 Hiromi Kubagawa1*
Hiromi Kubagawa1* Kazuhito Honjo2
Kazuhito Honjo2 Naganari Ohkura3
Naganari Ohkura3 Shimon Sakaguchi3
Shimon Sakaguchi3 Andreas Radbruch1
Andreas Radbruch1 Fritz Melchers1*
Fritz Melchers1* Peter K. Jani1*
Peter K. Jani1*- 1Deutsches Rheuma-Forschungszentrum, Berlin, Germany
- 2Department of Medicine, School of Medicine, University of Alabama at Birmingham, Birmingham, AL, United States
- 3Immunology Frontier Research Center, Osaka University, Osaka, Japan
It is now evident from studies of mice unable to secrete IgM that both non-immune “natural” and antigen-induced “immune” IgM are important for protection against pathogens and for regulation of immune responses to self-antigens. Since identification of its Fc receptor (FcμR) by a functional cloning strategy in 2009, the roles of FcμR in these IgM effector functions have begun to be explored. Unlike Fc receptors for switched Ig isotypes (e.g., FcγRs, FcεRs, FcαR, Fcα/μR, pIgR, FcRn), FcμR is selectively expressed by lymphocytes: B, T, and NK cells in humans and only B cells in mice. FcμR may have dual signaling ability: one through a potential as yet unidentified adaptor protein non-covalently associating with the FcμR ligand-binding chain via a His in transmembrane segment and the other through its own Tyr and Ser residues in the cytoplasmic tail. FcμR binds pentameric and hexameric IgM with a high avidity of ~10 nM in solution, but more efficiently binds IgM when it is attached to a membrane component via its Fab region on the same cell surface (cis engagement). Four different laboratories have generated Fcmr-ablated mice and eight different groups of investigators have examined the resultant phenotypes. There have been some clear discrepancies reported that appear to be due to factors including differences in the exons of Fcmr that were targeted to generate the knockouts. One common feature among these different mutant mice, however, is their propensity to produce autoantibodies of both IgM and IgG isotypes. In this review, we briefly describe recent findings concerning the functions of FcμR in both mice and humans and propose a model for how FcμR plays a regulatory role in B cell tolerance.
Introduction
Two forms of IgM exist that differ in the carboxyl terminus of the heavy chain (HC). Alternative splicing with a transmembrane exon (μm) generates monomeric membrane-bound IgM as a B cell receptor (BCR) for antigen and with a secretory exon (μs) polymeric IgM secreted by plasma cell as a component of humoral immunity. The secreted form of IgM consists mainly of J chain-containing pentamers. The existence of J chain-deficient hexamers has also been reported albeit at an unknown concentration. To determine the role of secreted IgM in immune responses, two different groups have independently disrupted the exon encoding the μs (μs KO) (1, 2). Such mutant mice normally express IgM and other Ig isotypes on the surface of B cells and secrete all Ig isotypes except for IgM. These mutant mice are unable to control infections, because of inefficient induction of a protective IgG antibody response (3–5). Paradoxically, the autoimmune pathology associated with IgG autoantibody is more severe in μs KO mice than in the control mice, possibly because of impaired clearance of autoantigen-containing apoptotic cells (6, 7). Yet, no studies have directly demonstrated such deficiency in removal of self-antigens. Thus, both natural and immune IgM are important for protection against pathogens as well as in regulation of immune responses to self-antigens (8).
A variety of secreted and cell surface proteins is involved in binding the Fc portion of antibody, thereby participating in its effector function, e.g., complement and various types of Fc receptors (FcRs). Classical FcRs for switched Ig isotypes (i.e., FcγRs, FcεRI, FcαR), the receptor for polymeric IgA and IgM (pIgR), the low affinity FcεRII/CD23, and the FcR for neonatal IgG (FcRn) have thus far extensively been characterized at both genetic and protein levels (9–17) (see also other articles in this issue), and much of the knowledge gained has now been translated to clinical practice (18, 19). On the other hand, the role of the IgM FcR (FcμR) as an effector molecule for IgM antibody, the first Ig isotype appearing during phylogeny, ontogeny and immune responses, has just begun to be explored, since the FCMR was identified in 2009 (20). Several FcμR review articles have recently been published elsewhere (21–25). Here we briefly reiterate the biochemical structure of the FcμR and its functional roles in the development of B cell subsets and plasma cells, describe the potential molecular bases for certain discrepancies observed among different Fcmr KO mice, and introduce our theoretical model for how FcμR is involved in B cell tolerance.
Unique Properties of FcμR
Dual Signaling Ability
FCMR is a single copy gene located on chromosome 1q32.2 adjacent to two other IgM-binding receptors PIGR and FCAMR (FcR for IgA and IgM) (20). The predicted human FcμR is a type I glycoprotein of 390 amino acids (aa) with a peptide core of ~41 kD, which consists of a signal peptide, a V-set Ig-like domain responsible for Fcμ binding, an additional extracellular region with unknown domain structure (termed the stalk region), a transmembrane (TM) segment containing a charged His residue (H253) and a relatively long cytoplasmic (CY) tail of 118 aa containing conserved, three Tyr and five Ser residues (see Figure 1A). Among these Tyr residues, the carboxyl terminal Y385 matches the Ig tail Tyr motif (DYxN; x indicates any aa) seen in IgG and IgE (26), but the other two do not correspond to any known Tyr-based signaling motifs, ITAM, ITIM or switch. Two carboxyl terminal Y366 and Y385 are involved in receptor-mediated endocytosis (27, 28) and the membrane proximal Y315 is predominantly involved in the FcμR-mediated protection from IgM anti-Fas monoclonal antibody (mAb)-induced apoptosis (28) (see below). An important role of the H253 residue in anchoring the receptor in the plasma membrane became evident when the fate of IgM bound to FcμR in cells stably expressing the wild type (WT) or H253F mutant form of receptor was examined by immunofluorescence microscopy; the mutant showed enhanced cap formation even at 4°C. IgM ligand-binding activity was found significantly increased in an FcμR mutant with a deletion of most of the CY tail compared to the WT receptor, despite comparable surface levels as determined by receptor-specific mAbs. Based on our preliminary data, this enhancement appears to result from the formation of an oligomeric FcμR as a consequence of its presumably mobile nature within the plasma membrane. This is different from our speculated inside-out regulation of FcμR ligand binding by its CY tail as seen in integrins. Ligation of FcμR with preformed soluble IgM immune complexes induced phosphorylation of both Tyr and Ser residues (20). Intriguingly, the phosphorylated FcμR migrated faster on SDS-PAGE than the unphosphorylated form, unlike most proteins that run slower when phosphorylated. Preliminary data with an epitope-tagged FcμR suggest that there could be cleavage of the CY tail of FcμR, but the precise molecular mechanisms for this cleavage and the functional role of the resultant FcμR stub still need to be elucidated. Collectively, these features of human FcμR suggest a dual signaling ability of FcμR: one via a potential as yet unidentified adaptor protein non-covalently associating with the FcμR via the H253 residue and the other from its own Tyr and Ser residues in the CY tail.
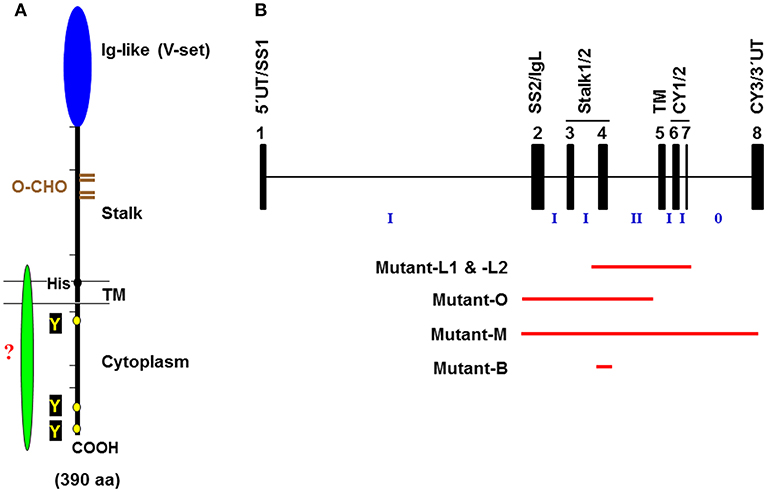
Figure 1. Schematic representation of the FcμR. (A) Predicted FcμR protein structure. The human FcμR cDNA encodes a type I transmembrane protein of 390 aa with a peptide core of ~41 kD that consists of a signal peptide (not shown), an Ig-like domain (V-set), remaining extracellular (stalk), transmembrane (TM; between two lines) and cytoplasmic region. Black and brown hatch marks indicate exon boundaries in the FCMR gene and O-glycosylation sites, respectively. Small black and yellow circles indicate a TM charged His residue and conserved Tyr residues, respectively. A green fusiform indicates a hypothetical adaptor protein non-covalently associating with the FcμR ligand-binding chain via the His residue. (B) Schematic representation of targeted exons in Fcmr-ablated mice. The exon (black closed boxes) organization of Fcmr is drawn along with intron phases (“phase 0” indicates between the codons; “phase I” between the first and second nucleotide of a codon; “phase II” between the second and third nucleotide). Exons encoding particular regions of the receptor are denoted as follows: the 5′ untranslated (5′UT), the signal peptide (SS1 and 2), the Ig-like domain (IgL), the uncharacterized extracellular (Stalk 1 and 2), the transmembrane (TM), the cytoplasmic (CY1-3), and the 3′ untranslated (3′UT) regions. Red lines indicate the exons targeted in each Fcmr knockout mouse strain (see text for details).
While mouse ortholog with 422 aa has relatively low homology (~54%) with human FcμR, the overall structural characteristics (a single Ig-like domain, a His residue in TM segment, and a long CY tail containing three Tyr and five Ser residues) are conserved. However, the analysis of its biochemical nature including the ligand binding is limited (22, 29).
