 Rajkumar Noubade
Rajkumar Noubade Sonia Majri-Morrison
Sonia Majri-Morrison Kristin V. Tarbell
Kristin V. Tarbell- Department of Inflammation and Oncology, Amgen Research, Amgen Inc., South San Francisco, CA, United States
Dendritic cells (DCs) efficiently process and present antigens to T cells, and by integrating environmental signals, link innate and adaptive immunity. DCs also control the balance between tolerance and immunity, and are required for T-cell mediated anti-tumor immunity. One subset of classical DCs, cDC1, are particularly important for eliciting CD8 T cells that can kill tumor cells. cDC1s are superior in antigen cross-presentation, a process of presenting exogenous antigens on MHC class I to activate CD8+ T cells. Tumor-associated cDC1s can transport tumor antigen to the draining lymph node and cross-present tumor antigens, resulting in priming and activation of cytotoxic T cells. Although cross-presenting cDC1s are critical for eliciting anti-tumor T cell responses, the role and importance of other DC subsets in anti-tumor immunity is not as well-characterized. Recent literature in other contexts suggests that critical crosstalk between DC subsets can significantly alter biological outcomes, and these DC interactions likely also contribute significantly to tumor-specific immune responses. Therefore, antigen presentation by cDC1s may be necessary but not sufficient for maximal immune responses against cancer. Here, we discuss recent advances in the understanding of DC subset interactions to maximize anti-tumor immunity, and propose that such interactions should be considered for the development of better DC-targeted immunotherapies.
Introduction
The interaction between various myeloid and lymphoid cell populations is crucial to initiate and orchestrate a robust anti-tumor response. By processing tumor associated antigens (TAAs) and migrating to draining lymph nodes (dLN), where T cell priming occurs, dendritic cells (DCs) are considered the most potent professional antigen presenting cells (APCs) to elicit adaptive anti-tumor immunity (1). In addition to presenting antigens, DCs use soluble molecules such as cytokines and chemokines as well as direct cell-cell contacts to prime and activate TAA-specific T cells. DCs were discovered by Ralph Steinman and Zanvil Cohn in 1973 as an APC population, distinct from macrophages, that initiate adaptive immune responses (2). As a result of more recent deep-phenotyping, DCs are now recognized to be a heterogenous population comprising several subsets distinguished by their development, phenotypic differences, localization, and functional specialization (2–6). This functional specialization of each subset allows DCs to initiate distinct immune responses in different immunological contexts (7). Here, we review literature supporting the hypothesis that, although one DC subset, conventional DC1(cDC1), has been shown to be crucial for anti-tumor immunity, multiple DC subsets, and interactions with other cells are needed for maximal responses.
DC Subsets Are Functionally Specialized
DCs are broadly classified as classical (or conventional) DCs (cDCs) and plasmacytoid DCs (pDCs), each with specialized functions. cDCs, specialized in antigen presentation to naïve T cells can be further segregated into cDC1s and cDC2s, excelling in MHC class I- and class II-mediated antigen-presentation, respectively (3, 6, 8–10). cDCs are found both as lymphoid and non-lymphoid tissue cells, the latter of which can migrate via the lymph to dLN to present tissue-derived antigens (3, 11). cDC1s, present at lower frequency compared to cDC2s, are identified by the expression of XCR1 (12), and in humans, also by the expression of CD141 (BDCA3) (5, 13, 14). cDC1s possess specialized mechanisms to mediate efficient antigen recognition, antigen transport to appropriate endosomal compartments and subsequent processing for the presentation to CD8 T cells in a process known as cross-presentation (15–18). cDC1s can also activate CD4 T cells through MHC class II antigen presentation and can polarize activated CD4 T cells toward a Th1 phenotype through the secretion of IL-12 (19).
cDC2s are specialized in MHC class II-mediated antigen presentation and are the most efficient APCs for activation and expansion of CD4 T cells (5, 13, 20). They are the most frequent DC population present in blood, lymphoid organs and tissues and promote a wide range of immune responses including Th1, Th2, and Th17 in specific contexts (13, 19, 21–25). Human cDC2s can be identified by their preferential expression of CD1c (BDCA1) and CD172a (SIRPα) (26). cDC2s are more heterogenous than cDC1s, and express various receptors that enable them to respond to broad spectrum of microbial products (22, 26–28). A subset of Notch2-dependent cDC2s specializes in IL-23 production and contributes to innate defense and adaptive immune responses (27, 29).
pDCs, distinguished by their ability to produce large amounts of type I IFN upon viral infection (30–33) are identified, in humans, by the expression of surface markers CD303 (BDCA-2), CD304 (BDCA-4/Neuropilin) and CD123 (5, 13). They are present mainly in lymphoid organs and can migrate to the LN through blood circulation (5, 34). Mature pDCs can also act as APCs and have distinct regulation of MHC class II surface expression that results in sustained membrane peptide-MHC complex and antigen presentation (30). A heterogeneity of pDCs is also described in terms of their ability to produce type I IFN and/or antigen presentation (35, 36).
Another related but developmentally distinct population, derived from monocytes, termed monocyte DCs (moDC) upregulates certain functional properties of DCs in some contexts and express tumor necrosis factor (TNF)- α and intracellular nitric oxide synthase (iNOS) (37). More commonly, the term moDCs refers to monocyte isolated from human peripheral blood mononuclear cells (PBMC) that are in vitro differentiated in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin (IL)-4 into cells sharing several phenotypic and functional features of DCs (26, 38, 39). moDCs are the most common in vitro model of DCs, yet are quite heterogeneous in both mouse and human, with unclear relationship to in vivo cell populations (40–42).
All DC subsets, including cDCs and pDCs, are found in the tumor microenvironment (TME) (30, 43–47) and among the cDCs, the cDC2s outnumber cDC1s, with cDC1s being the rarest APCs within the TME (43, 48). The role of pDCs in tumor immunity remains elusive and contradictory. Similarly, the precise role of cDC2s in anti-tumor immunity has been difficult to delineate due to lack of proper genetic tools. On the contrary, mounting evidence suggests cDC1s to be the critical antigen presenting DC subset for the generation of anti-tumor immunity. Here we summarize data supporting the importance of cDC1s in anti-tumor immunity, and then review the recent literature that documents DC crosstalk being necessary for effective immune responses, in other contexts such as anti-viral immune responses, and apply these principles to tumor immunity.
cDC1s Are Necessary for Anti-tumor Immunity
Since MHC class I molecules are expressed by every cell in the body (not just infected cells and cancer cells), to avoid bystander killing of healthy cells by CTLs, extracellular antigens do not enter the MHC class I-loading machinery (15, 18). Therefore, to generate an immune response, cancer cell antigens need special processing in APCs to be presented to naïve CD8 T cells. Moreover, naïve CD8 T cells primarily circulate through secondary lymphoid organs (15). Hence, cancer antigens must be brought to secondary lymphoid organs to be presented to naïve CD8 T cells. cDC1s fulfill both functions by patrolling tumor tissues, and by capturing, processing and presenting tumor-antigens on their surface through MHC class I molecules via antigen cross-presentation. cDC1s then migrate to dLN and deliver peptide/MHC class I signal to CD8 T cells which leads to their activation and the initiation of an immune response against tumor cells (15, 18).
Although other cell types have been reported to cross-present antigens (11, 49), this specialized function is mostly attributed to the cDC1 subset, owing to their unique adaptations of subcellular molecular machinery and vesicular trafficking (15, 18). Such adaptations include efficient antigen uptake of dying cells, delivery of cell-associated antigen to early endosomes, (15, 50–52), efficient phagosome-to-cytosol export of an ingested antigen possibly aided by ER-derived translocons and ER-associated degradation (ERAD) components such as Sec61, Derlin, p97 ATPase, Sec22 (15, 53–55), lower expression of lysosomal proteases (50) and antagonizing their degradative functions via NOX2-mediated ROS generation (56–60). The end result of such lower proteolysis, and therefore, increased antigen retention in cDC1s, is eventually an enhanced ability to carry the antigen all the way from peripheral tissues where the antigen is captured, to the dLN, where priming and activation of CD8 T cells occurs (56). The importance of cDC1s' ability to cross-present antigen in its immune functions is recently demonstrated using Wdfy4-deficient mice, which selectively lack cross-presentation (61).
Beyond their role in antigen cross-presentation, cDC1s are the major source of IL-12 production and thus influence anti-tumor immunity by activating NK cells and driving CD4 T cell responses toward Th1 responses (19, 62–64).
