
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 01 May 2019
Sec. Autoimmune and Autoinflammatory Disorders
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00853
This article is part of the Research Topic Autoantibodies in Kidney Diseases View all 11 articles
 Elisabetta Valoti1
Elisabetta Valoti1 Marta Alberti1
Marta Alberti1 Paraskevas Iatropoulos1
Paraskevas Iatropoulos1 Rossella Piras1
Rossella Piras1 Caterina Mele1
Caterina Mele1 Matteo Breno1Alessandra Cremaschi1Elena Bresin1Roberta Donadelli1Silvia Alizzi2
Matteo Breno1Alessandra Cremaschi1Elena Bresin1Roberta Donadelli1Silvia Alizzi2 Antonio Amoroso2
Antonio Amoroso2 Ariela Benigni1
Ariela Benigni1 Giuseppe Remuzzi1,3
Giuseppe Remuzzi1,3 Marina Noris1*
Marina Noris1*Atypical hemolytic uremic syndrome (aHUS) is a rare disease characterized by microangiopathic hemolytic anemia, thrombocytopenia and renal failure. It is caused by genetic or acquired defects of the complement alternative pathway. Factor H autoantibodies (anti-FHs) have been reported in 10% of aHUS patients and are associated with the deficiency of factor H-related 1 (FHR1). However, FHR1 deficiency is not enough to cause aHUS, since it is also present in about 5% of Caucasian healthy subjects. In this study we evaluated the prevalence of genetic variants in CFH, CD46, CFI, CFB, C3, and THBD in aHUS patients with anti-FHs, using healthy subjects with FHR1 deficiency, here defined “supercontrols,” as a reference group. “Supercontrols” are more informative than general population because they share at least one risk factor (FHR1 deficiency) with aHUS patients. We analyzed anti-FHs in 305 patients and 30 were positive. The large majority were children (median age: 7.7 [IQR, 6.6–9.9] years) and 83% lacked FHR1 (n = 25, cases) due to the homozygous CFHR3-CFHR1 deletion (n = 20), or the compound heterozygous CFHR3-CFHR1 and CFHR1-CFHR4 deletions (n = 4), or the heterozygous CFHR3-CFHR1 deletion combined with a frameshift mutation in CFHR1 that generates a premature stop codon (n = 1). Of the 960 healthy adult subjects 48 had the FHR1 deficiency (“supercontrols”). Rare likely pathogenetic variants in CFH, THBD, and C3 were found in 24% of cases (n = 6) compared to 2.1% of the “supercontrols” (P-value = 0.005). We also found that the CFH H3 and the CD46GGAAC haplotypes are not associated with anti-FHs aHUS, whereas these haplotypes are enriched in aHUS patients without anti-FHs, which highlights the differences in the genetic basis of the two forms of the disease. Finally, we confirm that common infections are environmental factors that contribute to the development of anti-FHs aHUS in genetically predisposed individuals, which fits with the sharp peak of incidence during scholar-age. Further studies are needed to fully elucidate the complex genetic and environmental factors underlying anti-FHs aHUS and to establish whether the combination of anti-FHs with likely pathogenetic variants or other risk factors influences disease outcome and response to therapies.
Atypical hemolytic uremic syndrome (aHUS) is a rare disease characterized by microangiopathic hemolytic anemia, thrombocytopenia and renal failure (1, 2). It is mainly caused by genetic or acquired defects of the alternative pathway (AP) of the complement system (3). Heterozygous likely pathogenic variants (LPVs) in genes encoding complement regulators, such as factor H (FH), factor I and CD46 (3–7), or components of the AP C3 convertase (C3 and factor B) (3, 8–11) were found in about half of aHUS patients. Moreover, genetic abnormalities in thrombomodulin (THBD), an endothelial anticoagulant protein that also regulates complement, were reported in 3–5% of aHUS cases (12).
Incomplete penetrance was observed in carriers of genetic abnormalities, indicating that although LPVs predispose to the development of aHUS, additional genetic and/or environmental hits are necessary for the disease to manifest (13). Indeed, in a large multinational study, 25% of patients with LPVs in CD46 or CFI, and 8–10% of those with LPVs in CFH, C3, and CFB, carried combined genetic abnormalities in other complement genes (13). Common variants and haplotypes in CFH and CD46 have been associated with aHUS and were reported to increase the penetrance of the disease in subjects carrying LPVs (14–16).
Factor H autoantibodies (anti-FHs) have been described in about 10% of aHUS patients and are strongly associated with the deficiency of factor H-related 1 (FHR1) (17–19). In most cases, the lack of FHR1 is due to the polymorphic homozygous deletion of the factor H related 3 and 1 genes (CFHR3-CFHR1) (20). In about 15% of patients the FHR1 deficiency is caused by compound heterozygous deletion of CFHR3-CFHR1 and CFHR1-CFHR4, while homozygous CFHR1-CFHR4 deletion is rare (21–23).
The pathogenic mechanism that links the FHR1 deficiency with the risk of the development of anti-FHs has not been clarified yet. Anti-FHs mainly target the C-terminal region of FH, and also cross react with FHR1 short consensus repeats (SCRs) 4–5, which have very high amino acid sequence identity with SCRs 19–20 of FH (24–27). It has recently been hypothesized that binding of certain proteins expressed by pathogens to the SCRs 19–20 of FH induces the exposure of a neoepitope in FH that is conformationally similar to that in SCRs 4–5 of FHR1, resulting in an autoimmune response in subjects with FHR1 deficiency (26).
Despite the above evidence, FHR1 deficiency is not enough to cause anti-FH associated aHUS, which is an ultra-rare disease with an estimated prevalence of 1:1,000,000 (1, 28). Indeed, the CFHR3-CFHR1 deletion is a rather common polymorphism, and is present in homozygosity in 3–10% of healthy European populations and in 7–30% of African populations, whereas it is very rare in Asia and South America (29). The combined CFHR3-CFHR1 and CFHR1-CFHR4 deletion is present in 0.9% of healthy European controls (22).
Autoimmune diseases are typically complex disorders in which multiple susceptibility genetic variants and environmental factors are involved (30, 31). The contribution of each genetic variant is usually small and only the presence of multiple variants favors the development of the disease (31). Notably, each single risk variant may be found in healthy subjects, although at a lower frequency than in patients, but it is the combination of multiple risk variants that clusters with the disease (30).
Previous studies reported that about 14.7% of aHUS patients with anti-FHs also carry LPVs in complement genes (18, 19, 21–23, 32); however the prevalence of LPVs in patients with anti-FHs should be compared to that in the general population or, even better, in a control group with FHR1 deficiency. This would support the hypothesis that LPVs have a role in anti-FHs aHUS.
In this study we investigated the genetic determinants of anti-FH-associated aHUS by comparing patients with anti-FHs and FHR1 deficiency (defined here as “cases”) and adult healthy subjects carrying the homozygous CFHR1 deletion (defined here as “supercontrols”) as a homogeneous, special control group, to optimize the power to detect differences. We hypothesized that the “supercontrols” were depleted for risk variants or even enriched in protective variants, since they evaded the development of aHUS despite carrying the known risk factor represented by the FHR1 deficiency.
Specifically, we compared the prevalence of rare LPVs in complement genes between cases and “supercontrols.” We also investigated the association with anti-FHs aHUS of common variants in complement genes, which in previous studies were reported to increase the risk of aHUS or of other immune-mediated glomerulopathies (33–38).
We found that rare LPVs but not common complement gene variants are enriched to a significant extent in cases compared to “supercontrols,” suggesting they are involved in the development of anti-FHs aHUS.
In all patients, atypical HUS was diagnosed based on microangiopathic hemolytic anemia and thrombocytopenia defined as hematocrit <30%, hemoglobin level <10 g/dl, serum lactate dehydrogenase level above 500 U/l, undetectable haptoglobin level, fragmented erythrocytes in peripheral blood smear, and platelets below 150 × 103/μl, associated with acute renal failure (serum creatinine >1.3 mg/dl for adults, >0.5 mg/dl for children under 5 years of age and >0.8 mg/dl for children aged 5–10 years old; and/or urinary protein/creatinine ratio >200 mg/g; or an increase of serum creatinine or urinary protein/creatinine ratio >15% compared to baseline levels). All aHUS patients (n = 305) were recruited through the International Registry of HUS/TTP (Ranica, Bergamo, Italy).
Healthy adult subjects (n = 960) were from internal donors and from the Piedmont Regional Registry of the Italian Bone Marrow Donors Registry (Azienda Ospedaliera–Universitaria, Città della Salute e della Scienza, Turin, Italy). Patient and healthy subject data were handled with respect for confidentiality and anonymity. All subjects provided informed written consent in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of Azienda Sanitaria Locale, Bergamo, Italy.
The presence of anti-FH IgGs was evaluated through an Enzyme-Linked Immunosorbent Assay (ELISA). Microtiter plates were coated with 100 μl of a solution of 1 μg/ml of purified human FH (0.1 μg, Calbiochem). After overnight incubation at 4°C, the plate was washed with Phosphate-Buffered Saline (PBS), 0.1% Tween20, and 0.2 M NaCl and blocked with PBS, 0.1% Tween 20, and 0.3% milk powder. After washes, 50 μl of plasma/serum samples at 1:100 dilution were added to duplicated wells, and incubated for 40 min. A parallel plate was set up in the absence of FH coating, in which only blocking solution was added to evaluate the presence of non-FH specific background, according to published recommendations (39). After washes, goat anti-human IgG antibody conjugated with horseradish peroxidase (HRP, 1:250, Sigma-Aldrich) was added and incubated for 1 h. Enzymatic activity was revealed using the 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. A positive control kindly gifted by Dr. Marie-Agnes Dragon-Durey, with 2,000 AU/ml titer was used at 1:100 dilution. The positive threshold was set up at the mean titer +2 standard deviations (SD) found in plasma samples of 98 healthy controls with 1 or 2 copies of CFHR1 (56 AU/ml). The sample concentrations expressed in arbitrary units/ml (AU/ml) were extrapolated from a sigmoidal curve and the background derived from wells without FH coating was subtracted. The ELISA was repeated in positive samples, adding 15 μg of FH into each sample to verify antibody specificity. Samples showing at least a 50% decrease of the ELISA absorbance in the assay performed in the presence of exogenous FH were considered as true positive.
This ELISA protocol was also used to evaluate the binding of autoantibodies to SCRs 1–5 and 15–20 FH fragments obtained by baculovirus transfection of Spodoptera frugiperda cells as reported (40). The plate was coated with molar equivalents of FH SCRs 1–5 and 15–20 and the results were expressed as absorbance at 450 nm. In a selected experiment, additional wells were coated with full length FH (positive control) or bovine serum albumin (BSA, negative control).