Lymphocyte-Restricted Distribution
Given the fact that IgM is the first Ig isotype to appear during phylogeny, ontogeny and immune responses, we initially thought that FcμR would have a broad cellular distribution, thereby serving as a first line of defense against pathogens. On the contrary, FcμR was found to be expressed by lymphocytes only: both B and T cells and, to a lesser extent, NK cells in humans, and only B cells in mice (20, 29–32). Unlike the phylogenetically broad distribution of IgM from jawed vertebrates onward (i.e., cartilaginous fish), computational analysis of existing genomic sequence databases unexpectedly reveals that FcμR appears probably in early reptiles and is found in all three major living (extant) groups of mammals (i.e., egg laying, marsupial and placental mammals) (33). FcμR is the only FcR constitutively expressed on human T cells, which are otherwise generally negative for FcRs, and for B cells, FcμR is the only IgM-binding FcR expressed. [In this regard, another IgM-binding receptor, Fcα/μR, was initially reported to be expressed by B cells, but subsequent analyses revealed that the major cell type expressing Fcα/μR in immune system is a follicular dendritic cell in both humans and mice (34).] During B-lineage differentiation, the cell surface expression of FcμR was detectable from pre-B/B transitional stage to plasmablasts, except for a transient down-modulation during germinal center reactions in both humans and mice (20, 29, 30, 32, 35). Collectively, the restriction of FcμR expression to adaptive immune cells is thus remarkable, because FcRs for switched Ig isotypes are expressed by various hematopoietic cells including myeloid cells as central mediators coupling innate and adaptive immune responses. Lymphocyte-specific FcμR may thus have a distinct function from myeloid cell FcRs.
Cis Engagement
Cell surface FcμR in humans is a sialoglycoprotein of ~60 kD and one third of the relative molecular mass of the mature FcμR is thus made up of O-linked glycans. It exclusively binds the Fc portion of pentameric and hexameric IgM with strikingly high avidity of ~10 nM as determined by Scatchard plot analysis with the assumption of a 1:1 stoichiometry of FcμR to IgM (20, 25) (Figure 2A). Much higher concentrations (>100-fold) are required for binding of monomeric IgM to FcμR-bearing cells, indicating the importance of IgM conformation. This in turn suggests that serum IgM, at its serum concentration of ~1 μM, constitutively binds to FcμR on the surface of lymphocytes. In addition to the high avidity for IgM in solution, a unique ligand-binding property of FcμR was observed when IgM mAbs to lymphocyte surface proteins were used as a ligand. When Fas death receptor is ligated with 10 pM agonistic IgM anti-Fas mAb, apoptosis-prone Jurkat cells undergo robust apoptosis within 1 day, but Jurkat cells stably expressing FcμR do not (Figures 2B,C). This finding is thus consistent with previously reported anti-apoptotic activity of Toso (the original name of FcμR) (36). [In this review we will only use “FcμR” as the name of the receptor, based on a recent nomenclature agreement (37).] However, ligation of Fas with agonistic IgG3 anti-Fas mAb or co-ligation of both Fas and FcμR with the corresponding mouse IgG mAbs plus an appropriate common secondary reagent [e.g., F(ab')2 fragments of anti-mouse γ antibody] had no inhibitory effects on the IgG3 Fas mAb-induced apoptosis (Figure 2D). This suggests that FcμR per se has no intrinsic activity to inhibit Fas-mediated apoptosis. The anti-apoptotic activity of FcμR depends on usage of the IgM Fas mAb and not on physical proximity of two receptors by artificial co-ligation as observed with ITIM containing receptors such as FcγRIIb and paired Ig-like receptor B (38, 39).
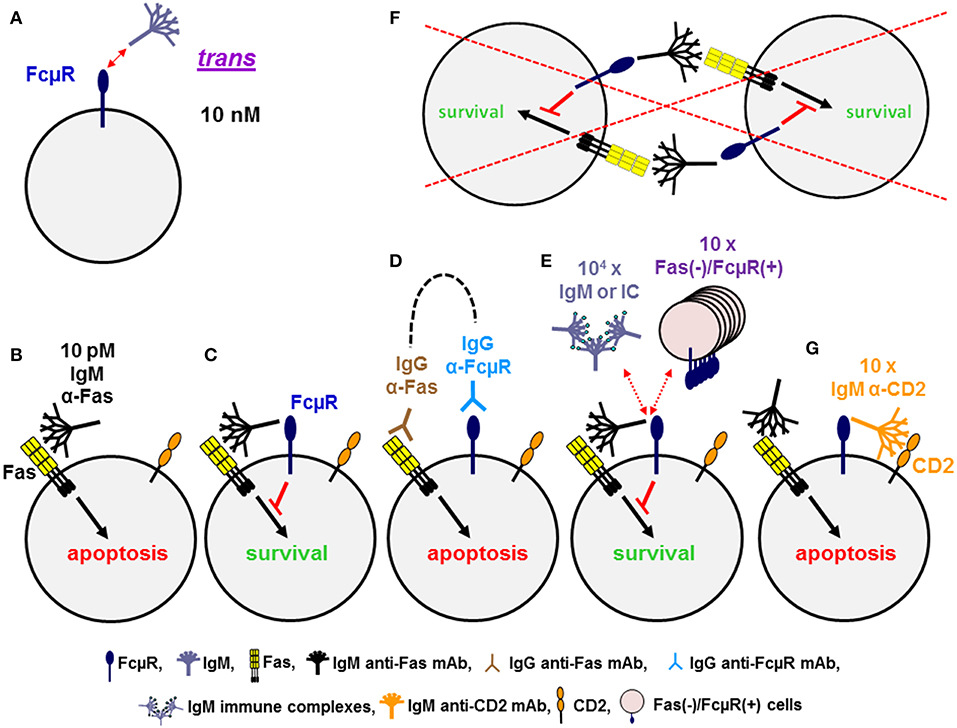
Figure 2. Dominant cis, rather than trans, interaction of FcμR. (A) FcμR positive cells bind IgM pentamers in solution in trans with high avidity of ~10 nM. (B–G) Ligation of Fas death receptor trimer with agonistic IgM anti-Fas mAb induces apoptosis in WT Jurkat cells (B), but not in FcμR-positive Jurkat cells (C). Co-ligation of Fas and FcμR with the corresponding IgG mAb plus a common secondary reagent (dotted line) has no inhibitory effects on the IgG Fas mAb-induced apoptosis (D). FcμR-mediated protection from IgM Fas mAb-induced apoptosis is not blocked by addition of 104 molar excess of IgM or its soluble immune complexes or 10-fold excess of Fas(–)/FcμR(+) cells, suggesting an efficient cis interaction of IgM Fas mAb and FcμR on the same cell surface (E), but not a trans interaction between neighboring cells (F). Addition of tenfold excess of IgM mAb reactive with CD2 on Jurkat cells can efficiently block the interaction of IgM Fas mAb and FcμR, resulting in apoptosis (G).
To determine whether the interaction of the Fc portion of IgM Fas mAb with FcμR occurs in cis or trans, a 10-fold excessive of Fas(–)/FcμR(+) cells as a potential competitive source of FcμR(+) cells was added into the assay but no inhibition of FcμR-mediated protection of Jurkat cells was observed (Figure 2E). This suggests that the interaction of the Fc portion of IgM Fas mAb with FcμR occurs in cis on the same surface of Jurkat cells (Figure 2E), but not in trans between neighboring cells (Figure 2F). Addition of >104 molar excess of IgM or its soluble immune complexes was required for partial, but significant blockade of such a cis interaction (Figure 2E), suggesting that the soluble IgM immune complexes are not potent competitors in the FcμR-mediated protection from IgM Fas mAb-induced apoptosis. However, when IgM mAb reactive with other surface proteins expressed on Jurkat cells (e.g., CD2 or TCR) was used as a potential competitor, a 10-fold excess of IgM anti-CD2 or anti-TCR mAb was sufficient to block the cis interaction, thereby permitting FcμR(+) cells to undergo apoptosis (Figure 2G) (28). Similar results with agonistic IgM vs. IgG3 Fas mAb were observed with Epstein Barr virus-transformed B cell lines simultaneously expressing endogenous FcμR and Fas on the cell surface (20). Furthermore, when BCR and FcμR on blood B cells were co-ligated with a mitogenic IgM anti-κ mAb in the presence of IgG2b anti-FcμR mAbs with blocking or non-blocking activity for IgM-ligand binding, Ca2+ mobilization was the same in the absence or presence of FcμR non-blocking mAb. By contrast, the FcμR blocking mAb significantly diminished Ca2+ mobilization by blood B cells, suggesting that FcμR provides stimulatory signals upon BCR cross-linkage with IgM mAbs (28). Collectively, these findings of human FcμR indicate that although FcμR binds soluble IgM pentamers and hexamers at a high avidity of ~10 nM, FcμR binds more efficiently to the Fc portion of IgM when it is attached to a membrane component via its Fab region on the same cell surface. FcμR expressed on lymphocytes may thus have a potential to modulate the function of target antigens or receptors when they are recognized by natural or immune IgM through its cis engagement.
In summary, FcμR: (i) is expressed by lymphocytes: B, T and NK cells in humans and only B cells in mice, suggesting that FcμR may have a distinct function compared to other FcRs, which are mainly expressed by myeloid cells, and potential species differences; (ii) may have dual signaling ability: one from a potential adaptor protein that non-covalently associates with FcμR ligand-binding chain via H253, and the other from its own Tyr/Ser residues in the CY tail; and (iii) binds more efficiently to the Fc portion of IgM when it is attached to a membrane component via the Fab region on the same cell surface (cis engagement), than to the Fc portion of IgM in solution/fluids.
Variant Results Observed in Different Fcmr-Deficient Mice
Despite the initial prediction of embryonic lethality of Fcmr ablation (40), there are now five different Fcmr KO mice that have been independently generated by four different groups of investigators [Lee et al. (mutant-L1 and -L2), Ohno et al. (mutant-O), Mak et al. (mutant-M), and Baumgarth et al. (mutant-B)]. Eight different groups of investigators have characterized these mutant mice with clear differences in reported phenotypes (29, 32, 35, 41–50) (see Table 1). This is an unusual case in the gene-targeting field. Several discrepancies could be in part due to the following: (i) Investigator's preconception of FcμR or Toso in terms of its cellular distribution (B cells vs. myeloid and T cells) (51, 52) and its function (binding IgM Fc vs. inhibiting Fas- or TNFα-mediated apoptosis) (53, 54). (ii) Differences in embryonic stem cells of C57BL/6 (mutant-L1, -L2, and -B) vs. 129/sv (mutant-O and -M) origin and the extent of the 129 mouse-origin DNA still present around the disrupted Fcmr gene after backcrossing onto C57BL/6 background; (iii) Differences in exon targeting strategies [exon 2-4 (mutant-O), 2-8 (mutant-M), 4 (mutant-B) vs. 4-7 (mutant-L1 and -L2) (Figure 1B)], global (mutant-O and -M) vs. conditional deletion (mutant-L1, -L2, and -B), and the Cd19 heterozygosity in the CD19-Cre-mediated deletion vs. the unmanipulated Cd19 in global deletion, and the presence (mutant-M) vs. absence (other mutants) of the Neo gene in the mouse genome; and/or (iv) other factors, e.g., ages of the mice examined, experimental procedures/conditions, environmental factors including intestinal microbiota, or reagents used.
