 Gerdien Mijnheer
Gerdien Mijnheer Femke van Wijk
Femke van Wijk
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 09 May 2019
Sec. Inflammation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00940
This article is part of the Research Topic Immunophysiology of Pediatric Rheumatic Diseases View all 18 articles
Chronic inflammatory diseases are characterized by a disturbed immune balance leading to recurring episodes of inflammation in specific target tissues, such as the joints in juvenile idiopathic arthritis. The tissue becomes infiltrated by multiple types of immune cell, including high numbers of CD4 and CD8 T-cells, which are mostly effector memory cells. Locally, these T-cells display an environment-adapted phenotype, induced by inflammation- and tissue-specific instructions. Some of the infiltrated T-cells may become tissue resident and play a role in relapses of inflammation. Adaptation to the environment may lead to functional (re)programming of cells and altered cellular interactions and responses. For example, specifically at the site of inflammation both CD4 and CD8 T-cells can become resistant to regulatory T-cell-mediated regulation. In addition, CD8 and CD4 T-cells show a unique profile with pro- and anti-inflammatory features coexisting in the same compartment. Also regulatory T-cells are neither homogeneous nor static in nature and show features of functional differentiation, and plasticity in inflammatory environments. Here we will discuss the recent insights in T-cell functional specialization, regulation, and clonal expansion in local (tissue) inflammation.
A typical hallmark of immune-mediated inflammatory diseases (IMIDs) is the intermittent presence of inflammation that manifest in specific target tissues. In affected tissues, activated antigen-specific T-cells with a memory phenotype are present. The combination of memory T-cell infiltrated at affected sites and genetic associations have pointed to T-cells as key players in the pathophysiology of chronic inflammatory diseases. Due to the ongoing inflammation in target tissues there is an increasing risk for collateral tissue damage. Especially in children this can have long-term effects and consequences. Understanding the biology of immune cells that are actively involved in the local inflammatory process is crucial for the development of therapeutic approaches. However, because of technical challenges, the vast majority of data in human studies comes from peripheral blood cells. With the emergence of novel techniques including (single-cell) RNA-sequencing and mass cytometry it is now possible to unravel T-cell signatures in a local (tissue) setting. Here, we will discuss the most recent findings on human T-cell functional programming and adaptation at the site of chronic inflammation with a focus on pediatric rheumatologic disease.
Inflammation is a process aimed at eliminating the triggering agent, but in chronic inflammation the (auto)antigens persist, inducing a sustained inflammation. When inflammation does resolve, a population of antigen-specific T-cells may remain in the tissue as memory cells and become tissue resident (1), the so called tissue-resident memory T-cells (Trm). This makes them unique compared to central memory, effector memory and naïve T-cells that recirculate within lymphoid organs and blood and/or lymphatics. A hallmark of Trm cells is their ability to respond quickly and robustly after re-encountering the antigen as well as the expression of inhibitory molecules to keep them in check. In steady-state, Trm play an important role in tissue homeostasis and protection. After immune re-activation of Trm, circulating immune cells are attracted to the site of inflammation as well, resulting in local accumulation of antigen-specific memory T-cells. So both Trm and infiltrating T-cells that might eventually become Trm themselves, may actively participate during the inflammatory response. Because of their vigilance and specific localization in tissues, Trm are suspected to play a dominant role in the typical relapsing remitting course of chronic inflammatory diseases. Not much is known about the dynamics and interaction between both memory subsets. An accumulation of both CD4+ and CD8+ T-cells are found at sites of chronic inflammation. However, research on the involvement of T-cells in disease pathogenesis has mainly focused on CD4+ T-cells, whereas CD8+ T-cells have been the focus of the Trm research field. In this review we summarize the findings on CD8+, CD4+, and FOXP3+ regulatory T-cell (Treg) subsets in the contexts of local (tissue) adaptation and functional differentiation. In line with the divarication of T helper cells, with each subset having a specified function in the immune system, it is now becoming apparent that specialization is also true for T-cells present in different tissues and types of inflammation. The local acquisition of additional or adjusted functions and phenotypes will be referred to as adaptation.
For a long time, CD4+ T have been recognized as central players in the immune-pathogenesis of autoimmune diseases, which is supported by strong associations of rheumatic diseases with MHC class II alleles (2). The CD4+ T-cell population is comprised of several T helper subsets that develop after the T-cell receptor (TCR) on naïve CD4+ T-cells interacts with activated antigen presenting cells (APCs). Next to this TCR stimulation, co-stimulation, subsequent signaling and cytokines in the micro-environment are important in determining the fate of a specific subset, by activating signaling molecules that establish a lineage-specific enhancer landscape and lead to the expression of master transcription factors. Those, together with a complex network of accessory transcription factors, can coordinate cellular differentiation programs committed toward a lineage, while simultaneously repressing the developmental programs of opposing Th lineages (3). For many years the only identified lineages were Th1 and Th2 subsets, with IFNγ producing Th1 cells being specialized in cell-mediated immune responses against intracellular bacteria and Th2 cells producing IL-4 and IL-13 targeting helminths. The discovery of other subsets, such as Th17, Th9, and T follicular helper (Tfh) cells has shifted the paradigm of two opposing lineages (4–6). Th17 cells, producing various cytokines including IL-17, IL-22, and GM-CSF, induce defense against fungi and extracellular bacteria and are crucial for the maintenance of mucosal homeostasis (7). Despite their relatively recent discovery, Th17 cells are implicated in the pathogenesis of many human (autoimmune-) diseases. This also accounts for Tfh cells that provide help to cognate B cells to produce high affinity antibodies and memory B cells and as such control humoral immunity (8). Recent reports indicate that Th9 cells, mainly producing IL-9, may also be involved in the pathogenesis of auto-immune diseases, possibly by promoting Th17 differentiation (4).
Many immune-mediated diseases are associated with aberrant Th responses. In the lamina propria of Crohn's disease, the synovial fluid (SF) of Juvenile Idiopathic Arthritis (JIA) and Rheumatoid Arthritis (RA) patients and renal tissue of Systemic Lupus Erythematosus (SLE) patients for example, Th1 cells are implicated in disease pathogenesis (9–12). In JIA, a mixed Th17/Th1 phenotype is also found in inflamed joints, capable of producing both IL-17 and IFNγ. The presence of this subset correlates with disease activity (13, 14) and its IL-17-producing capacity is associated with CD161 expression (15). Interestingly, this subset seems to be present specifically in inflammatory environments, and can be a transiently induced from Th17 cells upon exposure to IL-12 and/or TNFα (13, 14, 16). Whereas, the Th1 lineage is shown to be fairly stable, the Th17 lineage is known for its instability and is severely impacted by the environment (17). In line with this, the mixed Th1/Th17 phenotype likely derives from Th17 cells instead of Th1 cells (18). Next to pathogenic Th17 cells, non-pathogenic Th17 cells have been described in autoimmune diseases as well [reviewed in (19)]. A recent paper found delayed IL-10 production in about 25% of activated human Th17 clones in culture (20). This indicates that IL-10 production is an intrinsic property of a subset of Th17 cells after antigenic stimulation, perhaps to regulate and balance the immune response. Transcriptional analysis of the IL-10+ and IL-10− Th17 clones demonstrated immune-regulatory and tissue-resident properties of IL-10 producing Th17 cells, and a pro-inflammatory profile of IL-10− Th17 cells with high CCR7 expression, which may indicate circulatory properties (20). Local pathogenic Th17 cells have been described in multiple autoimmune disease [reviewed in (17)]. In muscle of JDM patients and affected kidneys of SLE patients, IL-17 producing cells are increased (21–23). One mechanism that explains the elevated production of IL-17 in JIA and SLE is the increased expression of the transcription factor cAMP-responsive element modulator (CREM)α. This induces repression of IL-2 transcription but also epigenetic changes of the IL-17A locus, resulting in enhanced IL-17A promoter activity (24, 25). Although evidence for involvement of the IL-17 signaling pathway in SLE pathogenesis is expanding, direct interference with IL-17 or it's receptor does not seem to be effective, at least in mouse models (26, 27). Also in JIA, IL-17 blockade is not part of standard treatment. This might be related to the pathogenic role of another CD4+ subset, Tfh cells that is increased in inflamed tissues of RA and SLE patients and located near B cells in affected kidneys in SLE patients (28, 29). Interestingly, STAT-3 and IL-21, signature molecules shared by Th17 and Tfh cells, are heavily implicated in SLE pathology and are capable of inducing autoreactive B cells (30, 31). Alternatively, the lack of IL-17 blockade efficacy in JIA might be explained by Th17 cells that can be polarized in the inflamed joints to shift toward the so called non-classic Th1 subset. These cells have been shown to lose the ability to produce IL-17 while maintaining both RORc and CD161 expression and produce high levels of IFNγ, in line with the mixed Th1/Th17 cells described above (18). Altogether, overactive CD4+ T-cells are present at the site of human inflammation and represent a mixed population of which especially Th17 cells show plasticity.