The critical role of cDC1s in anti-tumor immunity has been shown using mice deficient in basic leucine zipper transcription factor ATF-like 3 (Batf3), a transcription factor required for cDC1 differentiation (65). Batf3 knockout mice lack cDC1 cells but not other APCs and display impaired anti-tumor immunity in several models (43, 65–68). Expansion and activation of cDC1s using fms-related tyrosine kinase 3 ligand (Flt3L) and poly I:C leads to significant enhancement of antitumor responses (45). Immunotherapies such as PD1/PD-L1 blockade or CD137 agonists are ineffective in Batf3-deficient mice, highlighting the crucial role cDC1s in tumor immunity (68, 69). Furthermore, tumor-resident cDC1s are required for trafficking of adoptively transferred CD8 T cells into tumors through their ability to produce CXCL9 and CXCL10 (67, 70). DC-specific deletion of Sec22b leads not only to impaired cross-presentation of TAAs and reduced anti-tumor immune responses but also abolishes the efficacy of anti-PD1 therapy (53). In humans, the presence of cDC1s within the TME is associated with better prognosis and response to immunotherapy. Analysis of the cancer genome atlas (TCGA) dataset shows that a higher ratio of a cDC1 gene signature to a signature of all other myeloid cells (including monocyte/macrophage, and not just other DC subsets) is associated with better prognosis across human tumors (44, 71). Abundance of CD8 T cells positively correlates with cDC1 markers in pancreatic tumors (70). Taken together, these data show that cross-presenting cDC1s are crucial and necessary for the generation of an effective anti-tumor immunity.
cDC1 Are Not Sufficient for Maximal Anti-tumor Immunity: Potential Roles of Other DC Subsets
Tumor immunology is built upon the tenet that cytotoxic CD8 T cells (CTLs) eliminate tumor cells (72) and the prevailing dogma is that cDC1s are the most potent APCs for the CTL response against tumor. Because of the strong evidence for the importance of cDC1 in tumor immunity, as presented in the previous section, in one scenario it is possible that cDC1s are the sole DC subset sufficient for optimal anti-tumor CTL generation through antigen presentation via MHC class I as well as MHC class II (Figure 1A). A major driver of the current dogma is the studies conducted in mice genetically manipulated to lack cDC1 such as Batf3-deficient or Zbtb46-Cre mice. However, these tools are imperfect. For example, Batf3 is expressed in cDC2 and effector CD4 T cells (65, 73) and Zbtb46 is also expressed in DC2s as well as in endothelial cells (74–76), raising the possibility of contributions from additional DC subsets and other cell types. Hence, it is not clear whether the cDC1 subset alone is sufficient to provide the maximal immunity against tumor. Recent evidence in non-tumor settings has demonstrated that cDC1s require significant contributions from other DC subsets and are not sufficient for an optimal CTL response (77–79), pointing toward a role for the other cells in shaping a robust and durable anti-tumor immunity.
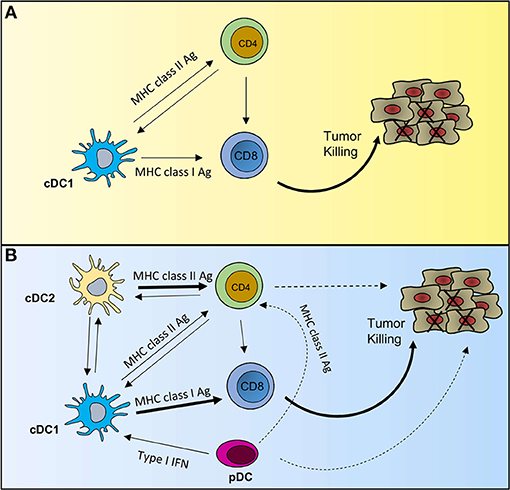
Figure 1. Potential scenarios of DC crosstalk in anti-tumor immunity. (A) Describes a scenario where an effective anti-tumor immune response would rely solely on cDC1 functions. cDC1s can activate both CD8 T cells and CD4 T cells through MHC class I- and MHC class II-mediated antigen presentation, respectively. Activated CD4 T cells provide licensing signal to cDC1s, which relay that help to CD8 T cells. Helped CD8 T cells have enhanced cytotoxic properties to efficiently kill tumor cells. (B) Describes multi-cellular interactions to achieve full-strength CTL responses against tumor. In this scenario, cDC1s predominantly activate CD8 T cells and cDC2s predominantly activate CD4 T cells. Activated CD4 T cells, in addition to providing help to maximize CTL responses can directly exhibit anti-tumor responses. Activated pDCs can modulate the TME mainly via type I IFN production, but can also activate CD4 T cells via MHC class II-mediated antigen presentation. Solid line indicates strong experimental evidence in tumor setting and dashed line indicates data in non-tumor setting. Thick line indicates predominant function.
Therefore, we describe a second scenario that includes possible roles of other DC subsets for a more robust anti-tumor immunity, directly and indirectly (Figure 1B). This scenario incorporates recent findings of spatiotemporal segregation of cDC1 and cDC2 activation within dLN to activate CD8 and CD4 T cells, respectively, during antiviral response. This robust CTL response requires interactions between multiple DC subsets, including cDCs and pDCs in a two-step priming process (77–79). Even though these responses are context dependent and are observed in anti-viral response, the general principals remain the same in anti-tumor immune response. Accordingly, in this scenario, the tumor-derived cDC1 primes CD8 T cells while tumor-derived cDC2 activates CD4 T cells in the first step of the CTL priming process and then in the second step, the activated CD4 T cells licenses a LN-resident cDC1 to relay the help for CTLs. Contributions of activated CD4 T cells to anti-tumor immunity can be more than just providing the help to CTLs, but also include activation of NK cells and macrophages through IFN-γ, modulation of tumor stroma and angiogenesis or direct cytolytic effects (80–83).
Additionally, during the two-step priming process, pDCs are recruited to cDC1-CD8 T cell priming sites, providing critical licensing signal to cDC1s through type 1 IFN. In this regard, lack of type 1 IFN receptor in cDC1s impairs their ability to reject tumors (84, 85). Furthermore, pDCs are usually weak APCs in the absence of activating signals but direct antigen presentation and T cell stimulation by pDCs has been described (30, 86). In fact, adoptive transfer of tumor-antigen-loaded pDCs induced potent anti-tumor T cell responses in melanoma patients (87), suggesting the possibility of anti-tumor immunity directly through APC functions by pDCs.
In the following sections, we mainly focus on this latter scenario of non-synchronous activation events by cDC1s and cDC2s and the reorganization of pDCs to the sites of CTL priming to describe the crosstalk between DC subsets and propose an integrated model of multi-DC subsets, multi-cell type interactions in achieving full-strength CTL responses in anti-tumor immunity.
Crosstalk Between DC Subsets
One of the goals of cancer immunotherapy is to promote tumor-antigen specific T cell responses. The current data supports the notion that cDC1s are well-suited for this purpose and that they are usually necessary for the generation of an anti-tumor response. However, as discussed below, they may not be sufficient for full-strength anti-tumor cytotoxic T cell responses and interactions with other DC subsets contribute to this process. In the following sections, we will review the interactions between each DC subsets separately.
cDC-pDC Crosstalk
cDCs and pDCs are co-localized in many immune contexts, e.g., non-inflamed LNs, skin biopsies from lupus erythematosus patients, thyroid glands from autoimmune thyroiditis patients and spleens of cancer patients (88–90). Such close-proximity of pDCs and cDCs suggests possible functional coordination. Indeed, local production of type I IFN by pDCs induces stimulatory molecules on cDCs driving their maturation during an effective immune response (79). Intravital two-photon microscopic analysis of DC subsets within dLN during vaccinia virus infection showed active, CCR5-mediated recruitment of pDCs to the site of CD8 T cell priming by virus-infected cDC1. The activated CD8 T cells also orchestrate, via XCL1, recruitment of resident, non-infected XCR1+cDC1s. pDCs produce type I IFN to induce upregulation of costimulatory molecules including CD40, CD80, and CD86 on non-infected resident-cDC1s (79), driving their maturation and antigen-presentation functions leading to robust CTL response. pDC help for CTL response, either through type I IFN or other costimulatory molecules such as CD40L has been described in other viral infection models (91–93). Depletion of pDCs results in impaired CTL responses in many viral infections, e.g., VSV infection (94), LCMV infection (95), and cutaneous herpes simplex virus (HSV) (92). In the LCMV infection model, pDC-mediated CD4 T cell activation was essential in providing help and generation of anti-viral CTL response (95). These observations underscore the pivotal role of the crosstalk between DC subsets in maximizing immune response against cell-associated antigens.
Similarly, in the context of anti-tumor immune responses, cooperation between pDCs and cDC1s and the resulting synergistic effects dependent on soluble factors such as type I IFN and/or cell-cell contact between the two DC subsets are described (11, 47). The potent anti-tumor T cell responses induced in melanoma patients by adoptive transfer of tumor-antigen-loaded pDCs (87) could be either a result of direct priming by pDCs or via interactions with other cells, including cDCs. However, tumor infiltrating pDCs exhibit an abnormal or hypofunctional state, most likely due to immuno-suppressive effects of the TME such as TGFβ (96). The presence of pDCs in tumors is associated with poor prognosis in cancers such as breast and ovarian cancers (97, 98). pDCs are generally thought to contribute to tolerance induction and tumor promotion in this setting, most likely due to Treg induction and expression of immunosuppressive factors such as indoleamine 2,3-dioxygenase (IDO) (98, 99). Thus, the role of pDCs in shaping adaptive tumor immunity remains elusive. It likely depends on their activation status and involves cooperativity with other cells but how pDCs are activated needs further investigation.
cDC1-cDC2 Crosstalk
The two cDC subsets communicate not only through soluble mediators such as IL-12 but also through a third cell viz., activated CD4 T cell. Even though both cDC subsets are adept in priming naïve T cells, cDC2s are more proficient in activating CD4 T cells than CD8 T cells while cDC1s are potent activators of CD8 T cells but present antigen to CD4 T cells less efficiently, both in vitro and in vivo (8, 20, 43). However, recent literature demonstrates that robust and maximal induction of cytotoxic CD8 T cell responses against cell-associated antigens not only requires interactions with cDC1s, but also interactions involving cDC2s (77, 100). Intravital microscopy demonstrated that, in the dLN, the two cDC subsets exhibit differential localization wherein cDC1s are largely segregated to the T cell zone in deep paracortical regions and cDC2s are more peripherally distributed (78, 100–103) and that CD8 T cells cluster with cDC1s and CD4 T cells cluster with cDC2s during step one of two-step T cell priming event in anti-viral immunity (78, 100, 104), suggesting parallel activation of the two T cell subsets by two different cDCs in an asynchronous manner. Such differential localization of the cDC subsets into non-overlapping T cell regions is also reported in the spleen (105).