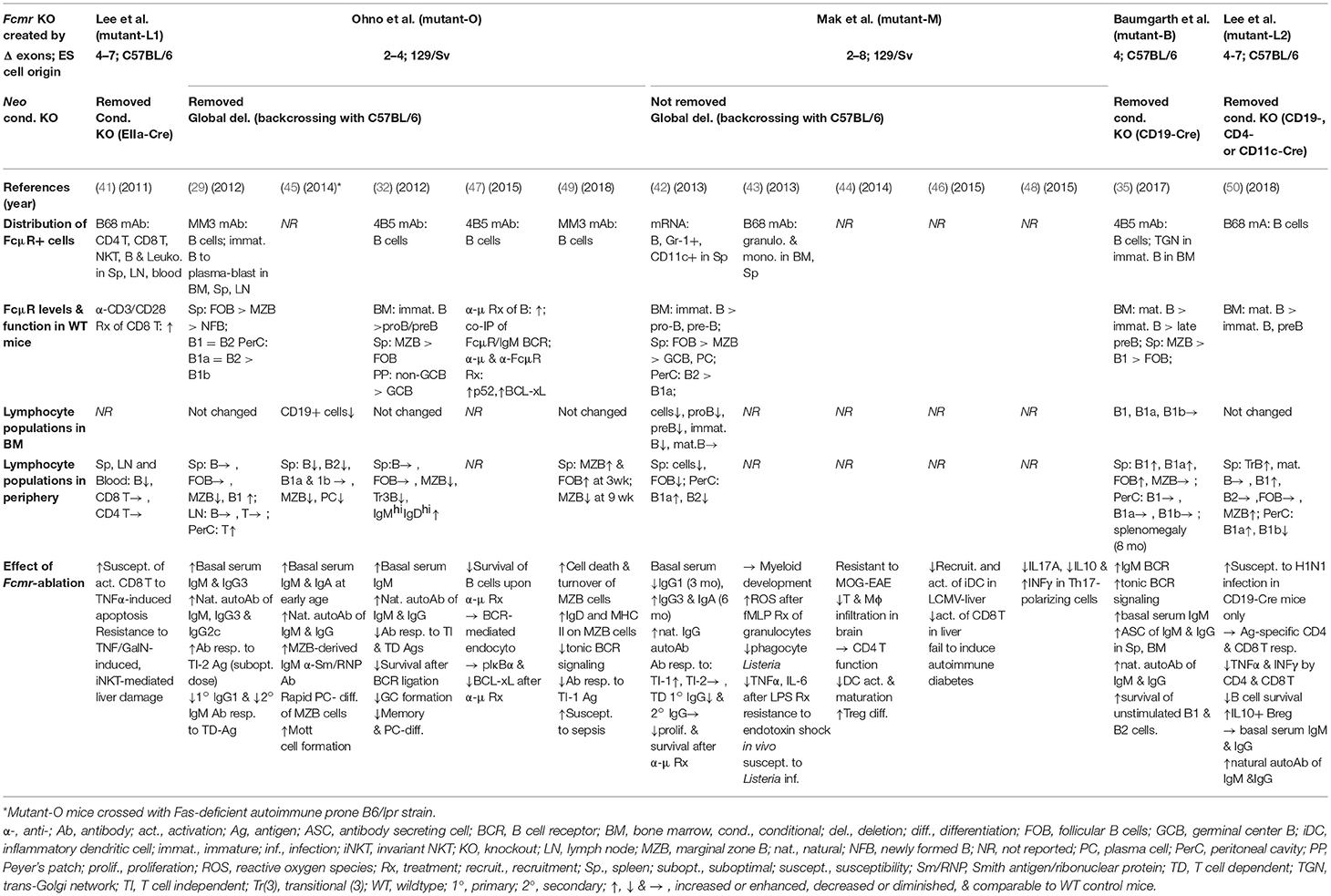
Table 1. Phenotypic comparison of five different Fcmr-deficient mice.
Another factor that could contribute to these discrepancies is the relative difficulty in assessing cell surface FcμR in mice by flow cytometry using receptor-specific mAbs, because of its relative low cell surface density as well as its sensitivity to extracellular IgM concentrations, tissue milieu and cellular activation status (20, 29, 35). This vulnerability could result in the discrepancy in reported cellular distribution of FcμR in mice. In fact, using the same receptor-specific rat mAb (B68 clone), FcμR was expressed by: mouse B cells (55), myeloid cells (43) or CD4 T, CD8 T, and B cells (41). This conflicted cellular distribution data about FcμR is a major reason why some investigators created additional Cre/loxP-mediated, cell type-specific Fcmr deletion systems (35, 50). In this regard, EIIa-Cre mediated Fcmr-deleted mutant-L1 (equivalent to global deletion) showed more TNFα-induced apoptosis of CD3/CD28-activated CD8 T cells than control mice (41). The abnormality was initially considered as an intrinsic T cell defect since this group originally reported that FcμR was expressed by T cells. However, subsequent results from conditional deletion clearly indicated no phenotypic differences between T cell-specific [or dendritic cell (DC)-specific] Fcmr deletion and control counterparts. The only differences were seen with B cell-specific Fcmr deletion. The authors thus concluded that FcμR on B cells might indirectly affect certain T cell functions (50), although it remains unclear how this would work. In this review, we will focus on the following aspects of B cell-related findings in Fcmr KO mice: (i) alterations in B cell subsets, (ii) IgM homeostasis, and (iii) dysregulated humoral immune responses.
Alteration in B Cell Subsets
The development of B-lineage cells in the bone marrow (BM) was unaffected in most Fcmr KO strains (29, 32, 35, 50) except for mutant-M where the numbers of pro-B, pre-B, and immature B cells were significantly diminished as compared to WT controls (42). Since the surface expression of FcμR begins to be detectable at the transitional stage of pre-B to B cells in differentiation, it seems conceivable that FcμR is dispensable in developing B-lineage cells in the BM. However, it is noteworthy that: (i) μs KO mice, which are deficient for secretion of IgM, have significantly altered B cell development from pre-B to the immature B cell transition (42); (ii) this alteration of early B cell development is corrected by administration of natural IgM (56); and (iii) many of the abnormalities observed in Fcmr KO mice mirror those seen in μs KO mice. Thus, despite the fact that FcμR is a key sensor of secreted IgM, it remains to be elucidated why, among five Fcmr KO mice, only mutant-M has an alteration in development of B cell precursors (42). In this regard, several human pre-B cell lines express FcμR transcripts but not FcμR protein on their cell surface at detectable levels unless stimulated with phorbol myrystate acetate (20, 57), suggesting the existence of post-transcriptional controls of FcμR.
Unlike in the BM, in peripheral lymphoid organs there were variable alterations in B cell subsets observed in these mutant mice but, as a general trend, Fcmr ablation was found to more profoundly affect innate-like B cells, B-1, and marginal zone (MZ) B cells, rather than the B-2 or follicular (FO) B cell compartment (see Table 1). Remarkably, an increase in B-1 B cell numbers, particularly in spleen accompanied by elevated levels of autoantibodies of both IgM and IgG isotypes, has been the sole result consistently observed in all five mutant mice. Thus, FcμR plays an important regulatory role in the homeostasis of B-1 B cell development and autoantibody production (see further discussion below).
For MZ B cells, the mutant-O had age-dependent alterations in their cell numbers, i.e., increase in young (3-wk) and marked decrease in old (>9-wk) mice (49). This age-dependent reduction of MZ B cells might result from their rapid differentiation into plasma cells in the absence of FcμR, as evidenced by the markedly elevated IgM autoantibodies to Smith antigen/ribonuclear protein, which are considered to be derived from MZ B cells (45). Alternatively, FcμR-deficient MZ B cells might undergo cell death due to lack of survival signals through FcμR upon BCR cross-linkage (49), as shown by cross-talk downstream of FcμR and BCR signaling via the non-canonical NFκB pathway (47). Notably, the reduction of MZ B cells was also observed with both Fas- and Fcmr-deficient, autoimmune-prone B6.MRL.Faslpr/lpr/Fcmr −/− mice (45). In mutant-M, unlike mutant-O, there were no changes in the MZ B cell compartment, whereas in both CD19-Cre-mediated deletion mutant-B and -L2, the number of MZ B cells was not reduced (for mutant-B) or enhanced (for mutant-L2) (35, 50). Since the number of MZ B cells in μs KO mice is increased by 3-fold and this increase can be normalized by passive administration of natural and polyclonal, but not monoclonal, IgM, we initially considered that the FcμR and its signals upon IgM-ligand binding might play an important regulatory role in the fate of MZ B cells. This simplistic view, however, may need further consideration based on the above conflicting results.
In addition to the aforementioned changes in cell numbers of B cell subsets, there were some differences in the density of certain cell surface markers (e.g., CD19, CD21, CD23, IgD, IgM) between Fcmr KO and WT controls (29, 49, 50). The surface levels of several co-receptors of the BCR complex such as CD21 and CD23 were diminished in certain B cell subsets from mutant-O compared to WT controls (29, 50). This was also the case with CD19 that was also significantly diminished on BM immature B cells, but not on BM recirculating and splenic B cells in mutant-O (our unpublished observation). Notably, the surface IgD levels were higher on splenic MZ B in mutant-O mice than WT controls (49). Indeed, our subsequent analysis of the same mutant mice also revealed higher expression of surface IgD on BM recirculating and splenic MZ B cells, but not on FO B cells, than WT controls (unpublished). The molecular basis for this elevated surface IgD density in mutant-O is unclear, but it has been shown that functionally hypo-responsive anergic B cells are characterized by high levels of IgD BCR and generally turn over rapidly when competing, non-tolerant B cells are present (58, 59).
