 Natalie E. Stevens
Natalie E. Stevens Allison J. Cowin
Allison J. Cowin Zlatko Kopecki
Zlatko Kopecki- Regenerative Medicine Laboratory, Future Industries Institute, University of South Australia, Adelaide, SA, Australia
One of the most important functions of the skin besides regulating internal body temperature includes formation of the barrier between the organism and the external environment, hence protecting against pathogen invasion, chemical and physical assaults and unregulated loss of water and solutes. Disruption of the protective barrier is observed clinically in blisters and erosions of the skin that form in autoimmune blistering diseases where the body produces autoantibodies against structural proteins of the epidermis or the epidermal-dermal junction. Although there is no cure for autoimmune skin blistering diseases, immune suppressive therapies currently available offer opportunities for disease management. In cases where no treatment is sought, these disorders can lead to life threatening complications and current research efforts have focused on developing therapies that target autoantibodies which contribute to disease symptoms. This review will outline the involvement of the skin barrier in main skin-specific autoimmune blistering diseases by describing the mechanisms underpinning skin autoimmunity and review current progress in development of novel therapeutic approaches targeting the underlying causes of autoimmune skin blistering diseases.
Introduction
The stratified squamous epithelium of the human epidermis forms a continuous barrier against the external environment and impairments in epithelial adhesions lead to disorders characterized by significant morbidity and/or mortality (1). The hallmark feature of autoimmune blistering diseases (AIBDs) is the disruption of the intact skin barrier as a consequence of blistering and erosions caused by production of autoantibodies against structural proteins in the epidermis or at the epidermal-dermal junction. AIBDs generally occur in the elderly, and often have substantial clinical and immunopathological overlap and polymorphic clinical presentation which can make diagnosis challenging (2). Immunologically, these conditions are driven by humoral and cellular autoimmune responses directed against distinct target antigens and can be classed in three main groups including pemphigoid and pemphigus diseases as well as dermatitis herpetiformis (DH) (3).
Over the past four decades, our knowledge of the pathophysiology of AIBDs has been greatly advanced by demonstrating that passive transfer of antibodies against skin antigens can disrupt the skin barrier and induce blisters in experimental animals models with clinical, histologic, and immunopathogenic responses similar to those observed in human disease (1). Each AIBD is characterized by the presence of specific autoantibodies targeting distinct antigens in the epidermis or at the dermal-epidermal junction. Intraepidermal blistering found in pemphigus disorders are caused by autoantibodies targeting cadherin proteins in desmosomes; subtypes pemphigus vulgaris and pemphigus foliaceus are associated with antibodies against desmoglein (dsg)-3 and−1, respectively. In bullous pemphigoid (BP), autoantibodies target two hemidesmosome components BP180 and BP230; and in epidermolysis bullosa acquisita (EBA) patients have autoantibodies target type VII collagen anchoring fibrils. In DH patients, autoantibodies target tissue and epidermal transglutaminase (eTG) proteins (3) however recently a case was reported where autoimmune intraepidermal and subepidermal blistering disease coexisted with a patient who was reported to have autoantibodies to both desmoglein (Dsg) 1 and BP230 (4).
AIBDs typically present with generalized blister eruption associated with itch however atypical presentations are often encountered. For example, 20% of BP patients present with “non-bullous” presentations, while anti-p200 pemphigoid patients that normally present with tense blisters with erythematosus often show normal skin resembling BP. Additionally, epidermolysis bullosa acquisita, an autoimmune disease associated with autoantibodies against type VII collagen, has several phenotypes including a classical form that mimics dystrophic epidermolysis bullosa, an inflammatory form that mimics BP, or a form more similar to mucous membrane pemphigoid-like lesions (2).
Diagnosis of AIBDs relies on direct immunofluorescence microscopy studies and immunoserological assays (5, 6). Multiple mechanisms of skin barrier disruption and blister formation in AIBDs have been described: in pemphigus disorders steric hindrance (the direct inhibition of protein-protein binding by autoantibodies) and cell signaling events cause desmosomal instability, while complement and inflammatory cell activation mediated through Fc-signaling cause keratinocyte death and blister formation in pemphigoid and epidermolysis bullosa acquisita (7–9). Development of targeted therapies and management of affected patients is often challenging due to frequent relapses, lack of efficacy and number of adverse events (10, 11). Current standard treatment options rely on non-specific immunosuppression, highlighting the need for development of targeted therapeutic approaches (12, 13). In this review we will focus on skin barrier involvement in mechanisms underpinning autoimmunity and describe the latest approaches for development of targeted therapeutics for the treatment of AIBDs.
Skin Barrier and Mechanisms Underpinning Autoimmune Skin Blistering
More than 2.5% of the world's population is affected by autoantibody driven autoimmune disease, including AIBDs (7). The principles of autoantibody generation and detection in AIBDs have been reviewed extensively (7). Technological advancement in the last two decades have allowed us to identify the sequence of specific nanostructural and functional changes in the skin barrier following the binding of autoantibodies and define critical pathways and processes responsible for autoimmune pathology (14). The pathogenesis of AIBDs can be divided into three phases: (i) the induction phase (loss of self-tolerance or the initiation of autoimmunity to the target antigen), (ii) the maintenance phase (maintained production of autoantibodies) and (iii) the effector phase (autoantibody-mediated tissue damage). Specific mechanisms relating to these phases have been described for AIBDs, including pemphigus disorders, BP, EBA, and DH.
Induction of Autoimmunity Against Skin Antigens
There are multiple theories that explain how the loss of tolerance to self-antigens initially occurs and it is understood that the majority of AIBDs are a product of several aberrant processes which disrupt skin barrier homeostasis. Genetic factors play an important role, as specific skin blistering diseases have varying prevalence in different populations and inherited human leukocyte antigen (HLA) types are associated with autoreactivity to specific autoantigens (15). Multiple HLA alleles have been identified which are associated with pemphigus vulgaris (15, 16), BP (17, 18), and EBA (19, 20). Genetic susceptibility is not limited to HLA types, as pemphigus vulgaris has been associated with mutations in ST18 (a gene encoding a pro-apoptotic transcription factor) in certain populations (21) and experimental models of EBA have identified non-HLA murine gene loci that confer susceptibility to disease development (22), however further studies are required to extrapolate these findings to clinical populations.
Cell damage has been proposed as a common “triggering factor” which causes development of pathogenic adaptive autoimmune reactions—cell damage due to surgical trauma (23), UV radiation (24), neurological disorders and other pre-existing conditions (25–29), viral infection (30–33), and radiotherapy (34–36) have all been associated with disrupted skin barrier function and development of AIBDs (37). Cell damage via necrosis or necroptosis releases a complex intracellular milieu into the extracellular space which serves as a source of sensitizing autoantigens (38); additionally cell death results in the release of damage associated molecular patterns which stimulate localized inflammation and wound healing processes (39, 40). Normal healing responses following trauma aiming to re-establish the skin barrier cause infiltration of dendritic cells and other antigen presenting cells which may also participate in autoimmune sensitization (41, 42) of AIBDs.
Epitope spreading is an inbuilt mechanism of the adaptive immune system that aids in protecting against changing pathogens (43), however spreading from pathogenic to autologous epitopes and molecular mimicry of similar epitopes may also contribute to the formation of AIBDs (44). Fogo selvage, an endemic form of pemphigus foliaceus found in Brazilian populations, is associated with a history of sand fly bites and characterized by autoantibodies against Dsg1. These autoantibodies have shown cross reactivity to proteins present in sand fly saliva (45), which may represent epitope spreading from foreign proteins to similar autoantigens. Epitope spreading is also thought to be involved in paraneoplastic (PNP) pemphigus (46) where tumor-associated antigens may become targeted in an effort to destroy the tumor, however similar antigens may also be shared by keratinocytes (47). PNP pemphigus is most commonly associated with lymphatic malignancies, including non-Hodgkin's lymphoma and chronic lymphocytic leukemia. These malignancies are associated with the production and release of cytokines which can lead to over-stimulation of humoral immunity and autoimmune reactions, including disruption of skin barrier and development of AIBDs. Findings of autoimmune skin blistering in carcinoma patients has fuelled speculation that these diseases may be triggered by an anti-tumor immune response (48–50), however further studies are required to determine the relationship between these findings (51, 52). Coeliac-disease associated skin blistering, known as DH, is caused by antibodies against gluten-induced digestive enzyme tissue transglutaminase which undergo epitope spreading to cross-react with epidermal transglutaminase (eTG) leading to the disruption of the skin barrier and subsequent skin blistering (53, 54). Epitope spreading may also contribute to the diversity of and disease progression of AIBDs, as epitope spreading to related autoantigens has been associated with atypical or altered disease presentations (53, 55, 56).
