpenoids from Plectranthus punctatus. Molecules. (2017) 22:1919. doi: 10.3390/molecules22111919
29. Nzogong RT, Nganou BK, Tedonkeu AT, Awouafack MD, Tene M, Ito T, et al. Three new abietane-type diterpenoids from plectranthus africanus and their antibacterial activities. Planta Medica. (2017) 84:59–64. doi: 10.1055/s-0043–114426
30. Cretton S, Saraux N, Monteillier A, Righi D, Marcourt L, Genta-Jouve G, et al. Anti-inflammatory and antiproliferative diterpenoids from Plectranthus scutellarioides. Phytochemistry. (2018) 10. 154:39–46. doi: 10.1016/j.phytochem.2018.06.012
31. Parravicini V, Gadina M, Kovarova M, Odom S, Gonzalezespinosa C, Furumoto Y, et al. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat Immunol. (2002) 3:741–8. doi: 10.1038/ni817
32. Park YH, Kim DK, Kim HW, Kim HS, Lee D, Lee M, et al. Repositioning of anti-cancer drug candidate, AZD7762, to an anti-allergic drug suppressing IgE-mediated mast cells and allergic responses via the inhibition of Lyn and Fyn. Biochem Pharmacol. (2018) 8:270–7. doi: 10.1016/j.bcp.2018.05.012
33. Kim MJ, Kim YY, Choi YA, Baek MC, Lee B, Park PH, et al. Elaeocarpusin inhibits mast cell-mediated allergic inflammation. Front Pharmacol. (2018) 9:591. doi: 10.3389/fphar.2018.00591
34. Sanderson MP, Wex E, Kono T, Uto K, Schnapp. Syk and Lyn mediate distinct Syk phosphorylation events in FcεRI-signal transduction: Implications for regulation of IgE-mediated degranulation. Mol Immunol. (2010) 48:171–8. doi: 10.1016/j.molimm.2010.08.012
35. Hwang SL, LiX, LuY, Jin Y, Jeong YT, Kim YD, et al. AMP-activated protein kinase negatively regulates FcεRI-mediated mast cell signaling and anaphylaxis in mice. J Allergy Clin Immunol. (2013) 132:729–736.e12. doi: 10.1016/j.jaci.2013.02.018
Keywords: antiallergy, FcεRIβ, Lyn, mast cell, scrodentoid A
Citation: Qian F, Zhang L, Lu S, Mao G, Guo F, Liu P, Xu J and Li Y (2019) Scrodentoid A Inhibits Mast Cell–Mediated Allergic Response by Blocking the Lyn–FcεRIβ Interaction. Front. Immunol. 10:1103. doi: 10.3389/fimmu.2019.01103
Received: 26 November 2018; Accepted: 30 April 2019;
Published: 16 May 2019.
Edited by:
Toshiaki Kawakami, La Jolla Institute for Immunology (LJI), United StatesReviewed by:
Ryo Suzuki, Kanazawa University, JapanCopyright © 2019 Qian, Zhang, Lu, Mao, Guo, Liu, Xu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinwen Xu, and4dTEwMDFAMTYzLmNvbQ==
Yiming Li, eW1saXVzQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.