For surface IgM in mutant-O, IgM staining with fluorochrome-labeled anti-μ mAb, which might include endogenous membrane-bound IgM plus cytophilic IgM bound via FcμR or other potential IgM-binding proteins/receptors, was indistinguishable in these B cell populations including BM immature B cells (29, 49). By contrast, in mutant-B the cell surface expression of IgM BCR was significantly increased as compared to control mice, but this phenotype was only demonstrable 3 days after transferring of Fcmr-deficient or control B cells into μs KO mice to avoid the influence of cytophilic IgM (35). The authors implied that this increase in IgM BCR in Fcmr KO mutant-B was due to the lack of FcμR-mediated constraints on the IgM BCR (see below), resulting in enhanced tonic BCR signaling, facilitating the spontaneous differentiation of B-1 B cells and the increase in autoantibody production. Stimulated emission-depletion microscopic analysis revealed a strong interaction of FcμR with membrane-bound IgM in the trans-Golgi network (TGN) of BM immature B cells, but a weak interaction with the IgM on the plasma membrane in mature B cells, thereby constraining transport of IgM to the plasma membrane. This effect on the exocytotic pathway was proposed to regulate surface expression of IgM and eventually limiting tonic IgM BCR signaling. When we examined the potential interaction of FcμR with IgM BCR on the plasma membrane by fluorescence resonance energy transfer, we also found a very low incidence of such an interaction. By contrast, another group showed the physical interaction of FcμR and IgM BCR on the plasma membrane of mature B cells by confocal microscopy (47) and that tonic BCR signaling was diminished in Fcmr KO mutant-O (49). Given the low avidity of FcμR for monomeric IgM in solution, it remains unclear how FcμR could interact with membrane-bound IgM in the TGN of BM immature B cells or on the plasma membrane of mature B cells.
Another remarkable finding related to this issue came from immunofluorescence confocal microscopic analysis: strong staining of intracellular FcμR in a region corresponding to the TGN in murine BM immature B cells (35). The results were in close agreement with the findings of FcμR-mediated endocytosis of IgM by chronic lymphocytic leukemia (CLL) B cells in humans (27). The bulk of the intracellular FcμR protein resided in the TGN and in small vesicles, probably sorting endosomes of CLL cells. While the major function of the TGN is to sort proteins destined for the plasma membrane, endosomal compartment or specialized secretory granules, retrograde transport in the endocytic route to the TGN has been demonstrated for several proteins (60). It is thus worth considering whether DNA- or RNA-containing autoantigens are engulfed into endosomes by IgM BCR on immature B cells in the BM, two thirds of which are known to be autoreactive at least in humans, followed by retrograde transport to the TGN where TLR9 or TLR7 recognizes the respective DNA or RNA/IgM BCR complexes and then FcμR binds the Cμ3/Cμ4 of the resultant oligomerized IgM BCRs in the TGN.
IgM Homeostasis
The pre-immune serum level of IgM or natural IgM was elevated in most Fcmr KO mice (29, 32, 35, 50) except for mutant-M (42) and this elevation correlated with the number of Fcmr null mutant alleles (Fcmr −/− > Fcmr +/− > Fcmr +/+) (32). The frequency of IgM-secreting cells in spleen and BM was significantly higher and the spot sizes in ELISPOT assays were also bigger in mutant-B than their control counterparts (35). FcμR was not expressed by phagocytic cells in spleen and liver including liver sinusoidal endothelial cells, which are thought to be the primary site of IgM catabolism at least in rat, as determined by both immuno-histological and RT-PCR analyses (29). The half-life of injected IgM was comparable between Fcmr KO (mutant-O) and WT mice. Thus, the increase in serum IgM levels in naive Fcmr KO mice is the consequence of lack of FcμR-mediated regulation of natural IgM production either at the B cell or plasmablast stage in innate-like B cells (29).
Dysregulated Humoral Immune Responses
Antibody responses to T cell-independent (TI) and T cell-dependent (TD) antigens were dysregulated in Fcmr KO mice as compared to WT controls, although there were some differences among mutant mice that might result from differences in mouse ages, antigen doses and forms, administration routes, kinetics, etc. Generally, mutant mice exhibited enhanced TI type 2 responses (involving multiple BCR cross-linkage) but impaired TD responses, especially at suboptimal doses. Since similar selective enhancement of TI-2 immune responses are also observed in μs KO mice (2) and mice deficient for components of the BCR complex such as CD19 (61) or CD81 (62), FcμR seems to regulate B cell responses to TI-2 and TD antigens by interacting differently with BCR complexes on the plasma membrane.
In summary, there are clear differences in reported phenotypes in five different Fcmr KO mice in terms of development of B cell subsets and plasma cells, IgM homeostasis and humoral immune responses. However, the increase in B-1 B cell compartment accompanied by elevated levels of autoantibodies of both IgM and IgG isotypes is the sole result consistently observed with all these mutant mice.
Epigenetic Findings in the Fcmr-Il10 Locus in Treg Cells
One of the biggest discrepancies in the field is the cellular distribution of FcμR in mice (B cells vs. non-B cells). While several groups of investigators described the predicted functions of FcμR in non-B cell populations, their actual evidence for the surface expression of FcμR by myeloid, dendritic and T cells was rather weak (41, 43, 51, 52). Most of their functional results came from the comparative analysis in chimeras adoptively transferred by a mixture of Fcmr KO and WT BM cells or the direct comparison of cellular function between Fcmr KO and WT controls (43, 44, 46, 48). This was the reason why the phrase “functional relevant expression of FcμR” by non-B cells was used (52). Nevertheless, several functional outcomes in non-B cells from some Fcmr KO mice could be worthy of consideration because of the clear-cut differences compared to WT controls, even though they might be indirect or bystander effects. For example, mutant-M were resistant to the induction of myelin oligodendrocyte glycoprotein (MOG)-induced autoimmune encephalomyelitis (EAE). The authors initially considered that this resistance was not due to an intrinsic impairment of mutant Th1 and Th17 cell functions (see different observations by another group of investigators below), but rather to the immature and tolerogenic nature of mutant DCs, as characterized by their weak inflammatory responses and increased induction of Treg cells (44). Intriguingly, administration of a recombinant soluble FcμR fusion protein, which consisted of the human FcμR ectodomain and human IgG1 Fc (lacking complement binding activity) (FcμR EC/IgG Fc), into EAE-susceptible WT mice resulted in delaying or ameliorating their disease, depending on the time points of injection. While its mode of action was not discussed, it might be possible that since IgM anti-MOG antibody also participates in the demyelination process in EAE, the soluble FcμR EC/IgG Fc could simply act as a decoy receptor.
By contrast, results from recent single-cell RNA sequencing analysis along with complex algorithmic assessments and its functional annotation indicated that FcμR is one of the four critical regulators of Th17 pathogenicity in MOG-induced EAE (48). [The other three included Gpr65 (G protein-coupled receptor 65), Plzp (promyelocytic leukemia zinc finger transcriptional repressor of the Th2 master regulator Gata3) and Cd5l (CD5-like antigen, apoptosis inhibitor expressed by macrophages [AIM], or soluble protein α [Spα]). Astonishingly, CD5L/AIM/Spα is a glycoprotein of ~45 kD secreted by macrophages, supports their survival and was originally identified as an IgM binding protein (63–65). Two out of four regulators identified for Th17-mediated EAE were thus capable of binding to IgM, although CD5L/AIM/Spα was annotated as a regulator of lipid biosynthesis (66).] Th17 cells polarized ex vivo by differentiation conditions with TGFß+IL-6 or IL-1ß+IL-23+IL-6 from Fcmr KO mutant-M were found to secrete significantly less IL-17A and IL-10 than those from control WT mice (48). Mutant naive CD4 T cells exhibited lower FOXP3 levels during Treg cell differentiation upon TGFß stimulation in vitro. The authors considered that FcμR could be a negative regulator in a non-pathologic state but a promoter of pathogenicity (48), although it was difficult to understand its mechanisms. Given our findings that none of the sorted T cells with the phenotype of IL-17+, INFγ+, or IL-17+/IFNγ+ expressed FcμR transcripts, as determined by gene array analysis (25), it is hard to imagine how such a minor population of Th17 cells expresses functional FcμR, possibly at low levels, on their surface and plays a major regulatory role in the pathogenesis of EAE.
To explore the molecular basis for the resistance of Fcmr KO mice to EAE as well as for the reduction of IL-10 production by their Th17 cells, a computational epigenetic analysis was performed. Since Fcmr and Il0 genes are ~139 kb apart from each other on chromosome 1, we analyzed the data of the histone post-translational modification by chromatin immunoprecipitation and sequencing and the assay for transposonase-accessible chromatin (ATAC) sequencing available for resting and activated Treg cells at the Fcmr-Il10 locus (67). These included marks of acetylation of histone H3 at lysine 27 (H3K27ac) as a predictor of enhancer activity (68, 69), albeit not exclusively, and of ATAC as an indication of open chromatin (70). As shown in Figure 3, the H3K27ac marks are selectively observed in three loci, i.e., 3′ site of Fcmr, 5′ upstream of Il10 and Il10, in activated Treg cells. The ATAC and H3K27ac marks coincided, suggesting that these loci were in an opened chromatin status, hence transcription factors would be highly accessible to these loci. Remarkably, the H3K27ac marks in the Fcmr gene of activated Treg cells were restricted to its 3′ region, i.e., exon 5 (TM) to exon 8 (encoding CY tail and 3′ UTR) and were absent in exon 2 (encoding the Ig-like domain responsible for IgM-ligand binding), consistent with the lack of functional FcμR expression by T cells. This 3′ Fcmr-restricted H3K27ac mark was not observed with resting Treg cells, suggesting that the potential enhancer activity of 3′ Fcmr in Treg cells was dependent on cell activation. By contrast, the H3K27ac marks in the 5′ upstream of Il10 were observed irrespective of cell activation. Notably, several regions besides exons in the 3′ Fcmr were conserved in 40 other placental mammalian Fcmr genes as determined by phastCons (not shown). The above H3K27ac marks were not observed in early B-lineage cells, i.e., pro-B cells of either young or old mice. Collectively, these three loci [3′ site of Fcmr, 5′ upstream of Il10, and Il10] could be involved in enhancing IL-10 expression by Treg cells upon cellular activation potentially through a chromatin loop formation.