AIBDs have been associated with the use of certain drugs which trigger pathogenesis through a variety of mechanisms. One of the most well-described etiologies is BP in diabetic patients taking dipeptidyl-peptidase 4 (DPP-4) inhibitors (57–59) which present with antibodies against the mid-portion of BP180. It has been suggested that DPP-4 inhibition reduces plasmin production and alters BP180 cleavage, resulting in altered antigenicity of BP180 (60) which is supported by the finding that symptoms generally subside after drug discontinuation. The use of immune checkpoint inhibitors for cancer therapy has been linked to secondary development of AIBD including mucous membrane pemphigoid and BP (61, 62), likely a consequence of nonspecific immune activation.
Maintained Autoantibody Production
Autoantibodies are often present in healthy individuals but are generally non-pathogenic IgM antibodies with low affinity present in low levels and do not alter skin barrier homeostasis. For autoantibodies to gain pathogenicity, class-switching to IgG or IgA subtypes, somatic mutation and increased production occur after exposure to self-antigen. Glycosylation and sialylation patterns on autoantibodies also contribute to pathogenicity and antibodies stimulated via T-cell interaction within germinal centers exhibit reduced sialyation patterns and are pro-inflammatory (63). In the context of AIBDs, the presence of self-antigen released by damaged keratinocytes may stimulate production of autoantibodies, which in turn bind to healthy tissue to stimulate skin barrier disruption and further tissue damage and promote more autoantibody generation in a self-perpetuating cycle. Continued production of pathogenic autoantibodies may be achieved by ongoing B cell activation and production of short-lived plasma cells (64, 65) or the production of long-lived plasma cells which are challenging to target (66–68). T-cell driven education within the germinal center appears to be a requirement for the development of long-lived plasma cells, which gives rise to both lasting immunity against pathogens and chronic autoimmunity. Murine models of EBA have also demonstrated the presence of plasma cells with “intermediate” lifespans which contribute to autoantibody persistence (69). Alterations in cellular immune networks also contribute to maintained autoantibody production and disease chronicity: Increased Th1 and Th17 cytokines and chemokines have been reported in patients with pemphigus disorders (70, 71), BP (72, 73), and DH (74) and changes in Treg populations are also associated with AIBDs (75, 76), with Tregs thought to be protective against pathogenic autoantibody production (77, 78). Higher frequencies of Th17 cells secreting IL-21 have been reported in pemphigus lesions which form a tertiary lymph node like structure within the skin and promote autoantibody production (79). Further investigation into cellular contributions to autoantibody production may reveal additional therapeutic targets which can be used to control AIBDs.
Autoantibody-induced Tissue Damage
Autoimmune diseases of the skin are the result of pathological processes caused by autoantibodies against skin antigens. A number of common antigens targeted by disease-associated autoantibodies have been discovered, including antigens present on desmosomes, hemidesmosomes and proteins expressed by keratinocytes. Transfer of patient autoantibodies against Dsg 1 (80, 81) and Dsg 3 (82) and type VII collagen (83) is sufficient to cause skin barrier disruption and epidermal blistering in mice consistent with the associated clinical disease, thus autoantibodies are considered instrumental to pathogenesis in many AIBDs. Figure 1 provides a diagram summary of blistering mechanisms and skin barrier disruption in prototypic AIBDs pemphigus disorders, BP, EBA, and DH. Inflammation triggered by autoantibody binding recruits and activates a number of myeloid and lymphoid cell subsets that participate in blister development and damage to the skin barrier, however these vary with specific disease and clinical context. Previous reviews have in details described the preclinical animal studies and some clinical evidence elucidating the contributions of eosinophils (84, 85), mast cells (86, 87), Th17 (70, 88), and Treg (89–91) cells to AIBD's, and these may represent novel therapeutic targets. Here we focus on contributions of autoantibodies to skin barrier disruption in different disease settings.
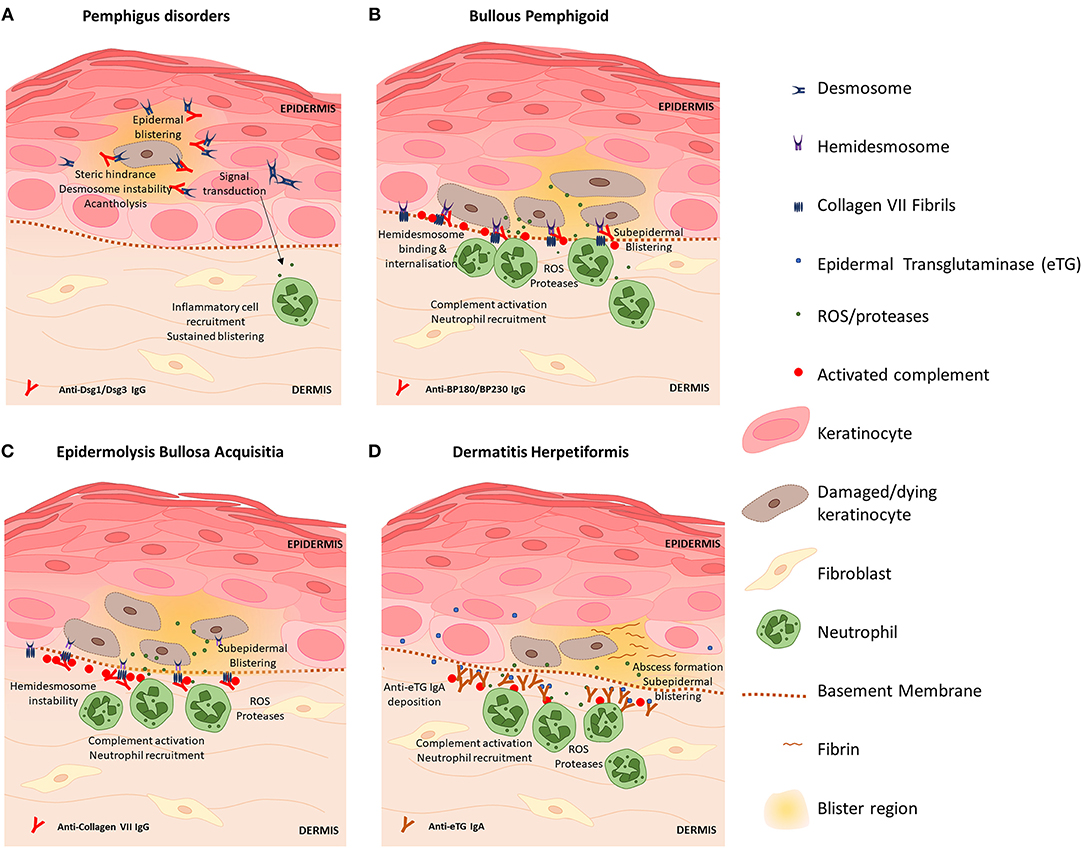
Figure 1. Blistering mechanisms of prototypic autoimmune skin blistering disorders result in skin barrier disruption. (A) Pemphigus disorders are caused by autoantibodies against desmoglein (Dsg) proteins Dsg1 and Dsg3. Binding of anti-Dsg destabilizes desmosomes to cause acantholysis of keratinocytes within the epidermis and triggers keratinocyte signal transduction events which promote inflammation, skin barrier disruption and further skin blistering. (B) Blisters in bullous pemphigoid are caused by anti-BP180 antibodies which bind hemidesmosomes on basal keratinocytes and trigger complement activation and inflammatory responses including ROS and protease release by neutrophils which directly kill keratinocytes. Skin barrier disruption and skin blistering is caused by keratinocyte death and sustained localized inflammation. (C) Epidermolysis bullosa acquisita is caused by anti-Collagen VII antibodies which bind fibrils that anchor hemidesmosomes to the basement membrane. Deposition of IgG induces complement activation via the classical pathway and activation of neutrophils. Basal keratinocytes sustain damage via action of neutrophil-derived ROS and proteases resulting in splitting at the dermal-epidermal junction and skin barrier disruption. (D) Dermatitis hepetiformis (DH) is caused by cross-reactive antibodies that bind epidermal transglutaminase (eTG). eTG is produced by keratinocytes and accumulates in the papillary dermis where it forms immunogenic immune complexes with anti-eTG IgA that trigger complement activation. Fibrin deposition and influx of leukocytes (which damage keratinocytes via release of ROS and proteases) cause the formation of neutrophilic abscesses which develop into fluid-filled subepidermal blisters that disrupt the intact skin barrier.
Unraveling the precise mechanisms of autoantibody-induced pathology has been the focus of much research in recent years. Autoantibodies against Dsg proteins, as found in pemphigus diseases, were initially thought to interfere with Dsg-Dsg interactions in desmosomes by steric hindrance (92) however evidence for direct effects of Dsg-anti-Dsg binding on intracellular signaling events was later discovered, including the p38 mitogen-activated protein kinase pathway leading to acantholysis (93, 94). Anti-Dsg may also reduce the number of desmosomes by clustering Dsg on the cell surface and interfering with normal turnover of desmosomal proteins, thereby depleting desmosomes of Dsg (95) and promoting acantholysis. In more recent years, other autoantibody species (96–98) and non-Dsg interactions (99) have been identified as contributing to pemphigus pathology and have prompted the hypothesis that multiple pathways may act synergistically (100) to cause classical pemphigus disease pathology (101).