Serum-induced C5b-9 deposition on a human microvascular endothelial cell line (HMEC-1) was analyzed as previously described, with minor modifications (41). HMEC-1 were plated on glass coverslips and used when confluent. Cells were activated with 10 μM ADP for 10 min and incubated for 4 h with serum from patients (ID: 18 and 20) or healthy controls diluted 1:2 with test medium (Hank's Balanced Salt Solution—HBSS +0.5% BSA). HMEC-1 were fixed and stained with rabbit anti-human C5b-9 antibody, followed by FITC-conjugated antibody. Fluorescent staining on cell surface was acquired in 15 fields through confocal microscopy and the staining area was evaluated using Image J software. Results were expressed as the percentage of staining in relation to a pooled control sera (n = 10 subjects) run in parallel. The sera from additional 35 controls were analyzed separately and the percentages of C5b-9 deposits vs. control serum pool were calculated to establish the normal range (mean ± 2SD of % C5b-9 deposits of the control sera vs. control serum pool: 60–149%).
FH levels were evaluated by ELISA. Microtiter plates were coated with 0.15 μg of purified sheep polyclonal anti-human factor H antibody (Abcam). After three washes with PBS, 0.05% Tween20, the plate was blocked with 200 μl of PBS, 1% BSA. After four washes with PBS, 0.05% Tween20, 100 μl of plasma samples at 1:10,000 dilution were added to duplicated wells, and incubated for 2 h. After the washes, 100 μl of mouse monoclonal anti-human factor H (OX-23, LifeSpan BioSciences), which recognizes the SCRs 1–4 of the FH N-terminal domain, were added at a 1:10,000 dilution in blocking solution. Notably, this anti-human factor H antibody also detected the FH-like 1 (FHL1) which shares the N-terminal domain with FH. The plate was washed and 100 μl of goat anti-mouse antibody conjugated with HRP (1:2,000, Thermo Fisher) were added and incubated for 1 h. Enzymatic activity was revealed using the TMB substrate.
FHR1 was studied by Western blotting in the serum of all patients with anti-FHs with at least one copy of CFHR1. Sera were diluted 1:40 in loading buffer (4X Laemmli Sample Buffer, Bio-Rad) in non-reducing conditions and analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE, Mini-Protean TGX Precast Gels, Bio-Rad). Proteins were transferred to polyvinylidene difluoride membrane (PVDF, Trans-Blot Turbo Midi PVDF Transfer; Bio-Rad), and blocked with 4% skim milk and 1% BSA. Mouse anti-human FHR1 IgGs (1:1,000, kindly given by Prof. Peter Zipfel) (42), which recognizes the N-terminal domains of FHR1, were added for 1 h. The binding of the antibody was detected by 1 h incubation with HRP goat anti-mouse IgG (1:5,000), followed by enhanced chemiluminescence (ECL) substrate (Amersham).
Multiplex-ligation dependent probe amplification (SALSA MLPA P236-A3 ARMD, MCR-Holland, Netherlands) was used to evaluate the presence of copy number variations (CNVs) in CFH, CFHR3, CFHR1, CFHR2, CFHR5 genes in the 30 aHUS patients with anti-FHs. The CFHR4 CNV was evaluated in aHUS patients with anti-FHs by a multiplex PCR, as reported by Moore et al. (22). To evaluate the presence of the homozygous deletion of CFHR1 in all 305 aHUS patients, and in the 960 healthy adult controls a multiplex PCR was set up that amplifies a 133 bp fragment in intron 3 of CFHR1 and a 83 bp fragment in the promoter of RNAseP.
Primers: CFHR1-For 5′-ATCACTACACATGGACCTGAAA-3′; CFHR1-Rev 5′-GATGTGGAAAAATAAAAGAAAATAAGTC-3′; RNAseP-For 5′-TAGATACCGTGTGCGTGCAT-3′; RNaseP-Rev 5′-GGGGTTCCAATTCCCAACTA-3′.
Through next-generation sequencing we performed genetic screening of all exons and flanking regions of CFH, CD46, CFI, CFB, C3, and THBD by highly multiplex PCR using the Ion AmpliSeq™ Library Kit on an Ion PGM Sequencer (Life Technologies) (34).
LPVs were defined as genetic variants with Minor Allelic Frequency (MAF) in the ExAC database ≤0.001 and a Combined Annotation Dependent Depletion (CADD) pathogenic score ≥20 (43). Considering the complexity of aHUS and the very low penetrance we also considered as likely pathogenic, variants with a MAF ≤0.01 for which there was evidence of functional effects in literature. In addition, we included as likely pathogenic, genetic variants with MAF ≤0.01 for which an association with aHUS was reported (P-value <0.001, vs. “ExAC all” in the database of complement gene variants, www.complement-DB.org).
The CFH promoter and the coding and flanking regions of CFHR1 were analyzed by direct sequencing.
All statistical tests were executed using MedCalc software. The Chi-square test or the Fisher's exact test were used to compare the allele frequencies between cases and “supercontrols,” as appropriate. The expectation maximization algorithm by Haploview software was used to estimate each CFH haplotype frequency. Findings were considered statistically significant at P-values <0.05 after Bonferroni correction. Odds ratio (OR) was reported with the 95% confidence interval. ANOVA or Kruskal-Wallis tests were used to compare the mean or the median of variables, as appropriate.
The presence of anti-FHs was assessed in 305 consecutive aHUS patients of the International Registry of HUS for whom serum or plasma was available. Thirty patients (9.8%) were positive for anti-FHs (anti-FHs titer threshold >56 AU/ml); in all of them the specificity of the result was confirmed in a replicated assay, in which an excess of FH was added to the serum/plasma sample and competed for the anti-FHs with FH coated on the well (Figure 1). The mean ± SD of anti-FH titers was 1,164.7 ± 1,189.5 AU/ml in samples collected during the acute phase of the disease (n = 4) and 521.8 ± 504.3 AU/ml in samples collected during remission (n = 26; ANOVA, P-value = 0.062).
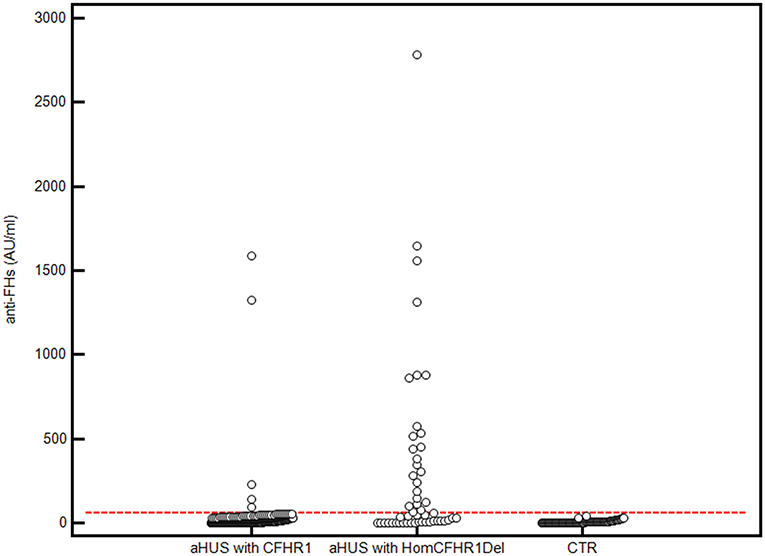
Figure 1. Anti-FHs in aHUS patients and healthy controls. 305 aHUS patients were analyzed for anti-FHs through ELISA: 254 were carriers of 1 or 2 copies of CFHR1 (aHUS with CFHR1), and 51 had 0 copies of CFHR1 (aHUS with HomCFHR1Del). Patient 23, carrying the heterozygous CFHR1Del and a frameshift variant in CFHR1 exon 2 (c.104delAfsX), which resulted in a complete deficiency of FHR1 has been also included in the group of patients defined as “aHUS with HomCFHR1Del.” Ninety-eight healthy controls with 1 or 2 copies of CFHR1 were also analyzed for anti-FHs (CTR). The positive threshold was set at mean +2SD of values recorded in the 98 controls (56 AU/mL) and is shown with the horizontal red dashed line.
In all 30 patients with anti-FHs aHUS, copy numbers were evaluated by MLPA for CFH, CFHR3, CFHR1, CFHR2, and CFHR5 and by a multiplex PCR for CFHR4. The homozygous CFHR3-CFHR1 deletion was observed in 20 patients (66.7%) and the heterozygous CFHR3-CFHR1 deletion combined with the heterozygous CFHR1-CFHR4 deletion was present in 4 patients (13.3%, Table 1). Overall, 24 patients had zero copies of CFHR1 (80%, Table 1). Four patients showed the heterozygous deletion of CFHR3 and CFHR1 (13.3%) and in two patients no CNV in CFHR genes were identified (6.7%, Table 1).
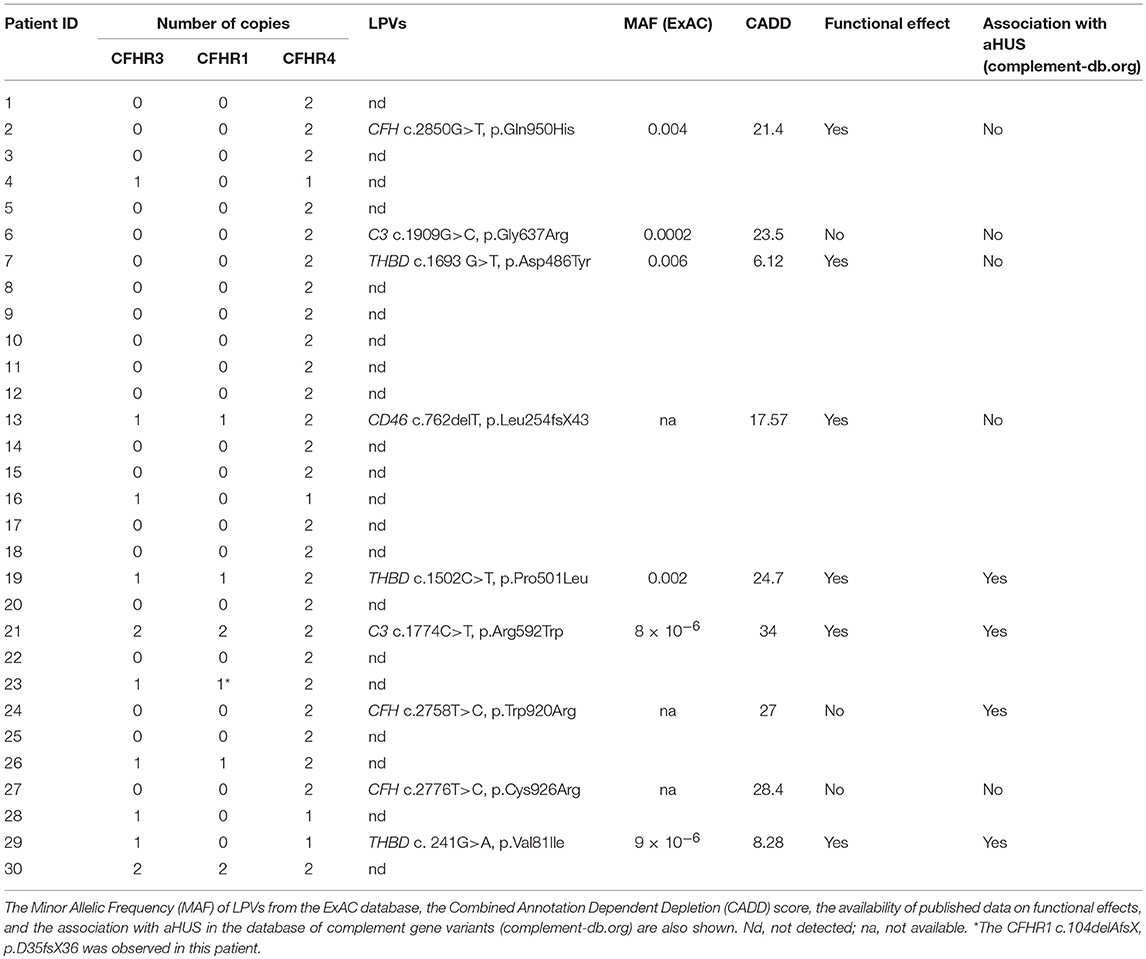
Table 1. Copy numbers of CFHR3, CFHR1, and CFHR4 and LPVs in complement genes observed in aHUS patients with anti-FHs.