The presence of mixed CD4+ phenotypes found in human sites of autoimmune inflammation is of great interest, but might not be inflammation-specific. Recent studies using novel technologies have revealed a continuum of cell fates rather than limited and distinct Th subsets in healthy tissues. Mass cytometry measuring T-cell trafficking receptor and cytokine expression in eight different human tissues revealed tissue-specific and unique T-cell phenotypes (32). This indicates that T-cells cannot be easily classified into separate lineages across human tissues. Furthermore, multi-color cytometry of peripheral blood cells of a healthy human cohort showed substantial subject-to-subject differences in T-cell populations that yet remained relatively stable for months within individuals (33). Age and disease associated genetic polymorphisms were identified as important factors in the identified variation. These publications highlight the importance of age and tissue influences on T cells [reviewed in (34)]. On top of this homeostatic variety, inflammation will probably add another layer of complexity by driving local tissue cells into an adapted phenotype.
Although long neglected in autoimmune diseases compared to CD4+ T-cells, CD8+ T-cells are equipped with different capacities by which they can contribute to the inflammatory process. For example, CD8+ T-cells have cytolytic activity, produce pro-inflammatory cytokines and can react to self-antigens upon cross-presentation (35). In several chronic inflammatory diseases CD8+ T-cells are described to be present in the inflamed tissues and increased CD8+ T-cell numbers are associated poor prognosis in several rheumatic diseases including JIA, RA and SLE (36–39). CD8+ T-cells form a diverse group of cells but in contrast to CD4+ T-cells subsets are less well-defined. The phenotype of CD8+ T-cells at the inflamed site of human autoimmune arthritis is heterogeneous, with both pro- and anti-inflammatory features [reviewed in (35)]. In SF of RA patients for example, CD8+ T-cells are characterized by increased expression of activation markers (CD80, CD86, CD25), pro-inflammatory cytokines like IL-6 and TNFα, and with a proliferative signature, but also by elevated levels of negative co-stimulatory markers, such as TIM-3 and PD-1 (36, 40, 41). In affected kidneys of SLE patients, the majority of CD8+ T-cells are located in the periglomerular regions where tissue damage occurs, and this infiltrate is correlated with renal injury (42). Despite multiple studies reporting CD8+ T-cells accumulation in SLE affected tissues, the phenotype of local CD8+ T-cells remains largely unexplored. One study reports a differentiated effector memory phenotype with loss of CD28 on infiltrating CD8+ T-cells, indicating active involvement of these cells in disease pathology (43). However, although counter-intuitive for effector cells in an autoimmune environment, a recent study shows that kidney-infiltrating T-cells are metabolically and functionally “exhausted” in three mouse models of lupus nephritis (44). The term “exhausted” stems from numerous studies on CD8+ T-cells in chronic viral infections and, to a lesser extent, in cancers. Chronic antigen exposure in these settings goes along with the upregulation of negative co-stimulatory markers, such as PD-1 in combination with reduced secretion of effector cytokines and proliferation. This has led to the hypothesis that these cells are terminally differentiated and severely functionally impaired (45, 46). However, recently this concept was challenged by the discovery that PD-1+ CD8+ T-cells are functionally adapted cells able to control the viral load or tumor cells without causing excessive immune pathology, and can be therapeutically reinvigorated by blocking PD-1/PD-L1 interaction (47, 48).
Not much is known about the functional profile of tissue-specific memory CD8+ T-cell in human chronic autoimmune diseases. Like CD8+ T-cells in infectious diseases and tumors, these cells are chronically activated. However, instead of resulting in strengthened regulation as is described for the aforementioned conditions, chronic stimulation in autoimmune diseases seems to lead toward overzealous and pathogenic effector functions. Previous observations from peripheral blood derived CD8+ T-cells show that the transcriptional signature reflecting exhaustion is associated with poor clearance of chronic viral infection, but conversely predicts better prognosis in multiple auto-immune diseases, including SLE (49). A recent paper addressing the enriched PD-1+ CD8+ T-cell population in SF of JIA patients is the first to study this cell subset locally, derived from the site of inflammation in humans (50). In this setting, PD-1 expressing CD8+ T-cells maintain their effector function, such as pro-inflammatory cytokine production, cytotoxic profile, and use of glycolysis as a metabolic pathway and thereby are suspected to have a detrimental role in autoimmune tissue damage. Strong inflammatory signals, and high levels of soluble PD-1 that block interaction with APCs (51), may overrule or hamper PD-1-signaling in CD8+ T-cells in SF of JIA patients. In line with this, Odorizzi et al. have shown that PD-1 expression is not a prerequisite for exhaustion to occur, by using a mouse model of chronic viral infection with genetic absence of PD-1. In this model, PD-1 prevented CD8+ T-cells overstimulation and apoptosis, as the absence of PD-1 led to more cytotoxic but terminally differentiated CD8+ T-cells (47). It is tempting to speculate that this mechanism may also play a role in local auto-immune inflammation, leading to survival of auto-antigen induced clonally expanding effector cells.
In line with this, other T-cell subsets have been described that may be induced by chronic stimulation and acquire a pathogenic role. The latter is reflected by the secretion of inflammatory cytokines and expression of granzymes and perforin. These include CD8+ T-cells expressing CD57 (52, 53) and CD4+/CD8+ T-cells that have downregulated CD28, so called CD28null cells (54, 55). Some reports have demonstrated that these cells are also present in SF of RA patients, whereas others failed to confirm this (56, 57). There are indications that these chronically activated CD4+ and CD8+ T-cells cells are induced by CMV infection in RA patients (58), although a causative linkage has not been established thus far (54, 59).