The peripheral DC subsets also exhibit different kinetics during their migration to dLN (106), with an implication that cDC2s might access CD4 T cells earlier. The CD4 T is cell activated in the first step of the priming process, then gets recruited to LN-resident, XCR1+ cDC1 during the second step of the priming process and delivers help signals to that cDC1. The receiver-cDC1 then transmits the help signal to CD8 T cell activated in the first step, resulting in a robust expansion of highly effective CTLs. In this regard, it is well-established that, in the absence of CD4 T cell help, CD8 T cell responses are weaker and insufficient to generate long-lasting memory (107–109). The CD4 T cell help includes molecules such as CD40L expressed on CD4 T cells, that induces expression of costimulatory molecules including CD70, CD80, CD86, and cytokines such as IL-12, IL-15 by cDC1 (66, 110–112). The molecular nature of CD4 T cell help in shaping the CTL response is recently reviewed (104) and will not be discussed here in detail. Signaling though type I IFN is critical for proper functioning of cDC1s (85) and cDC2s are one of the important sources of this cytokine, as shown by depletion of pDCs using anti-pDCA antibodies in Batf3-deficient mice (84).
cDC1s and cDC2s may also collaborate for optimal Th1 induction. In the context of leishmania infection, targeting antigen to either cDC1 or cDC2 can elicit IFNγ-producing T cells, but interestingly, the cDC2s require IL-12 produced by the cDC1s to induce Th1 responses, whereas the cDC1s induce Th1 responses via CD70, independent of IL-12 (19). Therefore, each DC subset provides different signals that can contribute to effector T cell responses. Among the activated CD4 T cells, Th1 cells excel in providing the help to cDC1s to prime and expand CTLs through of production large amounts of IFNγ (113), thus fostering an important crosstalk between the two cDCs.
The majority of the experimental data described above originates from studies in anti-viral immunity. However, where and how naïve cancer cell-specific CD4 T cells get activated in a tumor setting is less clear. Lessons learnt on the importance of MHC class II-restricted CD4 T cell responses in autoimmune pathogenesis may shed light on this question in anti-tumor responses as well, since the anti-tumor response is essentially a self-specific response (114). The highest genetic risk for autoimmunity is conferred by HLA class II genes, with odds ratios >6, suggesting that CD4 T cell responses are necessary for immunity against self. In the context of autoimmunity, although some priming in the target tissue may occur (115–117), most studies suggest that self-specific CD4 T cells are first primed in the dLN, suggesting that a similar phenomenon might be happening in the generation of an anti-tumor immune response.
Evidence for the Importance of Tumor-Derived cDC2s and Activation of CD4 T Cells in the Draining Lymph Node
A large body of literature shows that naïve CD8 T cell activation for the generation of anti-tumor immunity occurs in dLN and is mediated by DCs (118–121). Interestingly, requirement of CD4 T cell help for optimal CD8 T cell effector functions in the context of tumor immunity is also well-documented, including the ability of CTLs to infiltrate the tumors (8, 119, 122–127). Non-helped CD8 T cells exhibit dysfunctional state with high expression of exhaustion markers in metastatic lung tumor model (127). In this regard, it is also well-established that the TME contains both cDC1 and cDC2 subsets (43–46). But importantly, both cDC1s and cDC2s scavenge tumor antigens (44) and migrate to dLN in a CCR7-dependent manner (46). Under right conditions, cDC2s can induce CD4 T cell activation in response to cell-associated antigen (51). Consistent with this, tumor-derived and dLN-derived cDC2s stimulate CD4 T cells more efficiently, ex vivo, in Lewis lung carcinoma model expressing ova as a model antigen (43). Furthermore, in this experimental setting, while cDC1 efficiently primed CD8 T cells, cDC2s are the most efficient activators of CD4 T cells. In addition, vaccination with the activated cDC2s reduced tumor growth, similar to that observed with cDC1s (43). Delivery of tumor antigen to cDC2 using dendritic cell immunoreceptor 2 (Dcir2) leads to significant anti-tumor effects in a mouse melanoma model (128). In a lung adenocarcinoma mouse model engineered to express MHC class II-restricted cytosolic antigen, activated cDC2 are observed both in the tumor and dLN and antigen-specific naïve CD4 T are activated in the dLN (129). In breast cancer patients gene signature of cDC2s positively correlates with better survival, similar to that observed with cDC1s, (130) and MHC class II expression predicts response to anti-PD1/PD-L1 therapy in melanoma patients (131). Collectively, tumor-derived cDC2s are likely to contribute to CD4 T cell activation in the dLN.
Integrated Model of DC Crosstalk in Tumor Draining Lymph Node
The spatiotemporal nature of DC crosstalk suggests two distinct DC-mediated events for maximal CD8 T cell responses: one after the initial antigen capture and another after the antigen is transferred to dLN-resident cDC1 cells (8). This sequential CTL activation is demonstrated by the exclusive clustering of migratory cDC1s with CD8 T cells early on during the initiation of an antiviral immune response. Subsequent clustering of activated CD8 T cells with the LN-resident cDC1s acts as a platform for signal relay from pDCs and activated CD4 T cells (79). According to this “consecutive interaction” model (79, 112), the generation of maximal CTL response and therapeutic anti-tumor immunity requires a multicellular orchestration of events in the tumor dLN (Figure 2) wherein migratory cDC1s capture the antigen in tumors, migrate to the dLN and form the initial priming site to activate CD8 T cells. The activated CD8 T cells produce CCL3/CCL4 and XCL1 to mediate recruitment of CCR5+pDCs and XCR1+LN-resident cDC1s, respectively. The migratory cDC1s handoff antigen to resident cDC1s in a yet-to-be-described mechanism (44, 106). In parallel, migratory cDC2s that have captured the antigen also move from the tumor to dLN and activate CD4 T cells. The pDCs induce the maturation of newly recruited, LN-resident cDC1s and the activated CD4 T cells licenses them for superior CTL responses. The overall effect of such orchestration and functional-cooperativity of pathways between different DC subsets is the amplification of CTL responses against a given antigen, without potentially missing out on the critical help necessary for CTLs to function at their peak. In fact, vaccine-mediated induction of such coordinated efforts of multiple DC subsets is known to trigger sustained and potent CTL responses while inhibiting immunosuppressive pathways in preclinical models (132). Ex vivo analyses of individual DC subsets might fail to identify such cellular orchestration to appreciate the relative contribution of each interaction between the different DC subsets in the generation of potent immune response.
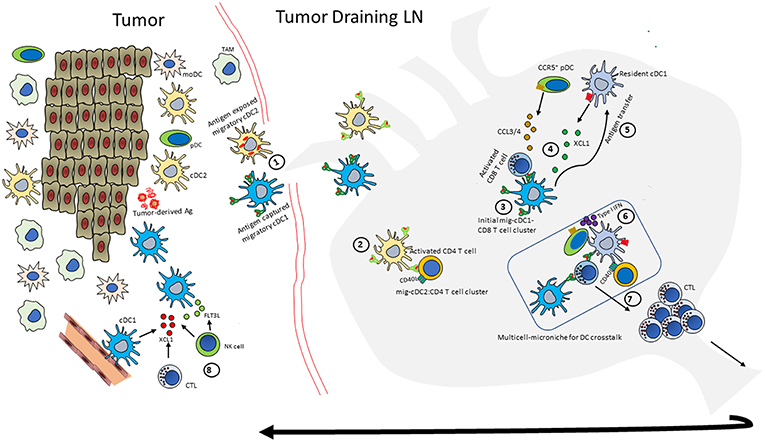
Figure 2. An integrated model of DC crosstalk for anti-tumor immunity. Outline of the multicellular orchestration of events that can contribute to a robust anti-tumor response. (Note: not all events happen in every context, and the order may also differ). (1) Intratumoral migratory-cDC1 and -cDC2s scavenge tumor-derived antigens and migrate to tumor dLN. (2) migratory-cDC2s (mig-cDC2) present MHC class II-restricted tumor antigen to CD4 T cells and induce expression of molecules such as CD40L (3) Migratory-cDC1s (mig-cDC1) prime and activate naïve CD8 T cells; (4) these activated CD8 T cells produce XCL1 and CCL3/4 to draw in XCR1+LN-resident-cDC1s and CCR5+ pDCs to the site of initial priming. (5) Mig-cDC1s can hand-off antigen to the newly recruited, LN-resident-cDC1. (6) pDCs produce type I IFN to mature cDCs. (7) The licensed CTL with enhanced effector functions undergoes clonal expansion and moves to the tumor to induce tumor cell killing. (8) The activated CD8 T cells and NK cells can mediate further increase in cDC1 numbers by producing XCL1 and FLT3L.
