 Selena Vigano1†
Selena Vigano1† Dimitrios Alatzoglou1†
Dimitrios Alatzoglou1† Melita Irving1*†
Melita Irving1*† Christine Ménétrier-Caux2
Christine Ménétrier-Caux2 Christophe Caux2
Christophe Caux2 Pedro Romero3George Coukos1*
Pedro Romero3George Coukos1*- 1Department of Oncology, Ludwig Institute for Cancer Research Lausanne, Lausanne University Hospital and University of Lausanne, Lausanne, Switzerland
- 2Department of Immunology Virology and Inflammation, INSERM 1052, CNRS 5286, Léon Bérard Cancer Center, Cancer Research Center of Lyon, University of Lyon, University Claude Bernard Lyon 1, Lyon, France
- 3Department of Oncology, University of Lausanne, Lausanne, Switzerland
T cells play a critical role in cancer control, but a range of potent immunosuppressive mechanisms can be upregulated in the tumor microenvironment (TME) to abrogate their activity. While various immunotherapies (IMTs) aiming at re-invigorating the T-cell-mediated anti-tumor response, such as immune checkpoint blockade (ICB), and the adoptive cell transfer (ACT) of natural or gene-engineered ex vivo expanded tumor-specific T cells, have led to unprecedented clinical responses, only a small proportion of cancer patients benefit from these treatments. Important research efforts are thus underway to identify biomarkers of response, as well as to develop personalized combinatorial approaches that can target other inhibitory mechanisms at play in the TME. In recent years, adenosinergic signaling has emerged as a powerful immuno-metabolic checkpoint in tumors. Like several other barriers in the TME, such as the PD-1/PDL-1 axis, CTLA-4, and indoleamine 2,3-dioxygenase (IDO-1), adenosine plays important physiologic roles, but has been co-opted by tumors to promote their growth and impair immunity. Several agents counteracting the adenosine axis have been developed, and pre-clinical studies have demonstrated important anti-tumor activity, alone and in combination with other IMTs including ICB and ACT. Here we review the regulation of adenosine levels and mechanisms by which it promotes tumor growth and broadly suppresses protective immunity, with extra focus on the attenuation of T cell function. Finally, we present an overview of promising pre-clinical and clinical approaches being explored for blocking the adenosine axis for enhanced control of solid tumors.
Introduction
IMT has led to unprecedented clinical success for some advanced cancer patients and has been accepted as a new pillar of cancer therapy (1). Thus, the identification of biomarkers predicting response to IMT, as well as the development of combinatorial strategies for increasing its effectiveness in more patients, and against a broader range of tumor-types, have become important areas of research (2). The nucleoside adenosine, involved in the regulation of multiple diverse physiological processes either as an intracellular metabolite of nucleic acid synthesis and energy-charge regulation or as an intercellular messenger in neurological, cardiovascular and immunological systems, has recently emerged as a major immuno-metabolomic checkpoint in tumors (3). Conditions of stress, such as hypoxia, lead to the accumulation of extracellular adenosine, predominantly derived from enzymatic ATP catabolism, which can act directly on tumor cells expressing adenosine receptors to promote their growth, survival and dissemination. In addition, adenosine, which under physiological conditions serves as an immuno-regulatory molecule to protect normal tissues from uncontrolled inflammation, can impair anti-tumor immunity, both through the attenuation of protective immune cells including T cells, NK cells, and dendritic cells (DCs), and by enhancing the suppressive capacity of T regulatory cells (Tregs), and myeloid-derived suppressor cells (MDSCs), amongst others. Here we review the targeting of the adenosine pathway to promote immune function and tumor control, with focus on T-cell activity, important experimental findings and an overview of clinical testing.
Regulation of Adenosine Levels in Healthy vs. Malignant Tissue
Extracellular adenosine, a nucleoside and derivative of ATP, is involved in the regulation of diverse physiological processes including vasodilation (4), kidney-exerted water reabsorption (5), pain perception (6), and fine-tuning of the sleep–wake cycle (7). Even though levels of extracellular adenosine within healthy tissues are negligible (8–11), upon injury this nucleoside sharply accumulates at the interstitium where it potently restricts immune responses (12) and directly promotes wound healing (13). Under homeostatic conditions in healthy tissues, the cytosolic concentration of ATP ranges from 1 to 10 mM (14), while its extracellular levels are negligible (15). This sharp gradient can be rapidly disrupted however upon breaches of the plasma membrane induced by necrosis, apoptosis or mechanical stress, as well as by regulated ATP efflux. The latter, induced by a variety of stimuli including hypoxia, ischemia and inflammation, has been shown to extensively occur via exocytosis, transmembrane transfer through ATP-binding cassette (ABC) transporters, as well as by diffusion through a variety of anion channels or non-selective plasma membrane pores formed by connexins, pannexin-1 or the ATP receptor P2X7R (16–18). For instance, stimulated T cells release ATP through pannexin-1 hemi-channels and via exocytosis (19, 20).
Once in the extracellular space, ATP undergoes rapid stepwise dephosphorylation by ecto-nucleotidases (21, 22) including the E-NTPDase CD39, which converts ATP or ADP to ADP or AMP, respectively, and the 5′-nucleotidase CD73, which dephosphorylates AMP to adenosine (18, 23) (Figure 1). Additional enzymes whose ecto-activity contributes toward extracellular adenosine generation are other E-NTPDases, members of the ecto-phosphodiesterase/pyrophosphatase (E-NPP) family, nicotinamide adenine dinucleotide (NAD+) glycohydrolases, the prostatic acid phosphatase (PAP), and the alkaline phosphatase (ALP) (21) (Figure 1). Briefly, the co-enzyme NAD+, another key cellular component whose extracellular concentration significantly rises in injured tissue (24, 25), is converted to adenosine diphosphate ribose (ADPR) by the NAD+ glycohydrolase CD38 (26), while ADPR as well as ATP are metabolized to AMP by the E-NPP CD203a (27). Moreover, PAP, which is predominantly, but non-exclusively, expressed in prostate tissue (28), is capable of converting extracellular AMP to adenosine (29), whereas ALP catalyzes the hydrolysis of ATP, ADP and AMP to adenosine (21). Finally, adenosine can also be produced intracellularly either by S-adenosylhomocysteine hydrolase (SAHH)-exerted hydrolysis of S-Adenosylhomocysteine (SAH), a metabolite of the transmethylation pathway, or due to soluble CD73-mediated catabolism of AMP, a nucleoside participating in multiple cellular processes and whose concentration rises within cells of low energy charge (30) (Figure 1). Intracellularly-generated adenosine can be secreted in a diffusion limited-manner through bidirectional equilibrative nucleoside transporters (ENTs) (31). However, although there is evidence suggesting that hypoxia can boost intracellular adenosine production (32, 33), the contribution of this pathway toward injury-caused interstitial adenosine buildup is considered minor due to concurrent hypoxia-induced downregulation of the aforementioned transporters (34, 35). Given its diverse effects, adenosine presence at the extracellular space is subject to tight spatiotemporal control (12, 13, 36). For instance, extracellular accumulation of adenosine is counteracted by its inward transfer through ENTs or concentrative, sodium gradient-dependent, symporters (31) as well as by the function of intra/extracellular adenosine deaminase (ADA) and of cytosolic adenosine kinase (ADK), which respectively convert adenosine to inosine or AMP (37) (Figure 1).
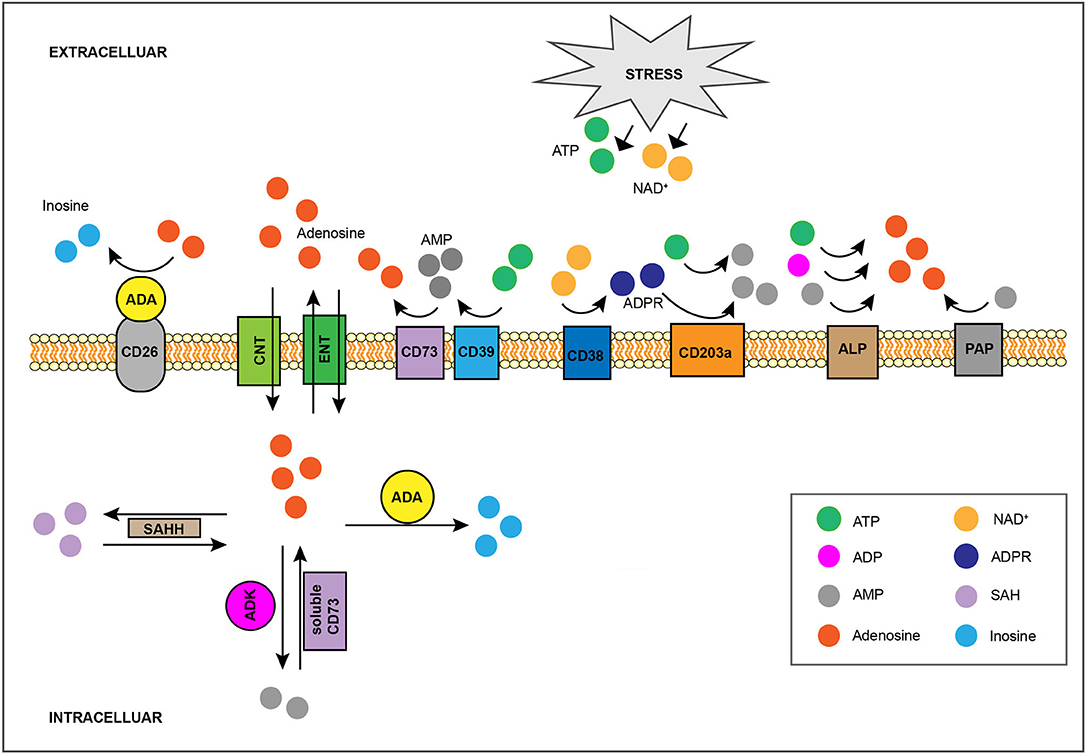
Figure 1. Regulation of interstitial adenosine levels in injured tissue. Stress-induced, extracellular buildup of ATP or NAD+ fuels catabolic adenosine-generating pathways, such as the one mediated by CD39 and CD73. The activity of other ecto-nucleotidases including CD38, CD203a, ALP, and PAP, also contribute toward extracellular adenosine accumulation. Adenosine can also be produced intracellularly by SAHH-exerted hydrolysis of SAH, as well as by soluble CD73-mediated catabolism of AMP, and it can be exported by ENTs in a diffusion-limited manner. On the flip side, the combination of CD26-bound ADA activity and of adenosine cellular uptake, either through equilibrative ENTs or via concentrative CNTs, limits interstitial adenosine levels. Intracellularly, adenosine can be eliminated via its conversion to SAH by SAHH, to AMP by ADK, or to inosine by ADA. SAHH, S-adenosylhomocysteine hydrolase; SAH, S-Adenosylhomocysteine; ENTs, equilibrative nucleoside transporters; CNTs, concentrative nucleoside transporters; ADK, adenosine kinase; ADA, adenosine deaminase.
In contrast to homeostatic conditions, ATP levels are highly elevated in the TME as a result of necrosis, apoptosis, hypoxia, and persistent inflammation (17, 18), and intra-tumoral adenosine levels can reach micromolar concentrations (9, 10, 38). ATP catabolism in tumors is primarily mediated by CD39 and CD73 (39–41), and high expression of these ecto-nucleotidases is strongly associated with poor clinical outcome for patients suffering a variety of cancer-types (3, 42, 43). In particular, CD39 and/or CD73 (over)expression has been detected on the surface of tumor cells (39, 44–51), cancer-associated fibroblasts (CAFs) (52–54), mesenchymal stem cells and stromal cells (55–57), endothelial cells (ECs) (45, 46, 51), myeloid derived suppressor cells (MDSCs) (58–60), tumor associated macrophages (TAMs) (53, 61), Tregs (46, 62–64), Th17 cells (65) and of antigen experienced/exhausted conventional CD4+ and CD8+ T cells (64, 66–68). In addition, CD39/CD73-bearing exosomes (69, 70), released by tumor cells (71), Tregs (72), and MDSCs (57, 73) further contribute to adenosine generation. Currently, hypoxia as well as incessant inflammation are considered to be the main drivers of intra-tumoral CD39 and CD73 overexpression. Namely, hypoxia-induced (74, 75) HIF1α (76–79) and Sp1 (80) activity promotes expression of these ecto-nucleotidases. Along the same lines, signaling pathways initiated by inflammation-associated molecules, such as IL-2 (81), IL-6 (66, 82), IL-1β (83), TNFα (83–85), type I IFNs (86, 87), IL-27 (66, 88), TGFβ (82, 89, 90) as well as by inducers of the Wnt (91, 92) or cAMP (83, 93–95) signaling pathways also boost CD39 (66, 81, 82, 88, 89, 95) and CD73 (81–87, 89–94) levels.
Although CD39 and CD73-mediated catabolism of extracellular ATP is considered to account for the bulk of intra-tumoral adenosine generation, expression levels of ecto-enzymes participating in alternative adenosine production pathways also rise in the advent of cancer. For instance, CD38 is frequently upregulated within neoplastic tissues (26, 96, 97) and sporadic evidence suggests that CD203a levels also increase on TME components (98, 99). Along the same lines, the serum concentration of PAP increases during prostate cancer progression (100) while others suggest it gets upregulated on cancerous tissue as well (28). Finally, several studies have demonstrated elevated levels of ALP on cancer cells (101, 102) as well as a correlation of serum ALP levels and disease stage (103–105). Critically, the relative contribution of these alternative adenosine-producing pathways toward intra-tumoral buildup of this nucleoside remains to be determined. Finally, along with aberrant production, defective uptake resulting from the down-modulation of equilibrative (106, 107) as well as concentrative (108–110) nucleoside transporters, also driven by hypoxia (34, 35, 111), further contributes to adenosine accumulation in the TME.
Adenosine Receptor Signaling
Four adenosine receptors (ARs), all coupled to G-proteins, have been identified; A1R, A2AR, A2BR, and A3R (112, 113). While A1, A2A, and A3 are described as high affinity adenosine receptors (EC50 in the range of 0.1–0.7 μM), A2BR is considered as low affinity because it is activated only in the presence of high concentrations of adenosine (EC50 of 15–25 μM), such as may be found in the TME or under other pathological conditions. Upon adenosine binding, these GPCRs induce the replacement of GDP bound by the heterotrimeric G proteins, a class of GTP hydrolases, with GTP thus resulting in the dissociation of the latter into Gα monomers and Gβγ dimers, now free to modulate downstream effectors before their GTP hydrolysis-induced re-association (114).
Of the four classes of Gα proteins characterized to date, namely Gαs, Gαi, Gαq/11, Gα12/13, only Gαs and Gαi directly influence the activity of adenylyl cyclases (AC), enzymes that catalyze the cyclization of intracellular ATP into cyclic adenosine monophosphate (cAMP) (114). In terms of function, triggering of the Gαs-coupled A2AR and A2BR promotes AC activity (115). In contrast, stimulation of the Gαi-paired A1R and A3R inhibits cAMP generation (115). Although modulation of intracellular cAMP content constitutes a crucial aspect of extracellular adenosine-exerted regulation, stimulation of its receptors induces a variety of cAMP-independent biochemical effects, such as A1R/Gαi, A2BR/Gαq/11, A3R/Gαq/11-induced stimulation of phospholipase C (PLC) activity and A1R/Gαi, A2AR, A2BR/Gq/11, A3R-mediated ERK activation (115). Finally, elevation of extracellular adenosine levels induces receptor-independent boosting of AMP-activated protein kinase (AMPK) via intracellular transfer of this nucleoside followed by its conversion to AMP (116, 117).
Adenosine-Induced Intracellular cAMP Accumulation Impairs T Cell-Mediated Antitumor Responses
It is now understood that T cells play a major role in tumor control (118–120). As will be discussed however, elevated levels of adenosine in the TME can potently impair T-cell function by inducing accumulation of intracellular cAMP.
Levels of Adenosine Receptors on the T Cell Surface
Murine (121–127) and human (128–132) T cells express all four ARs, and levels of A2AR (122, 124–127, 129), A2BR (126, 127, 130), and A3R (127, 131) increase upon T cell activation. However, the biology of T cells is primarily affected by the predominantly expressed A2AR (122, 123, 128, 132). Of note, similarly to CD39 and CD73, A2AR, and A2BR are upregulated due to hypoxia-induced HIF1α (133) transcriptional activity. Moreover, mRNA levels of both A2AR and A2BR are upregulated in T cells specifically upon provision of anergic stimulus (134). Validating these findings, adoptively transferred tumor-specific T cells isolated from tumors contained twice the A2AR mRNA levels than counterpart T cells isolated from spleens of tumor-bearing mice (135). Since triggering of the different ARs initiates diverse and even antagonistic signaling pathways, the net cellular effects of adenosine are determined by the relative surface expression of its receptors. It is clear, however, that treatment of human (136, 137) or murine (38, 126, 138, 139) T cells with adenosine or adenosine analogs induces A2AR- (38, 126, 137–139) as well as A2BR- (38, 136) mediated intracellular cAMP build-up.
The Mechanics of cAMP-Mediated T Cell Suppression
The secondary messenger of adenosine cAMP, also a derivative of ATP, is involved in a diverse range of cellular functions including metabolism, transcription, and growth, while oscillations of its levels within distinct cell populations are paramount for the regulation of multiple bodily functions, such as endocrine, cardiovascular, neuronal, and immune processes (140). The intracellular concentration of cAMP is determined by the antagonistic activities of ACs, and of cAMP-specific phosphodiesterases (PDEs), proteins that hydrolyze cAMP to 5′-AMP. Although cAMP can diffuse within the cytosol, the co-localization of the highly-targeted AC and PDE activities in particular subcellular regions results in the formation of distinct cAMP microdomains within which co-localized cAMP effectors are activated by in-situ generated cAMP before its swift degradation (141, 142). The formation of such microdomains is mediated by AKAPs, scaffold proteins shown to bind ACs, PDEs as well as effectors of the cAMP-signaling pathway (143, 144). Of the 10 currently identified AC isoforms, T cells express AC3, AC6, AC7 and AC9 (145, 146) with most cAMP production catalyzed by AC7 (146). As previously described, A2AR and A2BR are coupled to Gαs which stimulates the activity of ACs. Of the 11 PDE families characterized to date, isoforms belonging to the relatively strong-affinity (147) cAMP-binding families of PDE1 (145, 148), PDE3 (145, 149), PDE4 (145, 149), PDE7 (145, 149–151), PDE8 (145, 151, 152), and PDE11 (145) have been observed within T cells, with most cAMP hydrolysis carried out by PDE3 and PDE4 isoforms (148, 149, 153). Of note, cAMP levels in T cells can also be augmented by additional factors in the TME including prostaglandin E2 (PGE2) (154), norepinephrine (155), histamine (156), the neuropeptides VIP and PACAP (157, 158), and low pH (159). Additional phenomena contributing toward cAMP build-up within effector T cells include TCR triggering (160, 161) as well as direct cAMP transfer by tumor cells (162) or Tregs (163) via gap junctions.
Accumulation of cAMP within the T cell cytosol induces the activity of protein kinase A (PKA) and of exchange protein directly activated by cAMP (EPAC). PKA, the dominant effector of the cAMP signaling pathway (164) is an heterotetramer comprising two catalytic (C) subunits, maintained in an inactive state by tethering to two regulatory (R) subunits (165). Binding of cAMP to the R-subunits induces a conformational change resulting in the release of the C-subunits (166). As a result, liberated PKA C-subunits within T cells phosphorylate a wide variety of substrates affecting multiple signaling pathways (167). It is well established that sustained PKA activity disrupts signaling induced by triggering of the TCR, of the co-stimulatory receptor CD28 (168, 169) as well as by the IL-2 receptor (IL-2R) (170). Negative regulators of these signaling pathways, whose activity is bolstered by PKA, include Csk (171), SHP-1 (172), SHIP1 (173), HPK1 (174), and PP2A (175). Conversely, PLCγ1 (176, 177), Raf-1 (178, 179), JAK3 (170), RhoA (180, 181), VASP (182) as well as the transcription factors NFAT (183, 184) and NFkB (185, 186) constitute mediators or endpoint effectors of the aforementioned axes whose activity is dampened by PKA.
PKA activity also significantly affects cytoplasmic potassium concentration within T cells by inhibiting the activity of Kv1.3 (187) and KCa3.1 (188, 189), channels which are responsible for the bulk of potassium efflux by T cells (190). In a negative-feedback fashion, PKA induces reduction of the cytosolic cAMP concentration by directly phosphorylating AC6 in an inhibitory fashion (191) as well as isoforms of PDE3 (192), PDE4 (193, 194), PDE8A (195) in a stimulatory manner. At the transcriptional level, PKA augments the activity of CREB cAMP responsive element binding (CREB), cAMP responsive element modulator (CREM) and activating transcription factor-1 (ATF-1) (196), which induce or counteract the transcription of multiple inflammation-relevant genes such as IL-2 (197–199), IFNγ (200–202), IL-4 and IL-13 (203, 204), IL-17 (205–208), and FoxP3 (209, 210). Specifically, PKA promotes the transcriptional activity of CREB by phosphorylating it thus increasing its affinity for its co-activators CBP and p300 (211), and by promoting the nuclear localization of CRTC (212), another family of CREB co-activators. Finally, PKA directly phosphorylates and activates ATF-1 (213) as well as distinct CREM isoforms (214) in a way similar to CREB.
The guanine nucleotide exchange factor EPAC1 is another effector of cAMP in T cells (215, 216). cAMP binds to the cAMP-responsive N-terminal region of EPAC1 and induces an open conformation rendering its catalytic core accessible to its effectors (217, 218). The most heavily characterized EPAC1 effector in T cells is the anergy-associated GTPase Rap1 (219, 220) which in its GTP-bound form is targeted to the plasma membrane (221) where it inhibits TCR-induced MEK-ERK activation by sequestering Raf-1 (220, 222).
