
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 May 2019
Sec. Microbial Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.01011
 Tyler R. Tominello1†
Tyler R. Tominello1† Edson R. A. Oliveira2†
Edson R. A. Oliveira2† Shah S. Hussain2
Shah S. Hussain2 Amr Elfert2
Amr Elfert2 Jakob Wells1
Jakob Wells1 Brandon Golden1
Brandon Golden1 Nahed Ismail2*
Nahed Ismail2*Human monocytic ehrlichiosis (HME) is a potentially life-threatening tick-borne rickettsial disease (TBRD) caused by the obligate intracellular Gram-negative bacteria, Ehrlichia. Fatal HME presents with acute ailments of sepsis and toxic shock-like symptoms that can evolve to multi-organ failure and death. Early clinical and laboratory diagnosis of HME are problematic due to non-specific flu-like symptoms and limitations in the current diagnostic testing. Several studies in murine models showed that cell-mediated immunity acts as a “double-edged sword” in fatal ehrlichiosis. Protective components are mainly formed by CD4 Th1 and NKT cells, in contrast to deleterious effects originated from neutrophils and TNF-α-producing CD8 T cells. Recent research has highlighted the central role of the inflammasome and autophagy as part of innate immune responses also leading to protective or pathogenic scenarios. Recognition of pathogen-associated molecular patterns (PAMPS) or damage-associated molecular patterns (DAMPS) triggers the assembly of the inflammasome complex that leads to multiple outcomes. Recognition of PAMPs or DAMPs by such complexes can result in activation of caspase-1 and -11, secretion of the pro-inflammatory cytokines IL-1β and IL-18 culminating into dysregulated inflammation, and inflammatory cell death known as pyroptosis. The precise functions of inflammasomes and autophagy remain unexplored in infections with obligate intracellular rickettsial pathogens, such as Ehrlichia. In this review, we discuss the intracellular innate immune surveillance in ehrlichiosis involving the regulation of inflammasome and autophagy, and how this response influences the innate and adaptive immune responses against Ehrlichia. Understanding such mechanisms would pave the way in research for novel diagnostic, preventative and therapeutic approaches against Ehrlichia and other rickettsial diseases.
Ehrlichiosis is caused by Ehrlichia; an obligate intracellular Gram-negative bacteria that belongs to Anaplasmataceae family of the order Rickettsiales. Other genera within the family Anaplasmataceae are: Anaplasma, Neorickettsia, and Wolbachia (1). Ehrlichiae are maintained in natural cycles throughout persistently infected vertebrate hosts and tick vectors (2–4).
Ehrlichia infection is recognized as an important public health threat in tick-endemic areas including the south and south-central regions of the US, although a global threat also exists due to the prevalence of HME worldwide. Ehrlichia chaffeensis is the foremost etiologic agent of HME, however, other Ehrlichia species, including Ehrlichia canis, Ehrlichia muris and Ehrlichia muris-like agent (EMLA) are also isolated from patients with HME (5). The vector for E. chaffeensis is Amblyomma americanum, which is also vector for Rickettsia species such as R. amblyommii and R. parkeri (4).
Human monocytic ehrlichiosis presents as an acute febrile illness associated with fever, malaise, myalgia, and headache. Clinical manifestations of skin and gastrointestinal tract involvement such as rash, nausea, vomiting, diarrhea, and regional lymphadenopathy occurs in 20–40% of patients (4). Meningoencephalitis and pneumonia occur in 20% of patients who present with stiff neck, confusion, cough, and dyspnea. Life-threatening complications such as renal failure, adult respiratory distress syndrome, meningoencephalitis, multi-system organ failure, and toxic shock occur in a substantial portion of the patients who are hospitalized. HME is often undiagnosed or misdiagnosed as a result of non-specific clinical manifestations and lack of specific and sensitive diagnostic tests. Characteristic laboratory findings in HME patients are thrombocytopenia, leukopenia, neutropenia, and increased levels of hepatic transaminases (4, 6, 7). Diagnostic tests such as peripheral blood smear, in-vitro culture, PCR and serological testing are currently used to identify HME. However, each of these tests has potential limitations with suboptimal sensitivity or specificity at early stages of infection. Antibiotic treatment with doxycycline (drug of choice) is effective only if given early in infection. Failure to treat immunocompetent patients with doxycycline at the early stages of infection or in infected-immunocompromised individuals often results in serious and progressive disease that mimics septic or toxic shock-like syndrome and multi-organ failure with a case fatality rate of 3%. The clinical, diagnostic and therapeutic challenges in the management of patients with ehrlichiosis account for a high rate of hospitalization (40–63%) (4, 6). Thus, there is a critical need in creating new options for effective countermeasures (e.g., diagnostics, preventive and therapeutic measures) to control these pathogens. Understanding the immunopathogenesis of HME will enable us to develop new avenues for sensitive and specific diagnostic testing during early infection and immunotherapies for later disease management.
Ehrlichia canis is the major cause of canine ehrlichiosis, although other human Ehrlichia species such as E. chaffeensis and E. ewingii can also infect dogs. E. ewingii is transmitted by the lone star tick, A. americanum, while E. canis is transmitted by the brown dog tick, Rhipicephalus sanguineus. The bacteria are maintained through the tick life stages by transstadial, but not by transovarial, transmission (8, 9). Similar to E. chaffeensis, the natural host for E. canis is the white-tailed deer, although chronically infected dogs are also considered as reservoirs (10).
E. canis and E. chaffeensis primarily infect monocytes, thus causing canine monocytic ehrlichiosis, while E. ewingii infect granulocytes causing canine granulocytic ehrlichiosis. Dogs with acute canine ehrlichiosis may present with multi-system disease including lymphadenopathy, splenomegaly, ocular signs such as uveitis, retinitis, retinal hemorrhage or retinal detachment (11). Similar to HME, meningoencephalitis or cerebral hemorrhage may occur in 20% of infected dogs and present with stupor, ataxia, central or peripheral vestibular dysfunction, cerebellar dysfunction, convulsion, and tremors. Hematologic and immunologic abnormalities are commonly marked by the presence of petechiae, dermal ecchymosis, and autoimmunity including generation of anti-platelet antibodies that may account for thrombocytopenia, leukopenia, anemia, and hemolysis (10). Laboratory findings in dogs with either subacute or chronic monocytic or granulocytic ehrlichiosis include high serum levels of alkaline phosphatase and/or liver transaminases, hypocalcemia, hypokalemia, hyperglobulinemia, and seroconversion after 7–14 days post-infection (10, 11). Unlike HME, Ehrlichia infection in dogs can be self-limited even without antibiotic treatment but can cause persistent/chronic infection. Chronic infection is these animals may lead to the development of pancytopenia and potentially fatal hypoplastic bone marrow failure.
Ehrlichia species primarily infect macrophages and non-myeloid cells such as hepatocytes and endothelial cells. Ehrlichia exist in two forms within macrophages: (i) a small infectious nonreplicating dense core (0.4–0.6 μm), and (ii) a large, noninfectious reticulate form (0.4–0.6 μm × 0.7–1.9 μm) that undergoes binary fission within a cytoplasmic vacuole. The cytoplasmic vacuole contains Ehrlichia morulae (Latin for mulberry) which are visualized by Giemsa or Diff-Quick staining methods within infected monocytes or neutrophils from the peripheral blood smear.
Unlike other Gram-negative bacteria, Ehrlichia cell envelope lacks lipopolysaccharide (LPS) and peptidoglycan: two major PAMPS that are recognized by Toll-like receptors (TLRs) expressed by innate-immune and non-immune cells (12, 13). However, Ehrlichia cell outer membrane is enriched with proteins that express tandem repeat units (TRPs) (14–19). These TRPs are secreted into the target-cell cytosol via type I secretion system and are known to: (i) regulate host cell transcription factors involved in cell survival (20, 21); (ii) modulate cytoskeleton organization (21, 22); (iii) induce innate and adaptive immune responses; (23–27) and (iv) favor bacterial survival (28). Ehrlichia also express a 200 kDa protein resembling the host cell cytoskeletal protein, ankyrin, which is translocated to the host cell nucleus and binds to host cell chromatin (29, 30). Other outer membrane proteins include several members of the P28 family that mediate bacterial adhesion and invasion into macrophages (31, 32).
Internalization of Ehrlichia chaffeensis into macrophages is mediated by binding of the C terminus of an outer membrane entry-triggering protein (EtPE-C) to the mammalian glycosylphosphatidylinositol-anchored protein, DNase X, located on the host cell surface (33, 34). This binding triggers intracellular signaling via the transmembrane molecule, CD147, which recruits hnRNP-K to induce N-WASP activation (33, 35). Activation of N-WASP mediates actin polymerization and thus promotes bacterial internalization (36). Upon entry, E. chaffeensis is enclosed in endosome-like or phagosome-like compartments composed by lipid raft domains found in the host cell membrane that do not bind with lysosomes.
Mechanisms by which Ehrlichia spread from cell-to-cell at the early stages of infection, before host necrosis and/or pyroptosis are developed, are not completely understood. Studies have shown that Ehrlichia are transported through host cell filopodia during initial stages of infection but are released extracellularly during host cell necrosis at late stages of infection (37). Filopodia are cell membrane extensions that are organized by actin polymerization and continuous restructuring of filamentous actin (38). Inhibition of actin polymerization with cytochalasin D hindered the formation of filopodia and decreased bacterial burden (37). This indicated the requirement of actin polymerization for the spread and infection of new host cells during early infection (first 24 h). Transmission electron microscopy shows Ehrlichia cells migrating to new host cells within the confines of filopodia extensions without contact with the extracellular matrix (ECM) (3, 37). Thus, utilization of filopodia by Ehrlichia for cell-to-cell transmission may enable them to avoid exposure to the ECM, which is known to hinder bacterial nutrient acquisition, decrease bacterial replication, cause bacterial degradation and death via catabolic enzymes.
Initial models of HME were developed by infection of immunocompetent mice with E. chaffeensis (39) or infection of natural hosts such as dogs with E. canis (40). These studies defined several key parameters of the pathophysiology of the disease. However, E. chaffeensis is considered avirulent in immunocompetent mice as it causes an abortive infection that resolves approximately 10 days post-infection (39, 41). In addition, infection by E. chaffeensis in mice does not induce a measurable immune response or pathology that mimics ehrlichiosis in humans. In contrast, infection of immunocompromised mice with E. chaffeensis resulted in extensive tissue damage and persistent infection in different organs (liver, peritoneal cavity, brain, lung, and bone marrow) and mice became moribund within 24 days (42). Although utilization of immunocompromised mice has provided some information about the mechanisms of protective immunity and host resistance to ehrlichiosis, it did not address the mechanisms of host susceptibility to severe and potentially fatal ehrlichiosis. Therefore, other murine models of mild and fatal ehrlichiosis were developed by infection of immunocompetent C57BL/6 mice with mildly virulent Ehrlichia muris (E. muris) and highly virulent Ehrlichia species, Ixodes ovatus ticks (IOE), respectively (43–48). These murine models recapitulate the clinical outcome, the pathological aspects, and the laboratory findings in patients with HME.
