 Erika Méndez-Enríquez
Erika Méndez-Enríquez Jenny Hallgren
Jenny Hallgren- Department of Medical Biochemistry and Microbiology, Uppsala University, Uppsala, Sweden
Mast cells and their mediators have been implicated in the pathogenesis of asthma and allergy for decades. Allergic asthma is a complex chronic lung disease in which several different immune cells, genetic factors and environmental exposures influence the pathology. Mast cells are key players in the asthmatic response through secretion of a multitude of mediators with pro-inflammatory and airway-constrictive effects. Well-known mast cell mediators, such as histamine and bioactive lipids are responsible for many of the physiological effects observed in the acute phase of allergic reactions. The accumulation of mast cells at particular sites of the allergic lung is likely relevant to the asthma phenotype, severity and progression. Mast cells located in different compartments in the lung and airways have different characteristics and express different mediators. According to in vivo experiments in mice, lung mast cells develop from mast cell progenitors induced by inflammatory stimuli to migrate to the airways. Human mast cell progenitors have been identified in the blood circulation. A high frequency of circulating human mast cell progenitors may reflect ongoing pathological changes in the allergic lung. In allergic asthma, mast cells become activated mainly via IgE-mediated crosslinking of the high affinity receptor for IgE (FcεRI) with allergens. However, mast cells can also be activated by numerous other stimuli e.g. toll-like receptors and MAS-related G protein-coupled receptor X2. In this review, we summarize research with implications on the role and development of mast cells and their progenitors in allergic asthma and cover selected activation pathways and mast cell mediators that have been implicated in the pathogenesis. The review places an emphasis on describing mechanisms identified using in vivo mouse models and data obtained by analysis of clinical samples.
The Origin of Mast Cells in Mouse and Man
The development of mast cells has predominantly been studied in mice. The first phenotypic identification of mast cell progenitors (MCps) was made in fetal mouse blood, where isolated Thy-1lo c-kithi cells generated mast cells in vitro and could reconstitute mast cell deficient mice in vivo (1). In utero, mast cells originate from yolk sac-derived progenitors (2–4). Due to their slow turnover, yolk sac-derived mast cells to some extent remain while slowly being replaced by maturing bone-marrow derived mast cells in the adult (3, 4). In young naïve mice, mast cells in connective tissues such as in the skin and peritoneum are derived from fetal yolk sac/liver MCps (4). In other adult tissues, mast cells arise from committed MCps that under the influence of growth factors and transcriptional control differentiate into mast cells. The first identification of a mast cell precursor in adult mice was made by three independent labs (5–7). Chen et al. described a committed MCp population in the bone marrow by isolation of lineage (Lin)− c-kit+ Sca-1− Ly6c− FcεRI− CD27− integrin β7+ ST2+ cells using fluorescence-activated cell sorting (FACS) (6). Jamur et al. used immunomagnetic isolation by two monoclonal antibodies recognizing specific surface sites on rodent mast cells to isolate CD34+ CD13+ c-kit+ FcεRI− bone marrow cells, which developed to mast cells in vitro and in vivo (5). Meanwhile, Arinobu and colleagues demonstrated a committed MCp population in the intestine and a bipotent basophil–mast cell progenitor (BMCp) in the spleen (7). The close relationship between mast cells and basophils was supported by a study showing that isolated single granulocyte-monocyte progenitors (GMp) were capable of differentiating into both mast cells and basophils (8), which was recently confirmed by the demonstration of a BMCp population distinguished as Lin− Sca-1 − c-kit+ integrin β7hi CD16/32hi cells in mouse bone marrow using single cell RNA-sequencing (9). By taking advantage of the expression of GATA-1 in eosinophils, basophils and mast cells, Drissen et al. used Gata-1-EGFP mice to fractionate and to identify distinct myeloid progenitors by single cell sequencing (10). This study suggested that Gata-1+ progenitors, defined as Lin− c-kithi CD41− cells with variable expression of CD16/32, have the capacity to differentiate into eosinophils, mast cells or basophils.
While the main point of hematopoiesis in adults likely occurs in the bone marrow niche, white adipose tissue (WAT) has been demonstrated to contain not only adipocytes but also a quite large fraction of immature immune/hematopoietic cells, called the stroma-vascular fraction (SVF) (11). In agreement with this, the SVF also contains MCps and mast cells (12). In a model of acute myocardial infarction, WAT-derived MCps infiltrated the heart and gave rise to an increased mast cell population at this location (13). However, the differential contribution of bone marrow-derived vs. WAT-derived MCps creating increased pools of mast cells at different sites of the body during inflammatory conditions is currently unclear.
In homeostatic conditions, committed MCps can be detected by flow cytometry in the blood (14) and peripheral tissues of naïve laboratory mice (15). The MCp population in the blood is distinguished as Lin− c-kithi ST2+ integrin β7hi CD16/32hi cells (14). The MCps from the BALB/c and C57BL/6 strains differ in maturity (14). BALB/c mice have a higher proportion of FcεRI+ MCps in the circulation, but even the FcεRI− MCp differentiated into double positive c-kit+ FcεRI+ cells in vitro, whereas the blood MCps in C57BL/6 mice were largely FcεRI− and retained some basophil differentiation potency (14). The MCps are of lymphocyte size, contain none or a few granules and have a typical progenitor morphology. They do not stain, or stain weakly with basic dyes that typically stain mast cells metachromatically. The MCps are extremely rare, constituting only around 50 cells per 106 enriched mononuclear cells in the lung and peripheral blood. However, their existence in the periphery of naïve mice was predicted years ago by limiting dilution and clonal expansion assays (16, 17).
The development of MCps into mast cells in vivo is largely dependent on stem cell factor (SCF), which has effects on homing, proliferation, survival and function of mast cells and their progenitors. Interestingly, local administration of SCF promotes the expansion of mast cells in vivo (18). The importance of SCF in mast cells is underscored by the lack of mast cells in mice lacking the expression of a functional c-kit receptor, as in KitW/Wv (19) or KitW−sh/W−sh mice (20). Nevertheless, mouse mast cells can be derived in vitro by culture of hematopoietic cells with IL-3 alone (21, 22).
In 2016, we identified a human MCp population defined as Lin− CD34hi CD117int/hi (c-kit) FcεRI+ cells in the blood circulation (23). As with their mouse counterparts, the human MCps have an immature appearance, express mast cell specific genes and develop into mast cells in vitro. Interestingly, the frequency of circulating blood MCps was higher in individuals with a reduced lung function (23). For a comparison, see Table 1. When the human MCp population was investigated in patients receiving treatment inhibiting signaling through CD117 (imatinib) and depleting mast cells in vivo, the MCp population was intact (24). These results suggest that signaling through CD117 is dispensable for human MCps to develop and survive. However, for human MCps to develop into mast cells, SCF is required, as summarized in (25).
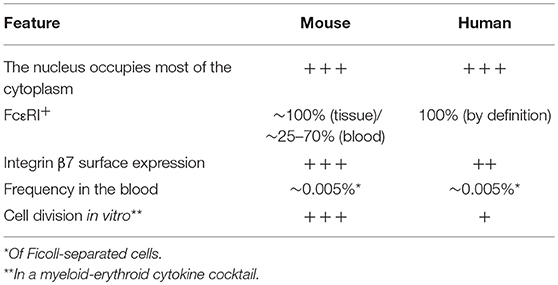
Table 1. Comparison of human and mouse mast cell progenitors.
Mast Cell Subtypes
Rodent mast cells are classically distinguished into two different phenotypes, connective tissue-type mast cells (CTMCs) and mucosal mast cells (MMCs). The division was originally based on their respective biochemical properties, which led to different staining patterns in response to histochemical dyes and corresponded to their location in the gut (26). CTMCs have heparin glycosaminoglycan-chains attached to the serglycin proteoglycan core protein, whereas in MMCs serglycin carries chondroitin sulfate-chains (27). Apart from the location in the submucosa of the gut, CTMCs are also found in the peritoneum and skin, while MMCs predominate in the intestinal mucosa. CTMCs express relatively high levels of mouse mast cell protease (mMCP)-4,-5 (chymases) and -6,-7 (tryptases), but not mMCP-1 and-2 (chymases), whereas MMCs express mMCP-1 and -2 and not mMCP-4,-5 and -6 (28, 29). Of note, the life span of CTMCs is extensively longer than that of MMCs (30), at least in the absence of ongoing inflammation. In accordance with this, CTMCs are constitutive and at least in the young mice they are mainly derived from fetal mast cells with self-generating capabilities (4). MMCs are induced and expand upon, e.g. inflammatory stimulus.
Similarly, human mast cells are divided into two subtypes. Since the mast cells in the human lung were shown to have both heparin and chondroitin sulfate proteoglycans (31), they were classified according to their protease content. Some human mast cell populations, e.g. skin mast cells, express both tryptase and chymase (MCTC) corresponding to the CTMCs in rodents, whereas other mast cell populations, e.g. in the bronchial/bronchiolar epithelium, predominately lack chymase expression (MCT), roughly corresponding to MMCs (32, 33). Nevertheless, the classification of mast cells into these two phenotypes is really an oversimplification for mast cells in the lung. In mice, both antibody-based methods (29) and microarray data from the ImmGen project (34) suggest that lung mast cells express a much wider range of proteases than previously thought. The constitutive mast cells in the trachea and lung of mice contained mast cells immunopositive for mMCP-1 and -2, mMCP-4,-5,-6 and -7 and carboxypeptidase A3 (CPA3), whereas the induced MMCs present in the epithelium of antigen-sensitized and challenged mice expressed mMCP-1,-2 and -6,-7 (29). In accordance with this study, constitutive tracheal mast cells express transcripts for mMCP-4,-5 and -6,-7 and CPA3 (34). In humans, studies of bronchial and transbronchial biopsies from non-smokers suggest a similar situation, where MCT and MCTC coexist in all compartments of the lung (35). Given that, MCT were more frequently found in the bronchi, bronchioles and alveolar parenchyma, whereas MCTC dominated in pulmonary vessels and pleura. The MCT and MCTC phenotypes could also be further divided into site-specific populations, which showed specific expression patterns of, for example, FcεRI, IL-9 receptor, histidine decarboxylase (HDC) and leukotriene C4 synthase (LTC4-S) (35). The most intriguing finding was the presence of alveolar mast cells that lack surface expression of FcεRI.
Mast Cell Accumulation in the Lung and the Mechanisms Behind
Mast Cell Accumulation in the Lung of Asthma Patients
Several studies suggest that the presence or accumulation of mast cells at certain compartments of the lung are pathological features of allergic asthma (Figure 1). An increased number of mast cells were found in the airway smooth muscle of asthma patients in comparison to controls or subjects with eosinophilic bronchitis (42). The number of mast cells was also higher in the smooth muscle of allergic asthmatics in relation to non-allergic asthmatics (43). In support of these studies, isolated human bronchi with the ability to contract in response to allergens had a higher number of smooth muscle-associated mast cells than unresponsive human bronchi isolates (46). Moreover, an increased number of mast cells was found in the distal airways of subjects with non-fatal and fatal asthma compared to non-asthmatic controls (44). In the distal airways, the greatest increase in mast cells was found within the smooth muscle and mucous glands. In another study, the same authors found an increased percentage of degranulated mast cells in the mucous glands from fatal asthma in comparison to non-fatal asthma and controls, suggesting that mast cells are highly activated in fatal asthma (47). In biopsies from patients with severe asthma, the number of MCTC and the MCTC/MCT ratio in the small airways were higher compared to normal subjects (48). However, a positive correlation between MCTC in the region of small airways/alveolar attachments and lung function was found (48), suggesting a protective role of this subtype of mast cells.
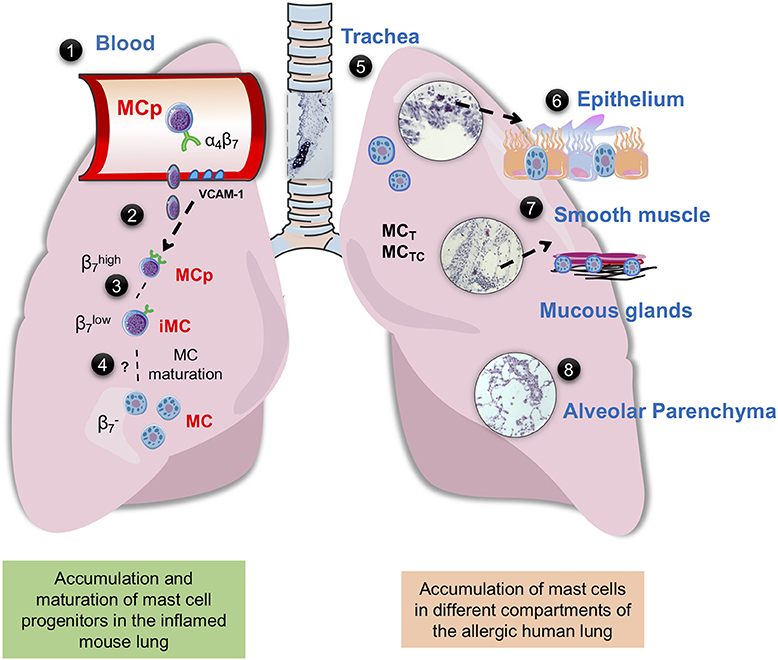
Figure 1. Mast cells in mouse and human lung. (1) Integrin-β7+ mast cell progenitors (MCps) are found in mouse and human peripheral blood (14, 23). (2) In mice with acute allergic airway inflammation, MCps are recruited to the lungs in a process dependent on α4β1 and α4β7 integrins on the MCp and on VCAM-1 expressed in the endothelium (36). (3) After the acute phase, three mast cell (MC) populations can be identified by flow cytometry, MCps expressing high levels of integrin β7, immature/induced MCs (iMCs) expressing intermediate levels of integrin β7, and mature MCs (37, 38). (4) The iMC gradually loses the expression of integrin β7 and mature, thereby expanding the resident lung MC population. (5) In the mouse trachea and in the proximal bronchi of unprovoked mouse airways, MCs express the MC proteases mMCP-1 and-2, mMCP-4,-5-6, and-7 and CPA3, while the MCs induced by allergic airway inflammation located in the bronchovascular bundles of the lung and the epithelial lining of the large bronchi express mMCP-1,-2 and -6,-7 (29). In the human lung MCT and MCTC coexist, MCT is more frequently found in the bronchi, bronchioles and alveolar parenchyma, whereas MCTC dominates in pulmonary vessels and pleura (35) (6) In the human bronchi, patients with “Th2-high” asthma have an increased number of intraepithelial MCs (39). Genetic analyses suggest that the MCs in this location mainly express tryptase and CPA3 (39–41). (7) The number of MCs are increased in the airway smooth muscle of asthma patients (42, 43). In diseased asthma patients, there are an increased number of mast cells in the distal airway, especially in the smooth muscle and mucous glands (44). (8) Uncontrolled atopic asthmatics have a high number of mast cells in the alveolar parenchyma (45). The histology pictures shown are from hematoxylin/eosin-stained lung sections of house dust mite-sensitized wild-type BALB/c mice obtained from our unpublished experiments.
Increased mast cell numbers are also found in other lung compartments of asthma patients. For example, sputum samples from asthmatics more often showed expression signatures of mast cell-specific protease genes (TPSAB1 and CPA3) and higher expression of these genes than samples from healthy subjects (49). In accordance with this, genetic analyses of samples from epithelial brushings have revealed increased gene expression of mast cell tryptase (TPSAB1, TPSB2) and CPA3 (but not CMA1, coding for chymase) in asthma patients (39–41). In the study by Singhania et al., a twofold increase in the number of intraepithelial mast cells was found in patients with “Th2-high” asthma compared to those characterized as “Th2-low” (39). Moreover, the presence of intraepithelial mast cells in the Th2-high asthma patients predicted responsiveness to inhaled corticosteroids. In a study of severe asthma, the proportion of MCTC among all mast cells was higher in patients with severe asthma compared to those with mild asthma (50). A higher proportion of MCTC was also found in the bronchi of uncontrolled atopic asthmatic subjects, and overall a higher number of mast cells was found in the alveolar parenchyma, paralleled by an increase in their expression of FcεRI (51). In a follow-up study, patients with mild atopic asthma were found to have an elevated number of alveolar mast cells with increased expression of FcεRI in comparison to healthy controls and non-asthmatic allergic rhinitis patients (45). Although the above-mentioned studies were performed on adults, another study demonstrated that submucosal mast cells are more frequent in bronchial biopsies from symptomatic children with severe asthma than in those with few symptoms (52). To summarize, mast cells accumulate in the smooth muscle, bronchial epithelium and alveolar parenchyma of patients with allergic and severe asthma, thereby presumably increasing the detrimental consequences of mast cell activation in the allergic lung. However, in some compartments of the lung, there might be an advantage to having chymase-expressing mast cells.
Identified Mechanisms and Factors Involved in Mast Cell Accumulation in the Lung
To address whether the increase in lung mast cells in patients with allergic asthma is due to the recruitment of MCps to the lung or a result of local proliferation of resident mast cells, mouse models have been used. In early studies, lung MCps were quantified by a limiting dilution and clonal expansion assay in an ovalbumin (OVA)-model of allergic airway inflammation (36). In OVA-sensitized and challenged mice, the number of lung MCps was around 30 times higher 1 day post-challenge than in control mice. The OVA-induced increase in lung MCps was absent in mice genetically deficient of endothelial VCAM-1 and in wild-type mice treated with blocking antibodies targeting VCAM-1, α4-, β1-, or β7-integrins during the challenge phase (36). This suggests that allergic airway inflammation induces the recruitment of MCps to the lung. After the acute phase, the accumulation of lung MCps gradually led to the appearance of toluidine blue+ mast cells in the epithelium of the trachea and the lung parenchyma (53, 54). Moreover, the OVA-induced recruitment of MCps to the lung was dependent on the presence of CD11c+ cells (54) and CD4+ cells (55), demonstrating that an adaptive immune response is required to stimulate this process.