DC Crosstalk in Tumors in situ
Accumulating evidence suggest that cross-priming by tumor-resident cDC1 in situ is also an important phenomenon in the generation of an anti-tumor immune response. Local T cell priming and activation within tumors were observed in mice that lacked LN, or when T cell recirculation was blocked (133–135). Furthermore, intratumoral cDCs are required for the tumor regression achieved with adoptively transferred T cells in an experimental setting where migration of T cells to dLN was prevented (44). Moreover, tumor-resident cDC1s are the predominant sources of CXCL9 and CXCL10 and mediate recruitment effector T cells into the tumor (67). Similar to the events described for the dLN in the previous section, activated CD8 T cells could potentially orchestrate events in situ in the tumor where LN-like structures known as tertiary lymphoid structures (TLS) are present. A hallmark of TLS is the presence of high endothelial venules (HEVs) and expression of CCL19 and CCL21, the ligands for CCR7 (136, 137). DCs migrate in a CCR7-dependent manner (43, 45, 46, 138, 139). Moreover, well-organized TLSs contain B cell and T cells areas with mature DC subsets including cDCs and pDCs. Such organization makes TLS an ideal place to sustain proximity and the crosstalk between various subsets, and orchestrating local events required for maximal tumor immunity (135, 136). In fact, tumor-associated TLSs are functional structures capable of recruiting antigen-specific T cells and facilitating their activation through interactions with DCs (140). Interestingly, TLSs have been observed in several human tumors and their presence, particularly the ones containing high amounts of DCs and Th1 cells within the TLS, is associated with better prognosis (137, 141, 142) and increased TLS density is associated with strong infiltration of effector and memory CD8 T cells within the tumors (141), reflecting the importance of crosstalk between DC subsets and, CD4 help in increased CTL trafficking. Lung cancer patients with intratumoral CD8 T cells but no TLS had poor survival, indicating the necessity of their in situ education within the TLS for better effector functions (141, 143). In a metastatic lung tumor model, administration of TLR9 activator leads to CD8 T cell infiltration concurrent with TLS formation. The presence of TLS in this model was completely dependent on CD4 help (127). Taken together, these data suggest that TLSs promote DC crosstalk and anti-tumor immunity. Thus, induction of TLS provides another opportunity to promote communication between DC subsets to augment the magnitude of protective immunity, particularly against neoantigens that arise during the later phases of tumor progression (121). Moreover, induction of simultaneous trafficking and activation of cDCs and pDCs, using a vaccination strategy that combined DC subset-specific adjuvants (e.g., CpG-ODN and GM-CSF) leads to local accumulation of CD8 T cells and superior anti-tumor responses (132) suggesting that, even in the absence of TLS, evoking appropriate DC-crosstalk within the tumor tissue has the potential to boost superior CTL responses than targeting a single DC subset.
Influence of DC Crosstalk With Other Cells in the TME on Anti-tumor Immunity
DCs can also engage with other immune cell types in the TME and lymphoid organs. Such interactions can enhance or dampen DC functions and anti-tumor immunity, depending on the cell types involved. For example, DCs interact with Treg cells, resulting in the suppression of CD8 T cell-mediated anti-tumor immunity (144). Two-photon laser-scanning microscopy analysis showed that Treg cells engage in prolonged physical interactions with DCs, six times longer than that of DC-CD8 T cell interaction in tumor. This extended physical contact between Treg cells and DCs results in upregulation of the immunosuppressive molecules such as IDO and lower maturation molecules on DC surface (144).
Interactions with other immune cell types such as natural killer (NK) cells with DCs can boost the immune response against tumors. It has long been established that, through the secretion of IL-12, cDC1s can license NK cells to kill tumor cells (145–147). However, recent studies have shown that NK cells can also influence DC functions in the context of tumors. In fact, NK cells produce XCL1 to recruit XCR+cDC1s to the TME (148). In addition, NK cells are one source of Flt3L within the tumor and dictate intratumoral accumulation of cDC1 cells by supporting DC survival, proliferation or development (71). Stimulation of NK cells with DC-derived factors such as IL-12, IL-15/IL-15Rα complex or contact–dependent interactions of OX40-OX40L augment NK cell functions to eliminate tumor cells (149–151). TCGA analysis suggests that NK cell/XCL1/cDC1 axis is associated with better survival in many cancer indications (148).
DCs also interact with NKT cells, the unconventional T lymphocytes expressing a semi-invariant T cell receptor (TCR) that recognize glycolipids presented by CD1d. (152). Although CD1d can be expressed by many hematopoietic cell types, DCs constitutively express CD1d and are the most potent APCs for exogenous glycolipids (153–155). The NKT cell ligand α-galactosylceramide (α-GalCer) acts as a potent in vivo adjuvant for DCs, resulting in increased expression of MHC class II and other costimulatory molecules (155). In addition, α-GalCer presented by DCs strongly activates NKT cells through CD40/CD40L interaction to induce IFN-γ production (156). Administration of α-GalCer was efficacious in preclinical tumor models (157) but not in patients (158), most likely due to soluble α-GalCer-induced anergy of NKT cell (159). Administration of α-GalCer, either soluble or loaded in DCs, is currently being explored to enhance anti-tumor immunity (160). Endogenous glycolipids are known to activate NKT cells (161) and CD1d expression is observed on tumor cells (162). In fact, the level of CD1d expression on tumor cells dictates NKT-mediated cytotoxicity (163).
Tumor-associated macrophages, in most carcinomas, are linked to poor prognosis primarily due to their immunosuppressive phenotype (164, 165). Macrophages produce IL-10 and in turn prevent IL-12 secretion of by DCs, resulting in dampened tumor-specific CD8 T cell activation (166). Among mononuclear phagocytes, monocyte-derived cells (including macrophages) are found at higher frequencies in tumors compared to DCs, and a higher monocyte-macrophage signature is associated with worse clinical prognosis (130, 167). These cells maintain a phenotype similar to in vitro M2 macrophages and contribute to the suppressive tumor microenvironment primarily via expression of anti-inflammatory mediators such as IL-10, TGF-β and IDO. Many of these signals dampen the ability of cDCs to present antigen in an immunogenic manner (164). However, in other contexts, macrophages can be inflammatory and effective APCs for eliciting T cell responses (168, 169). Thus, with the addition of the right signals, tumor macrophages have the potential to contribute to anti-tumor immunity.
Additionally, even though B cells have been described to play varied and often contrasting roles in the contexts of tumor immunity, emerging evidence suggests that B cells may also contribute to tumor immunity, both via antibody-mediated effects and by acting as APCs (170–172). Specifically, in terms of the crosstalk, DCs engage with B cells to promote their growth and differentiation, resulting in the production of antibodies. pDCs, through type I IFN production, can increase TLR7 expression and other activation markers on B cells (173). pDCs are specifically capable of inducing differentiation of activated B cells into Ig-secreting plasma cells through the secretion of type I IFN and IL-6 (174). Additionally, DCs dramatically enhance the secretion of IgG and IgA through the ligation of CD40 (175). B cells isolated from TLS-containing lung cancers showed significant antibody response against many TAAs (143, 176).
Finally, DC crosstalk with cancer cells has tremendous impact on the immune surveillance of the tumors. Cancer cells express several immunosuppressive factors such as PGE2, β-catenin and cytokines such as IL-10. PGE2 renders cDC1s unresponsive to XCL1 and CCL5 by downregulating XCR1 and CCR5 expression (148). β-catenin expression in cancer cells causes ATF3-mediated suppression of CCL4, the ligand for CCR5, leading to defective recruitment of cDC1 to the TME, and adversely affecting CD8 T cell priming against TAAs (177). Interestingly, PGE2 also induces the expression of β-catenin not only in tumor cells but also in stromal cells such as cancer associated fibroblasts (CAFs). CAFs respond to tumor-derived TNFα and IL-1β to secrete thymic stromal lymphopoietin (TSLP). TSLP is a strong driver of cDCs to activate Th2 CD4 T cells that are considered pro-tumorigenic (178). CAFs also produce stromal cell-derived factor 1 (SDF1) which drives cDCs toward tolerogenic DCs secreting IDO in a STAT3-dependent manner and promoting the recruitment and differentiation of Treg cells in tumors (179). However, co-targeting fibroblasts in combination with DC-based vaccine enhances the anti-tumor immune responses (180), suggesting that DC/stromal cell interactions can be manipulated to improve immunotherapies. Overall, with the property of bridging the innate and adaptive immune cells, DCs have a pivotal role in orchestrating an anti-tumor immune response by engaging interactions with many cell types within the TME.