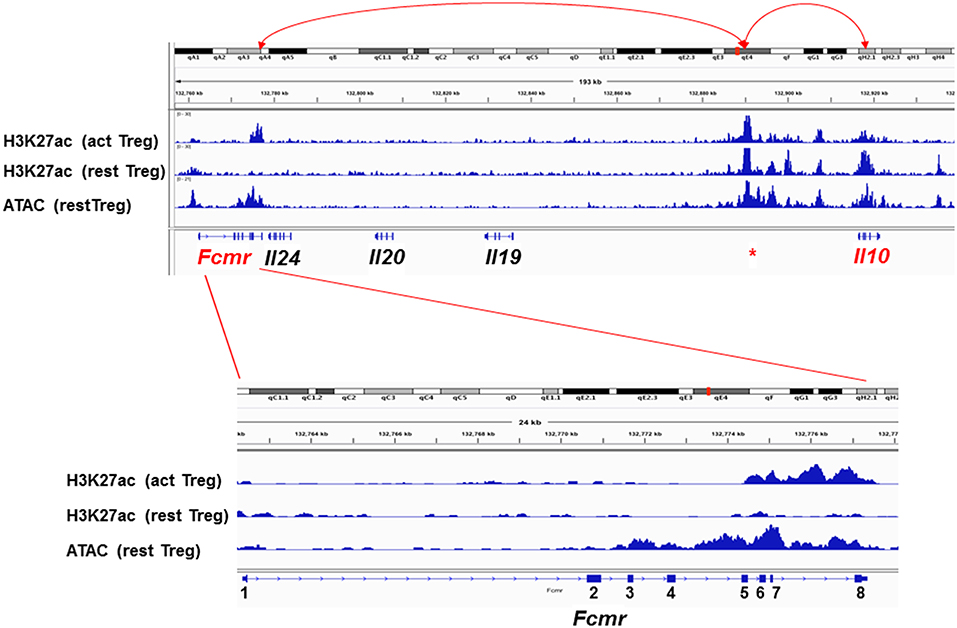
Figure 3. Epigenetic status of the Fcmr-Il10 locus in Treg cells. Top: Genomic locus (~180 kb) from Fcmr to Il10 is depicted along with the chromosome 1 site designation, distance, marks of the acetylation of histone H3 at lysine 27 (H3K27ac) in activated (act) or resting (rest) Treg cells, marks of the assay for transposonase-accessible chromatin (ATAC) and the exon (square) and intron (line) of indicated genes. Red * indicates the unique region with high H3K27ac marks in both activated and resting Treg cells and of ATAC at 5′ upstream of Il10 gene. Red arrow lines indicate potential association with the indicated loci by chromatin loop formation. Bottom: Enlarged illustration of Fcmr locus with coding exons numbered. 5′ and 3′ UTR regions are indicated by smaller squares.
While the above epigenetic results of the Fcmr-Il10 locus were derived from Treg cells, it remained to be elucidated whether a similar scenario was applicable for other cell types including Th17 cells. If so, Fcmr KO mutant-M, in which exons 2–8 were targeted, do not have this putative 3' Fcmr enhancer element for IL-10 in their genome, and this could account for the reduction of IL-10 production by Th17-polarizing cells (48). For Il17a, which is located at ~110 Mb upstream of Fcmr on chromosome 1, whether the 3′ Fcmr enhancer element is able to form such a long-range interaction with the Il17a promoter is an intriguing question. It is also unclear how absence of the 3′ Fcmr enhancer element contributes to the resistance to EAE in Fcmr KO mutant-M. Nevertheless, given the assumptions that in single-cell RNA sequencing analysis, most identified FcμR transcripts might be derived from its 3′ region and that only the resistance to EAE as the consequence of Fcmr-deficiency might be functionally annotated for FcμR, it is thus conceivable and very intriguing that FcμR could be one of the four important regulators of Th17 pathogenicity in EAE, despite the lack of expression of functional FcμR by such T cells (48). Collectively, some of the discrepancies observed in Fcmr KO mice could be attributed to differences in the exons disrupted.
In summary, the epigenetic analysis of Fcmr-Il10 locus reveals that three loci (3′ site of Fcmr, 5′ upstream of Il10, and Il10) may be involved in enhancing IL-10 expression by Treg cells upon cellular activation through chromatin loop formation. The epigenetic alteration selectively at the 3′ site of Fcmr may account for the functional abnormalities in non-B cell populations observed in certain Fcmr KO mice in conjunction with the exons targeted, even though functional FcμR is not expressed by such non-B cell populations.
FcμR in Central Deletion of Autoreactive B Cells Developing in Bone Marrow
The common feature among the different Fcmr KO mice is the propensity to produce autoantibodies of both IgM and IgG isotypes accompanied by increases in B-1 B cells, indicating an important regulatory role of FcμR in B cell development and central repertoire selection against those B cells expressing autoreactive BCRs. During B cell development in the BM, immature B cells are highly susceptible to deletion by BCR crosslinking. It has been estimated that ~90% of the newly generated BM B cells are deleted before entering the mature B cell compartment (71) and that approximately two thirds of the BM immature B cells in humans are self-reactive (72). During this development, the FcμR expression becomes detectable at the transition from BCR-non-expressing pre-B cells to BCR-expressing immature B cells. In three strains of mutant-O, -B and -L2 (29, 32, 35, 50), however, the sizes of the pro-, pre- and immature B cell compartments showed no alterations, when compared with WT control mice. Only one mutant-M had reduced pro-, pre-, and immature B cell compartments (42). Changes in sizes of BM B-lineage compartment might not become visible in such analyses, because such changes in the number of BCR+ B cells might occur, as the immature B cells exit the BM. Furthermore, the peripheral compartments of immature and mature B cells may fill by homeostasis to unaltered sizes, though with either non-autoreactive or autoreactive B cells.
It is noteworthy that μs KO mice, which are deficient for secreted pentameric IgM, the ligand of FcμR, have significantly altered B cell development at the transition from pre-B to immature B cells (42). This alteration of early B cell development, including the inability to centrally delete autoreactive B cells, can be corrected by administration of natural IgM (56). Therefore, ligation of the FcμR by its ligand, pentameric natural, polyclonal IgM in vivo contributes to the negative selection of autoreactive B cells. It remains to be elucidated in this experimental setting whether immature B cells in BM are the prime target of this correction. If so, it suggests that the provision of pentameric, natural, polyclonal IgM binding to FcμR on immature B cells allows cis-crosslinking of autoreactive BCRs with autoantigen presented by pentameric IgM ligated to FcμR (Figure 4). This crosslinking would be expected to connect signaling from the BCR (e.g., via PI3 kinase) (73) with signaling from FcμR. If FcμR-signaling would downregulate PI3 kinase activity, this could lead to upregulation of FOXO1, which, in turn, could upregulate RAG1/2 expression (74, 75). In this way the immature B cells could continue editing VL-JL-rearranged light chain (LC) gene loci (76, 77) to change the autoreactivity of the BCR. Any loss of autoreactivity would abolish cis-crosslinking with autoantigen-bound natural IgM/FcμR, thus terminate RAG expression and allow immature B cells to leave the BM.
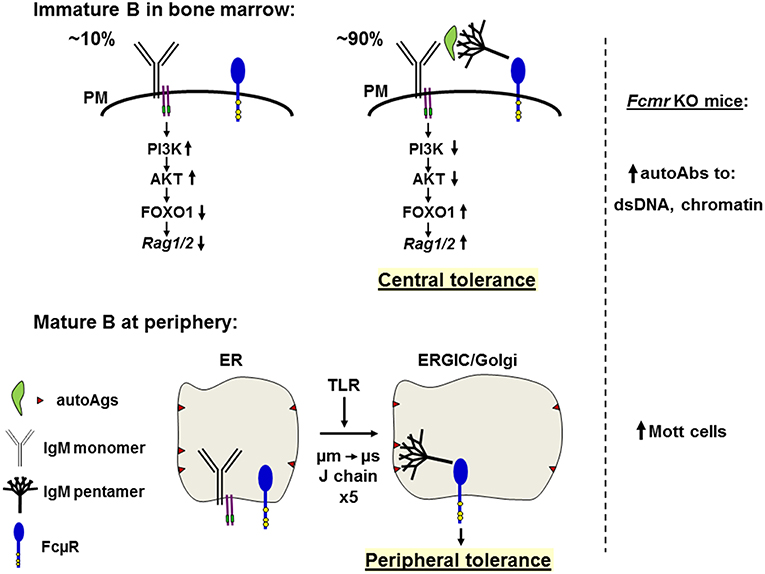
Figure 4. Hypothetical model of the role of FcμR in central deletion and B cell repertoire selection. Top: In the bone marrow, only small populations (~10%) of the newly generated B cells bearing monomeric IgM (Y shape with Igα/Igß, two purple lines carrying green ITAM) on the plasma membrane (PM) are survived by ligand-independent (or tonic) signals through activation of the PI3K-AKT pathway and suppression of the FOXO1-mediated Rag1/2 activity (left). By contrast, ~90% of the newly generated immature B cells have variable binding affinities for autoantigens (green leaf shape) and are subjected to negative selection by receptor editing or apoptosis (right). Autoantigens opsonized with natural pentameric IgM (broom shape) simultaneously bind the corresponding BCR and FcμR (blue tennis racket shape with three conserved Tyr residues in yellow). The resultant cross-linkage of BCR and FcμR on immature autoreactive B cells may inhibit the BCR-mediated PI3K-AKT pathway, resulting in relief of the AKT-mediated suppression of FOXO1 and leading to activation of Rag1/2 and receptor editing, thereby contributing to negative selection or central tolerance. In the absence of FcμR autoantibodies against dsDNA or chromatin are increased. Bottom: In peripheral lymphoid tissues, incompletely edited B cells express IgM BCR with self-reactivity to membrane components (brown triangles) present on ER membranes, but no interaction of monomeric IgM with the corresponding antigens occurs due to its low affinity. When cells receive signals from TLR, a switch from μm to μs exon usage occurs along with the synthesis of J chain during the translocation from ER to the ER Golgi intermediate compartment (ERGIC) or the Golgi and the resultant pentameric IgM is contained inside the vesicles where it binds membrane components via the Fab regions and its Fc portion binds FcμR. This cis engagement of self-antigen/secreted IgM/FcμR within the vesicles prevents further development of such autoreactive B cells, thereby contributing to peripheral tolerance. In the absence of FcμR, Mott cells containing intracellular Ig inclusion bodies are increased.
FcμR and Mott Cell Formation in the Control of Autoimmunity of B Cells
Another finding is the marked increase in Mott cells in mutant-O, even though it has only been described by our analysis (45). We propose that the FcμR may control autoantibody production by formation of Mott cells in the scenario described below. Mott cells are a variant form of plasma cells containing Ig inclusion bodies (called Russell bodies) that accumulate in dilated rough endoplasmic reticulum (ER). Mott cells are rarely observed in normal lymphoid tissues, but are found in various pathological conditions, such as Ig-associated neoplasms, chronic inflammatory diseases and autoimmune disorders (78–81). Several mechanisms for formation of Ig inclusions or for the defect in Ig secretion have been suggested, including (i) structural alteration of Ig HCs preventing their appropriate processing, (ii) impairment of Ig LCs in preventing Ig HC aggregation, and (iii) inability to degrade or to export Ig, leading to its aggregation. However, the most relevant mechanism associated with mutant-O seems to be that the Ig becomes stuck in the exocytotic pathway due to its autoreactivity with intracellular membrane components. Several precedents support this idea. (i) Two clonally unrelated IgM Mott cell hybridomas utilize germline Ig variable gene segments and have no obvious structural defects, suggesting their B-1 B cell origin (79). (ii) Ig inclusions are not generated when the Mott Ig μ HC or κ LC is by itself or is associated with a heterologous κ LC or μ HC, respectively. The inclusion body formation is only reconstituted when Mott VH and Mott Vκ genes are expressed with an IgM, but not IgG1, constant region, suggesting that both specificity and isotype are critical for Mott cell formation. (iii) LPS or IL-5 stimulation of sorted B-1 B cells from autoimmune mice (NZB/W F1) generates Mott cells ex vivo at a frequency of ~50 times higher than conventional B-2 B cells (81). (iv) In studies of autoantibody transgenic mice, incompletely edited B cells express multi-reactive IgM that accumulates in the Golgi and is released or detached from the membrane as insoluble amyloid-like immune complexes termed spherons reaching up to ~2 μm in diameter (82, 83).