In an effort to study a possible chemokine component in the antigen-induced recruitment of MCps to the lung, mice lacking CCR3 or CCR5 were used to test whether any of these chemokine receptors were involved in this process. However, CCR3−/− and CCR5−/− mice had an intact OVA-induced recruitment of MCps to the lung (53). Nevertheless, the OVA-induced recruitment of MCps to the lung was partly dependent on the presence of CXCR2 in stromal cells in the lung. Mice lacking CXCR2 had a reduced inflammation-induced upregulation of VCAM-1 on the lung endothelium, which could explain the reduced recruitment of MCp to the lung (53). CCR2-deficient mice also had a reduction in OVA-induced recruitment of MCps to the lung (56). Nonetheless, this was likely due to unidentified stromal defects and not to MCps lacking CCR2 per se (56). Therefore, any chemokine component required for the recruitment of MCps to the lung remains unknown.
The role of cytokines in OVA-induced recruitment of MCps to the lung has also been a matter of investigation. Interestingly, the OVA-induced recruitment of MCps to the lung occurs independently of genetic ablation of IL-4, IL-4Rα chain, STAT-6, IFN-γ, and IL-12 and antibody-mediated neutralization/blocking of IFN-γ, IL-3, IL-4, IL-5, IL-6, IL-13, IL-17A, IL-12p40, or IL-12p40Rβ1 during the challenge phase (55). However, IL-9 deficiency or IL-9 antibody neutralization efficiently prevented the OVA-induced recruitment of MCps to the lung. In an effort to identify the source of IL-9, we also found that genetic ablation of CD1d or blocking CD1d during the challenge phase inhibited the OVA-induced recruitment of MCps to the lung, but genetic ablation of invariant NKT cells (Jα18 deficient mice) had an intact infiltration of MCps to the lung (55). As blocking CD1d in IL-9-deficient mice or neutralizing CD1d in IL-9-deficient mice did not further inhibit the OVA-induced recruitment of MCp to the lung, type 2 NKT cells may provide or elicit IL-9 production (55). The importance of IL-9 in the accumulation of lung mast cells during allergic airway inflammation was also highlighted in a study where adoptive transfer of Th9 cells followed by challenge with OVA and TSLP increased the mast cell numbers estimated by histological analyses (57). Treatment with an anti-IL-9 antibody blocked the mast cell accumulation in both the adoptive transfer model and in an OVA sensitization and challenge model (57). In the same paper, decreased mast cell numbers were found in mice with PU.1-deficient T cells, which have reduced IL-9 levels in house dust mite (HDM)-induced allergic airway inflammation.
The importance of IgE for the survival of lung mast cells was demonstrated in a model of Aspergillus Fumigatus-induced allergic airway inflammation (58). However, no defect in the Aspergillus-induced recruitment of MCps to the lung could be detected in mice lacking IgE. Nevertheless, we found that the number of lung MCps increased significantly when sensitized wild-type mice were challenged with IgE-antigen immune complexes compared to control mice given the same dose antigen alone (59). The stimulating effect of IgE-immune complexes on the recruitment of MCps to the lung was lost in FcRγ-deficient mice, but not in CD23-deficient mice, indicating that MCp recruitment can be potentiated by Fc receptor-mediated activation (59). Thus, IgE-immune complex formation and IgE alone impacts the recruitment of MCps to the lung and the survival of lung mast cells, respectively.
The technical advances in multi-color flow cytometry have made it possible to distinguish different mast cell populations simultaneously. Using a prolonged protocol of OVA-induced allergic airway inflammation, three lung mast cell populations could be identified, MCps expressing high levels of integrin β7, resident mature mast cells and emerging/induced mast cells (37). In parallel, we demonstrated that influenza infection in mice induced the recruitment of MCps to the lung, which later gave rise to mast cells with intermediate expression of integrin β7 (immature/induced) and even later increased the number of mature mast cells with low expression of integrin β7 (38). While the recruitment of MCps induced by allergic lung inflammation was dependent on an adaptive immune response (54, 55), the influenza-induced MCp recruitment to the lung was due to the induction of innate immune responses (60). As respiratory infections commonly cause exacerbations of asthma symptoms, we think it is intriguing that influenza infections induces mast cell accumulation in the mouse lung.
Lung Physiology in Mouse and Humans
Mice have frequently been used to model allergic asthma with the aim of investigating the mechanism behind the disease. When interpreting the results from mouse models of allergic airway inflammation it is necessary to understand that the lungs of mice and humans have anatomical and physiological similarities and differences. The right lung of humans and mice consists of five lobes. The left lung in humans consists of two lobes, whereas mice only have a single lobe on the left side (61). Another difference is that mice have monopodial branching whereas humans have dichotomous branching of the airways. Further, the intrapulmonary bronchi in mice lack cartilage, which suggests that the difference between bronchi and bronchioles is less obvious in mice than in humans. The number of goblet cells and submucosal glands is lower in mice, at least in laboratory mice that have not been infected by pathogens or subjected to a disease model. Moreover, whereas smooth muscle cell bundles populate the connective tissue surrounding the respiratory bronchioles in humans, mice have very few respiratory bronchioles that lack a smooth muscle layer.
When comparing human and naïve mouse lungs there is also an apparent difference in the quantity of mast cells and their distribution. In human lungs, mast cells are located throughout the airways and in the parenchyma, while in naïve mice mast cells are mainly found in the trachea and in the central airways (62). However, in mouse models of allergic asthma and influenza infection, the induced lung mast cells accumulate at places where they are not usually found such as in the epithelium, surrounding bronchioles, in the perivascular space and in the alveolar parenchyma (38, 53, 54, 63). Although allergic airway inflammation in mice induced by e.g. OVA and HDM stimulates airway hyperresponsiveness (AHR) to methacholine in vivo, these models rarely induce antigen-induced bronchoconstriction that can be measured in vivo. Importantly, neither lung mast cell expansion nor lung function have been analyzed in the majority of published studies of allergic airway inflammation in mice. Nevertheless, a high dose of HDM (125μg) given intranasally on five consecutive days/week over 3 weeks to induce allergic airway inflammation also induced mast cell expansion and an increase in the mast cell specific mediator mMCP-1, along with HDM-induced bronchoconstriction (63). Importantly, the HDM-induced bronchoconstriction was abrogated in KitW−sh/W−sh mice.
In contrast, when isolated mouse trachea from mice with allergic airway inflammation is analyzed ex vivo, antigen-induced contractions can be measured using OVA as the antigen (64). The antigen-induced contractions ex vivo are also abrogated in KitW−sh/W−sh mice (64). A possible reason for the discrepancy between the lack of OVA-induced bronchoconstriction in vivo and the presence of OVA-induced contraction in isolated airways may be that the majority of mast cells are found around the central airways and hence it is easier to measure their responsiveness to antigen in isolation (ex vivo). Nevertheless, it is currently unclear whether the reason behind why it is so hard to observe antigen-induced bronchoconstriction in vivo is only due to a less expanded lung mast cell population with lower doses of antigen and more acute protocols, or whether there are other more profound differences in lung physiology between the species that also play a role.
More often in vivo studies of allergic airway inflammation in mice measure AHR with increasing doses of methacholine. There are examples of protocols that bypass the dependence of mast cells for AHR, e.g. (65–67), while other protocols find that mast cells, or a mast cell mediator, are necessary for a full-blown AHR, e.g. (68–71). Moreover, when Fuch et al. compared KitW−sh/W−sh mice with KitW−sh/W−sh mice reconstituted by bone marrow-derived mast cells (BMMCs) to wild type sensitized and challenged with OVA, the reconstituted mice had a higher density of mast cells which were distributed differently compared to the wild-type mice (72). This resulted in an increased AHR in the KitW−sh/W−sh mice reconstituted with BMMCs as compared to wild-type mice treated in parallel. We speculate that this conundrum reflects the situation in human asthma, i.e. that not all phenotypes of asthma have a significant mast cell component, while some do.
The Role of Mast Cells in Allergic Airway Inflammation
Numerous studies have shown how allergen challenge induces mast cell activation and changes in the lung function in sensitized mice. However, most of the studies have used OVA as an allergen. Presumably, HDM or other human allergens are more relevant to use for studies of allergic airway inflammation in mice since they are complex allergens that activate innate receptors and induce secretion of alarmins in addition to inducing adaptive immune responses.
FcγR-mediated activation of mast cells was demonstrated to be required for the development of AHR and inflammation in an OVA model of allergic airway inflammation using mast cell-deficient KitW/Wv or KitW−sh/W−sh mice (68, 69). Later, mast cell-derived TNF-α was implicated in mediating these features of allergic airway inflammation and AHR (70, 73). The KitW−sh/W−sh strain was also studied in a model of HDM-induced allergic airway inflammation (74). In this study, the KitW−sh/W−sh mice developed allergic airway inflammation but had reduced plasma IgE levels and bronchoalveolar lavage (BAL) eosinophils. However, lung function was not analyzed in this study (74). HDM has also been demonstrated to induce an increase in the levels of mMCP-1 in serum 30 min after a single intratracheal challenge (75). Prophylactic treatment with the mast cell stabilizer cromoglycate 1 h before HDM challenge suppressed the induced levels of mMCP-1, and when HDM was given repeatedly to induce allergic airway inflammation, cromoglycate-treatment before each HDM administration reduced the inflammatory response.
Possibly also indicating the involvement of IgE-antigen-mediated activation of mast cells in AHR, FcεRI-deficient mice had reduced AHR to methacholine in OVA-induced allergic airway inflammation (76). In a subsequent study, FcεRI-deficient mice exposed to nebulized OVA demonstrated diminished tracheal responses to electric field stimulation, which was normalized in FcεRI-deficient mice adoptively transferred with wild-type BMMCs but not after adoptive transfer of IL-13-deficient BMMCs (77). This suggests a role for IgE-antigen-mediated mast cell-derived IL-13 in antigen-induced bronchoconstriction. On the other hand, allergic airway inflammation and AHR were unperturbed in IgE-deficient mice in a model induced by repeated intranasal exposure to Aspergillus fumigatus extract (65). Still, pre-treatment with crosslinking monoclonal anti-mouse IgE enhanced the bronchoconstriction induced by methacholine in wild-type but not in mast cell-deficient (KitW/Wv) naive mice (78). However, as c-kit mediates early development of cell linages other than mast cells, the phenotype observed in Kit-dependent mouse models of mast cell deficiency cannot be securely associated only to mast cells (79, 80). The results obtained from Kit-dependent mast cell-deficient mouse models need to be re-evaluated using the new transgenic mouse strains that do not depend on a functional c-kit for their mast cell deficiency. There is a risk that the scientific community has overestimated the role of mast cells by trusting the data that have been generated using the Kit-dependent mouse strains. In addition, reconstitution experiments performed with BMMCs need to be interpreted with care as BMMCs do not fully replicate the natural lung mast cell phenotype. In re-constitution experiments of mast cell-deficient mice, BMMCs may end up at a higher (or lower) density and be routed to places other than where they normally exist in wild-type mice (72). A more thorough discussion about Kit-dependent mast cell-deficient models, their advantages and disadvantages and discussion about Kit-independent models can be found in (79, 80).
Few studies have used Kit-independent mouse models to study the role of mast cells in experimental asthma. The Mas-TRECK and Bas-TRECK mice carry a diphtheria toxin (DT)-based conditional deletion system using intron regions of the Il4 gene, which constitute enhancer elements that drive IL-4 production in mast cells and basophils (81). DT treatment in the Mas-TRECK mice results in loss of mast cells and basophils, whereas DT treatment in the Bas-TRECK mice leads to basophil-specific depletion. In a model of OVA-induced allergic airway inflammation, DT treatment before the challenge phase reduced AHR, which was accompanied by a remarkable reduction in histamine levels in Mas-TRECK but not in Bas-TRECK mice (81). Therefore, the study suggests that mast cells are critical for full-blown AHR while basophils are dispensable. There are also other recently constructed mouse strains which more specifically deplete mast cells but also reduce basophil numbers. One example is, the Cpa3Cre/+ strain, which takes advantage of the ability of Cre recombinase to induce toxicity if the expression is high as in CPA3-expressing mast cells (82). The Cpa3Cre/+ mice lack mast cells but have a normal number of other cell linages except for a reduction in the number of basophils. The Cpa3Cre/+ mice are currently used to re-evaluate the role of mast cells in allergic airway inflammation.
Nevertheless, c-kit (CD117) is also vital for human mast cell development and survival. Therefore, a recent proof-of-principal asthma study tested the effects of imatinib, a tyrosine kinase inhibitor designed to target ABL (Abelson murine leukemia viral oncogene homolog 1) for treatment of chronic myelogenous leukemia (CML), which also inhibits several other tyrosine kinases including c-kit (83). In this randomized, double-blind, placebo-controlled study, severe asthmatics were treated with imatinib for 24-weeks. The imatinib treatment reduced AHR and decreased the number of mast cells in endobronchial biopsies and tryptase levels in serum, suggesting that mast cells contribute to the pathogenesis of severe asthma and highlighting the importance of c-kit in human mast cells.
IgE-antigen-Mediated Activation
Mast cells and basophils express FcεRI as a complex consisting of an α-chain, a β-chain and two γ-chains. In humans, FcεRI is also present as a complex composed of the α chain and two γ-chains in Langerhans cells (84), dendritic cells (85), platelets and megakaryocytes (86), neutrophils (87), monocytes (88) eosinophils (89) and airway smooth muscle cells (90).
The trimeric receptor αγ2 on human blood dendritic cells and monocytes may function as a regulator of serum IgE levels by receptor internalization (91). In mice, the tetrameric form of FcεRI has also been demonstrated in mouse nerve cells (92, 93). However, the receptors were thought not to be expressed on the cell surface in the absence of the β chain (94, 95). Nevertheless, expression of the trimeric form of FcεRI has been found in dendritic cells after virus infection (96).
Activation of mast cells by IgE-antigen activates several intracellular signaling pathways that lead to the secretion of mediators which occur in waves and mediate potent biological effects (97, 98). The mast cell-mediators activate endothelial, epithelial, and smooth muscle cells, neurons and other immune cells, thereby inducing the influx of inflammatory cells and changes in lung function (99). The first wave of mast cell mediators released after IgE-antigen-mediated activation are preformed granule-associated mediators, which are released within 5 min after the antigen contact. The composition of granule-associated mediators varies between species and differs between mast cell subtypes and localization. However, they are generally composed of histamine and (or) serotonin, proteoglycans and proteases. In the second wave of mediators after IgE-antigen-mediated mast cell activation, newly formed lipid mediators generated from arachidonic acid, are released (100). Next, cytokines and chemokines, which require gene transcription and synthesis, are secreted hours after antigen contact. For a detailed description of the molecular events which follow IgE-antigen-mediated mast cell activation, see (101).
The presence of specific IgE in serum is a key feature of allergic asthma. Also, high total IgE levels in serum are strongly related to increased risk of asthma and are correlated with AHR (102, 103). Moreover, the success of omalizumab, a humanized monoclonal anti-IgE antibody, which leads to improvements in symptoms and quality of life, and reduces virus-induced exacerbations (104), stresses the significance of IgE in the pathogenesis of at least some subtypes of asthma (105). Importantly, omalizumab reduces mast cell degranulation and FcεRI-receptor expression on mast cells and basophils in patients with moderate to severe allergic asthma (106). IgE alone is also relevant for mast cells in the asthmatic lung. For example, monomeric IgE can induce the secretion of cytokines in BMMCs, which by an autocrine mechanism enhance their survival (107). However, individual IgE molecules vary in their ability to induce cytokine production and survival (108). Several studies demonstrating the effects of IgE alone on mast cells are summarized in (109). Interestingly, the cytokinergic activity of monomeric IgE is enhanced in the presence of cytokines such as IL-4 (110). Therefore, the capability of monomeric IgE to promote mast cell survival may be significant in the asthmatic lung.
IgE-independent Activation of Mast Cells in Asthma
Mast cells can be activated by several IgE-independent pathways. The possible activation mechanisms differ between mast cell subpopulations, their location in the body and the microenvironment. The IgE-independent stimuli include pathogen-associated molecular patterns (PAMPs) such as endotoxin, and products derived by the innate immune system such as complement components, cytokines and endogenous peptides (98). Here, we will summarize selected IgE-independent mast cell activation pathways which may be related to allergic asthma (Figure 2).
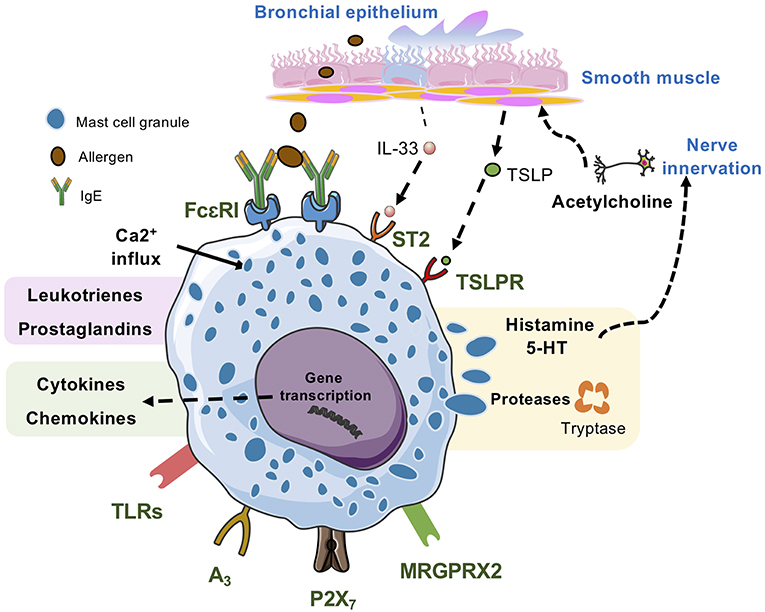
Figure 2. Lung mast cells can be activated by many kinds of stimuli. IgE/antigen-mediated activation of FcεRI triggers exocytosis of granular compounds, a rapid generation and release of lipid mediators such as leukotrienes and prostaglandins and the synthesis and release of cytokines and chemokines, which occurs hours after the activation event. Mast cell activation via MRGPRX2, the adenosine A3 receptor, and the ATP receptor P2X7 also triggers release of all three types of mast cell-derived compounds. Activation via TLRs generally triggers synthesis and release of cytokines and chemokines, and some TLRs may also trigger release of lipid mediators. However, this mode of mast cell activation does not induce degranulation. IL-33, which activates the ST2 receptor complex potentiates IgE/antigen-mediated degranulation in human mast cells but not in mouse mast cells. Alone, IL-33 triggers synthesis and release of cytokines and chemokines. In mice, TSLP acting via TSLPR promotes mast cell development, whereas in human mast cells TSLP potentiates IL-33-stimulated secretion of type 2 cytokines and chemokines.