In BP, autoantibodies against BP180 and BP230 components of hemidesmosomes produce blistering at the dermal-epidermal junction. Activation of the complement cascade via classical and alternative pathways has been demonstrated to contribute to skin barrier disruption and BP pathology (102) and C3 deposition at the epidermal basement membrane is a common clinical finding (103). Complement activation induces inflammation and damages keratinocytes via cytotoxic action of neutrophils leading to skin blistering and barrier disruption (104). Though complement-independent mechanisms of BP pathology have been since described (105, 106) including direct activation of neutrophils via immune complex-FcγR binding, complement activation is still a prevalent target for novel BP therapeutics (103, 107). Autoreactive IgE and eosinophilia are common findings in BP patients and IgE immune complexes binding and activating eosinophils thereby contributing to blister formation has been shown in in vitro and in vivo studies (108), implicating eosinophils as potential therapeutic targets in BP.
Disease pathology in EBA is caused by predominantly IgG1 and IgG3 autoantibodies that bind Collagen VII within anchoring fibrils at the dermal-epidermal junction. As a rare disease, much of what is known about EBA pathogenesis has been elucidated with experimental models. Like BP, complement activation is considered key to EBA pathogenesis (109), however the alternative pathway appears to be the dominant pathway behind experimental EBA pathology (110). Activation of complement induces inflammation, leukocyte extraversion, complement activation and subsequent tissue damage and disruption of skin barrier via release of ROS and proteases from neutrophils and other myeloid cells. Ex-vivo studies of patient serum incubated with healthy skin and donor neutrophils exhibit loss of epidermal adherence, hence clearly indicating that antibodies mediate clinical EBA blistering via neutrophil activation (111). The role of T cells in EBA pathology is yet to be fully elucidated, however murine studies show that NKT and γδT cells likely amplify tissue damage in EBA via interaction with immune complexes and neutrophils (112).
DH is characterized by accumulation of ant-eTG IgA antibodies within the papillary dermis, however eTG is primarily expressed within superficial epidermal layers (113). It is hypothesized that eTG may be released into the blood, where interaction with IgA occurs in nearby dermal vessels; alternatively eTG may be deposited along the basement membrane as a result of trauma (54, 114), however further research is required to confirm these hypotheses. Following IgA deposition, papillary abscesses characterized by a neutrophilic infiltrate and fibrin accumulation form which develop into a split at the basement membrane and subepidermal blistering. Patients with DH show reduced levels of anti-inflammatory IL-10 and reduced Treg cell numbers in lesional skin compared to healthy skin (115) which indicates the role of Tregs in modulating local inflammatory responses in DH and represents an attractive therapeutic target.
Targeted Approaches for Treatment of AIBDs
The use of different in-vitro systems and experimental animal models in recent years has significantly improved our understanding of AIBDs ultimately leading to novel diagnostic tools and differentiated therapeutic approaches for these disorders (6, 7, 75, 89, 109, 116–123). These approaches can be broadly grouped into: traditional and topical therapies; rituximab and intravenous immunoglobulins and other treatments in pre-clinical and clinical trials. In this review, we will provide a broad summary of traditional treatments and novel emerging therapies which are also summarized in Table 1.
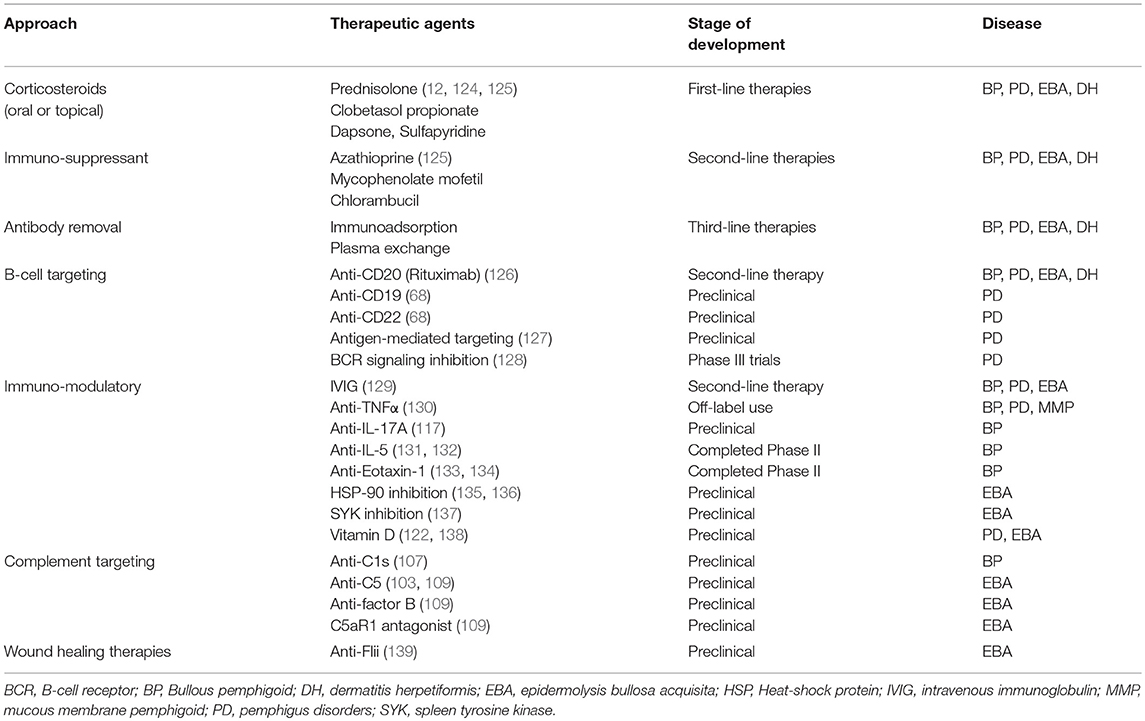
Table 1. Current and emerging therapeutic approaches in autoimmune skin blistering diseases.
First line therapies for AIBDs generally include systemic oral or intravenous corticosteroids (0.5–2.0 mg/kg/day) such as prednisolone (12, 29). Topical high potency corticosteroids such as clobetasol propionate have also been demonstrated to be efficient alternatives to oral prednisolone therapy in BP by reducing autoantibodies against BP180 and BP230 (2). For more severe patients unresponsive to topical therapy, oral prednisolone is combined with adjuvant immunosuppressant (azathioprine, mycophenolate, or rituximab). Unlike other AIBDs, oral corticosteroids do not normally have a dramatic effect in EBA and to date there are no randomized controlled trials providing level 1 evidence for EBA treatment (4). Milder forms of EBA and DH may respond well to topical steroids including dapsone or sulfapyridine, while rituximab has been reported to be effective for severe patients (12, 61, 124, 140, 141). Combining conventional systemic corticosteroids with rituximab treatment has also showed beneficial clinical outcomes in mucous membrane pemphigoid diseases (142).
Second line therapies include corticosteroid-sparing agents azathioprine, mycophenolate mofetil, or rituximab, which may be combined with intravenous immunoglobulin (IVIG) therapy. Clinically, IVIG administration has been shown to significantly improve BP disease symptoms for several weeks after infusion (129). Third line therapies are dependent on individual patient needs and include therapies in clinical trials: cyclophosphamide, IgE-targeted therapies, immunoadsorption to remove pathogenic autoantibodies, IVIG, methotrexate, and plasma exchange.
Preclinical studies have identified potential emerging therapies which target immunological mechanisms in AIBDs, which aim to reduce damaging inflammatory processes. Direct targeting of antibody-producing B cells is efficacious, as anti-CD20 antibody (Rituximab) has been successfully used in the clinic for multiple AIBDs, however not all patients respond equally. Targeting other B cell markers including anti-CD19 and anti-CD22, or antigen-specific B cell receptors may improve targeting of long-lived plasma cells which produce pathogenic autoantibody in patients that are refractory to rituximab. Therapies targeting components of the complement cascade have exhibited success in preclinical and clinical trials, however concerns with side effects associated with existing complement targeting therapies may limit the clinical utility of these approaches (143, 144). Previous studies have highlighted the roles of cytokines in mediating AIBD tissue damage and have identified multiple therapeutic targets including TNFα (130, 145), IL-5 (84), IL-17A(117), and IL-1 blockade (146) and administration of anti-inflammatory IL-10 (147). Immunomodulatory anti-cytokine therapies are in various stages of development, with existing TNF-α inhibitors showing efficacy in treating AIBDs and antibodies against IL-5 and eotaxin-1 having entered clinical trials for BP treatment (148–150). Like immunomodulatory therapies for other autoimmune diseases, these approaches are likely to be associated with known side effects of immunosuppression (151) thus further trials are needed to identify the safest and most efficacious dose regimens for specific diseases. Preclinical studies have identified cellular populations which may be exploited as therapeutic targets, such as Langerhans cells (152), dendritic cells (153), granulocytes (104, 111, 133, 154), and multiple T cell subsets including Th17 (117, 155), Treg (75), NKT (112), γδT (112), and CD8+ (156). Approaches to target specific immune cell functions are currently in preclinical development, including small molecule inhibition of spleen tyrosine kinase (137), an enzyme involves in proinflammatory Fc-receptor signal transduction in myeloid cells, and metabolites of Vitamin D which reduce myeloid cell activation and ROS production (122).