Given these precedents and the preferential cis engagement of FcμR, the following scenario would account for the high incidence of Mott cells in the absence of FcμR. Incompletely edited B cells migrate into peripheral lymphoid tissues and express membrane-bound IgM with self-reactivity to intracellular membrane components (e.g., glycans). The interaction of the monomeric IgM with self-antigens in the ER must be of low affinity. However, when cells receive certain signals such as from TLR4 to facilitate a switch in the usage of μm to μs exon along with J-chain synthesis during transition to the ER-Golgi intermediate compartment (ERGIC) or the Golgi, the resultant pentameric IgM is contained inside the ERGIC/Golgi vesicles and binds a self-antigen on intracellular membranes via its Fab region and simultaneously the FcμR via its Fc portion. This cis engagement may prevent further differentiation of such autoreactive B cells, thereby contributing to the peripheral tolerance to self-antigens located on intracellular membranes (Figure 4). Based on this hypothesis, Mott cell IgMs in Fcmr KO mice are anticipated to have autoantibody activity to intracellular membrane components.
Instead of IgM-opsonized self-antigens, it may be equally possible that DNA and DNA-associated autoantigens or RNA and RNA-associated autoantigens are recognized by the respective IgM on BM immature B cells and delivered to an endosomal or lysosomal compartment where TLR9 or TLR7 binds the corresponding ligand-containing IgM BCR. The resultant oligomerized IgM BCRs are transported via a retrograde route to the TGN where FcμR may bind the Cμ3/Cμ4 domain of the oligomeric IgM BCR. In summary, based on the findings of enhanced autoantibody production in all Fcmr KO mice and Mott cell formation in our mutant mice as well as the cis engagement of FcμR, we propose a model for how FcμR on B cells plays a regulatory role in central and peripheral tolerance.
FcμR in Diseases
The association of FcμR with human CLL has long been suggested, dating back to studies showing that CLL B cells could form rosettes with ox erythrocytes coated with IgM antibody (84, 85). By flow cytometric assays CLL B cells also exhibited specific IgM binding (57, 86). Subsequently, several investigators showed enhanced TOSO/FCMR gene expression in CLL and initially considered that this enhancement would contribute to increased resistance of CLL cells to apoptosis (87, 88). We also examined the surface expression of FcμR by B and T cells in CLL using receptor specific mAbs by flow cytometry. CLL B cells (CD19+/CD5+) expressed significantly much higher levels of surface FcμR than B cells from healthy donors. This enhancement was more evident in Ig HC variable region (IGHV)-mutated, better prognostic, CD38− or early Rai-stage CLL than in IGHV-unmutated, poor prognostic, CD38+ or advanced Rai-stage CLL (89). Intriguingly, surface FcμR levels were also significantly elevated in non-CLL B cells (CD19+/CD5−) and T cells (CD19−/CD5+), especially in patients with IGHV-mutated CLL, when compared with the corresponding populations in healthy individuals. This increase in FcμR expression on T cells in CLL was unique, because normal human T cells activated ex vivo with anti-CD3 mAb or PMA down-modulated surface FcμR, whereas normal B cells activated with anti-μ mAb or PMA up-regulated surface FcμR (20). Regarding the enhanced surface expression of FcμR on CLL B cells, CLL-derived BCRs, unlike those from other B cell malignancies, have been shown to ligate each other via interactions between Ig HC CDR3 of one BCR and the framework region 2 of another BCR irrespective of their IGHV mutation status, thereby providing antigen-independent cell-autonomous signaling (90, 91). This antigen-independent self-ligation of BCR on CLL cells could account for enhanced surface expression of FcμR as well as for the well-known phenomenon of reduced levels of surface IgM and IgD on CLL cells. It remains unclear, however, why surface FcμR levels were also elevated on non-CLL B and T cells in IGHV-mutated CLL patients.
Another remarkable finding was the marked elevation of serum titers of FcμR in CLL patients but not in healthy individuals (89). [One exception was an individual who was found 2 years later to have high serum autoantibody titers against dsDNA.] Detection of the serum FcμR was accomplished by sandwich ELISA using two different receptor-specific mAbs. It was resolved as an ~40 kD protein, distinct from the ~60 kD cell surface FcμR and found by proteomic analysis as a soluble form of the receptor (solFcμR), which was encoded by an alternative spliced FcμR transcript resulting from the direct splicing of exon 4 (stalk 2) to exon 6 (CY1), skipping exon 5 (TM). This splicing event resulted in a reading frame shift in exon 6 and generated a novel 70 aa hydrophilic carboxyl tail, thereby confirming the source of the solFcμR. The functional role of solFcμR in CLL and possibly in autoimmune disorders as observed with aforementioned exceptional control individual remains to be elucidated. In this regard it is noteworthy that administration of another form of solFcμR (FcμR EC/IgG Fc) into EAE-susceptible mice ameliorates the disease (44). Collectively, both membrane-bound and soluble forms of FcμR are elevated in patients with CLL.
Since among leukemia/lymphomas CLL uniquely expresses high levels of FcμR on their surface, two types of immunotherapy targeting for the receptor have thus been developed for CLL cells. One is an immunotoxin-coupled IgM Fc (Cμ2-Cμ4) and the other is chimeric antigen receptor-modified T cells using a single chain fragment-containing the variable region of an anti-FcμR mAb (6B10) (92, 93). In both cases, patient CLL B cells appear to be selectively eliminated in vitro without affecting the non-leukemic B and T cells. Apart from FcμR in hematologic malignancy, FCMR-deficiency has not yet been identified, but based on the data from Fcmr KO mice it may belong to hyper-IgM syndrome. Since FcμR is expressed by B, T, and NK cells in humans, the phenotypic abnormalities of FCMR deficiency in affected individuals are predicted to be more complex than those in Fcmr KO mice. In patients with selective IgM immunodeficiency, we initially predicted that surface FcμR levels might be high because of lack of ligand-induced down-modulation. Contrary to this assumption, cell surface FcμR levels on a particular circulating B cell subset with a MZ phenotype (IgM+/IgD+/CD27+) in such patients were significantly diminished as compared to age-matched controls, but the molecular basis for this reduction remains to be elucidated (94).
In summary, enhanced levels of both the membrane-bound and secretory forms of FcμR are evident in patients with CLL, possibly as the consequence of antigen-independent autonomous self-ligation of BCR on CLL cells.
Epilogue
It has been known for many years that passive administration of IgM antibody enhances the subsequent antibody responses to antigenic challenge, whereas passive administration of IgG antibody suppresses the response. Complement activation, but not its lytic activity, has so far been implicated as a mechanism for this IgM-mediated enhancement, and the inhibitory FcγR is involved in IgG-mediated suppression (95, 96). The existence of FcμR on a variety of cell types has also been suggested for nearly 50 years by many investigators including us, but the FcμR cDNA was identified just 10 years ago by a functional cloning strategy (20). However, since FcμR turned out to be identical to the Toso cDNA, which was also previously cloned by functional strategy as a potent inhibitor of Fas-mediated apoptosis, there have been lively debates regarding the real function of this receptor, IgM Fc binding vs. Fas-apoptosis inhibition. While we have now a general consensus that this is an authentic FcμR, there have been clear discrepancies in the phenotypic abnormalities reported in five different Fcmr KO mice. In this article, we have discussed potential molecular mechanisms underlying some of these discrepancies. One of the remarkable outcomes of our analysis is the finding of restricted H3K27ac and ATAC marks to the 3' Fcmr in activated, but not resting, Treg cells and could account for some puzzles in T cell function described in certain Fcmr KO mice. Given the fact that all Fcmr KO mice are prone to produce autoantibodies accompanied by increased B-1 B cells, we introduce our hypothetical model for how FcμR controls autoantibody production. We see that FcμR has a very important role in immature B cell in the BM to control against the development of autoreactivity in B cell repertoire. We hope that this short article may help to resolve many still existing puzzles and will open new avenues of investigation.
Author Contributions
PKJ performed the comparative analysis (Table 1). KH analyzed the FcμR ligand binding property (Figure 2) and the phenotype of mutant-O. NO and SS conducted the epigenetic analysis (Figure 3). AR intellectually contributed. HK and FM made the rest of figures (Figures 1, 4) and wrote the paper. All authors listed approved for publication. PKJ was a scholar of the Alexander von Humboldt Foundation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Peter Burrows for critical reading and Ms. Beate Löhr for literature support.
References
1. Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. (1998) 160:4776–87.