MAS-Related G Protein-Coupled Receptor X2 (MRGPRX2)
Pseudo-allergic reactions typically occur in response to basic substances such as compound 48/80 or cationic peptide drugs. These reactions share characteristics with allergic responses induced by IgE-mediated responses, but trigger-specific IgE molecules are not detected (111). In 2015, MRGPRX2 (MAS-related G protein-coupled receptor X2) and the mouse ortholog MRGPRB2 were described as the main receptors involved in pseudo-allergic reactions due to their interaction with cationic drugs, which led to mast cell activation (112). In mice, MrgprB2 transcripts were only found in CTMCs (112). The transcript levels of MRGPRX2 are higher in human skin mast cells than in lung mast cells (113, 114). Nevertheless, the expression of MRGPRX2 on mast cells and the number of MRGPRX2+ mast cells are higher in lung biopsies from patients who died from asthma-related causes than in lung biopsies from patients who died from other causes (115).
Substance P (SP) was one of the identified ligands of MRGPRX2 (112). SP is elevated in BAL and sputum in asthma patients compared to healthy controls (116, 117) and is further increased in BAL immediately after allergen provocation (116). Therefore, one possibility is that MRGPRX2 is involved in a positive feed-back loop where mast cells are activated via e.g. allergen release histamine, which activates neurons to produce cationic peptides such as SP, which amplifies mast cell activation. The activation of MRGPRX2 may also be related with exacerbations of asthma symptoms that occur in connection with viral respiratory infections (118). Several respiratory viruses trigger the secretion of anti-microbial peptides such as β-defensins from epithelial cells (119, 120). β-defensins activate mast cells trough MRGPRX2 (121) and may thus contribute to virus-induced asthma exacerbations. To conclude, MRGPRX2-mediated activation of mast cells may contribute to the pathogenesis of asthma.
Toll-Like Receptors (TLRs)
Mast cells are specialized in sensing external pathogens by recognition of pathogen-associated molecular patterns (PAMPs) and are equipped to respond to tissue damage by recognition of danger-associated molecular patterns (DAMPs) or alarmins (122). Interestingly, several allergens have the ability to stimulate toll-like receptors (TLRs), and these receptors may thus play a role in asthma development. For example, HDM extracts contain e.g. lipopolysaccharide (LPS) and proteins like Der p2 (Dermatophagoides pteronyssinus) mimicking MD-2, resulting in TLR4 activation (123). Mouse mast cells express several TLRs, and the expression pattern seems to differ between the mast cell subtypes, e.g. Tlr1-9 mRNA was detected in both MMC-like and CTMC-like BMMC but the level of Tlr3-5 expression differed (124). Human peripheral blood-derived mast cells express mRNA and protein for TLR1-4 and TLR6-8, and responded to double-stranded RNA stimulation via TLR3 by producing type I interferons (IFNs) (109). The expression of TLR2 was confirmed in isolated human lung mast cells by western blot, and in vitro stimulation with lipoteichoic acid (a TLR2 agonist) led to down regulation of FcεRI expression and decreased IgE-antigen-mediated mast cell degranulation (125). The expression of TLR4 has also been confirmed in human lung mast cells (126). In the same study, LPS stimulated release of TNF-α in peripheral blood-derived mast cells, and after pre-incubation with IFN-γ, LPS induced the expression of anti-viral genes (126). In vivo, TLR4-mediated mast cell activation was demonstrated to enhance eosinophilia and cytokine release in an OVA-model of allergic airway inflammation using mast cell-deficient (KitW/Wv) mice and reconstitution experiments (127). Furthermore, intranasal administrations of poly I:C expand the number of lung MCps in a TLR3-dependent fashion (60). To summarize, the activation of pattern recognition receptors may directly or indirectly induce or modulate mast cell responses in the allergic lung.
The Alarmins IL-33 and TSLP
Inhaled allergens and respiratory viruses induce the release of alarmins such as IL-33 and thymic stromal lymphopoietin (TSLP) from airway epithelial cells (128). IL-33 is a member of the IL-1 family of cytokines, which primarily binds to the IL-1 receptor family member ST2. However, the IL-33/ST2 complex assembles with the IL-1 receptor accessory protein (IL-1RAcP), which is needed for signal transduction (129). The loci for IL1RL1 (ST2 gene) and IL-33 contains a single nucleotide polymorphism that was associated with asthma in a large-scale genome-wide association study (130). Moreover, IL-33 expression was higher in lung samples from patients with severe asthma than in those with mild asthma and IL-33 expression was mainly localized to epithelial and endothelial cells, neutrophils, fibroblast and mast cells (131). Another source of IL-33 in the airways of asthma patients are the smooth muscle cells (132). Interestingly, there was a strong inverse correlation between the concentration of IL-33 in BAL and lung function (pre-bronchodilator FEV1) (133).
Human in vitro-derived mast cells from cord and peripheral blood express ST2 (134). In vitro, IL-33 accelerates mast cell maturation of CD34+ cells and induces the secretion of Th2 cytokines and chemokines (134). Moreover, pre-treatment with IL-33 increases the number and magnitude of degranulating in vitro-derived mast cells in response to a crosslinking anti-IgE antibody (135). IL-33 increases the survival of human skin mast cells by upregulation of the antiapoptotic protein, B-cell lymphoma-X large (BCLXL) (136). In cord blood-derived mast cells, IL-33 alone promoted adhesion to fibronectin and production of IL-8 and IL-13 (137). In mice, lung mast cells and their progenitors express ST2 protein on the cell surface and intranasal administrations of IL-33 induce an ST2-dependent increase in lung MCp (59, 60).
The role of IL-33 in the context of allergic airway inflammation has been studied in vivo. Generally, mice lacking ST2 and wild type mice administrated with ST2 blocking antibodies have reduced allergic airway inflammation (138). IL-33 induces an asthma-like phenotype in Rag2 −/− mice, which lack mature lymphocytes, demonstrating that adaptive immune responses are not required to induce an asthma-like phenotype (139). Further, in a new humanized mouse model (NOG IL-3/GM-CSF), administration of human IL-33 induced an asthma-like phenotype mediated by human IL-13 (140). The main producers of IL-13 in this setting were T cells and mast cells. Further, IL-33 given before the allergen challenge potentiated AHR in wild-type but not in mast cell-deficient KitW−sh/W−sh mice or mice treated with ketanserin (a nonselective 5-HT2R antagonist with high affinity also for H1R) in a OVA model of allergic airway inflammation (141). In contrast, in papain-induced airway inflammation, which promotes IL-33 production and increased mast cell numbers in the lung, mast cell-deficient (KitW−sh/W−sh) mice had an exacerbated type 2 inflammatory response (142). The suppressive effect of mast cells in this experimental set-up was explained by the observation that IL-33-activated mast cells produced the IL-2 necessary for the expansion of CD4+CD25+Foxp3+ regulatory T cells that were inhibiting the type 2 inflammation by production of IL-10. Altogether, there is substantial evidence that the IL-33-ST2 pathway is involved in allergic asthma and that this pathway interacts with mast cells.
TSLP is another alarmin released from epithelial cells as a DAMP signal after allergen exposure. This IL-7-like cytokine binds to the TSLP receptor (TSLPR), which shares the alpha chain with the IL-7 receptor (143, 144). The expression of TSLP was first associated with acute and chronic dermatitis (145). In human bronchial biopsies from asthmatics, TSLP transcripts were increased in epithelial cells and in the submucosa (146). Interestingly, the number of epithelial cells or submucosal cells expressing TSLP was inversely correlated with FEV1 (% of predicted) (146). TSLP was also increased in serum from asthmatics compared to healthy controls or patients with chronic obstructive lung disease (COPD) (147). In mice, the expression of TSLP is increased in allergic airway inflammation and TSLPR-deficient mice have an attenuated type 2 response (148). Moreover, overexpression of TSLP in epithelial cells in the mouse lung produced a spontaneous asthma-like phenotype (148). TSLP−/− mice have reduced numbers of mast cells in the intestine, kidney, nasal mucosa, skin, liver and lung, suggesting that TSLP is involved in mast cell development (149). In support of that, IL-3-induced proliferation and differentiation of BMMCs is blocked by neutralization of TSLP (149).
TSLP expression has also been demonstrated in human bronchial and submucosal mast cells as well as in the epithelium and airway smooth muscle (146, 150). Airway smooth muscle cells and human mast cells in this location expressed TSLPR (150). Further, the percentage of TSLP+ mast cells in bronchial samples was increased in asthmatic airways compared to healthy controls (151). In the same study, activation of human peripheral blood-derived mast cells by a crosslinking anti-IgE antibody induced TSLP expression, which was further enhanced by pre-incubation with IL-4. Moreover, TSLP potentiates the IL-33-stimulated secretion of type 2 cytokines and chemokines in human mast cells derived from peripheral blood or cord blood (134). The importance of TSLP in asthma is highlighted by clinical studies using a monoclonal anti-TSLP antibody. In 2014, an anti-TSLP antibody showed efficacy in several measures of allergen-induced early and late asthmatic responses including improved FEV1 (% of predicted) (152). Furthermore, anti-TSLP treatment has been shown to reduce the frequency of asthma exacerbations and improved FEV1 (% of predicted) in a randomized, double-blind, placebo-controlled trial of uncontrolled asthmatics with moderate to severe asthma (153). Altogether, the present data suggest that TSLP influences the development of mast cells and that mast cell-derived TSLP may contribute to allergic asthma.
Purinergic Signals (ATP, Adenosine)
Adenosine tri-phosphate (ATP) is a danger signal to the immune system when released to the extracellular milieu by many different cells types. Extracellular ATP is sensed by the class 2 purinergic P2Y and P2X receptors (154). In the extracellular space, ATP is quickly hydrolyzed by nucleoside triphosphate diphosphohydrolase (NTPDase or CD39) to adenosine mono-phosphate (AMP) via adenosine di-phosphate (155). Allergen challenge triggers an increase in ATP levels in BAL from asthma patients and mice with experimentally induced allergic airway inflammation (156). In the same study, neutralizing ATP by the ATP-hydrolyzing enzyme apyrase or inhibition with a broad-range inhibitor of P2 receptors before OVA challenge blocked type 2 inflammation and AHR.
That rodent mast cells are activated and degranulate in response to ATP has been known for several decades (157, 158). In an acute mouse model of colitis, ATP-mediated mast cell activation was demonstrated to occur through P2X7 receptors (159). A role for the P2X7 receptor in allergic airway inflammation and AHR was demonstrated using P2X7 deficient mice and treatment of wild type mice with a specific P2X7 antagonists given before each antigen challenge (160). Recently, CD203c or ecto-nucleotide pyrophosphatase-phosphodiesterase 3 (E-NNP3), a widely used activation marker of mouse mast cells and basophils, was demonstrated to negatively regulate IgE-antigen-mediated activation through hydrolyzation of extracellular ATP (161). Further, while E-NNP3−/− mice had exacerbated allergic airway inflammation, mice lacking both E-NNP3 and P2X7 had decreased responses to IgE-antigen-mediated activation of FcεRI. Therefore, ATP released by IgE-antigen-mediated activation of FcεRI stimulates mast cell activity through P2X7, whereas E-NNP3 decreases the ATP concentration and suppresses mast cell (and basophil) activity in vivo.
In human lung mast cells, the expression of the purinergic receptors (P2X1, P2X4, P2X7, P2Y1, P2Y2) has been confirmed by q-RT PCR and a gene array (162–164). An early study indicated that ATP stimulation of human lung mast cells did not directly induce degranulation but enhanced histamine release after anti-IgE mediated activation (162). The demonstration of P2X7 receptor on mast cells in the colon from patients with Crohn's disease (159) suggests that this receptor may regulate mast cell function also in human disease. However, a reduced risk of asthma and asthma severity was detected in children with loss-of-function mutations in the P2X7 receptor (165).
Adenosine is directly released from many cell types or generated from extracellular AMP by CD73 (ecto-5′-nucleotidase), and the extracellular levels increase under inflammatory conditions (166). In asthmatics and cigarette smokers, adenosine levels were increased in BAL (167, 168). Further, adenosine provoked bronchoconstriction in asthmatics (169, 170). Adenosine exerts its biological functions binding to four distinct G-protein coupled receptors: A1, A2a, A2b and A3 with different affinity (167). In human lung mast cells, adenosine potentiated IgE-antigen-mediated mast cell activation, thereby increasing the release of mediators such as LTC4 and histamine (171). Human and mouse lung mast cells express mRNA for A2a, A2b and A3, and activation via A3 induces histamine release (172–175). In mice, nebulization of an A3 agonist for only 5 min caused mast cell degranulation (175). Moreover, adenosine induced airway contraction, which is lost in mast cell deficient (KitW/Wv) and in A3−/− mice along with the loss of adenosine-induced mast cell degranulation (176). This suggests that mast cell activation via A3 is the major mechanism behind adenosine-induced bronchoconstriction. In a follow-up study, pre-exposure to aerosolized adenosine was demonstrated to increase methacholine-induced AHR in wild-type, but not in mast cell-deficient (KitW/Wv or KitW−sh/W−sh) or A3−/− mice (177). Further, the increased AHR after adenosine pre-treatment was regained by reconstitution of KitW−sh/W−sh mice with wild-type BMMCs but not with A3−/− BMMCs. These in vivo studies suggest that adenosine can promote bronchoconstriction or enhancing AHR by activating mast cells through the A3 receptor. In the context of human mast cells, adenosine or an A3-specific agonist potentiated FcεRI-induced activation of human lung mast cells (174). Altogether, ATP and adenosine may play an important role in promoting mast cell activation in the allergic lung.
The Role of Mast Cell Mediators in Allergic Asthma
Many mediators produced upon mast cell activation can be measured in the BAL and other fluids from asthmatic patients as residual levels or after local bronchial allergen challenge (178–181) (Table 2). Here, we summarize what is known about a selected number of mast cell mediators in the context of asthma and in vivo models thereof.
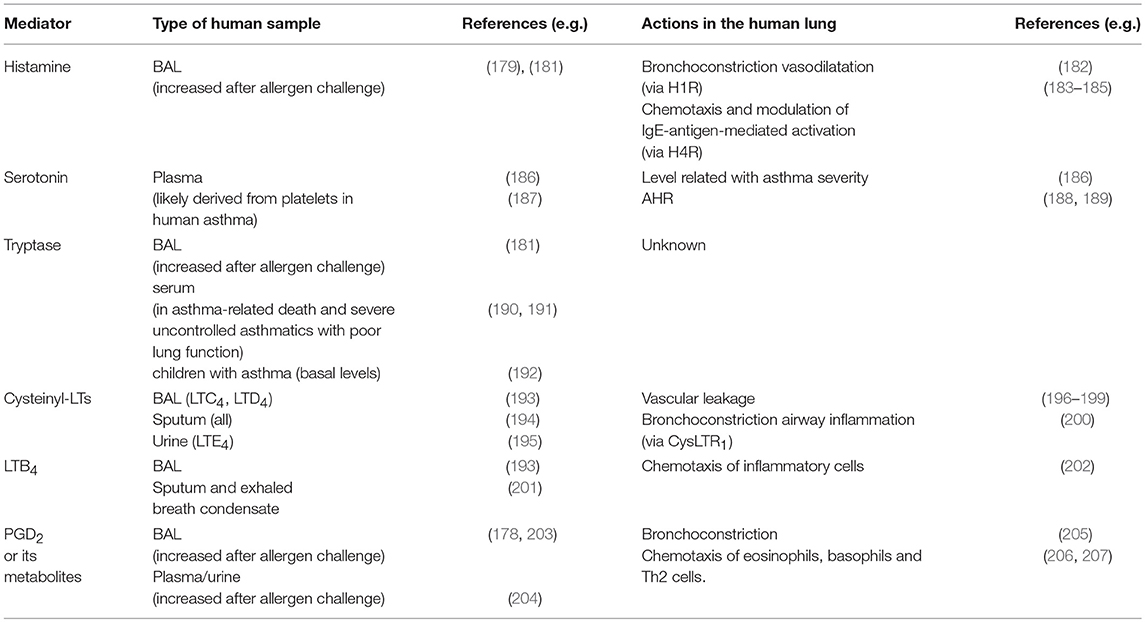
Table 2. Mast cell mediators detected in elevated levels in patients with asthma and their indicated action in the asthmatic lung.
Histamine
Histamine is a pivotal molecule involved in allergic reactions, and mast cells were recognized early as the main source of histamine (208). In patients with allergic asthma, allergen challenge via bronchoscope stimulates increased histamine levels in BAL (179). Histamine mediates its biological effects by binding to four histamine receptors (H1-4R), which are expressed in many different cell types such as immune cells, nerves and smooth muscle cells (209). In the human airways, the H1R mediates bronchoconstriction and increases vascular permeability (182). Despite this, H1R antagonists have demonstrated variable efficacy in clinical asthma trials (210). In OVA models of allergic airway inflammation, mice lacking H1R had reduced Th2 responses and AHR (211, 212).
Besides mediating the effect on acid production in the stomach, the histamine H2R is associated with the regulation of immune responses (213). A recent study demonstrated that genetic ablation or blocking of H2R in a model of OVA-induced allergic airway inflammation increased the eosinophilia in BAL, type 2 cytokines and mucous production, while an H2R agonist suppressed these responses (214). The exacerbated type 2 inflammation in H2R-deficient lungs was associated with increased numbers of CD1d+ dendritic cells and iNKT cells with an increased capacity to secrete cytokines in response to lipid antigens, suggesting that iNKT cell activity is controlled by histamine via H2Rs in the allergic lung.
The H3R is preferentially expressed in the nervous system and regulates its own release from neurons (215). The H4R is the most recently discovered histamine receptor, and like the H1R, this receptor mediates mostly pro-inflammatory effects. The H4R is mainly expressed by immune cells (216). In human eosinophils, histamine potentiated the chemotactic response to eotaxin via the H4R and directly mediated chemotaxis of human basophils in vitro (183, 184). In basophils, a specific H4R agonist reduced the mediator release after IgE-antigen-mediated activation (184). The H4R is also expressed in human cord blood-derived and purified lung mast cells (185, 217). In human lung mast cells an H4R-specific agonist induced chemotaxis (185), whereas in cord blood-derived mast cells another selective H4R agonist stimulated degranulation, and generation of lipid mediators and cytokines (217). The H4R is also expressed by mast cells, basophils and eosinophils in mice, and mediates histamine-induced chemotaxis in BMMCs (218). In OVA-induced models of allergic airway inflammation, H4R-deficient and wild-type mice treated with an H4R antagonist during either the sensitization or challenge phase had diminished type 2 inflammation (219, 220). The reduced type 2 lung inflammation in the absence of functional H4R was first attributed to a reduced ability of Th2 cells to produce cytokines (219), and more recently to the fact that dendritic cells cannot be fully activated in the absence of signals through the H4R and therefore the CD4+ T cell activation is reduced (220). For detailed information about the history of histamine, its receptors and the use of antagonists, see (221).