Direct sequencing of CFHR1 was performed in the 6 patients with anti-FHs and at least one copy of CFHR1. In patient 23, who carried the heterozygous CFHR1 deletion, a frameshift variant in CFHR1 exon 2 (c.104delAfsX) that resulted in a premature stop codon (p.D35fsX36) was observed. In all six patients, the presence of FHR1 in serum was verified by Western blot (Figure 2). The normal pattern of two FHR1 bands was observed in patients 13, 19, and 26 (all with one copy of CFHR1) and patients 21 and 30 (both with two copies of CFHR1). At variance, patient 23, who had one copy of CFHR1 plus the c.104delAfsX frameshift mutation did not have any FHR1 band, confirming that the mutation resulted in a non-secreted truncated protein, as expected. Thus, in our cohort, 25/30 (83.3%) patients with anti-FHs showed a complete deficiency of FHR1 and are defined as cases here.
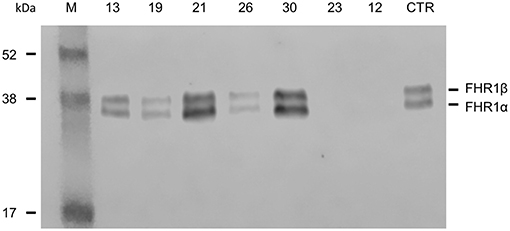
Figure 2. Detection of FHR1 by Western blotting in aHUS patients with anti-FHs and at least one copy of CFHR1. ID numbers 13, 19 and 26: patients with 1 copy of CFHR1; ID numbers 21 and 30: patients with 2 copies of CFHR1; ID number 23: patient with 1 copy of CFHR1 and a frameshift mutation in exon 2 (c.104delAfsX); ID number 12: patient with 0 copies of CFHR1; CTR: an healthy control with two copies of CFHR1.
We also evaluated the prevalence of the acidic and basic FHR1 isoforms in patients with anti-FHs and with at least one copy of CFHR1. In the basic FHR1 isoform that has been associated with the risk of aHUS (21), the amino acids 157His, 159Leu, and 175Glu are replaced by 157Tyr, 159Val, and 175Gln, which make the SCR3 of FHR1 identical to the SCR18 of FH. In patients with the heterozygous CFHR3-CFHR1 deletion, one exhibited the basic isoform and two the acidic isoform. Both patients with two copies of CFHR3 and CFHR1 were compound heterozygous for the two FHR1 isoforms. Thus, we did not find an enrichment of a specific FHR1 isoform in our aHUS patients with anti-FHs.
A multiplex PCR was used to identify subjects with the CFHR1 homozygous deletion in all 305 unrelated aHUS patients analyzed for anti-FHs and in a large cohort of healthy subjects (n = 960). Overall, 49 aHUS patients (16%) and 48 healthy subjects (5%) were found to be homozygous for the CFHR1 deletion (OR [95%CI] = 3.6 [2.4–5.5], P-value = 5.5 × 10−10, Table 2). The association of the homozygous CFHR1 deletion with aHUS strongly increased in the subgroup of patients with anti-FHs vs. healthy controls (OR [95%CI] = 76 [29.7–194.6], P-value = 3 × 10−52, Table 2).
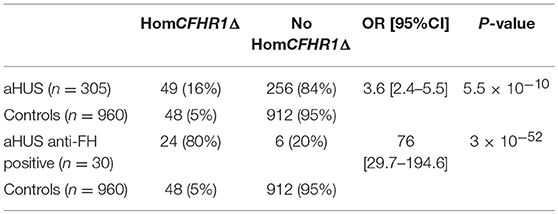
Table 2. Prevalence of homozygous CFHR1 deletion in aHUS patients (n = 305) and healthy controls (n = 960).
To discern which FH epitopes were recognized by anti-FHs, anti-FH ELISA was repeated using wells coated with N-terminal FH (SCRs 1–5) and C-terminal FH (SCRs 15–20) fragments (kindly given by Prof. Peter Zipfel). The results of samples from five patients with 1 or 2 copies of CFHR1 (patients 19, 13, 30, 21, and 26) and from seven patients carrying the homozygous CFHR1 deletion (patients 16, 14, 15, 10, 25, 18, and 8) are shown in Figure 3. Antibodies from patients 13, 19, and 30 (with 1, 1 and 2 copies of CFHR1, respectively) and from patients 8, 10, 14, 15, and 16 (with 0 copies of CFHR1) strongly bound to C-terminus but not to N-terminus of FH. In patient 25 with the homozygous deletion of CFHR1, we observed a less intense but selective binding to FH C-terminal fragment. Antibodies from patients 18 (with the homozygous deletion of CFHR1) and 21 (with two copies of CFHR1) bound weakly to both FH SCRs 1–5 and FH SCRs 15–20 and finally, antibodies from patient 26 (with one copy of CFHR1) did not specifically bind any FH fragments. It is possible that autoantibodies from patient 26 recognize either central FH epitopes or epitopes exposed only in the native, full length FH, as previously reported for other aHUS patients (22).
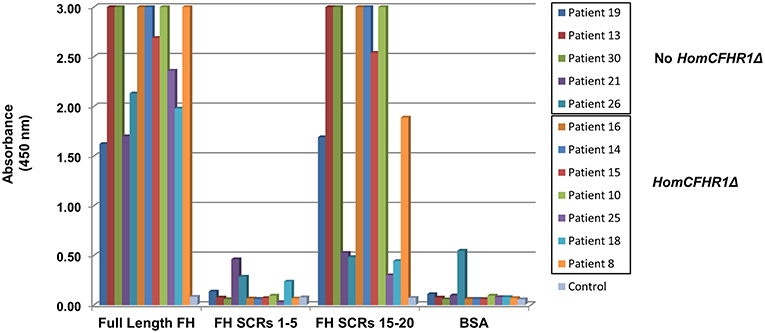
Figure 3. Binding site localization of anti-FHs detected in aHUS patients. Anti-FH binding to FH N-terminal fragment (SCRs 1–5) and FH C-terminal fragment (SCRs 15–20) evaluated in 12 aHUS patients. Seven patients were carriers of the homozygous CFHR1 deletion (HomCFHR1Δ) while 5 patients presented at least one copy of CFHR1 (No HomCFHR1Δ). BSA coating was used as negative control and full length FH coating as positive control. Absorbance of serum from a healthy subject was used as an additional control. The absorbance is shown on the ordinate axis. Data are representative of three experiments.
An ex-vivo test to evaluate complement activation at the endothelial cell level showed that serum from two patients (ID: 18 and 20, without LPVs and with homozygous CFHR1 deletion) taken after aHUS remission induced higher than normal (>149%) C5b-9 deposits on cultured ADP-activated human microvascular endothelial cells (Figure 4).
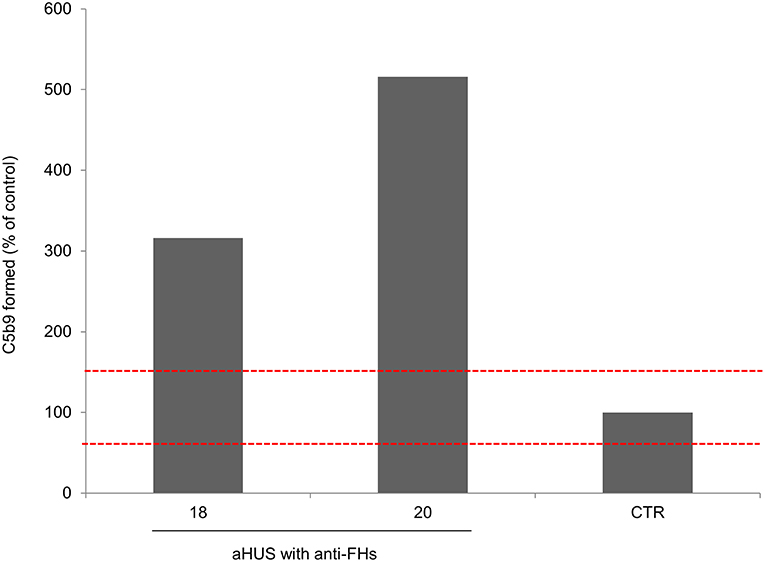
Figure 4. Complement activation on endothelial cells. Endothelial surface area covered by C5b-9 staining after incubation of ADP-activated HMEC-1 with serum from aHUS patients with anti-FHs studied at remission (patients 18 and 20). For each sample, values were expressed as the percentage of C5b-9 deposits induced by a pool of sera from 10 healthy controls run in parallel (reference 100%). The red dashed lines indicated the normal range (60–149%) determined by testing single sera from 35 different healthy controls.
CFH, CD46, CFI, CFB, C3, and THBD were sequenced in all patients with anti-FHs and 9 were found to be carriers of LPVs as defined in methods (30%, Table 1).
Of the 25 patients with anti-FHs and the FHR1 deficiency (cases), 6 had LPVs (24%, Table 1). Three cases carried LPVs in CFH (12%). Two Italian cases showed the heterozygous CFH LPV c.2850G>T, p.Gln950His in SCR16 (rs149474608, MAF (ExAC) = 0.004, CADD = 21.4) and the c.2758T>C, p.Trp920Arg (CADD = 27) in SCR15, respectively. These two patients had been previously reported by our group (3). Moreover, the p.Gln950His variant was previously described in another aHUS patient with anti-FHs (22), in other aHUS patients without anti-FHs (44–46), and in a patient who had developed TMA after renal transplantation (47). The third case was a Jewish patient who showed the heterozygous CFH c.2776C>T, p.Cys926Arg (CADD = 28.4) in SCR15. This variant is not present in public databases and has not been described in aHUS patients before. Notably, nuclear magnetic resonance studies (48) showed that the 3 LPVs involve amino acids that located close to each other in the three-dimensional structure of SCRs 15 and 16 of FH (Figure 5).