E. muris-infected mice develop mild and self-limited disease with all animals surviving to infection. Mild disease in E. muris-infected mice is characterized by hepatosplenomegaly, elevated serum levels of liver enzymes, and minimal hepatic apoptosis (44). Although intraperitoneal infection with E. muris results in disseminated infection, ehrlichiae are cleared by day 10 post-infection (49). In contrast, the outcome of infection with IOE is dose- and route-dependent. Intraperitoneal (i.p.) infection of wild type (WT) C57BL/6 mice with high doses of IOE (103–105 organisms per mouse) causes severe and fatal disease characterized by primary liver dysfunction, marked by elevated liver enzymes and development of focal areas of hepatic apoptosis and necrosis, followed by excessive cytokine and chemokine production, referred to as “cytokine storm,” cell death and immunosuppression. Finally, these mice succumb to infection due to toxic shock-like syndrome with multi-organ failure on days 8–10 post-infection. On the other hand, intradermal infection of C57BL/6 mice with high doses of IOE (103–105 organisms per mouse) causes a mild disease similar to that induced by i.p. infection with E. muris.
Primary infection with E. muris also induces strong cell-mediated immune responses characterized by the development of protective CD4 Th1 cells and type I CD8 T cells. Additionally, infection promotes humoral immunity characterized by the generation of cross-reactive Ehrlichia-specific IgG antibodies that can recognize other Ehrlichia species (45, 50). Primary infection of WT C57BL/6 mice with E. muris in mice induces both cell-mediated and humoral memory immune responses that renders heterologous protection of WT mice against re-challenge with a lethal dose of IOE (44, 50, 51). On the other hand, primary infection of WT C57BL/6 mice with a sublethal dose of IOE, that causes mild disease, fails to provide homologous protection against re-challenge with a lethal dose of IOE (50).
Given that Ehrlichia is an obligate intracellular pathogen, cell-mediated immunity is thought to be key in protecting the host against infection. Despite this traditional line of thinking, it was seen that in ehrlichiosis many of the cell-mediated-immune mechanisms are more inclined toward deleterious effects. The outcome of infection is determined by the host's ability to balance between protective and pathogenic immune responses. For example, while CD4 Th1 and NKT cells seem to be protective, mechanisms mediated by CD8 T cells or NK cells can contribute to a pathogenic outcome. We found that liver injury and excessive cytokine and chemokine production/release followed by lethal Ehrlichia infection are mediated by several innate and adaptive immune cells, which are detailed below and summarized in Figure 1.
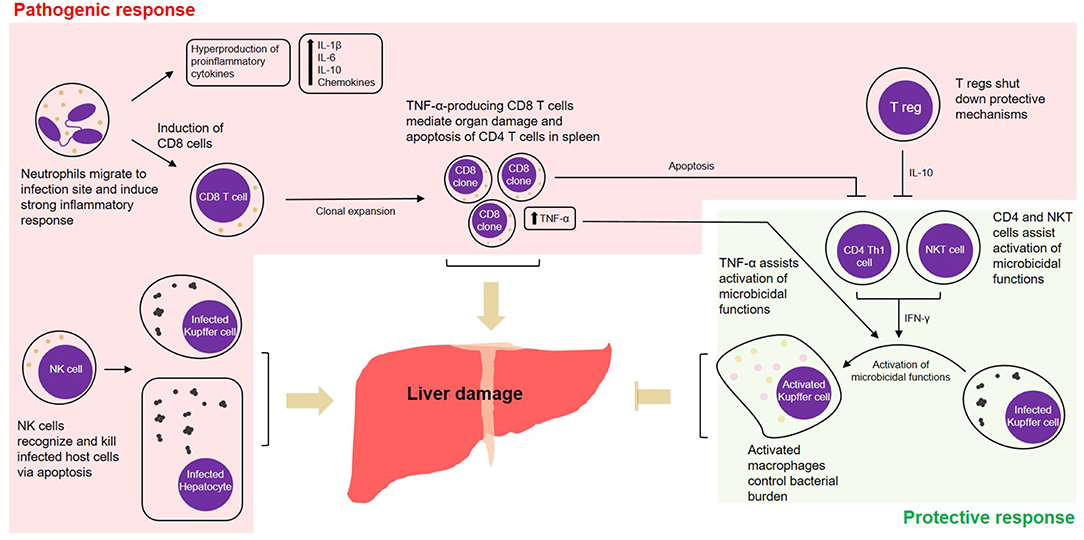
Figure 1. Cell-mediated pathogenic and protective responses in a model of fatal ehrlichiosis. Wild type C57BL/6 mice inoculated with virulent Ixodes Ovatus Ehrlichia (IOE) develop multi-organ failure and die between 8 and 10 days post-infection. Here we summarize protective and pathogenic mechanisms considering the cell-mediated immunity to Ehrlichia infection. Neutrophils migrate to infection site and induce strong pro-inflammatory response with hyperproduction of IL-1β, IL-6, IL-10, and chemokines. The role of neutrophils was also associated with induction of TNF-α-producing CD8 T cells that can either mediate organ damage or assist protective mechanisms. Another two important arms that mediate pathogenesis are the role of NK cells in eliminating infected cells in the target organs and the role of regulatory T cells (T regs), that end up shutting down protective mechanisms via IL-10. Protective mechanisms during infection by IOE include the induction of CD4 Th1 cells, that via IFN-γ activate on-site microbicidal functions of macrophages. NKT cells also play a role in stimulating such mechanism via IFN-γ.
Neutrophils serve as the frontline defenders against a variety of extracellular and intracellular pathogens including bacteria, fungi, and protozoa. Neutrophils are professional phagocytic cells that eliminate pathogens via phagocytosis and production of several antimicrobial peptides and molecules such as reactive oxygen species (ROS) (52). While neutrophils play a protective role during infections with other bacterial pathogens, our studies indicate that these cells are involved with a pathogenic outcome in ehrlichiosis. Depletion of neutrophils in IOE-infected mice enhanced protective immunity and resistance to fatal IOE infection (53). This was evidenced by attenuation of hepatic apoptosis and necrosis, two pivotal processes in the development of liver injury during infection with virulent IOE (53, 54). IOE-infected mice depleted of neutrophils showed reduced expansion of pathogenic CD8 T cells and their production of tumor necrosis factor-α (TNF-α). In this model, neutrophils were also associated with production of cytokines (IL-1β, IL-6, and IL-10) and several chemokines that are known to mediate migration of inflammatory and immune cells to sites of infection. The exact mechanism by which neutrophils promote the expansion of T cells is still unknown. However, as suggested by other studies, these cells could function as antigen presenting cells (APCs) to induce activation and clone expansion of T-cell subpopulations (55).
Cytotoxic CD8 T cells are considered a major cell subset that confers protection against intracellular pathogens. These cells are capable of recognizing and eliminating infected cells via perforin/granzyme B activities, as well as via death receptors such as FAS and TNF-α receptor I/II (56, 57). In addition, production of TNF-α and IFN-γ by CD8 and CD4 T cells mediates activation of microbicidal functions in phagocytic cells. These cytokine-induced bactericidal mechanisms are marked by the production of nitric oxide by nitric oxide synthase 2 (NOS2), tryptophan degradation to kynurenine by indoleamine 2, 3-dioxygenase (IDO), and reactive oxygen species. CD8 T cells contribute to protective immunity during infection of mice with E. muris. This was evidenced by higher susceptibility of MHC class I deficient mice to infection. In this case, nearly 80% of the MHC class I deficient animals succumbed to infection while all WT control mice survive (58). Adoptive transfer of CD8 T cells and CD4 T cells from E. muris-infected mice into naive mice confers protection of recipient mice against IOE infection. Depletion of IFN-γ or TNF-α resulted in lethality of 75% of these animals (44, 59).
In contrast to mild Ehrlichia infection, virulent IOE induces expansion of TNF-α-producing cytotoxic CD8 T cells. Notably, β2 microglobulin knockout mice and TAP knockout mice, that are CD8 T cell deficient, are more resistant to lethal IOE infection compared to WT mice. This observation suggested that CD8 T cells play a role in immunopathology during fatal ehrlichiosis (44, 47). Further studies in knockout mice indicated that immunopathology mediated by CD8 T cells is likely driven by TNF-α overproduction (46). Mice that do not express TNF-α receptors I/II, after infection with IOE are marked by prolonged survival, attenuated host cell death and reduced liver injury. However, these animals present higher bacterial load when compared to similarly-infected WT mice, indicating that TNF-α produced by CD8 T cells plays a dual role mediating hepatic pathology and control of intracellular ehrlichiae.
Our studies indicate that NK cells contribute to liver damage during severe Ehrlichia infection via secretion of several pro- and anti-inflammatory cytokines and production of granzyme B/perforins that activate cell death mechanisms (49). Depletion of NK cells enhanced mice survival and decreased tissue damage in IOE-infected mice suggesting a pathogenic role of NK cells in fatal ehrlichiosis. Considering persistent infection, which is the case of animals that are inoculated with E. muris, NK cells play a protective role during primary and recall immune responses. Primary infection with E. muris promoted polarization of memory CD4 Th1 cells and memory-like NK cells in the spleen and liver at day 28 post-infection (60).
This memory/recall response protected E. muris-primed mice against re-challenge with E. muris or lethal dose of virulent IOE. Interestingly, depletion of NK cells in E. muris-primed mice decreased the numbers of memory CD4 T cells and antibody-producing B cells, and made animals more susceptible to infection after re-challenge with IOE (60). These findings suggest that memory-like NK cells generated following primary E. muris infection are key for the development of effective long-term immunity to fatal Ehrlichia infection.
Similarly to what occurs in other infections by intracellular bacteria, IFN-γ-producing CD4 T helper-1 (Th1) cells and natural killer T cells (NKT) mediate protective immunity against Ehrlichia (53, 61, 62). Production of IFN-γ by these cells activates microbicidal functions of macrophages enhancing bacterial clearance. However, these protective immune cells undergo apoptosis during late stages of severe Ehrlichia infection.
NKT cells are a subset of T cells that in addition to expressing T cell receptors, also express several molecules typically expressed by NK cells. NKT cells are CD1d-restricted cells that contribute to host defense against various microbial pathogens. A study by Mattner et al. highlighted an alternative pathway by which Ehrlichia activate NKT cells compared to other Gram-negative bacteria (61). This study showed that Gram-negative intracellular bacteria, such as Salmonella, trigger activation of NKT cells via recognition of endogenous lysosomal glycosphingolipid, iGb3, presented by CD1d molecules on dendritic cells. In contrast, antigen-specific activation of human and murine NKT cells against LPS-negative E. muris occurs following recognition of cell wall glycosylceramide-like molecules by CD1d molecules.
Innate immune receptors play a central role in immune surveillance by sensing pathogens and initiating protective immune responses. However, under certain conditions, the innate immune system plays a deleterious role by overreacting to pathogens and causing excessive inflammation, immunopathology, cell death, and tissue damage. Recent studies have uncovered the complexity of innate immune receptors that sense Ehrlichia species, and downstream fundamental eukaryotic pathways, such as inflammasome activation, autophagy, and type I IFN response, which have multiple immunological effects on infection and immunity. This review highlights the cellular and molecular mechanisms by which Ehrlichia infection is sensed by different innate immune receptors, as well as the mechanisms of crosstalk between autophagy and inflammatory signaling cascades. We also discuss how these events mediate both host resistance to infection and the pathogenesis of fatal ehrlichiosis.