Serotonin
Serotonin (5-hydroxytryptamine, 5-HT) is stored in mast cell granules and released upon mast cell degranulation. However, while mouse mast cells contain large quantities of 5-HT, human mast cells contain none or very small amounts (222, 223). Instead, platelets are the main source of 5-HT in human airways (187). 5-HT has wide biological effects and acts by binding to seven different 5-HT receptor families (5-HT1−7R), the serotonin transporter (SERT) and by binding covalently to different effector proteins (224). Although many animal studies have demonstrated that 5-HT induces bronchoconstriction, 5-HT does not consistently induce bronchoconstriction in humans (187). Nevertheless, plasma levels of 5-HT are elevated in asthma patients and are associated with disease severity (186). Further, ketanserin decrease adenosine- and methacholine-induced bronchoconstriction in asthma patients (188, 189).
In mice, 5-HT plays a major role in airway contraction mainly via the activation of 5-HT2 receptors (225). 5-HT induces a ketanserin-sensitive depolarization-response in cholinergic neurons in the trachea, suggesting that 5-HT works by inducing secretion of acetylcholine from nerves that innervate the airways (64). Also, selective 5-HT2BR antagonists can block 5-HT-induced bronchoconstriction in mice ex vivo (226).
In human airways, 5-HT acts on airway smooth muscle via 5-HT2AR, which induces contraction, and via 5-HT1AR, which induces relaxation (227). Moreover, 5-HT may also work by inducing or augmenting the release of acetylcholine from cholinergic nerves via 5-HT3R and 5-HT7R (227). Nevertheless, 5-HT induced eosinophil migration via the 5-HT2AR in both mice and humans (228, 229). Mast cells express mRNA for several 5-HT receptors. In a study by Kushnir-Sukhov et al., transcripts for the 5-HT1A, 5-HT1B, 5-HT2A, 5-HT2B and 5-HT7 receptors were detected in BMMCs and peripheral blood-derived human mast cells (230). In addition, transcripts for the 5-HT1E, 5-HT2C, 5-HT3 and 5-HT4 receptors were also found in peripheral blood-derived human mast cells, while the BMMCs also expressed transcripts for 5-HT1DR, 5-HT6R and SERT. The same study revealed that 5-HT does not induce degranulation or cytokine release by in vitro-derived mast cells from mice or humans. Nevertheless, 5-HT stimulated adhesion to fibronectin and chemotaxis (230). In line with those findings, another study showed that intradermal injection of 5-HT induced 5-HT1AR-dependent accumulation of mast cells at the site of injection 48 h post-injection (230).
Mast Cell Proteases
Mast cell-specific proteases such as tryptase and chymase are stored inside the secretory granules in their active form. In addition, the mast cell granules contain other proteases such as CPA3, also expressed in basophils, as well as cathepsin C and G, found in e.g. neutrophils (231). Upon mast cell degranulation, the proteases are quickly released to the extracellular matrix. Tryptase, which is electrostatically bound to serglycin proteoglycans inside the acidic mast cell granules due to positively charged histidines, will gradually dissociate from the serglycin proteoglycans, possibly forming active monomers before losing its activity through the action of protease inhibitors (232–234). Chymase is also released as a complex with serglycin proteoglycans, but the association is not pH-dependent. Therefore, chymase may stay in complex with serglycin proteoglycan for an extended time and tends to remain close to the mast cell surface after degranulation (235, 236). As described earlier, the expression of tryptase and chymase has been used to classify human mast cells into subtypes. In the mouse, the major form of granule-stored tryptase is mMCP-6. In an acute OVA model of allergic airway inflammation, we found that mMCP-6-deficient mice had reduced methacholine-induced AHR but developed allergic airway inflammation (71). Moreover, the tryptase inhibitors nafamostat mesylate and gabexate mesylate reduced mast cell activation, AHR and eosinophil influx in an HDM model of allergic airway inflammation (237). A possible mechanism for the pro-inflammatory and broncho-constrictive effects of tryptase is cleavage and activation of protease activated receptor 2(PAR2). When trypsin-like proteases cleave PAR2 in the N-terminal, a tethered ligand sequence is exposed and the receptor auto-activates (238). For example, sensory neurons, which are frequently located in proximity to mast cells, express PAR2 and may thus be activated by mast cell tryptase (239). Moreover, immunostaining shows PAR2 expression in smooth muscle, epithelium, endothelium and glands of human bronchi (240). In the same study, tryptase and an artificial ligand peptide to PAR2 (SLIGKV-NH2) were demonstrated to stimulate contraction of isolated human bronchi (240). In mouse models, PAR2 deficiency or blocking PAR2 with a monoclonal antibody reduces allergic lung inflammation and AHR (241–243).
In asthma patients who died due to an asthma attack, tryptase levels in serum were increased compared to those who died from other causes (190). Further, a study of 60 asthmatics classified according to pathological findings and standard clinical parameters to define asthma severity by Bayesian network analysis combined with topological analysis revealed six disease clusters (191). Two of the clusters demonstrated higher serum tryptase levels than the others. One cluster consisted of asthma patients with severe asthma who were generally older, had high BMI, poor lung function and many symptoms, while the other group consisted predominately of patients who were female, obese, non-atopic, had later onset and poor lung function. In children, basal serum tryptase levels were higher in those with mild, moderate or severe persistent asthma than in those with mild intermittent asthma and healthy controls (192). Nevertheless, more studies are needed to clarify the importance of mast cell tryptase and the mechanisms behind its role in allergic and non-allergic asthma.
In mice, the functional homolog of human chymase, mMCP-4, has a protective effect on AHR and lung inflammation. This was shown in two studies, first by analyzing mMCP-4−/− mice in an OVA model of allergic airway inflammation and later in an HDM sensitization model (244, 245). The study by Waern et al. suggested that the protective effect of mMCP-4 is mediated by proteolytic cleavage of IL-33, leading to degradation of this important Type 2 cytokine (245). Moreover, mMCP-4 has the ability to suppress IL-13-induced enhancement of the contraction of the trachea in response to methacholine in vitro (246). The protective role of mast cell chymase is in accordance with a clinical study, mentioned earlier, where the number of chymase+ mast cells correlated positively with lung function in 20 severe asthmatics (48).
Lipid Mediators–Leukotrienes
Mast cells are a well-known source of lipid mediators derived from arachidonic acid metabolized by different enzymes in response to various stimuli. Leukotrienes are metabolized from arachidonic acid through the 5-lipoxygenase (5-LO) pathway that generates LTA4 as a main precursor of two different types of leukotrienes: the cysteinyl-leukotrienes (cysteinyl-LTs) LTC4, LTD4 and LTE4, and LTB4 (247).
The biological activities of the cysteinyl-LTs were discovered more than 30 years ago and included stimulation of smooth muscle contraction and vascular leakage (196–199). The levels of cysteinyl-LTs are increased in BAL (193) and sputum (194) of asthmatics, while LTE4 levels are increased in the urine after allergen challenge (195). Inhalation of LTC4 or LTD4 causes bronchoconstriction, which is 1,000-fold more potent than histamine or methacholine (197–199). The cysteinyl-LTs mediate their biological effects through the receptors CySLTR1, CySLTR2 and GPR99 (248). Two other receptors, P2Y12R and GPR17 are also involved in cysteinyl-LT and LTE4-elicited responses, respectively. LTD4 has a higher affinity for CySLTR1 than LTC4, whereas LTC4 and LTD4 have a similar affinity for CySLTR2. However, LTE4 has a high affinity for GPR99. CySLTR1 is expressed by many different immune cells such as mast cells, neutrophils and monocytes/macrophages, whereas CySLTR2 is expressed by eosinophils, macrophages and smooth muscle cells (248). The CysLTR1 antagonist, montelukast, attenuates CysLT-induced bronchoconstriction and airway inflammation (200), confirming many earlier studies in animal models (249, 250). CysLTR2 mediated vascular permeability in IgE-dependent passive cutaneous anaphylaxis and infiltration of macrophages and fibroblasts in a fibrosis model (251). Recently, a role for CysLTR2 was demonstrated in IL-33-dependent type 2 immunity (252). Using various knockout strains, CysLTR2 was shown to drive IL-33 expression induced by LTC4 in two models of allergic airway inflammation. Human cord blood-derived mast cells express both CysLTR1 and CysLTR2 (253). In response to CysLTR1 stimulation, human cord blood-derived mast cells proliferate in a process that is negatively regulated by CysLTR2 (254). Moreover, stimulation of the human mast cell line LAD-2 with LTD4 or LTE4 leads to chemokine and prostaglandin D2 (PGD2) release (255).
Inhaled LTE4 induces the influx of inflammatory cells in the bronchial mucosa of asthma patients more potently than LTD4, while they have similar bronchoconstrictive effects (256, 257). Nevertheless, LTE4 has only weak affinity for the earliest discovered cysteinyl-LT receptors, CysLTR1 and CysLTR2. In mice, P2Y12R and platelets were demonstrated to be required for LTE4-induced pulmonary inflammation, although LTE4 does not bind directly to P2Y12R (258). Recently, a placebo-controlled randomized double-blind study of asthmatic patients demonstrated that the P2Y12R antagonist prasugrel inhibited platelet reactivity but showed only a possible modest effect on mannitol-induced airway reactivity (259). After the discovery of the interaction of LTE4-induced responses with P2Y12R, another P2Y receptor family member called GPR17 was discovered to function as a negative regulator of CySLTR1-mediated responses (260). However, while GPR17 has a role in regulating cysLTR1-induced lung inflammation in a HDM model, it is unclear if this effect is directly mediated by Cys-LTs since different labs have conflicting data on whether Cys-LTs can activate GPR17 or not (261). Next, the P2Y receptor family member GPR99 was identified as the third high affinity cysteinyl-LT receptor, which preferentially binds LTE4 (262). GPR99 was recently demonstrated to mediate the release of mucin from epithelial cells and swelling of nasal mucosa in mice subjected to a single intranasal dose of Alternaria extract or LTE4 (263). However, in human asthmatics, LTE4-induced bronchoconstriction was completely blocked by the CySLTR1 inhibitor montelukast (264). The LTE4-induced bronchoconstriction was associated with increases in urinary PGD2 metabolites and other COX pathway products, which was also abrogated by montelukast, suggesting that LTE4 activates mast cells (and possibly other cell types) via CySLTR1 and that mast cell-derived products activate bronchial smooth muscle cells to constrict. These data suggest that CySLTR1 mediates LTE4-induced responses in the human lung or that montelukast also blocks GPR99.
The non-cysteinyl-LT, LTB4, is increased in exhaled breath condensate and sputum of asthmatics (201, 265). In a recent study categorizing asthma patients according to GINA guidelines (step 1–3), patients treated with short-acting beta-agonist (step 1) or inhaled corticosteroids plus long-acting beta-agonist (step 3) had higher LTB4 levels in sputum than those treated with inhaled corticosteroids (step 2) (266). This indicates that inhaled corticosteroids reduce LTB4 levels in mild-moderate asthma as previously described in (267), but not in more severe asthma. LTB4 is produced by various activated leukocytes including mast cells (268), and has a potent chemotactic effect on leukocytes such as neutrophils, T cells and immature BMMCs (202, 269, 270). LTB4 mediates its effect through BLT1R and BLT2R, which are G-protein-coupled receptors. BLT1R is a high affinity receptor for LTB4 and is responsible for LTB4-induced leukocyte migration (202, 248). In vivo, intradermal injection of LTB4 induced the accumulation of intravenously injected immature BMMCs at the injection site (269).
In experimental allergic airway inflammation, BLT1R mediates the infiltration of T cells to the lung (271) as well as AHR, eosinophilic inflammation and goblet cell hyperplasia (272). An indication of a role for mast cells in LTB4-mediated allergic airway inflammation was demonstrated in a study using mice lacking LTA4 hydrolase (LTA4H), which thereby were unable to produce LTB4 (273). The LTA4H−/− mice had reduced AHR and BAL eosinophilia compared to controls in an active systemic sensitization and challenge model, as well as when mice were passively sensitized and challenged. However, transfer of LTA4H+/+ BMMCs into LTA4H−/− mice normalized airway reactivity to methacholine assessed by electrical field stimulation of tracheal smooth muscle preparations, and partly restored eosinophilia in passively sensitized and challenged mice but not in mice subjected to active systemic sensitization and challenge (273). Thus, LTB4 plays a role in allergic airway inflammation, and LTB4 from mast cells may mediate some of these effects.
The low affinity LTB4 receptor BLT2R is expressed in the mouse lung. Intriguingly, BLT2R-deficient mice displayed enhanced eosinophilia in an OVA model of allergic airway inflammation (274). This might be explained by the fact that a cyclooxygenase metabolite (12(S)-hydroxyheptadeca-5Z,8E,10E-trienoic acid) was shown to have higher affinity than LTB4 for BLT2R (275). To summarize, leukotrienes and many of their receptors represent important targets in allergic asthma; for a recent detailed review, see (248).
Lipid Mediators–Prostaglandins
Prostaglandins are metabolites initially derived from the conversion of arachidonic acid by the cyclooxygenases (COX-1 and COX-2) to PGH2, which subsequently is a substrate of PGD synthase forming PGD2 or PGE synthase forming PGE2. There are also other primary prostaglandins (PGF2α, PGI2, and thromboxane A2). However, here we limit the discussion to PGD2, which is the major mast cell-derived prostaglandin produced in large amounts through the action of hematopoietic prostaglandin D synthase (HPGDS) in response to IgE-antigen-mediated activation (276). Nevertheless, mouse and human eosinophils also produce PGD2 (277). Extracellularly, PGD2 is metabolized to different compounds such as 9α,11β-PGF2, which can be measured in plasma as a possible indication of mast cell activation (204).
Allergen challenge in allergic asthmatics leads to an increased level of PGD2 in BAL, and PGD2 metabolites in plasma and urine (178, 203, 204). Inhalation of PGD2 leads to bronchoconstriction in normal subjects and to an even stronger bronchoconstrictive response in patients with asthma (205). Despite corticosteroid use, the PGD2 synthesis pathway is upregulated in asthmatics with severe and poorly controlled asthma (278). In the same study, the levels of HPGDS transcripts in epithelial brushings correlated strongly with the levels of TPSAB1/TPSAB2 (tryptase) transcripts, suggesting that mast cells are the main source of PGD2, at least at this site.
PGD2 acts via activation of the D-prostanoid receptors DP1 and DP2. Another name for DP2 is chemoattractant receptor-like protein expressed on Th2 cells, abbreviated CRTH2 (279, 280). The DP1 receptor is expressed on Th2 cells, dendritic cells, basophils, eosinophils, goblet cells and vascular endothelium (276). Recently, mast cell maturation was shown to be mediated by PGD2 acting on DP1, a process which was driven by phospholipase A2 group III (PLA2G3) secreted from mast cells, which activated fibroblasts to produce PGD2 by the action of lipocalin-type PGD2 synthase (281). An amplifying role of PGD2 in experimental asthma was discovered by comparing wild-type and mice lacking DP1 in an OVA-induced mouse model of allergic airway inflammation (282). In this study, DP1−/− mice had attenuated AHR and the production of type 2 cytokines was diminished. Further, pre-treatment of sensitized mice with aerosolized PGD2 before challenge amplified the type 2 response (283). In contrast, PGD2 acts in an anti-inflammatory fashion via DP1 expressed on dendritic cells during the sensitization phase. PGD2 or a DP1 agonist, but not a DP2 agonist, instilled intratracheally with FITC-OVA temporarily inhibited the migration of dendritic cells to the lung draining lymph nodes, which limited the expansion of adoptively transferred T cells (284). In a later study by Hammad et al. PGD2 or an agonist for DP1 or DP2 was intratracheally delivered 30 min before each OVA challenge in an OVA model of allergic airway inflammation (285). Interestingly, the DP1 agonist (but not PGD2 or the DP2 agonist) suppressed inflammation and AHR by a mechanism involving T regulatory cells producing IL-10. As the suppressive effect of the DP1 agonist could be mimicked by adoptively transferred dendritic cells pre-treated with the DP1 agonist. Hence, the DP1 agonist likely acted on lung dendritic cells which acquired a regulatory function by stimulating the expansion of T regulatory cells producing IL-10 (285). DP2/CRTH2 is expressed by various immune cells such as eosinophils, basophils, Th2 cells and ILC2s (286, 287). Moreover, Ptgdr2 transcripts have been found in BMMCs (288), and CRTH2 immunopositive human mast cells were found in nasal mucosa (289) and nasal polyps (290), although the expression seemed to be intracellular. Early studies demonstrated that CRTH2 mediates chemotaxis of human eosinophils, basophils and Th2 cells (206, 207). Further, patients with severe asthma have an increased level of CRTH2 transcripts in BAL cells compared to patients with mild to moderate asthma and heathy controls (278). In the same study, immunohistochemical analyses of CRTH2+ cells in BAL revealed a higher percentage of CRTH2+ cells in asthma patients compared to healthy controls, with the highest percentages found among those with severe asthma and as well as those with mild asthma but who did not have corticosteroid treatment (278).
In mice, sensitization with OVA followed by nebulization of a CRTH2 agonist before each challenge enhanced AHR and the eosinophilia in the BAL and lung tissue (291). In contrast, mice lacking Ptgdr2 have an enhanced BAL eosinophilia in OVA-induced allergic airway inflammation (292). This was likely due to the fact that T cells lacking CRTH2 produced higher levels of cytokines (292). However, a CRTH2 antagonist given in connection with OVA challenge in a model of allergic airway inflammation reduced eosinophilia and mucus hyperplasia (293). Similarly, mice sensitized and challenged with cockroach allergen given a CRTH2 antagonist before the final challenge had significantly reduced AHR, levels of type 2 cytokines in the lung and allergen-specific IgE and IgG2a in serum (294). For these reasons, CRTH2 antagonists have been tested as a treatment for asthma. Several different CRTH2 antagonists have been taken into clinical trials, but some of them have been discontinued for various reasons like poor effect or pharmacokinetics (295). However, other CRTH2 antagonists have been demonstrated to improve lung function, symptom scores and reduced sputum eosinophilia (296–298). Still more studies are needed to define what asthma phenotypes will benefit (most) from treatment with a CRTH2 antagonist.