Interestingly, the combined cytotoxic and regulatory profile of CD8+ T-cells, defined by increased expression of effector molecules, such as Granzyme B on the one hand, and of negative co-stimulatory molecules on the other hand, is also typical for Trm cells (60). In line with this, the PD-1+ CD8+ cells in SF were shown to be enriched for a Trm transcriptional profile compared to the PD-1− CD8+ T-cells from the same environment (50). Furthermore, Trm are defined by CD69 expression and downregulation of S1PR1, which is also found on PD-1+ CD8+ cells derived from SF of JIA patients. So local CD8+ T-cells from the site of autoimmune inflammation in JIA cannot be classified as exhausted, despite some overlapping features, but share much of their profile with Trm cells. Interestingly, tumor-infiltrating CD8+ T-cells in human cancers also display a Trm profile (61). Moreover, a recent paper describing the transcriptional, metabolic, and functional signatures of intra-tumoral PD-1+ CD8+ T-cells has revealed that, next to many commonalities, such as impaired cytokine production, these cells also differ from exhausted cells as described in chronic infections (62). The intra-tumoral CD8+ T-cells showed increased proliferation and glycolysis, and lack of enrichment of the exhausted T-cell gene signature (62), as was observed in SF of JIA patients. The interpretation now arises that CD8+ “exhausted” cells are a heterogeneous group of memory cells with diverse differentiation states but all driven by persistence of antigen that induces upregulation of inhibitory receptors, and with functional properties that are influenced by the environment (63, 64). This concept shares many features with the current view on Trm cells as they are also regarded as highly specialized cells with a tissue adapted profile, tightly regulated to prevent excessive tissue damage. The commonalities between CD8+ T-cells at inflammatory sites and the Trm profile suggest interplay between inflammation and tissue residency. Indeed, several studies indicate that inflammation is the trigger for initial homing of Trm cells, by providing the migratory signals needed to direct them to tissues (61). PD-1 expressing CD8+T-cells in an inflammatory exudate, such as SF of JIA could be tissue derived, but how they developmentally relate to Trm cells remains unknown. All in all, local CD8+ T-cells situated in the affected tissue of chronic inflammation are heterogeneous with both effector and regulatory responses that are highly influenced by chronic inflammation, possibly with disease specific profiles.
Overzealous CD4+ Th responses carry the risk of initiating detrimental pro-inflammatory responses that can result in collateral tissue damage. Thus, the maintenance of immune homeostasis and prevention of immunopathology requires tight regulatory mechanisms. Regulatory T-cells (Treg) are a subset of CD4+ T-cells with unique homeostatic functions. Absence of the Treg master transcription FOXP3 leads to fatal multi-organ autoimmunity in mice and men (65, 66). The capacity of Treg to dampen immune responses have made them attractive therapeutic targets in diverse settings, such as in autoimmune diseases, transplantation, and cancer. The best discriminative surface markers for Treg are high expression of CD25 (IL-2 receptor α) in combination with low expression of CD127 (IL-7 receptor α). Early studies often used solely high CD25 expression to identify or purify Treg, resulting in contamination with activated conventional CD4+ T-cells, and contradicting data.
Traditionally, the Treg lineage was considered as a homogeneous group of committed cells with suppressive capacities toward other immune cells. As it increasingly becoming apparent that most, if not all, immune cells have the capacity to adapt to their environment, data from the last decade demonstrated that Treg are perhaps the most heterogeneous in phenotype and function (67). Their high turnover and sensitivity to environmental signals leads to a large degree of adaptation that allows Treg to control diverse immune responses and even to exert tissue-specific functions. In that respect novel Treg phenotypes have been identified that differ markedly from their naïve recirculating counterparts (central Treg). Liston and Gray have proposed the model of environment-instructed effector Treg and polarized Treg differentiation (68). Effector Treg, or eTreg, are characterized in humans by Foxp3highCD25highCD45RAlow expression representing a small fraction of circulating Treg (69). They have signs of recent antigen encounter, have an heightened activation status and migratory potential, and express markers similar to activated conventional CD4+ T-cells while maintaining Treg functions (68). For example, increased expression of Foxp3, as well as typical functional markers, such as ICOS and CTLA4 is observed in eTreg (69, 70). Polarized (tissue-resident) Treg are present in non-lymphoid tissues, express specific homing receptors and exert tissue-specific functions and immune regulation. Treg do so by utilizing the transcription factor program of the population they are suppressing, or tissue specific transcription factors, respectively (67, 68, 71). For example, Treg that co-express T-BET next to FOXP3 can efficiently suppress Th1 responses (72), whereas the expression of adipose tissue-specific peroxisome proliferator-activated receptor gamma (PPARγ) is needed for Treg to control insulin sensitivity (73). In the latter case, Treg are tissue-resident in a physiological condition and there is an increasing list of tissue specific phenotypes of tissue-resident Treg exerting non-immunological but tissue-homeostatic functions [reviewed in (67)]. To what extend the profile of conventional Trm is applicable to tissue-resident Treg remains to be explored.
Using the proper gating strategy for Treg markers, the frequency of Treg in rheumatic diseases is increased at the site of inflammation in JIA and JDM (74–76). Whether Treg function is impaired in these diseases is still under debate, partly related to differences in phenotyping markers and conditions of in vitro assays used to test Treg functionality. Multiple studies have however shown that Treg derived from SF of JIA patients maintain their suppressive function and upregulate Treg functional markers, such as CD25, CTLA4, and GITR, rather pointing toward an eTreg profile (77–79) (Figure 1).
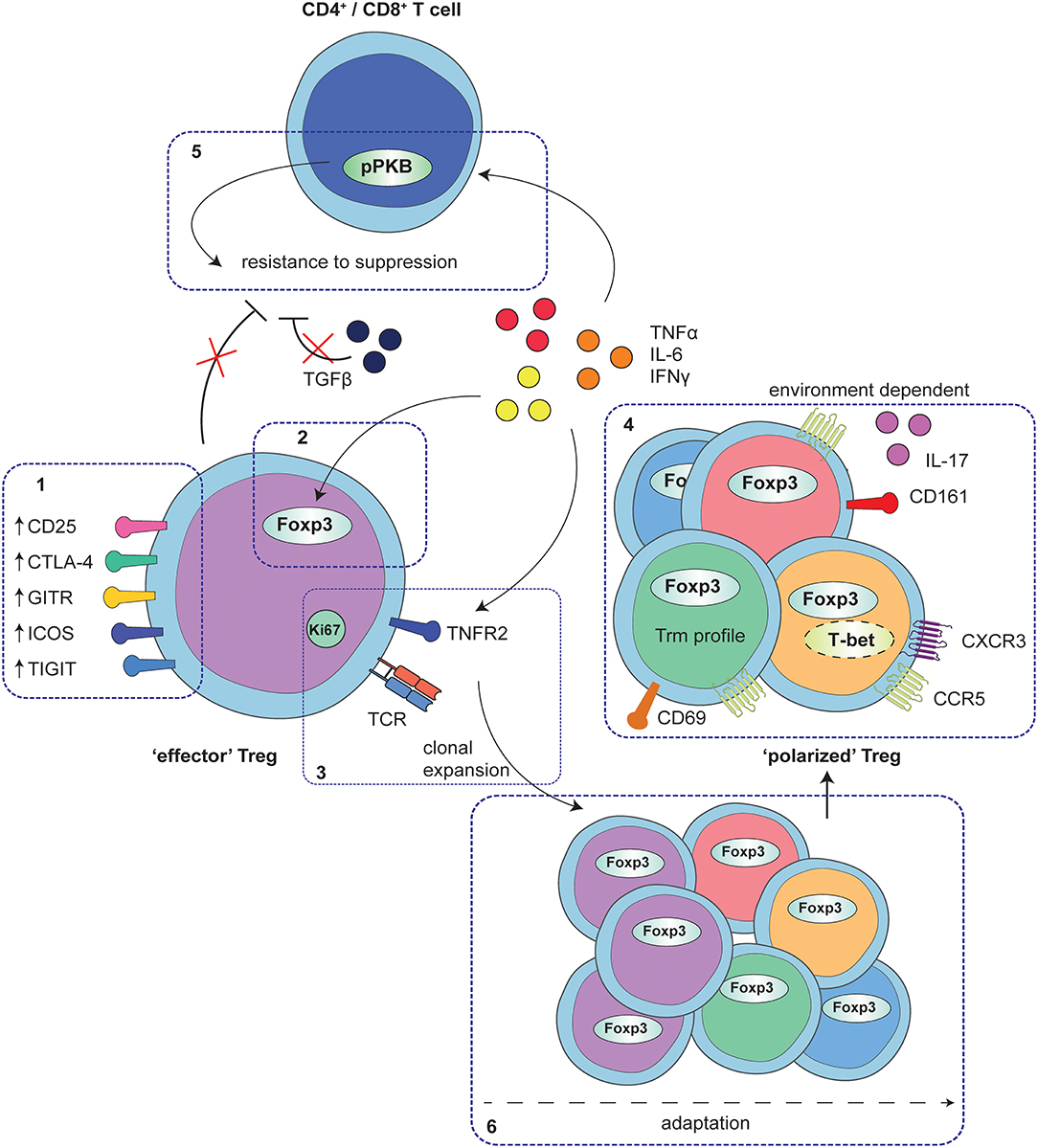
Figure 1. Adaptation of Treg to local auto-immune inflammation. At the site of human autoimmune-inflammation functional Treg are present that display an effector phenotype. This phenotype is characterized by increased expression of functional Treg markers, including CD25, CTLA4, GITR, ICOS, and TIGIT (1) and stable increased expression of Foxp3, and at least in part, instructed by local inflammatory signals (2). Furthermore, Treg are clonally expanding as is reflected by increased Ki67 expression and a local clonal TCR repertoire, possibly mediated by TNFα signaling via TNFR2 on Treg (3). In addition to the effector profile, Treg may also display a specific environment instructed profile, including e.g., expression of CD161 and IL-17 production, upregulation of Th1- and inflammation-associated markers and chemokine receptors and/or characteristics of a Trm profile (4). These polarization profiles are not exclusive but rather are overlapping, depending on the specific local conditions. Whereas, adapted Treg are functional, local cytokines produced by monocytes and fibroblasts also affect CD4+ and CD8+ T-cells, in part by hyper-phosphorylation of PKB/c-AKT, conferring resistance of local CD4+ and CD8+ T-cells to Treg suppression (5). Overall, the process of Treg adaptation in inflammatory settings is highly influenced by the local environment, most likely starting with an expanding effector population that can be further fine-tuned with environmental adaptations (6).