Understanding the mechanisms of autoantibody mediated tissue damage is critical in development of novel targeted therapies, however understanding the mechanisms behind resolution of blistering and inflammation in AIBDs can also offer insights into potential novel treatment modalities. Unresolved blistering can impact patient quality of life and increase risk of bleeding, infection and tumor development (157). Optimal healing is especially important for patients with extensive cutaneous blistering, or mucosal blistering which affects feeding, digestion and function of other organs (158, 159). Studies surrounding the role of Flightless protein (Flii) in skin blistering have revealed the first mechanism leading to the resolution of blistering and inflammation in antibody transfer induced EBA (8), and further research into similar pathways may reveal more potential therapeutic strategies where wound healing therapies may offer opportunities for decreasing the clinical symptoms associated with AIBDs.
In summary, there is a high unmet need for new targeted therapeutic approaches focussed on restoring the integrity of the skins' barrier 22and address both blistering mechanisms and clinical symptoms in systemic AIBDs. Innovative designs of randomized controlled trials using validated scales of assessment are needed to drive the development of novel therapeutic strategies for patients with AIBDs. Additionally, research efforts should focus on adapting immunomodulatory approaches that have been shown to be effective in other autoimmune diseases in order to target common pathogenic mechanisms and developing a better understanding of blister resolution and healing to improve patient symptoms.
Conclusion
As highlighted in this review, the contribution of the skin barrier to the mechanisms underpinning autoimmunity has greatly improve our understanding of AIBDs. Development of novel targeted therapeutics restoring skin barrier function and homeostasis will lead to improved treatment of patients with AIBDs.
Contribution to the Field Statement
Autoimmune skin blistering diseases are caused by pathogenic autoantibodies which trigger cellular, biochemical and immunological processes that disrupt the skin barrier and cause chronic blistering in patients. Understanding the mechanisms behind these processes has lead to the development of new targeted therapeutics which are in various stages of preclinical and clinical development. Current therapeutic approaches rely heavily on immunosuppressants and corticosteroids which are associated with adverse effects including risk of infection. Thus, new therapeutics are necessary to effectively control skin blistering and restore the skin barrier with fewer side effects. In this review, we highlight the different mechanisms behind autoimmune skin blistering disease development including initiation, maintenance and tissue damage. Additionally, we summarize current treatment and emerging therapeutics for autoimmune skin blistering diseases and highlight blistering mechanisms which may be exploited for development of novel targeted therapeutics.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
AC is supported by NHMRC Senior Research Fellowship (GNT#1102617) and ZK is supported by a Future Industries Institute Foundation Fellowship.
References
1. Yancey KB. The pathophysiology of autoimmune blistering diseases. J Clin Invest. (2005) 115:825–8. doi: 10.1172/JCI200524855
2. Amber KT, Murrell DF, Schmidt E, Joly P, Borradori L. Autoimmune subepidermal bullous diseases of the skin and mucosae: clinical features, diagnosis, and management. Clin Rev Allergy Immunol. (2018) 54:26–51. doi: 10.1007/s12016-017-8633-4
3. Vorobyev A, Ludwig RJ, Schmidt E. Clinical features and diagnosis of epidermolysis bullosa acquisita. Expert Rev Clin Immunol. (2017) 13:157–69. doi: 10.1080/1744666X.2016.1221343
4. Sugai T, Ujiie H, Nakamura H, Kikuchi K, Iwata H, Shimizu H. Case of autoimmune intraepidermal and subepidermal blistering disease in which autoantibodies to desmoglein 1 and BP230 coexist. J Dermatol. (2018). doi: 10.1111/1346-8138.14734. [Epub ahead of print].
5. Kopecki Z, Ludwig RJ, Cowin AJ. Cytoskeletal regulation of inflammation and its impact on skin blistering disease epidermolysis bullosa acquisita. Int J Mol Sci. (2016) 17:1116. doi: 10.3390/ijms17071116
6. Witte M, Zillikens D, Schmidt E. diagnosis of autoimmune blistering diseases. Front Med (Lausanne). (2018) 5:296. doi: 10.3389/fmed.2018.00296
7. Ludwig RJ, Vanhoorelbeke K, Leypoldt F, Kaya Z, Bieber K, McLachlan SM, et al. Mechanisms of autoantibody-induced pathology. Front Immunol. (2017) 8:603. doi: 10.3389/fimmu.2017.00603
8. Ludwig R. Immune mechanism-targeted treatment of experimental epidermolysis bullosa acquisita. Expert Rev Clin Immunol. (2015) 11:1365–78. doi: 10.1586/1744666X.2015.1085801
9. Kamaguchi M, Iwata H. The diagnosis and blistering mechanisms of mucous membrane pemphigoid. Front Immunol. (2019) 10:34. doi: 10.3389/fimmu.2019.00034
10. Murrell DF. Autoimmune diseases of the skin. Immunol Allerg Clin North Am. (2012) 32:xiii–iv. doi: 10.1016/j.iac.2012.04.015
11. Kasprick A, Bieber K, Ludwig RJ. Drug discovery for pemphigoid diseases. Curr Protoc Pharmacol. (2019) 84:e55. doi: 10.1002/cpph.55
12. Koga H, Prost-Squarcioni C, Iwata H, Jonkman MF, Ludwig RJ, Bieber K. Epidermolysis bullosa acquisita: the 2019 update. Front Med (Lausanne). (2018) 5:362. doi: 10.3389/fmed.2018.00362
13. Kasperkiewicz M, Schmidt E, Zillikens D. Current therapy of the pemphigus group. Clin Dermatol. (2012) 30:84–94. doi: 10.1016/j.clindermatol.2011.03.014
14. Seiffert-Sinha K, Yang R, Fung CK, Lai KW, Patterson KC, Payne AS, et al. Nanorobotic investigation identifies novel visual, structural and functional correlates of autoimmune pathology in a blistering skin disease model. PLoS ONE. (2014) 9:e106895. doi: 10.1371/journal.pone.0106895
15. Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Front Med (Lausanne). (2018) 5:226. doi: 10.3389/fmed.2018.00226
16. Li S, Zhang Q, Wang P, Li J, Ni J, Wu J, et al. Association between HLA-DQB1 polymorphisms and pemphigus vulgaris: a meta-analysis. Immunol Invest. (2018) 47:101–12. doi: 10.1080/08820139.2017.1385622
17. Fang H, Shen S, Zheng X, Dang E, Zhang J, Shao S, et al. Association of HLA class I and class II alleles with bullous pemphigoid in Chinese Hans. J Dermatol Sci. (2018) 89:258–62. doi: 10.1016/j.jdermsci.2017.11.014
18. Ujiie H, Muramatsu K, Mushiroda T, Ozeki T, Miyoshi H, Iwata H, et al. HLA-DQB1*03:01 as a biomarker for genetic susceptibility to bullous pemphigoid induced by DPP-4 inhibitors. J Invest Dermatol. (2018) 138:1201–4. doi: 10.1016/j.jid.2017.11.023
19. Zumelzu C, Le Roux-Villet C, Loiseau P, Busson M, Heller M, Aucouturier F, et al. Black patients of African descent and HLA-DRB1*15:03 frequency overrepresented in epidermolysis bullosa acquisita. J Invest Dermatol. (2011) 131:2386–93. doi: 10.1038/jid.2011.231
20. Lee CW, Kim SC, Han H. Distribution of HLA class II alleles in Korean patients with epidermolysis bullosa acquisita. Dermatology. (1996) 193:328–9. doi: 10.1159/000246282
21. Vodo D, Sarig O, Geller S, Ben-Asher E, Olender T, Bochner R, et al. Identification of a functional risk variant for pemphigus vulgaris in the ST18 gene. PLoS Genet. (2016) 12:e1006008. doi: 10.1371/journal.pgen.1006008
22. Ludwig RJ, Muller S, Marques A, Recke A, Schmidt E, Zillikens D, et al. Identification of quantitative trait loci in experimental epidermolysis bullosa acquisita. J Invest Dermatol. (2012) 132:1409–15. doi: 10.1038/jid.2011.466
23. Sen BB, Ekiz O, Rifaioglu EN, Sen T, Atik E, Dogramaci AC. Localized bullous pemphigoid occurring on surgical scars. Indian J Dermatol Venereol Leprol. (2013) 79:554. doi: 10.4103/0378-6323.113111
24. Sanchez-Palacios C, Chan LS. Development of pemphigus herpetiformis in a patient with psoriasis receiving UV-light treatment. J Cutan Pathol. (2004) 31:346–9. doi: 10.1111/j.0303-6987.2004.0188.x
25. Khosravani S, Handjani F, Alimohammadi R, Saki N. Frequency of neurological disorders in bullous pemphigoid patients: a cross-sectional study. Int Sch Res Notices. (2017) 2017:6053267. doi: 10.1155/2017/6053267
26. Gambichler T, Segert H, Hoxtermann S, Schmitz L, Altmeyer P, Teegen B. Neurological disorders in patients with bullous pemphigoid: clinical and experimental investigations. J Eur Acad Dermatol Venereol. (2015) 29:1758–62. doi: 10.1111/jdv.12995
27. Bastuji-Garin S, Joly P, Lemordant P, Sparsa A, Bedane C, Delaporte E, et al. Risk factors for bullous pemphigoid in the elderly: a prospective case-control study. J Invest Dermatol. (2011) 131:637–43. doi: 10.1038/jid.2010.301
28. Varpuluoma O, Jokelainen J, Forsti AK, Timonen M, Huilaja L, Tasanen K. Dermatitis herpetiformis and celiac disease increase the risk of bullous pemphigoid. J Invest Dermatol. (2019) 139:600–4. doi: 10.1016/j.jid.2018.10.010
29. Bech R, Kibsgaard L, Vestergaard C. Comorbidities and treatment strategies in bullous pemphigoid: an appraisal of the existing litterature. Front Med (Lausanne). (2018) 5:238. doi: 10.3389/fmed.2018.00238
30. Baroero L, Coppo P, Bertolino L, Maccario S, Savino F. Three case reports of post immunization and post viral Bullous Pemphigoid: looking for the right trigger. BMC Pediatr. (2017) 17:60. doi: 10.1186/s12887-017-0813-0
31. Jang H, Jin YJ, Yoon CH, Kim CW, Kim L. Bullous pemphigoid associated with chronic hepatitis C virus infection in a hepatitis B virus endemic area: a case report. Medicine (Baltimore). (2018) 97:e0377. doi: 10.1097/MD.0000000000010377
32. Fernandes NC, Rampinelli H, Souza LM, Guimaraes M. Refractory pemphigus foliaceus associated with herpesvirus infection: case report. Rev Inst Med Trop Sao Paulo. (2017) 59:e41. doi: 10.1590/s1678-9946201759041
33. Gee SN, Velez NF, Sepehr A, Burgin S. Two distinct viral infections complicating pemphigus foliaceus. Dermatol Online J. (2012) 18:3.