We previously showed that fatal IOE infection differentially triggers upregulation of several PRRs in hepatic cells when compared to infection with E. muris. Liver tissues from IOE-infected mice express significantly higher levels of nucleotide-binding oligomerization domain-containing protein 2 (NOD2), a cytosolic PRR that senses microbial ligands such as peptidoglycan, and TLR2, a surface TLR that recognizes LPS and lipoproteins. Both NOD2 and TLR2 signal via MyD88 to induce activation of NF-κb and production of several cytokines and chemokines. We have shown that TLR2 deficient mice are more susceptible to severe and fatal IOE infection as indicated by higher mortality, increased bacterial burden, and presence of a significantly higher number of inflammatory foci and necrotic hepatocytes and macrophages 7 days after infection when compared to infected WT mice (54). In contrast, NOD2 deficient mice are more resistant to fatal ehrlichiosis when compared to infected WT controls (54). IOE-infected NOD2 deficient mice have less hepatic apoptosis and necrosis and were able to effectively clear ehrlichiae compared to infected WT controls. Enhanced resistance of NOD2 deficient mice to fatal ehrlichiosis was associated with restoration of T cells and NKT cell numbers, increased IFN-γ production, as well as decreased frequency of pathogenic CD8 T cells, suggesting that NOD2, at least in part, mediates disease progression following IOE infection.
We recently examined the role of MyD88 in the immune response to lethal IOE infection. We showed that MyD88-signaling functions under a “double-edged sword” manner, with MyD88 signaling playing protective and pathogenic roles in fatal ehrlichiosis by regulating two key innate immune events in macrophages: autophagy and inflammasome activation (63). As a host-protective mechanism, activation of MyD88 signaling by IOE attenuates bacterial survival and replication by inhibiting induction of autophagy, a host innate response that promotes survival and/or replication of Ehrlichia as suggested by studies from us and other investigators (63–66). Livers of IOE-infected MyD88 deficient mice or primary bone marrow derived macrophages (BMM) isolated from these animals and infected with IOE, show higher autophagy induction as characterized by autophagosome formation compared to similarly infected WT mice or WT-BMM. This effect was associated with increased bacterial burden in vivo or increased numbers of intracellular bacteria in in-vitro cultured BMM. Further, pharmacological enhancement of autophagy increased bacterial replication in IOE-infected WT-BMM compared to untreated and infected BMM. As a host-pathogenic mechanism, we found that MyD88 signaling blocks autophagy flux (i.e., autophagosome-lysosomal fusion) in macrophages following lethal IOE infection, which leads to inhibition of mitochondrial autophagy (i.e., mitophagy) as well as blockage of ehrlichial degradation via lysosome due to lack of colocalization of autophagosomes with lysosomes. Defective mitophagy and the blocking of autophagic flux in IOE-infected macrophages led to the accumulation of mitochondrial DAMPS (e.g., ROS and mitochondrial DNA) and PAMPs, which in turn resulted in activation of canonical and non-canonical inflammasome pathways. As discussed below, inflammasome activation plays a deleterious role in the pathogenesis of fatal ehrlichiosis as it promotes excessive and dysregulated inflammation, development of pathogenic innate and adaptive immune responses (mediated by NK cells, neutrophils, and CD8 T cells), and finally cell death and multi-organ dysfunction. This conclusion is supported by data showing that lack of MyD88-attenuated liver pathology, decreased the production of NF-κB-dependent pro-inflammatory cytokines (e.g., TNF-α) and inflammasome-dependent cytokines (IL-1α, IL-1β), and reduced the frequency of cytotoxic CD8 T cells. Attenuated immunopathology in MyD88-deficient mice was associated with enhanced survival and host resistance to fatal ehrlichiosis (63). Indeed, the lack of correlation between bacterial burden and survival in MyD88-deficient mice reinforces our previous conclusion using WT mice that severe and fatal ehrlichiosis is not due to an overwhelming infection, but rather due to immunopathology.
Further analysis of the upstream TLRs that signal via MyD88 revealed TLR9 as the major TLR that mediates MyD88 effector functions during fatal Ehrlichia infection. We also explored whether other extracellular and endosomal TLRs such as TLR2 and TLR7, respectively, are linked to MyD88 activation and subsequent inflammasome promotion in the liver. Both TLR7−/− and TLR9−/− mice produced lower amounts of IL-1β, with TLR9−/− mice having significantly lower levels compared to WT and TLR7−/− (63). Importantly, 85% of TLR9−/− mice infected with lethal IOE survived until 60 days post-infection while all infected WT mice died between 10 and 12 days post-infection (63). These data support that the TLR9-MyD88 axis mediates host susceptibility and pathogenic responses during fatal ehrlichiosis. The results from the IOE model are consistent with observations from Rikihisa and coworkers using E. chaffeensis strain Wakulla, a virulent strain known to induce diffuse hepatitis in immunodeficient mice. E. chaffeensis infection induced TNF-α and IL-1β expression in the liver of immunodeficient mice and in isolated BMM (67). The expression of these cytokines was MyD88-dependent, but not dependent on TRIF, TLR2/4 or IL-1 receptor 1/IL-18 receptor 1. In vitro infection using human THP-1 cells (leukemia cell line) indicated that E. chaffeensis induces upregulation of IL-8, IL-1β, and TNF-α mRNAs as well as the extracellular regulated kinase 2 (ERK2) activation. Hence, E. chaffeensis Wakulla strain may induce inflammatory responses through MyD88-dependent NF-κB and ERK pathways, independent of TRIF and TLR2/4. Similarly, infection with Anaplasma phagocytophilum, another obligate intracellular bacterium that infects neutrophils and causes human anaplasmosis, triggers TLR2 signals which induce secretion of proinflammatory cytokines via NF-κB (68).
The role of MyD88 in host resistance and susceptibility to infections with other Rickettsial pathogens, such as Rickettsia conorii or Rickettsia australis; Spotted fever group (SFG) rickettsiae has also been examined. MyD88 deficient mice infected with either R. conorri or R. australis were more susceptible to infection than WT mice, suggesting a protective role of MyD88 in the immune response against SFG Rickettsiae (69, 70). Similar to its role in ehrlichiosis, MyD88 signaling mediated clearance of intracellular rickettsiae within macrophages and dendritic cells. Mechanistically, MyD88 signaling promoted IFN-γ production in mice, which is known to be critical for activation of microbicidal functions of macrophages. In addition, MyD88 signaling enhanced inflammasome activation, which was found to be a host-protective mechanism during Rickettsia infection.
Inflammasomes are high molecular weight protein complexes that consist of intracellular NOD-like receptors (NLRs), programmed cell death ASC (apoptosis-associated speck like protein) adaptor molecules and pro-caspases. Activation of inflammasomes leads to the activation of caspases which subsequently induce secretion of IL-1β and IL-18 proinflammatory cytokines (71–75). Inflammasome complexes include NLRP1, NLRP2, NLRP3, NLRP4, NRLP6, NRLP7, and NLRP12 (75). The canonical inflammasome activation pathway requires a priming event via triggering of a PRR, such as Toll-like receptors (TLRs), by a microbial or host ligand or by binding of a pro-inflammatory cytokine to its receptor, such as binding of TNF-α to TNF-α receptors (74–78). This ligand-receptor binding causes the MyD88-mediated activation of NF-κB leading to upregulation of pro-IL-1β, pro-IL-18, and several inflammasome complexes, such as NLRP3 (75). Next, assembly of the inflammasome complexes occurs upon binding to PAMPS or DAMPS (e.g., bacterial toxins, DNA, bacterial RNA and flagella, viral protein, host DNA and RNA, and host-derived ATP, glucose, cholesterol crystals, calcium pyrophosphate dihydrate, mitochondrial ROS and DNA, and amyloid β), which leads to activation of caspase 1, cleavage of IL-1β and IL-18, as well as pyroptosis. Recently, a non-canonical inflammasome pathway has been described in which cytosolic LPS activates the inflammasome via activation of caspase-11 which promotes caspase 1-dependent secretion of IL-1β and IL-18 as well as pyroptosis and release of IL-1α and high mobility group box 1 (HMGB1) (3, 73, 79–83). The inflammasome could function as a host protective mechanism to clear the infection and promote induction of protective adaptive immune responses against infectious agents, but it can also produce tissue injury, excessive inflammation, and immunopathology under dysregulated circumstances (75).
Employing murine models of ehrlichiosis, we have shown that LPS-negative IOE infection differentially induces upregulation of several inflammasome complexes including NLRP1, NLRP3, NLRC4, AIM2, and NLRP12 (54). Unlike infections with other intracellular bacteria, such as Legionella and Mycobacterium tuberculosis, inflammasome activation plays a deleterious role in the host response against Ehrlichia. This point is supported by several studies showing a strong link between the production of inflammasome-dependent cytokines, IL-18 and IL-1β, and the induction of pathogenic adaptive immune responses and liver damage. IL-18R knockout mice are more resistant to fatal ehrlichiosis than WT mice as marked by prolonged survival and decreased bacterial burden (48). A lack of IL-18/IL-18R signaling enhanced bacterial clearance, attenuated liver injury, decreased the production of pro-inflammatory cytokines (such as TNF-α), and decreased expansion of pathogenic TNF-producing CD8 T cells and NK cells following IOE infection. Further studies have shown that production of IL-1β is mediated by NLRP3, caspase 1, and caspase 11, indicating activation of both canonical and non-canonical inflammasome pathways (54, 63, 84). Notably, IOE-infected NRLP3-deficient mice effectively cleared Ehrlichia, but still displayed acute mortality and liver injury compared to IOE-infected WT mice. This suggests that pathogenic inflammasome activation during fatal IOE infection is only partially mediated by NLRP3 (84). The fact that virulent Ehrlichia infection causes caspase-11 production is eye-catching considering that the non-canonical inflammasome pathway is best-known to be activated by LPS directly binding to pro-caspase-11 in the cytosol, yet Ehrlichia species lack LPS as described above (85). Regardless, IOE-induced activation of caspase-1 and -11 production contributes to the development of pyronecrosis, liver injury, and excessive inflammatory responses (63).
The number of studies showing the involvement of other related bacterial species and inflammasome activation is minimal. In a study using mouse and human macrophages by Smalley and coworkers, it was shown that infection with Rickettsia australis activates NRLP3 inflammasomes in an ASC-dependent manner. ASC-inflammasome activation was characterized by significantly higher concentrations of IL-1β, IL-18, and mature caspase-1, however, BMM isolated from ASC−/− mice displayed an attenuated inflammasome response with significantly reduced levels of pro-inflammatory cytokines and caspases (86). This work suggested that NLRP3 inflammasome contributes to cytosolic recognition of R. australis.
Similar to Ehrlichia, infection with Anaplasma, another tick-borne obligate intracellular bacteria that belongs to the family Anaplasmataecae (1, 2), triggers inflammasome activation (87). Even though sensing of Anaplasma spp. by PRRs remains mostly unidentified, it was seen that infection by A phagocytophilum, the etiologic agent of human granulocytic anaplasmosis, induces activation of the NLRC4 inflammasome. During A. phagocytophilum infection, cytosolic phospholipase A2 metabolizes arachidonic acid from phospholipids, which is converted to the eicosanoid prostaglandin E2 (PGE2) via cyclooxygenase 2 (COX2) and prostaglandin mPGES-1 activity. PGE2-EP3 receptor signaling culminates in the activation of the NLRC4 inflammasome and secretion of IL-1β and IL-18 (87).