Instability of Treg has long been suspected to play a role in disease pathology. Instability is defined by loss of FOXP3 expression and suppressive function, with a concomitant acquisition of an effector phenotype. The stability of Treg is a contentious issue, with contradicting data from several studies (80). Multiple mouse models, including genetic fate-mapping models that allow tracking Foxp3 expressing cells, revealed that Treg are fairly stable in vivo with a small proportion of cells that lose Foxp3 expression (81–84).
At the site of autoimmune inflammation in humans, FOXP3-expressing Treg that produce pro-inflammatory cytokines have been described (85, 86). In specific tissues however, it is unknown if aberrant adapted Treg add to disease pathogenesis. In this regard, it is important to distinguish between functional plasticity/adaptability and lineage instability. In JIA, a small fraction of SF Treg expresses CD161 and is capable of producing pro-inflammatory cytokines. At the same time, FOXP3 expression remains high and suppressive capacity is also maintained (87, 88). Another paper studying Treg stability in SF of JIA patients, demonstrated that the T-cell receptor (TCR) repertoires of Treg is very distinct from conventional T-cells in SF, indicating a different origin and thus excluding a large degree of instability of Treg (89). On top of that, the same paper showed that Treg need inflammatory signals present in SF to maintain their FOXP3 expression, supporting the idea that local signals in an inflammatory environment can stabilize or even enhance the Treg phenotype.
Systemic administration of IL-2 as a therapy to maintain and possibly expand Treg is currently being tested for SLE patients. A recent paper reports on reduced CD25 expression on peripheral blood Treg of SLE patients, that correlates to the reduced production of IL-2 from circulating memory T-cells (90). Since the increased expression of CREMα leads to reduced IL-2 production of effector T-cells, and IL-2 receptor (CD25) signaling via STAT5 is pivotal for maintained Foxp3 expression in Treg, impaired Treg function could be a consequence (91). This provides a rationale for Treg targeted therapy by low dose IL-2 administration (92). However, it is not known whether the reduced CD25 expression on Treg also occurs at the site of inflammation. Moreover, in peripheral blood of active SLE patients enhanced levels of functional, non-cytokine producing Helios+ Treg have been identified that positively correlate with disease activity (93, 94). In addition, these cells were shown to express CXCR3 and CCR4, allowing them to migrate to inflammatory sites (93).
The stability of Treg is regulated on multiple levels. The acquisition of a specific epigenetic landscape is a strong determinant (95), as well as transcriptional and post-transcriptional regulation of FOXP3. In vitro studies have shown that environmental cues can modulate these processes. For example ubiquitination of FOXP3, that targets its proteosomal degradation, is highly regulated by the ubiquitin ligase Stub1 and the deubiquitinating enzyme USP7. In vitro, inflammatory stimuli allow Stub1 to bind FOXP3 and promote its degradation, which is further facilitated by the downregulation of USP7 (96, 97). However, ex vivo gene expression analysis of SF Treg from JIA patients shows stable expression of both proteins (Mijnheer et al., unpublished data). This illustrates that in vivo regulation of FOXP3 and Treg function is a highly complex organized process in which multiple proteins are involved. Also, depending on the environmental conditions, different proteins can take part in this network, allowing fine-tuning of the cells to a specific environment and further polarization (81). Altogether, there are no indications for Treg instability on a large scale in vivo, but inflammation does seem to impact Treg by differentiation toward an eTreg phenotype.
Unresponsiveness of T-cells to Treg suppression is most likely a normal transient phenomenon during the assembly of an immune response to clear an infectious threat. However, in auto-immune diseases this resistance of local effector T-cells to suppression contributes to a sustained inflammatory response and subsequent disease pathogenesis (98). In JIA, both CD4+ and CD8+ T-cells from the synovial fluid of affected joints have been found to be intrinsically resistant to suppression (78, 79, 99). The same has been described for CD4+ T-cells from SLE patients (100). The resistance to suppression is at least partly mediated by protein kinase B (PKB)/c-akt hyper-activation, induced by local cytokines. TNFα and IL-6 were found to induce resistance in CD4+ T-cells, whereas TNFα and autocrine release of IFNγ were responsible for the intrinsic resistance in CD8+ T-cells (79, 99). The hyperphosphorylation of PKB/c-akt is interesting in the light of PD-1 signaling, since PD-1 is a strong negative regulator of PKB/c-akt (101). Apparently PD-1 signaling in SF T-cells is not sufficient enough to downregulate this signaling pathway. Blockade of TNFα can restore the susceptibility of CD4 and CD8 T-cells to suppression, which is in line with the effectiveness of therapies targeting TNFα (99, 102). In IBD, resistance of lamina propria T-cells to Treg-mediated suppression has been described as well. In this setting high levels of Smad7, causing insensitivity to TGFβ, have been related to the resistance of effector cells (103). Thus, impaired regulation of the local immune response involves resistance of effector cells, possibly despite functional Tregs.
When activated T-cells encounter their cognate antigen in the context of co-stimulation and cytokines, specific clones will expand to initiate a robust adaptive immune response. A diverse repertoire of T-cell receptors (TCRs) of conventional T-cells allows a response to a multitude of possible pathogens. Also thymic derived Treg need a diverse TCR repertoire to regulate auto-immune responses. The TCR repertoire of Treg, representing only a small fraction of the total T-cell pool, is equally diverse as the larger effector pool (104). The need for a diverse (auto-antigen specific) TCR repertoire of Treg has been illustrated by studies using transgenic mice with a restricted TCR repertoire of Treg. In these models, a loss of tolerance toward commensal bacteria and develop autoimmune diseases has been observed (105–108).