Overview of the Inhibitory Effects of cAMP on T-Cell Biology
A variety of molecules, including cAMP analogs, direct AC activators (e.g., forskolin and cholera toxin) and PDE inhibitors have been used to elucidate the diverse effects of intracellular cAMP accumulation on T-cell biology. In the presence of such molecules (223–228) as well as by A2AR triggering (125, 126, 229) the capacity of previously unstimulated T cells, CD4+ or unfractionated, to differentiate post-activation toward cells that produce Th1 (125, 126, 223–225, 229) or Th2 (226–229)-signature cytokines is drastically diminished. This occurs in a PKA-dependent fashion (230, 231) through multi-level disruption of TCR- or CD28-induced signaling (122, 232). Intriguingly, A2AR agonist-induced impairment of IFNγ production remains evident even when A2AR agonist-pretreated T cells are re-stimulated in the absence of this agent (139). Furthermore, agents that directly activate the cAMP pathway (233–235), as well as adenosine (122, 138, 232, 236, 237), have been shown to restrict stimulation-induced AKT activation (122, 232, 233, 238) and to induce stabilization of β-catenin, which restricts maturation toward terminally differentiated effector cells (239). Moreover, such agents can prevent FasL upregulation, thus averting FasL-mediated activation-induced cell death (AICD) (127, 138, 235, 237). Finally, such molecules abolish mitogenic-stimulus-induced T cell proliferation, in a PKA-dependent manner (240), by downmodulating the transmission of TCR/ CD28- and IL-2 (241)-initiated signaling, as well as IL-2 production (126, 229, 231) and IL-2Ra expression (242).
Forskolin, cAMP analogs, PDE inhibitors (152, 243–245) and adenosine (188, 246–248) also diminish T cell adherence (152, 243, 246, 248) by down-modulating the expression levels of ICAM-1 (249, 250) as well as of the integrins α4 (251, 252) and β2 (251, 253), components of VLA-4 and LFA-1, respectively. Such agents also impair T-cell migration (188, 244, 245, 247) by inducing KCa3.1 inhibition (188, 189). In addition, cAMP-mediated signaling (230, 254, 255) or the presence of A2AR agonists (139, 168, 230, 231) diminishes T cell cytotoxicity, in a PKA-dependent manner (168, 230, 231), probably as a result of impaired TCR signaling, motility/adhesion, granule exocytosis (138), as well as due to decreased expression of FasL, Granzyme B (GzB), and perforin (127).
Lastly, cholera toxin (256), PDE inhibitors (257–259), forskolin (157) and A2AR agonists (126, 260) not only skew T cells toward the Treg lineage via induction of FoxP3 expression (126, 256–258, 260), but also enhance the capacity of Treg cells to suppress responder T cells (258–260), at least in part by upregulating CTLA-4 levels (157, 260). Thus, cAMP can potently diminish the differentiation and effector activities of CD4+ and CD8+ T cells, while promoting the differentiation toward Tregs, as well as their suppressive capacity.
The Pleiotropic Effects of Adenosine in the Tumor Microenvironment
Along with T cells, many other cell types in the TME including other protective or suppressive immune infiltrates, tumor-associated fibroblasts, endothelial cells and cancer cells also express functional ARs (3, 261–266). Here we briefly describe the effects of adenosine-induced signaling on them (Figure 2).
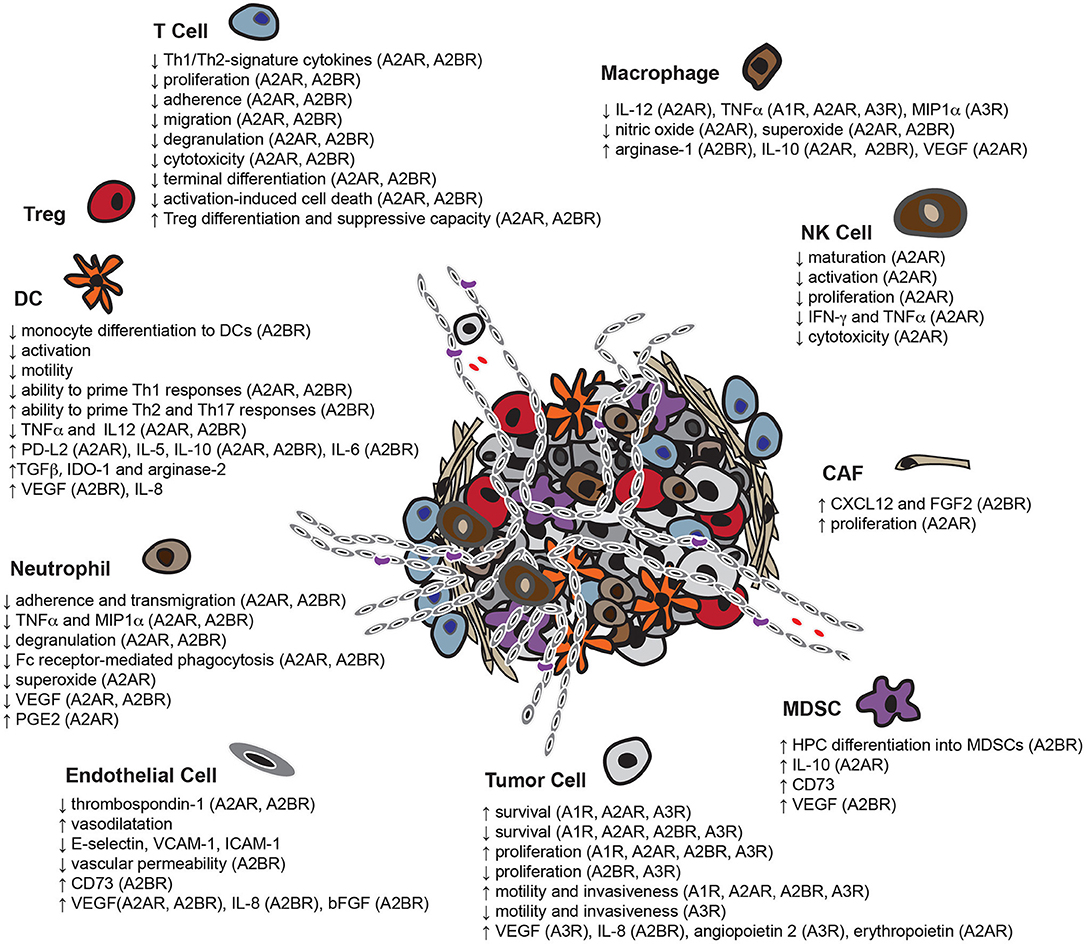
Figure 2. Overview of the pleiotropic effects of adenosine in the TME. Adenosine enables tumor cells to escape immune-surveillance by limiting the functionality of multiple potentially protective immune infiltrates including T cells, DCs, NK cells, macrophages and neutrophils, while enhancing the activity of immunosuppressive cell-types, such as MDSCs and Tregs. In addition, adenosine not only assists tumor cells in co-opting adjacent fibroblasts for support, but also induces the formation of new blood vessels. Adenosine also affects the capacity of some tumor cell-types to survive, proliferate, migrate and invade neighboring tissues (HPC, bone marrow-derived hematopoietic progenitor cells).
Dendritic Cells
The biology of DCs, specialized antigen presenting cells (APCs) and critical messengers between the innate and adaptive immune system, can be severely impaired by adenosinergic signaling. For example, it has been reported that adenosine binding to A2BR (267) halts the differentiation of monocytes to DCs (267, 268). In addition, adenosine averts inflammatory stimulus-induced DC activation (269), whereas A2AR (270) and A2BR triggering (267, 271, 272) diminishes the capacity of DCs to prime Th1 immune responses (267, 270, 271) but rather prompts DCs to skew naïve T cell differentiation toward Th2 (267, 271) and Th17 (272) lineages. Adenosine-treated DCs exhibit decreased expression or secretion of TNFα and IL-12 (268–271, 273) and enhanced production of IL-5 (270), IL-10 (267, 268, 270, 273), IL-6 (267, 272) and TGFβ (267). Moreover, such DCs are less motile due to chemokine receptor downregulation (274), and have a tolerogenic effect on the TME due to overexpression of TGFβ (267), IL-10, IDO-1 (267), arginase-2 (267, 275), as well as A2AR-mediated upregulation of PD-L2 (276). Finally, adenosine compels DCs to secrete the proangiogenic factors VEGF (267, 275) in an A2BR-dependent manner as well as IL-8 (267).
Macrophages
Stimulation of adenosine receptors hinders the differentiation of monocytes to macrophages, probably through cAMP accumulation (277). Moreover, by engaging A1R (278), A2AR (278–282), A3R (281, 283) or setting off Gαs-paired ARs (284), adenosine reduces the pro-inflammatory activity of macrophages by dampening their ability to produce IL-12 (279), TNFα (278–280, 282, 283), macrophage inflammatory protein-1α (MIP1α) (281), nitric oxide (278, 285) and superoxide (284). In addition, by triggering A2AR (282, 286, 287), A2BR (288, 289) or unidentified ARs, adenosine promotes an M2 polarization of macrophages by inducing upregulation of arginase-1 (288, 290), IL-10 (279, 286, 289) and VEGF production (282, 287).
NK Cells
A2AR stimulation by adenosine not only restricts the NK maturation (291), but also their capacity for stimulus-induced CD69 upregulation (292, 293), proliferation (291, 294) as well as IFNγ (292, 293) and TNFα (294, 295) production. Furthermore, largely via A2AR triggering, adenosine diminishes target cell killing by NK cells (292, 294, 296–298).
Neutrophils
Adenosine exerts a variety of inhibitory effects on neutrophils. For example, triggering of A2AR (299–303), A3R (304), non-specified A2Rs (304–307) or ARs dampens their ability to adhere (299, 305, 308, 309), transmigrate (310), secrete TNFα and MIP1α (300, 306), degranulate (301, 302, 304, 311), perform Fc receptor-mediated phagocytosis (307) and produce superoxide (299, 301–303). Interestingly, others claim that A2AR and A2BR signaling has been shown to suppress VEGF production (310). Finally, A2AR stimulation prompts neutrophils to secrete higher levels of PGE2 (312).
MDSCs
A2BR-mediated signaling boosts differentiation of bone marrow hematopoietic progenitors toward a tolerogenic myeloid-derived cell subset, the MDSCs (313). Moreover, A2AR activation promotes IL-10 production by MDSCs (314) and treatment with an adenosine analog results in increased expression of CD73 (313). Finally, it has also been shown that A2BR stimulation on MDSCs augments VEGF production (315).
Stromal Cells
Adenosine, along with critically contributing to the establishment of a tolerogenic TME, also enables tumors to subvert fibroblasts into supporting them and to induce formation of new blood vessels, processes essential to their growth and dissemination. CAFs, for example, are stromal cells that support tumors by secreting the pro-metastatic and angiogenic (316) chemokine CXCL12 (317), as well as the mitogenic (318) fibroblast growth factor 2 (FGF2) (319). Triggering of A2BR on the surface of CAFs boosts expression of both CXCL12 and FGF2 (320) whereas A2AR-induced signaling stimulates their proliferation (54). As previously mentioned, adenosine can stimulate VEGF secretion by multiple cell types found within the TME, which in turn promotes angiogenesis by supporting the survival, migration and proliferation of endothelial cells (321, 322). It has also been shown that A2AR (323) and A2BR (66) stimulation diminishes production of the anti-angiogenic factor thrombospondin-1 by endothelial cells. Furthermore, adenosine not only augments the rate of intra-tumoral nutrient delivery by inducing vasodilatation (324), but also hinders leukocyte extravasation (325) through downregulation of adhesion molecules, such as E-selectin (326, 327) VCAM-1 (326, 327) and ICAM-1 (327, 328) on the surface of endothelial cells, as well as by limiting vascular permeability (325, 327, 329–331) through A2BR activation (329–331). Finally, signaling initiated by triggering of A2AR (332, 333), A2BR (334–336) or non-specified ARs prompts endothelial cells to overexpress CD73 (334) as well as the proangiogenic factors VEGF (332, 333, 335, 336), IL-8 (335) and basic fibroblast growth factor (bFGF) (335, 336).
Tumor Cells
Adenosine binding to ARs on the surface of cancer cells has a profound impact on their biology. For example, the triggering of A1R (337, 338), A2AR (54, 339, 340), and A3R (339, 341–343) induces a variety of cellular responses that augment cancer cell survival such as AKT and ERK1/2 stimulation, as well as Bad inactivation (342). Additional responses to AR signaling contributing to bolstered cancer cell survival include upregulation of Bcl2 (343), downregulation of p53 (338) and Bax (343) as well as aversion of caspase-9 (343) and caspase-3 (54, 343, 344) activation. Paradoxically, extracellular adenosine has also been demonstrated to cause cancer cell death either by setting off A1R (345, 346), A2AR (341, 347), A2BR (348, 349), and A3R (339, 350–354) or via induction of AMPK activation upon its cellular uptake and subsequent conversion to AMP (345).
Moreover, A1R (337, 355), A2AR (341, 356), A2BR (344, 357–359), and A3R (343, 360) stimulation augments cancer cell proliferation through activation of PLC (356), protein kinase C-delta (PKC-δ) (356), AKT (356, 357), ERK1/2 (356–360), JNK (356, 358), and p38 (358). Furthermore, triggering of the ARs leads to upregulation of cyclins A (343), B (358), D (343, 358), E (337, 343, 358), estrogen receptor-α (355) as well as downregulation of the cell-cycle inhibitors p27 (337) and p21 (343, 358). Surprisingly, though, activation of A2BR (349) and A3R (341, 350, 353, 361–363) has also been reported to result in a potent cytostatic effect.
Motility (358, 359, 364–369) and invasiveness (358, 359, 367, 370) are additional features of cancer cells that are boosted upon engagement of A1R (364, 365), A2AR (366), A2BR (358, 359, 367, 368), and A3R (369, 370). In terms of mechanisms, signaling initiated by these receptors promotes filopodia formation (367) as well as expression of matrix metalloproteases (MMPs) (358, 359, 370) and FXYD5 (359), a cell membrane glycoprotein known to drive metastasis by reducing cell adhesion (371). In contrast, others claim that A3R triggering hinders the motility and invasiveness of cancer cells (372, 373). Finally, A2AR (374), A2BR (369, 375), and A3R (369, 375–377) stimulation on the surface of cancer cells promotes angiogenesis by boosting secretion of the pro-angiogenic factors VEGF (369, 375, 377), IL-8 (369, 375), angiopoietin 2 (376), and erythropoietin (374).
The contrasting consequences of triggering particular ARs, on the survival, proliferation or migration and invasiveness of tumor cells most probably occur due to the heterogeneity between cells and/or experimental settings employed to assess them. For instance, two different cancer cell lines of distinct tissue origin could have profoundly diverse AR expression profiles as well as different ability to transmit/terminate signaling initiated by these receptors. Moreover, they might have different capacity to produce adenosine, which once released into the medium can trigger ARs in an autocrine fashion. Finally, different concentrations used between experiments, as well as limited specificity of the AR agonists/antagonists, probably constitute additional factors contributing to the observed discrepancies.
Targeting Adenosinergic Signaling in Cancer Immunotherapy
Adenosine confers potent immunosuppressive as well as direct tumor-promoting effects in the TME. Thus, approaches to both blocking its generation and hindering binding to its receptors have become important areas of research (Figure 3). Indeed, extensive pre-clinical experimentation has firmly established that targeting the adenosinergic signaling on its own (Table 1) or in combination with emerging IMTs or established cancer treatments (Table 2) shows important promise and soundly supports the clinical evaluation (Table 3) of these concepts. Here we present an overview of such pre-clinical and clinical studies.
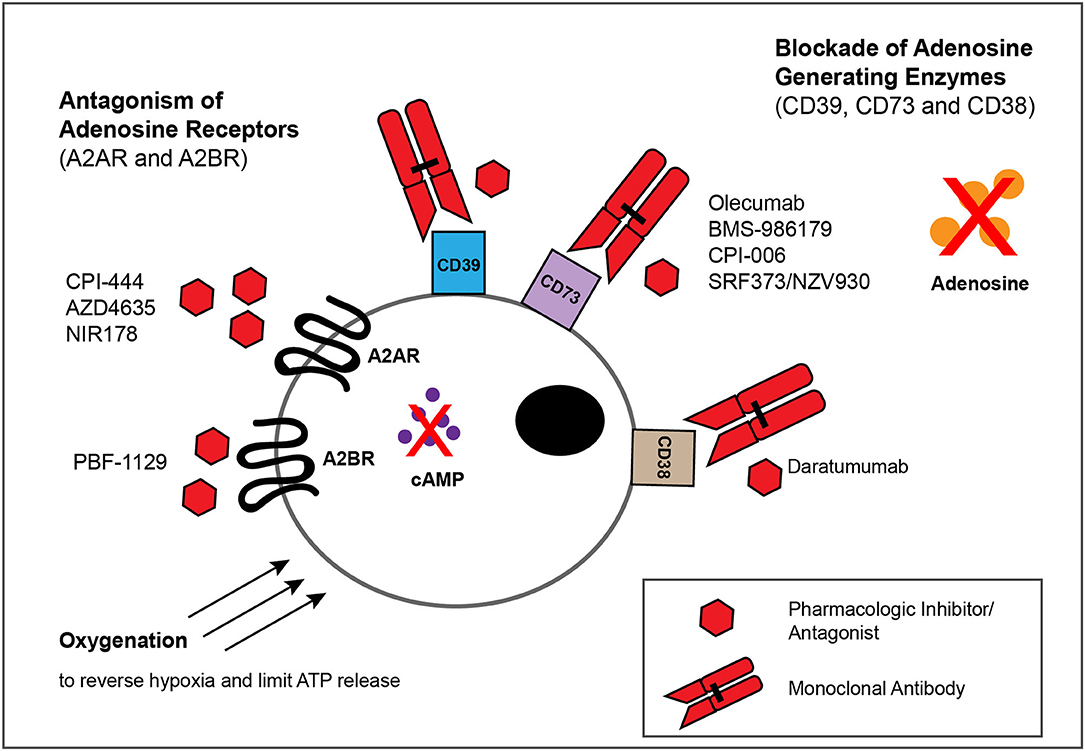
Figure 3. Approaches for blocking adenosinergic signaling in the TME. The inhibitory effects of adenosine in the TME can be circumvented by administration of mAbs or small molecules that target enzymes involved in the catabolism of ATP and NAD, such as CD39,CD73 and CD38, as well as by pharmacologic antagonists of A2AR and A2BR to block adenosine-mediated signaling. Whereas multiple such mAbs and pharmacologic inhibitors/antagonists display antitumor activity within murine models of solid tumors (Tables 1, 2), depicted are only those currently evaluated in patients with solid tumor malignancies (Table 3). Finally, treatments that reduce the extracellular export of ATP, such as oxygenation to reverse hypoxia, can attenuate adenosinergic signaling.

Table 1. Evaluation of adenosine-axis blockade in murine models of solid malignancies.
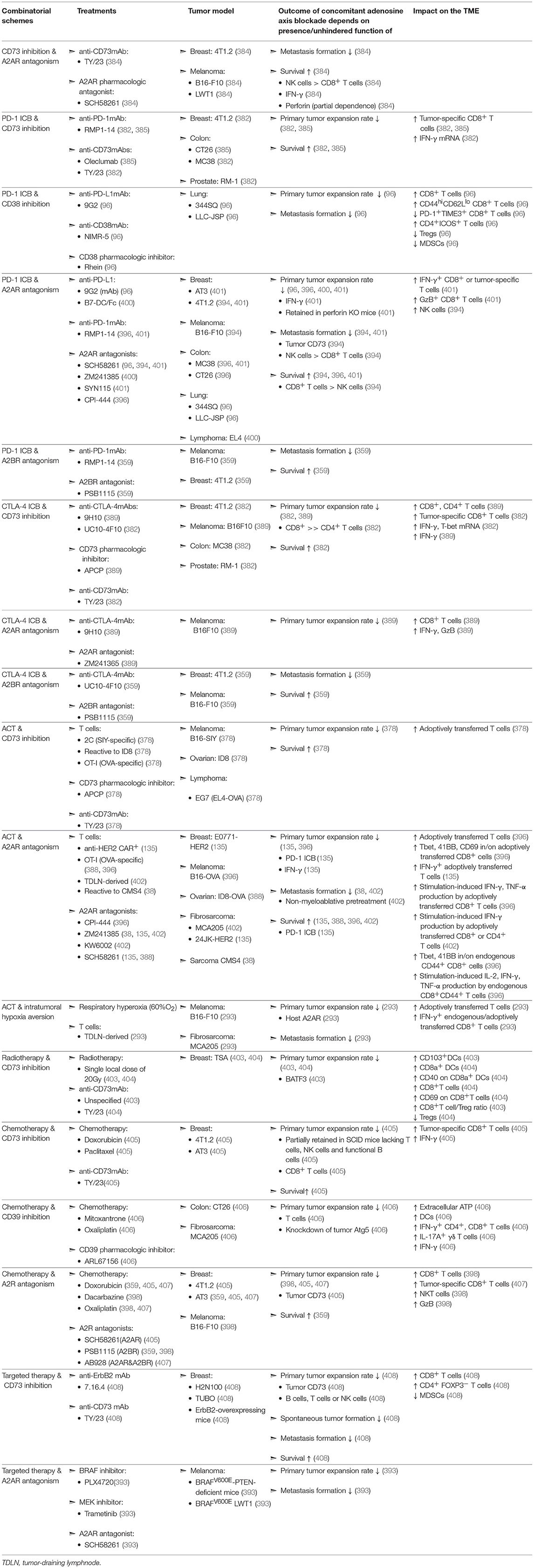
Table 2. Evaluation of concomitant adenosine-axis blockade in murine models of solid malignancies.