Lipid Mediators–Sphingosine-1-Phosphate
A more unknown lipid mediator that is generated and released from mast cells after IgE-antigen-mediated activation is sphingosine-1-phosphate (S1P) (299). S1P is produced as a result of sphingosine (Sph) phosphorylation by two Sph kinases (SphK1 and SphK2). In mast cells, IgE-antigen-mediated crosslinking of FcεRI induces the activation and translocation of SphK1 to the plasma membrane and leads to increased levels of S1P, which then becomes secreted extracellularly (299). S1P is produced by most cell types but is usually degraded or dephosphorylated intracellularly, and the level in the body is tightly regulated (300). S1P binds to five different receptors (S1PR1-S1PR5) expressed by both innate and adaptive immune cells. The main outcome of S1P binding to these receptors seems to be directed migration. In patients with asthma, the S1P concentration is increased in BAL after allergen challenge (301). In the same study, stimulation of human airway smooth muscle cells by the S1P-activated signaling pathways was involved in contraction, proliferation and stimulated IL-6 release. In addition, S1P induced contraction of human airway smooth muscle cells embedded in collagen matrices (302).
In mice, an inhibitor of Sph kinases given before each OVA challenge in a model of allergic airway inflammation reduced inflammatory BAL cells and type 2 cytokines, AHR and mucus production (303). In isolated bronchi from mice, S1P enhanced acetylcholine-induced contraction (304). Further, S1P induced contraction of OVA-sensitized lungs but not in naïve lungs as demonstrated both in isolated bronchi and in vivo measured by whole-body plethysmography (304). In a follow-up study, Roviezzo et al. demonstrated that subcutaneous administration of S1P day 0 and day 7 dose-dependently increased bronchial responsiveness in vivo and ex vivo (305). The effect was time-dependent, with the greatest effect found 21 days after S1P administration. At this time point the number of mast cells quantified in BAL was twice that found in the vehicle controls. Using the same protocol of S1P-induced asthma-like inflammation, the same authors recently demonstrated that while LPS potentiated S1P-induced AHR, TLR4-defective mice (C3H/HeJ) or BALB/c mice pre-treated with an TLR4 blocking antibody were protected from S1P-induced AHR (306). Moreover, S1P induced higher expression of TLR4 in the proximity of the bronchi, and immunoprecipitation revealed an increased association between S1PR1 and TLR4. Together, this study suggests a functional interaction between S1PR1 and TLR4 that amplifies allergic airway inflammation and airway reactivity.
Mast cells were implicated in S1P-mediated responses in vivo in a model of IgE-mediated systemic anaphylaxis. In this study, pre-treatment with a S1P neutralizing antibody delayed and reduced peri-vascular lung inflammation along with BAL tryptase and histamine serum levels in mice injected intraperitoneally with dinitrophenyl (DNP)-specific IgE 12 h before injection of DNP coupled to human serum albumin (307). In the same model, mast cell deficient mice (KitW−sh/W−sh) or mice pre-treated with an S1PR2 antagonist or an anti-S1P antibody had reduced early peri-vascular lung inflammation. This suggests that mast cells providing and/or responding to S1P via S1PR2 are responsible for early peri-vascular lung inflammation observed in the model of IgE-mediated systemic anaphylaxis. In humans, an inhibitor of all S1PRs except S1PR2 (FTY720, fingolimod) is approved as a drug to treat multiple sclerosis (308). This drug was tested on moderate asthmatics in a double-blind, placebo-controlled 10-day study (309). Patients that received high doses of fingolimod showed only mild reductions in some lung function parameters and needed more short acting beta agonists, perhaps suggesting that although S1PR2 may be a target for asthma, the other S1P-receptors should not be targeted.
Cytokines
Cytokines play a crucial role in coordinating and maintaining allergic inflammation in the asthmatic lung. For a recent update on the general importance of cytokines in asthma, please see (310). Mast cells have the ability to secrete a broad spectrum of cytokines e.g. IL-1β, IL-2,-3,-4,-5,-6,-9,-10,-11,-12,-13, TNF-α, IFN-γ, GM-CSF, SCF, and TGF-β, as recently reviewed (311). In asthma patients, mast cells have been demonstrated to produce and release IL-4 (312). Interesting to note is that mouse MCp also have the ability to produce IL-4 (313). Possibly, this Th2-skewing cytokine derived from mast cells may promote or support early Th2-differentiation, although the contribution of mast cell-derived IL-4 in asthmatic lungs is unknown. Tryptase+ mast cells producing IL-4, as well as IL-5, IL-6 and TNF, were found in the bronchial mucosa from both healthy and asthmatic subjects (314). Moreover, isolated human mast cells produce IL-13 upon IgE-crosslinking (315), and IL-13 and IL-4 expressing mast cells are found within the airway smooth muscle of asthma patients (316). In mice, a role for IL-13, FcεRI and mast cells in the development of AHR was demonstrated in a model of OVA-induced allergic airway inflammation (77). Human lung mast cells also produce TNF-α (317). As already described, mast cell-derived TNF-α has been suggested to contribute to the development of allergic airway inflammation, Th2/Th17 cytokine production and AHR in mice (70, 73).
IL-9 is a pleiotropic cytokine, produced by many types of immunes cells including mast cells (318). Genetic studies have revealed that the IL-9 receptor is associated with asthma susceptibility (319, 320), and elevated IL-9 mRNA and IL-9 immunoreactive cells are found in lungs from asthmatics compared to patients with chronic bronchitis, sarcoidosis and healthy controls (321). In mouse models, lung selective IL-9 overexpression leads to mast cell accumulation, along with massive airway inflammation and AHR in the absence of an antigen (322). Nevertheless, as reviewed in (323), there is some conflicting evidence for the significance of IL-9 in mouse models of asthma, indicating that an alternative pathway exists. In the context of allergic asthma, mast cells responding to IL-9 signals are likely more important than mast cells as a source of this cytokine, as discussed earlier and demonstrated in different experimental set-ups of allergic lung inflammation in mice. In principle, IL-9 seems to be required for antigen-induced accumulation of lung mast cells in mice. Interestingly, a humanized anti-IL-9 monoclonal antibody (MEDI-528) has been tested on 36 mild asthmatics (injected subcutaneously) (324). The results demonstrated a trend toward a protective effect of MEDI-528 on lung function (324). However, in a randomized, controlled trial of 329 patients in which MEDI-528 (or placebo) was added to their usual asthma treatment over 24 weeks, no improvements in quality of life, asthma exacerbation rates or FEV1 values (pre-bronchodilator) were found (325). Thus, the future of targeting IL-9 in asthma is unclear although it is still possible that this pathway may be a valid target for a subgroup of asthma patients. To summarize, mast cells can produce many different cytokines that have been demonstrated to play significant roles in allergic asthma. However, whether or not the mast cell-produced cytokines play a major critical role in the human disease is currently unclear.
Conclusion
To conclude, asthma is a heterogenous disease highlighted by the differences in, e.g. time of on-set, severity, inflammatory pattern and responsiveness to corticosteroid-treatment. Similarly, we imagine that asthma patients differ in whether or not mast cells play a major or minor role in the pathogenesis. Given the plentitude of mediators that mast cells secrete, the limited beneficial effect of histamine receptor antagonists it is not surprising. It will likely be impossible to efficiently treat allergic asthmatics by targeting another single mast cell mediator. Still, the success of Omalizumab as a treatment for a subgroup of allergic asthmatics with severe or persistent symptoms indicates an IgE-mediated mast cell-involvement in their disease (104–106). Thus, finding out what other triggers of mast cell activation that may cause symptoms in different sub-groups of allergic asthmatics would be a better strategy for the development of novel drugs targeting mast cells.
We find that recent advances suggest that resident CTMCs are mainly self-sustained and of fetal origin. In the lung of patients with allergic asthma, mast cells accumulate in the smooth muscles, bronchial epithelium and alveolar parenchyma. The unique location of these lung mast cells can be replicated using mouse models of allergic asthma, and in these models the accumulation of mast cells is preceded by the recruitment of MCps to the lung. Although studies using mast cell-deficient mice subjected to allergic airway inflammation protocols have defined many new possible mechanisms that may occur in the human disease, these mechanisms need to be re-evaluated using the new Kit-independent mast cell-deficient strains in an attempt to clarify the role of mast cells in allergic airway inflammation.
Author Contributions
EM-E and JH conceptualized, wrote, and edited the manuscript. EM-E designed the study. JH edited the figures.
Funding
Our research is funded by grants to JH from the Swedish Research Council, the Swedish Heart-Lung foundation, Malin and Lennart Philipson Foundation, Ruth and Nils-Erik Stenbäck Foundation, Gustav V's 80-year Foundation, Konsul ThC Bergh's Foundation and the Knut and Alice Wallenberg Foundation, and to EM-E from Consejo Nacional de Ciencia y Tecnología (CONACyT) México.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The figures were created with images adapted from Servier Medical Art (smart.servier.com/), which is licensed under a Creative Commons Attribution 3.0 Unported License. This review is dedicated to the memory of Mike Gurish. He was a truly inspiring scientist and a warm and genuine person.
References
1. Rodewald HR, Dessing M, Dvorak AM, Galli SJ. Identification of a committed precursor for the mast cell lineage. Science. (1996) 271:818–22. doi: 10.1126/science.271.5250.818
2. Sonoda T, Hayashi C, Kitamura Y. Presence of mast cell precursors in the yolk sac of mice. Dev Biol. (1983) 97:89. doi: 10.1016/0012-1606(83)90066-0
3. Gentek R, Ghigo C, Hoeffel G, Bulle MJ, Msallam R, Gautier G, et al. Hemogenic endothelial fate mapping reveals dual developmental origin of mast cells. Immunity. (2018) 48:1160–71.e5. doi: 10.1016/j.immuni.2018.04.025
4. Li Z, Liu S, Xu J, Zhang X, Han D, Liu J, et al. Adult connective tissue-resident mast cells originate from late erythro-myeloid progenitors. Immunity. (2018) 49:640–53 e5. doi: 10.1016/j.immuni.2018.09.023
5. Jamur MC, Grodzki AC, Berenstein EH, Hamawy MM, Siraganian RP, Oliver C. Identification and characterization of undifferentiated mast cells in mouse bone marrow. Blood. (2005) 105:4282–9. doi: 10.1182/blood-2004-02-0756
6. Chen CC, Grimbaldeston MA, Tsai M, Weissman IL, Galli SJ. Identification of mast cell progenitors in adult mice. Proc Natl Acad Sci USA. (2005) 102:11408–13. doi: 10.1073/pnas.0504197102
7. Arinobu Y, Iwasaki H, Gurish MF, Mizuno S, Shigematsu H, Ozawa H, et al. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc Natl Acad Sci USA. (2005) 102:18105–10. doi: 10.1073/pnas.0509148102
8. Qi X, Hong J, Chaves L, Zhuang Y, Chen Y, Wang D, et al. Antagonistic regulation by the transcription factors C/EBPalpha and MITF specifies basophil and mast cell fates. Immunity. (2013) 39:97–110. doi: 10.1016/j.immuni.2013.06.012
9. Dahlin JS, Hamey FK, Pijuan-Sala B, Shepherd M, Lau WWY, Nestorowa S, et al. A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood. (2018) 131:e1-e11. doi: 10.1182/blood-2017-12-821413
10. Drissen R, Buza-Vidas N, Woll P, Thongjuea S, Gambardella A, Giustacchini A, et al. Distinct myeloid progenitor-differentiation pathways identified through single-cell RNA sequencing. Nat Immunol. (2016) 17:666–76. doi: 10.1038/ni.3412
11. Han J, Koh YJ, Moon HR, Ryoo HG, Cho CH, Kim I, et al. Adipose tissue is an extramedullary reservoir for functional hematopoietic stem and progenitor cells. Blood. (2010) 115:957–64. doi: 10.1182/blood-2009-05-219923
12. Poglio SF, De Toni-Costes Arnaud E, Laharrague P, Espinosa E, Casteilla L, Cousin B. Adipose tissue as a dedicated reservoir of functional mast cell progenitors. Stem Cells. (2010) 28:2065–72. doi: 10.1002/stem.523
13. Ngkelo A, Richart A, Kirk JA, Bonnin P, Vilar J, Lemitre M, et al. Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med. (2016) 213:1353–74. doi: 10.1084/jem.20160081
14. Dahlin JS, Heyman B, Hallgren J. Committed mast cell progenitors in mouse blood differ in maturity between Th1 and Th2 strains. Allergy. (2013) 68:1333–7. doi: 10.1111/all.12223
15. Dahlin JS, Ding Z, Hallgren J. Distinguishing mast cell progenitors from mature mast cells in Mice. Stem Cells Dev. (2015) 24:1703–11. doi: 10.1089/scd.2014.0553
16. Crapper RM, Schrader JW. Frequency of mast cell precursors in normal tissues determined by an in vitro assay: antigen induces parallel increases in the frequency of P cell precursors and mast cells. J Immunol. (1983) 131:923–8.
17. Gurish MF, Tao H, Abonia JP, Arya A, Friend DS, Parker CM, et al. Intestinal mast cell progenitors require CD49dbeta7 (alpha4beta7 integrin) for tissue-specific homing. J Exp Med. (2001) 194:1243–52. doi: 10.1084/jem.194.9.1243
18. Tsai M, Shih LS, Newlands GF, Takeishi T, Langley KE, Zsebo KM, et al. The rat c-kit ligand, stem cell factor, induces the development of connective tissue-type and mucosal mast cells in vivo. Analysis by anatomical distribution, histochemistry, and protease phenotype. J Exp Med. (1991) 174:125. doi: 10.1084/jem.174.1.125
19. Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. (1978) 52:447.
20. Grimbaldeston MA, Chen CC, Piliponsky AM, Tsai M, Tam SY, Galli SJ. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am J Pathol. (2005) 167:835–48. doi: 10.1016/S0002-9440(10)62055-X
21. Schrader JW. In in vitro production and cloning of the P cell, a bone marrow-derived null cell that expresses H−2 and Ia-antigens, has mast cell-like granules, and is regulated by a factor released by activated T cells. J Immunol. (1981) 126:452–8.
22. Ihle JN, Keller J, Oroszlan S, Henderson LE, Copeland TD, Fitch F, et al. Biologic properties of homogeneous interleukin 3. I. Demonstration of WEHI−3 growth factor activity, mast cell growth factor activity, p cell-stimulating factor activity, colony-stimulating factor activity, and histamine-producing cell-stimulating factor activity. J Immunol. (1983) 131:282–7.
23. Dahlin JS, Malinovschi A, Ohrvik H, Sandelin M, Janson C, Alving K, et al. Lin- CD34hi CD117int/hi FcepsilonRI+ cells in human blood constitute a rare population of mast cell progenitors. Blood. (2016) 127:383–91. doi: 10.1182/blood-2015-06-650648
24. Dahlin JS, Ekoff M, Grootens J, Lof L, Amini RM, Hagberg H, et al. KIT signaling is dispensable for human mast cell progenitor development. Blood. (2017) 130:1785–94. doi: 10.1182/blood-2017-03-773374
25. Ribatti D. The development of human mast cells. An historical reappraisal. Exp Cell Res. (2016) 342:210–5. doi: 10.1016/j.yexcr.2016.03.013
26. Enerback L. Mast cells in rat gastrointestinal mucosa. 2. Dye-binding and metachromatic properties. Acta Pathol Microbiol Scand. (1966) 66:303. doi: 10.1111/apm.1966.66.3.303
27. Ronnberg E, Melo FR, Pejler G. Mast cell proteoglycans. J Histochem Cytochem. (2012) 60:950–62. doi: 10.1369/0022155412458927
28. Reynolds DS, Stevens RL, Lane WS, Carr MH, Austen KF, Serafin WE. Different mouse mast cell populations express various combinations of at least six distinct mast cell serine proteases. Proc Natl Acad Sci USA. (1990) 87:3230–4. doi: 10.1073/pnas.87.8.3230
29. Xing W, Austen KF, Gurish MF, Jones TG. Protease phenotype of constitutive connective tissue and of induced mucosal mast cells in mice is regulated by the tissue. Proc Natl Acad Sci USA. (2011) 108:14210–5. doi: 10.1073/pnas.1111048108
30. Fukuzumi T, Waki N, Kanakura Y, Nagoshi J, Hirota S, Yoshikawa K, et al. Differences in irradiation susceptibility and turnover between mucosal and connective tissue-type mast cells of mice. Exp Hematol. (1990) 18:843–7.
31. Stevens RL, Fox CC, Lichtenstein LM, Austen KF. Identification of chondroitin sulfate E proteoglycans and heparin proteoglycans in the secretory granules of human lung mast cells. Proc Natl Acad Sci USA. (1988) 85:2284–7. doi: 10.1073/pnas.85.7.2284
32. Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci USA. (1986) 83:4464–8. doi: 10.1073/pnas.83.12.4464
33. Schechter NM, Choi JK, Slavin DA, Deresienski DT, Sayama S, Dong G, et al. Identification of a chymotrypsin-like proteinase in human mast cells. J Immunol. (1986) 137:962–70.