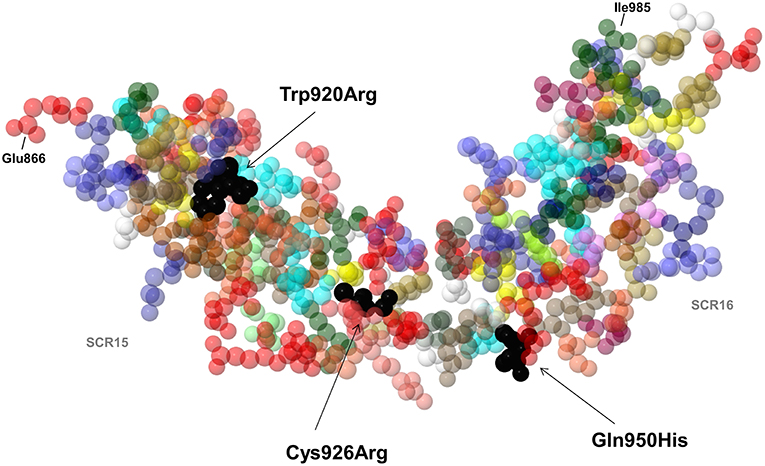
Figure 5. 3D structure by nuclear magnetic resonance of SCR15 and SCR16 of FH (PDB code: 1HFH). Black arrows indicate the FH amino acid residues Trp920, Cys926 (SCR15), and Gln950 (SCR16) (colored in black) which are substituted in three aHUS patients with anti-FHs due to the Trp920Arg, Cys926Arg, and Gln950His heterozygous mutations. Each amino acid is represented with a specific color. The first amino acid of SCR15 (Glu866) and the last amino acid of SCR16 (Ile985) are also indicated. Cysteine residues are shown in yellow color. Created by Jmol, an open-source Java viewer for chemical structures in 3D (http://www.jmol.org).
In an Italian case we found the C3 heterozygous c.1909G>C, p.Gly637Arg (rs149850773, MAF (ExAC) = 0.0002, CADD = 23.5), which is located in the Linker domain of the C3 molecule and has not been reported in patients with aHUS before.
We identified two THBD heterozygous LPVs: the c.1693G>T, p.Asp486Tyr (rs41348347, MAF (ExAC) = 0.006, CADD = 6.12) was identified in an Italian case and the c.241G>A, p.Val81Ile (rs772288987, MAF (ExAC) = 9 × 10−6, CADD = 8.28) was found in a case from Yemen. Both THBD variants have already been reported in patients with aHUS (3, 49).
Of the 48 adult healthy “supercontrols” with the homozygous deletion of CFHR1, only one carried a LPV, namely the heterozygous CFI c.949 G>A, p.Arg317Trp [rs121964917, MAF (ExAC) = 9.9 × 10−5, CADD = 16.27]. The FI 317Trp variant has previously been reported to have 30% C3b and C4b cofactor activity compared to wild type FI, but conflicting results have been found by other authors (50, 51).
Overall, in cases the prevalence of LPVs was significantly higher than in “supercontrols” (24 vs. 2.1%, Fisher's exact test, P-value = 0.005).
We then investigated whether common genetic variants in CFH contribute to determining susceptibility to anti-FHs mediated aHUS by comparing the prevalence of CFH c.1-332C>T (rs3753394), c.184G>A p.Val62Ile (rs800292), c.1204T>C p.Tyr402His (rs1061170), c.2016A>G Gln572Gln (rs3753396), c.2237-543G>A (rs1410996), c.2808G>T Glu936Asp (rs1065489) in the 25 cases and in the 48 “supercontrols.” As shown in Table 3, allele frequencies did not differ between cases and “supercontrols.”
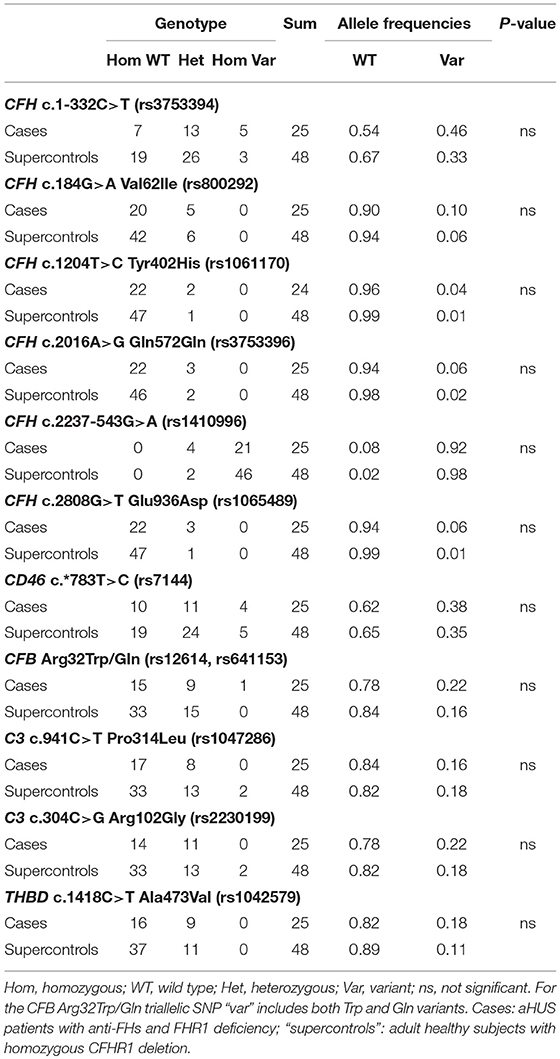
Table 3. Prevalence of common variants in CFH, CD46, CFB, C3, and THBD in cases (n = 25) and “supercontrols” (n = 48).
CFH haplotypes were also studied in cases and “supercontrols.” The minimal informative SNPs within the CFH gene considered for this analysis were rs3753394 (promoter), rs800292 (exon 2), rs1061170 (exon 9), rs3753396 (exon 14), rs1410996 (intron 15), and rs1065489 (exon 19). The most frequent CFH haplotype was the CGTAAG (H4a haplotype), with a frequency of 0.492 in cases and 0.638 in “supercontrols,” followed by the TGTAAG (H4b haplotype, Table 4), with no difference between cases (0.338) and “supercontrols” (0.279). On the contrary, the frequency of the H3 CFH haplotype (TGTGGT), which had previously been associated with aHUS (15) was very low and estimated to be 0.024 in cases and 0.01 in “supercontrols” (P-value = 0.04, Table 4). Notably, the frequency of the CFH H3 haplotype in aHUS patients without anti-FHs of our cohort (n = 275) was 0.292 and was significantly higher compared to cases (P-value = 0.0002), “supercontrols” (P-value = 5.6 × 10−8) as well as the general population (H3 haplotype frequency = 0.203, among 2,504 controls from 1,000 genomes project, P-value = 1.85 × 10−6).
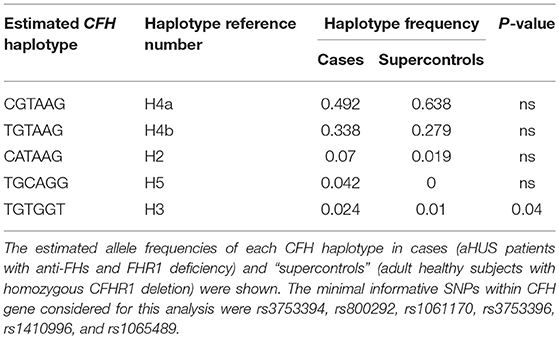
Table 4. Estimated CFH haplotypes in the 25 cases and the 48 “supercontrols.”
In summary, no significant association has been found between CFH haplotypes and anti-FHs mediated aHUS, using “supercontrols” as the reference group.
In our cohort of aHUS patients with anti-FHs, the H4a and H4b CFH haplotypes were in strong LD with the CFHR3-CFHR1 deletion (Table 5), which is consistent with published data (16), while the H5 and H3 haplotypes were associated with the CFHR1-CFHR4 deletion (Table 5).
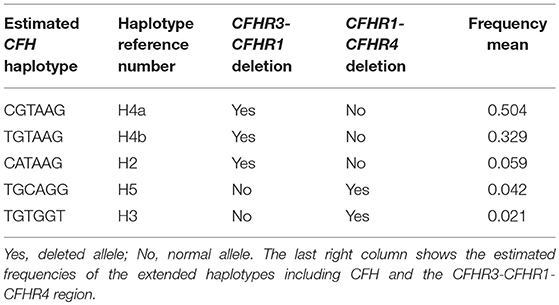
Table 5. Linkage disequilibrium between CFH haplotypes and CFHR3-CFHR1 or CFHR1-CFHR4 deletions in the 30 aHUS patients with anti-FHs.
The frequency of the CD46 c.*783C allele (rs7144) that tags the CD46GGAAC aHUS risk haplotype (52), did not differ between cases (0.38) and “supercontrols” (0.35, Table 3). On the contrary, the frequency of the C allele was significantly higher in our aHUS patients without anti-FHs (0.45) as compared with the 2,504 controls (allele frequency: 0.35) from the 1,000 genomes project (P-value = 0.001).
Moreover, we studied the prevalence of common polymorphisms in C3 c.941C>T, p.Pro314Leu (rs1047286), C3 c.304C>G, p.Arg102Gly (rs2230199), CFB c.94C>T, p.Arg32Trp (rs12614), CFB c.95G>A, p.Arg32Gln (rs641153) and THBD c.1418C>T, p.Ala473Val (rs1042579), which were previously reported in association with other immune-mediated diseases, including C3 glomerulopathy (33, 34), IgA nephropathy (35), and age-related macular degeneration (36–38). No significant difference was found with regard to allele frequencies of any of the above single nucleotide polymorphisms (SNPs) between cases and “supercontrols” (Table 3).
Finally, we analyzed the combination of the three variants FH Val62, FB Arg32, and C3 102Gly, which in functional studies (53, 54) have been associated with higher C3 convertase activity (risk complotype), and we did not find any difference in the prevalence of the risk complotype between cases (0.24) and “supercontrols” (0.17, P-value = 0.39).
We then wondered whether rare variants in the CFH promoter that could affect FH expression in the thymus may predispose to a lack of central tolerance toward FH and to the development of anti-FHs HUS. The analysis through Matinspector software (Genomatix) revealed the presence of two sequences predicted with a good score as consensus motifs for the “autoimmune regulator” transcription factor (AIRE, Figure 6), a DNA binding molecule that is involved in central thymic tolerance by promoting the expression of tissue-specific antigens in medullary thymic epithelial cells. We sequenced the c.1-1070 region upstream the CFH gene, including the two predicted AIRE consensus regions, in our aHUS patients with anti-FHs.