2. Ehrenstein MR, O'Keefe TL, Davies SL, Neuberger MS. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc. Natl. Acad. Sci. USA. (1998) 95:10089–93. doi: 10.1073/pnas.95.17.10089
3. Boes M, Prodeus AP, Schmidt T, Carroll MC, Chen J. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J. Exp. Med. (1998) 188:2381–6. doi: 10.1084/jem.188.12.2381
4. Baumgarth N, Herman OC, Jager GC, Brown LE, Herzenberg LA, Chen J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. (2000) 192:271–80. doi: 10.1084/jem.192.2.271
5. Subramaniam KS, Datta K, Quintero E, Manix C, Marks MS, Pirofski LA. The absence of serum IgM enhances the susceptibility of mice to pulmonary challenge with Cryptococcus neoformans. J. Immunol. (2010) 184:5755–67. doi: 10.4049/jimmunol.0901638
6. Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. USA. (2000) 97:1184–9. doi: 10.1073/pnas.97.3.1184
7. Ehrenstein MR, Cook HT, Neuberger MS. Deficiency in serum immunoglobulin (Ig) M predisposes to development of IgG autoantibodies. J. Exp. Med. (2000) 191:1253–8. doi: 10.1084/jem.191.7.1253
8. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat. Rev. Immunol. (2010) 10:778–86. doi: 10.1038/nri2849
9. Ravetch JV, Kinet JP. Fc receptors. Annu. Rev. Immunol. (1991) 9:457–92. doi: 10.1146/annurev.iy.09.040191.002325
10. Daëron M. Fc receptor biology. Annu. Rev. Immunol. (1997) 15:203–34. doi: 10.1146/annurev.immunol.15.1.203
11. Turner H, Kinet JP. Signalling through the high-affinity IgE receptor FceRI. Nature. (1999) 402:B24–30. doi: 10.1038/35037021
12. Ravetch JV, Bolland S. IgG Fc receptors. Annu. Rev. Immunol. (2001) 19:275–90. doi: 10.1146/annurev.immunol.19.1.275
13. Conrad DH. FcεRII/CD23: the low affinity receptor for IgE. Annu. Rev. Immunol. (1990) 8:623–45. doi: 10.1146/annurev.iy.08.040190.003203
14. Monteiro RC, Van De Winkel JG. IgA Fc receptors. Annu. Rev. Immunol. (2003) 21:177–204. doi: 10.1146/annurev.immunol.21.120601.141011
15. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgRIV: a novel FcR with distinct IgG subclass specificity. Immunity. (2005) 23:41–51. doi: 10.1016/j.immuni.2005.05.010
16. Kaetzel CS, Mostov K. Immunoglobulin transport and the polymeric immunoglobulin receptor. In: Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, Mayer L, editors. Mucosal Immunology. Volume 1. San Diego, CA: Elsevier Academic Press (2005). p. 211–250. doi: 10.1016/B978-012491543-5/50016-4
17. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat. Rev. Immunol. (2007) 7:715–25. doi: 10.1038/nri2155
18. Ravetch JV, Nimmerjahn F. Fc receptors and their role in immune regulation and inflammation. In: Paul WE, editor. Fundamental Immunology. Philadelphia, PA: Lippincott Williams & Wilkins. (2008) p. 684–705.
19. Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat. Rev. Immunol. (2013) 13:176–89. doi: 10.1038/nri3401
20. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, et al. Identity of the elusive IgM Fc receptor (FcμR) in humans. J. Exp. Med. (2009) 206:2779–93. doi: 10.1084/jem.20091107
21. Klimovich VB. IgM and its receptors: structural and functional aspects. Biochemistry. (2011) 76:534–49. doi: 10.1134/S0006297911050038
22. Kubagawa H, Oka S, Kubagawa Y, Torii I, Takayama E, Kang DW, et al. The long elusive IgM Fc receptor, FcmR. J. Clin. Immunol. (2014) 34(Suppl. 1):S35–45. doi: 10.1007/s10875-014-0022-7
23. Kubagawa H, Kubagawa Y, Jones D, Nasti TH, Walter MR, Honjo K. The old but new IgM Fc receptor (FcmR). Curr. Top. Microbiol. Immunol. (2014) 382:3–28. doi: 10.1007/978-3-319-07911-0_1
24. Wang H, Coligan JE, Morse HC III. Emerging functions of natural IgM and its Fc receptor FCMR in immune homeostasis. Front. Immunol. (2016) 7:99. doi: 10.3389/fimmu.2016.00099
25. Kubagawa H, Skopnik CM, Zimmermann J, Durek P, Chang HD, Yoo E, et al. Authentic IgM Fc Receptor (FcmR). Curr. Top. Microbiol. Immunol. (2017) 408:25–45. doi: 10.1007/82_2017_23
26. Engels N, Wienands J. The signaling tool box for tyrosine-based costimulation of lymphocytes. Curr. Opin. Immunol. (2011) 23:324–9. doi: 10.1016/j.coi.2011.01.005
27. Vire B, David A, Wiestner A. TOSO, the Fcμ receptor, is highly expressed on chronic lymphocytic leukemia B cells, internalizes upon IgM binding, shuttles to the lysosome, and is downregulated in response to TLR activation. J. Immunol. (2011) 187:4040–50. doi: 10.4049/jimmunol.1100532
28. Honjo K, Kubagawa Y, Kearney JF, Kubagawa H. Unique ligand-binding property of the human IgM Fc receptor. J. Immunol. (2015) 194:1975–82. doi: 10.4049/jimmunol.1401866
29. Honjo K, Kubagawa Y, Jones DM, Dizon B, Zhu Z, Ohno H, et al. Altered Ig levels and antibody responses in mice deficient for the Fc receptor for IgM (FcmR). Proc. Natl. Acad. Sci. USA. (2012) 109:15882–7. doi: 10.1073/pnas.1206567109
30. Shima H, Takatsu H, Fukuda S, Ohmae M, Hase K, Kubagawa H, et al. Identification of TOSO/FAIM3 as an Fc receptor for IgM. Int. Immunol. (2010) 22:149–56. doi: 10.1093/intimm/dxp121
31. Murakami Y, Narayanan S, Su S, Childs R, Krzewski K, Borrego F, et al. Toso, a functional IgM receptor, is regulated by IL-2 in T and NK cells. J. Immunol. (2012) 189:587–97. doi: 10.4049/jimmunol.1200840
32. Ouchida R, Mori H, Hase K, Takatsu H, Kurosaki T, Tokuhisa T, et al. Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc. Natl. Acad. Sci. USA. (2012) E2699–706. doi: 10.1073/pnas.1210706109
33. Akula S, Mohammadamin S, Hellman L. Fc receptors for immunoglobulins and their appearance during vertebrate evolution. PLoS ONE. (2014) 9:e96903. doi: 10.1371/journal.pone.0096903
34. Kikuno K, Kang DW, Tahara K, Torii I, Kubagawa HM, Ho KJ, et al. Unusual biochemical features and follicular dendritic cell expression of human Fca/m receptor. Eur. J. Immunol. (2007) 37:3540–50. doi: 10.1002/eji.200737655
35. Nguyen TT, Klasener K, Zurn C, Castillo PA, Brust-Mascher I, Imai DM, et al. The IgM receptor FcmR limits tonic BCR signaling by regulating expression of the IgM BCR. Nat. Immunol. (2017) 18:321–33. doi: 10.1038/ni.3677
36. Hitoshi Y, Lorens J, Kitada SI, Fisher J, LaBarge M, Ring HZ, et al. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. (1998) 8:461–71. doi: 10.1016/S1074-7613(00)80551-8
37. Kubagawa H, Carroll MC, Jacob CO, Lang KS, Lee KH, Mak T, et al. Nomenclature of Toso, Fas apoptosis inhibitory molecule 3, and IgM FcR. J. Immunol. (2015) 194:4055–7. doi: 10.4049/jimmunol.1500222
38. Ravetch JV, Lanier LL. Immune inhibitory receptors. Science. (2000) 290:84–9. doi: 10.1126/science.290.5489.84
39. Bléry M, Kubagawa H, Chen CC, Vely F, Cooper MD, Vivier E. The paired Ig-like receptor PIR-B is an inhibitory receptor that recruits the protein-tyrosine phosphatase SHP-1. Proc. Natl. Acad. Sci. USA. (1998) 95:2446–51. doi: 10.1073/pnas.95.5.2446
40. Song Y, Jacob CO. The mouse cell surface protein TOSO regulates Fas/Fas ligand-induced apoptosis through its binding to Fas-associated death domain. J. Biol. Chem. (2005) 280:9618–26. doi: 10.1074/jbc.M413609200
41. Nguyen XH, Lang PA, Lang KS, Adam D, Fattakhova G, Foger N, et al. Toso regulates the balance between apoptotic and nonapoptotic death receptor signaling by facilitating RIP1 ubiquitination. Blood. (2011) 118:598–608. doi: 10.1182/blood-2010-10-313643
42. Choi SC, Wang H, Tian L, Murakami Y, Shin DM, Borrego F, et al. Mouse IgM Fc receptor, FCMR, promotes B cell development and modulates antigen-driven immune responses. J. Immunol. (2013) 190:987–96. doi: 10.4049/jimmunol.1202227
43. Lang KS, Lang PA, Meryk A, Pandyra AA, Boucher LM, Pozdeev VI, et al. Involvement of Toso in activation of monocytes, macrophages, and granulocytes. Proc. Natl. Acad. Sci. USA. (2013) 110:2593–8. doi: 10.1073/pnas.1222264110
44. Brenner D, Brustle A, Lin GH, Lang PA, Duncan GS, Knobbe-Thomsen CB, et al. Toso controls encephalitogenic immune responses by dendritic cells and regulatory T cells. Proc. Natl. Acad. Sci. USA. (2014) 111:1060–5. doi: 10.1073/pnas.1323166111
45. Honjo H, Kubagawa Y, Suzuki Y, Takagi M, Ohno H, Bucy RP, et al. Enhanced autoantibody and Mott cell formation in FcmR-decient autoimmune mice. Int. Immunol. (2014) 26:659–72. doi: 10.1093/intimm/dxu070
46. Lang PA, Meryk A, Pandyra AA, Brenner D, Brustle A, Xu HC, et al. Toso regulates differentiation and activation of inflammatory dendritic cells during persistence-prone virus infection. Cell Death Differ. (2015) 22:164–73. doi: 10.1038/cdd.2014.138
47. Ouchida R, Lu Q, Liu J, Li Y, Chu Y, Tsubata T, et al. FcmR interacts and cooperates with the B cell receptor to promote B cell survival. J. Immunol. (2015) 194:3096–101. doi: 10.4049/jimmunol.1402352
48. Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. (2015) 163:1400–12. doi: 10.1016/j.cell.2015.11.009
49. Liu J, Zhu H, Qian J, Xiong E, Zhang L, Wang YQ, et al. Fcm receptor promotes the survival and activation of marginal zone B cells and protects mice against bacterial sepsis. Front. Immunol. (2018) 9:160. doi: 10.3389/fimmu.2018.00160
50. Yu J, Duong VHH, Westphal K, Westphal A, Suwandi A, Grassl GA, et al. Surface receptor Toso controls B cell-mediated regulation of T cell immunity. J. Clin. Invest. (2018) 128:1820–36. doi: 10.1172/JCI97280