34. Dwyer DF, Barrett NA, Austen KF. Immunological Genome ProjectExpression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat Immunol. (2016) 17:878–87. doi: 10.1038/ni.3445
35. Andersson CK, Mori M, Bjermer L, Lofdahl CG, Erjefalt JS. Novel site-specific mast cell subpopulations in the human lung. Thorax. (2009) 64:297–305. doi: 10.1136/thx.2008.101683
36. Abonia JP, Hallgren J, Jones T, Shi T, Xu Y, Koni P, et al. Alpha−4 integrins and VCAM−1, but not MAdCAM−1, are essential for recruitment of mast cell progenitors to the inflamed lung. Blood. (2006) 108:1588–94. doi: 10.1182/blood-2005-12-012781
37. Bankova LG, Dwyer DF, Liu AY, Austen KF, Gurish MF. Maturation of mast cell progenitors to mucosal mast cells during allergic pulmonary inflammation in mice. Mucosal Immunol. (2015) 8:596–606. doi: 10.1038/mi.2014.91
38. Zarnegar B, Mendez-Enriquez E, Westin A, Soderberg C, Dahlin JS, Gronvik KO, et al. Influenza infection in mice induces accumulation of lung mast cells through the recruitment and maturation of mast cell progenitors. Front Immunol. (2017) 8:310. doi: 10.3389/fimmu.2017.00310
39. Dougherty RH, Sidhu SS, Raman K, Solon M, Solberg OD, Caughey GH, et al. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J Allergy Clin Immunol. (2010) 125:1046–53 e8. doi: 10.1016/j.jaci.2010.03.003
40. Woodruff PG, Boushey HA, Dolganov GM, Barker CS, Yang YH, Donnelly S, et al. Genome-wide profiling identifies epithelial cell genes associated with asthma and with treatment response to corticosteroids. Proc Natl Acad Sci USA. (2007) 104:15858–63. doi: 10.1073/pnas.0707413104
41. Singhania A, Rupani H, Jayasekera N, Lumb S, Hales P, Gozzard N, et al. Altered epithelial gene expression in peripheral airways of severe asthma. PLoS ONE. (2017) 12:e0168680. doi: 10.1371/journal.pone.0168680
42. Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. (2002) 346:1699–705. doi: 10.1056/NEJMoa012705
43. Amin K, Janson C, Boman G, Venge P. The extracellular deposition of mast cell products is increased in hypertrophic airways smooth muscles in allergic asthma but not in nonallergic asthma. Allergy. (2005) 60:1241–7. doi: 10.1111/j.1398-9995.2005.00823.x
44. Carroll NG, Mutavdzic S, James AL. Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur Respir J. (2002) 19:879. doi: 10.1183/09031936.02.00275802
45. Andersson CK, Tufvesson E, Aronsson D, Bergqvist A, Mori M, Bjermer L, et al. Alveolar mast cells shift to an FcepsilonRI-expressing phenotype in mild atopic asthma: a novel feature in allergic asthma pathology. Allergy. (2011) 66:1590–7. doi: 10.1111/j.1398-9995.2011.02723.x
46. Ammit AJ, Bekir SS, Johnson PR, Hughes JM, Armour CL, Black JL. Mast cell numbers are increased in the smooth muscle of human sensitized isolated bronchi. Am J Respir Crit Care Med. (1997) 155:1123–9. doi: 10.1164/ajrccm.155.3.9116997
47. Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax. (2002) 57:677–82. doi: 10.1136/thorax.57.8.677
48. Balzar S, Chu HW, Strand M, Wenzel S. Relationship of small airway chymase-positive mast cells and lung function in severe asthma. Am J Respir Crit Care Med. (2005) 171:431–9. doi: 10.1164/rccm.200407-949OC
49. Wang G, Baines KJ, Fu JJ, Wood LG, Simpson JL, McDonald VM, et al. Sputum mast cell subtypes relate to eosinophilia and corticosteroid response in asthma. Eur Respir J. (2016) 47:1123–33. doi: 10.1183/13993003.01098-2015
50. Balzar S, Fajt ML, Comhair SA, Erzurum SC, Bleecker E, Busse WW, et al. Mast cell phenotype, location, and activation in severe asthma. . Am J Respir Crit Care Med. (2011) 183:299–309. doi: 10.1164/rccm.201002-0295OC
51. Andersson CK, Bergqvist A, Mori M, Mauad T, Bjermer L, Erjefalt JS. Mast cell-associated alveolar inflammation in patients with atopic uncontrolled asthma. J Allergy Clin Immunol. (2011) 127:905–12 e1–7. doi: 10.1016/j.jaci.2011.01.022
52. Lezmi G, Galmiche-Rolland L, Rioux S, Jaubert F, Tillie-Leblond I, Scheinmann P, et al. Mast cells are associated with exacerbations and eosinophilia in children with severe asthma. Eur Respir J. (2016) 48:1320–8. doi: 10.1183/13993003.00947-2016
53. Hallgren J, Jones TG, Abonia JP, Xing W, Humbles A, Austen KF, et al. Pulmonary CXCR2 regulates VCAM−1 and antigen-induced recruitment of mast cell progenitors. Proc Natl Acad Sci USA. (2007) 104:20478–83. doi: 10.1073/pnas.0709651104
54. Dahlin JS, Feinstein R, Cui Y, Heyman B, Hallgren J. CD11c+ cells are required for antigen-induced increase of mast cells in the lung. J Immunol. (2012) 189:3869–77. doi: 10.4049/jimmunol.1201200
55. Jones TG, Hallgren J, Humbles A, Burwell T, Finkelman FD, Alcaide P, et al. Antigen-induced increases in pulmonary mast cell progenitor numbers depend on IL−9 and CD1d-restricted NKT cells. J Immunol. (2009) 183:5251–60. doi: 10.4049/jimmunol.0901471
56. Collington SJ, Hallgren J, Pease JE, Jones TG, Rollins BJ, Westwick J, et al. The role of the CCL2/CCR2 axis in mouse mast cell migration in vitro and in vivo. J Immunol. (2010) 184:6114–23. doi: 10.4049/jimmunol.0904177
57. Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, et al. TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J Allergy Clin Immunol. (2015) 136:433–40e1. doi: 10.1016/j.jaci.2015.01.021
58. Mathias CB, Freyschmidt EJ, Caplan B, Jones T, Poddighe D, Xing W, et al. IgE influences the number and function of mature mast cells, but not progenitor recruitment in allergic pulmonary inflammation. J Immunol. (2009) 182:2416–24. doi: 10.4049/jimmunol.0801569
59. Dahlin JS, Ivarsson MA, Heyman B, Hallgren J. IgE immune complexes stimulate an increase in lung mast cell progenitors in a mouse model of allergic airway inflammation. PLoS ONE. (2011) 6:e20261. doi: 10.1371/journal.pone.0020261
60. Zarnegar B, Westin A, Evangelidou S, Hallgren J. Innate immunity induces the accumulation of lung mast cells during influenza infection. Front Immunol. (2018) 9:2288. doi: 10.3389/fimmu.2018.02288
61. Suarez CS. D'intzis, Frevert C. Respiratory. In Comparative Anatomy and Histology, Chapter 9. Cambridge, MA: Academic Press (2012), 121–34.
62. Lei Y, Gregory JA, Nilsson GP, Adner M. Insights into mast cell functions in asthma using mouse models. Pulm Pharmacol Ther. (2013) 26:532–9. doi: 10.1016/j.pupt.2013.03.019
63. Li S, Aliyeva M, Daphtary N, Martin RA, Poynter ME, Kostin SF, et al. Antigen-induced mast cell expansion and bronchoconstriction in a mouse model of asthma. Am J Physiol Lung Cell Mol Physiol. (2014) 306:L196–206. doi: 10.1152/ajplung.00055.2013
64. Weigand LA, Myers AC, Meeker S, Undem BJ. Mast cell-cholinergic nerve interaction in mouse airways. J Physiol. (2009) 587(Pt 13):3355–62. doi: 10.1113/jphysiol.2009.173054
65. Mehlhop PD, van de Rijn M, Goldberg AB, Brewer JP, Kurup VP, Martin TR, et al. Allergen-induced bronchial hyperreactivity and eosinophilic inflammation occur in the absence of IgE in a mouse model of asthma. Proc Natl Acad Sci USA. (1997) 94:1344–9. doi: 10.1073/pnas.94.4.1344
66. Nogami M, Suko M, Okudaira H, Miyamoto T, Shiga J, Ito M, et al. Experimental pulmonary eosinophilia in mice by Ascaris suum extract. Am Rev Respir Dis. (1990) 141(5 Pt 1):1289–95. doi: 10.1164/ajrccm/141.5_Pt_1.1289
67. Takeda K, Hamelmann E, Joetham A, Shultz LD, Larsen GL, Irvin CG, et al. Development of eosinophilic airway inflammation and airway hyperresponsiveness in mast cell-deficient mice. J Exp Med. (1997) 186:449–54. doi: 10.1084/jem.186.3.449
68. Williams CM, Galli SJ. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J Exp Med. (2000) 192:455–62. doi: 10.1084/jem.192.3.455
69. Yu M, Tsai M, Tam SY, Jones C, Zehnder J, Galli SJ. Mast cells can promote the development of multiple features of chronic asthma in mice. J Clin Invest. (2006) 116:1633–41. doi: 10.1172/JCI25702
70. Reuter S, Heinz A, Sieren M, Wiewrodt R, Gelfand EW, Stassen M, et al. Mast cell-derived tumour necrosis factor is essential for allergic airway disease. Eur Respir J. (2008) 31:773–82. doi: 10.1183/09031936.00058907
71. Cui Y, Dahlin JS, Feinstein R, Bankova LG, Xing W, Shin K, et al. Mouse mast cell protease−6 and MHC are involved in the development of experimental asthma. J Immunol. (2014) 193:4783–9. doi: 10.4049/jimmunol.1302947
72. Fuchs B, Sjoberg L, Moller Westerberg C, Ekoff M, Swedin L, Dahlen SE, et al. Mast cell engraftment of the peripheral lung enhances airway hyperresponsiveness in a mouse asthma model. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L1027–36. doi: 10.1152/ajplung.00227.2012
73. Nakae S, Ho LH, Yu M, Monteforte R, Iikura M, Suto H, et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J Allergy Clin Immunol. (2007) 120:48–55. doi: 10.1016/j.jaci.2007.02.046
74. de Boer JD, Yang J, van den Boogaard FE, Hoogendijk AJ, de Beer R, van der Zee JS, et al. Mast cell-deficient kit mice develop house dust mite-induced lung inflammation despite impaired eosinophil recruitment. J Innate Immun. (2014) 6:219–26. doi: 10.1159/000354984
75. Yu CK, Chen CL. Activation of mast cells is essential for development of house dust mite Dermatophagoides farinae-induced allergic airway inflammation in mice. J Immunol. (2003) 171:3808. doi: 10.4049/jimmunol.171.7.3808
76. Mayr SI, Zuberi RI, Zhang M, de Sousa-Hitzler J, Ngo K, Kuwabara Y, et al. IgE-dependent mast cell activation potentiates airway responses in murine asthma models. J Immunol. (2002) 169:2061–8. doi: 10.4049/jimmunol.169.4.2061
77. Taube C, Wei X, Swasey CH, Joetham A, Zarini S, Lively T, et al. Mast cells, Fc epsilon RI, and IL−13 are required for development of airway hyperresponsiveness after aerosolized allergen exposure in the absence of adjuvant. J Immunol. (2004) 172:6398–406. doi: 10.4049/jimmunol.172.10.6398
78. Martin TR, Takeishi T, Katz HR, Austen KF, Drazen JM, Galli SJ. Mast cell activation enhances airway responsiveness to methacholine in the mouse. J Clin Invest. (1993) 91:1176–82. doi: 10.1172/JCI116277
79. Rodewald HR, Thorsten Feyerabend B. Widespread immunological functions of mast cells: fact or fiction? Immunity. (2012) 37:13–24. doi: 10.1016/j.immuni.2012.07.007
80. Reber LL, Marichal T, Galli SJ. New models for analyzing mast cell functions in vivo. Trends Immunol. (2012) 33:613–25. doi: 10.1016/j.it.2012.09.008
81. Sawaguchi M, Tanaka S, Nakatani Y, Harada Y, Mukai K, Matsunaga Y, et al. Role of mast cells and basophils in IgE responses and in allergic airway hyperresponsiveness. J Immunol. (2012) 188:1809–18. doi: 10.4049/jimmunol.1101746
82. Feyerabend TB, Weiser A, Tietz A, Stassen M, Harris N, Kopf M, et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity. (2011) 35:832–44. doi: 10.1016/j.immuni.2011.09.015
83. Cahill KN, Katz HR, Cui J, Lai J, Kazani S, Crosby-Thompson A, et al. KIT Inhibition by imatinib in patients with severe refractory asthma. N Engl J Med. (2017) 376:1911–20. doi: 10.1056/NEJMoa1613125
84. Bieber T, de la Salle H, Wollenberg A, Hakimi J, Chizzonite R, Ring J, et al. Human epidermal Langerhans cells express the high affinity receptor for immunoglobulin E (Fc epsilon RI). J Exp Med. (1992) 175:1285–90. doi: 10.1084/jem.175.5.1285
85. Maurer D, Fiebiger S, Ebner C, Reininger B, Fischer GF, Wichlas S, et al. Peripheral blood dendritic cells express Fc epsilon RI as a complex composed of Fc epsilon RI alpha- and Fc epsilon RI gamma-chains and can use this receptor for IgE-mediated allergen presentation. J Immunol. (1996) 157:607–616.
86. Joseph M, Gounni AS, Kusnierz JP, Vorng H, Sarfati M, Kinet JP, et al. Expression and functions of the high-affinity IgE receptor on human platelets and megakaryocyte precursors. Eur J Immunol. (1997) 27:2212–8. doi: 10.1002/eji.1830270914
87. Gounni AS, Lamkhioued B, Koussih L, Ra C, Renzi PM, Hamid Q. Human neutrophils express the high-affinity receptor for immunoglobulin E (Fc epsilon RI): role in asthma. FASEB J. (2001) 15:940–9. doi: 10.1096/fj.00-0378com
88. Maurer D, Fiebiger E, Reininger B, Wolff-Winiski B, Jouvin MH, Kilgus O, et al. Expression of functional high affinity immunoglobulin E receptors (Fc epsilon RI) on monocytes of atopic individuals. J Exp Med. (1994) 179:745–750. doi: 10.1084/jem.179.2.745
89. Rajakulasingam K, Till S, Ying S, Humbert M, Barkans J, Sullivan M, et al. Increased expression of high affinity IgE (FcepsilonRI) receptor-alpha chain mRNA and protein-bearing eosinophils in human allergen-induced atopic asthma. Am J Respir Crit Care Med. (1998) 158:233–40. doi: 10.1164/ajrccm.158.1.9708106
90. Gounni AS, Wellemans V, Yang J, Bellesort F, Kassiri K, Gangloff S, et al. Human airway smooth muscle cells express the high affinity receptor for IgE (FcεRI): a critical role of fcεri in human airway smooth muscle cell function. J Immunol. (2005) 175:2613–2621. doi: 10.4049/jimmunol.175.4.2613
91. Greer AM, Wu N, Putnam AL, Woodruff PG, Wolters P, Kinet JP, et al. Serum IgE clearance is facilitated by human FcεRI internalization. J Clin Invest. (2014) 124:1187–1198. doi: 10.1172/JCI68964
92. van der Kleij H, Charles N, Karimi K, Mao YK, Foster J, Janssen L, et al. Evidence for neuronal expression of functional Fc (ε and γ) receptors. J Allergy Clin Immunol. (2010) 125:757–760. doi: 10.1016/j.jaci.2009.10.054
93. Andoh T, Kuraishi Y. Expression of Fc epsilon receptor I on primary sensory neurons in mice. Neurorep. (2004) 15:2029–31. doi: 10.1097/00001756-200409150-00007
94. Ra C, Jouvin MH, Kinet JP. Complete structure of the mouse mast cell receptor for IgE (Fc epsilon RI) and surface expression of chimeric receptors (rat-mouse-human) on transfected cells. J Biol Chem. (1989) 264:15323–7.