Potential Therapeutic Applications of Tumor DC-Crosstalk
The field of cancer immunotherapy, energized by the effect of T cell checkpoint inhibitors (CPI) in some patients, is beginning to focus on ways to treat “cold” tumors that lack T cells which can be activated with an anti-PD1 or other CPI. There is a large unmet medical need to increase the proportion of patients who respond to immunotherapy. Enhancing innate immunity, and DC function in particular, is one way to make tumors “warmer” that has tremendous potential. To date, most cell-based DC cancer therapies have utilized moDCs and have shown limited efficacy (121, 181, 182). With our current knowledge of both the importance of cross-presenting cDC1s for tumor immunity and the plasticity of monocyte-derived cells, moDCs are likely not the best cell type to use for inducing optimal clinical outcomes against cancer. Most studies show that moDCs have limited capacity for both cross-presentation and migration to draining LN compared to Batf3-dependent cells (43, 183). In addition, most monocyte-derived cells in the TME are immunosuppressive, and even if ex vivo moDCs can be activated to sustain cDC1-like properties, these are not likely maintained in the TME (121, 181). Therefore, moDC-based vaccines may not be the answer, and a new generation of DC-focused cancer immunotherapies are needed.
Increasing cDC1 function is one important goal, but as described here, some of this can occur indirectly via the cooperative interactions with other cells. In addition, both cDCs and pDCs have the potential to directly activate T cells that can kill cancer cells if exposed to the right activating signals (Figure 1B). Therefore, targeting maturation signals specifically to just cDC1s may not be the optimal therapy, and delivering signals that can enhance the function of all DC subsets may enhance efficacy and durability. For example, although tumor pDCs often correlate with poor prognosis, they are the most efficient producers of type 1 IFN and have the capacity for sustained MHC class II expression; these functions together may inflame the tumor and elicit strong T cell help that in turn could be sustained by newly matured cDC1s. Therefore, identifying signals that target and activate all DC subsets, and the cells that crosstalk with them will help provide novel insights into the cellular and molecular nature of tumor-specific CTL priming. The goal is to design therapies that build a site of sustained, immunostimulatory tumor-antigen presentation and increase the magnitude of anti-tumor immunity, so we can successfully treat a broader set of patients.
Author Contributions
RN, SM-M, and KT designed and wrote the manuscript.
Funding
The authors, employees of Amgen Inc., declare that this study received funding from Amgen Inc. in its entirety. Amgen Inc. did not have a role in the study design.
Conflict of Interest Statement
RN, SM-M, and KT are full-time employees of Amgen Inc.
Acknowledgments
We thank Carlos Briseno, Michael Gonzalez, and Jackson G. Egen for critical reading of the manuscript and helpful discussions.
References
1. Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease. Nat Rev Immunol. (2017) 17:30–48. doi: 10.1038/nri.2016.116
2. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med. (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
3. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Ann Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
4. Cosenza H, Leserman LD, Rowley DA. The third cell type required for the immune response of spleen cells in vitro. J Immunol. (1971) 107:414–21.
5. Anderson DA 3rd., Murphy KM, Briseno CG. Development, diversity, and function of dendritic cells in mouse and human. Cold Spring Harb Perspect Biol. (2018) 10:a028613. doi: 10.1101/cshperspect.a028613
6. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. (2014) 14:571–8. doi: 10.1038/nri3712
7. Dress RJ, Wong AY, Ginhoux F. Homeostatic control of dendritic cell numbers and differentiation. Immunol Cell Biol. (2018) 96:463–76. doi: 10.1111/imcb.12028
8. Theisen D, Murphy K. The role of cDC1s in vivo: CD8 T cell priming through cross-presentation. F1000Res. (2017) 6:98. doi: 10.12688/f1000research.9997.1
9. Devi KS, Anandasabapathy N. The origin of DCs and capacity for immunologic tolerance in central and peripheral tissues. Seminars Immunopathol. (2017) 39:137–52. doi: 10.1007/s00281-016-0602-0
10. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseno CG, Iwata A, et al. Transcriptional control of dendritic cell development. Ann Rev Immunol. (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
11. Nierkens S, Tel J, Janssen E, Adema GJ. Antigen cross-presentation by dendritic cell subsets: one general or all sergeants? Trends Immunol. (2013) 34:361–70. doi: 10.1016/j.it.2013.02.007
12. Scott CL, Soen B, Martens L, Skrypek N, Saelens W, Taminau J, et al. The transcription factor Zeb2 regulates development of conventional and plasmacytoid DCs by repressing Id2. J Exp Med. (2016) 213:897–911. doi: 10.1084/jem.20151715
13. Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. (2018) 154:3–20. doi: 10.1111/imm.12888
14. Poulin LF, Salio M, Griessinger E, Anjos-Afonso F, Craciun L, Chen JL, et al. Characterization of human DNGR-1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med. (2010) 207:1261–71. doi: 10.1084/jem.20092618
15. Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Ann Rev Immunol. (2017) 35:149–76. doi: 10.1146/annurev-immunol-041015-055254
16. Haniffa M, Shin A, Bigley V, McGovern N, Teo P, See P, et al. Human tissues contain CD141hi cross-presenting dendritic cells with functional homology to mouse CD103+ nonlymphoid dendritic cells. Immunity. (2012) 37:60–73. doi: 10.1016/j.immuni.2012.04.012
17. Bachem A, Guttler S, Hartung E, Ebstein F, Schaefer M, Tannert A, et al. Superior antigen cross-presentation and XCR1 expression define human CD11c+CD141+ cells as homologues of mouse CD8+ dendritic cells. J Exp Med. (2010) 207:1273–81. doi: 10.1084/jem.20100348
18. Blander JM. Regulation of the cell biology of antigen cross-presentation. Ann Rev Immunol. (2018) 36:717–53. doi: 10.1146/annurev-immunol-041015-055523
19. Soares H, Waechter H, Glaichenhaus N, Mougneau E, Yagita H, Mizenina O, et al. A subset of dendritic cells induces CD4+ T cells to produce IFN-gamma by an IL-12-independent but CD70-dependent mechanism in vivo. J Exp Med. (2007) 204:1095–106. doi: 10.1084/jem.20070176
20. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. (2007) 315:107–11. doi: 10.1126/science.1136080
21. Guilliams M, van de Laar L. A Hitchhiker's Guide to Myeloid Cell Subsets: practical implementation of a novel mononuclear phagocyte classification system. Front Immunol. (2015) 6:406. doi: 10.3389/fimmu.2015.00406
22. Tussiwand R, Everts B, Grajales-Reyes GE, Kretzer NM, Iwata A, Bagaitkar J, et al. Klf4 expression in conventional dendritic cells is required for T helper 2 cell responses. Immunity. (2015) 42:916–28. doi: 10.1016/j.immuni.2015.04.017
23. Gao Y, Nish SA, Jiang R, Hou L, Licona-Limon P, Weinstein JS, et al. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity. (2013) 39:722–32. doi: 10.1016/j.immuni.2013.08.028
24. Schlitzer A, McGovern N, Teo P, Zelante T, Atarashi K, Low D, et al. IRF4 transcription factor-dependent CD11b+ dendritic cells in human and mouse control mucosal IL-17 cytokine responses. Immunity. (2013) 38:970–83. doi: 10.1016/j.immuni.2013.04.011
25. Persson EK, Uronen-Hansson H, Semmrich M, Rivollier A, Hagerbrand K, Marsal J, et al. IRF4 transcription-factor-dependent CD103(+)CD11b(+) dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. (2013) 38:958–69. doi: 10.1016/j.immuni.2013.03.009
26. Alcantara-Hernandez M, Leylek R, Wagar LE, Engleman EG, Keler T, Marinkovich MP, et al. High-dimensional phenotypic mapping of human dendritic cells reveals interindividual variation and tissue specialization. Immunity. (2017) 47:1037–50 e6. doi: 10.1016/j.immuni.2017.11.001
27. Satpathy AT, Briseno CG, Lee JS, Ng D, Manieri NA, Kc W, et al. Notch2-dependent classical dendritic cells orchestrate intestinal immunity to attaching-and-effacing bacterial pathogens. Nat Immunol. (2013) 14:937–48. doi: 10.1038/ni.2679
28. Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science. (2017) 356:eaah4573. doi: 10.1126/science.aah4573
29. Lewis KL, Caton ML, Bogunovic M, Greter M, Grajkowska LT, Ng D, et al. Notch2 receptor signaling controls functional differentiation of dendritic cells in the spleen and intestine. Immunity. (2011) 35:780–91. doi: 10.1016/j.immuni.2011.08.013
30. Colonna M, Trinchieri G, Liu YJ. Plasmacytoid dendritic cells in immunity. Nat Immunol. (2004) 5:1219–26. doi: 10.1038/ni1141