Figure 6. CFH promoter sequence. Exon 1 (yellow), basal promoter (light gray), proximal promoter (dark gray), and two AIRE consensus sequences predicted by Matinspector software–Genomatix (underlined) are shown.
We did not find any rare variant (with MAF ≤ 0.01) in the two DNA sequences predicted as AIRE consensus regions or in the entire sequenced CFH promoter region.
Clinical and biochemical features at onset for all the 30 patients with anti-FHs are reported in Table 6.
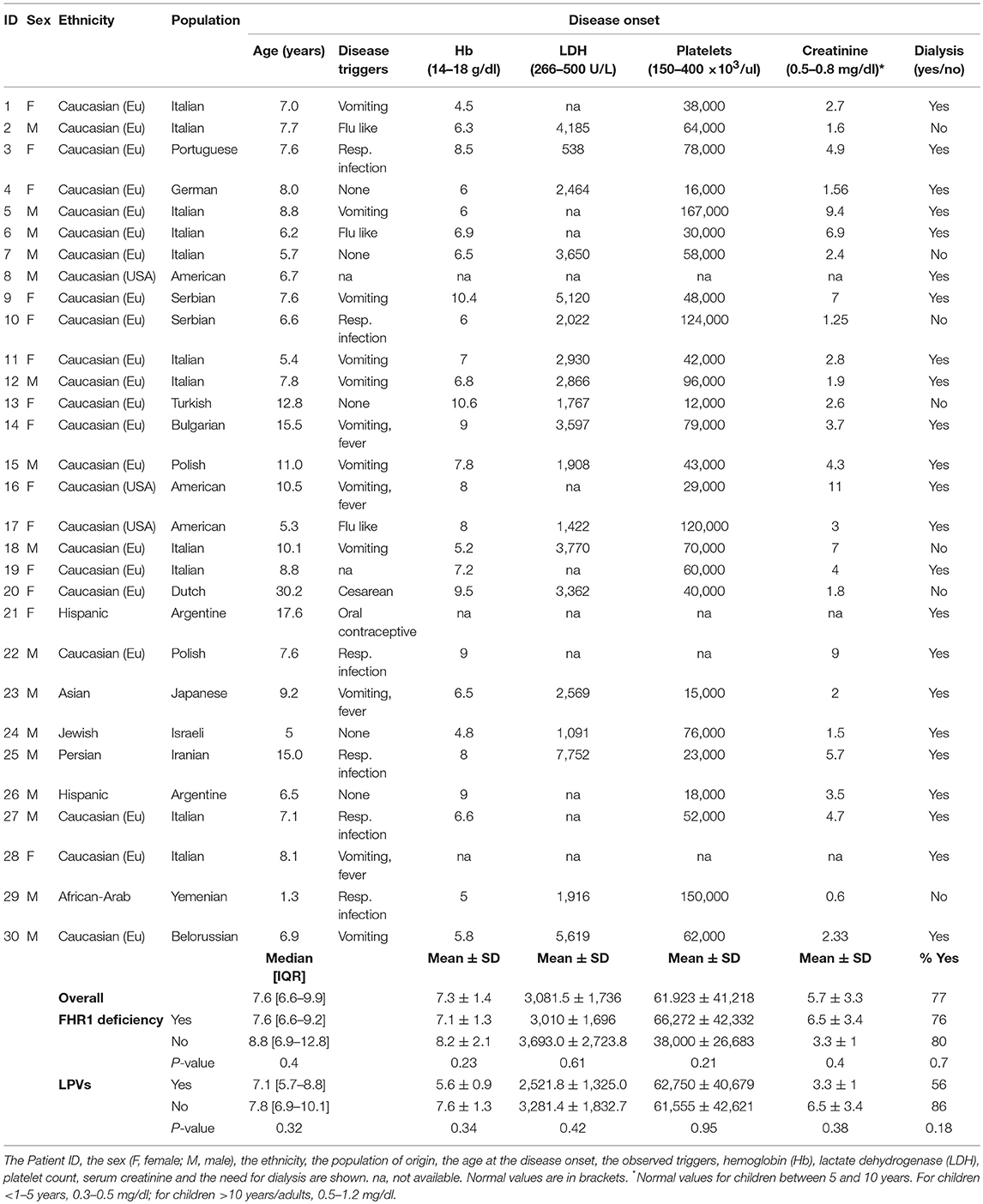
Table 6. Clinical and biological data at onset of the 30 patients with anti-FH associated aHUS.
The large majority of patients with anti-FHs in our cohort were Caucasian (53% males, Table 6). All but one developed aHUS during childhood or adolescence (96.7%). The median age of disease onset was 7.6 [6.6–9.9] years (Table 6). The outliers were a woman who developed anti-FHs aHUS at 30 years of age after a cesarean delivery and a child who manifested aHUS at 16 months (he also carried a THBD LPV). The distribution of the age at disease onset in patients with anti-FHs was unimodal, with a sharp incidence peak around 8 years of age (Figure 7A), which was significantly different from aHUS patients negative for anti-FHs who exhibited a bimodal distribution, with an early peak before 4 years of age, and a late peak during adulthood (Figure 7B). Similar bimodal distribution was observed when we considered only patients negative for anti-FHs and carrying complement gene abnormalities (in CFH, MCP, CFI, THBD, C3, and CFB; Figure 7C).
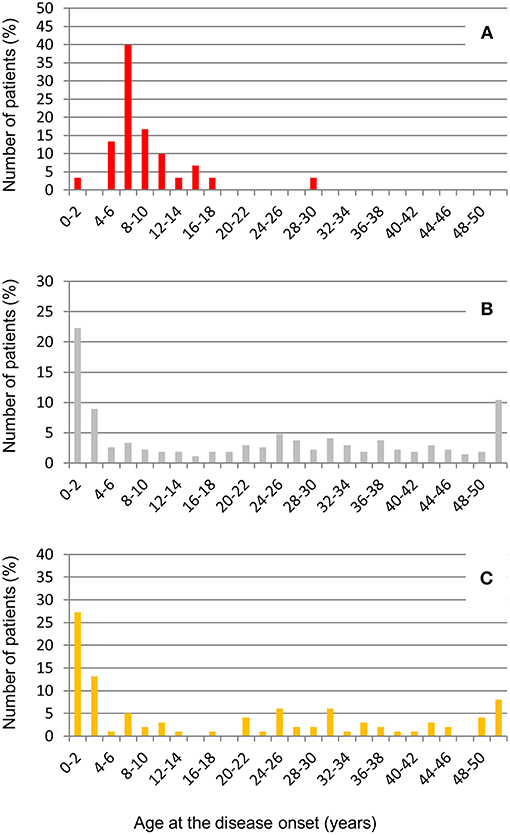
Figure 7. Distribution of age at disease onset in aHUS patients analyzed for anti-FHs. (A) Patients with anti-FHs (n = 30); (B) Patients without anti-FHs (n = 275), and (C) Patients without anti-FHs and carrying LPVs in complement genes (n = 99).
Through logistic regression we estimated that the homozygous deletion of CFHR1 and age at disease onset between 4 and 12 years were strongly associated with the risk for an aHUS patient to present anti-FHs. Twenty-four out of 30 aHUS patients with anti-FHs (80%) vs. 25 out of 275 aHUS patients without anti-FHs (9%) had the homozygous deletion of CFHR1 (OR [95% CI] = 40 [14.9–107.1], P-value = 1.4 × 10−22, Table 7). Twenty-four out of 30 aHUS patients with anti-FHs (80%) vs. 27 out of 275 aHUS patients without anti-FHs (9.8%) developed the disease between 4 and 12 years of age (OR [95% CI] = 37 [13.8–97.9], P-value = 1.7 × 10−21, Table 7). The combination of the homozygous CFHR1 deletion and age at disease onset between 4 and 12 years further increased the risk to have anti-FHs aHUS (OR [95% CI] = 108 [33.7–346.5], P-value = 6.9 × 10−33, Table 7).
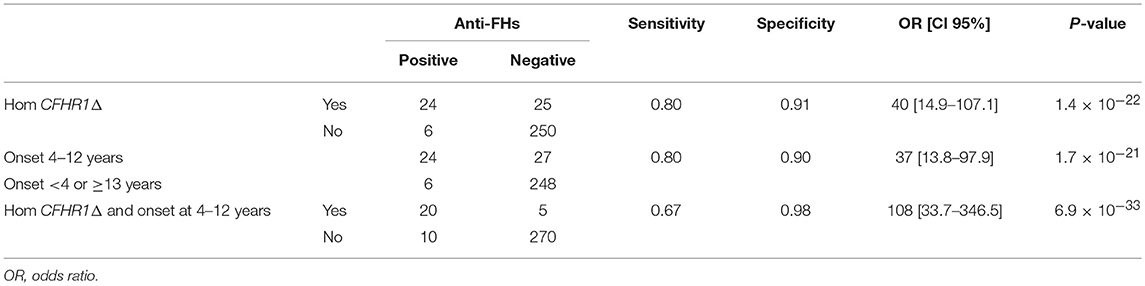
Table 7. Estimate of the risk of an aHUS patient having anti-FHs in the presence of CFHR1 homozygous deletion and/or age at disease onset between 4 and 12 years.
Prodromal signs were observed in most (70%) anti-FH positive patients: twelve patients (40%) exhibited gastrointestinal symptoms (four of them with fever), and nine patients (30%) had upper respiratory tract infections (including flu like symptoms). One patient developed aHUS post-partum and another while on oral contraception. Five patients did not have any prodromal signs or triggers and for two patients trigger data were not available (Table 6). At disease onset, all patients showed anemia, thrombocytopenia, high LDH levels, and high serum creatinine. Twenty-three (77%) patients developed kidney failure requiring dialysis (Table 6).
Among patients with anti-FHs, we did not find any significant difference in the age of onset and in clinical complement parameters between cases with FHR1 deficiency and patients with one or two copies of CFHR1 (Table 6), as well as between patients with or without LPVs (Table 6).
Data on serum C3 and C4 levels at the time of anti-FH antibody assay were available for 28 patients (Table 8). Fourteen patients (46.7%) had lower than normal serum C3 levels, in most of them C3 was measured during convalescence (Table 8). C4 levels, on the contrary, were normal in all patients (Table 8). Plasma FH plus FHL1 levels were in the normal range in all patients (Table 8) with the only exception being patient 14 (studied in remission and with a high antibody titer, 1,557.3 AU/ml) who had 80 mg/l FH (Table 8). Plasma FH plus FHL1 levels did not differ between patients studied during the acute episode and patients studied in remission (acute: 340.3 ± 49.3 mg/l n = 3; vs. remission: 344.7 ± 117.1 mg/l, n = 21; ANOVA, P-value = 0.951) and did not correlate with the antibody titer (r2 = 0.0085, P-value = ns). The latter finding would indicate that the antibodies used in our FH ELISA assay were detecting total FH, including free FH and immune-complexed FH (55), which is consistent with previously published studies (17, 56).