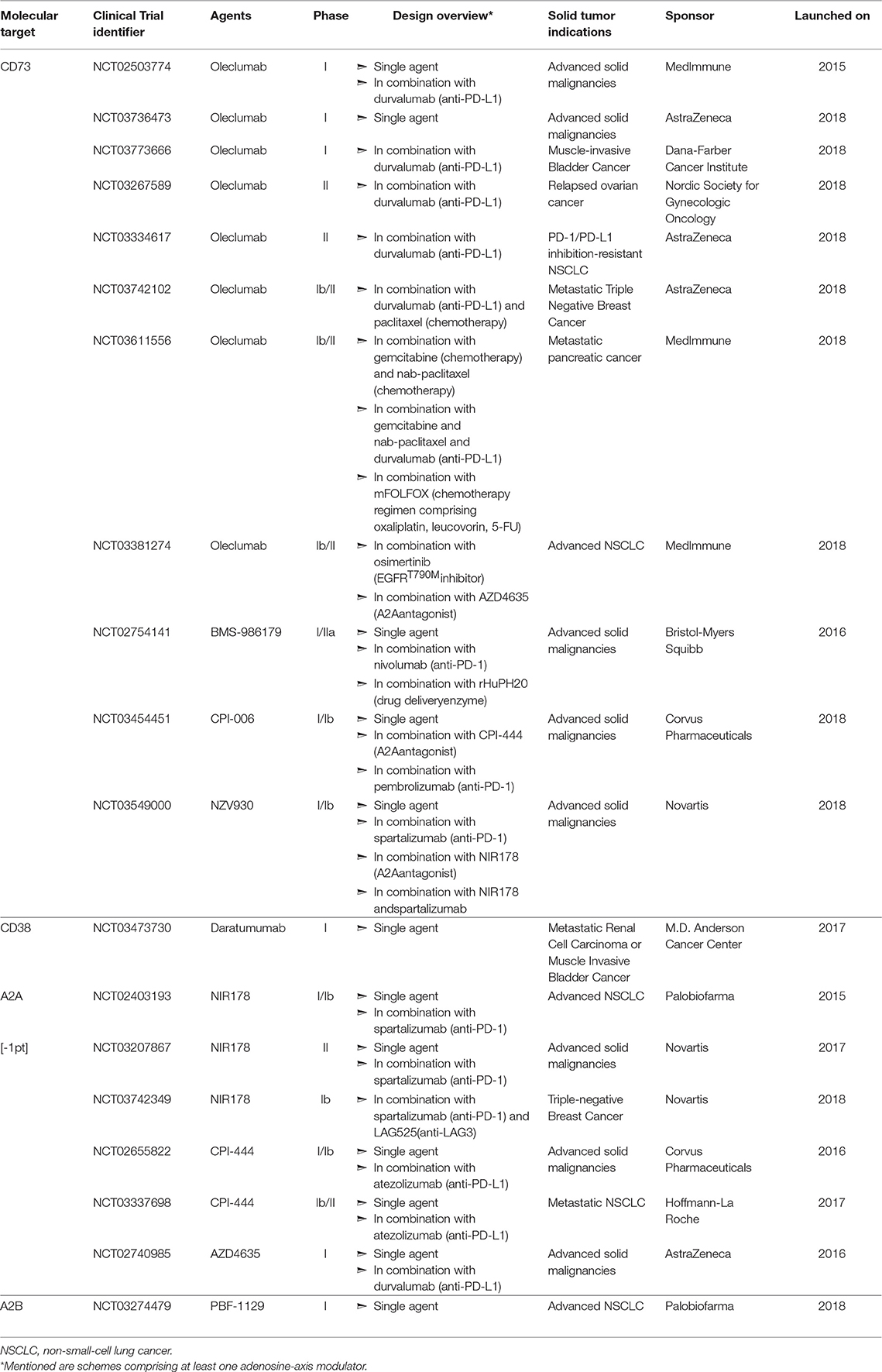
Table 3. Clinical evaluation of adenosine-axis targeting in patients with solid tumors.
Blockade of Adenosine Generation
As previously described, CD73 is an nucleotidase that converts AMP, generated from CD39- or CD38/CD203-mediated catabolism of ATP or NAD respectively, to adenosine. Its central role in adenosine generation is underscored by the fact that CD73-deficient mice display drastically decreased interstitial levels of adenosine, not only at steady state, but also upon induction of trauma or hypoxia (409, 410). CD73 knock-out mice exhibit hindered tumor growth and metastatic spreading (378–380, 387) and mice inoculated with tumor cells lacking CD73 survive longer than mice inoculated with tumor cells expressing this ecto-enzyme (378, 388). Indeed, administration of anti-CD73 monoclonal antibodies (mAb) (368, 378–386) or of a CD73-specific pharmacologic inhibitor (378, 379, 381, 383, 384, 387–389) impairs tumor growth (368, 378–382, 385, 387, 389) and metastasis (368, 379, 380, 384, 386) while increasing survival (378, 382, 384, 388). Of note, CD73 can also act as an adhesion/signaling molecule to promote metastasis in a catalytic-activity independent manner (386, 411, 412). Mechanistically, the aforementioned treatments have been shown to promote intra-tumoral accumulation of CD8+ T cells (381, 382, 385, 389), B cells (381) as well as of Th1- and Th17-associated cytokines (381) while decreasing the levels of intra-tumoral VEGF (383) and the presence of Tregs (389). Of note, even though metastasis can be modestly inhibited by anti-CD73 therapy in an immune-system independent fashion (368, 386), most of the antitumor effect of CD73 blockade is due to alleviation of A2AR-mediated immunosuppression (368).
No doubt encouraged by these pre-clinical studies, four anti-CD73 mAbs are currently being evaluated as monotherapies in small scale trials targeting a variety of solid tumors. In July 2015, MedImmune launched a first in-human trial (NCT02503774) evaluating the human anti-CD73 mAb Oleclumab, which allosterically prevents CD73 from assuming its catalytically active conformation (413). In June 2016, Bristol-Myers Squibb (BMS) launched a Phase I/IIa trial (NCT02754141) to assess the efficacy of BMS-986179, a human IgG2-IgG1 hybrid mAb that not only inhibits CD73-exerted AMP hydrolysis but also induces CD73 internalization (414). In April 2018, Corvus Pharmaceuticals initiated clinical evaluation (NCT03454451) of their humanized anti-CD73 mAb, CPI-006, which directly competes with AMP for the CD73 active site (415). Finally, in July 2018, Novartis listed a Phase I/Ib trial (NCT03549000) evaluating the efficacy of SRF373/NZV930, a human mAb that impedes CD73 activity via a currently undisclosed mechanism, and was pre-clinically developed by Surface Oncology before being exclusively licensed to Novartis for further clinical development.
CD39 also critically contributes to the generation of extracellular adenosine from ATP as evidenced by the fact that deficiency of this enzyme results in significantly decreased adenosine content in tissues, not only at steady state, but also upon ischemia induction (80). Similar to studies with CD73-deficient mice, tumor growth and metastasis are reduced in CD39-null mice (391, 416). In addition, intraperitoneal delivery of a CD39 inhibitor in immunocompetent mice reduces tumor growth rates (391). Administration of an anti-CD39 mAb increased the survival of immuno-deficient mice inoculated with patient-derived tumors (390), indicating that CD39 can also promote tumor growth or metastasis in an immune system independent manner. In terms of mechanisms, several studies have demonstrated that in vitro inhibition of CD39 activity by pharmacologic inhibitors (45, 47, 62) or blocking mAbs (45, 417, 418) results in enhanced functionality of T cells (45, 47, 62, 418) and NK cells (45, 47, 418), as well as decreased Treg-mediated suppression of T cell proliferation (47, 417). Even though restriction of CD39 activity in vitro conclusively alleviates adenosine-induced immunosuppression, a surprisingly small number of studies demonstrate effectiveness of this approach within tumor-bearing mice. Finally, while humanized mAbs targeting CD39, such as IPH52 (Innate Pharma) have been developed, clinical studies exploring CD39 blockade/inhibition have not been launched.
As previously mentioned, the concerted activity of CD38 and CD203a, can functionally replace CD39 toward the generation of extracellular adenosine. Further substantiating the soundness of CD38-blockade as a cancer treatment, immunocompetent CD38-null mice display reduced tumor growth (419) whereas tumors devoid of this ectonucleotidase grow slower both in immuno-competent (96) as well as in immuno-deficient mice (97). Indeed, administration of CD38 mAbs retards tumor growth (96, 420). Interestingly, tumors derived from anti-CD38 mAb-treated mice encompass more CD8+ T cells and less Tregs and MDSCs (96). Moreover, increased fraction of CD8+ T cells infiltrating these tumors display an effector memory phenotype while less of these cells are double positive for the exhaustion markers PD-1 and TIM3 (96). Three anti-CD38 mAbs, Daratumumab (Janssen Biotech), Isatuximab (Sanofi), and MOR202 (Morphosys) are being clinically evaluated. Daratumumab was FDA-approved in 2015 for treating multiple myeloma patients, while to date the most advanced testing of Isatuximab and MOR202 as monotherapies are respectively the Phase II trials NCT01084252, NCT02960555, and NCT02812706, as well as the Phase I/IIa trial NCT01421186. Of note, in addition to modulating the enzymatic activity of CD38, these mAbs also have the capacity to induce cytotoxicity through diverse mechanisms, such as induction of complement activation, Ab-dependent cellular cytotoxicity (ADCC) or phagocytosis, and programmed cell death (420). Albeit extensive clinical experience of utilizing the aforementioned mAbs against CD38-overexpressing hematologic malignancies, the recently launched trial NCT03473730 constitutes the first application of a CD38-specific mAb in patients with solid tumor malignancies.
Another approach for limiting the intratumoral interstitial adenosine is the oxygenation of the TME (293). As mentioned, hypoxia promotes build-up of extracellular adenosine at least by inducing upregulation of CD39 and CD73 as well as downregulation of adenosine transporters. Indeed, in pre-clinical models, respiratory hyperoxia (60% oxygen) lowers intra-tumoral adenosine levels (9), tumor growth rates (9), metastasis formation (293) and increases survival of tumor-bearing mice (9, 293). Mechanistically, this treatment boosts MHC-I levels on the tumor-cell surface (9), the presence of CD8+, CD69+, or CD44+ cells within the TME (293) and reduces the presence of Tregs (293) as well as the latter's capacity to express CD39, CD73, CTLA-4, or FoxP3 (293). Moreover, increased oxygenation of tumors not only averts angiogenesis through reduction of VEGF concentration (9), but also dampens expression of molecules associated with immune dysfunction, such as TGF-β, CD39, CD73, A2AR, A2BR and COX-2 (9, 293), the rate-limiting enzyme of PGE2 biosynthesis, while increasing the mRNA levels of pro-inflammatory agents, such as IL-2, and IL-12a (293).
Blockade of Adenosine Receptor Binding
Along with blocking adenosine production with small molecules or mAbs, another approach to inhibit adenosine-induced signaling is to directly block binding to its receptors A2AR and A2BR. Underscoring the potent protumoral effect of A2AR-trigerring, mice devoid of this receptor present reduced rates of tumor growth and metastasis, and in some instances tumors undergo complete rejection (38, 292, 400, 402). In addition, administration of pharmacologic A2AR antagonists recapitulates the anti-tumor effects of A2AR-deletion since it results to reduced primary tumor expansion (38, 54, 389, 392, 393, 396) and metastasis formation (292, 384, 393, 394, 397) ultimately leading to prolonged survival (384, 396). Mechanistically, tumors derived from A2AR-antagonist-treated mice are more heavily infiltrated by CD8+ T cells (389, 392, 393) as well as NK cells (389, 392, 393) and encompass fewer Tregs (389, 392, 393). In addition, in vivo A2AR antagonism leads to increased expression of CD69 (393), T-bet (396), and 4-1BB (396) as well as production of IFNγ and TNFα (392, 396) by intra-tumoral CD8+ T cells. Furthermore, this intervention increases the fraction of intra-tumoral NK cells producing GzB (292) and reduces the expression of PD-1, LAG3, FoxP3 and A2AR by tumor-infiltrating Tregs (392, 396). Interestingly, the A2AR antagonists ZM241385 and SCH58261 exhibit the capacity to curb primary tumor growth even in a T cell-independent manner (54). Notably, A2A antagonism in vivo increases activation induced cell death (AICD) of intra-tumoral T cells (395), a finding corroborating observations that cAMP-accumulation in the T cell cytosol averts terminal effector differentiation and AICD (421, 422). Three A2AR antagonists are currently being evaluated as single agents in Phase I/II trials to treat cancer patients bearing solid tumors. In particular, Corvus Pharmaceuticals, AstraZeneca, and Novartis have undertaken the clinical development of CPI-444 (NCT02655822), AZD4635 (NCT02740985), and NIR178 (NCT02403193, NCT03207867), respectively.
As for A2AR, genetic deletion of A2BR reduces tumor growth rate (399, 423) while A2BR−/− tumor cells display reduced metastatic potential (359, 367). Notably, administration of A2BR antagonists in tumor-bearing mice reduces tumor growth (315, 398, 399) and metastasis (292, 359, 368) eventually prolonging their survival (359). Mechanistically, antagonism of A2BR in vivo augments the intra-tumoral presence of CD8+ T cells (315, 398), NKT (315, 398) as well as the mRNA levels of IFNγ and CXCL10 (399) and the concentration of TNFα, IFNγ, and GzB (398) in the TME. This intervention further results in decreased accumulation of MDSCc (315, 398) and IL-10 (398), as well as reduced levels of VEGF and angiogenesis (315). Based on encouraging preclinical results, Palobiofarma recently launched a dose escalation Phase I study (NCT03274479) administering PBF-1129, a selective A2BR inhibitor, in patients with advanced Non-Small Cell Lung Cancer (NSCLC).
Combinatorial Treatment Approaches
Since multiple ecto-enzymes with redundant functions contribute toward extracellular adenosine production and both A2AR and A2BR triggering mediate the majority of adenosine's pro-tumoral effects, monotherapies may not be sufficient to block the adenosine-signaling axis. In addition, there is strong rationale for combination with IMTs, such as ICB of PD-1/PDL-1 or CTLA-4, as well as ACT, radiotherapy and chemotherapy, to further unleash the cytotoxic capacity of T cells, which, as will be discussed, can become highly sensitized to adenosine-mediated immunosuppression.
Combinations of Adenosine-Axis Blockade Agents
Concurrent mAb-mediated (418) or pharmacologic (47) inhibition of CD39 and CD73 failed to potentiate CD73-blockade-induced suppression of adenosine production by Tregs and ovarian cancer cell lines. These findings are corroborated by the observation that skin biopsies derived from CD39−/−CD73−/− mice have identical capacity to produce adenosine upon injury induction with counterpart biopsies derived from CD73−/− mice (424).
Alone the same lines, others addressed whether simultaneous blockade of CD73 and of A2AR would result in higher anti-tumor efficacy. Of note, CD73−/−A2AR−/− mice present superior tumor control as compared to single knockout mice (384). Moreover, tumors in A2AR-null mice express twice as much CD73 at their core when compared to tumors formed in wild-type mice (384). Indeed, dual therapy with an anti-CD73 mAb and an A2AR agonist confers superior tumor protection as compared to either one as a monotherapy (384). However, this additive effect is lost when CD73 is targeted with a pharmacologic inhibitor, thus underscoring the capacity of CD73 to promote tumor progression in a catalytic activity-independent manner (384). In light of these studies, Evotec and Exscientia have partnered to develop A2AR/CD73 bi-specific inhibitory molecules (425), whereas NCT03454451, NCT03549000 as well as the Phase Ib/II clinical trial NCT03381274 sponsored by MedImmune all include solid tumor-bearing patient cohorts scheduled to be treated with combinations of an anti-CD73 mAb along with a pharmacologic A2AR antagonist.
Adenosine-Axis and PD-1 Blockade
Briefly, PD-1 is an immunosuppressive receptor that upon binding to its ligands, PDL-1 and PDL-2, dampens T-cell activity thereby enabling tumors to evade immune-destruction. Blockade of the PD-1-PDL-1/2 signaling axis results in durable complete responses in the clinic for a fraction of treated patients (1), and many pre-clinical and clinical studies have explored concomitant inhibition of adenosine production, or antagonism of A2AR and A2BR, to improve response rates.
It has been demonstrated that CD73+ tumor cells are resistant to PD-1 ICB (401) and that simultaneous mAb-mediated blockade of CD73 and PD-1 synergistically enhances tumor control and survival in mice (382, 385). Mechanistically, the dual therapy augments intra-tumoral CD8+ tumor-specific T cells (382, 385) and IFNγ mRNA levels (382) as compared to single-agent treatments. Several clinical trials assessing anti-CD73 mAb treatment along with anti-PD-1 mAb (NCT03454451, NCT03549000) or anti-PDL-1 mAb (NCT02503774, NCT03773666, NCT03267589, NCT03334617) of advanced solid tumors are recruiting or underway. Intra-tumoral upregulation of CD38 and subsequent adenosine production was recently identified as a mechanism of acquired resistance to PD-1/PD-L1 blockade and mAb-mediated or pharmacologic inhibition of CD38 was shown to significantly improve the anti-tumor efficacy of an anti-PDL-1 mAb (96). In terms of mechanisms, tumors from mice receiving the combinatorial therapy displayed higher accumulation of CD8+ T cells, effector memory CD8+ T cells, ICOS+ CD4+ T cells and lower levels of MDSCs and Tregs as compared to tumors from single-agent treated mice (96).
The potential for synergy between the co-administration of A2R antagonists with anti-PD-1 mAb is underscored by the observations that PD-1 blockade enhances A2AR expression on tumor-infiltrating CD8+ T cells (401), as well as that PD-1 blockade is more efficacious, in terms of increasing the survival of tumor-bearing mice, when these mice lack the A2AR (400). Vice versa, A2AR triggering on the surface of CD8+ T cells derived from tumor tissue (382), tumor draining lymph nodes or spleen (396) promotes PD-1 expression suggesting that simultaneous PD-1 blockade would boost the anti-tumor efficacy of A2A antagonism. Indeed, several groups demonstrated that concurrent provision of PD-1 checkpoint inhibitors along with A2AR antagonists is more effective than single-agent treatments at reducing tumor growth rate (96, 396, 400, 401) and metastasis formation (394, 401), as well as at improving survival (394, 396, 401). Moreover, the combination enables increased production of IFNγ and GzB by CD8+ tumor infiltrating T cells (401) while augmenting the intra-tumoral presence of NK cells (394). Five clinical trials for the treatment of solid-tumor patient cohorts with A2AR antagonists along with anti-PD-1 Ab (NCT02403193, NCT03207867) or anti-PD-L1 Ab (NCT02655822, NCT03337698, NCT02740985) are ongoing. Finally, dual therapy comprising A2BR antagonism and PD-1 blockade is superior to either monotherapy at decreasing metastasis and improving survival of tumor-bearing mice (359). However, no clinical trials have been launched to date to explore this combination in human cancer patients.
Adenosine-Axis and CLTA-4 Blockade
The blockade of CTLA-4, an immune checkpoint receptor predominantly expressed by T cells and which competes with the co-stimulatory receptor CD28 for binding to CD80/CD86 on the surface of antigen presenting cells (APCs), has also generated durable clinical responses in advanced cancer patients (1). Tumor-bearing mice receiving CTLA-4 blockade and pharmacologic (389) or Ab-mediated (382) inhibition of CD73 display superior tumor control (382, 389) and overall survival (382) than counterparts receiving single agent treatments. Mechanistically, these dual therapies are more effective than corresponding monotherapies at increasing the intra-tumoral presence of tumor-specific CD8+ T cells (382), CD4+FoxP3neg T cells (389) as well as the levels of IFNγ (389) and of mRNA coding for IFNγ and T-bet (382). Likewise, concomitant provision of CTLA-4 ICB and antagonists of either A2AR (389) or A2BR (359) leads to decreased tumor growth (389) and metastasis formation (359), as well as to higher survival of tumor-bearing mice (359) when compared to single treatments. In terms of mechanisms, combining CTLA-4 ICB with an A2AR antagonist augments intratumoral CD8+ T cell presence as well as IFNγ and GzmB levels (389).
Adenosine-Axis Blockade and Adoptive T Cell Therapy
There are two main approaches to ACT. Either autologous tumor-reactive T cells are expanded from tumor biopsies prior to patient re-infusion [i.e., tumor infiltrating lymphocyte (TIL) therapy], or peripheral blood T cells are gene-engineered to express a tumor-specific T cell receptor (TCR), or a so-called chimeric antigen receptor (CAR; a fusion protein that links scFv-mediated tumor antigen-binding with intracellular endo-domains associated with T cell activation). Cancer patients are typically lymphodepleted prior to ACT, and following infusion they receive high doses of IL-2, both of which support T cell engraftment (426). TIL therapy has achieved robust and durable responses in advanced melanoma patients, while CAR therapy targeting CD19 has yielded unprecedented clinical responses against a variety of advanced, treatment-refractory B cell malignancies (118, 427, 428).
Synergy has been demonstrated between strategies limiting adenosine production blockade and ACT within tumor-bearing mice. Indeed, ACT confers increased control of tumors lacking CD73 expression (388) and dual therapy of ACT and pharmacologic or mAb-mediated inhibition of CD73 was more robust than single treatments at augmenting tumor control and overall survival (378). Mechanistically, pharmacologic inhibition of CD73 potentiated the anti-tumor efficacy of ACT at least by boosting the homing of the adoptively transferred tumor-specific T cells at the tumor sites (378). Likewise, respiratory hyperoxia in mice increased the ability of adoptively transferred T cells to curb primary tumor expansion and metastasis formation by augmenting their capacity to accumulate in the TME and produce IFNγ (293).
Similarly, A2AR deficiency (402) or siRNA-mediated suppression of A2AR and A2BR expression (38) on the surface of adoptively transferred T cells leads to enhanced prevention of metastatic spreading (38, 402) and improved survival of tumor-bearing mice (38). Several groups have validated these observations by demonstrating that ACT and concomitant administration of A2AR antagonists is superior to single treatments in terms of decreasing tumor growth (135, 396), hindering metastasis formation (38, 402) and ultimately improving survival (135, 388, 396, 402). Interestingly, others claim that A2AR antagonism improves the efficacy of adoptively transferred CAR+ T cells only if PD1 ICB is co-administered (135). In terms of mechanisms, concomitant A2AR antagonism not only increases intra-tumoral presence of adoptively transferred T cells (396) but also elevates their activation status. In particular, when A2AR antagonists were co-administered, tumor-derived, adoptively transferred or endogenous CD44+ CD8+ T cells, exhibit heightened expression levels of T-bet, 4-1BB, and CD69 (396) while demonstrating increased capacity to produce IFNγ and TNFα (135, 396, 402).