31. Grouard G, Rissoan MC, Filgueira L, Durand I, Banchereau J, Liu YJ. The enigmatic plasmacytoid T cells develop into dendritic cells with interleukin (IL)-3 and CD40-ligand. J Exp Med. (1997) 185:1101–11. doi: 10.1084/jem.185.6.1101
32. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. (1999) 5:919–23. doi: 10.1038/11360
33. Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. (1999) 284:1835–7. doi: 10.1126/science.284.5421.1835
34. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. (2008) 135:37–48. doi: 10.1016/j.cell.2008.09.016
35. Alculumbre SG, Saint-Andre V, Di Domizio J, Vargas P, Sirven P, Bost P, et al. Diversification of human plasmacytoid predendritic cells in response to a single stimulus. Nat Immunol. (2018) 19:63–75. doi: 10.1038/s41590-017-0012-z
36. Rodrigues PF, Alberti-Servera L, Eremin A, Grajales-Reyes GE, Ivanek R, Tussiwand R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat Immunol. (2018) 19:711–22. doi: 10.1038/s41590-018-0136-9
37. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. (2003) 19:59–70. doi: 10.1016/S1074-7613(03)00171-7
38. Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
39. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. (2007) 449:419–26. doi: 10.1038/nature06175
40. Helft J, Bottcher J, Chakravarty P, Zelenay S, Huotari J, Schraml BU, et al. GM-CSF mouse bone marrow cultures comprise a heterogeneous population of CD11c(+)MHCII(+) macrophages and dendritic cells. Immunity. (2015) 42:1197–211. doi: 10.1016/j.immuni.2015.05.018
41. Sander J, Schmidt SV, Cirovic B, McGovern N, Papantonopoulou O, Hardt AL, et al. Cellular differentiation of human monocytes is regulated by time-dependent interleukin-4 signaling and the transcriptional regulator NCOR2. Immunity. (2017) 47:1051–66 e12. doi: 10.1016/j.immuni.2017.11.024
42. Wu X, Briseno CG, Durai V, Albring JC, Haldar M, Bagadia P, et al. Mafb lineage tracing to distinguish macrophages from other immune lineages reveals dual identity of Langerhans cells. J Exp Med. (2016) 213:2553–65. doi: 10.1084/jem.20160600
43. Laoui D, Keirsse J, Morias Y, Van Overmeire E, Geeraerts X, Elkrim Y, et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat Commun. 2016;7:13720. doi: 10.1038/ncomms13720
44. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell. 2014;26(6):938. doi: 10.1016/j.ccell.2014.11.010
45. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103(+) dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity. (2016) 44:924–38. doi: 10.1016/j.immuni.2016.03.012
46. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
47. Lou Y, Liu C, Kim GJ, Liu YJ, Hwu P, Wang G. Plasmacytoid dendritic cells synergize with myeloid dendritic cells in the induction of antigen-specific antitumor immune responses. J Immunol. (2007) 178:1534–41. doi: 10.4049/jimmunol.178.3.1534
48. Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell. (2017) 169:750–65 e17. doi: 10.1016/j.cell.2017.04.014
49. Tang-Huau TL, Gueguen P, Goudot C, Durand M, Bohec M, Baulande S, et al. Human in vivo-generated monocyte-derived dendritic cells and macrophages cross-present antigens through a vacuolar pathway. Nat Commun. 2018;9(1):2570. doi: 10.1038/s41467-018-04985-0
50. Cohn L, Chatterjee B, Esselborn F, Smed-Sorensen A, Nakamura N, Chalouni C, et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J Exp Med. (2013) 210:1049–63. doi: 10.1084/jem.20121251
51. Kretzer NM, Theisen DJ, Tussiwand R, Briseno CG, Grajales-Reyes GE, Wu X, et al. RAB43 facilitates cross-presentation of cell-associated antigens by CD8alpha+ dendritic cells. J Exp Med. (2016) 213:2871–83. doi: 10.1084/jem.20160597
52. Chatterjee B, Smed-Sorensen A, Cohn L, Chalouni C, Vandlen R, Lee BC, et al. Internalization and endosomal degradation of receptor-bound antigens regulate the efficiency of cross presentation by human dendritic cells. Blood. (2012) 120:2011–20. doi: 10.1182/blood-2012-01-402370
53. Alloatti A, Rookhuizen DC, Joannas L, Carpier JM, Iborra S, Magalhaes JG, et al. Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. J Exp Med. (2017) 214:2231–41. doi: 10.1084/jem.20170229
54. Wu SJ, Niknafs YS, Kim SH, Oravecz-Wilson K, Zajac C, Toubai T, et al. A critical analysis of the role of SNARE protein SEC22B in antigen cross-presentation. Cell Rep. (2017) 19:2645–56. doi: 10.1016/j.celrep.2017.06.013
55. Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell. (2011) 147:1355–68. doi: 10.1016/j.cell.2011.11.021
56. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. (2006) 126:205–18. doi: 10.1016/j.cell.2006.05.035
57. Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, et al. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood. (2008) 112:4712–22. doi: 10.1182/blood-2008-01-134791
58. Savina A, Peres A, Cebrian I, Carmo N, Moita C, Hacohen N, et al. The small GTPase Rac2 controls phagosomal alkalinization and antigen crosspresentation selectively in CD8(+) dendritic cells. Immunity. (2009) 30:544–55. doi: 10.1016/j.immuni.2009.01.013
59. Jancic C, Savina A, Wasmeier C, Tolmachova T, El-Benna J, Dang PM, et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat Cell Biol. (2007) 9:367–78. doi: 10.1038/ncb1552
60. Ding Y, Guo Z, Liu Y, Li X, Zhang Q, Xu X, et al. The lectin Siglec-G inhibits dendritic cell cross-presentation by impairing MHC class I-peptide complex formation. Nat Immunol. (2016) 17:1167–75. doi: 10.1038/ni.3535
61. Theisen DJ, Davidson JTt, Briseno CG, Gargaro M, Lauron EJ, Wang Q, et al. WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science. (2018) 362:694–9. doi: 10.1126/science.aat5030
62. Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, et al. CD8α+ dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. (2011) 35:249–59. doi: 10.1016/j.immuni.2011.08.008
63. Martinez-Lopez M, Iborra S, Conde-Garrosa R, Sancho D. Batf3-dependent CD103+ dendritic cells are major producers of IL-12 that drive local Th1 immunity against Leishmania major infection in mice. Eur J Immunol. (2015) 45:119–29. doi: 10.1002/eji.201444651
64. Everts B, Tussiwand R, Dreesen L, Fairfax KC, Huang SC, Smith AM, et al. Migratory CD103+ dendritic cells suppress helminth-driven type 2 immunity through constitutive expression of IL-12. J Exp Med. (2016) 213:35–51. doi: 10.1084/jem.20150235
65. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. (2008) 322:1097–100. doi: 10.1126/science.1164206
66. Byrne KT, Vonderheide RH. CD40 Stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. (2016) 15:2719–32. doi: 10.1016/j.celrep.2016.05.058
67. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. (2017) 31:711–23 e4. doi: 10.1016/j.ccell.2017.04.003
68. Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat Commun. (2018) 9:1250. doi: 10.1038/s41467-018-03600-6
69. Sanchez-Paulete AR, Cueto FJ, Martinez-Lopez M, Labiano S, Morales-Kastresana A, Rodriguez-Ruiz ME, et al. Cancer Immunotherapy with immunomodulatory anti-CD137 and anti-PD-1 monoclonal antibodies requires BATF3-dependent dendritic cells. Cancer Discov. (2016) 6:71–9. doi: 10.1158/2159-8290.CD-15-0510
70. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity. (2018) 49:178–93 e7. doi: 10.1016/j.immuni.2018.06.006
71. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1158/2326-6074.CRICIMTEATIAACR18-PR04
72. Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell. (2018) 33:563–9. doi: 10.1016/j.ccell.2018.03.008
73. Lee W, Kim HS, Hwang SS, Lee GR. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med. (2017) 49:e393. doi: 10.1038/emm.2017.157
74. Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. (2012) 209:1153–65. doi: 10.1084/jem.20112675
75. Robbins SH, Walzer T, Dembele D, Thibault C, Defays A, Bessou G, et al. Novel insights into the relationships between dendritic cell subsets in human and mouse revealed by genome-wide expression profiling. Genome Biol. (2008) 9:R17. doi: 10.1186/gb-2008-9-1-r17
76. Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. (2012) 209:1135–52. doi: 10.1084/jem.20120030
77. Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust anti-viral immunity requires multiple distinct T cell-dendritic cell interactions. Cell. (2015) 162:1322–37. doi: 10.1016/j.cell.2015.08.004
78. Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally distinct interactions with dendritic cell subsets facilitates CD4+ and CD8+ T cell activation to localized viral infection. Immunity. (2015) 43:554–65. doi: 10.1016/j.immuni.2015.07.020
79. Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, et al. CD8+ T cells orchestrate pDC-XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity. (2017) 46:205–19. doi: 10.1016/j.immuni.2017.01.003
80. Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4+ T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. (2010) 207:637–50. doi: 10.1084/jem.20091918