34. Furukawa F, Ozaki M, Imamura S, Hirose S. Bullous pemphigoid associated with radiotherapy for esophageal carcinoma. Dermatologica. (1981) 162:451–4. doi: 10.1159/000250316
35. Melani L, Giomi B, Antiga E, Torchia D, Caproni M, Fabbri P. Radiation therapy as a trigger factor for initially localized bullous pemphigoid. Breast J. (2005) 11:485–6. doi: 10.1111/j.1075-122X.2005.00150.x
36. Nguyen T, Kwan JM, Ahmed AR. Relationship between radiation therapy and bullous pemphigoid. Dermatology. (2014) 229:88–96. doi: 10.1159/000362208
37. Fujinami RS, von Herrath MG, Christen U, Whitton JL. Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev. (2006) 19:80–94. doi: 10.1128/CMR.19.1.80-94.2006
38. MacKay IR, Leskovsek NV, Rose NR. Cell damage and autoimmunity: a critical appraisal. J Autoimmun. (2008) 30:5–11. doi: 10.1016/j.jaut.2007.11.009
39. Land WG. The role of damage-associated molecular patterns in human diseases: part I - promoting inflammation and immunity. Sultan Qaboos Univ Med J. (2015) 15:e9–21.
40. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. (2017) 22:1189–204. doi: 10.1007/s10495-017-1413-z
41. Saadeh D, Kurban M, Abbas O. Update on the role of plasmacytoid dendritic cells in inflammatory/autoimmune skin diseases. Exp Dermatol. (2016) 25:415–21. doi: 10.1111/exd.12957
42. Ma L, Chan K, Trendell-Smith N, Lo C, To C, Huang F. Necrotic cells induce systemic autoimmune disease in vivo by activation of dendritic cells (DCs). J Allergy Clin Immunol. (2005) 115:891. doi: 10.1016/j.jaci.2005.01.047
43. Powell AM, Black MM. Epitope spreading: protection from pathogens, but propagation of autoimmunity? Clin Exp Dermatol. (2001) 26:427–33. doi: 10.1046/j.1365-2230.2001.00852.x
44. Aoki V, Millikan RC, Rivitti EA, Hans-Filho G, Eaton DP, Warren SJ, et al. Environmental risk factors in endemic pemphigus foliaceus (fogo selvagem). J Investig Dermatol Symp Proc. (2004) 9:34–40. doi: 10.1111/j.1087-0024.2004.00833.x
45. Qian Y, Jeong JS, Maldonado M, Valenzuela JG, Gomes R, Teixeira C, et al. Cutting Edge: Brazilian pemphigus foliaceus anti-desmoglein 1 autoantibodies cross-react with sand fly salivary LJM11 antigen. J Immunol. (2012) 189:1535–9. doi: 10.4049/jimmunol.1200842
46. Wieczorek M, Czernik A. Paraneoplastic pemphigus: a short review. Clin Cosmet Investig Dermatol. (2016) 9:291–5. doi: 10.2147/CCID.S100802
47. Chan LS. Epitope spreading in paraneoplastic pemphigus: autoimmune induction in antibody-mediated blistering skin diseases. Arch Dermatol. (2000) 136:663–4. doi: 10.1001/archderm.136.5.663
48. Fernandes J, Barad P, Shukla P. Association of bullous pemphigoid with malignancy: a myth or reality? Indian J Dermatol. (2014) 59:390–3. doi: 10.4103/0019-5154.135493
49. Das A, Das S, Das SK, Basuthakur S. A case of paraneoplastic bullous pemphigoid in association with squamous cell carcinoma of lung. J Postgrad Med. (2015) 61:197–9. doi: 10.4103/0022-3859.150906
50. Noguchi K, Kawamura H, Ishizu H, Okada K. Dramatic resolution of bullous pemphigoid after surgery for gastric cancer: a case report. Int J Surg Case Rep. (2014) 5:212–4. doi: 10.1016/j.ijscr.2014.02.008
51. Balestri R, Magnano M, La Placa M, Patrizi A, Angileri L, Tengattini V, et al. Malignancies in bullous pemphigoid: a controversial association. J Dermatol. (2016) 43:125–33. doi: 10.1111/1346-8138.13079
52. Cai SC, Allen JC, Lim YL, Tan SH, Tang MB. Association of bullous pemphigoid and malignant neoplasms. JAMA Dermatol. (2015) 151:665–7. doi: 10.1001/jamadermatol.2014.5263
53. Didona D, Di Zenzo G. Humoral epitope spreading in autoimmune bullous diseases. Front Immunol. (2018) 9:779. doi: 10.3389/fimmu.2018.00779
54. Clarindo MV, Possebon AT, Soligo EM, Uyeda H, Ruaro RT, Empinotti JC. Dermatitis herpetiformis: pathophysiology, clinical presentation, diagnosis and treatment. An Bras Dermatol. (2014) 89:865–75; quiz: 76–7. doi: 10.1590/abd1806-4841.20142966
55. Jonkman MF, Schuur J, Dijk F, Heeres K, de Jong MC, van der Meer JB, et al. Inflammatory variant of epidermolysis bullosa acquisita with IgG autoantibodies against type VII collagen and laminin alpha3. Arch Dermatol. (2000) 136:227–31. doi: 10.1001/archderm.136.2.227
56. Di Zenzo G, Thoma-Uszynski S, Calabresi V, Fontao L, Hofmann SC, Lacour JP, et al. Demonstration of epitope-spreading phenomena in bullous pemphigoid: results of a prospective multicenter study. J Invest Dermatol. (2011) 131:2271–80. doi: 10.1038/jid.2011.180
57. Kridin K, Bergman R. Association of bullous pemphigoid with dipeptidyl-peptidase 4 inhibitors in patients with diabetes: estimating the risk of the new agents and characterizing the patients. JAMA Dermatol. (2018) 154:1152–8. doi: 10.1001/jamadermatol.2018.2352
58. Kridin K, Ludwig RJ. The growing incidence of bullous pemphigoid: overview and potential explanations. Front Med (Lausanne). (2018) 5:220. doi: 10.3389/fmed.2018.00220
59. Mendonca FM, Martin-Gutierrez FJ, Rios-Martin JJ, Camacho-Martinez F. Three cases of bullous pemphigoid associated with dipeptidyl peptidase-4 inhibitors - one due to linagliptin. Dermatology. (2016) 232:249–53. doi: 10.1159/000443330
60. Izumi K, Nishie W, Mai Y, Wada M, Natsuga K, Ujiie H, et al. Autoantibody profile differentiates between inflammatory and noninflammatory bullous pemphigoid. J Invest Dermatol. (2016) 136:2201–10. doi: 10.1016/j.jid.2016.06.622
61. Sibaud V. Dermatologic reactions to immune checkpoint inhibitors : skin toxicities and immunotherapy. Am J Clin Dermatol. (2018) 19:345–61. doi: 10.1007/s40257-017-0336-3
62. Zumelzu C, Alexandre M, Le Roux C, Weber P, Guyot A, Levy A, et al. Mucous membrane pemphigoid, bullous pemphigoid, and anti-programmed death-1/ programmed death-ligand 1: a case report of an elderly woman with mucous membrane pemphigoid developing after pembrolizumab therapy for metastatic melanoma and review of the literature. Front Med (Lausanne). (2018) 5:268. doi: 10.3389/fmed.2018.00268
63. Hess C, Winkler A, Lorenz AK, Holecska V, Blanchard V, Eiglmeier S, et al. T cell-independent B cell activation induces immunosuppressive sialylated IgG antibodies. J Clin Invest. (2013) 123:3788–96. doi: 10.1172/JCI65938
64. Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, et al. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med. (2004) 199:1577–84. doi: 10.1084/jem.20040168
65. Colliou N, Picard D, Caillot F, Calbo S, Le Corre S, Lim A, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. (2013) 5:175ra30. doi: 10.1126/scitranslmed.3005166
66. Hiepe F, Dorner T, Hauser AE, Hoyer BF, Mei H, Radbruch A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat Rev Rheumatol. (2011) 7:170–8. doi: 10.1038/nrrheum.2011.1
67. Hiepe F, Radbruch A. Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat Rev Nephrol. (2016) 12:232–40. doi: 10.1038/nrneph.2016.20