Degradation of cytoplasmic components is achieved through several pathways including macroautophagy, microautophagy and chaperone-mediated autophagy (88). Macroautophagy (standardly referred to as autophagy) is a highly conserved process by which cells recycle organelles and intracellular debris via degradation in the lysosomes. Autophagy is characterized by the regulated formation of double-membrane compartments known as phagophores. Phagophores encapsulate tagged intracellular materials, as well as intracellular pathogens for host defense purposes. Autophagic flux involves the maturation of phagophores into autophagosomes, which then fuse with lysosomes to form single-membrane autolysosomes, where degradation of autophagic cargo, recycling of proteins and ATP synthesis occur. Several autophagy-promoting molecules including, ATG5, ATG12, ATG16, ATG8, and Beclin-1, mediate the induction of autophagy (89–92). Any marked decrease in production of these proteins or in lipidation of ATG8/LC3 (LC3 is the mammalian homolog of yeast ATG8) attenuates the formation of autophagosomes and impairs the autophagy process overall. Autophagy has been implicated in many fundamental biological processes including aging, immunity, cell development and differentiation by regulating inflammation.
Autophagy vitally combats pathogens during most infection processes. However, several intracellular pathogens evade the host innate immune defense system by exploiting autophagy as a pathway to obtain nutrients, fatty acids, and carbohydrates required for intracellular survival and replication (93). Recent studies suggested that autophagy promotes survival and replication of obligate intracellular bacteria, such as Ehrlichia chaffeensis (64), Anaplasma phagocytophilum (94) and SFG Rickettsia (95). These bacteria would be examples of microorganisms capable of capturing nutrients through autophagy to promote bacterial growth and replication (17, 96–98). E. chaffeensis exploits autophagy proteins to obtain nutrients by secreting the protein ETF-1 (Ehrlichia translocated factor-1). ETF-1 targets the endosomal protein, RAB5, which is associated with an early endosome-like membrane-bound compartment that contains Ehrlichia but lacks bactericidal functions (i.e., ehrlichial inclusions). Binding of E. chaffeensis ETF-1 to RAB5 and autophagy-initiating class III phosphatidylinositol 3 kinase (PtdIns3K) is followed by Beclin-1, VPS34, and ATG5 recruitment to form an autophagosome that binds to the ehrlichial inclusion (64, 97, 99). Thus, E. chaffeensis hijacks the RAB5 autophagy pathway to create a non-microbicidal phagosomal complex to capture and deliver nutrients from already degraded cellular debris within the cytoplasm. In other words, the RAB5-associated phagosomal complex acts as an intracellular transport vesicle, in which captured cytosolic nutrients are delivered to E. chaffeensis. E. chaffeensis remains sheltered in endosome-like inclusions that were formed upon initial entry and internalization into the host cell (100).
Using a murine model of fatal ehrlichiosis, we have shown that IOE also exploits autophagy to survive and replicate within macrophages. Like E. chaffeensis, IOE induces autophagy in macrophages (the primary target cells) to survive and replicate within these phagocytic cells. Enhanced autophagy in IOE-infected MyD88−/− BMM enhanced intracellular bacterial survival and replication (63). As a counteracting protective mechanism, signaling via MyD88 negatively regulates autophagy induction. Thus, MyD88 signaling during IOE infection plays a protective role by attenuating bacterial survival and replication via inhibition of autophagy induction (63). As mentioned above, MyD88 not only blocks autophagy induction, but also inhibits autophagosome-lysosomal fusion and, consequently, also inhibits the autophagic flux. Blocking autophagy induction with inhibitor compounds attenuated bacterial survival. The MyD88-mediated blocking of autophagic flux also resulted in defective elimination of mitochondrial DAMPs (i.e., defective mitophagy).
Mechanistically, we found that MyD88 negatively regulates the autophagy process in IOE-infected macrophages via activation of mammalian target of rapamycin complex 1 (mTORC1), a member of the phosphoinositide 3-kinase (PI3K) family (63). The mTORC1 pathway is a well-known negative regulator of autophagy induction as it inhibits the binding of Beclin-1 to ULK: a vital first step in the formation of autophagosomes (101). It is not clear how MyD88 induces activation of mTORC1 in macrophages following infection with virulent IOE. The mTORC1 pathway is activated under homeostatic conditions because it promotes cell survival and proliferation. In contrast, mTORC1 is inhibited by amino acid starvation and cellular stress. Cell starvation activates mechanisms that cause upregulation of the autophagy process during times of critical metabolic need. It is possible that metabolic dysregulation during Ehrlichia infection may cause mTORC1 activation. Although mTORC1 mediates inhibition of autophagy, which works as a source of nutrients to Ehrlichia, it promotes host cell survival and masks Ehrlichia within the host cell. This paradoxical interaction between mTORC1 and autophagy is an important immune evasion mechanism that enables survival and replication of Ehrlichia. A model of the canonical inflammasome activation involving regulation of autophagy by the infection is described in Figure 2.
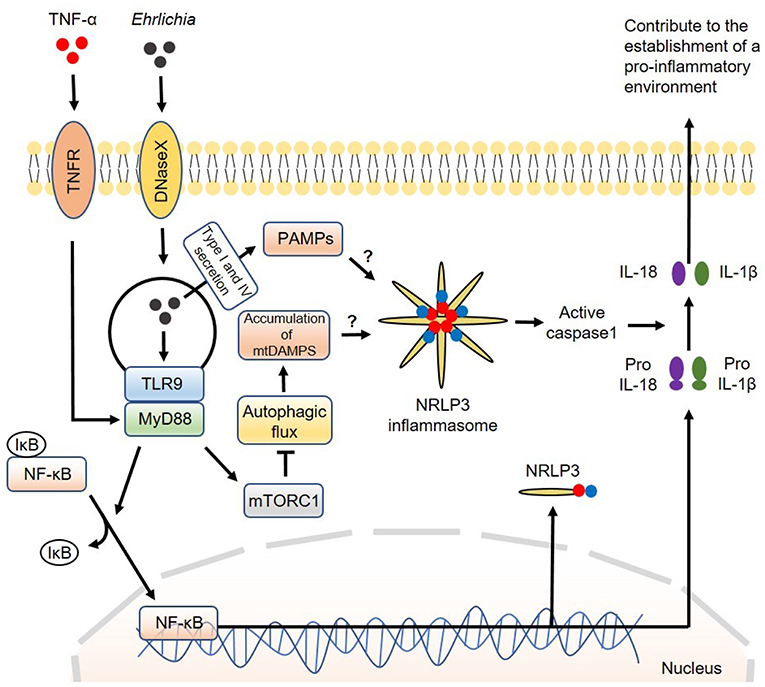
Figure 2. Model of canonical inflammasome activation involving regulation of autophagy induced by Ehrlichia. The canonical iflammasome activation pathway proposed in Ehrlichia infection involves two steps. In the first step, Ehrlichia invade the target cell and induce TLR9/MyD88 downstream targets to activate NF-κB. Activated NF-κB function as a transcription factor to upregulate NRLP3 complexes, pro-IL1β and pro-IL-18. In the second step, NRLP3 inflammasomes would be activated after recognition of Ehrlichial PAMPS which are secreted to the cytosol via type I and IV secretion systems. Accumulation of mtDAMPS originated by inhibition of autophagy via MyD88/mTORC1 signaling would also induce inflammasome activation. Inflammasome activation results in activation of caspase-1 that subsequently induces cleavage of pro-IL1β and pro-IL-18 into their mature forms. Mature IL-1β and IL-18 are then secreted to the extracellular space to contribute to the establishment of a pro-inflammatory environment. Direct arrow: positive regulation; blocking arrow: negative regulation; question mark (?) means hypothetical, and not experimentally proven.
Similar to Ehrlichia infection and ehrlichiosis, autophagy appears to play a major role in the pathogenesis of other rickettsial diseases. Earlier studies in Rickettsia infection in mice suggested a role of autophagy in host defense against SFG Rickettsia. Endothelial cells infected with Rickettsia conorii displayed strong antirickettsial effect upon cytokine-induced nitric oxide (NO) stimulation. Ultrastructural analysis of infected endothelial cells revealed double membrane structures that appeared to be derived from the endoplasmic reticulum and contained rickettsiae. This has been described as phagolysosomes, which was postulated to be a process by which cytokine-activated endothelial cells eliminate intracellular Rickettsia (102). The anti-microbial role of autophagy in host response against Rickettsia was also suggested by a study comparing the growth kinetics of pathogenic and nonpathogenic rickettsiae in Vero and Hela cells. Cells infected with the non-pathogenic strain, R. montanensis, displayed the formation of autophagosomes, which was associated with a low number of intracellular organisms. On the other hand, superinfection of R. montanensis-infected cells with the pathogenic species, R. japonica, failed to control replication of both strains. This fact was associated with restriction or inhibition of autophagosome formation under superinfection conditions (103, 104). By employing computational biology techniques, Gong and coworkers showed that tissues or cells infected with R. conorii displayed the presence of tRNA-derived RNA fragments (tRFs) which may interact with transcripts associated with autophagy (105). Moreover, a recent study using ATG5flox/flox mice suggested that autophagy promotes Rickettsia australis infection. Tissues or BMM from ATG5flox/flox infected with R. australis harbor a higher number of Rickettsia compared to their counterparts of ATG5flox/flox Lyz-Cre mice (in which macrophages only are deficient of ATG5, one of the autophagy genes that are essential for initiation of autophagosome). These data suggest that autophagy enhances rickettsial survival and/or replication. Notably, treatment of infected macrophages from ATG5flox/flox mice with recombinant IL-1β attenuated rickettsial replication, indicating a protective function of IL-1β and inflammasome activation in the host response against Rickettsia. Importantly, these data suggest that autophagy induction following R. australis infection negatively regulates inflammasome activation and IL-1β production, a consistent conclusion with the occurrence of cross-talk between autophagy and inflammasome during Ehrlichia infection.
Similar to Ehrlichia and Rickettsia, autophagy induction and formation of autophagosomes support the survival and/or growth of Anaplasma phagocytophilum. This conclusion is supported by earlier studies by Galindo and colleagues showing that blood samples from pigs infected with A. phagocytophilum express higher levels of several genes including GJA1, integrin alpha-8, TSP-4, formin 1, Rho GTPase activating protein 5, keratin associated protein 26–1, calponin 3 and laminin receptor 1 (106). These genes are involved in cytoskeletal rearrangement and actin polymerization. Since autophagy requires cytoskeleton rearrangement for the formation of phagophore and internalization of invading microbes (107), this study suggested that infection with A. phagocytophilum induces modulation of autophagy. Elegant studies by Rikihisa and co-workers revealed the key mechanism by which A. phagocytophilum exploit autophagy to obtain nutrients for their survival. A. phagocytophilum were initially found to replicate in double-lipid bilayer membrane compartments that colocalize with LC3 and Beclin 1. Stimulation of autophagy by rapamycin favored A. phagocytophilum infection, and inhibition of the autophagosomal pathway impaired bacterial growth (94). Mechanistically, it was found that A. phagocytophilum induces autophagy through binding of Anaplasma translocated substrate 1 (Ats-1), type IV secretion system effector to Beclin 1-ATG14L pathway. This binding of the bacterial secreted molecule to the autophagosome enables Anaplasma to obtain nutrients and essential amino acids for their survival and replication (65, 66, 108).