81. Qin Z, Blankenstein T. CD4+ T cell–mediated tumor rejection involves inhibition of angiogenesis that is dependent on IFN gamma receptor expression by nonhematopoietic cells. Immunity. (2000) 12:677–86. doi: 10.1016/S1074-7613(00)80218-6
82. Kim HJ, Cantor H. CD4 T-cell subsets and tumor immunity: the helpful and the not-so-helpful. Cancer Immunol Res. (2014) 2:91–8. doi: 10.1158/2326-6066.CIR-13-0216
83. Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. (2008) 222:129–44. doi: 10.1111/j.1600-065X.2008.00616.x
84. Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. (2011) 208:2005–16. doi: 10.1084/jem.20101159
85. Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. (2011) 208:1989–2003. doi: 10.1084/jem.20101158
86. Fonteneau JF, Gilliet M, Larsson M, Dasilva I, Munz C, Liu YJ, et al. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. (2003) 101:3520–6. doi: 10.1182/blood-2002-10-3063
87. Tel J, Aarntzen EH, Baba T, Schreibelt G, Schulte BM, Benitez-Ribas D, et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. (2013) 73:1063–75. doi: 10.1158/0008-5472.CAN-12-2583
88. Perez-Cabezas B, Naranjo-Gomez M, Ruiz-Riol M, Bastos-Amador P, Fernandez MA, Carmona F, et al. TLR-activated conventional DCs promote gamma-secretase-mediated conditioning of plasmacytoid DCs. J Leukoc Biol. (2012) 92:133–43. doi: 10.1189/jlb.0911452
89. Nascimbeni M, Perie L, Chorro L, Diocou S, Kreitmann L, Louis S, et al. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood. (2009) 113:6112–9. doi: 10.1182/blood-2008-07-170803
90. Vermi W, Lonardi S, Morassi M, Rossini C, Tardanico R, Venturini M, et al. Cutaneous distribution of plasmacytoid dendritic cells in lupus erythematosus. Selective tropism at the site of epithelial apoptotic damage. Immunobiology. (2009) 214:877–86. doi: 10.1016/j.imbio.2009.06.013
91. Rogers GL, Shirley JL, Zolotukhin I, Kumar SRP, Sherman A, Perrin GQ, et al. Plasmacytoid and conventional dendritic cells cooperate in crosspriming AAV capsid-specific CD8+ T cells. Blood. (2017) 129:3184–95. doi: 10.1182/blood-2016-11-751040
92. Yoneyama H, Matsuno K, Toda E, Nishiwaki T, Matsuo N, Nakano A, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med. (2005) 202:425–35. doi: 10.1084/jem.20041961
93. Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. (2001) 2:1144–50. doi: 10.1038/ni736
94. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8+ T cell accrual. Immunity. (2010) 33:955–66. doi: 10.1016/j.immuni.2010.11.020
95. Cervantes-Barragan L, Lewis KL, Firner S, Thiel V, Hugues S, Reith W, et al. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci USA. (2012) 109:3012–7. doi: 10.1073/pnas.1117359109
96. Terra M, Oberkampf M, Fayolle C, Rosenbaum P, Guillerey C, Dadaglio G, et al. Tumor-derived TGFbeta alters the ability of plasmacytoid dendritic cells to respond to innate immune signaling. Cancer Res. (2018) 78:3014–26. doi: 10.1158/0008-5472.CAN-17-2719
97. Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clinical Cancer Res. (2004) 10:7466–74. doi: 10.1158/1078-0432.CCR-04-0684
98. Conrad C, Gregorio J, Wang YH, Ito T, Meller S, Hanabuchi S, et al. Plasmacytoid dendritic cells promote immunosuppression in ovarian cancer via ICOS costimulation of Foxp3+ T-regulatory cells. Cancer Res. (2012) 72:5240–9. doi: 10.1158/0008-5472.CAN-12-2271
99. Mellor AL, Munn DH. IDO expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4:762–74. doi: 10.1038/nri1457
100. Kitano M, Yamazaki C, Takumi A, Ikeno T, Hemmi H, Takahashi N, et al. Imaging of the cross-presenting dendritic cell subsets in the skin-draining lymph node. Proc Natl Acad Sci USA. (2016) 113:1044–9. doi: 10.1073/pnas.1513607113
101. Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity. (2015) 42:172–85. doi: 10.1016/j.immuni.2014.12.024
102. Gerner MY, Kastenmuller W, Ifrim I, Kabat J, Germain RN. Histo-cytometry: a method for highly multiplex quantitative tissue imaging analysis applied to dendritic cell subset microanatomy in lymph nodes. Immunity. (2012) 37:364–76. doi: 10.1016/j.immuni.2012.07.011
103. Gerner MY, Casey KA, Kastenmuller W, Germain RN. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J Exp Med. (2017) 214:3105–22. doi: 10.1084/jem.20170335
104. Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4+ T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. (2018) 18:635–47. doi: 10.1038/s41577-018-0044-0
105. Calabro S, Liu D, Gallman A, Nascimento MS, Yu Z, Zhang TT, et al. Differential intrasplenic migration of dendritic cell subsets tailors adaptive immunity. Cell Rep. (2016) 16:2472–85. doi: 10.1016/j.celrep.2016.07.076
106. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. (2006) 25:153–62. doi: 10.1016/j.immuni.2006.04.017
107. Amigorena S. Helping the help for CD8+ T cell responses. Cell. (2015) 162:1210–2. doi: 10.1016/j.cell.2015.08.051
108. Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. (2003) 421:852–6. doi: 10.1038/nature01441
109. Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. (2003) 300:337–9. doi: 10.1126/science.1082305
110. Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. (1998) 393:478–80. doi: 10.1038/30996
111. Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. (1998) 393:480–3. doi: 10.1038/31002
112. Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. (1998) 393:474–8. doi: 10.1038/30989
113. Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med. (1999) 190:617–27. doi: 10.1084/jem.190.5.617
114. Tarbell KV, Egen JG. Breaking self-tolerance during autoimmunity and cancer immunity: myeloid cells and type I IFN response regulation. J Leukoc Biol. (2018) 103:1117–29. doi: 10.1002/JLB.3MIR1017-400R
115. Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med. (2002) 196:369–77. doi: 10.1084/jem.20011353
116. Hoglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med. (1999) 189:331–9. doi: 10.1084/jem.189.2.331
117. Unanue ER. Perspectives on anti-CD47 antibody treatment for experimental cancer. Proc Natl Acad Sci USA. (2013) 110:10886–7. doi: 10.1073/pnas.1308463110
118. Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. (2012) 21:402–17. doi: 10.1016/j.ccr.2012.01.008
119. Gerner MY, Casey KA, Mescher MF. Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. J Immunol. (2008) 181:155–64. doi: 10.4049/jimmunol.181.1.155
120. Anderson MJ, Shafer-Weaver K, Greenberg NM, Hurwitz AA. Tolerization of tumor-specific T cells despite efficient initial priming in a primary murine model of prostate cancer. J Immunol. (2007) 178:1268–76. doi: 10.4049/jimmunol.178.3.1268
121. Gardner A, Ruffell B. Dendritic cells and cancer immunity. Trends Immunol. (2016) 37:855–65. doi: 10.1016/j.it.2016.09.006
122. Zhu Z, Cuss SM, Singh V, Gurusamy D, Shoe JL, Leighty R, et al. CD4+ T cell help selectively enhances high-avidity tumor antigen-specific CD8+ T cells. J Immunol. (2015) 195:3482–9. doi: 10.4049/jimmunol.1401571
123. Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res. (2010) 70:8368–77. doi: 10.1158/0008-5472.CAN-10-1322
124. Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4+ and CD8+ T cells during the effector phase. J Exp Med. (2010) 207:2469–77. doi: 10.1084/jem.20092450
125. Marzo AL, Kinnear BF, Lake RA, Frelinger JJ, Collins EJ, Robinson BW, et al. Tumor-specific CD4+ T cells have a major “post-licensing” role in CTL mediated anti-tumor immunity. J Immunol. (2000) 165:6047–55. doi: 10.4049/jimmunol.165.11.6047
126. Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. CD4+ T cell help confers a cytotoxic T cell effector program including coinhibitory receptor downregulation and increased tissue invasiveness. Immunity. (2017) 47:848–61 e5. doi: 10.1016/j.immuni.2017.10.009
127. Gallotta M, Assi H, Degagne E, Kannan SK, Coffman RL, Guiducci C. Inhaled TLR9 agonist renders lung tumors permissive to PD-1 blockade by promoting optimal CD4+ and CD8+ T-cell Interplay. Cancer Res. (2018) 78:4943–56. doi: 10.1158/0008-5472.CAN-18-0729
128. Neubert K, Lehmann CH, Heger L, Baranska A, Staedtler AM, Buchholz VR, et al. Antigen delivery to CD11c+CD8- dendritic cells induces protective immune responses against experimental melanoma in mice in vivo. J Immunol. (2014) 192:5830–8. doi: 10.4049/jimmunol.1300975
129. Alonso R, Flament H, Lemoine S, Sedlik C, Bottasso E, Peguillet I, et al. Induction of anergic or regulatory tumor-specific CD4+ T cells in the tumor-draining lymph node. Nat Commun. (2018) 9:2113. doi: 10.1038/s41467-018-04524-x
130. Michea P, Noel F, Zakine E, Czerwinska U, Sirven P, Abouzid O, et al. Adjustment of dendritic cells to the breast-cancer microenvironment is subset specific. Nat Immunol. (2018) 19:885–97. doi: 10.1038/s41590-018-0145-8
131. Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi: 10.1038/ncomms10582
132. Ali OA, Emerich D, Dranoff G, Mooney DJ. In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci Transl Med. (2009) 1:8ra19. doi: 10.1126/scitranslmed.3000359
133. Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med. (2010) 207:1791–804. doi: 10.1084/jem.20092454
134. Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. (2013) 38:729–41. doi: 10.1016/j.immuni.2013.03.003
135. Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, et al. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat Immunol. (2004) 5:141–9. doi: 10.1038/ni1029
136. Pitzalis C, Jones GW, Bombardieri M, Jones SA. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat Rev Immunol. (2014) 14:447–62. doi: 10.1038/nri3700
137. Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. (2014) 35:571–80. doi: 10.1016/j.it.2014.09.006
138. Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. (1999) 189:451–60. doi: 10.1084/jem.189.3.451
139. Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, et al. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. (1999) 99:23–33. doi: 10.1016/S0092-8674(00)80059-8
140. Joshi NS, Akama-Garren EH, Lu Y, Lee DY, Chang GP, Li A, et al. Regulatory T cells in tumor-associated tertiary lymphoid structures suppress anti-tumor T cell responses. Immunity. (2015) 43:579–90. doi: 10.1016/j.immuni.2015.08.006
141. Goc J, Germain C, Vo-Bourgais TK, Lupo A, Klein C, Knockaert S, et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. (2014) 74:705–15. doi: 10.1158/0008-5472.CAN-13-1342
142. Dieu-Nosjean MC, Antoine M, Danel C, Heudes D, Wislez M, Poulot V, et al. Long-term survival for patients with non-small-cell lung cancer with intratumoral lymphoid structures. J Clin Oncol. (2008) 26:4410–7. doi: 10.1200/JCO.2007.15.0284
143. Remark R, Becker C, Gomez JE, Damotte D, Dieu-Nosjean MC, Sautes-Fridman C, et al. The non-small cell lung cancer immune contexture. A major determinant of tumor characteristics and patient outcome. Am J Respir Crit Care Med. (2015) 191:377–90. doi: 10.1164/rccm.201409-1671PP
144. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. (2017) 20:558–71. doi: 10.1016/j.celrep.2017.06.062
145. Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nat Med. (1999) 5:405–11. doi: 10.1038/7403
146. Borg C, Jalil A, Laderach D, Maruyama K, Wakasugi H, Charrier S, et al. NK cell activation by dendritic cells (DCs) requires the formation of a synapse leading to IL-12 polarization in DCs. Blood. (2004) 104:3267–75. doi: 10.1182/blood-2004-01-0380
147. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nat Rev Cancer. (2016) 16:7–19. doi: 10.1038/nrc.2015.5
148. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37 e14. doi: 10.1016/j.cell.2018.01.004
149. Ferlazzo G, Pack M, Thomas D, Paludan C, Schmid D, Strowig T, et al. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc Natl Acad Sci USA. (2004) 101:16606–11. doi: 10.1073/pnas.0407522101
150. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. (2007) 26:503–17. doi: 10.1016/j.immuni.2007.03.006
151. Liu C, Lou Y, Lizee G, Qin H, Liu S, Rabinovich B, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest. (2008) 118:1165–75. doi: 10.1172/JCI33583
152. Godfrey DI, Uldrich AP, McCluskey J, Rossjohn J, Moody DB. The burgeoning family of unconventional T cells. Nat Immunol. (2015) 16:1114–23. doi: 10.1038/ni.3298
153. Fujii S, Shimizu K, Kronenberg M, Steinman RM. Prolonged IFN-gamma-producing NKT response induced with alpha-galactosylceramide-loaded DCs. Nat Immunol. (2002) 3:867–74. doi: 10.1038/ni827
154. Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. (2004) 199:1607–18. doi: 10.1084/jem.20040317
155. Fujii S, Shimizu K, Smith C, Bonifaz L, Steinman RM. Activation of natural killer T cells by alpha-galactosylceramide rapidly induces the full maturation of dendritic cells in vivo and thereby acts as an adjuvant for combined CD4 and CD8 T cell immunity to a coadministered protein. J Exp Med. (2003) 198:267–79. doi: 10.1084/jem.20030324
156. Kitamura H, Iwakabe K, Yahata T, Nishimura S, Ohta A, Ohmi Y, et al. The natural killer T (NKT) cell ligand alpha-galactosylceramide demonstrates its immunopotentiating effect by inducing interleukin (IL)-12 production by dendritic cells and IL-12 receptor expression on NKT cells. J Exp Med. (1999) 189:1121–8. doi: 10.1084/jem.189.7.1121
157. Kobayashi E, Motoki K, Uchida T, Fukushima H, Koezuka Y. KRN7000, a novel immunomodulator, and its antitumor activities. Oncol Res. (1995) 7:529–34.
158. Giaccone G. Clinical impact of novel treatment strategies. Oncogene. (2002) 21:6970–81. doi: 10.1038/sj.onc.1205565
159. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. (2005) 115:2572–83. doi: 10.1172/JCI24762
160. King LA, Lameris R, de Gruijl TD, van der Vliet HJ. CD1d-Invariant natural killer T cell-based cancer immunotherapy: alpha-galactosylceramide and beyond. Front Immunol. (2018) 9:1519. doi: 10.3389/fimmu.2018.01519
161. Mattner J, Debord KL, Ismail N, Goff RD, Cantu C III, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. (2005) 434:525–9. doi: 10.1038/nature03408
162. Nair S, Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. (2017) 8:1178. doi: 10.3389/fimmu.2017.01178
163. Haraguchi K, Takahashi T, Nakahara F, Matsumoto A, Kurokawa M, Ogawa S, et al. CD1d expression level in tumor cells is an important determinant for anti-tumor immunity by natural killer T cells. Leuk Lymphoma. (2006) 47:2218–23. doi: 10.1080/10428190600682688
164. Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. (2002) 196:254–65. doi: 10.1002/path.1027
165. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
166. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
167. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. (2014) 344:921–5. doi: 10.1126/science.1252510
168. Pozzi LA, Maciaszek JW, Rock KL. Both dendritic cells and macrophages can stimulate naive CD8 T cells in vivo to proliferate, develop effector function, and differentiate into memory cells. J Immunol. (2005) 175:2071–81. doi: 10.4049/jimmunol.175.4.2071
169. Unanue ER. Antigen-presenting function of the macrophage. Ann Rev Immunol. (1984) 2:395–428. doi: 10.1146/annurev.iy.02.040184.002143
170. Joseph CG, Darrah E, Shah AA, Skora AD, Casciola-Rosen LA, Wigley FM, et al. Association of the autoimmune disease scleroderma with an immunologic response to cancer. Science. (2014) 343:152–7. doi: 10.1126/science.1246886
171. Tsou P, Katayama H, Ostrin EJ, Hanash SM. The Emerging Role of B Cells in Tumor Immunity. Cancer Res. (2016) 76:5597–601. doi: 10.1158/0008-5472.CAN-16-0431
172. Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer. (2016) 2:747–57. doi: 10.1016/j.trecan.2016.10.010
173. Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. (2005) 174:4043–50. doi: 10.4049/jimmunol.174.7.4043
174. Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. (2003) 19:225–34. doi: 10.1016/S1074-7613(03)00208-5
175. Dubois B, Vanbervliet B, Fayette J, Massacrier C, Van Kooten C, Briere F, et al. Dendritic cells enhance growth and differentiation of CD40-activated B lymphocytes. J Exp Med. (1997) 185:941–51. doi: 10.1084/jem.185.5.941
176. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med. (2014) 189:832–44. doi: 10.1164/rccm.201309-1611OC
177. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
178. Liu YJ, Soumelis V, Watanabe N, Ito T, Wang YH, Malefyt Rde W, et al. TSLP: an epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Ann Rev Immunol. (2007) 25:193–219. doi: 10.1146/annurev.immunol.25.022106.141718
179. Cheng JT, Deng YN, Yi HM, Wang GY, Fu BS, Chen WJ, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. (2016) 5:e198. doi: 10.1038/oncsis.2016.7
180. Ohshio Y, Teramoto K, Hanaoka J, Tezuka N, Itoh Y, Asai T, et al. Cancer-associated fibroblast-targeted strategy enhances antitumor immune responses in dendritic cell-based vaccine. Cancer Sci. (2015) 106:134–42. doi: 10.1111/cas.12584
181. Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. (2013) 39:38–48. doi: 10.1016/j.immuni.2013.07.004
182. Constantino J, Gomes C, Falcao A, Cruz MT, Neves BM. Antitumor dendritic cell-based vaccines: lessons from 20 years of clinical trials and future perspectives. Transl Res. (2016) 168:74–95. doi: 10.1016/j.trsl.2015.07.008
Keywords: dendritic cells, cDC1, cDC2, crosstalk, cancer immunity
Citation: Noubade R, Majri-Morrison S and Tarbell KV (2019) Beyond cDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 10:1014. doi: 10.3389/fimmu.2019.01014
Received: 30 August 2018; Accepted: 23 April 2019;
Published: 09 May 2019.
Edited by:
Diana Dudziak, Universitätsklinikum Erlangen, GermanyReviewed by:
Joke M. M. Den Haan, VU University Medical Center, NetherlandsRoxane Tussiwand, Universität Basel, Switzerland
Copyright © 2019 Noubade, Majri-Morrison and Tarbell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajkumar Noubade, cm5vdWJhZGVAYW1nZW4uY29t