68. Musette P, Bouaziz JD. B cell modulation strategies in autoimmune diseases: new concepts. Front Immunol. (2018) 9:622. doi: 10.3389/fimmu.2018.00622
69. Tiburzy B, Szyska M, Iwata H, Chrobok N, Kulkarni U, Hirose M, et al. Persistent autoantibody-production by intermediates between short-and long-lived plasma cells in inflamed lymph nodes of experimental epidermolysis bullosa acquisita. PLoS ONE. (2013) 8:e83631. doi: 10.1371/journal.pone.0083631
70. Timoteo RP, da Silva MV, Miguel CB, Silva DA, Catarino JD, Rodrigues Junior V, et al. Th1/Th17-related cytokines and chemokines and their implications in the pathogenesis of pemphigus vulgaris. Mediators Inflamm. (2017) 2017:7151285. doi: 10.1155/2017/7151285
71. Lee SH, Hong WJ, Kim SC. Analysis of serum cytokine profile in pemphigus. Ann Dermatol. (2017) 29:438–45. doi: 10.5021/ad.2017.29.4.438
72. Pickford WJ, Gudi V, Haggart AM, Lewis BJ, Herriot R, Barker RN, et al. T cell participation in autoreactivity to NC16a epitopes in bullous pemphigoid. Clin Exp Immunol. (2015) 180:189–200. doi: 10.1111/cei.12566
73. Gounni Abdelilah S, Wellemans V, Agouli M, Guenounou M, Hamid Q, Beck LA, et al. Increased expression of Th2-associated chemokines in bullous pemphigoid disease. Role of eosinophils in the production and release of these chemokines. Clin Immunol. (2006) 120:220–31. doi: 10.1016/j.clim.2006.03.014
74. Makino T, Yoshihisa Y, Mizawa M, Tsutsui K, Nishijima C, Inaoki M, et al. Increased serum levels of Th2-type cytokines and eotaxin in fibrillar-type dermatitis herpetiformis. Acta Derm Venereol. (2017) 97:642–3. doi: 10.2340/00015555-2604
75. Bieber K, Sun S, Witte M, Kasprick A, Beltsiou F, Behnen M, et al. Regulatory T cells suppress inflammation and blistering in pemphigoid diseases. Front Immunol. (2017) 8:1628. doi: 10.3389/fimmu.2017.01628
76. Sugiyama H, Matsue H, Nagasaka A, Nakamura Y, Tsukamoto K, Shibagaki N, et al. CD4+CD25high regulatory T cells are markedly decreased in blood of patients with pemphigus vulgaris. Dermatology. (2007) 214:210–20. doi: 10.1159/000099585
77. Veldman C, Hohne A, Dieckmann D, Schuler G, Hertl M. Type I regulatory T cells specific for desmoglein 3 are more frequently detected in healthy individuals than in patients with pemphigus vulgaris. J Immunol. (2004) 172:6468–75. doi: 10.4049/jimmunol.172.10.6468
78. Yokoyama T, Matsuda S, Takae Y, Wada N, Nishikawa T, Amagai M, et al. Antigen-independent development of Foxp3+ regulatory T cells suppressing autoantibody production in experimental pemphigus vulgaris. Int Immunol. (2011) 23:365–73. doi: 10.1093/intimm/dxr020
79. Yuan H, Zhou S, Liu Z, Cong W, Fei X, Zeng W, et al. Pivotal role of lesional and perilesional T/B lymphocytes in pemphigus pathogenesis. J Invest Dermatol. (2017) 137:2362–70. doi: 10.1016/j.jid.2017.05.032
80. Anhalt GJ, Labib RS, Voorhees JJ, Beals TF, Diaz LA. Induction of pemphigus in neonatal mice by passive transfer of IgG from patients with the disease. N Engl J Med. (1982) 306:1189–96. doi: 10.1056/NEJM198205203062001
81. Ding X, Diaz LA, Fairley JA, Giudice GJ, Liu Z. The anti-desmoglein 1 autoantibodies in pemphigus vulgaris sera are pathogenic. J Invest Dermatol. (1999) 112:739–43. doi: 10.1046/j.1523-1747.1999.00585.x
82. Amagai M, Nishikawa T, Nousari HC, Anhalt GJ, Hashimoto T. Antibodies against desmoglein 3 (pemphigus vulgaris antigen) are present in sera from patients with paraneoplastic pemphigus and cause acantholysis in vivo in neonatal mice. J Clin Invest. (1998) 102:775–82. doi: 10.1172/JCI3647
83. Woodley DT, Ram R, Doostan A, Bandyopadhyay P, Huang Y, Remington J, et al. Induction of epidermolysis bullosa acquisita in mice by passive transfer of autoantibodies from patients. J Invest Dermatol. (2006) 126:1323–30. doi: 10.1038/sj.jid.5700254
84. Amber KT, Valdebran M, Kridin K, Grando SA. The role of eosinophils in bullous pemphigoid: a developing model of eosinophil pathogenicity in mucocutaneous disease. Front Med (Lausanne). (2018) 5:201. doi: 10.3389/fmed.2018.00201
85. de Graauw E, Sitaru C, Horn M, Borradori L, Yousefi S, Simon HU, et al. Evidence for a role of eosinophils in blister formation in bullous pemphigoid. Allergy. (2017) 72:1105–13. doi: 10.1111/all.13131
86. Fang H, Zhang Y, Li N, Wang G, Liu Z. The autoimmune skin disease bullous pemphigoid: the role of mast cells in autoantibody-induced tissue injury. Front Immunol. (2018) 9:407. doi: 10.3389/fimmu.2018.00407
87. Yu X, Kasprick A, Hartmann K, Petersen F. The role of mast cells in autoimmune bullous dermatoses. Front Immunol. (2018) 9:386. doi: 10.3389/fimmu.2018.00386
88. Eyerich K, Dimartino V, Cavani A. IL-17 and IL-22 in immunity: driving protection and pathology. Eur J Immunol. (2017) 47:607–14. doi: 10.1002/eji.201646723
89. Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E, et al. Regulatory T-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. (2018) 142:1831–42.e7. doi: 10.1016/j.jaci.2018.04.006
90. Hashimoto T, Takahashi H, Sakaguchi S. Regulatory T-cell deficiency and autoimmune skin disease: Beyond the scurfy mouse and immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol. (2018) 142:1754–6. doi: 10.1016/j.jaci.2018.08.028
91. Yang CH, Tian L, Ling GS, Trendell-Smith NJ, Ma L, Lo CK, et al. Immunological mechanisms and clinical implications of regulatory T cell deficiency in a systemic autoimmune disorder: roles of IL-2 versus IL-15. Eur J Immunol. (2008) 38:1664–76. doi: 10.1002/eji.200838190
92. Amagai M. Autoimmunity against desmosomal cadherins in pemphigus. J Dermatol Sci. (1999) 20:92–102. doi: 10.1016/S0923-1811(99)00016-X
93. Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J Clin Invest. (2005) 115:3157–65. doi: 10.1172/JCI23475
94. Berkowitz P, Chua M, Liu Z, Diaz LA, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am J Pathol. (2008) 173:1628–36. doi: 10.2353/ajpath.2008.080391
95. Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol. (1999) 112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x
96. Chen Y, Chernyavsky A, Webber RJ, Grando SA, Wang PH. Critical role of the neonatal Fc receptor (FcRn) in the pathogenic action of antimitochondrial autoantibodies synergizing with anti-desmoglein autoantibodies in pemphigus vulgaris. J Biol Chem. (2015) 290:23826–37. doi: 10.1074/jbc.M115.668061
97. Saleh MA, Salem H, El Azizy H. Autoantibodies other than anti-desmogleins in pemphigus vulgaris patients. Indian J Dermatol. (2017) 62:47–51. doi: 10.4103/0019-5154.198032
98. Amber KT, Valdebran M, Grando SA. Non-desmoglein antibodies in patients with pemphigus vulgaris. Front Immunol. (2018) 9:1190. doi: 10.3389/fimmu.2018.01190
99. Nguyen VT, Ndoye A, Grando SA. Novel human alpha9 acetylcholine receptor regulating keratinocyte adhesion is targeted by Pemphigus vulgaris autoimmunity. Am J Pathol. (2000) 157:1377–91. doi: 10.1016/S0002-9440(10)64651-2
100. Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity. (2012) 45:7–35. doi: 10.3109/08916934.2011.606444
101. Sajda T, Sinha AA. Autoantibody signaling in pemphigus vulgaris: development of an integrated model. Front Immunol. (2018) 9:692. doi: 10.3389/fimmu.2018.00692
102. Chiorean RM, Baican A, Mustafa MB, Lischka A, Leucuta DC, Feldrihan V, et al. Complement-activating capacity of autoantibodies correlates with disease activity in bullous pemphigoid patients. Front Immunol. (2018) 9:2687. doi: 10.3389/fimmu.2018.02687
103. Kushner CJ, Payne AS. Increasing the complement of therapeutic options in bullous pemphigoid. J Invest Dermatol. (2018) 138:246–8. doi: 10.1016/j.jid.2017.09.026
104. Lin L, Hwang BJ, Culton DA, Li N, Burette S, Koller BH, et al. Eosinophils mediate tissue injury in the autoimmune skin disease bullous pemphigoid. J Invest Dermatol. (2018) 138:1032–43. doi: 10.1016/j.jid.2017.11.031
105. Ujiie H, Sasaoka T, Izumi K, Nishie W, Shinkuma S, Natsuga K, et al. Bullous pemphigoid autoantibodies directly induce blister formation without complement activation. J Immunol. (2014) 193:4415–28. doi: 10.4049/jimmunol.1400095
106. Iwata H, Kamio N, Aoyama Y, Yamamoto Y, Hirako Y, Owaribe K, et al. IgG from patients with bullous pemphigoid depletes cultured keratinocytes of the 180-kDa bullous pemphigoid antigen (type XVII collagen) and weakens cell attachment. J Invest Dermatol. (2009) 129:919–26. doi: 10.1038/jid.2008.305
107. Kasprick A, Holtsche MM, Rose EL, Hussain S, Schmidt E, Petersen F, et al. The anti-C1s antibody TNT003 prevents complement activation in the skin induced by bullous pemphigoid autoantibodies. J Invest Dermatol. (2018) 138:458–61. doi: 10.1016/j.jid.2017.08.030
108. Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. (2016) 11:175–97. doi: 10.1146/annurev-pathol-012615-044313
109. Mihai S, Hirose M, Wang Y, Thurman JM, Holers VM, Morgan BP, et al. Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Front Immunol. (2018) 9:535. doi: 10.3389/fimmu.2018.00535
110. Mihai S, Chiriac MT, Takahashi K, Thurman JM, Holers VM, Zillikens D, et al. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J Immunol. (2007) 178:6514–21. doi: 10.4049/jimmunol.178.10.6514
111. Sitaru C, Kromminga A, Hashimoto T, Brocker EB, Zillikens D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am J Pathol. (2002) 161:301–11. doi: 10.1016/S0002-9440(10)64182-X
112. Bieber K, Witte M, Sun S, Hundt JE, Kalies K, Drager S, et al. T cells mediate autoantibody-induced cutaneous inflammation and blistering in epidermolysis bullosa acquisita. Sci Rep. (2016) 6:38357. doi: 10.1038/srep38357
113. Valencia-Guerrero A, Dresser K, Cornejo K. The utility of tissue and epidermal transglutaminase immunohistochemistry in dermatitis herpetiformis. Indian J Dermatopathol Diagn Dermatol. (2018) 5:97–100. doi: 10.4103/ijdpdd.ijdpdd_22_18
114. Collin P, Salmi TT, Hervonen K, Kaukinen K, Reunala T. Dermatitis herpetiformis: a cutaneous manifestation of coeliac disease. Ann Med. (2017) 49:23–31. doi: 10.1080/07853890.2016.1222450
115. Antiga E, Quaglino P, Pierini I, Volpi W, Lami G, Bianchi B, et al. Regulatory T cells as well as IL-10 are reduced in the skin of patients with dermatitis herpetiformis. J Dermatol Sci. (2015) 77:54–62. doi: 10.1016/j.jdermsci.2014.11.003
116. Nigam R, Levitt J. Where does rituximab fit in the treatment of autoimmune mucocutaneous blistering skin disease? J Drugs Dermatol. (2012) 11:622–5.
117. Chakievska L, Holtsche MM, Kunstner A, Goletz S, Petersen BS, Thaci D, et al. IL-17A is functionally relevant and a potential therapeutic target in bullous pemphigoid. J Autoimmun. (2019) 96:104–12. doi: 10.1016/j.jaut.2018.09.003
118. Kasperkiewicz M, Sadik CD, Bieber K, Ibrahim SM, Manz RA, Schmidt E, et al. Epidermolysis bullosa acquisita: from pathophysiology to novel therapeutic options. J Invest Dermatol. (2016) 136:24–33. doi: 10.1038/JID.2015.356
119. Koga H, Kasprick A, Lopez R, Auli M, Pont M, Godessart N, et al. Therapeutic effect of a novel phosphatidylinositol-3-kinase delta inhibitor in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:1558. doi: 10.3389/fimmu.2018.01558
120. Lee J, Werth VP, Hall RP III, Eming R, Fairley JA, Fajgenbaum DC, et al. Perspective from the 5th international pemphigus and pemphigoid foundation scientific conference. Front Med (Lausanne). (2018) 5:306. doi: 10.3389/fmed.2018.00306
121. Ludwig RJ. Signalling and targeted therapy of inflammatory cells in epidermolysis bullosa acquisita. Exp Dermatol. (2017) 26:1179–86. doi: 10.1111/exd.13335
122. Tukaj S, Bieber K, Witte M, Ghorbanalipoor S, Schmidt E, Zillikens D, et al. Calcitriol treatment ameliorates inflammation and blistering in mouse models of epidermolysis bullosa acquisita. J Invest Dermatol. (2018) 138:301–9. doi: 10.1016/j.jid.2017.09.009
123. Witte M, Koga H, Hashimoto T, Ludwig RJ, Bieber K. Discovering potential drug-targets for personalized treatment of autoimmune disorders - what we learn from epidermolysis bullosa acquisita. Expert Opin Ther Targets. (2016) 20:985–98. doi: 10.1517/14728222.2016.1148686
124. Harman KE, Brown D, Exton LS, Groves RW, Hampton PJ, Mohd Mustapa MF, et al. British Association of Dermatologists' guidelines for the management of pemphigus vulgaris 2017. Br J Dermatol. (2017) 177:1170–201. doi: 10.1111/bjd.15930
125. Venning VA, Taghipour K, Mohd Mustapa MF, Highet AS, Kirtschig G. British Association of Dermatologists' guidelines for the management of bullous pemphigoid 2012. Br J Dermatol. (2012) 167:1200–14. doi: 10.1111/bjd.12072
126. Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. (2017) 389:2031–40. doi: 10.1016/S0140-6736(17)30070-3
127. Proby CM, Ota T, Suzuki H, Koyasu S, Gamou S, Shimizu N, et al. Development of chimeric molecules for recognition and targeting of antigen-specific B cells in pemphigus vulgaris. Br J Dermatol. (2000) 142:321–30. doi: 10.1046/j.1365-2133.2000.03328.x
128. Kridin K. Emerging treatment options for the management of pemphigus vulgaris. Therap Clin Risk Manage. (2018) 14:757–78. doi: 10.2147/TCRM.S142471
129. Amagai M, Ikeda S, Hashimoto T, Mizuashi M, Fujisawa A, Ihn H, et al. A randomized double-blind trial of intravenous immunoglobulin for bullous pemphigoid. J Dermatol Sci. (2017) 85:77–84. doi: 10.1016/j.jdermsci.2016.11.003
130. Hirose M, Kasprick A, Beltsiou F, Dieckhoff Schulze K, Schulze FS, Samavedam UK, et al. Reduced skin blistering in experimental epidermolysis bullosa acquisita after anti-TNF treatment. Mol Med. (2017) 22:918–26. doi: 10.2119/molmed.2015.00206
131. Diny NL, Rose NR, Cihakova D. Eosinophils in autoimmune diseases. Front Immunol. (2017) 8:484. doi: 10.3389/fimmu.2017.00484