At the site of human autoimmune inflammation in both JIA and SLE, clonal T-cell expansions of CD4+ T-cell, CD8+ T-cell and FOXP3+ Treg populations are found (43, 50, 89, 109, 110). Remarkably, especially Treg were found to be hyper-expanded, and to express high levels of Ki67. It is possible that dominant auto-antigens present at the affected sites induce this proliferation (79, 89). Local TNFα can also act as a contributing factor to Treg expansion, as TNFα induces Treg proliferation and effector Treg differentiation via TNFR2 signaling [(111–114); Mijnheer et al., unpublished data]. Since hyper-expanded Treg clones were demonstrated to display a very distinct repertoire compared to conventional CD4+ T-cells, local induction of Treg from conventional T-cells is unlikely (89). Tissue resident Treg also show a considerable oligoclonality regarding their TCR repertoire, supporting the notion that tissue Treg and Treg from the site of autoimmune inflammation share typical features (115, 116) (Figure 1). Interestingly, in refractory JIA and JDM patients undergoing hematopoietic stem cell transplantation the TCR repertoire of circulating Treg prior to transplantation was also found to be highly clonal. After transplantation, the Treg TCR repertoire became more diverse over time, except for one patient that experienced a relapse (117). These dates suggest that Treg TCR repertoire abnormalities may contribute to disease pathogenesis, possibly by limiting the chance of antigen encounter, thereby being outcompeted by the less restricted Teff.
Recent data from multiple affected joints in RA show that total TCR repertoires are substantially overlapping (118). In JIA patients, especially PD-1+ CD8+ T-cells were shown to have a clonal repertoire, with a high overlap in dominant clones between different affected joints (35, 62). The overlap may be explained by the presence of common antigens that drive the disease at multiple sites, and/or (re)circulation of dominant T-cell clones. In affected kidneys of SLE patients expanded CD8+ T-cells clones were found to be present for years in sequential biopsies (43), suggesting long-term persistence of dominant T-cell clones. It is tempting to speculate that these dominant T-cell clones play a role in disease relapses, but further studies are need to conclude this.
The expanding field of T-cells, including the discovery of multiple Th subsets as well as observations of mixed phenotypes in inflamed tissues, has made the classification of subsets increasingly complex (5). This complexity, however, likely represents human immunity that is continuously exposed to multiple microorganisms and environmental conditions, in contrast to highly controlled mouse models that have contributed to most of the current knowledge (32, 119). A more nuanced view on T-cells in tissues now arises, with fine-tuning of immune cells to the local environment allowing tailored responses explaining the observed diversity in phenotypes. When inflammation becomes chronic this fine-tuning of T-cells might be unrestrained or insufficient, and as a result cause or contribute to pathogenesis. Potential determinants in this process could be the strength and frequency of TCR stimulation, as well as the presence of absence of CD4 help or co-stimulation, whereas local metabolites also seem to be important. How this is regulated exactly, and what the importance is of different factors remains to be answered, as well as the question what is different in affected human tissue in disease vs. healthy human tissue. The possibilities of gaining new insight are enormous, with newly developed high-throughput technologies that require only small numbers of cells and that allow for analysis of rare T-cell populations from small tissue samples. Single cell sequencing combined with TCR sequencing and mass cytometry on whole tissues will give a deeper understanding about the heterogeneity of T-cells present in human autoimmunity. This, together with smart use of patients samples, such as sequential sampling and sampling from multiple affected sites, will provide novel insights and undoubtedly improve the therapeutic options for patients with rheumatic diseases.
GM and FvW have designed the structure and discussed the content of the manuscript. GM has searched the literature and has drafted the text and figure. FvW has critically edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Moro-García MA, Mayo JC, Sainz RM, Alonso-Arias R. Influence of inflammation in the process of T lymphocyte differentiation: proliferative, metabolic, and oxidative changes. Front Immunol. (2018) 9:339. doi: 10.3389/fimmu.2018.00339
2. Okada Y, Kishikawa T, Sakaue S, Hirata J. Future directions of genomics research in rheumatic diseases. Rheum Dis Clin N Am. (2017) 43:481–7. doi: 10.1016/j.rdc.2017.04.009
3. Loo TT, Gao Y, Lazarevic V. Transcriptional regulation of CD4+ TH cells that mediate tissue inflammation. J. Leukoc. Biol. (2018) 104:1069–85. doi: 10.1002/JLB.1RI0418-152RR
4. Deng Y, Wang Z, Chang C, Lu L, Lau CS, Lu Q. Th9 cells and IL-9 in autoimmune disorders: Pathogenesis and therapeutic potentials. Hum Immunol. (2017) 78:120–8. doi: 10.1016/j.humimm.2016.12.010
5. Palmer MT, Weaver CT. Autoimmunity: increasing suspects in the CD4+ T cell lineup. Nat Immunol. (2010) 11:36–40. doi: 10.1038/ni.1802
6. Skapenko A, Leipe J, Lipsky PE, Schulze-Koops H. The role of the T cell in autoimmune inflammation. Arthritis Res Ther. (2005) 7(Suppl. 2):S4. doi: 10.1186/ar1703
7. Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. Immunodeficiencies. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. (2015) 349:606–13. doi: 10.1126/science.aaa4282
8. Gensous N, Charrier M, Duluc D, Contin-Bordes C, Truchetet M-E, Lazaro E, et al. T follicular helper cells in autoimmune disorders. Front Immunol. (2018) 9:1637. doi: 10.3389/fimmu.2018.01637
9. Masutani K, Akahoshi M, Tsuruya K, Tokumoto M, Ninomiya T, Kohsaka T, et al. Predominance of Th1 immune response in diffuse proliferative lupus nephritis. Arthritis Rheum. (2001) 44:2097–106. doi: 10.1002/1529-0131(200109)44:9<2097::AID-ART360>3.0.CO;2-6
10. Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn's disease. J Exp Med. (2002) 195:1129–43. doi: 10.1084/jem.20011956
11. Wedderburn LR, Robinson N, Patel A, Varsani H, Woo P. Selective recruitment of polarized T cells expressing CCR5 and CXCR3 to the inflamed joints of children with juvenile idiopathic arthritis. Arthritis Rheum. (2000) 43:765–74. doi: 10.1002/1529-0131(200004)43:4<765::AID-ANR7>3.0.CO;2-B
12. Yamada H, Nakashima Y, Okazaki K, Mawatari T, Fukushi J, Oyamada A, et al. Preferential accumulation of activated Th1 cells not only in rheumatoid arthritis but also in osteoarthritis joints. J Rheumatol. (2011) 38:1569–75. doi: 10.3899/jrheum.101355
13. Cosmi L, Cimaz R, Maggi L, Santarlasci V, Capone M, Borriello F, et al. Evidence of the transient nature of the Th17 phenotype of CD4+CD161+ T cells in the synovial fluid of patients with juvenile idiopathic arthritis. Arthritis Rheum. (2011) 63:2504–15. doi: 10.1002/art.30332
14. Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W, et al. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA. (2010) 107:14751–6. doi: 10.1073/pnas.1003852107
15. Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol. (2010) 40:2174–81. doi: 10.1002/eji.200940257
16. Maggi L, Cimaz R, Capone M, Santarlasci V, Querci V, Simonini G, et al. Brief report: etanercept inhibits the tumor necrosis factor α-driven shift of Th17 lymphocytes toward a nonclassic Th1 phenotype in juvenile idiopathic arthritis. Arthritis Rheumatol. (2014) 66:1372–7. doi: 10.1002/art.38355
17. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J Autoimmun. (2018) 87:1–15. doi: 10.1016/j.jaut.2017.12.007
18. Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F. Th17 and non-classic Th1 cells in chronic inflammatory disorders: two sides of the same coin. Int Arch Allergy Immunol. (2014) 164:171–7. doi: 10.1159/000363502
19. Wu X, Tian J, Wang S. Insight into non-pathogenic Th17 cells in autoimmune diseases. Front Immunol. (2018) 9:1112. doi: 10.3389/fimmu.2018.01112
20. Aschenbrenner D, Foglierini M, Jarrossay D, Hu D, Weiner HL, Kuchroo VK, et al. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells. Nat. Immunol. (2018) 19:1126–36. doi: 10.1038/s41590-018-0200-5
21. Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. (2008) 181:8761–6. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/19050297
22. De Pasquale L, D'Amico A, Verardo M, Petrini S, Bertini E, De Benedetti F. Increased muscle expression of interleukin-17 in Duchenne muscular dystrophy. Neurology. (2012) 78:1309–14. doi: 10.1212/WNL.0b013e3182518302
23. Zickert A, Amoudruz P, Sundström Y, Rönnelid J, Malmström V, Gunnarsson I. IL-17 and IL-23 in lupus nephritis–association to histopathology and response to treatment. BMC Immunol. (2015) 16:7. doi: 10.1186/s12865-015-0070-7
24. Ohl K, Nickel H, Moncrieffe H, Klemm P, Scheufen A, Föll D, et al. The transcription factor CREM drives an inflammatory phenotype of T cells in oligoarticular juvenile idiopathic arthritis. Pediatr Rheumatol. (2018) 16:39. doi: 10.1186/s12969-018-0253-x
25. Rauen T, Hedrich CM, Juang Y-T, Tenbrock K, Tsokos GC. cAMP-responsive element modulator (CREM)α protein induces interleukin 17A expression and mediates epigenetic alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem. (2011) 286:43437–46. doi: 10.1074/jbc.M111.299313
26. Katsuyama T, Tsokos GC, Moulton VR. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front Immunol. (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
27. Schmidt T, Paust H-J, Krebs CF, Turner J-E, Kaffke A, Bennstein SB, et al. Function of the Th17/interleukin-17A immune response in murine lupus nephritis. Arthritis Rheumatol. (2015) 67:475–87. doi: 10.1002/art.38955
28. Liarski VM, Kaverina N, Chang A, Brandt D, Yanez D, Talasnik L, et al. Cell distance mapping identifies functional T follicular helper cells in inflamed human renal tissue. Sci Transl Med. (2014) 6:230ra46. doi: 10.1126/scitranslmed.3008146
29. Penatti A, Facciotti F, De Matteis R, Larghi P, Paroni M, Murgo A, et al. Differences in serum and synovial CD4+ T cells and cytokine profiles to stratify patients with inflammatory osteoarthritis and rheumatoid arthritis. Arthritis Res Ther. (2017) 19:103. doi: 10.1186/s13075-017-1305-1
30. Wang S, Wang J, Kumar V, Karnell JL, Naiman B, Gross PS, et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat Commun. (2018) 9:1758. doi: 10.1038/s41467-018-03750-7
31. Zhang C-J, Wang C, Jiang M, Gu C, Xiao J, Chen X, et al. Act1 is a negative regulator in T and B cells via direct inhibition of STAT3. Nat Commun. (2018) 9:2745. doi: 10.1038/s41467-018-04974-3
32. Wong MT, Ong DEH, Lim FSH, Teng KWW, McGovern N, Narayanan S, et al. A high-dimensional atlas of human T cell diversity reveals tissue-specific trafficking and cytokine signatures. Immunity. (2016) 45:442–56. doi: 10.1016/j.immuni.2016.07.007
33. Lu Y, Biancotto A, Cheung F, Remmers E, Shah N, McCoy JP, et al. Systematic analysis of cell-to-cell expression variation of T lymphocytes in a human cohort identifies aging and genetic associations. Immunity. (2016) 45:1162–75. doi: 10.1016/j.immuni.2016.10.025
34. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
35. Petrelli A, van Wijk F. CD8+ T cells in human autoimmune arthritis: the unusual suspects. Nat Rev Rheumatol. (2016) 12:421–8. doi: 10.1038/nrrheum.2016.74
36. Cho B-A, Sim JH, Park JA, Kim HW, Yoo W-H, Lee S-H, et al. Characterization of effector memory CD8+ T cells in the synovial fluid of rheumatoid arthritis. J Clin Immunol. (2012) 32:709–20. doi: 10.1007/s10875-012-9674-3
37. Costello P, Bresnihan B, O'Farrelly C, FitzGerald O. Predominance of CD8+ T lymphocytes in psoriatic arthritis. J Rheumatol. (1999) 26:1117–24. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/10332977
38. Hunter PJ, Nistala K, Jina N, Eddaoudi A, Thomson W, Hubank M, et al. Biologic predictors of extension of oligoarticular juvenile idiopathic arthritis as determined from synovial fluid cellular composition and gene expression. Arthritis Rheum. (2010) 62:896–907. doi: 10.1002/art.27284
39. McKinney EF, Lyons PA, Carr EJ, Hollis JL, Jayne DRW, Willcocks LC, et al. A CD8+ T cell transcription signature predicts prognosis in autoimmune disease. Nat Med. (2010) 16:586–91, 1p following 591. doi: 10.1038/nm.2130
40. Carvalheiro H, Duarte C, Silva-Cardoso S, da Silva JAP, Souto-Carneiro MM. CD8+ T cell profiles in patients with rheumatoid arthritis and their relationship to disease activity. Arthritis Rheum. (2015) 67:363–71. doi: 10.1002/art.38941
41. Li S, Peng D, He Y, Zhang H, Sun H, Shan S, et al. Expression of TIM-3 on CD4+ and CD8+ T cells in the peripheral blood and synovial fluid of rheumatoid arthritis. APMIS. (2014) 122:899–904. doi: 10.1111/apm.12228
42. Couzi L, Merville P, Deminière C, Moreau J-F, Combe C, Pellegrin J-L, et al. Predominance of CD8+ T lymphocytes among periglomerular infiltrating cells and link to the prognosis of class III and class IV lupus nephritis. Arthritis Rheum. (2007) 56:2362–70. doi: 10.1002/art.22654
43. Winchester R, Wiesendanger M, Zhang H-Z, Steshenko V, Peterson K, Geraldino-Pardilla L, et al. Immunologic characteristics of intrarenal T cells: trafficking of expanded CD8+ T cell β-chain clonotypes in progressive lupus nephritis. Arthritis Rheum. (2012) 64:1589–600. doi: 10.1002/art.33488
44. Tilstra JS, Avery L, Menk AV, Gordon RA, Smita S, Kane LP, et al. Kidney–infiltrating T cells in murine lupus nephritis are metabolically and functionally exhausted. J Clin Invest. (2018) 128:4884–97. doi: 10.1172/JCI120859
45. Gallimore A, Glithero A, Godkin A, Tissot AC, Plückthun A, Elliott T, et al. Induction and exhaustion of lymphocytic choriomeningitis virus-specific cytotoxic T lymphocytes visualized using soluble tetrameric major histocompatibility complex class I-peptide complexes. J Exp Med. (1998) 187:1383–93. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/9565631
46. Moskophidis D, Lechner F, Pircher H, Zinkernagel RM. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature. (1993) 362:758–61. doi: 10.1038/362758a0
47. Odorizzi PM, Pauken KE, Paley MA, Sharpe A, Wherry EJ. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. (2015) 212:1125–37. doi: 10.1084/jem.20142237