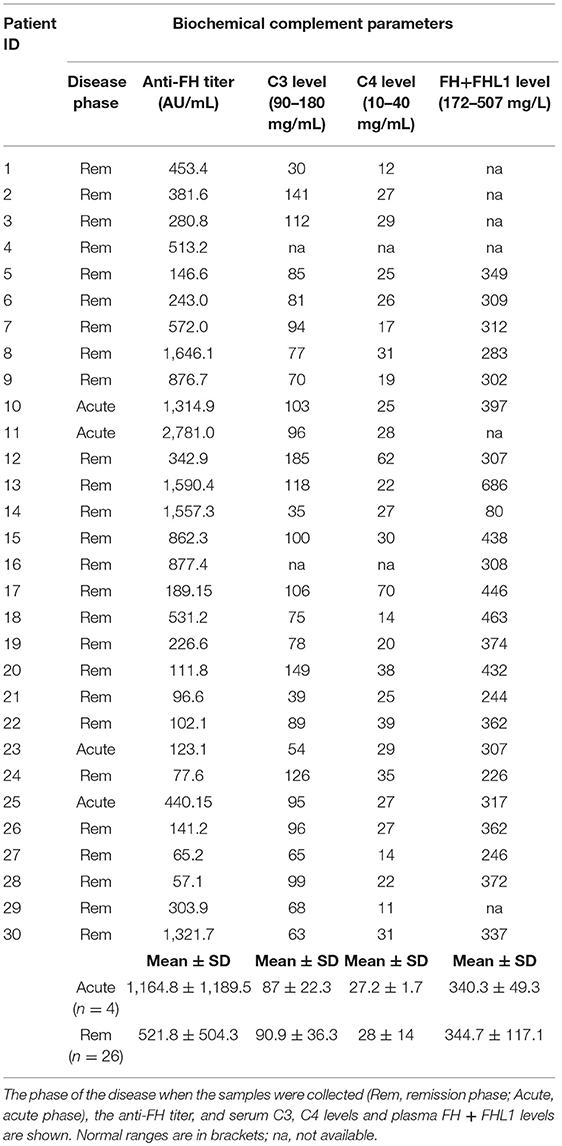
Table 8. Serum C3, C4, and plasma FH + FHL1 levels evaluated at the time of anti-FH measurement.
During a median of 36 months follow-up (IQR = 12–72 months), 11 patients with anti-FHs experienced relapses of the disease (37%): 9 had the FHR1 deficiency, one the FHR1 deficiency plus a LPV in CFH and one a LPV in CD46 and 1 copy of CFHR1. Overall, 15 patients (50%) developed end stage renal disease (ESRD): 8 had the FHR1 deficiency, 2 the FHR1 deficiency plus a LPV in C3 and CFH, respectively and 2 a LPV in THBD (and 1 copy of CFHR1) and C3 (and 2 copies of CFHR1), respectively. The prevalence of relapses or ESRD during the follow-up did not differ statistically between patients with or without LPVs (relapses: patients with LPVs 22%, without LPVs 43%; P-value = 0.42, ESRD: patients with LPVs 44%, without LPVs 52%; P-value = 1.0).
Four patients received kidney transplantation and aHUS recurrence was observed in one of them, who also carried the C3 heterozygous LPV p.Arg592Trp. Another patient died following the transplant due to clinical complications unrelated to aHUS.
Here we report the results of a retrospective study in a large cohort of patients with aHUS in which we described the clinical and genetic features of 30 patients with anti-FHs.
Through combined CNV analysis and CFHR1 sequencing we confirm that the FHR1 deficiency is strongly associated with anti-FHs aHUS. Moreover, through an approach based on the inclusion of “supercontrols,” we demonstrate that patients with anti-FHs are enriched in complement gene LPVs, while common complement gene variants known to increase the risk of aHUS or other immune -mediated diseases do not significantly contribute to anti-FHs aHUS.
The prevalence of anti-FHs in our cohort of aHUS patients (10%) is consistent with data from other European cohorts (5–13%) (28), but is lower than that observed in an Indian aHUS cohort in which the percentage of anti-FH positive patients was dramatically higher up to 56.1% (57).
Finding that 83% of our aHUS patients with anti-FHs had a complete FHR1 deficiency due to the homozygous CFHR3-CFHR1 deletion or, more rarely, to compound heterozygous CFHR3-CFHR1 and CFHR1-CFHR4 deletions is consistent with earlier published studies in other cohorts (21, 22). Interestingly, we describe a FHR1-deficient patient carrying the CFHR1 deletion in one allele and a frameshift CFHR1 mutation causing an early termination of protein translation in the other allele. To the best of our knowledge only one other aHUS patient with anti-FHs and a truncating CFHR1 mutation has been described in a published study (21). However, we found 5 patients with anti-FHs carrying at least one normal copy of CFHR1, indicating that autoantibodies against FH can also be formed in the presence of FHR1.
In agreement with previous reports (24–27, 58), anti-FHs from 92% of our patients predominantly targeted the C terminus of FH, which is a mutational hot spot in aHUS and a crucial domain involved in the regulation of the complement AP on cell surface (59). Our results, showing that serum from two patients with anti-FHs and no LPVs in complement genes induced higher than normal C5b-9 deposits on cultured endothelial cells, support a pathogenetic role of anti-FHs in causing complement dysregulation on endothelium (58, 60). This finding is in line with previous studies showing that aHUS-associated anti-FH autoantibodies impaired FH capability to protect rabbit or sheep erythrocytes from complement-mediated lysis (25, 61).
The mechanisms that link the lack of FHR1 to the development of anti-FHs have not been clarified yet. Evidence that the FH C-terminal domain is homologous to FHR1 C-terminus (62) and that most anti-FHs cross-react with SCRs 4–5 of FHR1 (22, 26) suggested that the absence of FHR1 plays a role in the loss of tolerance to FH (28). In the CFH promoter region we identified two sequences predicted as possible consensus site for the binding of AIRE, a transcription factor that is crucial for the expression of tissue-specific antigens in the thymus and the maintenance of central tolerance (63–65). However, the failure to find any variant in the CFH promoter in our patients with anti-FHs aHUS does not support the hypothesis that there is a defect of central tolerance related to AIRE in anti-FHs development.
Of relevance to understand the immunologic basis of anti-FHs, are data that the large majority of patients with anti-FHs of our cohort had prodromal infectious illnesses, most commonly upper respiratory tract and gastrointestinal infections, and developed the disease between 4 and 12 years of age, which corresponds to the peak of incidence of common infections. These findings, together with published reports showing a high prevalence of respiratory tract infections (19, 66), and gastrointestinal pathogens in aHUS patients with anti-FHs (67), would support a “two-hit” model according to which the autoimmunity toward FH could develop as a result of an infection in subjects with genetic predisposing background (28). Bhattacharjee et al. (26) identified an anti-FH autoantibody epitope cluster within SCR20, which includes amino acids used by microbes to bind FH and evade the immune system (68–70). The authors proposed that binding of this domain with certain microbes induces a conformation change in FH SCR20 generating a neoepitope similar to FHR1, which might predispose to the development of the autoantibodies against FH in subjects with the FHR1 deficiency.
Anti-FHs aHUS is an ultra-rare disease with a prevalence of about 1/1,000,000 people in Europe and the USA (1, 28) which does not fit with the ~5% prevalence of FHR1 deficiency in European Caucasian populations (29), and with bacterial infections that commonly occur during school-age (67). From these observations we infer that multiple genetic and/or environmental risk factors are required for the disease to manifest, as reported for other autoimmune disorders (71).
Recently, as a new strategy to identify genetic variants associated with complex traits, patients were compared with centenarians, as supercontrols, who have evaded all the common diseases associated with aging and are expected to be depleted in disease predisposing genetic variants or enriched in protective variants (72, 73). Inspired by these reports, to identify other anti-FHs aHUS genetic determinants, here we adopted a similar approach using as “supercontrols” adults subjects with the homozygous CFHR1 deletion who did not get anti-FHs aHUS despite carrying the known genetic risk factor for the disease (i.e., FHR1 deficiency) and likely having been exposed to common infections during childhood. We hypothesized that the comparison of our patients with anti-FHs aHUS and FHR1 deficiency (cases) with “supercontrols” could allow us detecting genetic variants with clinical value at relatively small sample size. With this approach we demonstrate that cases are significantly enriched in CFH, THBD and C3 LPVs compared to “supercontrols.” Other authors have screened patients with anti-FHs aHUS for complement gene defects, but there was no comparison with a control group and the results were conflicting (18, 19, 21–23, 32). Our data support the hypothesis that rare complement gene variants are involved in predisposing to anti-FHs aHUS. CFH LPVs are the most abundant in our cases, two of which are not reported in public control databases and are predicted to be strongly damaging in silico. The third CFH LPV, the p.Gln950His, was also found in 4 patients without anti-FHs of our aHUS cohort and in other published patients (22, 44–46). The functional relevance of this variant was documented by Mohlin and colleagues (44) who showed that the 950His variant is less effective in inhibiting complement-mediated sheep erythrocyte lysis than the wild type Gln950. As for the THBD variants, both have been found to be less effective in promoting C3b inactivation to iC3b on the cell surface (12). Finally, the C3 p.Gly637Arg has not been described in patients with aHUS, it is predicted to be strongly damaging by in silico analysis but functional data are not available. We would speculate that complement LPVs contribute to aHUS in subjects with FHR1 deficiency by synergizing with anti-FHs in inducing AP activation on cell surfaces. Since complement activation products can enhance the adaptive immune response (74), the possibility that AP dysregulation associated with LPVs per se could favor the proliferation of the self-reactive T cell clones and the formation of anti-FHs is worth investigating. However, further studies are required to clarify the mechanisms through which LPVs predispose to anti-FHs aHUS.
We found only one LPV in the “supercontrol” group, namely the p.Arg317Trp in FI. This variant was previously reported to have a lower cofactor activity compared to wild type FI (50). However, later on Nilsson and colleagues found that the activity of the 317Trp variant was not impaired in any functional assay, rather this variant was more efficient than wild type FI in C3b cleavage on the surface of endothelial cells (51).