Adenosine-Axis Blockade Combined With Radiotherapy, Chemotherapy or Targeted Therapies
It is well documented that radiotherapy (RT) as well as several chemotherapeutic (CT) drugs have the capacity to induce ATP release (406, 429–433). Since such regimens also elevate the expression levels of CD39 (405, 407, 434) and CD73 (405, 407, 435–437), it is possible that the concentration of interstitial adenosine in the TME rises sharply upon application of these treatments. Therefore, several investigators have explored whether concomitant provision of agents targeting the adenosine axis increase the anti-tumor efficacy of RT or of various CT agents.
Indeed, mAb-mediated inhibition of CD73 increased the anti-tumor efficacy of RT (403, 404) and this synergistic effect was even more apparent upon concurrent CTLA-4-blockade (404). Mechanistically, CD73 inhibition increases the presence of CD8+ T cells as well as of CD8α+ or CD103+ DCs within irradiated tumors while decreasing Tregs (403, 404). Moreover, concomitant CD73 blockade was shown to increase the activation status of CD8+ T cells and CD8α+ DCs within irradiated tumors as evidenced by the elevated expression levels of CD69 and CD40, respectively (404). Likewise, concurrent mAb-mediated inhibition of CD73 (405) or pharmacologic blockade of CD39 activity (406) boosted the tumor control (405, 406) and survival (405) of mice treated with the CT drugs Doxorubicin (405), Paclitaxel (405), and Mitoxantrone (406). Of note, such dual therapies were shown to not only augment intra-tumoral presence of DCs (406) and tumor-specific CD8+ T cells (405) but also the fraction of intra-tumoral CD4+ or CD8+ T cells producing IFNγ (406) as well as the levels of IFNγ in the TME (405, 406). In light of such observations, the clinical trials NCT03611556 and NCT03742102 are set to decipher the potency of CT regimens when provided in combination with the CD73-blocking Ab Oleclumab, supplemented or not by PD-1 blockade.
Along the same lines, others explored if direct antagonism of A2AR and A2BR would augment the antitumor effects of CT agents. Indeed, tumor-bearing mice treated with Doxorubicin (359, 405, 407), Dacarbazine (398), or Oxaliplatin (398, 407) in combination with A2AR (405), A2BR (359, 398), or dual A2AR/A2BR antagonists (407) displayed superior tumor control (398, 405, 407) or survived longer (359). Of note, tumors derived from mice treated with the combination of Dacarbazine and PSB1115, an A2BR antagonist, were more heavily infiltrated by CD8+ T cells as well as NKT cells and contained higher levels of GzB than tumors derived from counterpart mice subjected to Dacarbazine monotherapy (398). Likewise, concomitant administration of AB928, a dual A2AR and A2BR antagonist, along with Doxorubicin or Oxaliplatin increased the intra-tumoral detection of tumor-specific CD8+ T cells (407).
Finally, others have sought to decipher whether adenosine axis blockade enhances the anti-tumor efficacy of particular targeted therapies. For instance, it has been recently demonstrated that high expression levels of CD73 in tumors derived from breast cancer patients are associated with resistance to Trastuzumab, an anti-HER2/ErbB2 mAb, and that artificial CD73 overexpression promotes resistance to Trastuzumab-like therapy in immunocompetent murine models of breast cancer (408). Subsequently, the authors moved on to show that when such mice receive dual therapy comprising anti-CD73 and anti-ERB2 mAbs they exhibit inferior tumor expansion rate as well as reduced metastatic spreading and survive longer than counterpart mice treated with either single agent treatments (408). In terms of mechanisms, the combinatorial therapy significantly increases the intra-tumoral presence of CD8+ and CD4+FoxP3neg T cells while decreasing MDSCs (408). In addition, melanoma patients harboring BRAF-mutant tumors exhibit a trend for elevated expression of CD73 whereas co-administration of an A2AR antagonist in mice bearing BRAF-mutant tumors increased the therapeutic benefit achieved either by BRAF inhibition or by the combination of BRAF and MEK inhibitors (393). Finally, CD73 and A2AR are overexpressed in NSCLCs harboring EGFR mutations (438) and even though preclinical studies demonstrating increased efficacy of concomitant inhibition of EGFR and A2AR are not currently publicly available, the clinical trial NCT03381274 includes a cohort of patients with advanced NSCLC that will receive both an EGFR inhibitor and an A2AR antagonist.
Summary and Future Perspectives
Adenosine is critically involved in a range of physiologic processes including wound healing, and its levels are tightly regulated under homeostatic conditions. In solid tumors, however, adenosine concentration is significantly elevated, predominantly due to stress-induced ATP release coupled with the overexpression of nucleotidases, such as CD39 and CD73 that contribute to its catabolism. Primarily by engaging A2AR and A2BR, also overexpressed in the TME as a result of hypoxia and inflammation, adenosine diminishes the activity of protective immune infiltrates, such as T cells, NK cells and DCs, while boosting the inhibitory capacity of immunosuppressive subsets, including Tregs and MDSCs. For instance, A2AR and A2BR-induced cAMP accumulation within T cells blunts their differentiation, proliferation, cytokine production and target cell killing, predominantly through PKA activation. Along with establishing an anti-inflammatory and tolerogenic TME, adenosine also promotes blood vessel formation and assists tumors in subverting adjacent fibroblasts to further support tumor growth and metastasis.
Administration of small molecules or mAbs with the aim to block adenosine-signaling, either by limiting its production or its binding to ARs, has yielded important tumor control in various pre-clinical tumor models. Moreover, simultaneous blockade of adenosine production and receptor binding, achieved by an anti-CD73 mAb co-administered with an A2AR antagonist, for example, have demonstrated it synergy. Given the potent suppression of T cells by adenosine, it comes as no surprise that increases in tumor control and survival conferred by ICB (anti-PD-1 and anti-CTLA-4 mAbs) or ACT, is significantly enhanced by concomitant administration of agents countering the adenosine axis. Synergy of such adenosine axis modulators has further been shown with RT, as well as CTs, schemes known to promote immunogenic cell death (i.e., ATP is released), as well as with some targeted therapies.
While blockade of adenosine production and A2AR/A2BR antagonism are being tested in the clinic as monotherapies, increasing numbers of clinical trials combining adenosine-signaling blockade with IMTs or classic treatment approaches (i.e., RT, CT and targeted therapies) are recruiting and/or underway. Given the important responses achieved by a proportion of patients to immunotherapeutic-regimens, and the tremendous levels of immunosuppression mediated by adenosine, the development of existing or new agents targeting this axis, along with further testing of combinatorial strategies, is warranted. Indeed, targeting the adenosine axis holds great promise in the improved treatment of cancer patients.
Author Contributions
GC, MI, DA, and SV conceived the manuscript. DA, SV, and MI drafted the manuscript. GC, PR, CM-C, and CC reviewed the manuscript and provided feedback, and MI revised the manuscript. MI, DA, and SV made the figures, and DA assembled the tables.
Funding
This project was supported by the Ludwig Institute for Cancer Research, the ISREC Foundation, an ERC Advanced Grant to GC (1400206AdG-322875), the Biltema Foundation, and a Kummer fellowship to DA.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. (2018) 8:1–14. doi: 10.3389/fonc.2018.00086
2. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. (2017) 31:311–25. doi: 10.1016/j.ccell.2017.02.008
3. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:709–24. doi: 10.1038/nrc.2017.86
4. Layland J, Carrick D, Lee M, Oldroyd K, Berry C. Adenosine: physiology, pharmacology, and clinical applications. JACC Cardiovasc Interv. (2014) 7:581–91. doi: 10.1016/j.jcin.2014.02.009
5. Oyarzún C, Garrido W, Alarcón S, Yáñez A, Sobrevia L, Quezada C, et al. Adenosine contribution to normal renal physiology and chronic kidney disease. Mol Aspects Med. (2017) 55:75–89. doi: 10.1016/j.mam.2017.01.004
6. Zylka MJ. Pain-relieving prospects for adenosine receptors and ectonucleotidases. Trends Mol Med. (2011) 17:188–96. doi: 10.1016/j.molmed.2010.12.006
7. Kreutzmann JC, Havekes R, Abel T, Meerlo P. Sleep deprivation and hippocampal vulnerability: changes in neuronal plasticity, neurogenesis and cognitive function. Neuroscience. (2015) 309:173–90. doi: 10.1016/j.neuroscience.2015.04.053
8. Lonnroth P, Jansson PA, Fredholm BB, Smith U. Microdialysis of intercellular adenosine concentration in subcutaneous tissue in humans. Am J Physiol. (1989) 256:E250–5. doi: 10.1152/ajpendo.1989.256.2.E250
9. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med. (2014) 92:1283–92. doi: 10.1007/s00109-014-1189-3
10. Blay J, White TD, Hoskin DW. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. (1997) 57:2602–5.
11. Pedata F, Corsi C, Melani A, Bordoni F, Latini S. Adenosine extracellular brain concentrations and role of A2A receptors in ischemia. Ann N Y Acad Sci. (2001) 939:74–84. doi: 10.1111/j.1749-6632.2001.tb03614.x
12. Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. (2016) 16:177–92. doi: 10.1038/nri.2016.4
13. Feoktistov I, Biaggioni I, Cronstein BN. Adenosine receptors in wound healing, fibrosis and angiogenesis. Handb Exp Pharmacol. (2009) 193:383–97. doi: 10.1007/978-3-540-89615-9_13
14. Milo R, Jorgensen P, Moran U, Weber G, Springer M. BioNumbers–the database of key numbers in molecular and cell biology. Nucleic Acids Res. (2010) 38:D750–3. doi: 10.1093/nar/gkp889
15. Falzoni S, Donvito G, Di Virgilio F. Detecting adenosine triphosphate in the pericellular space. Interface Focus. (2013) 3:20120101. doi: 10.1098/rsfs.2012.0101
16. Sabirov RZ, Okada Y. ATP release via anion channels. Purinergic Signal. (2005) 1:311–28. doi: 10.1007/s11302-005-1557-0
17. Di Virgilio F, Sarti AC, Falzoni S, de Marchi E, Adinolfi E. Extracellular ATP and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer. (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
18. Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. (2017) 276:121–44. doi: 10.1111/imr.12528
19. Schenk U, Westendorf AM, Radaelli E, Casati A, Ferro M, Fumagalli M, et al. Purinergic control of T cell activation by ATP released through pannexin-1 hemichannels. Sci Signal. (2008) 1:3–8. doi: 10.1126/scisignal.1160583
20. Tokunaga A, Tsukimoto M, Harada H, Moriyama Y, Kojima S. Involvement of SLC17A9-dependent vesicular exocytosis in the mechanism of ATP release during T cell activation. J Biol Chem. (2010) 285:17406–16. doi: 10.1074/jbc.M110.112417
21. Zimmermann H, Zebisch M, Sträter N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. (2012) 8:437–502. doi: 10.1007/s11302-012-9309-4
22. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta Mol Cell Res. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
23. Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. (2000) 362:299–309. doi: 10.1007/s002100000309
24. Adriouch S, Haag F, Boyer O, Seman M, Koch-Nolte F. Extracellular NAD+: a danger signal hindering regulatory T cells. Microbes Infect. (2012) 14:1284–92. doi: 10.1016/j.micinf.2012.05.011
25. Haag F, Adriouch S, Braß A, Jung C, Möller S, Scheuplein F, et al. Extracellular NAD and ATP: Partners in immune cell modulation. Purinergic Signal. (2007) 3:71–81. doi: 10.1007/s11302-006-9038-7
26. Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The pharmacology of CD38/NADase: an emerging target in cancer and diseases of aging. Trends Pharmacol Sci. (2018) 39:424–36. doi: 10.1016/j.tips.2018.02.001
27. Horenstein AL, Chillemi A, Zaccarello G, Bruzzone S, Quarona V, Zito A, et al. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology. (2013) 2:e26246. doi: 10.4161/onci.26246
28. Graddis TJ, McMahan CJ, Tamman J, Page KJ, Trager JB. Prostatic acid phosphatase expression in human tissues. Int J Clin Exp Pathol. (2011) 4:295–306.
29. Zimmermann H. Prostatic acid phosphatase, a neglected ectonucleotidase. Purinergic Signal. (2009) 5:273–5. doi: 10.1007/s11302-009-9157-z
30. Camici M, Garcia-Gil M, Tozzi MG. The inside story of adenosine. Int J Mol Sci. (2018) 19:1–14. doi: 10.3390/ijms19030784
31. Pastor-Anglada M, Pérez-Torras S. Who is who in adenosine transport. Front Pharmacol. (2018) 9:627. doi: 10.3389/fphar.2018.00627
32. Xu Y, Wang Y, Yan S, Zhou Y, Yang Q, Pan Y, et al. Intracellular adenosine regulates epigenetic programming in endothelial cells to promote angiogenesis. EMBO Mol Med. (2017) 9:1263–78. doi: 10.15252/emmm.201607066
33. Decking UK, Schlieper G, Kroll K, Schrader J. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ Res. (1997) 81:154–64. doi: 10.1161/01.RES.81.2.154
34. Eltzschig HK, Abdulla P, Hoffman E, Hamilton KE, Daniels D, Schönfeld C, et al. HIF-1-dependent repression of equilibrative nucleoside transporter (ENT) in hypoxia. J Exp Med. (2005) 202:1493–505. doi: 10.1084/jem.20050177
35. Morote-Garcia JC, Rosenberger P, Nivillac NMI, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. (2009) 136:607–18. doi: 10.1053/j.gastro.2008.10.037
36. Klabunde RE. Dipyridamole inhibition of adenosine metabolism in human blood. Eur J Pharmacol. (1983) 93:21–6. doi: 10.1016/0014-2999(83)90026-2
37. Allard B, Beavis PA, Darcy PK, Stagg J. Immunosuppressive activities of adenosine in cancer. Curr Opin Pharmacol. (2016) 29:7–16. doi: 10.1016/j.coph.2016.04.001
38. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci USA. (2006) 103:13132–7. doi: 10.1073/pnas.0605251103
39. Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. (2010) 29:5346–58. doi: 10.1038/onc.2010.292
40. Zhao H, Bo C, Kang Y, Li H. What else can CD39 tell us? Front Immunol. (2017) 8:727. doi: 10.3389/fimmu.2017.00727
41. Sorrentino R, Pinto A, Morello S. The adenosinergic system in cancer: key therapeutic target. Oncoimmunology. (2013) 2:1–3. doi: 10.4161/onci.22448
42. Jiang T, Xu X, Qiao M, Li X, Zhao C, Zhou F, et al. Comprehensive evaluation of NT5E / CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer. (2018) 18:1–10. doi: 10.1186/s12885-018-4073-7
43. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. (2018) 6:57. doi: 10.1186/s40425-018-0360-8
44. Aliagas E, Vidal A, Texidó L, Ponce J, Condom E, Martín-Satué M. High expression of ecto-nucleotidases CD39 and CD73 in human endometrial tumors. Mediators Inflamm. (2014) 2014:509027. doi: 10.1155/2014/509027
45. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C, et al. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. (2015) 3:254–65. doi: 10.1158/2326-6066.CIR-14-0018
46. Cai XY, Ni XC, Yi Y, He HW, Wang JX, Fu YP, et al. Overexpression of CD39 in hepatocellular carcinoma is an independent indicator of poor outcome after radical resection. Medicine. (2016) 95:e4989. doi: 10.1097/MD.0000000000004989
47. Häusler SF, Montalbán del Barrio I, Strohschein J, Chandran PA, Engel JB, Hönig A, et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother. (2011) 60:1405–18. doi: 10.1007/s00262-011-1040-4
48. Dzhandzhugazyan KN, Kirkin AF, Thor Straten P, Zeuthen J. Ecto-ATP diphosphohydrolase/CD39 is overexpressed in differentiated human melanomas. FEBS Lett. (1998) 430:227–30. doi: 10.1016/S0014-5793(98)00603-6
49. Stella J, Bavaresco L, Braganhol E, Rockenbach L, Farias PF, Wink MR, et al. Differential ectonucleotidase expression in human bladder cancer cell lines. Urol Oncol Semin Orig Investig. (2010) 28:260–7. doi: 10.1016/j.urolonc.2009.01.035
50. Monteiro I, Vigano S, Faouzi M, Treilleux I, Michielin O, Ménétrier-Caux C, et al. CD73 expression and clinical significance in human metastatic melanoma. Oncotarget. (2018) 9:26659–69. doi: 10.18632/oncotarget.25426
51. Allard B, Turcotte M, Stagg J. CD73-generated adenosine: orchestrating the tumor-stroma interplay to promote cancer growth. J Biomed Biotechnol. (2012) 2012:485156. doi: 10.1155/2012/485156
52. Yu M, Guo G, Huang L, Mellor A, Yan C. CD73 expression in cancer-associated fibroblasts exacerbates immune suppression and promotes tumor progression via augmenting adenosine accumulation in the tumor microenvironment. J Immunol. (2017) 198:76.29.
53. Montalbán Del Barrio I, Penski C, Schlahsa L, Stein RG, Diessner J, Wöckel A, et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages–a self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J Immunother Cancer. (2016) 4:49. doi: 10.1186/s40425-016-0154-9
54. Mediavilla-Varela M, Luddy K, Noyes D, Khalil FK, Neuger AM, Soliman H, et al. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol Ther. (2013) 14:860–8. doi: 10.4161/cbt.25643
55. de Lourdes Mora-García M, García-Rocha R, Morales-Ramírez O, Montesinos JJ, Weiss-Steider B, Hernández-Montes J, et al. Mesenchymal stromal cells derived from cervical cancer produce high amounts of adenosine to suppress cytotoxic T lymphocyte functions. J Transl Med. (2016) 14:302. doi: 10.1186/s12967-016-1057-8
56. Hernanda PY, Pedroza-Gonzalez A, van der Laan LJ, Bröker ME, Hoogduijn MJ, Ijzermans JN, et al. Tumor promotion through the mesenchymal stem cell compartment in human hepatocellular carcinoma. Carcinogenesis. (2013) 34:2330–40. doi: 10.1093/carcin/bgt210
57. Kerkelä E, Laitinen A, Räbinä J, Valkonen S, Takatalo M, Larjo A, et al. Adenosinergic immunosuppression by human mesenchymal stromal cells requires co-operation with T cells. Stem Cells. (2016) 34:781–90. doi: 10.1002/stem.2280
58. Li L, Wang L, Li J, Fan Z, Yang L, Zhang Z, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. (2018) 78:1779–91. doi: 10.1158/0008-5472.CAN-17-2460
59. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. CD39/CD73 upregulation on myeloid-derived suppressor cells via TGF-β-mTOR-HIF-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. (2017) 6:e1320011. doi: 10.1080/2162402X.2017.1320011
60. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS ONE. (2013) 8:e57114. doi: 10.1371/journal.pone.0057114
61. d'Almeida SM, Kauffenstein G, Roy C, Basset L, Papargyris L, Henrion D, et al. The ecto-ATPDase CD39 is involved in the acquisition of the immunoregulatory phenotype by M-CSF-macrophages and ovarian cancer tumor-associated macrophages: regulatory role of IL-27. Oncoimmunology. (2016) 5:e1178025. doi: 10.1080/2162402X.2016.1178025
62. Hilchey SP, Kobie JJ, Cochran MR, Secor-Socha S, Wang JC, Hyrien O, et al. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J Immunol. (2009) 183:6157–66. doi: 10.4049/jimmunol.0900475
63. Sim GC, Martin-Orozco N, Jin L, Yang Y, Wu S, Washington E, et al. IL-2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest. (2014) 124:99–110. doi: 10.1172/JCI46266
64. Gourdin N, Bossennec M, Rodriguez C, Vigano S, Machon C, Jandus C, et al. Autocrine adenosine regulates tumor polyfunctional CD73(+)CD4(+) effector T cells devoid of immune checkpoints. Cancer Res. (2018) 78:3604–18. doi: 10.1158/0008-5472.CAN-17-2405
65. Thibaudin M, Chaix M, Boidot R, Végran F, Derangère V, Limagne E, et al. Human ectonucleotidase-expressing CD25highTh17 cells accumulate in breast cancer tumors and exert immunosuppressive functions. Oncoimmunology. (2016) 5:e1055444. doi: 10.1080/2162402X.2015.1055444
66. Canale FP, Ramello MC, Núñez N, Araujo Furlan CL, Bossio SN, Gorosito Serrán M, et al. CD39 expression defines cell exhaustion in tumor-infiltrating CD8(+) T cells. Cancer Res. (2018) 78:115–28. doi: 10.1158/0008-5472.CAN-16-2684
67. Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. (2018) 557:575–9. doi: 10.1038/s41586-018-0130-2
68. Fang F, Yu M, Cavanagh MM, Hutter Saunders J, Qi Q, Ye Z, et al. Expression of CD39 on activated T cells impairs their survival in older individuals. Cell Rep. (2016) 14:1218–31. doi: 10.1016/j.celrep.2016.01.002
69. Schuler PJ, Saze Z, Hong CS, Muller L, Gillespie DG, Cheng D, et al. Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol. (2014) 177:531–43. doi: 10.1111/cei.12354
70. Ludwig S, Floros T, Theodoraki MN, Hong CS, Jackson EK, Lang S, et al. Suppression of lymphocyte functions by plasma exosomes correlates with disease activity in patients with head and neck cancer. Clin Cancer Res. (2017) 23:4843–54. doi: 10.1158/1078-0432.CCR-16-2819
71. Clayton A, Al-Taei S, Webber J, Mason MD, Tabi Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J Immunol. (2011) 187:676–83. doi: 10.4049/jimmunol.1003884
72. Smyth LA, Ratnasothy K, Tsang JY, Boardman D, Warley A, Lechler R, et al. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. (2013) 43:2430–40. doi: 10.1002/eji.201242909
73. Yang Y, Bucan V, Baehre H, von der Ohe J, Otte A, Hass R. Acquisition of new tumor cell properties by MSC-derived exosomes. Int J Oncol. (2015) 47:244–52. doi: 10.3892/ijo.2015.3001
74. Dengler VL, Galbraith MD, Espinosa JM. Transcriptional regulation by hypoxia inducible factors. Crit Rev Biochem Mol Biol. (2014) 49:1–15. doi: 10.3109/10409238.2013.838205
75. Koizume S, Miyagi Y. Diverse mechanisms of Sp1-dependent transcriptional regulation potentially involved in the adaptive response of cancer cells to oxygen-deficient conditions. Cancers (Basel). (2015) 8:2. doi: 10.3390/cancers8010002
76. Tak E, Jung DH, Kim SH, Park GC, Jun DY, Lee J, et al. Protective role of hypoxia-inducible factor-1α-dependent CD39 and CD73 in fulminant acute liver failure. Toxicol Appl Pharmacol. (2017) 314:72–81. doi: 10.1016/j.taap.2016.11.016
77. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5′-nucleotidase. (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. (2002) 110:993–1002. doi: 10.1172/JCI15337
78. Sarkar K, Cai Z, Gupta R, Parajuli N, Fox-Talbot K, Darshan MS, et al. Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc Natl Acad Sci USA. (2012) 109:10504–9. doi: 10.1073/pnas.1208314109
79. Poth JM, Brodsky K, Ehrentraut H, Grenz A, Eltzschig HK. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J Mol Med. (2013) 91:183–93. doi: 10.1007/s00109-012-0988-7
80. Eltzschig HK, Köhler D, Eckle T, Kong T, Robson SC, Colgan SP. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood. (2009) 113:224–32. doi: 10.1182/blood-2008-06-165746
81. Zhang H, Xia Y, Ye Q, Yu F, Zhu W, Li P, et al. In vivo expansion of regulatory T cells with IL-2/IL-2 antibody complex protects against transient ischemic stroke. J Neurosci. (2018) 3411–17. doi: 10.1523/JNEUROSCI.3411-17.2018
82. Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. (2012) 36:362–73. doi: 10.1016/j.immuni.2011.12.019
83. Savic V, Stefanovic V, Ardaillou N, Ardaillou R. Induction of ecto-5′-nucleotidase of rat cultured mesangial cells by interleukin-1 beta and tumour necrosis factor-alpha. Immunology. (1990) 70:321–6.