48. Speiser DE, Utzschneider DT, Oberle SG, Münz C, Romero P, Zehn D. T cell differentiation in chronic infection and cancer: functional adaptation or exhaustion? Nat Rev Immunol. (2014) 14:768–74. doi: 10.1038/nri3740
49. McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. (2015) 523:612–6. doi: 10.1038/nature14468
50. Petrelli A, Mijnheer G, van Konijnenburg DPH, van der Wal MM, Giovannone B, Mocholi E, et al. PD-1+CD8+ T cells are clonally expanding effectors in human chronic inflammation. J Clin Invest. (2018) 128:4669–81. doi: 10.1172/JCI96107
51. Bommarito D, Hall C, Taams LS, Corrigall VM. Inflammatory cytokines compromise programmed cell death-1 (PD-1)-mediated T cell suppression in inflammatory arthritis through up-regulation of soluble PD-1. Clin Exp Immunol. (2017) 188:455–66. doi: 10.1111/cei.12949
52. Chiang SCC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood. (2013) 121:1345–56. doi: 10.1182/BLOOD-2012-07-442558
53. Le Priol Y, Puthier D, Lécureuil C, Combadière C, Debré P, Nguyen C, et al. High cytotoxic and specific migratory potencies of senescent CD8+ CD57+ cells in HIV-infected and uninfected individuals. J Immunol. (2006) 177:5145–54. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/17015699
54. Dumitriu IE. The life (and death) of CD4+ CD28 null T cells in inflammatory diseases. Immunology. (2015) 146:185–93. doi: 10.1111/imm.12506
55. Kared H, Martelli S, Ng TP, Pender SLF, Larbi A. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol Immunother. (2016) 65:441–52. doi: 10.1007/s00262-016-1803-z
56. Fasth AER, Snir O, Johansson AAT, Nordmark B, Rahbar A, Af Klint E, et al. Skewed distribution of proinflammatory CD4+CD28null T cells in rheumatoid arthritis. Arthritis Res Ther. (2007) 9:R87. doi: 10.1186/ar2286
57. Schmidt D, Martens PB, Weyand CM, Goronzy JJ. The repertoire of CD4+ CD28− T cells in rheumatoid arthritis. Mol Med. (1996) 2:608–18. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/8898376
58. Hooper M, Kallas EG, Coffin D, Campbell D, Evans TG, Looney RJ. Cytomegalovirus seropositivity is associated with the expansion of CD4+CD28− and CD8+CD28− T cells in rheumatoid arthritis. J Rheumatol. (1999) 26:1452–57. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/10405929
59. Halenius A, Hengel H. Human cytomegalovirus and autoimmune disease. Biomed Res Int. (2014) 2014:1–15. doi: 10.1155/2014/472978
60. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
61. Amsen D, van Gisbergen KPJM, Hombrink P, van Lier RAW. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol. (2018) 19:538–46. doi: 10.1038/s41590-018-0114-2
62. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. (2018) 24:994–1004. doi: 10.1038/s41591-018-0057-z
63. Bengsch B, Ohtani T, Khan O, Setty M, Manne S, O'Brien S, et al. Epigenomic-guided mass cytometry profiling reveals disease-specific features of exhausted CD8 T cells. Immunity. (2018) 48:1029–45.e5. doi: 10.1016/j.immuni.2018.04.026
64. Utzschneider DT, Alfei F, Roelli P, Barras D, Chennupati V, Darbre S, et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med. (2016) 213:1819–34. doi: 10.1084/jem.20150598
65. Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. (2001) 27:68–73. doi: 10.1038/83784
66. Ochs HD, Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. (2001) 27:20–1. doi: 10.1038/83713
67. Sharma A, Rudra D. Emerging functions of regulatory T cells in tissue homeostasis. Front Immunol. (2018) 9:883. doi: 10.3389/fimmu.2018.00883
68. Liston A, Gray DHD. Homeostatic control of regulatory T cell diversity. Nat Rev Immunol. (2014) 14:154–65. doi: 10.1038/nri3605
69. Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. (2009) 30:899–911. doi: 10.1016/j.immuni.2009.03.019
70. Cretney E, Kallies A, Nutt SL. Differentiation and function of Foxp3+ effector regulatory T cells. Trends Immunol. (2013) 34:74–80. doi: 10.1016/j.it.2012.11.002
71. Panduro M, Benoist C, Mathis D. Tissue Tregs. Annu Rev Immunol. (2016) 34:609–33. doi: 10.1146/annurev-immunol-032712-095948
72. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. (2009) 10:595–602. doi: 10.1038/ni.1731
73. Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, et al. PPAR-? Is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature. (2012) 486:549. doi: 10.1038/nature11132
74. de Kleer IM, Wedderburn LR, Taams LS, Patel A, Varsani H, Klein M, et al. CD4+CD25bright regulatory T cells actively regulate inflammation in the joints of patients with the remitting form of juvenile idiopathic arthritis. J Immunol. (2004) 172:6435–43. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/15128835
75. Ohl K, Tenbrock K. Regulatory T cells in systemic lupus erythematosus. Eur J Immunol. (2015) 45:344–55. doi: 10.1002/eji.201344280
76. Vercoulen Y, Bellutti Enders F, Meerding J, Plantinga M, Elst EF, Varsani H, et al. Increased presence of FOXP3+ regulatory T cells in inflamed muscle of patients with active juvenile dermatomyositis compared to peripheral blood. PLoS ONE. (2014) 9:e105353. doi: 10.1371/journal.pone.0105353
77. Chandrasekaran U, Yi W, Gupta S, Weng C-H, Giannopoulou E, Chinenov Y, et al. Regulation of effector Treg cells in murine lupus. Arthritis Rheum. (2016) 68:1454–66. doi: 10.1002/art.39599
78. Haufe S, Haug M, Schepp C, Kuemmerle-Deschner J, Hansmann S, Rieber N, et al. Impaired suppression of synovial fluid CD4+CD25− T cells from patients with juvenile idiopathic arthritis by CD4+CD25+ Treg cells. Arthritis Rheum. (2011) 63:3153–62. doi: 10.1002/art.30503
79. Wehrens EJ, Mijnheer G, Duurland CL, Klein M, Meerding J, van Loosdregt J, et al. Functional human regulatory T cells fail to control autoimmune inflammation due to PKB/c-akt hyperactivation in effector cells. Blood. (2011) 118:3538–48. doi: 10.1182/blood-2010-12-328187
80. Lee W, Lee GR. Transcriptional regulation and development of regulatory T cells. Exp Mol Med. (2018) 50:e456. doi: 10.1038/emm.2017.313
81. Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. (2018) 19:665–73. doi: 10.1038/s41590-018-0120-4
82. Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Heterogeneity of natural Foxp3+ T cells: a committed regulatory T-cell lineage and an uncommitted minor population retaining plasticity. Proc Natl Acad Sci USA. (2009) 106:1903–8. doi: 10.1073/pnas.0811556106
83. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Science. (2010) 329:1667–71. doi: 10.1126/science.1191996
84. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. (2009) 10:1000–7. doi: 10.1038/ni.1774
85. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
86. MacDonald KG, Dawson NAJ, Huang Q, Dunne JV, Levings MK, Broady R. Regulatory T cells produce profibrotic cytokines in the skin of patients with systemic sclerosis. J Allergy Clin Immunol. (2015) 135:946–55.e9. doi: 10.1016/j.jaci.2014.12.1932
87. Duurland CL, Brown CC, O'Shaughnessy RFL, Wedderburn LR. CD161+ Tconv and CD161+ Treg share a transcriptional and functional phenotype despite limited overlap in TCRβ repertoire. Front Immunol. (2017) 8:103. doi: 10.3389/fimmu.2017.00103
88. Pesenacker AM, Bending D, Ursu S, Wu Q, Nistala K, Wedderburn LR. CD161 defines the subset of FoxP3+ T cells capable of producing proinflammatory cytokines. Blood. (2013) 121:2647–58. doi: 10.1182/blood-2012-08-443473