Classical studies have focused on the role of type I interferons (IFN-I) in the induction of antiviral host defense (109, 110), however, others have demonstrated that IFN-I are also induced during infections with non-viral pathogens such as bacteria, mycobacterium, and parasites (87, 111–115). Despite this understanding, the mechanism of action of IFN-I in host defense against bacterial infections is still poorly elucidated. It looks like IFN-I exert disparate/dual roles depending on the type of bacterial infection. For example, infection with Gram-negative intracellular Chlamydia trachomatis can induce IFN-I, which restrict bacterial growth (116–118). Similarly, IFN-I are induced in mice infected with Gram-positive bacteria, such as Group B streptococcus and Listeria monocytogenes. In these cases, IFN-I are protective as evidenced by a higher susceptibility to infection in IFNAR−/− mice compared to WT (119–122). On the other hand, IFN-I play a deleterious role during infection with other pathogens by promoting dysregulated inflammation and host cell death. For example, induction of IFN-I response in macrophages causes necroptosis upon infection with Salmonella (80). Additionally, IFN-I have also strong pro-inflammatory activities that contribute to high mortality rates in cases of septic shock (123). Factors that determine the protective or pathogenic function of IFN-I during infections with diverse pathogens are not entirely understood. However, studies suggested that the effector function of IFN-I depend on multiple bacterial and host factors including route and site of infection, bacterial virulence, and infectious dose. Table 1 summarizes the diverse contributions of IFN-I to host responses against several pathogens and the potential mechanism(s) by which they promote protective or deleterious roles.

Table 1. IFN-I response in bacterial infections.
We and others have shown that infection of WT mice with virulent Ehrlichia species (IOE) induces upregulation of IFN-I mRNA in liver tissues and secretion of IFN-I by plasmacytoid dendritic cells and macrophages in the spleen. IFNAR signaling plays a deleterious role in ehrlichiosis as evidenced by increased resistance of IFNAR−/− mice to fatal IOE infection. IFN-I mediate host cell death and suppression of protective adaptive immune responses provided by CD4 Th1 cells. IFNAR signaling suppresses IFN-γ signaling, which impairs induction of antimicrobial pathways crucial for clearance of intracellular Ehrlichia (130, 131). Both murine IFNAR deficiency and neutralization of IFN-α and IFN-β individually decreased bacterial burden while correlating with increased IFN-γ production (130). Notably, increased IFN-γ in IOE-infected IFNAR−/− mice was not essential for protection against fatal Ehrlichia infection (130). Studies from our laboratory suggest that attenuated inflammasome activation and enhanced autophagy are potential mechanisms that afford protection against fatal ehrlichiosis in IFNAR−/− mice (54, 84). Deficiency of IFNAR resulted in attenuation of caspase-11 mediated-noncanonical inflammasome activation during IOE infection, also leading to reduced secretion of IL-1β and minimal cell death. IFN-I signaling also contributed to a pathogenic immune response during fatal Ehrlichia infection due to induction of cytotoxic TNF-α-producing pathogenic NK cells, neutrophils, and CD8 T cells that cause tissue damage. Additionally, IFNAR signaling induced pyroptosis or pyronecrosis in ehrlichiosis, which is consistent with other infection models. Expression of IFNAR on non-hematopoietic, but not on hematopoietic cells, is likely to be an important point of modulation of immunopathology during fatal Ehrlichia infection. Based on the above studies, we propose that endothelial cells and/or hepatocytes are major cellular sources of deleterious IFNAR signaling for the following reasons: (i) although the primary target cells are macrophages, Ehrlichia species infect other parenchymal cells, such as hepatocytes and endothelial cells, as shown by immunohistochemistry staining of infected liver tissues; (ii) fatal ehrlichiosis in humans and mice are associated with hepatic apoptosis and necrosis which correlate with IFN-I response in mice suggesting that hepatocytes play a role in the pathogenesis of the disease; and (iii) Ehrlichia exhibit tropism for microvascular endothelium leading to vascular inflammation and dysfunction/damage; this represents one of the key features of Ehrlichial pathogenesis, especially in patients with meningitis or encephalitis.
How IFN-I regulate canonical or non-canonical inflammasome activation is not completely understood. The major inflammasome complex that is regulated by IFN-β is NLRP3. Several studies showed that TLR4-TRIF axis regulates caspase-11 expression and non-canonical NLRP3 inflammasome-mediated host defense against enteropathogens, such as Escherichia coli, Citrobacter rodentium and Salmonella Typhimurium (132). Further studies indicated that IRF3 and IFNAR signaling during infections with intracellular bacterial pathogens that reside within phagosomes are required for caspase-11 expression and activation of the NLRP3 inflammasome and pyroptosis (80, 133). Recent studies have demonstrated that IFN-I promote caspase-11 activation by inducing several genes which are members of the IRG and GBP families of IFN-inducible GTPases. These GBP proteins are found to be essential for the activation of caspase-11–dependent pyroptosis in response to infections with Legionella pneumophila as they induce disruption of vacuoles containing Legionella causing the release of bacterial LPS into the cytosol (72, 117, 134–136). Cytosolic LPS is a known PAMP that triggers activation of caspase-11. Other studies have shown that IFN-I-induced GBP protein expression is required for the full induction of pyroptosis by LPS delivered to the cytoplasm independent of infection. Pyroptosis has been widely associated with cleavage of gasdermin D with release of its N-terminal portion that can destabilize the membrane creating pores leading to cell death (137). Collectively, these studies support that GBP proteins play a role in the detection of cytoplasmic LPS and/or the subsequent activation of the noncanonical inflammasome leading to pyroptosis (116, 117). Interestingly, while the induction of pyroptosis by cytoplasmic L. pneumophila LPS appears to be strictly dependent on GBP proteins, cytoplasmic LPS derived from Enterobacteriaceae can trigger pyroptosis in the absence of GBP proteins, however, with diminished efficiency (117, 124). This discrepancy could be due to the difference in the structures of LPS and TLR ligands among these bacterial species. Nevertheless, whether IFN-I-induced GBP proteins contribute to activation of caspase-11 by Ehrlichia that reside within vacuole or phagosome remains elusive. A model for the participation of IFN-I in the regulation of inflammasome activation in Ehrlichia infection is proposed in Figure 3.
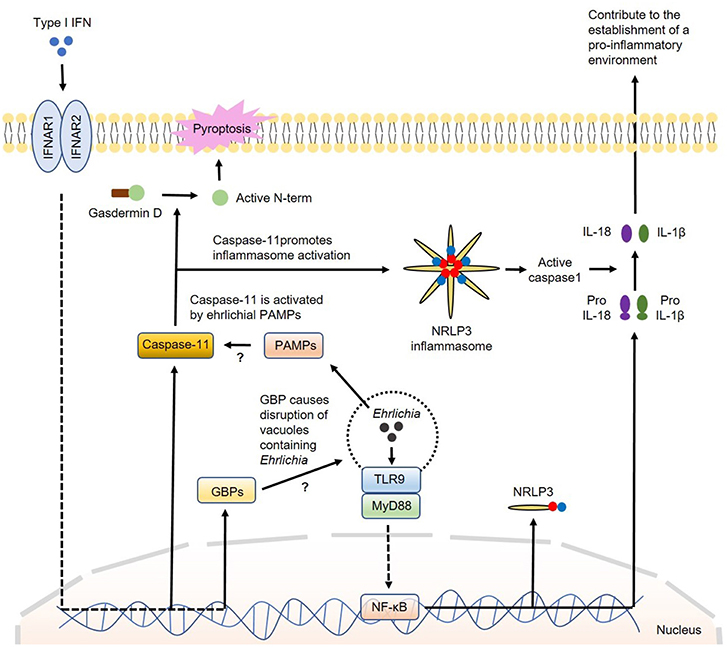
Figure 3. Model of regulation of inflammasome activation by type I IFNs in Ehrlichia infection. During Ehrlichia infection, type I IFNs are thought to regulate inflammasome activation under a non-canonical fashion. Initially, Ehrlichia invade the target cell and induce TLR9/MyD88 signaling to upregulate NRLP3 complexes, pro-IL1β and pro-IL-18 via NF-κB. Type I IFNs from autocrine or paracrine sources would signal through IFNAR to upregulate GBPs and caspase-11. GBPs would disrupt vesicles containing Ehrlichia allowing the escape of ehrlichial PAMPs to the cytosol. Ehrlichial PAMPs would bind caspase-11 and subsequently induce inflammasome activation followed by secretion of mature IL-1β and IL-18 (as in the canonical pathway) and cleavage of gasdermin D resulting in pyroptosis. Direct arrow: positive regulation; blocking arrow: negative regulation; question mark (?) means hypothetical, and not experimentally proven.
The last decades were marked by several advances in the description of how Ehrlichia-host interactions are established. Animal models have provided valuable information regarding the cell-mediated mechanisms that govern protection and immuomopathogenesis. Even though there is no vaccination available against Ehrlichia, understanding the mechanisms that protect the host against infection will assist us in the development of an effective vaccine. In terms of cell-mediated immunity, we currently know the importance of CD4 Th1 and NKT cell responses in protecting the host. Therefore, vaccine prototypes that prioritize such responses would represent promising tools within the vaccine development pipeline. Regarding the intracellular innate immune mechanisms and their nuances in modulating Ehrlichia infection, targeting autophagy and inflammasome activation can also represent promising options in drug development. By inhibiting autophagy and attenuating inflammasome activation via modulation of MyD88 and IFN-I signaling it would be possible to control the disease, once these mechanisms were categorically highlighted as key in Ehrlichia pathogenesis. Despite the current understanding, several gaps in knowledge concerning Ehrlichia-host interaction are yet to be solved for better planning of translational strategies against the disease.
TT wrote the original draft of the manuscript. EO created the figures, captions, and wrote part of the manuscript. SH reviewed the literature, draw the table and wrote a section of the review. JW contributed to a section of the review. BG wrote a section of the review and edited section written by TT. TT, EO, SH, AE, JW, BG, and NI reviewed and edited the manuscript.
This study was funded by grant NIH-NIAID-R56A1097679-01A to NI and the startup fund from the Department of Pathology at University of Illinois at Chicago.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Muhamuda Kader from the University of Pittsburgh, Dr. Abdeljabar El Andaloussi and Mohamed Haloul from the University of Illinois at Chicago for insights and scientific discussion toward the manuscript.