At variance with LPVs, common genetic variants in complement genes predisposing to aHUS or to other immune-mediated diseases (as C3 glomerulopathy, age-related macular degeneration, and IgA nephropathy) (33–38) were not more abundant in our cases compared to “supercontrols.” Specifically, the CFH H3 haplotype, which has been strongly associated with an increased risk of aHUS (15), was rare both in cases and in “supercontrols,” although it was slightly more prevalent in cases. As expected, this haplotype was enriched in our patients without anti-FHs. These results do not surprise, because most cases and “supercontrols” share the CFH H4 haplotypes that are in LD with the CFHR3-CFHR1 deletion. As for the frequency of the CD46 c.*783C allele (rs7144) variant that tags the CD46GGAAC haplotype, described in strong association with aHUS (52), we did not observe any difference between cases and “supercontrols,” whereas the frequency of this allele was significantly enriched in our aHUS patients without anti-FHs. Altogether our data demonstrate that the CFH H3 and the CD46GGAAC haplotypes are risk factors for aHUS without anti-FHs but do not contribute to development of anti-FHs aHUS.
The marked difference we observed in the age of onset between patients with anti-FHs and those negative for anti-FHs further supports the hypothesis that diverse predisposing factors underlying the two forms of the disease are involved.
Importantly, within the entire aHUS cohort reported here we have shown that a disease onset between ages 4 and 12, identifies the presence of anti-FHs with a specificity of 90%. Patients with the above age of onset should be screened carefully for anti-FHs. Prompt identification of anti-FHs is of great clinical relevance since anti-FHs can be removed by successfully plasma exchange, which also supplements free FH and provides FHR1, which might act as a decoy for the antibodies (75). Patients should be followed up carefully long-term to monitor the re-emergence of anti-FHs, since this form of aHUS is associated with a high prevalence of relapses, as documented by published (28) and present data. Relapses can be prevented by maintenance therapy with immunosuppressive agents that inhibit the further production of anti-FH antibodies (28), but these drugs may have serious side effects particularly in children.
In conclusion, we have confirmed in our cohort of patients, the strong association between FHR1 deficiency and aHUS with anti-FH autoantibodies. Through an innovative approach based on the comparison with “supercontrols” carrying the homozygous CFHR1 deletion, identified by screening a large number of healthy adult subjects, we have documented that patients with anti-FHs aHUS are enriched in complement gene LPVs. This observation indicates that the pathogenesis of anti-FHs aHUS is complex and multiple “hits” are required for its clinical manifestation. We also document that the CFH H3 and the CD46GGAAC haplotypes are not associated with anti-FHs aHUS, whereas these haplotypes are enriched in aHUS patients without anti-FHs, which highlights the differences in the genetic basis of the two forms of the disease. Finally, we confirm the role of common infections as environmental factors that contribute to the development of anti-FHs aHUS in genetically predisposed individuals. The latter finding fits with the sharp peak of disease onset during scholar-age. Further studies are needed to fully elucidate the genetic and environmental factors underlying anti-FHs aHUS and to establish whether the combination of anti-FHs with LPVs or other risk factors influences the course of the disease and the response to therapies.
All subjects provided informed written consent in accordance with the Declaration of Helsinki. The study was approved by the Ethics Committee of Azienda Sanitaria Locale, Bergamo, Italy.
EV, MN, and GR designed research, interpreted data, and wrote the paper. EV, MA, PI, RP, CM, MB, AC, RD, and SA performed the research and analyzed the data. EB provided detailed clinical information of patients. AB and AA analyzed the data and critically revised the manuscript.
MN has received honoraria from Alexion Pharmaceuticals for giving lectures, and for participating in advisory boards and research grants from Omeros, Alnylam, and Chemocentryx. GR has consultancy agreements with AbbVie*, Alexion Pharmaceuticals*, Bayer Healthcare*, Reata Pharmaceuticals*, Novartis Pharma*, AstraZeneca*, Otsuka Pharmaceutical Europe*, Concert Pharmaceuticals*.
*No personal remuneration is accepted, compensation is paid to his institution for research and educational activities. None of these activities have had any influence on the results or interpretations in this article.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Drs. Paola Cuccarolo and Flavio Gaspari for biochemical evaluation of FH, C3, and C4 in plasma/serum samples, Sara Gastoldi for the evaluation of serum-induced C5b-9 deposits on HMEC-1, Kerstin Mierke for editing the manuscript and Manuela Passera for secretarial assistance.
This work was partially supported by the FP7 EURenOmics grant. EV and MB are the recipients of a fellowship from Fondazione Aiuti per la Ricerca sulle Malattie Rare ARMR ONLUS (Bergamo, Italy). RP is the recipient of a research contract from Progetto DDD Onlus—Associazione per la lotta alla DDD.
The funding sources had no role in study design, or in the collection, analysis, and interpretation of data, nor in the writing of the report or in the decision to submit the paper for publication.
1. Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 361:1676–87. doi: 10.1056/NEJMra0902814
2. Kavanagh D, Goodship TH. Atypical hemolytic uremic syndrome. Curr Opin Hematol. (2010) 17:432–8. doi: 10.1097/MOH.0b013e32833cae86
3. Noris M, Caprioli J, Bresin E, Mossali C, Pianetti G, Gamba S, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. (2010) 5:1844–59. doi: 10.2215/CJN.02210310
4. Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. (2003) 100:12966–71. doi: 10.1073/pnas.2135497100
5. Noris M, Brioschi S, Caprioli J, Todeschini M, Bresin E, Porrati F, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. (2003) 362:1542–7. doi: 10.1016/S0140-6736(03)14742-3
6. Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, Vigneau C, Kuypers D, Boudailliez B, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. (2004) 41:e84. doi: 10.1136/jmg.2004.019083
7. Rodriguez E, Rallapalli PM, Osborne AJ, Perkins SJ. New functional and structural insights from updated mutational databases for complement factor H, Factor I, membrane cofactor protein and C3. Biosci Rep. (2014) 34:e00146. doi: 10.1042/BSR20140117
8. Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J, Brown AL, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. (2008) 112:4948–52. doi: 10.1182/blood-2008-01-133702
9. Kavanagh D, Richards A, Fremeaux-Bacchi V, Noris M, Goodship T, Remuzzi G, et al. Screening for complement system abnormalities in patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. (2007) 2:591–6. doi: 10.2215/CJN.03270906
10. Sartz L, Olin AI, Kristoffersson AC, Stahl AL, Johansson ME, Westman K, et al. A novel C3 mutation causing increased formation of the C3 convertase in familial atypical hemolytic uremic syndrome. J Immunol. (2012) 188:2030–7. doi: 10.4049/jimmunol.1100319
11. Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, Carreras L, Arranz EA, Garrido CA, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. (2007) 104:240–5. doi: 10.1073/pnas.0603420103
12. Delvaeye M, Noris M, De Vriese A, Esmon CT, Esmon NL, Ferrell G, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. (2009) 361:345–57. doi: 10.1056/NEJMoa0810739
13. Bresin E, Rurali E, Caprioli J, Sanchez-Corral P, Fremeaux-Bacchi V, Rodriguez de Cordoba S, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. (2013) 24:475–86. doi: 10.1681/ASN.2012090884
14. Caprioli J, Castelletti F, Bucchioni S, Bettinaglio P, Bresin E, Pianetti G, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. (2003) 12:3385–95. doi: 10.1093/hmg/ddg363
15. de Cordoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol. (2008) 151:1–13. doi: 10.1111/j.1365-2249.2007.03552.x
16. Bernabeu-Herrero ME, Jimenez-Alcazar M, Anter J, Pinto S, Sanchez Chinchilla D, Garrido S, et al. Complement factor H, FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. (2015) 67(2 Pt B):276–86. doi: 10.1016/j.molimm.2015.06.021
17. Dragon-Durey MA, Loirat C, Cloarec S, Macher MA, Blouin J, Nivet H, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. (2005) 16:555–63. doi: 10.1681/ASN.2004050380
18. Hofer J, Janecke AR, Zimmerhackl LB, Riedl M, Rosales A, Giner T, et al. Complement factor H-related protein 1 deficiency and factor H antibodies in pediatric patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. (2013) 8:407–15. doi: 10.2215/CJN.01260212
19. Dragon-Durey MA, Sethi SK, Bagga A, Blanc C, Blouin J, Ranchin B, et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. (2010) 21:2180–7. doi: 10.1681/ASN.2010030315
20. Jozsi M, Licht C, Strobel S, Zipfel SL, Richter H, Heinen S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. (2008) 111:1512–4. doi: 10.1182/blood-2007-09-109876
21. Abarrategui-Garrido C, Martinez-Barricarte R, Lopez-Trascasa M, de Cordoba SR, Sanchez-Corral P. Characterization of complement factor H-related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. (2009) 114:4261–71. doi: 10.1182/blood-2009-05-223834
22. Moore I, Strain L, Pappworth I, Kavanagh D, Barlow PN, Herbert AP, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. (2010) 115:379–87. doi: 10.1182/blood-2009-05-221549
23. Brocklebank V, Johnson S, Sheerin TP, Marks SD, Gilbert RD, Tyerman K, et al. Factor H autoantibody is associated with atypical hemolytic uremic syndrome in children in the United Kingdom and Ireland. Kidney Int. (2017) 92:1261–71. doi: 10.1016/j.kint.2017.04.028
24. Jozsi M, Strobel S, Dahse HM, Liu WS, Hoyer PF, Oppermann M, et al. Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood. (2007) 110:1516–8. doi: 10.1182/blood-2007-02-071472
25. Blanc C, Roumenina LT, Ashraf Y, Hyvarinen S, Sethi SK, Ranchin B, et al. Overall neutralization of complement factor H by autoantibodies in the acute phase of the autoimmune form of atypical hemolytic uremic syndrome. J Immunol. (2012) 189:3528–37. doi: 10.4049/jimmunol.1200679
26. Bhattacharjee A, Reuter S, Trojnar E, Kolodziejczyk R, Seeberger H, Hyvarinen S, et al. The major autoantibody epitope on factor H in atypical hemolytic uremic syndrome is structurally different from its homologous site in factor H-related protein 1, supporting a novel model for induction of autoimmunity in this disease. J Biol Chem. (2015) 290:9500–10. doi: 10.1074/jbc.M114.630871
27. Nozal P, Bernabeu-Herrero ME, Uzonyi B, Szilagyi A, Hyvarinen S, Prohaszka Z, et al. Heterogeneity but individual constancy of epitopes, isotypes and avidity of factor H autoantibodies in atypical hemolytic uremic syndrome. Mol Immunol. (2016) 70:47–55. doi: 10.1016/j.molimm.2015.12.005
28. Durey MA, Sinha A, Togarsimalemath SK, Bagga A. Anti-complement-factor H-associated glomerulopathies. Nat Rev Nephrol. (2016) 12:563–78. doi: 10.1038/nrneph.2016.99
29. Holmes LV, Strain L, Staniforth SJ, Moore I, Marchbank K, Kavanagh D, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS ONE. (2013) 8:e60352. doi: 10.1371/journal.pone.0060352
30. Rosenblum MD, Remedios KA, Abbas AK. Mechanisms of human autoimmunity. J Clin Invest. (2015) 125:2228–33. doi: 10.1172/JCI78088
31. Zenewicz LA, Abraham C, Flavell RA, Cho JH. Unraveling the genetics of autoimmunity. Cell. (2010) 140:791–7. doi: 10.1016/j.cell.2010.03.003
32. Geerdink LM, Westra D, van Wijk JA, Dorresteijn EM, Lilien MR, Davin JC, et al. Atypical hemolytic uremic syndrome in children: complement mutations and clinical characteristics. Pediatr Nephrol. (2012) 27:1283–91. doi: 10.1007/s00467-012-2131-y
33. Finn JE, Mathieson PW. Molecular analysis of C3 allotypes in patients with nephritic factor. Clin Exp Immunol. (1993) 91:410–4.