95. Blank U, Ra C, Miller L, White K, Metzger H, Kinet JP. Complete structure and expression in transfected cells of high affinity IgE receptor. Nature. (1989) 337:187–9. doi: 10.1038/337187a0
96. Grayson MH, Cheung D, Rohlfing MM, Kitchens R, Spiegel DE, Tucker J, et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J Exp Med. (2007) 204:2759–69. doi: 10.1084/jem.20070360
97. Siraganian RP. Mast cell signal transduction from the high-affinity IgE receptor. Curr Opin Immunol. (2003) 15:639–46. doi: 10.1016/j.coi.2003.09.010
98. Galli SJ. The mast cell-IgE paradox: from homeostasis to anaphylaxis. Am J Pathol. (2016) 186:212–24. doi: 10.1016/j.ajpath.2015.07.025
99. Bradding P, Arthur G. Mast cells in asthma–state of the art. Clin Exp Allergy. (2016) 46:194–263. doi: 10.1111/cea.12675
100. Boyce JA. Mast cells and eicosanoid mediators: a system of reciprocal paracrine and autocrine regulation. Immunol Rev. (2007) 217:168–85. doi: 10.1111/j.1600-065X.2007.00512.x
101. Rivera J, Fierro NA, Olivera A, Suzuki R. New insights on mast cell activation via the high affinity receptor for IgE. Adv Immunol. (2008) 98:85–120. doi: 10.1016/S0065-2776(08)00403-3
102. Burrows B, Martinez FD, Halonen M, Barbee RA, Cline MG. Association of asthma with serum IgE levels and skin-test reactivity to allergens. N Engl J Med. (1989) 320:271–7. doi: 10.1056/NEJM198902023200502
103. Sears MR, Burrows B, Flannery EM, Herbison GP, Hewitt CJ, Holdaway MD. Relation between airway responsiveness and serum IgE in children with asthma and in apparently normal children. N Engl J Med. (1991) 325:1067–71. doi: 10.1056/NEJM199110103251504
104. Busse W, Corren J, Lanier BQ, McAlary M, Fowler-Taylor A, Cioppa GD, et al. Omalizumab, anti-IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol. (2001) 108:184. doi: 10.1067/mai.2001.117880
105. Tabatabaian F, Ledford DK. Omalizumab for severe asthma: toward personalized treatment based on biomarker profile and clinical history. J Asthma Allergy. (2018) 11:53–61. doi: 10.2147/JAA.S107982
106. Holgate S, Smith N, Massanari M, Jimenez P. Effects of omalizumab on markers of inflammation in patients with allergic asthma. Allergy. (2009) 64:1728–36. doi: 10.1111/j.1398-9995.2009.02201.x
107. Kalesnikoff J, Huber M, Lam V, Damen JE, Zhang J, Siraganian RP, et al. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity. (2001) 14:801–11. doi: 10.1016/S1074-7613(01)00159-5
108. Kitaura J, Song J, Tsai M, Asai K, Maeda-Yamamoto M, Mocsai A, et al. Evidence that IgE molecules mediate a spectrum of effects on mast cell survival and activation via aggregation of the FcepsilonRI. Proc Natl Acad Sci USA. (2003) 100:12911–6. doi: 10.1073/pnas.1735525100
109. Bax HJ, Keeble AH, Gould HJ. Cytokinergic IgE action in mast cell activation. Front Immunol. (2012) 3:229. doi: 10.3389/fimmu.2012.00229
110. Matsuda K, Piliponsky AM, Iikura M, Nakae S, Wang EW, Dutta SM, et al. Monomeric IgE enhances human mast cell chemokine production: IL−4 augments and dexamethasone suppresses the response. J Allergy Clin Immunol. (2005) 116:1357–63. doi: 10.1016/j.jaci.2005.08.042
111. Wang H, Wang HS, Liu ZP. Agents that induce pseudo-allergic reaction. Drug Discov Ther. (2011) 5:211–9. doi: 10.5582/ddt.2011.v5.5.211
112. McNeil BD, Pundir P, Meeker S, Han L, Undem BJ, Kulka M, et al. Identification of a mast cell specific receptor crucial for pseudo-allergic drug reactions. Nature. (2015) 519:237–41. doi: 10.1038/nature14022
113. Fujisawa D, Kashiwakura J, Kita H, Kikukawa Y, Fujitani Y, Sasaki-Sakamoto T, et al. Expression of Mas-related gene X2 on mast cells is upregulated in the skin of patients with severe chronic urticaria. J Allergy Clin Immunol. (2014) 134:622–33 e9. doi: 10.1016/j.jaci.2014.05.004
114. Kajiwara N, Sasaki T, Bradding P, Cruse G, Sagara H, Ohmori K, et al. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunol. (2010) 125:1137–45.e6. doi: 10.1016/j.jaci.2010.01.056
115. Manorak W, Idahosa C, Gupta K, Roy S, Panettieri R, Ali H. Upregulation of mas-related G Protein coupled receptor X2 in asthmatic lung mast cells and its activation by the novel neuropeptide hemokinin−1. Respir Res. (2018) 19:1. doi: 10.1186/s12931-017-0698-3
116. Nieber K, Baumgarten CR, Rathsack R, Furkert J, Oehme P, Kunkel G. Substance P and beta-endorphin-like immunoreactivity in lavage fluids of subjects with and without allergic asthma. J Allergy Clin Immunol. (1992) 90(4 Pt 1):646–52. doi: 10.1016/0091-6749(92)90138-R
117. Tomaki M, Ichinose M, Miura M, Hirayama Y, Yamauchi H, Nakajima N, et al. Elevated substance P content in induced sputum from patients with asthma and patients with chronic bronchitis. Am J Respir Crit Care Med. (1995) 151(3_pt_1):613–7. doi: 10.1164/ajrccm/151.3_Pt_1.613
118. Subramanian H, Gupta K, Ali H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J Allergy Clin Immunol. (2016) 138:700–10. doi: 10.1016/j.jaci.2016.04.051
119. Duits LA, Nibbering PH, van Strijen E, Vos JB, Mannesse-Lazeroms SP, van Sterkenburg MA, et al. Rhinovirus increases human beta-defensin−2 and −3 mRNA expression in cultured bronchial epithelial cells. FEMS Immunol Med Microbiol. (2003) 38:59–64. doi: 10.1016/S0928-8244(03)00106-8
120. Proud D, Sanders SP, Wiehler S. Human rhinovirus infection induces airway epithelial cell production of human beta-defensin 2 both in vitro and in vivo. J Immunol. (2004) 172:4637–45. doi: 10.4049/jimmunol.172.7.4637
121. Subramanian H, Gupta K, Lee D, Bayir AK, Ahn H, Ali H. beta-Defensins activate human mast cells via Mas-related gene X2. J Immunol. (2013) 191:345–52. doi: 10.4049/jimmunol.1300023
122. Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. (2010) 10:440–52. doi: 10.1038/nri2782
123. Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. (2009) 457:585–8. doi: 10.1038/nature07548
124. Saluja R, Delin I, Nilsson GP, Adner M. FcepsilonR1-mediated mast cell reactivity is amplified through prolonged Toll-like receptor-ligand treatment. PLoS ONE. (2012) 7:e43547. doi: 10.1371/journal.pone.0043547
125. Yoshioka M, Fukuishi N, Iriguchi S, Ohsaki K, Yamanobe H, Inukai A, et al. Lipoteichoic acid downregulates FcepsilonRI expression on human mast cells through Toll-like receptor 2. J Allergy Clin Immunol. (2007) 120:452–61. doi: 10.1016/j.jaci.2007.03.027
126. Okumura S, Kashiwakura J, Tomita H, Matsumoto K, Nakajima T, Saito H, et al. Identification of specific gene expression profiles in human mast cells mediated by Toll-like receptor 4, FcepsilonRI. Blood. (2003) 102:2547–54. doi: 10.1182/blood-2002-12-3929
127. Nigo YI, Yamashita M, Hirahara K, Shinnakasu R, Inami M, Kimura M, et al. Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc Natl Acad Sci USA. (2006) 103:2286–91. doi: 10.1073/pnas.0510685103
128. Al-Sajee D, Oliveria JP, Sehmi R, Gauvreau GM. Antialarmins for treatment of asthma: future perspectives. Curr Opin Pulm Med. (2018) 24:32–41. doi: 10.1097/MCP.0000000000000443
129. Lingel A, Weiss TM, Niebuhr M, Pan B, Appleton BA, Wiesmann C, et al. Structure of IL−33 and its interaction with the ST2 and IL−1RAcP receptors–insight into heterotrimeric IL−1 signaling complexes. Structure. (2009) 17:1398–410. doi: 10.1016/j.str.2009.08.009
130. Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. (2010) 363:1211–21. doi: 10.1056/NEJMoa0906312
131. Wang W, Li Y, Lv Z, Chen Y, Li Y, Huang K, et al. Bronchial allergen challenge of patients with atopic asthma triggers an alarmin (IL−33, TSLP, and IL−25) response in the airways epithelium and submucosa. J Immunol. (2018) 201:2221–31. doi: 10.4049/jimmunol.1800709
132. Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, et al. Increased Expression of IL−33 in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. (2009) 183:5094–5103. doi: 10.4049/jimmunol.0802387
133. Li Y, Wang W, Lv Z, Li Y, Chen Y, Huang K, et al. Elevated Expression of IL−33 and TSLP in the airways of human asthmatics in vivo: a potential biomarker of severe refractory disease. J Immunol. (2018) 200:2253–62. doi: 10.4049/jimmunol.1701455
134. Allakhverdi Z, Smith DE, Comeau MR, Delespesse G. Cutting edge: The ST2 ligand IL−33 potently activates and drives maturation of human mast cells. J Immunol. (2007) 179:2051–4. doi: 10.4049/jimmunol.179.4.2051
135. Joulia R, L'Faqihi FE, Valitutti S, Espinosa E. IL−33 fine tunes mast cell degranulation and chemokine production at the single-cell level. J Allergy Clin Immunol. (2017) 140:497–509 e10. doi: 10.1016/j.jaci.2016.09.049
136. Wang JX, Kaieda S, Ameri S, Fishgal N, Dwyer D, Dellinger A, et al. IL−33/ST2 axis promotes mast cell survival via BCLXL. Proc Natl Acad Sci USA. (2014) 111:10281–6. doi: 10.1073/pnas.1404182111
137. Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, et al. IL−33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. (2007) 87:971–8. doi: 10.1038/labinvest.3700663
138. Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med. (1999) 190:895–902. doi: 10.1084/jem.190.7.895
139. Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL−33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. (2008) 20:791–800. doi: 10.1093/intimm/dxn037
140. Ito R, Maruoka S, Soda K, Katano I, Kawai K, Yagoto M, et al. A humanized mouse model to study asthmatic airway inflammation via the human IL−33/IL−13 axis. JCI Insight. (2018) 3:121580. doi: 10.1172/jci.insight.121580
141. Sjöberg LC, Gregory JA, Dahlén SE, Nilsson GP, Adner M. Interleukin−33 exacerbates allergic bronchoconstriction in the mice via activation of mast cells. Allergy. (2015) 70:514–521. doi: 10.1111/all.12590
142. Morita H, Arae K, Unno H, Miyauchi K, Toyama S, Nambu A, et al. An interleukin−33-mast cell-interleukin−2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. (2015) 43:175–86. doi: 10.1016/j.immuni.2015.06.021
143. Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, et al. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. (2000) 1:59–64. doi: 10.1038/76923
144. Park LS, Martin U, Garka K, Gliniak B, Di Santo JP, Muller W, et al. Cloning of the murine thymic stromal lymphopoietin (TSLP) receptor: formation of a functional heteromeric complex requires interleukin 7 receptor. J Exp Med. (2000) 192:659–70. doi: 10.1084/jem.192.5.659
145. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. (2002) 3:673–80. doi: 10.1038/ni805
146. Ying S, O'Connor B, Ratoff J, Meng Q, Mallett K, Cousins D, et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J Immunol. (2005) 174:8183–8190. doi: 10.4049/jimmunol.174.12.8183
147. Skrgat S, Marc Malovrh M, Sarc I, Silar M, Dimitric V, Korosec P. TSLP as biomarker in asthma patients. Eur Respir J. (2015) 46(suppl. 59). doi: 10.1183/13993003.congress-2015.PA3868
148. Zhou B, Comeau MR, Smedt TD, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. (2005) 6:1047. doi: 10.1038/ni1247
149. Han NR, Oh HA, Nam SY, Moon PD, Kim DW, Kim HM, et al. TSLP induces mast cell development and aggravates allergic reactions through the activation of MDM2 and STAT6. J Invest Dermatol. (2014) 134:2521–30. doi: 10.1038/jid.2014.198
150. Kaur D, Doe C, Woodman L, Heidi Wan WY, Sutcliffe A, Hollins F, et al. Mast cell-airway smooth muscle crosstalk: the role of thymic stromal lymphopoietin. Chest. (2012) 142:76–85. doi: 10.1378/chest.11-1782
151. Okayama Y, Okumura S, Sagara H, Yuki K, Sasaki T, Watanabe N, et al. FcϵRI-mediated thymic stromal lymphopoietin production by IL−4-primed human mast cells. Eur Respir J. (2009) doi: 10.1183/09031936.00121008
152. Gauvreau GM, O'Byrne PM, Boulet LP, Wang Y, Cockcroft D, Bigler J, et al. Effects of an anti-TSLP antibody on allergen-induced asthmatic responses. N Engl J Med. (2014) 370:2102–10. doi: 10.1056/NEJMoa1402895
153. Corren J, Parnes JR, Wang L, Mo M, Roseti SL, Griffiths JM, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. (2017) 377:936–46. doi: 10.1056/NEJMoa1704064
154. Idzko M, Ferrari D, Eltzschig HK. Nucleotide signalling during inflammation. Nature. (2014) 509:310–7. doi: 10.1038/nature13085
155. Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta. (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
156. Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, Muskens F, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. (2007) 13:913. doi: 10.1038/nm1617
157. Dahlquist R, Diamant B. Interaction of ATP and calcium on the rat mast cell: effect on histamine release. Acta Pharmacol Toxicol (Copenh). (1974) 34:368–84. doi: 10.1111/j.1600-0773.1974.tb03533.x
158. Cockcroft S, Gomperts BD. The ATP4- receptor of rat mast cells. Biochem J. (1980) 188:789–98. doi: 10.1042/bj1880789
159. Kurashima Y, Amiya T, Nochi T, Fujisawa K, Haraguchi T, Iba H, et al. Extracellular ATP mediates mast cell-dependent intestinal inflammation through P2X7 purinoceptors. Nat Commun. (2012) 3:1034. doi: 10.1038/ncomms2023
160. Muller T, Vieira RP, Grimm M, Durk T, Cicko S, Zeiser R, et al. A potential role for P2X7R in allergic airway inflammation in mice and humans. Am J Respir Cell Mol Biol. (2011) 44:456–64. doi: 10.1165/rcmb.2010-0129OC
161. Tsai SH, Kinoshita M, Kusu T, Kayama H, Okumura R, Ikeda K, et al. The ectoenzyme E-NPP3 negatively regulates ATP-dependent chronic allergic responses by basophils and mast cells. Immunity. (2015) 42:279–293. doi: 10.1016/j.immuni.2015.01.015
162. Schulman ES, Glaum MC, Post T, Wang Y, Raible DG, Mohanty J, et al. ATP modulates anti-IgE-induced release of histamine from human lung mast cells. Am J Respir Cell Mol Biol. (1999) 20:530–7. doi: 10.1165/ajrcmb.20.3.3387
163. Bradding P, Okayama Y, Kambe N, Saito H. Ion channel gene expression in human lung, skin, and cord blood-derived mast cells. J Leukoc Biol. (2003) 73:614–20. doi: 10.1189/jlb.1202602
164. Wareham K, Vial C, Wykes RC, Bradding P, Seward EP. Functional evidence for the expression of P2X1, P2X4 and P2X7 receptors in human lung mast cells. Br J Pharmacol. (2009) 157:1215–24. doi: 10.1111/j.1476-5381.2009.00287.x
165. Manthei DM, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Gern JE, et al. Protection from asthma in a high-risk birth cohort by attenuated P2X(7) function. J Allergy Clin Immunol. (2012) 130:496–502. doi: 10.1016/j.jaci.2012.05.040
166. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med. (2012) 367:2322–33. doi: 10.1056/NEJMra1205750
167. Spicuzza L, Di Maria G, Polosa R. Adenosine in the airways: implications and applications. Eur J Pharmacol. (2006) 533:77–88. doi: 10.1016/j.ejphar.2005.12.056
168. Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. (1993) 148:91–7. doi: 10.1164/ajrccm/148.1.91
169. Cushley MJ, Holgate ST. Adenosine-induced bronchoconstriction in asthma: role of mast cell-mediator release. J Allergy Clin Immunol. (1985) 75:272–8. doi: 10.1016/0091-6749(85)90057-0
170. Cushley MJ, Tattersfield AE, Holgate ST. Adenosine-induced bronchoconstriction in asthma. Antagonism by inhaled theophylline. Am Rev Respir Dis. (1984) 129:380–4.