1. Rikihisa Y. Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nat Rev Microbiol. (2010) 8:328–39. doi: 10.1038/nrmicro2318
2. Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nat Rev Microbiol. (2008) 6:375–86. doi: 10.1038/nrmicro1866
3. Ismail N, Bloch KC, McBride JW. Human ehrlichiosis and anaplasmosis. Clin Lab Med. (2010) 30:261–92. doi: 10.1016/j.cll.2009.10.004
4. Ismail N, McBride JW. Tick-Borne Emerging Infections. Clin Lab Med. (2017) 37:317–40. doi: 10.1016/j.cll.2017.01.006
5. Rikihisa Y. Molecular pathogenesis of Ehrlichia chaffeensis infection. Ann Rev Microbiol. (2015) 69:283–304. doi: 10.1146/annurev-micro-091014-104411
6. Dumler JS, Madigan JE, Pusterla N, Bakken JS. Ehrlichioses in humans: epidemiology, clinical presentation, diagnosis, and treatment. Clin Infect Dis. (2007) 45(Suppl.1):S45–51. doi: 10.1086/518146
7. Paris DH, Dumler JS. State of the art of diagnosis of rickettsial diseases. Curr Opin Infect Dis. (2016) 29:433–9. doi: 10.1097/qco.0000000000000298
8. Harrus S, Waner T. Diagnosis of canine monocytotropic ehrlichiosis (Ehrlichia canis): an overview. Vet J. (2011) 187:292–6. doi: 10.1016/j.tvjl.2010.02.001
9. Mylonakis M, Harrus S, Breitschwerdt E. An update on the treatment of canine monocytic ehrlichiosis (Ehrlichia canis). Vet J. (2019) 246:45–53. doi: 10.1016/j.tvjl.2019.01.015
10. Little SE. Ehrlichiosis and anaplasmosis in dogs and cats. Vet Clin North Am. (2010) 40:1121–40. doi: 10.1016/j.cvsm.2010.07.004
12. Lee EH, Rikihisa Y. Absence of tumor necrosis factor alpha, interleukin-6 (IL-6), and granulocyte-macrophage colony-stimulating factor expression but presence of IL-1beta, IL-8, and IL-10 expression in human monocytes exposed to viable or killed Ehrlichia chaffeensis. Infect Immun. (1996) 64:4211–9.
13. Lin M, Rikihisa Y. Ehrlichia chaffeensis and anaplasma phagocytophilum lack genes for Lipid A Biosynthesis and Incorporate Cholesterol for Their Survival. Infect Immun. (2003) 71:5324–31. doi: 10.1128/iai.71.9.5324-5331.2003
14. Mavromatis K, Doyle CK, Lykidis A, Ivanova N, Francino MP, Chain P, et al. The genome of the obligately intracellular bacterium Ehrlichia canis reveals themes of complex membrane structure and immune evasion strategies. J Bacteriol. (2006) 188:4015–23. doi: 10.1128/jb.01837-05
15. McBride JW, Zhang X, Wakeel A, Kuriakose JA. Tyrosine-phosphorylated Ehrlichia chaffeensis and Ehrlichia canis tandem repeat orthologs contain a major continuous cross-reactive antibody epitope in lysine-rich repeats. Infect Immun. (2011) 79:3178–87. doi: 10.1128/iai.01347-10
16. Dunphy PS, Luo T, McBride JW. Ehrlichia chaffeensis exploits host SUMOylation pathways to mediate effector-host interactions and promote intracellular survival. Infect Immun. (2014) 82:4154–68. doi: 10.1128/iai.01984-14
17. Lina TT, Luo T, Velayutham TS, Das S, McBride JW. Ehrlichia activation of Wnt-PI3K-mTOR signaling inhibits autolysosome generation and autophagic destruction by the mononuclear phagocyte. Infect Immun. (2017) 85:e00690–17. doi: 10.1128/iai.00690-17
18. Klema VJ, Sepuru KM, Füllbrunn N, Farris TR, Dunphy PS, McBride JW, et al. Ehrlichia chaffeensis TRP120 nucleomodulin binds DNA with disordered tandem repeat domain. PLoS ONE. (2018) 13:e0194891. doi: 10.1371/journal.pone.0194891
19. Farris TR, Zhu B, Wang JY, McBride JW. Ehrlichia chaffeensis TRP32 nucleomodulin function and localization is regulated by NEDD4L-Mediated ubiquitination. Front Cell Infect Microbiol. (2018) 7:534. doi: 10.3389/fcimb.2017.00534
20. Wakeel A, Kuriakose JA, McBride JW. An Ehrlichia chaffeensis tandem repeat protein interacts with multiple host targets involved in cell signaling, transcriptional regulation, and vesicle trafficking. Infect Immun. (2009) 77:1734–45. doi: 10.1128/iai.00027-09
21. Luo T, Kuriakose JA, Zhu B, Wakeel A, McBride JW. Ehrlichia chaffeensis TRP120 interacts with a diverse array of eukaryotic proteins involved in transcription, signaling, and cytoskeleton organization. Infect Immun. (2011) 79:4382–91. doi: 10.1128/iai.05608-11
22. Luo T, Mitra S, McBride JW. Ehrlichia chaffeensis TRP75 interacts with host cell targets involved in homeostasis, cytoskeleton organization, and apoptosis regulation to promote infection. mSphere. (2018) 3:e00147–18. doi: 10.1128/msphere.00147-18
23. Doyle CK, Nethery KA, Popov VL, McBride JW. Differentially expressed and secreted major immunoreactive protein orthologs of Ehrlichia canis and E. chaffeensis elicit early antibody responses to epitopes on glycosylated tandem repeats. Infect Immun. (2005) 74:711–20. doi: 10.1128/iai.74.1.711-720.2006
24. Luo T, Zhang X, Wakeel A, Popov VL, McBride JW. A variable-length PCR target protein of Ehrlichia chaffeensis contains major species-specific antibody epitopes in acidic serine-rich tandem repeats. Infect Immun. (2008) 76:1572–80. doi: 10.1128/iai.01466-07
25. Luo T, Zhang X, McBride JW. Major species-specific antibody epitopes of the Ehrlichia chaffeensis p120 and E. canis p140 orthologs in surface-exposed tandem repeat regions. Clin Vac Immunol. (2009) 16:982–90. doi: 10.1128/cvi.00048-09
26. McBride JW, Walker DH. Molecular and cellular pathobiology of Ehrlichia infection: targets for new therapeutics and immunomodulation strategies. Expert Rev Mol Med. (2011) 13:e3. doi: 10.1017/s1462399410001730
27. Kuriakose JA, Zhang X, Luo T, McBride JW. Molecular basis of antibody mediated immunity against Ehrlichia chaffeensis involves species-specific linear epitopes in tandem repeat proteins. Microbes Infect. (2012) 14:1054–63. doi: 10.1016/j.micinf.2012.05.012
28. Luo T, McBride JW. Ehrlichia chaffeensis TRP32 interacts with host cell targets that influence intracellular survival. Infect Immun. (2012) 80:2297–306. doi: 10.1128/iai.00154-12
29. Wakeel A, den Dulk-Ras A, Hooykaas PJJ, McBride JW. Ehrlichia chaffeensis tandem repeat proteins and Ank200 are type 1 secretion system substrates related to the repeats-in-toxin exoprotein family. Front Cell Infect Microbiol. (2011) 1:22. doi: 10.3389/fcimb.2011.00022
30. Dunphy PS, Luo T, McBride JW. Ehrlichia moonlighting effectors and interkingdom interactions with the mononuclear phagocyte. Microbes Infect. (2013) 15:1005–16. doi: 10.1016/j.micinf.2013.09.011
31. Crocquet-Valdes PA, Thirumalapura NR, Ismail N, Yu X, Saito TB, Stevenson HL, et al. Immunization with Ehrlichia P28 outer membrane proteins confers protection in a mouse model of ehrlichiosis. Clin Vac Immunol. (2011) 18:2018–25. doi: 10.1128/cvi.05292-11
32. McClure EE, Chávez ASO, Shaw DK, Carlyon JA, Ganta RR, Noh SM, et al. Engineering of obligate intracellular bacteria: progress, challenges and paradigms. Nat Rev Microbiol. (2017) 15:544–58. doi: 10.1038/nrmicro.2017.59
33. Kumar DM, Lin M, Xiong Q, Webber MJ, Kural C, Rikihisa Y. EtpE binding to DNase X induces ehrlichial entry via CD147 and hnRNP-K recruitment, followed by mobilization of N-WASP and actin. mBio (2015) 6:e01541–15. doi: 10.1128/mbio.01541-15
34. Teymournejad O, Lin M, Rikihisa Y. Ehrlichia chaffeensis and Its invasin EtpE block reactive oxygen species generation by macrophages in a DNase X-Dependent manner. mBio. (2017) 8:e01551–17. doi: 10.1128/mbio.01551-17
35. Kumar DM, Yamaguchi M, Miura K, Lin M, Los M, Coy JF, et al. Ehrlichia chaffeensis uses its surface protein EtpE to bind GPI-anchored protein DNase X and trigger entry into mammalian cells. PLoS Pathogens. (2013) 9:e1003666. doi: 10.1371/journal.ppat.1003666
36. Dutta D, Dutta S, Veettil MV, Roy A, Ansari MA, Iqbal J, et al. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-β responses. PLoS Pathogens. (2015) 11:e1005030. doi: 10.1371/journal.ppat.1005030
37. Alnemri ES. Sensing cytoplasmic danger signals by the inflammasome. J Clin Immunol. (2010) 30:512–9. doi: 10.1007/s10875-010-9419-0
38. Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. (2008) 9:446–54. doi: 10.1038/nrm2406
39. Winslow G, Yager E, Shilo K, Collins D, Chu F. Infection of the laboratory mouse with the intracellular pathogen Ehrlichia chaffeensis. Infect Immun. (1998) 66:3892–9.
40. Gaunt S, Corstvet R, Berry C, Brennan B. Isolation of Ehrlichia canis from dogs following subcutaneous inoculation. J Clin Microbiol. (1996) 34:1429–32.