34. Iatropoulos P, Noris M, Mele C, Piras R, Valoti E, Bresin E, et al. Complement gene variants determine the risk of immunoglobulin-associated MPGN and C3 glomerulopathy and predict long-term renal outcome. Mol Immunol. (2016) 71:131–42. doi: 10.1016/j.molimm.2016.01.010
35. Rambausek M, van den Wall Bake AW, Schumacher-Ach R, Spitzenberg R, Rother U, van Es LA, et al. Genetic polymorphism of C3 and Bf in IgA nephropathy. Nephrol Dial Transplant. (1987) 2:208–11.
36. Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. (2007) 357:553–61. doi: 10.1056/NEJMoa072618
37. Spencer KL, Olson LM, Anderson BM, Schnetz-Boutaud N, Scott WK, Gallins P, et al. C3 R102G polymorphism increases risk of age-related macular degeneration. Hum Mol Genet. (2008) 17:1821–4. doi: 10.1093/hmg/ddn075
38. Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. (2006) 38:458–62. doi: 10.1038/ng1750
39. Watson R, Lindner S, Bordereau P, Hunze EM, Tak F, Ngo S, et al. Standardisation of the factor H autoantibody assay. Immunobiology. (2014) 219:9–16. doi: 10.1016/j.imbio.2013.06.004
40. Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol. (1995) 155:5663–70.
41. Noris M, Galbusera M, Gastoldi S, Macor P, Banterla F, Bresin E, et al. Dynamics of complement activation in aHUS and how to monitor eculizumab therapy. Blood. (2014) 124:1715–26. doi: 10.1182/blood-2014-02-558296
42. Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol. (2013) 56:170–80. doi: 10.1016/j.molimm.2013.06.001
43. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–94. doi: 10.1093/nar/gky1016
44. Mohlin FC, Nilsson SC, Levart TK, Golubovic E, Rusai K, Muller-Sacherer T, et al. Functional characterization of two novel non-synonymous alterations in CD46 and a Q950H change in factor H found in atypical hemolytic uremic syndrome patients. Mol Immunol. (2015) 65:367–76. doi: 10.1016/j.molimm.2015.02.013
45. Neumann HP, Salzmann M, Bohnert-Iwan B, Mannuelian T, Skerka C, Lenk D, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet. (2003) 40:676–81. doi: 10.1136/jmg.40.9.676
46. Maga TK, Nishimura CJ, Weaver AE, Frees KL, Smith RJ. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. (2010) 31:E1445–60. doi: 10.1002/humu.21256
47. Le Quintrec M, Lionet A, Kamar N, Karras A, Barbier S, Buchler M, et al. Complement mutation-associated de novo thrombotic microangiopathy following kidney transplantation. Am J Transplant. (2008) 8:1694–701. doi: 10.1111/j.1600-6143.2008.02297.x
48. Barlow PN, Steinkasserer A, Norman DG, Kieffer B, Wiles AP, Sim RB, et al. Solution structure of a pair of complement modules by nuclear magnetic resonance. J Mol Biol. (1993) 232:268–84. doi: 10.1006/jmbi.1993.1381
49. Matsumoto T, Fan X, Ishikawa E, Ito M, Amano K, Toyoda H, et al. Analysis of patients with atypical hemolytic uremic syndrome treated at the Mie University Hospital: concentration of C3 p.I1157T mutation. Int J Hematol. (2014) 100:437–42. doi: 10.1007/s12185-014-1655-2
50. Kavanagh D, Richards A, Noris M, Hauhart R, Liszewski MK, Karpman D, et al. Characterization of mutations in complement factor I (CFI) associated with hemolytic uremic syndrome. Mol Immunol. (2008) 45:95–105. doi: 10.1016/j.molimm.2007.05.004
51. Nilsson SC, Kalchishkova N, Trouw LA, Fremeaux-Bacchi V, Villoutreix BO, Blom AM. Mutations in complement factor I as found in atypical hemolytic uremic syndrome lead to either altered secretion or altered function of factor I. Eur J Immunol. (2010) 40:172–85. doi: 10.1002/eji.200939280
52. Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, Carreras Berges L, Lopez-Trascasa M, Sanchez-Corral P, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. (2005) 14:703–12. doi: 10.1093/hmg/ddi066
53. Heurich M, Martinez-Barricarte R, Francis NJ, Roberts DL, Rodriguez de Cordoba S, Morgan BP, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. (2011) 108:8761–6. doi: 10.1073/pnas.1019338108
54. Harris CL, Heurich M, Rodriguez de Cordoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. (2012) 33:513–21. doi: 10.1016/j.it.2012.06.001
55. Ansari M, McKeigue PM, Skerka C, Hayward C, Rudan I, Vitart V, et al. Genetic influences on plasma CFH and CFHR1 concentrations and their role in susceptibility to age-related macular degeneration. Hum Mol Genet. (2013) 22:4857–69. doi: 10.1093/hmg/ddt336
56. Nozal P, Garrido S, Alba-Dominguez M, Espinosa L, Pena A, Cordoba SR, et al. An ELISA assay with two monoclonal antibodies allows the estimation of free factor H and identifies patients with acquired deficiency of this complement regulator. Mol Immunol. (2014) 58:194–200. doi: 10.1016/j.molimm.2013.11.021
57. Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. (2014) 85:1151–60. doi: 10.1038/ki.2013.373
58. Guo WY, Song D, Liu XR, Chen Z, Xiao HJ, Ding J, et al. Immunological features and functional analysis of anti-CFH autoantibodies in patients with atypical hemolytic uremic syndrome. Pediatr Nephrol. (2019) 34:269–81. doi: 10.1007/s00467-018-4074-4
59. Manuelian T, Hellwage J, Meri S, Caprioli J, Noris M, Heinen S, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest. (2003) 111:1181–90. doi: 10.1172/JCI16651
60. Strobel S, Hoyer PF, Mache CJ, Sulyok E, Liu WS, Richter H, et al. Functional analyses indicate a pathogenic role of factor H autoantibodies in atypical haemolytic uraemic syndrome. Nephrol Dial Transplant. (2010) 25:136–44. doi: 10.1093/ndt/gfp388
61. Blanc C, Togarsimalemath SK, Chauvet S, Le Quintrec M, Moulin B, Buchler M, et al. Anti-factor H autoantibodies in C3 glomerulopathies and in atypical hemolytic uremic syndrome: one target, two diseases. J Immunol. (2015) 194:5129–38. doi: 10.4049/jimmunol.1402770
62. Medjeral-Thomas N, Pickering MC. The complement factor H-related proteins. Immunol Rev. (2016) 274:191–201. doi: 10.1111/imr.12477
63. Abramson J, Goldfarb Y. AIRE: From promiscuous molecular partnerships to promiscuous gene expression. Eur J Immunol. (2016) 46:22–33. doi: 10.1002/eji.201545792
64. Passos GA, Speck-Hernandez CA, Assis AF, Mendes-da-Cruz DA. Update on Aire and thymic negative selection. Immunology. (2018) 153:10–20. doi: 10.1111/imm.12831
65. Kumar PG, Laloraya M, Wang CY, Ruan QG, Davoodi-Semiromi A, Kao KJ, et al. The autoimmune regulator (AIRE) is a DNA-binding protein. J Biol Chem. (2001) 276:41357–64. doi: 10.1074/jbc.M104898200
66. Kim JJ, McCulloch M, Marks SD, Waters A, Noone D. The clinical spectrum of hemolytic uremic syndrome secondary to complement factor H autoantibodies. Clin Nephrol. (2015) 83:49–56. doi: 10.5414/CN107777
67. Togarsimalemath SK, Si-Mohammed A, Puraswani M, Gupta A, Vabret A, Liguori S, et al. Gastrointestinal pathogens in anti-FH antibody positive and negative Hemolytic Uremic Syndrome. Pediatr Res. (2018) 84:118–24. doi: 10.1038/s41390-018-0009-9
68. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. (2008) 6:132–42. doi: 10.1038/nrmicro1824
69. Bhattacharjee A, Oeemig JS, Kolodziejczyk R, Meri T, Kajander T, Lehtinen MJ, et al. Structural basis for complement evasion by Lyme disease pathogen Borrelia burgdorferi. J Biol Chem. (2013) 288:18685–95. doi: 10.1074/jbc.M113.459040
70. Meri T, Amdahl H, Lehtinen MJ, Hyvarinen S, McDowell JV, Bhattacharjee A, et al. Microbes bind complement inhibitor factor H via a common site. PLoS Pathog. (2013) 9:e1003308. doi: 10.1371/journal.ppat.1003308
71. Abbas AK, Lichtman AH, Pillai S editors. Cellular and Molecular Immunology. 8th ed. Philadelphia: Elsevier Saunders (2015).
72. Garagnani P, Giuliani C, Pirazzini C, Olivieri F, Bacalini MG, Ostan R, et al. Centenarians as super-controls to assess the biological relevance of genetic risk factors for common age-related diseases: a proof of principle on type 2 diabetes. Aging (Albany NY). (2013) 5:373–85. doi: 10.18632/aging.100562
74. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
Keywords: autoantibodies, atypical hemolytic uremic syndrome, factor H, factor H related 1, complement, genetic variants, supercontrols
Citation: Valoti E, Alberti M, Iatropoulos P, Piras R, Mele C, Breno M, Cremaschi A, Bresin E, Donadelli R, Alizzi S, Amoroso A, Benigni A, Remuzzi G and Noris M (2019) Rare Functional Variants in Complement Genes and Anti-FH Autoantibodies-Associated aHUS. Front. Immunol. 10:853. doi: 10.3389/fimmu.2019.00853
Received: 10 December 2018; Accepted: 02 April 2019;
Published: 01 May 2019.
Edited by:
Kevin James Marchbank, Newcastle University, United KingdomReviewed by:
Mihály Józsi, Eötvös Loránd University, HungaryCopyright © 2019 Valoti, Alberti, Iatropoulos, Piras, Mele, Breno, Cremaschi, Bresin, Donadelli, Alizzi, Amoroso, Benigni, Remuzzi and Noris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina Noris, bWFyaW5hLm5vcmlzQG1hcmlvbmVncmkuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.