132. Jaunzems AE, Woods AE, Staples A. Electron microscopy and morphometry enhances differentiation of epidermolysis bullosa subtypes. With normal values for 24 parameters in skin. Arch Dermatol Res. (1997) 289:631–9. doi: 10.1007/s004030050252
133. Gunther C, Wozel G, Meurer M, Pfeiffer C. Up-regulation of CCL11 and CCL26 is associated with activated eosinophils in bullous pemphigoid. Clin Exp Immunol. (2011) 166:145–53. doi: 10.1111/j.1365-2249.2011.04464.x
134. Koivukangas V, Oikarinen A. Suction blister model of wound healing. Methods Mol Med. (2003) 78:255–61. doi: 10.1385/1-59259-332-1:255
135. Tukaj S, Bieber K, Kleszczynski K, Witte M, Cames R, Kalies K, et al. Topically applied Hsp90 blocker 17AAG inhibits autoantibody-mediated blister-inducing cutaneous inflammation. J Invest Dermatol. (2017) 137:341–9. doi: 10.1016/j.jid.2016.08.032
136. Kasperkiewicz M, Muller R, Manz R, Magens M, Hammers CM, Somlai C, et al. Heat-shock protein 90 inhibition in autoimmunity to type VII collagen: evidence that nonmalignant plasma cells are not therapeutic targets. Blood. (2011) 117:6135–42. doi: 10.1182/blood-2010-10-314609
137. Samavedam UK, Mitschker N, Kasprick A, Bieber K, Schmidt E, Laskay T, et al. Whole-genome expression profiling in skin reveals SYK As a key regulator of inflammation in experimental epidermolysis bullosa acquisita. Front Immunol. (2018) 9:249. doi: 10.3389/fimmu.2018.00249
138. Costa Arantes DA, Guimarães JM, Batista AC. Therapeutic success of vitamin D replacement in oral pemphigus vulgaris: a case report. Oral Surg Oral Med Oral Pathol Oral Radiol. (2018) 126:e52. doi: 10.1016/j.oooo.2018.02.085
139. Kopecki Z, Ruzehaji N, Turner C, Iwata H, Ludwig RJ, Zillikens D, et al. Topically applied flightless i neutralizing antibodies improve healing of blistered skin in a murine model of epidermolysis bullosa acquisita. J Invest Dermatol. (2012) 133:1008–16. doi: 10.1038/jid.2012.457
140. Albers LN, Zone JJ, Stoff BK, Feldman RJ. Rituximab treatment for recalcitrant dermatitis herpetiformis. JAMA Dermatol. (2017) 153:315–8. doi: 10.1001/jamadermatol.2016.4676
141. Antiga E, Caproni M. The diagnosis and treatment of dermatitis herpetiformis. Clin Cosmet Investig Dermatol. (2015) 8:257–65. doi: 10.2147/CCID.S69127
142. Maley A, Warren M, Haberman I, Swerlick R, Kharod-Dholakia B, Feldman R. Rituximab combined with conventional therapy versus conventional therapy alone for the treatment of mucous membrane pemphigoid (MMP). J Am Acad Dermatol. (2016) 74:835–40. doi: 10.1016/j.jaad.2016.01.020
143. Benamu E, Montoya JG. Infections associated with the use of eculizumab: recommendations for prevention and prophylaxis. Curr Opin Infect Dis. (2016) 29:319–29. doi: 10.1097/QCO.0000000000000279
144. Rathbone J, Kaltenthaler E, Richards A, Tappenden P, Bessey A, Cantrell A. A systematic review of eculizumab for atypical haemolytic uraemic syndrome (aHUS). BMJ Open. (2013) 3:e003573. doi: 10.1136/bmjopen-2013-003573
145. Berookhim B, Fischer HD, Weinberg JM. Treatment of recalcitrant pemphigus vulgaris with the tumor necrosis factor alpha antagonist etanercept. Cutis. (2004) 74:245-7.
146. Sadeghi H, Lockmann A, Hund AC, Samavedam UK, Pipi E, Vafia K, et al. Caspase-1-independent IL-1 release mediates blister formation in autoantibody-induced tissue injury through modulation of endothelial adhesion molecules. J Immunol. (2015) 194:3656–63. doi: 10.4049/jimmunol.1402688
147. Toto P, Feliciani C, Amerio P, Suzuki H, Wang B, Shivji GM, et al. Immune modulation in pemphigus vulgaris: role of CD28 and IL-10. J Immunol. (2000) 164:522–9. doi: 10.4049/jimmunol.164.1.522
148. Konig A, Bruckner-Tuderman L. Transforming growth factor-beta promotes deposition of collagen VII in a modified organotypic skin model. Lab Invest. (1994) 70:203–9.
149. Ryynanen J, Sollberg S, Olsen DR, Uitto J. Transforming growth factor-beta up-regulates type VII collagen gene expression in normal and transformed epidermal keratinocytes in culture. Biochem Biophys Res Commun. (1991) 180:673–80. doi: 10.1016/S0006-291X(05)81118-0
150. Sawamura D, McMillan JR, Akiyama M, Shimizu H. Epidermolysis bullosa: directions for future research and new challenges for treatment. Arch Dermatol Res. (2003) 295(Suppl. 1):S34–42. doi: 10.1007/s00403-002-0370-7
151. Rider P, Carmi Y, Cohen I. Biologics for targeting inflammatory cytokines, clinical uses, and limitations. Int J Cell Biol. (2016) 2016:9259646. doi: 10.1155/2016/9259646
152. Kitashima DY, Kobayashi T, Woodring T, Idouchi K, Doebel T, Voisin B, et al. Langerhans cells prevent autoimmunity via expansion of keratinocyte antigen-specific regulatory T cells. EBioMedicine. (2018) 27:293–303. doi: 10.1016/j.ebiom.2017.12.022
153. Iwata H, Bieber K, Tiburzy B, Chrobok N, Kalies K, Shimizu A, et al. B cells, dendritic cells, and macrophages are required to induce an autoreactive CD4 helper T cell response in experimental epidermolysis bullosa acquisita. J Immunol. (2013) 191:2978–88. doi: 10.4049/jimmunol.1300310
154. Samavedam UK, Iwata H, Muller S, Schulze FS, Recke A, Schmidt E, et al. GM-CSF modulates autoantibody production and skin blistering in experimental epidermolysis bullosa acquisita. J Immunol. (2014) 192:559–71. doi: 10.4049/jimmunol.1301556
155. Zebrowska A, Wozniacka A, Juczynska K, Ociepa K, Waszczykowska E, Szymczak I, et al. Correlation between IL36alpha and IL17 and Activity of the disease in selected autoimmune blistering diseases. Mediators Inflamm. (2017) 2017:8980534. doi: 10.1155/2017/8980534
156. Giurdanella F, Fania L, Gnarra M, Toto P, Di Rollo D, Sauder DN, et al. A possible role for CD8+ T lymphocytes in the cell-mediated pathogenesis of pemphigus vulgaris. Mediators Inflamm. (2013) 2013:764290. doi: 10.1155/2013/764290
157. Roujeau JC, Lok C, Bastuji-Garin S, Mhalla S, Enginger V, Bernard P. High risk of death in elderly patients with extensive bullous pemphigoid. Arch Dermatol. (1998) 134:465–9. doi: 10.1001/archderm.134.4.465
158. Zhao CY, Murrell DF. Autoimmune blistering diseases in females: a review. Int J Womens Dermatol. (2015) 1:4–12. doi: 10.1016/j.ijwd.2015.01.002
Keywords: skin barrier, autoimmunity, autoantibody, skin blistering diseases, therapy, pemphigus, pemphigoid, epidermolysis bullosa acquisita
Citation: Stevens NE, Cowin AJ and Kopecki Z (2019) Skin Barrier and Autoimmunity—Mechanisms and Novel Therapeutic Approaches for Autoimmune Blistering Diseases of the Skin. Front. Immunol. 10:1089. doi: 10.3389/fimmu.2019.01089
Received: 29 March 2019; Accepted: 29 April 2019;
Published: 14 May 2019.
Edited by:
Ralf J. Ludwig, Universität zu Lübeck, GermanyReviewed by:
Unni Samavedam, University of Cincinnati, United StatesAlina Sesarman, Babes-Bolyai University, Romania
Stefan Tukaj, University of Gdansk, Poland
Copyright © 2019 Stevens, Cowin and Kopecki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zlatko Kopecki, emxhdGtvLmtvcGVja2lAdW5pc2EuZWR1LmF1