84. Reinhardt J, Landsberg J, Schmid-Burgk JL, Ramis BB, Bald T, Glodde N, et al. MAPK signaling and inflammation link melanoma phenotype switching to induction of CD73 during immunotherapy. Cancer Res. (2017) 77:4697–709. doi: 10.1158/0008-5472.CAN-17-0395
85. Pagnotta SM, Laudanna C, Pancione M, Sabatino L, Votino C, Remo A, et al. Ensemble of gene signatures identifies novel biomarkers in colorectal cancer activated through PPARγ and TNFα signaling. PLoS ONE. (2013) 8:1–12. doi: 10.1371/journal.pone.0072638
86. Niemela J, Henttinen T, Yegutkin GG, Airas L, Kujari AM, Rajala P, et al. IFN-a induced adenosine production on the endothelium: a mechanism mediated by CD73 (ecto-5′-nucleotidase) up-regulation. J Immunol. (2004) 172:1646–53. doi: 10.4049/jimmunol.172.3.1646
87. Bellingan G, Maksimow M, Howell DC, Stotz M, Beale R, Beatty M, et al. The effect of intravenous interferon-beta-1a. (FP-1201) on lung CD73 expression and on acute respiratory distress syndrome mortality: an open-label study. Lancet Respir Med. (2014) 2:98–107. doi: 10.1016/S2213-2600(13)70259-5
88. Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. (2013) 14:1054–63. doi: 10.1038/ni.2695
89. Ryzhov SV, Pickup MW, Chytil A, Gorska AE, Zhang Q, Owens P, et al. Role of TGF-β signaling in generation of CD39+ CD73+ myeloid cells in tumors. J Immunol. (2014) 193:3155–64. doi: 10.4049/jimmunol.1400578
90. Regateiro FS, Howie D, Nolan KF, Agorogiannis EI, Greaves DR, Cobbold SP, et al. Generation of anti-inflammatory adenosine byleukocytes is regulated by TGF-β. Eur J Immunol. (2011) 41:2955–65. doi: 10.1002/eji.201141512
91. Spychala J, Kitajewski J. Wnt and β-catenin signaling target the expression of ecto-5′-nucleotidase and increase extracellular adenosine generation. Exp Cell Res. (2004) 296:99–108. doi: 10.1016/j.yexcr.2003.11.001
92. Spychala J, Zimmermann AG, Mitchell BS. Tissue-specific regulation of the ecto-5′-nucleotidase promoter. Role of the cAMP response element site in mediating repression by the upstream regulatory region. J Biol Chem. (1999) 274:22705–12. doi: 10.1074/jbc.274.32.22705
93. Al-Taei S, Salimu J, Spary LK, Clayton A, Lester JF, Tabi Z. Prostaglandin E2-mediated adenosinergic effects on CD14+cells: self-amplifying immunosuppression in cancer. Oncoimmunology. (2017) 6:1–10. doi: 10.1080/2162402X.2016.1268308
94. Salimu J, Webber J, Gurney M, Al-Taei S, Clayton A, Tabi Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J Extracell Vesicles. (2017) 6:1368823. doi: 10.1080/20013078.2017.1368823
95. Liao H, Hyman MC, Baek AE, Fukase K, Pinsky DJ. cAMP/CREB-mediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. J Biol Chem. (2010) 285:14791–805. doi: 10.1074/jbc.M110.116905
96. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. (2018) 8:1156–1175. doi: 10.1158/2159-8290.CD-17-1033
97. Bu X, Kato J, Hong JA, Merino MJ, Schrump DS, Lund FE, et al. CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis. (2018) 39:242–51. doi: 10.1093/carcin/bgx137
98. Belfiore A, Costantino A, Frasca F, Pandini G, Mineo R, Vigneri P, et al. Overexpression of membrane glycoprotein PC-1 in MDA-MB231 breast cancer cells is associated with inhibition of insulin receptor tyrosine kinase activity. Mol Endocrinol. (1996) 10:1318–26. doi: 10.1210/me.10.11.1318
99. Quarona V, Ferri V, Chillemi A, Bolzoni M, Mancini C, Zaccarello G, et al. Unraveling the contribution of ectoenzymes to myeloma life and survival in the bone marrow niche. Ann N Y Acad Sci. (2015) 1335:10–22. doi: 10.1111/nyas.12485
100. Kong HY, Byun J. Emerging roles of human prostatic acid phosphatase. Biomol Ther. (2013) 21:10–20. doi: 10.4062/biomolther.2012.095
101. Rao SR, Snaith AE, Marino D, Cheng X, Lwin ST, Orriss IR, et al. Tumour-derived alkaline phosphatase regulates tumour growth, epithelial plasticity and disease-free survival in metastatic prostate cancer. Br J Cancer. (2017) 116:227–36. doi: 10.1038/bjc.2016.402
102. Hofmann MC, Millán JL. Developmental expression of alkaline phosphatase genes; reexpression in germ cell tumours and in vitro immortalized germ cells. Eur Urol. (1993) 23:38–44; discussion 45. doi: 10.1159/000474568
103. Singh AK, Pandey A, Tewari M, Kumar R, Sharma A, Singh KA, et al. Advanced stage of breast cancer hoist alkaline phosphatase activity: risk factor for females in India. 3 Biotech. (2013) 3:517–20. doi: 10.1007/s13205-012-0113-1
104. Ren HY, Sun LL, Li HY, Ye ZM. Prognostic significance of serum alkaline phosphatase level in osteosarcoma: a meta-analysis of published data. Biomed Res Int. (2015) 2015:160835. doi: 10.1155/2015/160835
105. Alexander D, Wicox CM, Saif MW, Alexander D, Wicox CM. Serum alkaline phosphatase level as a prognostic tool in colorectal cancer: a study of 105 patients. J Appl Res. (2005) 5:88–95.
106. Pennycooke M, Chaudary N, Shuralyova I, Zhang Y, Coe IR. Differential expression of human nucleoside transporters in normal and tumor tissue. Biochem Biophys Res Commun. (2001) 280:951–9. doi: 10.1006/bbrc.2000.4205
107. Pastor-Anglada M, Pérez-Torras S. Emerging roles of nucleoside transporters. Front Pharmacol. (2018) 9:606. doi: 10.3389/fphar.2018.00606
108. Farré X, Guillén-Gómez E, Sánchez L, Hardisson D, Plaza Y, Lloberas J, et al. Expression of the nucleoside-derived drug transporters hCNT1, hENT1 and hENT2 in gynecologic tumors. Int J Cancer. (2004) 112:959–66. doi: 10.1002/ijc.20524
109. Bhutia YD, Hung SW, Patel B, Lovin D, Govindarajan R. CNT1 expression influences proliferation and chemosensitivity in drug-resistant pancreatic cancer cells. Cancer Res. (2011) 71:1825–35. doi: 10.1158/0008-5472.CAN-10-2736
110. Lane J, Martin TA, McGuigan C, Mason MD, Jiang WG. The differential expression of hCNT1 and hENT1 i n breast cancer and the possible impact on breast cancer therapy. J Exp Ther Oncol. (2010) 8:203–10.
111. Medina-Pulido L, Molina-Arcas M, Justicia C, Soriano E, Burgaya F, Planas AM, et al. Hypoxia and P1 receptor activation regulate the high-affinity concentrative adenosine transporter CNT2 in differentiated neuronal PC12 cells. Biochem J. (2013) 454:437–45. doi: 10.1042/BJ20130231
112. Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Muller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors–an update. Pharmacol Rev. (2011) 63:1–34. doi: 10.1124/pr.110.003285
113. de Lera Ruiz M, Lim Y, Zheng J. Adenosine A 2A receptor as a drug discovery target. J Med Chem. (2014) 57:3623–50. doi: 10.1021/jm4011669
114. McCudden CR, Hains MD, Kimple RJ, Siderovski DP, Willard FS. G-protein signaling: back to the future. Cell Mol Life Sci. (2005) 62:551–77. doi: 10.1007/s00018-004-4462-3
115. Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. (2001) 53:527–52.
116. da Silva CG, Jarzyna R, Specht A, Kaczmarek E. Extracellular nucleotides and adenosine independently activate AMP-activated protein kinase in endothelial cells: involvement of P2 receptors and adenosine transporters. Circ Res. (2006) 98:e39–47. doi: 10.1161/01.RES.0000215436.92414.1d
117. Saitoh M, Nagai K, Nakagawa K, Yamamura T, Yamamoto S, Nishizaki T. Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-activated protein kinase. Biochem Pharmacol. (2004) 67:2005–11. doi: 10.1016/j.bcp.2004.01.020
118. Lanitis E, Dangaj D, Irving M, Coukos G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann Oncol. (2017) 28:xii18–32. doi: 10.1093/annonc/mdx238
119. Ostroumov D, Fekete-Drimusz N, Saborowski M, Kühnel F, Woller N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol Life Sci. (2018) 75:689–713. doi: 10.1007/s00018-017-2686-7
120. Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. (2017) 117:451–60. doi: 10.1038/bjc.2017.220
121. Yang A, Mucsi AD, Desrosiers MD, Chen JF, Schnermann JB, Blackburn MR, et al. Adenosine mediated desensitization of cAMP signaling enhances T-cell responses. Eur J Immunol. (2010) 40:449–59. doi: 10.1002/eji.200939586
122. Cekic C, Sag D, Day YJ, Linden J. Extracellular adenosine regulates naive T cell development and peripheral maintenance. J Exp Med. (2013) 210:2693–706. doi: 10.1084/jem.20130249
123. Lukashev DE, Smith PT, Caldwell CC, Ohta A, Apasov SG, Sitkovsky MV. Analysis of A2a receptor-deficient mice reveals no significant compensatory increases in the expression of A2b, A1, and A3 adenosine receptors in lymphoid organs. Biochem Pharmacol. (2003) 65:2081–90. doi: 10.1016/S0006-2952(03)00158-8
124. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
125. Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-g production in murine CD4+ T cells. J Immunol. (2005) 174:1073–80. doi: 10.4049/jimmunol.174.2.1073
126. Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J, et al. A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood. (2008) 111:251–9. doi: 10.1182/blood-2007-03-081646
127. Hoskin DW, Butler JJ, Drapeau D, Haeryfar SM, Blay J. Adenosine acts through an A3 receptor to prevent the induction of murine anti-CD3-activated killer T cells. Int J Cancer. (2002) 99:386–95. doi: 10.1002/ijc.10325
128. Longhi MS, Moss A, Bai A, Wu Y, Huang H, Cheifetz A, et al. Characterization of human CD39+ Th17 cells with suppressor activity and modulation in inflammatory bowel disease. PLoS ONE. (2014) 9:e87956. doi: 10.1371/journal.pone.0087956
129. Koshiba M, Rosin DL, Hayashi N, Linden J, Sitkovsky MV. Patterns of A2A extracellular adenosine receptor expression in different functional subsets of human peripheral T cells. Flow cytometry studies with anti-A2A receptor monoclonal antibodies. Mol Pharmacol. (1999) 55:614–24.
130. Mirabet M, Herrera C, Cordero OJ, Mallol J, Lluis C, Franco R. Expression of A2B adenosine receptors in human lymphocytes: their role in T cell activation. J Cell Sci. (1999) 112(Pt 4):491–502.
131. Gessi S, Varani K, Merighi S, Cattabriga E, Avitabile A, Gavioli R, et al. Expression of A3 adenosine receptors in human lymphocytes: up-regulation in T cell activation. Mol Pharmacol. (2004) 65:711–9. doi: 10.1124/mol.65.3.711
132. Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, et al. A2Aadenosine receptor. (AR) activation inhibits pro-inflammatory cytokine production by human CD4+helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol. (2009) 2:232–42. doi: 10.1038/mi.2009.4
133. Hilaire CST, Carroll SH, Chen H, Ravid K. Mechanisms of induction of adenosine receptor genes and its functional significance. J Cell Physiol. (2009) 218:35–44. doi: 10.1002/jcp.21579
134. Safford M, Collins S, Lutz MA, Allen A, Huang CT, Kowalski J, et al. Egr-2 and Egr-3 are negative regulators of T cell activation. Nat Immunol. (2005) 6:472–80. doi: 10.1038/ni1193
135. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
136. Hinz S, Navarro G, Borroto-Escuela D, Seibt BF, Ammon YC, de Filippo E, et al. Adenosine A2Areceptor ligand recognition and signaling is blocked by A2Breceptors. Oncotarget. (2018) 9:13593–611. doi: 10.18632/oncotarget.24423
137. Duhant X, Schandené L, Bruyns C, Gonzalez NS, Goldman M, Boeynaems JM, et al. Extracellular adenine nucleotides inhibit the activation of human CD4+ T lymphocytes. J Immunol. (2002) 169:15–21. doi: 10.4049/jimmunol.169.1.15
138. Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A 2A purinergic receptor-mediated signaling in murine T cells. J Biol Chem. (1997) 272:25881–9. doi: 10.1074/jbc.272.41.25881
139. Ohta A, Ohta A, Madasu M, Kini R, Subramanian M, Goel N, et al. A2A adenosine receptor may allow expansion of T cells lacking effector functions in extracellular adenosine-rich microenvironments. J Immunol. (2009) 183:5487–93. doi: 10.4049/jimmunol.0901247
140. Yan K, Gao LN, Cui YL, Zhang Y, Zhou X. The cyclic AMP signaling pathway: Exploring targets for successful drug discovery. Mol Med Rep. (2016) 13:3715–23. doi: 10.3892/mmr.2016.5005
141. Arumugham VB, Baldari CT. cAMP: a multifaceted modulator of immune synapse assembly and T cell activation. J Leukoc Biol. (2017) 101:1301–16. doi: 10.1189/jlb.2RU1116-474R
142. Johnstone TB, Agarwal SR, Harvey RD, Ostrom RS. cAMP signaling compartmentation: adenylyl cyclases as anchors of dynamic signaling complexes. Mol Pharmacol. (2017) 92:270–6. doi: 10.1124/mol.117.110825
143. Dessauer CW. Adenylyl cyclase–a-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. (2009) 76:935–41. doi: 10.1124/mol.109.059345
144. Pidoux G, Taskén K. Specificity and spatial dynamics of protein kinase a signaling organized by A-kinase-anchoring proteins. J Mol Endocrinol. (2010) 44:271–84. doi: 10.1677/JME-10-0010
145. Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+T cells. Mol Immunol. (2010) 47:678–84. doi: 10.1016/j.molimm.2009.10.032
146. Duan B, Davis R, Sadat EL, Collins J, Sternweis PC, Yuan D, et al. Distinct roles of adenylyl cyclase VII in regulating the immune responses in mice. J Immunol. (2010) 185:335–44. doi: 10.4049/jimmunol.0903474
147. Keravis T, Lugnier C. Cyclic nucleotide phosphodiesterase. (PDE) isozymes as targets of the intracellular signalling network: benefits of PDE inhibitors in various diseases and perspectives for future therapeutic developments. Br J Pharmacol. (2012) 165:1288–305. doi: 10.1111/j.1476-5381.2011.01729.x
148. Kanda N, Watanabe S. Regulatory roles of adenylate cyclase and cyclic nucleotide phosphodiesterases 1 and 4 in interleukin-13 production by activated human T cells. Biochem Pharmacol. (2001) 62:495–507. doi: 10.1016/S0006-2952(01)00688-8
149. Giembycz MA, Corrigan CJ, Seybold J, Newton R, Barnes PJ. Identification of cyclic AMP phosphodiesterases 3, 4 and 7 in human CD4+ and CD8+ T-lymphocytes: role in regulating proliferation and the biosynthesis of interleukin-2. Br J Pharmacol. (1996) 118:1945–58. doi: 10.1111/j.1476-5381.1996.tb15629.x
150. Li L, Yee C, Beavo JA. CD3- and CD28-dependent induction of PDE7 required for T cell activation. Science. (1999) 283:848–9. doi: 10.1126/science.283.5403.848
151. Glavas NA, Ostenson C, Schaefer JB, Vasta V, Beavo JA. T cell activation up-regulates cyclic nucleotide phosphodiesterases 8A1 and 7A3. Proc Natl Acad Sci USA. (2001) 98:6319–24. doi: 10.1073/pnas.101131098
152. Vang AG, Ben-Sasson SZ, Dong H, Kream B, DeNinno MP, Claffey MM, et al. PDE8 regulates rapid Teff cell adhesion and proliferation independent of ICER. PLoS ONE. (2010) 5:e12011. doi: 10.1371/journal.pone.0012011
153. Tenor H, Staniciu L, Schudt C, Hatzelmann A, Wendel A, Djukanović R, et al. Cyclic nucleotide phosphodiesterases from purified human CD4+ and CD8+ T lymphocytes. Clin Exp Allergy. (1995) 25:616–24. doi: 10.1111/j.1365-2222.1995.tb01109.x
154. Bartik MM, Brooks WH, Roszman TL. Modulation of T cell proliferation by stimulation of the β-adrenergic receptor: lack of correlation between inhibition of T cell proliferation and cAMP accumulation. Cell Immunol. (1993) 148:408–21. doi: 10.1006/cimm.1993.1122
155. Cazaux CA, Sterin-Borda L, Gorelik G, Cremaschi GA. Down-regulation of β-adrenergic receptors induced by mitogen activation of intracellular signaling events in lymphocytes. FEBS Lett. (1995) 364:120–4. doi: 10.1016/0014-5793(95)00366-H
156. Roszkowski W, Plaut M, Lichtenstein L. Selective display of histamine receptors on lymphocytes. Science. (1977) 195:683–5. doi: 10.1126/science.190677
157. Anderson P, Gonzalez-Rey E. Vasoactive intestinal peptide induces cell cycle arrest and regulatory functions in human T cells at multiple levels. Mol Cell Biol. (2010) 30:2537–51. doi: 10.1128/MCB.01282-09
158. Delgado M, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit expression of Fas ligand in activated T lymphocytes by regulating c-Myc, NF-B, NF-AT, and early growth factors 2/3. J Immunol. (2001) 166:1028–40. doi: 10.4049/jimmunol.166.2.1028
159. Radu CG, Nijagal A, McLaughlin J, Wang L, Witte ON. Differential proton sensitivity of related G protein-coupled receptors T cell death-associated gene 8 and G2A expressed in immune cells. Proc Natl Acad Sci USA. (2005) 102:1632–7. doi: 10.1073/pnas.0409415102
160. Abrahamsen H, Baillie G, Ngai J, Vang T, Nika K, Ruppelt A, et al. TCR- and CD28-mediated recruitment of phosphodiesterase 4 to lipid rafts potentiates TCR signaling. J Immunol. (2004) 173:4847–58. doi: 10.4049/jimmunol.173.8.4847
161. Bjørgo E, Solheim SA, Abrahamsen H, Baillie GS, Brown KM, Berge T, et al. Cross talk between phosphatidylinositol 3-kinase and cyclic AMP (cAMP)-protein kinase A signaling pathways at the level of a protein kinase B/-Arrestin/cAMP Phosphodiesterase 4 complex. Mol Cell Biol. (2010) 30:1660–72. doi: 10.1128/MCB.00696-09
162. Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S, et al. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med. (2014) 6:1294–311. doi: 10.15252/emmm.201403918
163. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. (2007) 204:1303–10. doi: 10.1084/jem.20062129
164. Wehbi VL, Taskén K. Molecular mechanisms for cAMP-mediated immunoregulation in T cells–role of anchored protein kinase a signaling units. Front Immunol. (2016) 7:222. doi: 10.3389/fimmu.2016.00222
165. Skalhegg BS, Tasken K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front Biosci. (2000) 5:D678–93. doi: 10.2741/A543
166. Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem Rev. (2001) 101:2243–70. doi: 10.1021/cr000226k
167. Giansanti P, Stokes MP, Silva JC, Scholten A, Heck AJR. Interrogating cAMP-dependent kinase signaling in Jurkat T cells via a protein kinase A targeted immune-precipitation phosphoproteomics approach. Mol Cell Proteomics. (2013) 12:3350–9. doi: 10.1074/mcp.O113.028456
168. Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol Res. (2016) 4:541–51. doi: 10.1158/2326-6066.CIR-15-0263
169. Oberprieler NG, Lemeer S, Kalland ME, Torgersen KM, Heck AJ, Taskén K. High-resolution mapping of prostaglandin E 2–dependent signaling networks identifies a constitutively active PKA signaling node in CD8+CD45RO+ T cells. Methods. (2010) 116:2–4. doi: 10.1182/blood-2010-01-266650
170. Rodriguez G, Ross JA, Nagy ZS, Kirken RA. Forskolin-inducible cAMP pathway negatively regulates T-cell proliferation by uncoupling the interleukin-2 receptor complex. J Biol Chem. (2013) 288:7137–46. doi: 10.1074/jbc.M112.408765
171. Vang T, Torgersen KM, Sundvold V, Saxena M, Levy FO, Skålhegg BS, et al. Activation of the COOH-terminal Src kinase. (Csk) by cAMP-dependent protein kinase inhibits signaling through the T cell receptor. J Exp Med. (2001) 193:497–507. doi: 10.1084/jem.193.4.497
172. Cerny O, Kamanova J, Masin J, Bibova I, Skopova K, Sebo P. Bordetella pertussis adenylate cyclase toxin blocks induction of bactericidal nitric oxide in macrophages through cAMP-dependent activation of the SHP-1 phosphatase. J Immunol. (2015) 194:4901–13. doi: 10.4049/jimmunol.1402941
173. Zhang J, Ravichandran KS, Garrison JC. A key role for the phosphorylation of Ser440by the cyclic AMP-dependent protein kinase in regulating the activity of the Src homology 2 domain-containing inositol 5-phosphatase (SHIP1). J Biol Chem. (2010) 285:34839–49. doi: 10.1074/jbc.M110.128827
174. Sawasdikosol S, Pyarajan S, Alzabin S, Matejovic G, Burakoff SJ. Prostaglandin E2 activates HPK1 kinase activity via a PKA-dependent pathway. J Biol Chem. (2007) 282:34693–9. doi: 10.1074/jbc.M707425200
175. Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, Nairn AC. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci USA. (2007) 104:2979–84. doi: 10.1073/pnas.0611532104
176. Park DJ, Min HK, Rhee SG. Inhibition of CD3-linked phospholipase C by phorbol ester and by cAMP is associated with decreased phosphotyrosine and increased phosphoserine contents of PLC-γ1. J Biol Chem. (1992) 267:1496–501.