51. Honjo K, Kubagawa Y, Kubagawa H. Is Toso/IgM Fc receptor (FcmR) expressed by innate immune cells? Proc. Natl. Acad. Sci. USA. (2013) 110:E2540–1. doi: 10.1073/pnas.1304904110
52. Lang KS, Lang PA, Meryk A, Pandyra AA, Merches K, Lee KH, et al. Reply to Honjo et al.: functional relevant expression of Toso on granulocytes. Proc. Natl. Acad. Sci. U.S.A. (2013) 110:E2542–3. doi: 10.1073/pnas.1306422110
53. Honjo K, Kubagawa Y, Kubagawa H. Is Toso an antiapoptotic protein or an Fc receptor for IgM? Blood. (2012) 119:1789–90. doi: 10.1182/blood-2011-09-380782
54. Nguyen XH, Fattakhova G, Lang PA, Lang KS, Adam D, Foger N, et al. Antiapoptotic function of Toso (Faim3) in death receptor signaling. Blood. (2012) 119:1790–1. doi: 10.1182/blood-2011-11-386839
55. Lapke N, Tartz S, Lee KH, Jacobs T. The application of anti-Toso antibody enhances CD8(+) T cell responses in experimental malaria vaccination and disease. Vaccine. (2015) 33:6763–70. doi: 10.1016/j.vaccine.2015.10.065
56. Nguyen TT, Elsner RA, Baumgarth N. Natural IgM prevents autoimmunity by enforcing B cell central tolerance induction. J. Immunol. (2015) 194:1489–502. doi: 10.4049/jimmunol.1401880
57. Ohno T, Kubagawa H, Sanders SK, Cooper MD. Biochemical nature of an Fcm receptor on human B-lineage cells. J. Exp. Med. (1990) 172:1165–75. doi: 10.1084/jem.172.4.1165
58. Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. (1988) 334:676–82. doi: 10.1038/334676a0
59. Cyster JG, Hartley SB, Goodnow CC. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature. (1994) 371:389–95. doi: 10.1038/371389a0
60. Bonifacino JS, Rojas R. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. (2006) 7:568–79. doi: 10.1038/nrm1985
61. Sato S, Steeber DA, Tedder TF. The CD19 signal transduction molecule is a response regulator of B-lymphocyte differentiation. Proc. Natl. Acad. Sci. USA. (1995) 92:11558–62. doi: 10.1073/pnas.92.25.11558
62. Tsitsikov EN, Gutierrez-Ramos JC, Geha RS. Impaired CD19 expression and signaling, enhanced antibody response to type II T independent antigen and reduction of B-1 cells in CD81-deficient mice. Proc. Natl. Acad. Sci. USA. (1997) 94:10844–9. doi: 10.1073/pnas.94.20.10844
63. Tissot JD, Sanchez JC, Vuadens F, Scherl A, Schifferli JA, Hochstrasser DF, et al. IgM are associated to Spa (CD5 antigen-like). Electrophoresis. (2002) 23:1203–6. doi: 10.1002/1522-2683(200204)23:7/8<1203::AID-ELPS1203>3.0.CO;2-1
64. Martinez VG, Moestrup SK, Holmskov U, Mollenhauer J, Lozano F. The conserved scavenger receptor cysteine-rich superfamily in therapy and diagnosis. Pharmacol.Rev. (2011) 63:967–1000. doi: 10.1124/pr.111.004523
65. Miyazaki T, Kurokawa J, Arai S. AIMing at metabolic syndrome. - towards the development of novel therapies for metabolic diseases via apoptosis inhibitor of macrophage (AIM). Circ. J. (2011) 75:2522–31. doi: 10.1253/circj.CJ-11-0891
66. Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, et al. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell. (2015) 163:1413–27. doi: 10.1016/j.cell.2015.10.068
67. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of regulatory T cell development by Satb1-dependent super-enhancer establishment. Nat. Immunol. (2017) 18:173–83. doi: 10.1038/ni.3646
68. Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA. (2010) 107:21931–6. doi: 10.1073/pnas.1016071107
69. Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. (2011) 470:279–83. doi: 10.1038/nature09692
70. Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. (2014) 345:943–9. doi: 10.1126/science.1256271
71. Melchers F. Checkpoints that control B cell development. J. Clin. Invest. (2015) 125:2203–10. doi: 10.1172/JCI78083
72. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. (2003) 301:1374–7. doi: 10.1126/science.1086907
73. Srinivasan L, Sasaki Y, Calado DP, Zhang B, Paik JH, DePinho RA, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. (2009) 139:573–86. doi: 10.1016/j.cell.2009.08.041
74. Ochodnicka-Mackovicova K, Bahjat M, Maas C, van der Veen A, Bloedjes TA, de der Veen AM, et al. The DNA damage response regulates RAG1/2 expression in Pre-B cells through ATM-FOXO1 signaling. J. Immunol. (2016) 197:2918–29. doi: 10.4049/jimmunol.1501989
75. Benhamou D, Labi V, Novak R, Dai I, Shafir-Alon S, Weiss A, et al. A c-Myc/miR17-92/Pten axis controls PI3K-mediated positive and negative selection in B cell development and reconstitutes CD19 deficiency. Cell Rep. (2016) 16:419–31. doi: 10.1016/j.celrep.2016.05.084
76. Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J. Exp. Med. (1993) 177:999–1008. doi: 10.1084/jem.177.4.999
77. Nemazee D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. (2017) 17:281–94. doi: 10.1038/nri.2017.19
78. Shultz LD, Coman DR, Lyons BL, Sidman CL, Taylor S. Development of plasmacytoid cells with Russell bodies in autoimmune “viable motheaten” mice. Am. J. Pathol. (1987) 127:38–50.
79. Tarlinton D, Forster I, Rajewsky K. An explanation for the defect in secretion of IgM Mott cells and their predominant occurrence in the Ly-1 B cell compartment. Eur. J. Immunol. (1992) 22:531–9. doi: 10.1002/eji.1830220236
80. Jäck HM, Beck-Engeser G, Sloan B, Wong ML, Wabl M. A different sort of mott cell. Proc. Natl. Acad. Sci. USA. (1992) 89:11688–91. doi: 10.1073/pnas.89.24.11688
81. Jiang Y, Hirose S, Hamano Y, Kodera S, Tsurui H, Abe M, et al. Mapping of a gene for the increased susceptibility of B1 cells to Mott cell formation in murine autoimmune disease. J. Immunol. (1997) 158:992–7.
82. Khan SN, Cox JV, Nishimoto SK, Chen C, Fritzler MJ, Hendershot LM, et al. Intra-Golgi formation of IgM-glycosaminoglycan complexes promotes Ig deposition. J. Immunol. (2011) 187:3198–207. doi: 10.4049/jimmunol.1101336
83. Radic M, Weigert MG, Khan SN, Han J, Kalinina O, Luning Prak ET. Antibodies that bind complex glycosaminoglycans accumulate in the Golgi. Proc. Natl. Acad. Sci. USA. (2013) 110:11958–63. doi: 10.1073/pnas.1308620110
84. Pichler WJ, Knapp W. Receptors for IgM-coated erythrocytes on chronic lymphatic leukemia cells. J. Immunol. (1977) 118:1010–5.
85. Ferrarini M, Hoffman T, Fu SM, Winchester R, Kunkel HG. Receptors for IgM on certain human B lymphocytes. J. Immunol. (1977) 119:1525–9.
86. Sanders SK, Kubagawa H, Suzuki T, Butler JL, Cooper MD. IgM binding protein expressed by activated B cells. J. Immunol. (1987) 139:188–93.
87. Proto-Siqueira R, Panepucci RA, Careta FP, Lee A, Clear A, Morris K, et al. SAGE analysis demonstrates increased expression of TOSO contributing to Fas-mediated resistance in CLL. Blood. (2008) 112:394–7. doi: 10.1182/blood-2007-11-124065
88. Pallasch CP, Schulz A, Kutsch N, Schwamb J, Hagist S, Kashkar H, et al. Overexpression of TOSO in CLL is triggered by B-cell receptor signaling and associated with progressive disease. Blood. (2008) 112:4213–9. doi: 10.1182/blood-2008-05-157255
89. Li FJ, Kubagawa Y, McCollum MK, Wilson L, Motohashi T, Bertoli LF, et al. Enhanced levels of both membrane-bound and soluble forms of IgM Fc receptor (FcmR) in patients with chronic lymphocytic leukemia. Blood. (2011) 118:4902–9. doi: 10.1182/blood-2011-04-350793
90. Duhren-von MM, Ubelhart R, Schneider D, Wossning T, Bach MP, Buchner M, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. (2012) 489:309–12. doi: 10.1038/nature11309
91. Minici C, Gounari M, Ubelhart R, Scarfo L, Duhren-von Minden M, Schneider D, et al. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat. Commun. (2017) 8:15746. doi: 10.1038/ncomms15746
92. Vire B, Skarzynski M, Thomas JD, Nelson CG, David A, Aue G, et al. Harnessing the Fcm receptor for potent and selective cytotoxic therapy of chronic lymphocytic leukemia. Cancer Res. (2014) 74:7510–20. doi: 10.1158/0008-5472.CAN-14-2030
93. Faitschuk E, Hombach AA, Frenzel LP, Wendtner CM, Abken H. Chimeric antigen receptor T cells targeting Fcμ receptor selectively eliminate CLL cells while sparing healthy B cells. Blood. (2016) 128:1711–22. doi: 10.1182/blood-2016-01-692046
94. Gupta S, Agrawal S, Gollapudi S, Kubagawa H. FcμR in human B cell subsets in primary selective IgM deficiency, and regulation of FcμR and production of natural IgM antibodies by IGIV. Hum. Immunol. (2016) 77:1194–201. doi: 10.1016/j.humimm.2016.10.003
95. Henry C, Jerne NK. Competition of 19S and 7S antigen receptors in the regulation of the primary immune response. J. Exp. Med. (1968) 128:133–52. doi: 10.1084/jem.128.1.133
Keywords: FcμR, autoantibody, natural IgM, tolerance, Mott cell, epigenetics
Citation: Kubagawa H, Honjo K, Ohkura N, Sakaguchi S, Radbruch A, Melchers F and Jani PK (2019) Functional Roles of the IgM Fc Receptor in the Immune System. Front. Immunol. 10:945. doi: 10.3389/fimmu.2019.00945
Received: 22 January 2019; Accepted: 12 April 2019;
Published: 03 May 2019.
Edited by:
Gestur Vidarsson, Sanquin Research, NetherlandsReviewed by:
Kristi Baker, University of Alberta, CanadaJeanette Leusen, University Medical Center Utrecht, Netherlands
Copyright © 2019 Kubagawa, Honjo, Ohkura, Sakaguchi, Radbruch, Melchers and Jani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fritz Melchers, ZnJpdHoubWVsY2hlcnNAdW5pYmFzLmNo
Peter K. Jani, amFuaS5wZXRlci5rQGdtYWlsLmNvbQ==
Hiromi Kubagawa, aGlyb21pLmt1YmFnYXdhQGRyZnouZGU=