89. Bending D, Giannakopoulou E, Lom H, Wedderburn LR. Synovial regulatory T cells occupy a discrete TCR niche in human arthritis and require local signals to stabilize FOXP3 protein expression. J Immunol. (2015) 195:5616–24. doi: 10.4049/jimmunol.1500391
90. Costa N, Marques O, Godinho SI, Carvalho C, Leal B, Figueiredo AM, et al. Two separate effects contribute to regulatory T cell defect in systemic lupus erythematosus patients and their unaffected relatives. Clin Exp Immunol. (2017) 189:318–30. doi: 10.1111/cei.12991
91. Solomou EE, Juang YT, Gourley MF, Kammer GM, Tsokos GC. Molecular basis of deficient IL-2 production in T cells from patients with systemic lupus erythematosus. J Immunol. (2001) 166:4216–22. Retrieved from: http://www.ncbi.nlm.nih.gov/pubmed/11238674
92. Mizui M, Tsokos GC. Targeting regulatory T cells to treat patients with systemic lupus erythematosus. Front Immunol. (2018) 9:786. doi: 10.3389/fimmu.2018.00786
93. Alexander T, Sattler A, Templin L, Kohler S, Groß C, Meisel A, et al. Foxp3+ Helios+ regulatory T cells are expanded in active systemic lupus erythematosus. Ann Rheum Dis. (2013) 72:1549–58. doi: 10.1136/annrheumdis-2012-202216
94. Golding A, Hasni S, Illei G, Shevach EM. The percentage of FoxP3+Helios+ Treg cells correlates positively with disease activity in systemic lupus erythematosus. Arthritis Rheum. (2013) 65:2898–906. doi: 10.1002/art.38119
95. Morikawa H, Sakaguchi S. Genetic and epigenetic basis of Treg cell development and function: from a FoxP3-centered view to an epigenome-defined view of natural Treg cells. Immunol Rev. (2014) 259:192–205. doi: 10.1111/imr.12174
96. Chen Z, Barbi J, Bu S, Yang H-Y, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity. (2013) 39:272–85. doi: 10.1016/j.immuni.2013.08.006
97. van Loosdregt J, Fleskens V, Fu J, Brenkman AB, Bekker CPJ, Pals CEGM, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity. (2013) 39:259–71. doi: 10.1016/j.immuni.2013.05.018
98. Wehrens EJ, Prakken BJ, van Wijk F. T cells out of control—impaired immune regulation in the inflamed joint. Nat Rev Rheumatol. (2012) 9:34–42. doi: 10.1038/nrrheum.2012.149
99. Petrelli A, Wehrens EJ, Scholman RC, Prakken BJ, Vastert SJ, van Wijk F. Self-sustained resistance to suppression of CD8+ Teff cells at the site of autoimmune inflammation can be reversed by tumor necrosis factor and interferon-γ blockade. Arthritis Rheumatol. (2016) 68:229–36. doi: 10.1002/art.39418
100. Venigalla RKC, Tretter T, Krienke S, Max R, Eckstein V, Blank N, et al. Reduced CD4+, CD25− T cell sensitivity to the suppressive function of CD4+, CD25high, CD127−/low regulatory T cells in patients with active systemic lupus erythematosus. Arthritis Rheum. (2008) 58:2120–30. doi: 10.1002/art.23556
101. Riley JL. PD-1 signaling in primary T cells. Immunol Rev. (2009) 229:114–25. doi: 10.1111/j.1600-065X.2009.00767.x
102. Wehrens EJ, Vastert SJ, Mijnheer G, Meerding J, Klein M, Wulffraat NM, et al. Anti-tumor necrosis factor α targets protein kinase B/c-Akt-induced resistance of effector cells to suppression in juvenile idiopathic arthritis. Arthritis Rheum. (2013) 65:3279–84. doi: 10.1002/art.38132
103. Fantini MC, Rizzo A, Fina D, Caruso R, Sarra M, Stolfi C, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. (2009) 136:1308–16.e1–3. doi: 10.1053/j.gastro.2008.12.053
104. Wing JB, Sakaguchi S. TCR diversity and Treg cells, sometimes more is more. Eur J Immunol. (2011) 41:3097–100. doi: 10.1002/eji.201142115
105. Adeegbe D, Matsutani T, Yang J, Altman NH, Malek TR. CD4(+) CD25(+) Foxp3(+) T regulatory cells with limited TCR diversity in control of autoimmunity. J Immunol. (2010) 184:56–66. doi: 10.4049/jimmunol.0902379
106. Föhse L, Suffner J, Suhre K, Wahl B, Lindner C, Lee C-W, et al. High TCR diversity ensures optimal function and homeostasis of Foxp3+ regulatory T cells. Eur J Immunol. (2011) 41:3101–13. doi: 10.1002/eji.201141986
107. Nishio J, Baba M, Atarashi K, Tanoue T, Negishi H, Yanai H, et al. Requirement of full TCR repertoire for regulatory T cells to maintain intestinal homeostasis. Proc Natl Acad Sci USA. (2015) 112:12270–5. doi: 10.1073/pnas.1516617112
108. Yu H, Paiva R, Flavell RA. Harnessing the power of regulatory T cells to control autoimmune diabetes: overview and perspective. Immunology. (2017) 153:161–70. doi: 10.1111/imm.12867
109. Henderson LA, Volpi S, Frugoni F, Janssen E, Kim S, Sundel RP, et al. Next-generation sequencing reveals restriction and clonotypic expansion of Treg cells in juvenile idiopathic arthritis. Arthritis Rheumatol. (2016) 68:1758–68. doi: 10.1002/art.39606
110. Rossetti M, Spreafico R, Consolaro A, Leong JY, Chua C, Massa M, et al. TCR repertoire sequencing identifies synovial Treg cell clonotypes in the bloodstream during active inflammation in human arthritis. Ann Rheum Dis. (2016) 76:135–41. doi: 10.1136/annrheumdis-2015-208992
111. Okubo Y, Mera T, Wang L, Faustman DL. Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep. (2013) 3:3153. doi: 10.1038/srep03153
112. Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, et al. The TNF receptor superfamily-NF-κB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep. (2017) 20:2906–20. doi: 10.1016/j.celrep.2017.08.068
113. Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, et al. TNFR2 ligation in human T regulatory cells enhances IL2-induced cell proliferation through the non-canonical NF-κB pathway. Sci Rep. (2018) 8:12079. doi: 10.1038/s41598-018-30621-4
114. Zaragoza B, Chen X, Oppenheim JJ, Baeyens A, Gregoire S, Chader D, et al. Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med. (2016) 22:16–7. doi: 10.1038/nm.4019
115. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
116. Sanchez Rodriguez R, Pauli ML, Neuhaus IM, Yu SS, Arron ST, Harris HW, et al. Memory regulatory T cells reside in human skin. J Clin Invest. (2014) 124:1027–36. doi: 10.1172/JCI72932
117. Delemarre EM, van den Broek T, Mijnheer G, Meerding J, Wehrens EJ, Olek S, et al. Autologous stem cell transplantation benefits autoimmune patients through functional renewal and TCR diversification of the regulatory T cell compartment. Blood. (2015) 127:91–101. doi: 10.1182/blood-2015-06-649145
118. Musters A, Klarenbeek PL, Doorenspleet ME, Balzaretti G, Esveldt REE, van Schaik BDC, et al. In rheumatoid arthritis, synovitis at different inflammatory sites is dominated by shared but patient-specific T cell clones. J Immunol. (2018) 201:417–22. doi: 10.4049/jimmunol.1800421
Keywords: T-cell adaptation, autoimmune-inflammation, regulatory T-cell, CD8 T-cell, tissue-resident memory T-cell, clonal expansion, JIA, SLE
Citation: Mijnheer G and van Wijk F (2019) T-Cell Compartmentalization and Functional Adaptation in Autoimmune Inflammation: Lessons From Pediatric Rheumatic Diseases. Front. Immunol. 10:940. doi: 10.3389/fimmu.2019.00940
Received: 29 November 2018; Accepted: 11 April 2019;
Published: 09 May 2019.
Edited by:
Philippe Saas, INSERM U1098 Interactions Hôte-Greffon-Tumeur & Ingénierie Cellulaire et Génique, FranceReviewed by:
Alejandra Pera, Universidad de Córdoba, SpainCopyright © 2019 Mijnheer and van Wijk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Femke van Wijk, Zi52YW53aWprQHVtY3V0cmVjaHQubmw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.