177. Granja C, Lin LL, Yunis EJ, Relias V, Dasgupta JD. PLCgamma1, a possible mediator of T cell receptor function. J Biol Chem. (1991) 266:16277–80.
178. Dumaz N, Marais R. Protein kinase A blocks Raf-1 activity by stimulating 14-3-3 binding and blocking Raf-1 interaction with Ras. J Biol Chem. (2003) 278:29819–23. doi: 10.1074/jbc.C300182200
179. Ramstad C, Sundvold V, Johansen HK, Lea T. cAMP-dependent protein kinase. (PKA) inhibits T cell activation by phosphorylating Ser-43 of Raf-1 in the MAPK/ERK pathway. Cell Signal. (2000) 12:557–63. doi: 10.1016/S0898-6568(00)00097-8
180. Dong JM, Leung T, Manser E, Lim L. cAMP-induced morphological changes are counteracted by the activated RhoA small GTPase and the Rho kinase ROKα. J Biol Chem. (1998) 273:22554–62. doi: 10.1074/jbc.273.35.22554
181. Ellerbroek SM, Wennerberg K, Burridge K. Serine phosphorylation negatively regulates RhoA in vivo. J Biol Chem. (2003) 278:19023–31. doi: 10.1074/jbc.M213066200
182. Barzik M, Kotova TI, Higgs HN, Hazelwood L, Hanein D, Gertler FB, et al. Ena/VASP proteins enhance actin polymerization in the presence of barbed end capping proteins. J Biol Chem. (2005) 280:28653–62. doi: 10.1074/jbc.M503957200
183. Chow CW, Davis RJ. Integration of calcium and cyclic AMP signaling pathways by 14-3-3. Mol Cell Biol. (2000) 20:702–12. doi: 10.1128/MCB.20.2.702-712.2000
184. Sheridan CM, Heist EK, Beals CR, Crabtree GR, Gardner P. Protein kinase A negatively modulates the nuclear accumulation of NF-ATc1 by priming for subsequent phosphorylation by glycogen synthase kinase-3. J Biol Chem. (2002) 277:48664–76. doi: 10.1074/jbc.M207029200
185. Minguet S, Huber M, Rosenkranz L, Schamel WW, Reth M, Brummer T. Adenosine and cAMP are potent inhibitors of the NF-κB pathway downstream of immunoreceptors. Eur J Immunol. (2005) 35:31–41. doi: 10.1002/eji.200425524
186. Takahashi N, Tetsuka T, Uranishi H, Okamoto T. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur J Biochem. (2002) 269:4559–65. doi: 10.1046/j.1432-1033.2002.03157.x
187. Kuras Z, Kucher V, Gordon SM, Neumeier L, Chimote AA, Filipovich AH, et al. Modulation of Kv1.3 channels by protein kinase A I in T lymphocytes is mediated by the disc large 1-tyrosine kinase Lck complex. Am J Physiol Cell Physiol. (2012) 302:C1504–12. doi: 10.1152/ajpcell.00263.2011
188. Chimote AA, Hajdu P, Kucher V, Boiko N, Kuras Z, Szilagyi O, et al. Selective inhibition of KCa3.1 channels mediates adenosine regulation of the motility of human T cells. J Immunol. (2013) 191:6273–80. doi: 10.4049/jimmunol.1300702
189. Wong R, Schlichter LC. PKA reduces the rat and human KCa3.1 current CaM binding and Ca2+ signaling which requires Ser332/334 in the CaM-binding C terminus. J Neurosci. (2014) 34:13371–83. doi: 10.1523/JNEUROSCI.1008-14.2014
190. Cahalan MD, Chandy KG. The functional network of ion channels in T lymphocytes. Immunol Rev. (2009) 231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x
191. Chen Y, Harry A, Li J, Smit MJ, Bai X, Magnusson R, et al. Adenylyl cyclase 6 is selectively regulated by protein kinase A phosphorylation in a region involved in Galphas stimulation. Proc Natl Acad Sci USA. (1997) 94:14100–4. doi: 10.1073/pnas.94.25.14100
192. Palmer D, Jimmo SL, Raymond DR, Wilson LS, Carter RL, Maurice DH. Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J Biol Chem. (2007) 282:9411–9. doi: 10.1074/jbc.M606936200
193. MacKenzie SJ, Baillie GS, McPhee I, MacKenzie C, Seamons R, McSorley T, et al. Long PDE4 cAMP specific phosphodiesterases are activated by protein kinase A-mediated phosphorylation of a single serine residue in upstream conserved region 1 (UCR1). Br J Pharmacol. (2002) 136:421–33. doi: 10.1038/sj.bjp.0704743
194. Alvarez R, Sette C, Yang D, Eglen RM, Wilhelm R, Shelton ER, et al. Activation and selective inhibition of a cyclic AMP-specific phosphodiesterase, PDE-4D3. Mol Pharmacol. (1995) 48:616–22.
195. Brown KM, Lee LCY, Findlay JE, Day JP, Baillie GS. Cyclic AMP-specific phosphodiesterase, PDE8A1, is activated by protein kinase A-mediated phosphorylation. FEBS Lett. (2012) 586:1631–7. doi: 10.1016/j.febslet.2012.04.033
196. Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. (1999) 68:821–61. doi: 10.1146/annurev.biochem.68.1.821
197. Solomou EE, Juang YT, Tsokos GC. Protein kinase C-θ participates in the activation of cyclic AMP-responsive element-binding protein and its subsequent binding to the−180 site of the IL-2 promoter in normal human T lymphocytes. J Immunol. (2001) 166:5665–74. doi: 10.4049/jimmunol.166.9.5665
198. Hedrich CM, Rauen T, Tsokos GC. cAMP-responsive element modulator (CREM)α protein signaling mediates epigenetic remodeling of the human interleukin-2 gene: Implications in systemic lupus erythematosus. J Biol Chem. (2011) 286:43429–36. doi: 10.1074/jbc.M111.299339
199. Butscher W, Powers C, Vinson C, Gardner K. Coordinate transactivation of the cd28 response element of the interleukin-2 promoter by c-rel and ATF-I/CREB2. FASEB J. (1997) 11:552–60. doi: 10.1074/jbc.273.1.552
200. Penix LA, Sweetser MT, Weaver WM, Hoeffler JP, Kerppola TK, Wilson CB. The proximal regulatory element of the interferon-gamma promoter mediates selective expression in T cells. J Biol Chem. (1996) 271:31964–72. doi: 10.1074/jbc.271.50.31964
201. Samten B, Townsend JC, Weis SE, Bhoumik A, Klucar P, Shams H, et al. CREB, ATF, and AP-1 transcription factors regulate IFN-γ secretion by human T cells in response to mycobacterial antigen. J Immunol. (2008) 181:2056–64. doi: 10.4049/jimmunol.181.3.2056
202. De Araujo-Souza PS, Hanschke SCH, Viola JPB. Epigenetic control of interferon-gamma expression in CD8 T cells. J Immunol Res. (2015) 2015:849573. doi: 10.1155/2015/849573
203. Strempel JM, Vercelli D. Functional dissection identifies a conserved noncoding sequence-1 core that mediates IL13 and IL4 transcriptional enhancement. J Biol Chem. (2007) 282:3738–46. doi: 10.1074/jbc.M606615200
204. Verjans E, Ohl K, Reiss LK, van Wijk F, Toncheva AA, Wiener A, et al. The cAMP response element modulator. (CREM) regulates TH2 mediated inflammation. Oncotarget. (2015) 6:38538–51. doi: 10.18632/oncotarget.6041
205. Hernandez JB, Chang C, LeBlanc M, Grimm D, Le Lay J, Kaestner KH, et al. The CREB/CRTC2 pathway modulates autoimmune disease by promoting Th17 differentiation. Nat Commun. (2015) 6:1–9. doi: 10.1038/ncomms8216
206. Wang X, Ni L, Chang D, Lu H, Jiang Y, Kim BS, et al. Cyclic AMP-responsive element-binding protein (CREB) is critical in autoimmunity by promoting Th17 but inhibiting Treg cell differentiation. EBio Med. (2017) 25:165–74. doi: 10.1016/j.ebiom.2017.10.010
207. Hedrich CM, Rauen T, Kis-toth K, Kyttaris VC, Tsokos GC. cAMP-responsive element modulator alpha (CREMalpha) suppresses IL-17F protein expression in T lymphocytes from patients with systemic lupus erythematosus (SLE). J Biol Chem. (2012) 287:4715–25. doi: 10.1074/jbc.M111.323261
208. Rauen T, Hedrich CM, Juang YT, Tenbrock K, Tsokos GC. cAMP-responsive element modulator (CREM)a induces interleukin 17A expression and mediates epigenetic ? Protein alterations at the interleukin-17A gene locus in patients with systemic lupus erythematosus. J Biol Chem. (2011) 286:43437–46. doi: 10.1074/jbc.M111.299313
209. Kim H-P, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. (2007) 204:1543–51. doi: 10.1084/jem.20070109
210. Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T, et al. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol Cell Biol. (2014) 34:3993–4007. doi: 10.1128/MCB.00919-14
211. Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. (2010) 185:6413–9. doi: 10.4049/jimmunol.1001829
212. Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. (2011) 12:141–51. doi: 10.1038/nrm3072
213. Kobayashi M, Shimomura A, Hagiwara M, Kawakami K. Phosphorylation of ATF-1 enhances its DNA binding and transcription of the Na, K-ATPase alpha 1 subunit gene promoter. Nucleic Acids Res. (1997) 25:877–82. doi: 10.1093/nar/25.4.877
214. Fimia GM, De Cesare D, Sassone-Corsi P. Mechanisms of activation by CREB and CREM: phosphorylation, CBP, and a novel coactivator, ACT. Cold Spring Harb Symp Quant Biol. (1998) 63:631–42. doi: 10.1101/sqb.1998.63.631
215. Gerlo S, Verdood P, Hooghe-Peters EL, Kooijman R. Multiple cAMP-induced signaling cascades regulate prolactin expression in T cells. Cell Mol Life Sci. (2006) 63:92–9. doi: 10.1007/s00018-005-5433-4
216. Vang AG, Housley W, Dong H, Basole C, Ben-Sasson SZ, Kream BE, et al. Regulatory T-cells and cAMP suppress effector T-cells independently of PKA-CREM/ICER: a potential role for Epac. Biochem J. (2013) 456:463–73. doi: 10.1042/BJ20130064
217. Cheng X, Ji Z, Tsalkova T, Mei F. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin. (2008) 40:651–62. doi: 10.1111/j.1745-7270.2008.00438.x
218. Parnell E, Palmer TM, Yarwood SJ. The future of EPAC-targeted therapies: agonism versus antagonism. Trends Pharmacol Sci. (2015) 36:203–14. doi: 10.1016/j.tips.2015.02.003
219. de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. (1998) 396:474. doi: 10.1038/24884
220. Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. (1997) 278:124–8. doi: 10.1126/science.278.5335.124
221. Bivona TG, Wiener HH, Ahearn IM, Silletti J, Chiu VK, Philips MR. Rap1 up-regulation and activation on plasma membrane regulates T cell adhesion. J Cell Biol. (2004) 164:461–70. doi: 10.1083/jcb.200311093
222. Carey KD, Dillon TJ, Schmitt JM, Baird AM, Holdorf AD, Straus DB, et al. CD28 and the tyrosine kinase lck stimulate mitogen-activated protein kinase activity in T cells via inhibition of the small G protein Rap1. Mol Cell Biol. (2000) 20:8409–19. doi: 10.1128/MCB.20.22.8409-8419.2000
223. Katamura K, Shintaku N, Yamauchi Y, Fukui T, Ohshima Y, Mayumi M, et al. Prostaglandin E2 at priming of naive CD4+ T cells inhibits acquisition of ability to produce IFN-gamma and IL-2, but not IL-4 and IL-5. J Immunol. (1995) 155:4604–12.
224. Aandahl EM, Moretto WJ, Haslett PA, Vang T, Bryn T, Tasken K, et al. Inhibition of antigen-specific T cell proliferation and cytokine production by protein kinase A type I. J Immunol. (2002) 169:802–8. doi: 10.4049/jimmunol.169.2.802
225. Tsai H-C, Wu R. Cholera toxin directly enhances IL-17A production from human CD4+ T cells. J Immunol. (2013) 191:4095–102. doi: 10.4049/jimmunol.1301079
226. Tang H, Sun L, Xin Z, Ganea D. Down-regulation of cytokine expression in murine lymphocytes by PACAP and VIP. Ann N Y Acad Sci. (1996) 805:768–78. doi: 10.1111/j.1749-6632.1996.tb17555.x
227. Borger P, Kauffman HF, Postma DS, Vellenga E. Interleukin-4 gene expression in activated human T lymphocytes is regulated by the cyclic adenosine monophosphate-dependent signaling pathway. Blood. (1996) 87:691–8.
228. Gerlo S, Verdood P, Kooijman R. Modulation of cytokine production by cyclic adenosine monophosphate analogs in human leukocytes. J Interferon Cytokine Res. (2010) 30:883–91. doi: 10.1089/jir.2009.0021
229. Csóka B, Himer L, Selmeczy Z, Vizi ES, Pacher P, Ledent C, et al. Adenosine A2A receptor activation inhibits T helper 1 and T helper 2 cell development and effector function. FASEB J. (2008) 22:3491–9. doi: 10.1096/fj.08-107458
230. Su Y, Huang X, Raskovalova T, Zacharia L, Lokshin A, Jackson E, et al. Cooperation of adenosine and prostaglandin E2(PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother. (2008) 57:1611–23. doi: 10.1007/s00262-008-0494-5
231. Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, et al. Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine-protein kinase A type I signaling. Cancer Res. (2007) 67:5949–56. doi: 10.1158/0008-5472.CAN-06-4249
232. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology. (2009) 128:e728–37. doi: 10.1111/j.1365-2567.2009.03075.x
233. Grader-Beck T, van Puijenbroek A, Nadler LM, Boussiotis VA. cAMP inhibits both Ras and Rap1 activation in primary human T lymphocytes, but only Ras inhibition correlates with blockade of cell cycle progression. Blood. (2003) 101:998–1006. doi: 10.1182/blood-2002-06-1665
234. Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of β-catenin by cyclic AMP-dependent protein kinase stabilizes β-catenin through inhibition of its ubiquitination. Mol Cell Biol. (2005) 25:9063–72. doi: 10.1128/MCB.25.20.9063-9072.2005
235. Bodor J, Bodorova J, Bare C, Hodge DL, Young HA, Gress RE. Differential inducibility of the transcriptional repressor ICER and its role in modulation of Fas ligand expression in T and NK lymphocytes. Eur J Immunol. (2002) 32:203–12. doi: 10.1002/1521-4141(200201)32:1<203::AID-IMMU203>3.0.CO;2-C
236. Shaikh G, Zhang J, Perez-Aso M, Mediero A, Cronstein B. Adenosine A 2A receptor promotes collagen type III synthesis via β-catenin activation in human dermal fibroblasts. Br J Pharmacol. (2016) 173:3279–91. doi: 10.1111/bph.13615
237. Himer L, Csóka B, Selmeczy Z, Koscsó B, Pócza T, Pacher P, et al. Adenosine A(2A) receptor activation protects CD4(+) T lymphocytes against activation-induced cell death. FASEB J. (2010) 24:2631–40. doi: 10.1096/fj.10-155192
238. Bono MR, Fernández D, Flores-Santibá-ez F, Rosemblatt M, Sauma D. CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett. (2015) 589:3454–60. doi: 10.1016/j.febslet.2015.07.027
239. Gattinoni L, Ji Y, Restifo NP. Wnt/β-catenin signaling in T-cell immunity and cancer immunotherapy. Clin Cancer Res. (2010) 16:4695–701. doi: 10.1158/1078-0432.CCR-10-0356
240. Sakowicz-Burkiewicz M, Kocbuch K, Grden M, Szutowicz A, Pawelczyk T. Diabetes-induced decrease of adenosine kinase expression impairs the proliferation potential of diabetic rat T lymphocytes. Immunology. (2006) 118:402–12. doi: 10.1111/j.1365-2567.2006.02380.x
241. Zhang H, Conrad DM, Butler JJ, Zhao C, Blay J, Hoskin DW. Adenosine acts through A2 receptors to inhibit IL-2-induced tyrosine phosphorylation of STAT5 in T lymphocytes: role of cyclic adenosine 3′,5′-monophosphate and phosphatases. J Immunol. (2004) 173:932–44. doi: 10.4049/jimmunol.173.2.932
242. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. (1997) 90:1600–10.
243. MacKenzie WM, Hoskin DW, Blay J. The adhesion of anti-CD3-activated mouse T cells to syngeneic colon adenocarcinoma cells is differentially regulated by protein tyrosine kinase-, protein kinase C-, and cAMP-dependent pathways in the effector cell. Biochem Biophys Res Commun. (1999) 255:460–5. doi: 10.1006/bbrc.1999.0232
244. Oppenheimer-Marks N, Kavanaugh AF, Lipsky PE. Inhibition of the transendothelial migration of human T lymphocytes by prostaglandin E2. J Immunol. (1994) 152:5703–13.
245. Paccani SR, Dal Molin F, Benagiano M, Ladant D, D'Elios MM, Montecucco C, et al. Suppression of T-lymphocyte activation and chemotaxis by the adenylate cyclase toxin of Bordetella pertussis. Infect Immun. (2008) 76:2822–32. doi: 10.1128/IAI.00200-08
246. MacKenzie WM, Hoskin DW, Blay J. Adenosine suppresses α4β7integrin-mediated adhesion of T lymphocytes to colon adenocarcinoma cells. Exp Cell Res. (2002) 276:90–100. doi: 10.1006/excr.2002.5514
247. Hesdorffer CS, Malchinkhuu E, Biragyn A, Mabrouk OS, Kennedy RT, Madara K, et al. Distinctive immunoregulatory effects of adenosine on T cells of older humans. FASEB J. (2012) 26:1301–10. doi: 10.1096/fj.11-197046
248. MacKenzie WM, Hoskin DW, Blay J. Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adenocarcinoma cells through an A3 receptor. Cancer Res. (1994) 54:3521–6.
249. An BS, You SH, Kim H, Kwon OY, Ro HK, Cho BY, et al. Hormonal regulation of ICAM-1 gene expression in thyroid cells, FRTL-5. Exp Mol Med. (1997) 29:45. doi: 10.1038/emm.1997.7
250. Johnston A, Gudjonsson JE, Sigmundsdottir H, Runar Ludviksson B, Valdimarsson H. The anti-inflammatory action of methotrexate is not mediated by lymphocyte apoptosis, but by the suppression of activation and adhesion molecules. Clin Immunol. (2005) 114:154–63. doi: 10.1016/j.clim.2004.09.001
251. Jung EY, Ohshima Y, Shintaku N, Sumimoto S, Heike T, Katamura K, et al. Effects of cyclic AMP on expression of LFA-1, Mac-1, and VLA-4 and eosinophilic differentiation of a human leukemia cell line, EoL-1. Eur J Haematol. (1994) 53:156–62. doi: 10.1111/j.1600-0609.1994.tb00664.x
252. Sullivan GW, Lee DD, Ross WG, DiVietro JA, Lappas CM, Lawrence MB, et al. Activation of A 2A adenosine receptors inhibits expression of α4/β1 integrin (very late antigen-4) on stimulated human neutrophils. J Leukoc Biol. (2004) 75:127–34. doi: 10.1189/jlb.0603300
253. Thiel M, Chambers JD, Chouker A, Fischer S, Zourelidis C, Bardenheuer HJ, et al. Effect of adenosine on the expression of beta(2) integrins and L-selectin of human polymorphonuclear leukocytes in vitro. J Leukoc Biol. (1996) 59:671–82. doi: 10.1002/jlb.59.5.671
254. Valitutti S, Dessing M, Lanzavecchia A. Role of cAMP in regulating cytotoxic T lymphocyte adhesion and motility. Eur J Immunol. (1993) 23:790–5. doi: 10.1002/eji.1830230403
255. Takayama H, Sitkovsky MV. Potential use of an antagonist of cAMP-dependent protein kinase to block inhibition and modulate T-cell receptor-triggered activation of cytotoxic T-lymphocytes. J Pharm Sci. (1989) 78:8–10. doi: 10.1002/jps.2600780104
256. Alexander KL, Katz J, Elson CO. CBirTox is a selective antigen-specific agonist of the Treg-IgA-microbiota homeostatic pathway. PLoS ONE. (2017) 12:e0181866. doi: 10.1371/journal.pone.0181866
257. Emmons T, Wetzel S. The role of estrogen in atrazine-mediated inhibition of CD4+ T cell function and induction of CD4+CD25+Foxp3+ regulatory T cells (IRC4P.479). J Immunol. (2014) 192:60.6.