41. Winslow G, Yager E, Shilo K, Volk E, Reilly A, Chu F. Antibody-mediated elimination of the obligate intracellular bacterial pathogen Ehrlichia chaffeensis during active infection. Infect Immun. (2000) 68:2187–95. doi: 10.1128/IAI.68.4.2187-2195.2000
42. Loftis AD, Nicholson WL, Levin ML. Evaluation of immunocompetent and immunocompromised mice (Mus musculus) for infection with Ehrlichia chaffeensis and transmission to Amblyomma americanum ticks. Vector Borne Zoonotic Dis. (2004) 4:323–33. doi: 10.1089/vbz.2004.4.323
43. Sotomayor EA, Popov VL, Feng HM, Walker DH, Olano JP. Animal model of fatal human monocytotropic Ehrlichiosis. Am J Pathol. (2001) 158:757–69. doi: 10.1016/s0002-9440(10)64018-7
44. Ismail N, Soong L, McBride JW, Valbuena G, Olano JP, Feng HM, et al. Overproduction of TNF-alpha by CD8+ type 1 cells and down-regulation of IFN-gamma production by CD4+ Th1 cells contribute to toxic shock-like syndrome in an animal model of fatal monocytotropic ehrlichiosis. J Immunol. (2004) 172:1786–800. doi: 10.4049/jimmunol.172.3.1786
45. Olano JP, Wen G, Feng HM, McBride JW, Walker DH. Histologic, serologic, and molecular analysis of persistent ehrlichiosis in a murine model. Am J Pathol. (2004) 165:997–1006. doi: 10.1016/s0002-9440(10)63361-5
46. Ismail N, Stevenson HL, Walker DH. Role of tumor necrosis factor alpha (TNF-α) and interleukin-10 in the pathogenesis of severe murine monocytotropic ehrlichiosis: increased resistance of TNF receptor p55- and p75-deficient mice to fatal ehrlichial infection. Infect Immun. (2006) 74:1846–56. doi: 10.1128/iai.74.3.1846-1856.2006
47. Ismail N, Crossley EC, Stevenson HL, Walker DH. Relative importance of T-Cell subsets in monocytotropic ehrlichiosis: a novel effector mechanism involved in Ehrlichia-induced immunopathology in murine ehrlichiosis. Infect Immun. (2007) 75:4608–20. doi: 10.1128/iai.00198-07
48. Ghose P, Ali AQ, Fang R, Forbes D, Ballard B, Ismail N. The interaction between IL-18 and IL-18 receptor limits the magnitude of protective immunity and enhances pathogenic responses following infection with intracellular bacteria. J Immunol. (2011) 187:1333–46. doi: 10.4049/jimmunol.1100092
49. Stevenson HL, Estes MD, Thirumalapura NR, Walker DH, Ismail N. Natural killer cells promote tissue injury and systemic inflammatory responses during fatal Ehrlichia-Induced toxic shock-like syndrome. Am J Pathol. (2010) 177:766–76. doi: 10.2353/ajpath.2010.091110
50. Thirumalapura NR, Stevenson HL, Walker DH, Ismail N. Protective heterologous immunity against fatal ehrlichiosis and lack of protection following homologous challenge. Infect Immun. (2008) 76:1920–30. doi: 10.1128/iai.01293-07
51. Thirumalapura NR, Crossley EC, Walker DH, Ismail N. Persistent infection contributes to heterologous protective immunity against fatal ehrlichiosis. Infect Immun. (2009) 77:5682–9. doi: 10.1128/iai.00720-09
52. Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li Y, et al. Neutrophil extracellular traps promote macrophage pyroptosis in sepsis. Cell Death Dis. (2018) 9:597. doi: 10.1038/s41419-018-0538-5
53. Yang Q, Ghose P, Ismail N. Neutrophils mediate immunopathology and negatively regulate protective immune responses during fatal bacterial infection-induced toxic shock. Infect Immun. (2013) 81:1751–63. doi: 10.1128/iai.01409-12
54. Chattoraj P, Yang Q, Khandai A, Al-Hendy O, Ismail N. TLR2 and Nod2 mediate resistance or susceptibility to fatal intracellular Ehrlichia infection in murine models of ehrlichiosis. PLoS ONE. (2013) 8:e58514. doi: 10.1371/journal.pone.0058514
55. Vono M, Lin A, Norrby-Teglund A, Koup RA, Liang F, Loré K. Neutrophils acquire the capacity for antigen presentation to memory CD4 T cells in vitro and ex vivo. Blood. (2017) 129:1991–2001. doi: 10.1182/blood-2016-10-744441
56. Walker DH, Dumler JS. The role of CD8 T lymphocytes in rickettsial infections. Semin Immunopathol. (2015) 37:289–99. doi: 10.1007/s00281-015-0480-x
57. Laidlaw BJ, Craft JE, Kaech SM. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat Rev Immunol. (2016) 16:102–11. doi: 10.1038/nri.2015.10
58. de Sousa R. The presence of eschars, but not greater severity, in portuguese patients infected with israeli spotted fever. Ann N Y Acad Sci. (2005) 1063:197–202. doi: 10.1196/annals.1355.032
59. Feng HM, Walker DH. Mechanisms of immunity to Ehrlichia muris: a model of monocytotropic ehrlichiosis. Infect Immun. (2004) 72:966–71. doi: 10.1128/iai.72.2.966-971.2004
60. Habib S, Andaloussi AE, Hisham A, Ismail N. NK cell-mediated regulation of protective memory responses against intracellular ehrlichial pathogens. PLoS ONE. (2016) 11:e0153223. doi: 10.1371/journal.pone.0153223
61. Mattner J, DeBord KL, Ismail N, Goff RD, Cantu C, Zhou D, et al. Exogenous and endogenous glycolipid antigens activate NKT cells during microbial infections. Nature. (2005) 434:525–9. doi: 10.1038/nature03408
62. Stevenson HL, Crossley EC, Thirumalapura N, Walker DH, Ismail N. Regulatory roles of CD1d-Restricted NKT cells in the induction of toxic shock-like syndrome in an animal model of fatal ehrlichiosis. Infect Immun. (2008) 76:1434–44. doi: 10.1128/iai.01242-07
63. Kader M, Alaoui-EL-Azher M, Vorhauer J, Kode BB, Wells JZ, Stolz D, et al. MyD88-dependent inflammasome activation and autophagy inhibition contributes to Ehrlichia-induced liver injury and toxic shock. PLoS Pathogens. (2017) 13:e1006644. doi: 10.1371/journal.ppat.1006644
64. Rikihisa Y. Subversion of RAB5-regulated autophagy by the intracellular pathogen Ehrlichia chaffeensis. Small GTPases. (2017) doi: 10.1080/21541248.2017.1332506. [Epub ahead of print].
65. Niu H, Xiong Q, Yamamoto A, Hayashi-Nishino M, Rikihisa Y. Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc Natl Acad Sci USA. (2012) 109:20800–7. doi: 10.1073/pnas.1218674109
66. Niu H, Rikihisa Y. Ats-1: a novel bacterial molecule that links autophagy to bacterial nutrition. Autophagy. (2013) 9:787–8. doi: 10.4161/auto.23693
67. Miura K, Matsuo J, Rahman MA, Kumagai Y, Li X, Rikihisa Y. Ehrlichia chaffeensis induces monocyte inflammatory responses through MyD88, ERK, and NF-kB but Not through TRIF, interleukin-1 receptor 1 (IL-1R1)/IL-18R1, or toll-like receptors. Infect Immun. (2011) 79:4947–56. doi: 10.1128/iai.05640-11
68. Choi KS, Scorpio DG, Dumler JS. Anaplasma phagocytophilumLigation to toll-like receptor (TLR) 2, but Not to TLR4, activates macrophages for nuclear Factor–kB nuclear translocation. J Infect Dis. (2004) 189:1921–5. doi: 10.1086/386284
69. Sahni SK, Narra HP, Sahni A, Walker DH. Recent molecular insights into rickettsial pathogenesis and immunity. Future Microbiol. (2013) 8:1265–88. doi: 10.2217/fmb.13.102
70. Osterloh A. Immune response against rickettsiae: lessons from murine infection models. Med Microbiol Immunol. (2017) 206:403–17. doi: 10.1007/s00430-017-0514-1
71. Abdelaziz DHA, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, et al. Asc-dependent and independent mechanisms contribute to restriction of legionella pneumophila infection in murine macrophages. Front Microbiol. (2011) 2:18. doi: 10.3389/fmicb.2011.00018
72. Akhter A, Caution K, Khweek AA, Tazi M, Abdulrahman BA, Abdelaziz DHA, et al. Caspase-11 promotes the fusion of phagosomes harboring pathogenic bacteria with lysosomes by modulating actin polymerization. Immunity. (2012) 37:35–47. doi: 10.1016/j.immuni.2012.05.001
73. Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr Opin Microbiol. (2013) 16:319–26. doi: 10.1016/j.mib.2013.04.004
74. Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, et al. Canonical inflammasomes drive IFN-γ to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe. (2015) 18:320–32. doi: 10.1016/j.chom.2015.07.016
75. Abboud A, Namas RA, Ramadan M, Mi Q, Almahmoud K, Abdul-Malak O, et al. Computational analysis supports an early, Type 17 cell-associated divergence of blunt trauma survival and mortality. Crit Care Med. (2016) 44:e1074–81. doi: 10.1097/ccm.0000000000001951
76. Anand PK, Malireddi RKS, Lukens JR, Vogel P, Bertin J, Lamkanfi M, et al. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature. (2012) 488:389–93. doi: 10.1038/nature11250
77. Cunha LD, Silva ALN, Ribeiro JM, Mascarenhas DPA, Quirino GFS, Santos LL, et al. AIM2 engages active but unprocessed caspase-1 to induce noncanonical activation of the NLRP3 inflammasome. Cell Rep. (2017) 20:794–805. doi: 10.1016/j.celrep.2017.06.086
78. Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, et al. IRF8 regulates transcription of Naip s for NLRC4 inflammasome activation. Cell. (2018) 173:920–33.e13. doi: 10.1016/j.cell.2018.02.055
79. Bauer EM, Shapiro R, Billiar TR, Bauer PM. High mobility group Box 1 inhibits human pulmonary artery endothelial cell migration via a toll-like receptor 4- and interferon response factor 3-dependent mechanism(s). J Biol Chem. (2012) 288:1365–73. doi: 10.1074/jbc.m112.434142
80. Broz P, Ruby T, Belhocine K, Bouley DM, Kayagaki N, Dixit VM, et al. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nature. (2012) 490:288–91. doi: 10.1038/nature11419
81. Broz P, Monack DM. Noncanonical inflammasomes: caspase-11 activation and effector mechanisms. PLoS Pathogens. (2013) 9:e1003144. doi: 10.1371/journal.ppat.1003144
82. qiang Bao G, He L, Lee D, D'Angelo J, chao Wang H. An ongoing search for potential targets and therapies for lethal sepsis. Military Med Res. (2015) 2:20. doi: 10.1186/s40779-015-0047-0
83. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
84. Yang Q, Stevenson HL, Scott MJ, Ismail N. Type I interferon contributes to noncanonical inflammasome activation, mediates immunopathology, and impairs protective immunity during fatal infection with lipopolysaccharide-negative ehrlichiae. Am J Pathol. (2015) 185:446–61. doi: 10.1016/j.ajpath.2014.10.005
85. Asciolla JJ, Renault TT, Chipuk JE. Examining BCL-2 family function with large unilamellar vesicles. J Vis Exp. (2012) 68:4291. doi: 10.3791/4291
86. Smalley C, Bechelli J, Rockx-Brouwer D, Saito T, Azar SR, Ismail N, et al. Rickettsia australis activates inflammasome in human and murine macrophages. PLoS ONE. (2016) 11:e0157231. doi: 10.1371/journal.pone.0157231
87. Wang X, Shaw DK, Hammond HL, Sutterwala FS, Rayamajhi M, Shirey KA, et al. The prostaglandin E2-EP3 receptor axis regulates anaplasma phagocytophilum-mediated NLRC4 inflammasome activation. PLoS Pathogens. (2016) 12:e1005803. doi: 10.1371/journal.ppat.1005803
88. Klionsky DJ, Abdelmohsen K, Abe A. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
90. Abdulrahman BA, Khweek AA, Akhter A, Caution K, Kotrange S, Abdelaziz DHA, et al. Autophagy stimulation by rapamycin suppresses lung inflammation and infection byBurkholderia cenocepaciain a model of cystic fibrosis. Autophagy. (2011) 7:1359–70. doi: 10.4161/auto.7.11.17660
91. Seveau S, Turner J, Gavrilin MA, Torrelles JB, Hall-Stoodley L, Yount JS, et al. Checks and balances between autophagy and inflammasomes during infection. J Mol Biol. (2018) 430:174–92. doi: 10.1016/j.jmb.2017.11.006
92. Khweek AA, Caution K, Akhter A, Abdulrahman BA, Tazi M, Hassan H, et al. A bacterial protein promotes the recognition of theLegionella pneumophilavacuole by autophagy. Eur J Immunol. (2013) 43:1333–44. doi: 10.1002/eji.201242835
93. Abdelaziz DHA, Khalil H, Cormet-Boyaka E, Amer AO. The cooperation between the autophagy machinery and the inflammasome to implement an appropriate innate immune response: do they regulate each other? Immunol Rev. (2015) 265:194–204. doi: 10.1111/imr.12288
94. Niu H, Yamaguchi M, Rikihisa Y. Subversion of cellular autophagy by anaplasma phagocytophilum. Cell Microbiol. (2008) 10:593–605. doi: 10.1111/j.1462-5822.2007.01068.x
95. Bechelli J, Vergara L, Smalley C, Buzhdygan TP, Bender S, Zhang W, et al. Atg5 supports rickettsia australis infection in macrophages in vitro and in vivo. Infect Immun. (2018) 87:e00651–18. doi: 10.1128/iai.00651-18
96. Lin M, Liu H, Xiong Q, Niu H, Cheng Z, Yamamoto A, et al. Ehrlichia secretes Etf-1 to induce autophagy and capture nutrients for its growth through RAB5 and class III phosphatidylinositol 3-kinase. Autophagy. (2016) 12:2145–66. doi: 10.1080/15548627.2016.1217369
97. Takahama M, Akira S, Saitoh T. Autophagy limits activation of the inflammasomes. Immunol Rev. (2017) 281:62–73. doi: 10.1111/imr.12613
98. Corradetti MN, Guan KL. Upstream of the mammalian target of rapamycin: do all roads pass through mTOR? Oncogene. (2006) 25:6347–60. doi: 10.1038/sj.onc.1209885
99. Sharma P, Teymournejad O, Rikihisa Y. Peptide nucleic acid knockdown and intra-host cell complementation of Ehrlichia Type IV secretion system effector. Front Cell Infect Microbiol. (2017) 7:228. doi: 10.3389/fcimb.2017.00228
100. Kumagai Y, Huang H, Rikihisa Y. Expression and porin activity of P28 and OMP-1F during intracellular Ehrlichia chaffeensis development. J Bacteriol. (2008) 190:3597–605. doi: 10.1128/jb.02017-07
101. Ahn H, Kim J, Lee MJ, Kim YJ, Cho YW, Lee GS. Methylsulfonylmethane inhibits NLRP3 inflammasome activation. Cytokine. (2015) 71:223–31. doi: 10.1016/j.cyto.2014.11.001
102. Walker D, Popov V, Crocquet-Valdes P, Welsh C, Feng H. Cytokine-induced, nitric oxide-dependent, intracellular antirickettsial activity of mouse endothelial cells. Lab Invest. (1997) 76:129–38.