258. Feng G, Nadig SN, Bäckdahl L, Beck S, Francis RS, Schiopu A, et al. Functional regulatory T cells produced by inhibiting cyclic nucleotide phosphodiesterase type 3 prevent allograft rejection. Sci Transl Med. (2011) 3:1–11. doi: 10.1126/scitranslmed.3002099
259. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol. (2009) 182:4017–24. doi: 10.4049/jimmunol.0803310
260. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4+ CD25+ FoxP3+ regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol. (2012) 3:190. doi: 10.3389/fimmu.2012.00190
261. Morello S, Pinto A, Blandizzi C, Antonioli L. Myeloid cells in the tumor microenvironment: role of adenosine. Oncoimmunology. (2016) 5:e1108515. doi: 10.1080/2162402X.2015.1108515
262. Haskó G, Pacher P. Regulation of macrophage function by adenosine. Arterioscler Thromb Vasc Biol. (2012) 32:865–9. doi: 10.1161/ATVBAHA.111.226852
263. Hoskin DW, Mader JS, Furlong SJ, Conrad DM, Blay J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (Review). Int J Oncol. (2008) 32:527–35. doi: 10.3892/ijo.32.3.527
264. Barletta KE, Ley K, Mehrad B. Regulation of neutrophil function by adenosine. Arterioscler Thromb Vasc Biol. (2012) 32:856–64. doi: 10.1161/ATVBAHA.111.226845
265. Cronstein BN, Sitkovsky M. Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol. (2017) 13:41–51. doi: 10.1038/nrrheum.2016.178
266. Kazemi MH, Raoofi Mohseni S, Hojjat-Farsangi M, Anvari E, Ghalamfarsa G, Mohammadi H, et al. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J Cell Physiol. (2018) 233:2032–57. doi: 10.1002/jcp.25873
267. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. (2008) 112:1822–31. doi: 10.1182/blood-2008-02-136325
268. Challier J, Bruniquel D, Sewell AK, Laugel B. Adenosine and cAMP signalling skew human dendritic cell differentiation towards a tolerogenic phenotype with defective CD8+ T-cell priming capacity. Immunology. (2013) 138:402–10. doi: 10.1111/imm.12053
269. Desrosiers MD, Cembrola KM, Fakir MJ, Stephens LA, Jama FM, Shameli A, et al. Adenosine deamination sustains dendritic cell activation in inflammation. J Immunol. (2007) 179:1884–92. doi: 10.4049/jimmunol.179.3.1884
270. Panther E, Corinti S, Idzko M, Herouy Y, Napp M, la Sala A, et al. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T-cell stimulatory capacity of human dendritic cells. Blood. (2003) 101:3985–90. doi: 10.1182/blood-2002-07-2113
271. Yang M, Ma C, Liu S, Shao Q, Gao W, Song B, et al. HIF-dependent induction of adenosine receptor A2b skews human dendritic cells to a Th2-stimulating phenotype under hypoxia. Immunol Cell Biol. (2010) 88:165–71. doi: 10.1038/icb.2009.77
272. Wilson JM, Kurtz CC, Black SG, Ross WG, Alam MS, Linden J, et al. The A2B adenosine receptor promotes Th17 differentiation via stimulation of dendritic cell IL-6. J Immunol. (2011) 186:6746–52. doi: 10.4049/jimmunol.1100117
273. Wilson JM, Ross WG, Agbai ON, Frazier R, Figler RA, Rieger J, et al. The A2B adenosine receptor impairs the maturation and immunogenicity of dendritic cells. J Immunol. (2009) 182:4616–23. doi: 10.4049/jimmunol.0801279
274. Hofer S, Ivarsson L, Stoitzner P, Auffinger M, Rainer C, Romani N, et al. Adenosine slows migration of dendritic cells but does not affect other aspects of dendritic cell maturation. J Invest Dermatol. (2003) 121:300–7. doi: 10.1046/j.1523-1747.2003.12369.x
275. Ben AA, Lefort A, Hua X, Libert F, Communi D, Ledent C, et al. Modulation of murine dendritic cell function by adenine nucleotides and adenosine: involvement of the A2B receptor. Eur J Immunol. (2008) 38:1610–20. doi: 10.1002/eji.200737781
276. Li L, Huang L, Ye H, Song SP, Bajwa A, Lee SJ, et al. Dendritic cells tolerized with adenosine A2AR agonist attenuate acute kidney injury. J Clin Invest. (2012) 122:3931–42. doi: 10.1172/JCI63170
277. Najar HM, Ruhl S, Bru-Capdeville AC, Peters JH. Adenosine and its derivatives control human monocyte differentiation into highly accessory cells versus macrophages. J Leukoc Biol. (1990) 47:429–39. doi: 10.1002/jlb.47.5.429
278. Haskó G, Szabó C, Németh ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine receptor agonists differentially regulate IL-10, TNF-alpha, and nitric oxide production in RAW 264.7 macrophages and in endotoxemic mice. J Immunol. (1996) 157:4634–40.
279. Haskó G, Kuhel DG, Chen JF, Schwarzschild MA, Deitch EA, Mabley JG, et al. Adenosine inhibits IL-12 and TNF-α production via adenosine A 2a receptor-dependent and independent mechanisms. FASEB J. (2000) 14:2065–74. doi: 10.1096/fj.99-0508com
280. Haschemi A, Wagner O, Marculescu R, Wegiel B, Robson SC, Gagliani N, et al. Cross-regulation of carbon monoxide and the adenosine A2a receptor in macrophages. J Immunol. (2007) 178:5921–9. doi: 10.4049/jimmunol.178.9.5921
281. Szabó C, Scott GS, Virág L, Egnaczyk G, Salzman AL, Shanley TP, et al. Suppression of macrophage inflammatory protein (MIP)-1 α production and collagen-induced arthritis by adenosine receptor agonists. Br J Pharmacol. (1998) 125:379–87. doi: 10.1038/sj.bjp.0702040
282. Pinhal-Enfield G, Ramanathan M, Hasko G, Vogel SN, Salzman AL, Boons GJ, et al. An angiogenic switch in macrophages involving synergy between toll-like receptors 2, 4, 7, and 9 and adenosine A2A receptors. Am J Pathol. (2003) 163:711–21. doi: 10.1016/S0002-9440(10)63698-X
283. Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS. Inhibition of TNF-alpha expression by adenosine: role of A3 adenosine receptors. J Immunol. (1996) 156:3435–42.
284. Si Q, Nakamura Y, Kataoka K. Adenosine inhibits superoxide production in rat peritoneal macrophages via elevation of cAMP level. Immunopharmacology. (1997) 36:1–7. doi: 10.1016/S0162-3109(96)00158-0
285. Costales MG, Alam MS, Cavanaugh C, Williams KM. Extracellular adenosine produced by ecto-5′-nucleotidase. (CD73) regulates macrophage pro-inflammatory responses, nitric oxide production, and favors Salmonella persistence. Nitric Oxide. (2018) 72:7–15. doi: 10.1016/j.niox.2017.11.001
286. Csoka B, Németh ZH, Virág L, Gergely P, Leibovich SJ, Pacher P, et al. A2A adenosine receptors and C/EBP are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. (2007) 110:2685–95. doi: 10.1182/blood-2007-01-065870
287. Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A 2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell. (2007) 18:14–23. doi: 10.1091/mbc.e06-07-0596
288. Csóka B, Selmeczy Z, Koscsó B, Németh ZH, Pacher P, Murray PJ, et al. Adenosine promotes alternative macrophage activation via A 2A and A 2B receptors. FASEB J. (2012) 26:376–86. doi: 10.1096/fj.11-190934
289. Németh ZH, Lutz CS, Csóka B, Deitch EA, Leibovich SJ, Gause WC, et al. Adenosine augments IL-10 production by macrophages through an A2B receptor-mediated posttranscriptional mechanism. J Immunol. (2005) 175:8260–70. doi: 10.4049/jimmunol.175.12.8260
290. Ferrante CJ, Pinhal-Enfield G, Elson G, Cronstein BN, Hasko G, Outram S, et al. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation. (2013) 36:921–31. doi: 10.1007/s10753-013-9621-3
291. Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. (2018) 78:1003–16. doi: 10.1158/0008-5472.CAN-17-2826
292. Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, et al. Blockade of A 2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA. (2013) 110:14711–6. doi: 10.1073/pnas.1308209110
293. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. (2015) 7:277ra30. doi: 10.1126/scitranslmed.aaa1260
294. Miller JS, Cervenka T, Lund J, Okazaki IJ, Moss J. Purine metabolites suppress proliferation of human NK cells through a lineage-specific purine receptor. J Immunol. (1999) 162:7376–82.
295. Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine-mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res. (2006) 66:7758–65. doi: 10.1158/0008-5472.CAN-06-0478
296. Priebe T, Platsoucas CD, Nelson JA. Adenosine receptors and modulation of natural killer cell activity by purine nucleosides. Cancer Res. (1990) 50:4328–31.
297. Williams BA, Manzer A, Blay J, Hoskin DW. Adenosine acts through a novel extracellular receptor to inhibit granule exocytosis by natural killer cells. Biochem Biophys Res Commun. (1997) 231:264–9. doi: 10.1006/bbrc.1997.6077
298. Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. (2005) 175:4383–91. doi: 10.4049/jimmunol.175.7.4383
299. Yago T, Tsukamoto H, Liu Z, Wang Y, Thompson LF, McEver RP. Multi-inhibitory effects of A2A adenosine receptor signaling on neutrophil adhesion under flow. J Immunol. (2015) 195:3880–9. doi: 10.4049/jimmunol.1500775
300. McColl SR, St-Onge M, Dussault A-A, Laflamme C, Bouchard L, Boulanger J, et al. Immunomodulatory impact of the A 2A adenosine receptor on the profile of chemokines produced by neutrophils. FASEB J. (2006) 20:187–9. doi: 10.1096/fj.05-4804fje
301. Visser SS, Theron AJ, Ramafi G, Ker JA, Anderson R. Apparent involvement of the A2A subtype adenosine receptor in the anti-inflammatory interactions of CGS. (2000) 21680:cyclopentyladenosine, and IB-MECA with human neutrophils. Biochem Pharmacol. 60:993–9. doi: 10.1016/S0006-2952(00)00414-7
302. Anderson R, Visser SS, Ramafi G, Theron AJ. Accelerated resequestration of cytosolic calcium and suppression of the pro-inflammatory activities of human neutrophils by CGS 21680 in vitro. Br J Pharmacol. (2000) 130:717–24. doi: 10.1038/sj.bjp.0703344
303. Fredholm BB, Zhang Y, van der Ploeg I. Adenosine A2A receptors mediate the inhibitory effect of adenosine on formyl-Met-Leu-Phe-stimulated respiratory burst in neutrophil leucocytes. Naunyn Schmiedebergs Arch Pharmacol. (1996) 354:262–7. doi: 10.1007/BF00171056
304. Bouma MG, Jeunhomme TM, Boyle DL, Dentener MA, Voitenok NN, van den Wildenberg FA, et al. Adenosine inhibits neutrophil degranulation in activated human whole blood: involvement of adenosine A2 and A3 receptors. J Immunol. (1997) 158:5400–8.
305. Cronstein BN, Levin RI, Philips M, Hirschhorn R, Abramson SB, Weissmann G. Neutrophil adherence to endothelium is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J Immunol. (1992) 148:2201–6.
306. Thiel M, Chouker A. Acting via A2 receptors, adenosine inhibits the production of tumor necrosis factor-alpha of endotoxin-stimulated human polymorphonuclear leukocytes. J Lab Clin Med. (1995) 126:275–82.
307. Zalavary S, Stendahl O, Bengtsson T. The role of cyclic AMP, calcium and filamentous actin in adenosine modulation of Fc receptor-mediated phagocytosis in human neutrophils. Biochim Biophys Acta. (1994) 1222:249–56. doi: 10.1016/0167-4889(94)90176-7
308. Firestein GS, Bullough DA, Erion MD, Jimenez R, Ramirez-Weinhouse M, Barankiewicz J, et al. Inhibition of neutrophil adhesion by adenosine and an adenosine kinase inhibitor. The role of selectins. J Immunol. (1995) 154:326–34.
309. Kilian JG, Nakhla S, Sieveking DP, Celermajer DS. Adenosine prevents neutrophil adhesion to human endothelial cells after hypoxia/reoxygenation. Int J Cardiol. (2005) 105:322–6. doi: 10.1016/j.ijcard.2005.03.010
310. Wakai A, Wang JH, Winter DC, Street JT, O'Sullivan RG, Redmond HP. Adenosine inhibits neutrophil vascular endothelial growth factor release and transendothelial migration via A2Breceptor activation. Shock. (2001) 15:297–301. doi: 10.1097/00024382-200115040-00008
311. Richter J. Effect of adenosine analogues and cAMP-raising agents on TNF-, GM-CSF-, and chemotactic peptide-induced degranulation in single adherent neutrophils. J Leukoc Biol. (1992) 51:270–5. doi: 10.1002/jlb.51.3.270
312. Pouliot M, Fiset M-E, Masse M, Naccache PH, Borgeat P. Adenosine up-regulates cyclooxygenase-2 in human granulocytes: impact on the balance of eicosanoid generation. J Immunol. (2002) 169:5279–86. doi: 10.4049/jimmunol.169.9.5279
313. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of CD11b+Gr1+ cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
314. Cekic C, Day Y-J, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. (2014) 74:7250–9. doi: 10.1158/0008-5472.CAN-13-3583
315. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Myeloid-derived suppressor cells contribute to A2B adenosine receptor-induced VEGF production and angiogenesis in a mouse melanoma model. Oncotarget. (2015) 6:27478–89. doi: 10.18632/oncotarget.4393
316. Domanska UM, Kruizinga RC, Nagengast WB, Timmer-Bosscha H, Huls G, de Vries EG, et al. A review on CXCR4/CXCL12 axis in oncology: No place to hide. Eur J Cancer. (2013) 49:219–30. doi: 10.1016/j.ejca.2012.05.005
317. Tao L, Huang G, Song H, Chen Y, Chen L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol Lett. (2017) 14:2611–20. doi: 10.3892/ol.2017.6497
318. Akl MR, Nagpal P, Ayoub NM, Tai B, Prabhu SA, Capac CM, et al. Molecular and clinical significance of fibroblast growth factor 2 (FGF2 /bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget. (2016) 7:44735–62. doi: 10.18632/oncotarget.8203
319. Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans. (2017) 45:229–36. doi: 10.1042/BST20160387
320. Sorrentino C, Miele L, Porta A, Pinto A, Morello S. Activation of the A2B adenosine receptor in B16 melanomas induces CXCL12 expression in FAP-positive tumor stromal cells, enhancing tumor progression. Oncotarget. (2016) 7:11729. doi: 10.18632/oncotarget.11729
321. Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol. (2016) 17:611–25. doi: 10.1038/nrm.2016.87
322. Nowak-Sliwinska P, Alitalo K, Allen E, Anisimov A, Aplin AC, Auerbach R, et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis. (2018) 21:425–532. doi: 10.1007/s10456-018-9613-x
323. Desai A, Victor-Vega C, Gadangi S, Montesinos MC, Chu CC, Cronstein BN. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol Pharmacol. (2005) 67:1406–13. doi: 10.1124/mol.104.007807
324. Tabrizchi R, Bedi S. Pharmacology of adenosine receptors in the vasculature. Pharmacol Ther. (2001) 91:133–47. doi: 10.1016/S0163-7258(01)00152-8
325. Henttinen T, Jalkanen S, Yegutkin GG. Adherent leukocytes prevent adenosine formation and impair endothelial barrier function by ecto-5′-nucleotidase/CD73-dependent mechanism. J Biol Chem. (2003) 278:24888–95. doi: 10.1074/jbc.M300779200
326. Bouma MG, van den Wildenberg FA, Buurman WA. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am J Physiol. (1996) 270:C522–9. doi: 10.1152/ajpcell.1996.270.2.C522
327. Grünewald JK, Ridley AJ. CD73 represses pro-inflammatory responses in human endothelial cells. J Inflamm. (2010) 7:10. doi: 10.1186/1476-9255-7-10
328. Walker G, Langheinrich AC, Dennhauser E, Bohle RM, Dreyer T, Kreuzer J, et al. 3-deazaadenosine prevents adhesion molecule expression and atherosclerotic lesion formation in the aortas of C57BL/6J mice. Arterioscler Thromb Vasc Biol. (1999) 19:2673–9. doi: 10.1161/01.ATV.19.11.2673
329. Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. (2008) 111:2024–35. doi: 10.1182/blood-2007-10-117044
330. Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. FASEB J. (2002) 16:583–5. doi: 10.1096/fj.01-0739fje
331. Srinivas SP, Satpathy M, Gallagher P, Lariviere E, Van Driessche W. Adenosine induces dephosphorylation of myosin II regulatory light chain in cultured bovine corneal endothelial cells. Exp Eye Res. (2004) 79:543–51. doi: 10.1016/j.exer.2004.06.027
332. Acurio J, Herlitz K, Troncoso F, Aguayo C, Bertoglia P, Escudero C. Adenosine A2Areceptor regulates expression of vascular endothelial growth factor in feto-placental endothelium from normal and late-onset pre-eclamptic pregnancies. Purinergic Signal. (2017) 13:51–60. doi: 10.1007/s11302-016-9538-z
333. Khoa ND, Montesinos MC, Williams AJ, Kelly M, Cronstein BN. Th1 cytokines regulate adenosine receptors and their downstream signaling elements in human microvascular endothelial cells. J Immunol. (2003) 171:3991–8. doi: 10.4049/jimmunol.171.8.3991
334. Narravula S, Lennon PF, Mueller BU, Colgan SP. Regulation of endothelial CD73 by adenosine: paracrine pathway for enhanced endothelial barrier function. J Immunol. (2000) 165:5262–8. doi: 10.4049/jimmunol.165.9.5262
335. Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno-Yasenetskaya T, et al. Differential expression of adenosine receptors in human endothelial cells. Circ Res. (2002) 90:531–8. doi: 10.1161/01.RES.0000012203.21416.14
336. Grant MB, Tarnuzzer RW, Caballero S, Ozeck MJ, Davis MI, Spoerri PE, et al. Adenosine receptor activation induces vascular endothelial growth factor in human retinal endothelial cells. Circ Res. (1999) 85:699–706. doi: 10.1161/01.RES.85.8.699
337. Mirza A, Basso A, Black S, Malkowski M, Kwee L, Pachter JA, et al. RNA interference targeting of A1 receptor-overexpressing breast carcinoma cells leads to diminished rates of cell proliferation and induction of apoptosis. Cancer Biol Ther. (2005) 4:1355–60. doi: 10.4161/cbt.4.12.2196
338. Dastjerdi MN, Valiani A, Mardani M, Ra MZ. Adenosine A1 receptor modifies P53 expression and apoptosis in breast cancer cell line Mcf-7. Bratisl Lek Listy. (2016) 117:242–6. doi: 10.4149/BLL_2016_046
339. Sakowicz-Burkiewicz M, Kitowska A, Grden M, Maciejewska I, Szutowicz A, Pawelczyk T. Differential effect of adenosine receptors on growth of human colon cancer HCT 116 and HT-29 cell lines. Arch Biochem Biophys. (2013) 533:47–54. doi: 10.1016/j.abb.2013.02.007
340. Kuzumaki N, Suzuki A, Narita M, Hosoya T, Nagasawa A, Imai S, et al. Multiple analyses of G-protein coupled receptor. (GPCR) expression in the development of gefitinib-resistance in transforming non-small-cell lung cancer. PLoS ONE. (2012) 7:1–11. doi: 10.1371/journal.pone.0044368
341. Merighi S, Mirandola P, Milani D, Varani K, Gessi S, Klotz KN, et al. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J Invest Dermatol. (2002) 119:923–33. doi: 10.1046/j.1523-1747.2002.00111.x
342. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, et al. Hypoxia inhibits paclitaxel-induced apoptosis through adenosine-mediated phosphorylation of bad in glioblastoma cells. Mol Pharmacol. (2007) 72:162–72. doi: 10.1124/mol.106.031849
343. Kim H, Kang JW, Lee S, Choi WJ, Jeong LS, Yang Y, et al. A3 adenosine receptor antagonist, truncated Thio-Cl-IB-MECA, induces apoptosis in T24 human bladder cancer cells. Anticancer Res. (2010) 30:2823–30.
344. Wei Q, Costanzi S, Balasubramanian R, Gao Z-G, Jacobson KA. A2B adenosine receptor blockade inhibits growth of prostate cancer cells. Purinergic Signal. (2013) 9:271–80. doi: 10.1007/s11302-012-9350-3
345. Sai K, Yang D, Yamamoto H, Fujikawa H, Yamamoto S, Nagata T, et al. A1 adenosine receptor signal and AMPK involving caspase-9/-3 activation are responsible for adenosine-induced RCR-1 astrocytoma cell death. Neurotoxicology. (2006) 27:458–67. doi: 10.1016/j.neuro.2005.12.008
346. Saito M, Yaguchi T, Yasuda Y, Nakano T, Nishizaki T. Adenosine suppresses CW2 human colonic cancer growth by inducing apoptosis via A1 adenosine receptors. Cancer Lett. (2010) 290:211–5. doi: 10.1016/j.canlet.2009.09.011
347. Yasuda Y, Saito M, Yamamura T, Yaguchi T, Nishizaki T. Extracellular adenosine induces apoptosis in Caco-2 human colonic cancer cells by activating caspase-9/-3 via A2a adenosine receptors. J Gastroenterol. (2009) 44:56–65. doi: 10.1007/s00535-008-2273-7
348. Long JS, Schoonen PM, Graczyk D, O'Prey J, Ryan KM. p73 engages A2B receptor signalling to prime cancer cells to chemotherapy-induced death. Oncogene. (2015) 34:5152–62. doi: 10.1038/onc.2014.436
349. Jafari SM, Joshaghani HR, Panjehpour M, Aghaei M. A2B adenosine receptor agonist induces cell cycle arrest and apoptosis in breast cancer stem cells via ERK1/2 phosphorylation. Cell Oncol. (2018) 41:61–72. doi: 10.1007/s13402-017-0359-z
350. Aghaei M, Panjehpour M, Karami-Tehrani F, Salami S. Molecular mechanisms of A3 adenosine receptor-induced G1 cell cycle arrest and apoptosis in androgen-dependent and independent prostate cancer cell lines: involvement of intrinsic pathway. J Cancer Res Clin Oncol. (2011) 137:1511–23. doi: 10.1007/s00432-011-1031-z
351. Chung H, Jung JY, Cho SD, Hong KA, Kim HJ, Shin DH, et al. The antitumor effect of LJ-529, a novel agonist to A3 adenosine receptor, in both estrogen receptor-positive and estrogen receptor-negative human breast cancers. Mol Cancer Ther. (2006) 5:685–92. doi: 10.1158/1535-7163.MCT-05-0245
352. Otsuki T, Kanno T, Fujita Y, Tabata C, Fukuoka K, Nakano T, et al. A3 adenosine receptor-mediated p53-dependent apoptosis in Lu-65 human lung cancer cells. Cell Physiol Biochem. (2012) 30:210–20. doi: 10.1159/000339058
353. Varani K, Maniero S, Vincenzi F, Targa M, Stefanelli A, Maniscalco P, et al. A 3 receptors are overexpressed in pleura from patients with mesothelioma and reduce cell growth via Akt/nuclear factor-κB pathway. Am J Respir Crit Care Med. (2011) 183:522–30. doi: 10.1164/rccm.201006-0980OC
354. Kanno T, Nakano T, Fujita Y, Gotoh A, Nishizaki T. Adenosine induces apoptosis in SBC-3 human lung cancer cells through A 3 adenosine receptor-dependent AMID upregulation. Cell Physiol Biochem. (2012) 30:666–76. doi: 10.1159/000341447
355. Lin Z, Yin P, Reierstad S, O'Halloran M, Coon VJ, Pearson EK, et al. Adenosine A1 receptor, a target and regulator of estrogen receptorα action, mediates the proliferative effects of estradiol in breast cancer. Oncogene. (2010) 29:1114–22. doi: 10.1038/onc.2009.409
356. Gessi S, Bencivenni S, Battistello E, Vincenzi F, Colotta V, Catarzi D, et al. Inhibition of A2A adenosine receptor signaling in cancer cells proliferation by the novel antagonist TP455. Front Pharmacol. (2017) 8:1–13. doi: 10.3389/fphar.2017.00888
357. Kasama H, Sakamoto Y, Kasamatsu A, Okamoto A, Koyama T, Minakawa Y, et al. Adenosine A2b receptor promotes progression of human oral cancer. BMC Cancer. (2015) 15:1–12. doi: 10.1186/s12885-015-1577-2
358. Zhou Y, Chu X, Deng F, Tong L, Tong G, Yi Y, et al. The adenosine A2b receptor promotes tumor progression of bladder urothelial carcinoma by enhancing MAPK signaling pathway. Oncotarget. (2017) 8:48755–68. doi: 10.18632/oncotarget.17835
359. Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. (2016) 76:4372–82. doi: 10.1158/0008-5472.CAN-16-0544
360. Gessi S, Merighi S, Varani K, Cattabriga E, Benini A, Mirandola P, et al. Adenosine receptors in colon carcinoma tissues and colon tumoral cell lines: focus on the A3 adenosine subtype. J Cell Physiol. (2007) 211:826–36. doi: 10.1002/jcp.20994
361. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, et al. A 3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem. (2005) 280:19516–26. doi: 10.1074/jbc.M413772200
362. Ohana G, Bar-Yehuda S, Arich A, Madi L, Dreznick Z, Rath-Wolfson L, et al. Inhibition of primary colon carcinoma growth and liver metastasis by the A3 adenosine receptor agonist CF101. Br J Cancer. (2003) 89:1552–8. doi: 10.1038/sj.bjc.6601315
363. Fishman P, Bar-Yehuda S, Ohana G, Pathak S, Wasserman L, Barer F, et al. Adenosine acts as an inhibitor of lymphoma cell growth: a major role for the A3 adenosine receptor. Eur J Cancer. (2000) 36:1452–8. doi: 10.1016/S0959-8049(00)00130-1
364. Woodhouse EC, Amanatullah DF, Schetz JA, Liotta LA, Stracke ML, Clair T. Adenosine receptor mediates motility in human melanoma cells. Biochem Biophys Res Commun. (1998) 246:888–94. doi: 10.1006/bbrc.1998.8714
365. Zhou Y, Tong L, Chu X, Deng F, Tang J, Tang Y, et al. The adenosine A1 receptor antagonist DPCPX inhibits tumor progression via the ERK/JNK pathway in renal cell carcinoma. Cell Physiol Biochem. (2017) 43:733–42. doi: 10.1159/000481557
366. Zhou JZ, Riquelme MA, Gao X, Ellies LG, Sun LZ, Jiang JX. Differential impact of adenosine nucleotides released by osteocytes on breast cancer growth and bone metastasis. Oncogene. (2015) 34:1831–42. doi: 10.1038/onc.2014.113
367. Desmet CJ, Gallenne T, Prieur A, Reyal F, Visser NL, Wittner BS, et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc Natl Acad Sci USA. (2013) 110:5139–44. doi: 10.1073/pnas.1222085110
368. Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. (2010) 107:1547–52. doi: 10.1073/pnas.0908801107
369. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Simioni C, et al. Caffeine inhibits adenosine-induced accumulation of hypoxia-inducible factor-1, vascular endothelial growth factor, and interleukin-8 expression in hypoxic human colon cancer cells. Mol Pharmacol. (2007) 72:395–406. doi: 10.1124/mol.106.032920
370. Gessi S, Sacchetto V, Fogli E, Merighi S, Varani K, Baraldi PG, et al. Modulation of metalloproteinase-9 in U87MG glioblastoma cells by A3 adenosine receptors. Biochem Pharmacol. (2010) 79:1483–95. doi: 10.1016/j.bcp.2010.01.009
371. Raman P, Purwin T, Pestell R, Tozeren A. FXYD5 is a marker for poor prognosis and a potential driver for metastasis in ovarian carcinomas. Cancer Inform. (2015) 14:CIN.S30565. doi: 10.4137/CIN.S30565
372. Jajoo S, Mukherjea D, Watabe K, Ramkumar V. Adenosine A3 receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia. (2009) 11:1132-IN5. doi: 10.1593/neo.09744
373. Ledderose C, Hefti MM, Chen Y, Bao Y, Seier T, Li L, et al. Adenosine arrests breast cancer cell motility by A3 receptor stimulation. Purinergic Signal. (2016) 12:673–85. doi: 10.1007/s11302-016-9531-6
374. Fisher JW, Brookins J. Adenosine A(2A) and A(2B) receptor activation of erythropoietin production. Am J Physiol Renal Physiol. (2001) 281:F826–32. doi: 10.1152/ajprenal.0083.2001
375. Merighi S, Simioni C, Gessi S, Varani K, Mirandola P, Tabrizi MA, et al. A(2B) and A(3) adenosine receptors modulate vascular endothelial growth factor and interleukin-8 expression in human melanoma cells treated with etoposide and doxorubicin. Neoplasia. (2009) 11:1064–73. doi: 10.1593/neo.09768
376. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, et al. A3 adenosine receptors modulate hypoxia-inducible factor-1alpha expression in human A375 melanoma cells. Neoplasia. (2005) 7:894–903. doi: 10.1593/neo.05334
377. Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, et al. Adenosine modulates vascular endothelial growth factor expression via hypoxia-inducible factor-1 in human glioblastoma cells. Biochem Pharmacol. (2006) 72:19–31. doi: 10.1016/j.bcp.2006.03.020
378. Wang L, Fan J, Thompson LF, Zhang Y, Shin T, Curiel TJ, et al. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J Clin Invest. (2011) 121:2371–82. doi: 10.1172/JCI45559
379. Stagg J, Divisekera U, Duret H, Sparwasser T, Teng MW, Darcy PK, et al. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. (2011) 71:2892–900. doi: 10.1158/0008-5472.CAN-10-4246
380. Stagg J, Beavis PA, Divisekera U, Liu MC, Möller A, Darcy PK, et al. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. (2012) 72:2190–6. doi: 10.1158/0008-5472.CAN-12-0420
381. Forte G, Sorrentino R, Montinaro A, Luciano A, Adcock IM, Maiolino P, et al. Inhibition of CD73 improves B cell-mediated anti-tumor immunity in a mouse model of melanoma. J Immunol. (2012) 189:2226–33. doi: 10.4049/jimmunol.1200744
382. Allard B, Pommey S, Smyth MJ, Stagg J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res. (2013) 19:5626–35. doi: 10.1158/1078-0432.CCR-13-0545
383. Allard B, Turcotte M, Spring K, Pommey S, Royal I, Stagg J. Anti-CD73 therapy impairs tumor angiogenesis. Int J Cancer. (2014) 134:1466–73. doi: 10.1002/ijc.28456
384. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
385. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
386. Terp MG, Olesen KA, Arnspang EC, Lund RR, Lagerholm BC, Ditzel HJ, et al. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J Immunol. (2013) 191:4165–73. doi: 10.4049/jimmunol.1301274
387. Yegutkin GG, Marttila-Ichihara F, Karikoski M, Niemelä J, Laurila JP, Elima K, et al. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur J Immunol. (2011) 41:1231–41. doi: 10.1002/eji.201041292
388. Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. (2010) 70:2245–55. doi: 10.1158/0008-5472.CAN-09-3109
389. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Adenosine limits the therapeutic effectiveness of anti-CTLA4 mAb in a mouse melanoma model. Am J Cancer Res. (2014) 4:172–81.
390. Hayes GM, Cairns B, Levashova Z, Chinn L, Perez M, Theunissen JW, et al. CD39 is a promising therapeutic antibody target for the treatment of soft tissue sarcoma. Am J Transl Res. (2015) 7:1181–8.
391. Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Müller CE, et al. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology. (2010) 139:1030–40. doi: 10.1053/j.gastro.2010.05.007
392. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer. (2017) 16:99. doi: 10.1186/s12943-017-0665-0
393. Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, et al. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. (2017) 77:4684–96. doi: 10.1158/0008-5472.CAN-17-0393
394. Mittal D, Young A, Stannard K, Yong M, Teng MW, Allard B, et al. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. (2014) 74:3652–8. doi: 10.1158/0008-5472.CAN-14-0957
395. Cekic C, Linden J. Adenosine A2A receptors intrinsically regulate CD8+ T cells in the tumor microenvironment. Cancer Res. (2014) 74:7239–49. doi: 10.1158/0008-5472.CAN-13-3581
396. Leone RD, Sun IM, Oh MH, Sun IH, Wen J, Englert J, et al. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol Immunother. (2018) 67:1271–84. doi: 10.1007/s00262-018-2186-0
397. Mediavilla-Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor-infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia. (2017) 19:530–6. doi: 10.1016/j.neo.2017.02.004
398. Iannone R, Miele L, Maiolino P, Pinto A, Morello S. Blockade of A2b adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia. (2013) 15:1400-IN10. doi: 10.1593/neo.131748
399. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol. (2012) 188:198–205. doi: 10.4049/jimmunol.1101845
400. Waickman AT, Alme A, Senaldi L, Zarek PE, Horton M, Powell JD. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol Immunother. (2012) 61:917–26. doi: 10.1007/s00262-011-1155-7
401. Beavis PA, Milenkovski N, Henderson MA, John LB, Allard B, Loi S, et al. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol Res. (2015) 3:506–17. doi: 10.1158/2326-6066.CIR-14-0211
402. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A 2A adenosine receptor gene deletion or synthetic A 2A antagonist liberate tumor-reactive CD8+ T cells from tumor-induced immunosuppression. J Immunol. (2018) 201:782–91. doi: 10.4049/jimmunol.1700850
403. Wennerberg E, Cronstein B, Formenti SC, Demaria S. Adenosine generation limits radiation-induced tumor immunogenicity by abrogating recruitment and activation of CD103+ DCs. J Immunol. (2017) 198:154.6. doi: 10.1158/2326-6074.TUMIMM17-B05
404. Wennerberg E, Kawashima N, Demaria S. Adenosine regulates radiation therapy-induced anti-tumor immunity. J Immunother Cancer. (2015) 3:P378. doi: 10.1186/2051-1426-3-S2-P378
405. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci USA. (2013) 110:11091–6. doi: 10.1073/pnas.1222251110
406. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. (2011) 334:1573–7. doi: 10.1126/science.1208347
407. Schindler U, Seitz L, Ashok D, Piovesan D, Tan J, DiRenzo D, et al. AB928, a dual antagonist of the A 2a R and A 2b R adenosine receptors, leads to greater immune activation and reduced tumor growth when combined with chemotherapy. Eur J Cancer. (2018) 92:S14–5. doi: 10.1016/j.ejca.2018.01.037
408. Turcotte M, Allard D, Mittal D, Bareche Y, Buisseret L, José V, et al. CD73 promotes resistance to HER2/ErbB2 antibody therapy. Cancer Res. (2017) 77:5652–63. doi: 10.1158/0008-5472.CAN-17-0707
409. Volmer JB, Thompson LF, Blackburn MR. Ecto-5′-nucleotidase (CD73)-mediated adenosine production is tissue protective in a model of bleomycin-induced lung injury. J Immunol. (2006) 176:4449–58. doi: 10.4049/jimmunol.176.7.4449
410. Ozüyaman B, Ding Z, Buchheiser A, Koszalka P, Braun N, Gödecke A, et al. Adenosine produced via the CD73/ecto-5′-nucleotidase pathway has no impact on erythropoietin production but is associated with reduced kidney weight. Pflügers Arch Eur J Physiol. (2006) 452:324–31. doi: 10.1007/s00424-006-0045-x
411. Zhi X, Chen S, Zhou P, Shao Z, Wang L, Ou Z, et al. RNA interference of ecto-5′-nucleotidase (CD73) inhibits human breast cancer cell growth and invasion. Clin Exp Metastasis. (2007) 24:439–48. doi: 10.1007/s10585-007-9081-y
412. Sadej R, Skladanowski AC. Dual, enzymatic and non-enzymatic, function of ecto-5′-nucleotidase. (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochim Pol. (2012) 59:647–52. doi: 10.18388/abp.2012_2105
413. Geoghegan JC, Diedrich G, Lu X, Rosenthal K, Sachsenmeier KF, Wu H, et al. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs. (2016) 8:454–67. doi: 10.1080/19420862.2016.1143182
414. Barnhart BC, Sega E, Yamniuk A, Hatcher S, Lei M, Ghermazien H, et al. Abstract 1476: A therapeutic antibody that inhibits CD73 activity by dual mechanisms. Cancer Res. (2016) 76:1476. doi: 10.1158/1538-7445.AM2016-1476
415. Piccione EC, Mikesell G, Daine-Matsuoka B, Walter K, Miller R, McCaffery I. Abstract 5577: A novel CD73-blocking antibody reduces production of immunosuppressive adenosine and restores T cell function. Cancer Res. (2017) 77:5577. doi: 10.1158/1538-7445.AM2017-5577
416. Jackson SW, Hoshi T, Wu Y, Sun X, Enjyoji K, Cszimadia E, et al. Disordered purinergic signaling inhibits pathological angiogenesis in cd39/Entpd1-null mice. Am J Pathol. (2007) 171:1395–404. doi: 10.2353/ajpath.2007.070190
417. Nikolova M, Carriere M, Jenabian MA, Limou S, Younas M, Kök A, et al. CD39/adenosine pathway is involved in AIDS progression. PLoS Pathog. (2011) 7:e1002110. doi: 10.1371/journal.ppat.1002110
418. Häusler SF, Del Barrio IM, Diessner J, Stein RG, Strohschein J, Hönig A, et al. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am J Transl Res. (2014) 6:129–39.
419. Levy A, Blacher E, Vaknine H, Lund FE, Stein R, Mayo L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro Oncol. (2012) 14:1037–49. doi: 10.1093/neuonc/nos121
420. van de Donk NW, Janmaat ML, Mutis T, Lammerts van Bueren JJ, Ahmadi T, Sasser AK, et al. Monoclonal antibodies targeting CD38 in hematological malignancies and beyond. Immunol Rev. (2016) 270:95–112. doi: 10.1111/imr.12389
421. Vig M, George A, Sen R, Durdik J, Rath S, Bal V. Commitment of activated T cells to secondary responsiveness is enhanced by signals mediated by cAMP-dependent protein kinase A-I. Mol Pharmacol. (2002) 62:1471–81. doi: 10.1124/mol.62.6.1471
422. Suarez A, Mozo L, Gutierrez C. Generation of CD4+CD45RA+ effector T cells by stimulation in the presence of cyclic adenosine 5′-monophosphate-elevating agents. J Immunol. (2002) 169:1159–67. doi: 10.4049/jimmunol.169.3.1159
423. Ryzhov S, Novitskiy SV, Zaynagetdinov R, Goldstein AE, Carbone DP, Biaggioni I, et al. Host A2B adenosine receptors promote carcinoma growth. Neoplasia. (2008) 10:987–95. doi: 10.1593/neo.08478
424. Fernández P, Perez-Aso M, Smith G, Wilder T, Trzaska S, Chiriboga L, et al. Extracellular generation of adenosine by the ectonucleotidases CD39 and CD73 promotes dermal fibrosis. Am J Pathol. (2013) 183:1740–6. doi: 10.1016/j.ajpath.2013.08.024
425. Fons P, Esquerré M, Versluys S, Mambrini G, Paillasse M, Bell A, et al. Abstract 3970: Targeting the adenosine immunosuppressive pathway for cancer immunotherapy with small molecule agents. Cancer Res. (2017) 77:3970. doi: 10.1158/1538-7445.AM2017-3970
426. Wolf B, Zimmermann S, Arber C, Irving M, Trueb L, Coukos G. Safety and tolerability of adoptive cell therapy in cancer. Drug Saf. (2019) 42:315–34. doi: 10.1007/s40264-018-0779-3
427. Lim WA, June CH. The principles of engineering immune cells to treat cancer. Cell. (2017) 168:724–40. doi: 10.1016/j.cell.2017.01.016
428. Irving M, Vuillefroy de Silly R, Scholten K, Dilek N, Coukos G. Engineering chimeric antigen receptor T-cells for racing in solid tumors: don't forget the fuel. Front Immunol. (2017) 8:267. doi: 10.3389/fimmu.2017.00267
429. Hu ZI, McArthur HL, Ho AY. The abscopal effect of radiation therapy: what is it and how can we use it in breast cancer? Curr Breast Cancer Rep. (2017) 9:45–51. doi: 10.1007/s12609-017-0234-y
430. Kojima S, Ohshima Y, Nakatsukasa H, Tsukimoto M. Role of ATP as a key signaling molecule mediating radiation-induced biological effects. Dose Response. (2017) 15:155932581769063. doi: 10.1177/1559325817690638
431. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. (2014) 3:e28518. doi: 10.4161/onci.28518
432. Martins I, Tesniere A, Kepp O, Michaud M, Schlemmer F, Senovilla L, et al. Chemotherapy induces ATP release from tumor cells. Cell Cycle. (2009) 8:3723–8. doi: 10.4161/cc.8.22.10026
433. Lee M, Kim SW, Nam EJ, Cho H, Kim JH, Kim YT, et al. ATP-based chemotherapy response assay in primary or recurrent ovarian and peritoneal cancer. Yonsei Med J. (2014) 55:1664. doi: 10.3349/ymj.2014.55.6.1664
434. Sheth S, Bleibel W, Thukral C, A-Rahim Y, Beldi G, Csizmadia E, et al. Heightened NTPDase-1/CD39 expression and angiogenesis in radiation proctitis. Purinergic Signal. (2009) 5:321–6. doi: 10.1007/s11302-009-9154-2
435. Wirsdörfer F, de Leve S, Cappuccini F, Eldh T, Meyer AV, Gau E, et al. Extracellular adenosine production by ecto-5′-nucleotidase (CD73) enhances radiation-induced lung fibrosis. Cancer Res. (2016) 76:3045–56. doi: 10.1158/0008-5472.CAN-15-2310
436. Nevedomskaya E, Perryman R, Solanki S, Syed N, Mayboroda OA, Keun HC. A systems oncology approach identifies NT5E as a key metabolic regulator in tumor cells and modulator of platinum sensitivity. J Proteome Res. (2016) 15:280–90. doi: 10.1021/acs.jproteome.5b00793
437. Samanta D, Park Y, Ni X, Li H, Zahnow CA, Gabrielson E, et al. Chemotherapy induces enrichment of CD47+/CD73+/PDL1+ immune evasive triple-negative breast cancer cells. Proc Natl Acad Sci USA. (2018) 115:E1239–48. doi: 10.1073/pnas.1718197115
Keywords: adenosine, cAMP, CD73, CD39, cancer immunotherapy, T cells, tumor microenvironment
Citation: Vigano S, Alatzoglou D, Irving M, Ménétrier-Caux C, Caux C, Romero P and Coukos G (2019) Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front. Immunol. 10:925. doi: 10.3389/fimmu.2019.00925
Received: 12 January 2019; Accepted: 10 April 2019;
Published: 06 June 2019.
Edited by:
Anil Shanker, Meharry Medical College, United StatesReviewed by:
Per Thor Straten, Herlev Hospital, DenmarkMikhail M. DIkov, The Ohio State University, United States
Copyright © 2019 Vigano, Alatzoglou, Irving, Ménétrier-Caux, Caux, Romero and Coukos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melita Irving, bWVsaXRhLmlydmluZ0B1bmlsLmNo; George Coukos, Z2VvcmdlLmNvdWtvc0BjaHV2LmNo
†These authors have contributed equally to this work