103. Uchiyama T, Kishi M, Ogawa M. Restriction of the growth of a nonpathogenic spotted fever group rickettsia. FEMS Immunol Med Microbiol. (2012) 64:42–47. doi: 10.1111/j.1574-695x.2011.00879.x
104. Uchiyama T. Tropism and Pathogenicity of Rickettsiae. Front Microbiol. (2012) 3:230. doi: 10.3389/fmicb.2012.00230
105. Gong B, Lee YS, Lee I, Shelite TR, Kunkeaw N, Xu G, et al. Compartmentalized, functional role of angiogenin during spotted fever group rickettsia-induced endothelial barrier dysfunction: evidence of possible mediation by host tRNA-derived small noncoding RNAs. BMC Infect Dis. (2013) 13:285. doi: 10.1186/1471-2334-13-285
106. Galindo RC, Ayllón N, Smrdel K, Boadella M, Beltrán-Beck B, Mazariegos M, et al. Gene expression profile suggests that pigs (Sus scrofa) are susceptible to Anaplasma phagocytophilum but control infection. Parasites Vectors. (2012) 5:181. doi: 10.1186/1756-3305-5-181
107. Sanjuan MA, Dillon CP, Tait SWG, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signaling in macrophages links the autophagy pathway to phagocytosis. Nature. (2007) 450:1253–57. doi: 10.1038/nature06421
108. Rikihisa Y. Role and function of the Type IV secretion system in anaplasma and Ehrlichia Species. In: Current Topics in Microbiology and Immunology. Heidelberg: Springer International Publishing (2017). p. 297–321.
109. Katze MG, He Y, Gale M. Viruses and interferon: a fight for supremacy. Nat Rev Immunol. (2002) 2:675–87. doi: 10.1038/nri888
110. Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. (2015) 15:231–42. doi: 10.1038/nri3806
111. Decker T, Müller M, Stockinger S. The Yin and Yang of type I interferon activity in bacterial infection. Nat Rev Immunol. (2005) 5:675–87. doi: 10.1038/nri1684
112. Carrero JA. Confounding roles for type I interferons during bacterial and viral pathogenesis. Int Immunol. (2013) 25:663–9. doi: 10.1093/intimm/dxt050
113. Malireddi RKS, Kanneganti TD. Role of type I interferons in inflammasome activation, cell death, and disease during microbial infection. Front Cell Infect Microbiol. (2013) 3:77. doi: 10.3389/fcimb.2013.00077
114. Storek KM, Gertsvolf NA, Ohlson MB, Monack DM. cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella Infection. J Immunol. (2015) 194:3236–45. doi: 10.4049/jimmunol.1402764
115. Saeij JP, Frickel EM. Exposing Toxoplasma gondii hiding inside the vacuole: a role for GBPs, autophagy and host cell death. Curr Opin Microbiol. (2017) 40:72–80. doi: 10.1016/j.mib.2017.10.021
116. Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, et al. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in chlamydia-infected macrophages. Infect Immun. (2015) 83:4740–9. doi: 10.1128/iai.00856-15
117. Man SM, Place DE, Kuriakose T, Kanneganti TD. Interferon-inducible guanylate-binding proteins at the interface of cell-autonomous immunity and inflammasome activation. J Leukocyte Biol. (2016) 101:143–50. doi: 10.1189/jlb.4mr0516-223r
118. Snyder DT, Hedges JF, Jutila MA. Getting “Inside” Type I IFNs: Type I IFNs in intracellular bacterial infections. J Immunol Res. (2017) 2017:1–17. doi: 10.1155/2017/9361802
119. Agbayani G, Wachholz K, Murphy SP, Sad S, Krishnan L. Type I interferons differentially modulate maternal host immunity to infection by Listeria monocytogenes and Salmonella enterica serovar Typhimurium during pregnancy. Am J Reprod Immunol. (2018) 81:e13068. doi: 10.1111/aji.13068
120. Carrero JA, Calderon B, Unanue ER. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J Exp Med. (2004) 200:535–40. doi: 10.1084/jem.20040769
121. O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. (2004) 200:437–45. doi: 10.1084/jem.20040712
122. Pitts MG, Myers-Morales T, D'Orazio SEF. Type I IFN does not promote susceptibility to foodborne listeria monocytogenes. J Immunol. (2016) 196:3109–16. doi: 10.4049/jimmunol.1502192
123. Lasseaux C, Fourmaux MP, Chamaillard M, Poulin LF. Type I interferons drive inflammasome-independent emergency monocytopoiesis during endotoxemia. Sci Rep. (2017) 7:16935. doi: 10.1038/s41598-017-16869-2
124. Franco MMC, Marim F, Guimarães ES, Assis NRG, Cerqueira DM, Alves-Silva J, et al. Brucella abortus triggers a cGAS-independent STING pathway to induce host protection that involves guanylate-binding proteins and inflammasome activation. J Immunol. (2017) 200:607–22. doi: 10.4049/jimmunol.1700725
125. Ka MB, Mezouar S, Amara AB, Raoult D, Ghigo E, Olive D, et al. Coxiella burnetii induces inflammatory interferon-like signature in plasmacytoid dendritic cells: a new feature of immune response in Q fever. Front Cell Infect Microbiol. (2016) 6:70. doi: 10.3389/fcimb.2016.00070
126. Hedges JF, Robison A, Kimmel E, Christensen K, Lucas E, Ramstead A, et al. Type I interferon counters or promotes coxiella burnetii replication dependent on tissue. Infect Immun. (2016) 84:1815–25. doi: 10.1128/iai.01540-15
127. Richard K, Vogel SN, Perkins DJ. Type I interferon licenses enhanced innate recognition and transcriptional responses to Franciscella tularensis live vaccine strain. Innate Immun. (2016) 22:363–72. doi: 10.1177/1753425916650027
128. Meunier E, Broz P. Interferon-induced guanylate-binding proteins promote cytosolic lipopolysaccharide detection by caspase-11. DNA Cell Biol. (2015) 34:1–5. doi: 10.1089/dna.2014.2701
129. Skyberg JA, Lacey CA. Hematopoietic MyD88 and IL-18 are essential for IFN-γ–dependent restriction of type A Francisella tularensis infection. J Leukocyte Biol. (2017) 102:1441–50. doi: 10.1189/jlb.4a0517-179r
130. Zhang Y, Thai V, McCabe A, Jones M, MacNamara KC. Type I interferons promote severe disease in a mouse model of lethal ehrlichiosis. Infect Immun. (2014) 82:1698–709. doi: 10.1128/iai.01564-13
131. Cai J, Yuan H, Wang Q, Yang H, Al-Abed Y, Hua Z, et al. HMGB1-Driven inflammation and intimal hyperplasia after arterial injury involves cell-specific actions mediated by TLR4. Arterioscler Thromb Vasc Biol. (2015) 35:2579–93. doi: 10.1161/atvbaha.115.305789
132. Sharma D, Kanneganti TD. Inflammatory cell death in intestinal pathologies. Immunol Rev. (2017) 280:57–73. doi: 10.1111/imr.12602
133. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. (2012) 150:606–19. doi: 10.1016/j.cell.2012.07.007
134. Abdelaziz DH, Gavrilin MA, Akhter A, Caution K, Kotrange S, Khweek AA, et al. Apoptosis-associated speck-like protein (ASC) controls Legionella pneumophila infection in human monocytes. J Biol Chem. (2010) 286:3203–8. doi: 10.1074/jbc.m110.197681
135. Amer AO, Swanson MS. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell Microbiol. (2005) 7:765–778. doi: 10.1111/j.1462-5822.2005.00509.x
136. Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. (2010) 11:1136–42. doi: 10.1038/ni.1960
Keywords: ehrlichiosis, inflammasome, autophagy, pathogenesis, innate immunity, mechanism
Citation: Tominello TR, Oliveira ERA, Hussain SS, Elfert A, Wells J, Golden B and Ismail N (2019) Emerging Roles of Autophagy and Inflammasome in Ehrlichiosis. Front. Immunol. 10:1011. doi: 10.3389/fimmu.2019.01011
Received: 28 February 2019; Accepted: 23 April 2019;
Published: 08 May 2019.
Edited by:
J. Stephen Dumler, Uniformed Services University of the Health Sciences, United StatesReviewed by:
Rong Fang, The University of Texas Medical Branch at Galveston, United StatesCopyright © 2019 Tominello, Oliveira, Hussain, Elfert, Wells, Golden and Ismail. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nahed Ismail, aXNtYWlsN0B1aWMuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.