171. Peachell PT, Columbo M, Kagey-Sobotka A, Lichtenstein LM, Marone G. Adenosine potentiates mediator release from human lung mast cells. Am Rev Respir Dis. (1988) 138:1143–51. doi: 10.1164/ajrccm/138.5.1143
172. Sereda MJ, Bradding P, Vial C. Adenosine potentiates human lung mast cell tissue plasminogen activator activity. J Immunol. (2011) 186:1209–17. doi: 10.4049/jimmunol.1001563
173. Buceta M, Dominguez E, Castro M, Brea J, Alvarez D, Barcala J, et al. A new chemical tool (C0036E08) supports the role of adenosine A(2B) receptors in mediating human mast cell activation. Biochem Pharmacol. (2008) 76:912–21. doi: 10.1016/j.bcp.2008.07.011
174. Gomez G, Zhao W, Schwartz LB. Disparity in FcepsilonRI-induced degranulation of primary human lung and skin mast cells exposed to adenosine. J Clin Immunol. (2011) 31:479–87. doi: 10.1007/s10875-011-9517-7
175. Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol. (2003) 171:338–45. doi: 10.4049/jimmunol.171.1.338
176. Tilley SL, Tsai M, Williams CM, Wang ZS, Erikson CJ, Galli SJ, et al. Identification of A3 receptor- and mast cell-dependent and -independent components of adenosine-mediated airway responsiveness in mice. J Immunol. (2003) 171:331–7. doi: 10.4049/jimmunol.171.1.331
177. Hua X, Chason KD, Fredholm BB, Deshpande DA, Penn RB, Tilley SL. Adenosine induces airway hyperresponsiveness through activation of A3 receptors on mast cells. J Allergy Clin Immunol. (2008) 122:107–13, 113.e1–7. doi: 10.1016/j.jaci.2008.03.026
178. Murray JJ, Tonnel AB, Brash AR, Roberts LJ 2nd, Gosset P, Workman R, et al. Release of prostaglandin D2 into human airways during acute antigen challenge. N Engl J Med. (1986) 315:800–4. doi: 10.1056/NEJM198609253151304
179. Casale TB, Wood D, Richerson HB, Zehr B, Zavala D, Hunninghake GW. Direct evidence of a role for mast cells in the pathogenesis of antigen-induced bronchoconstriction. J Clin Invest. (1987) 80:1507–11. doi: 10.1172/JCI113234
180. Wenzel SE, Larsen GL, Johnston K, Voelkel NF, Westcott JY. Elevated levels of leukotriene C4 in bronchoalveolar lavage fluid from atopic asthmatics after endobronchial allergen challenge. Am Rev Respir Dis. (1990) 142:112–9. doi: 10.1164/ajrccm/142.1.112
181. Wenzel SE, Fowler AA 3rd, Schwartz LB. Activation of pulmonary mast cells by bronchoalveolar allergen challenge. In vivo release of histamine and tryptase in atopic subjects with and without asthma. Am Rev Respir Dis. (1988) 137:1002–8. doi: 10.1164/ajrccm/137.5.1002
182. Togias A. H1-receptors: localization and role in airway physiology and in immune functions. J Allergy Clin Immunol. (2003) 112(4 Suppl.):S60–8. doi: 10.1016/S0091-6749(03)01878-5
183. Buckland KF, Williams TJ, Conroy DM. Histamine induces cytoskeletal changes in human eosinophils via the H(4) receptor. Br J Pharmacol. (2003) 140:1117–27. doi: 10.1038/sj.bjp.0705530
184. Mommert S, Kleiner S, Gehring M, Eiz-Vesper B, Stark H, Gutzmer R, et al. Human basophil chemotaxis and activation are regulated via the histamine H4 receptor. Allergy. (2016) 71:1264–73. doi: 10.1111/all.12875
185. Kay LJ, Suvarna SK, Peachell PT. Histamine H4 receptor mediates chemotaxis of human lung mast cells. Eur J Pharmacol. (2018) 837:38–44. doi: 10.1016/j.ejphar.2018.08.028
186. Lechin F, van der Dijs B, Orozco B, Lechin M, Lechin AE. Increased levels of free serotonin in plasma of symptomatic asthmatic patients. Annal Allergy Asth Immunol. (1996) 77:245–53. doi: 10.1016/S1081-1206(10)63263-2
187. Cazzolau M, Matera MG, D'Amato G, Rossi F. Effects of serotonin on airways: recent developments. Allergy. (1995) 50:1–10. doi: 10.1111/j.1398-9995.1995.tb02476.x
188. Cazzola M, Assogna G, Lucchetti G, Cicchitto G, D'Amato G. Effect of ketanserin, a new blocking agent of the 5-HT2 receptor, on airway responsiveness in asthma. Allergy. (1990) 45:151–153. doi: 10.1111/j.1398-9995.1990.tb00473.x
189. Cazzola M, Matera MG, Santangelo G, Assogna G, D'Amato G, Rossi F, et al. Effect of the selective 5-HT2 antagonist ketanserin on adenosme-mduced bronchoconstriction in asthmatic subjects. Immunopharmacology. (1992) 23:21–28. doi: 10.1016/0162-3109(92)90005-W
190. Scarpelli MP, Keller S, Tran L, Palmiere C. Postmortem serum levels of IgE and mast cell tryptase in fatal asthma. Forensic Sci Int. (2016) 269:113–118. doi: 10.1016/j.forsciint.2016.11.001
191. Hinks TS, Zhou X, Staples KJ, Dimitrov BD, Manta A, Petrossian T, et al. Innate and adaptive T cells in asthmatic patients: Relationship to severity and disease mechanisms. J Allergy Clin Immunol. (2015) 136:323–33. doi: 10.1016/j.jaci.2015.01.014
192. Gao S, Fan J, Wang Z. Diagnostic value of serum baseline tryptase levels in childhood asthma and its correlation with disease severity. Int Arch Allergy Immunol. (2016) 171:194–202. doi: 10.1159/000452624
193. Wardlaw AJ, Hay H, Cromwell O, Collins JV, Kay AB. Leukotrienes, LTC4 and LTB4, in bronchoalveolar lavage in bronchial asthma and other respiratory diseases. J Allergy Clin Immunol. (1989) 84:19–26. doi: 10.1016/0091-6749(89)90173-5
194. Pavord ID, Ward R, Woltmann G, Wardlaw AJ, Sheller JR, Dworski R. Induced sputum eicosanoid concentrations in asthma. Am J Respir Crit Care Med. (1999) 160:1905–9. doi: 10.1164/ajrccm.160.6.9903114
195. Taylor GW, Taylor I, Black P, Maltby NH, Turner N, Fuller RW, et al. Urinary leukotriene E4 after antigen challenge and in acute asthma and allergic rhinitis. Lancet. (1989) 1:584–8. doi: 10.1016/S0140-6736(89)91611-5
196. Soter NA, Lewis RA, Corey EJ, Austen KF. Local effects of synthetic leukotrienes (LTC4, LTD4, LTE4, and LTB4) in human skin. J Invest Dermatol. (1983) 80:115–9. doi: 10.1111/1523-1747.ep12531738
197. Griffin M, Weiss JW, Leitch AG, McFadden ER Jr, Corey EJ, Austen KF, et al. Effects of leukotriene D on the airways in asthma. N Engl J Med. (1983) 308:436–9. doi: 10.1056/NEJM198302243080807
198. Weiss JW, Drazen JM, Coles N, McFadden ER Jr, Weller PF, Corey EJ, et al. Bronchoconstrictor effects of leukotriene C in humans. Science. (1982) 216:196–8. doi: 10.1126/science.7063880
199. Holroyde MC, Altounyan RE, Cole M, Dixon M, Elliott EV. Bronchoconstriction produced in man by leukotrienes CD, Lancet. (1981) 2:17–8. doi: 10.1016/S0140-6736(81)90254-3
200. Takeda K, Shiraishi Y, Matsubara S, Miyahara N, Matsuda H, Okamoto M, et al. Effects of combination therapy with montelukast and carbocysteine in allergen-induced airway hyperresponsiveness and airway inflammation. Br J Pharmacol. (2010) 160:1399–407. doi: 10.1111/j.1476-5381.2010.00797.x
201. Kostikas K, Gaga M, Papatheodorou G, Karamanis T, Orphanidou D, Loukides S. Leukotriene B4 in exhaled breath condensate and sputum supernatant in patients with COPD and asthma. Chest. (2005) 127:1553–9. doi: 10.1378/chest.127.5.1553
202. Ford-Hutchinson AW, Bray MA, Doig MV, Shipley ME, Smith MJ. Leukotriene B. A potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature. (1980) 286:264–5. doi: 10.1038/286264a0
203. Wenzel SE, Westcott JY, Smith HR, Larsen GL. Spectrum of prostanoid release after bronchoalveolar allergen challenge in atopic asthmatics and in control groups. An alteration in the ratio of bronchoconstrictive to bronchoprotective mediators. Am Rev Respir Dis. (1989) 139:450–7. doi: 10.1164/ajrccm/139.2.450
204. Bochenek G, Nizankowska E, Gielicz A, Swierczynska M, Szczeklik A. Plasma 9alpha,11beta-PGF2, a PGD2 metabolite, as a sensitive marker of mast cell activation by allergen in bronchial asthma. Thorax. (2004) 59:459–64. doi: 10.1136/thx.2003.013573
205. Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The bronchoconstrictor effect of inhaled prostaglandin D2 in normal and asthmatic men. N Engl J Med. (1984) 311:209–13. doi: 10.1056/NEJM198407263110401
206. Monneret G, Gravel S, Diamond M, Rokach J, Powell WS. Prostaglandin D2 is a potent chemoattractant for human eosinophils that acts via a novel DP receptor. Blood. (2001) 98:1942–8. doi: 10.1182/blood.V98.6.1942
207. Hirai H, Tanaka K, Takano S, Ichimasa M, Nakamura M, Nagata K. Cutting edge: agonistic effect of indomethacin on a prostaglandin D2 receptor, CRTH2. J Immunol. (2002) 168:981–5. doi: 10.4049/jimmunol.168.3.981
208. Riley JF, West GB. The presence of histamine in tissue mast cells. J Physiol. (1953) 120:528–537. doi: 10.1113/jphysiol.1953.sp004915
209. Thangam EB, Jemima EA, Singh H, Baig MS, Khan M, Mathias CB, et al. The role of histamine and histamine receptors in mast cell-mediated allergy and inflammation: the hunt for new therapeutic targets. Front Immunol. (2018) 9:1873. doi: 10.3389/fimmu.2018.01873
210. Nelson HS. Prospects for antihistamines in the treatment of asthma. J Allergy Clin Immunol. (2003) 112(4 Suppl.):S96–100. doi: 10.1016/S0091-6749(03)01883-9
211. Bryce PJ, Mathias CB, Harrison KL, Watanabe T, Geha RS, Oettgen HC. The H1 histamine receptor regulates allergic lung responses. J Clin Invest. (2006) 116:1624–32. doi: 10.1172/JCI26150
212. Miyamoto K, Iwase M, Nyui M, Arata S, Sakai Y, Gabazza EC, et al. Histamine type 1 receptor deficiency reduces airway inflammation in a murine asthma model. Int Arch Allergy Immunol. (2006) 140:215–22. doi: 10.1159/000093246
213. Jutel M, Watanabe T, Klunker S, Akdis M, Thomet OA, Malolepszy J, et al. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature. (2001) 413:420–5. doi: 10.1038/35096564
214. Ferstl R, Frei R, Barcik W, Schiavi E, Wanke K, Ziegler M, et al. Histamine receptor 2 modifies iNKT cell activity within the inflamed lung. Allergy. (2017) 72:1925–35. doi: 10.1111/all.13227
215. Nieto-Alamilla G, Marquez-Gomez R, Garcia-Galvez AM, Morales-Figueroa GE, Arias-Montano JA. The Histamine H3 Receptor: Structure, Pharmacology, et al. Mol Pharmacol. (2016) 90:649–73. doi: 10.1124/mol.116.104752
216. Thurmond RL. The histamine H4 receptor: from orphan to the clinic. Front Pharmacol. (2015) 6:65. doi: 10.3389/fphar.2015.00065
217. Jemima EA, Prema A, Thangam EB. Functional characterization of histamine H4 receptor on human mast cells. Mol Immunol. (2014) 62:19–28. doi: 10.1016/j.molimm.2014.05.007
218. Hofstra CL, Desai PJ, Thurmond RL, Fung-Leung WP. Histamine H4 receptor mediates chemotaxis and calcium mobilization of mast cells. J Pharmacol Exp Ther. (2003) 305:1212–21. doi: 10.1124/jpet.102.046581
219. Dunford PJ, O'Donnell N, Riley JP, Williams KN, Karlsson L, Thurmond RL. The histamine H4 receptor mediates allergic airway inflammation by regulating the activation of CD4+ T cells. J Immunol. (2006) 176:7062–70. doi: 10.4049/jimmunol.176.11.7062
220. Hartwig C, Munder A, Glage S, Wedekind D, Schenk H, Seifert R, et al. The histamine H4-receptor (H4R) regulates eosinophilic inflammation in ovalbumin-induced experimental allergic asthma in mice. Eur J Immunol. (2015) 45:1129–40. doi: 10.1002/eji.201445179
221. Tiligada E, Ennis M. Histamine pharmacology: from Sir Henry Dale to the 21st century. Br J Pharmacol. (2018). doi: 10.1111/bph.14524
222. Sjoerdsma A, Waalkes TP, Weissbach H. Serotonin and histamine in mast cells. Science. (1957) 125:1202–3. doi: 10.1126/science.125.3259.1202
223. Kushnir-Sukhov NM, Brown JM, Wu Y, Kirshenbaum A, Metcalfe DD. Human mast cells are capable of serotonin synthesis and release. J Allergy Clin Immunol. (2007) 119:498–9. doi: 10.1016/j.jaci.2006.09.003
224. Herr N, Bode C, Duerschmied D. The effects of serotonin in immune cells. Front Cardiovasc Med. (2017) 4:48. doi: 10.3389/fcvm.2017.00048
225. Martin TR, Cohen ML, Drazen JM. Serotonin-induced pulmonary responses are mediated by the 5-HT2 receptor in the mouse. J Pharmacol Exp Ther. (1994) 268:104–9.
226. Lofdahl A, Wenglen C, Rydell-Tormanen K, Westergren-Thorsson G, Larsson-Callerfelt AK. Effects of 5-hydroxytryptamine class 2 receptor antagonists on bronchoconstriction and pulmonary remodeling processes. Am J Pathol. (2018) 188:1113–9. doi: 10.1016/j.ajpath.2018.01.006
227. Cazzola M, Matera MG. 5-HT modifiers as a potential treatment of asthma. Trend Pharmacol Sci. (2000) 21:13–16. doi: 10.1016/S0165-6147(99)01408-X
228. Kang BN, Ha SG, Bahaie NS, Hosseinkhani MR, Ge XN, Blumenthal MN, et al. Regulation of serotonin-induced trafficking and migration of eosinophils. PLoS ONE. (2013) 8:e54840. doi: 10.1371/journal.pone.0054840
229. Boehme SA, Lio FM, Sikora L, Pandit TS, Lavrador K, Rao SP, et al. Cutting edge: serotonin is a chemotactic factor for eosinophils and functions additively with eotaxin. J Immunol. (2004) 173:3599–603. doi: 10.4049/jimmunol.173.6.3599
230. Kushnir-Sukhov NM, Gilfillan AM, Coleman JW, Brown JM, Bruening S, Toth M, et al. 5-hydroxytryptamine induces mast cell adhesion and migration. J Immunol. (2006) 177:6422–32. doi: 10.4049/jimmunol.177.9.6422
231. Caughey GH. Mast cell proteases as pharmacological targets. Eur J Pharmacol. (2016) 778:44–55. doi: 10.1016/j.ejphar.2015.04.045
232. Hallgren J, Backstrom S, Estrada S, Thuveson M, Pejler G. Histidines are critical for heparin-dependent activation of mast cell tryptase. J Immunol. (2004) 173:1868–75. doi: 10.4049/jimmunol.173.3.1868
233. Hallgren J, Spillmann D, Pejler G. Structural requirements and mechanism for heparin-induced activation of a recombinant mouse mast cell tryptase, mouse mast cell protease−6: formation of active tryptase monomers in the presence of low molecular weight heparin. J Biol Chem. (2001) 276:42774–81. doi: 10.1074/jbc.M105531200
234. Fajardo I, Pejler G. Formation of active monomers from tetrameric human beta-tryptase. Biochem J. (2003) 369(Pt 3):603–10. doi: 10.1042/bj20021418
235. Schwartz LB, Riedel C, Caulfield JP, Wasserman SI, Austen KF. Cell association of complexes of chymase, heparin proteoglycan, and protein after degranulation by rat mast cells. J Immunol. (1981) 126:2071–8.
236. Pejler G, Abrink M, Wernersson S. Serglycin proteoglycan: regulating the storage and activities of hematopoietic proteases. Biofactors. (2009) 35:61–8. doi: 10.1002/biof.11
237. Chen CL, Wang SD, Zeng ZY, Lin KJ, Kao ST, Tani T, et al. Serine protease inhibitors nafamostat mesilate and gabexate mesilate attenuate allergen-induced airway inflammation and eosinophilia in a murine model of asthma. J Allergy Clin Immunol. (2006) 118:105–12. doi: 10.1016/j.jaci.2006.02.047
238. Rothmeier AS, Ruf W. Protease-activated receptor 2 signaling in inflammation. Semin Immunopathol. (2012) 34:133–49. doi: 10.1007/s00281-011-0289-1
239. Steinhoff M, Vergnolle N, Young SH, Tognetto M, Amadesi S, Ennes HS, et al. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. (2000) 6:151–8. doi: 10.1038/72247
240. Schmidlin F, Amadesi S, Vidil R, Trevisani M, Martinet N, Caughey G, et al. Expression and function of proteinase-activated receptor 2 in human bronchial smooth muscle. Am J Respir Crit Care Med. (2001) 164:1276–81. doi: 10.1164/ajrccm.164.7.2101157
241. Schmidlin F, Amadesi S, Dabbagh K, Lewis DE, Knott P, Bunnett NW, et al. Protease-activated receptor 2 mediates eosinophil infiltration and hyperreactivity in allergic inflammation of the airway. J Immunol. (2002) 169:5315–21. doi: 10.4049/jimmunol.169.9.5315
242. Takizawa T, Tamiya M, Hara T, Matsumoto J, Saito N, Kanke T, et al. Abrogation of bronchial eosinophilic inflammation and attenuated eotaxin content in protease-activated receptor 2-deficient mice. J Pharmacol Sci. (2005) 98:99–102. doi: 10.1254/jphs.SCZ050138
243. Asaduzzaman M, Davidson C, Nahirney D, Fiteih Y, Puttagunta L, Vliagoftis H. Proteinase-activated receptor−2 blockade inhibits changes seen in a chronic murine asthma model. Allergy. (2018) 73:416–420. doi: 10.1111/all.13313
244. Waern I, Jonasson S, Hjoberg J, Bucht A, Abrink M, Pejler G, et al. Mouse mast cell protease 4 is the major chymase in murine airways and has a protective role in allergic airway inflammation. J Immunol. (2009) 183:6369–76. doi: 10.4049/jimmunol.0900180
245. Waern I, Lundequist A, Pejler G, Wernersson S. Mast cell chymase modulates IL−33 levels and controls allergic sensitization in dust-mite induced airway inflammation. Mucosal Immunol. (2013) 6:911–20. doi: 10.1038/mi.2012.129
246. Sugimoto K, Kudo M, Sundaram A, Ren X, Huang K, Bernstein X, et al. The alphavbeta6 integrin modulates airway hyperresponsiveness in mice by regulating intraepithelial mast cells. J Clin Invest. (2012) 122:748–58. doi: 10.1172/JCI58815
247. Hallstrand TS, Henderson WR. An update on the role of leukotrienes in asthma. Curr Opin Allergy Clin Immunol. (2010) 10:60–6. doi: 10.1097/ACI.0b013e32833489c3
248. Yokomizo T, Nakamura M, Shimizu T. Leukotriene receptors as potential therapeutic targets. J Clin Invest. (2018) 128:2691–701. doi: 10.1172/JCI97946
249. Ikeda G, Miyahara N, Koga H, Fuchimoto Y, Waseda K, Kurimoto E, et al. Effect of a cysteinyl leukotriene receptor antagonist on experimental emphysema and asthma combined with emphysema. Am J Respir Cell Mol Biol. (2014) 50:18–29. doi: 10.1165/rcmb.2012-0418OC
250. Wu AY, Chik SC, Chan AW, Li Z, Tsang KW, Li W. Anti-inflammatory effects of high-dose montelukast in an animal model of acute asthma. Clin Exp Allergy. (2003) 33:359–66. doi: 10.1046/j.1365-2222.2003.01615.x
251. Beller TC, Maekawa A, Friend DS, Austen KF, Kanaoka Y. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J Biol Chem. (2004) 279:46129–34. doi: 10.1074/jbc.M407057200
252. Liu T, Barrett NA, Kanaoka Y, Yoshimoto E, Garofalo D, Cirka H, et al. Type 2 cysteinyl leukotriene receptors drive IL−33-dependent type 2 immunopathology and aspirin sensitivity. J Immunol. (2018) 200:915–27. doi: 10.4049/jimmunol.1700603
253. Mellor EA, Maekawa A, Austen KF, Boyce JA. Cysteinyl leukotriene receptor 1 is also a pyrimidinergic receptor and is expressed by human mast cells. Proc Natl Acad Sci USA. (2001) 98:7964. doi: 10.1073/pnas.141221498
254. Jiang Y, Borrelli LA, Kanaoka Y, Bacskai BJ, Boyce JA. CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene dependent mitogenic responses of mast cells. Blood. (2007) 110:3263–70. doi: 10.1182/blood-2007-07-100453
255. Paruchuri S, Jiang Y, Feng C, Francis SA, Plutzky J, Boyce JA. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J Biol Chem. (2008) 283:16477–87. doi: 10.1074/jbc.M705822200
256. Laitinen LA, Laitinen A, Haahtela T, Vilkka V, Spur BW, Lee TH. Leukotriene E4 and granulocytic infiltration into asthmatic airways. Lancet. (1993) 341:989–90. doi: 10.1016/0140-6736(93)91073-U
257. Gauvreau GM, Parameswaran KN, Watson RM, O'Byrne PM. Inhaled leukotriene E(4), but not leukotriene D(4), increased airway inflammatory cells in subjects with atopic asthma. Am J Respir Crit Care Med. (2001) 164(8 Pt 1):1495–500. doi: 10.1164/ajrccm.164.8.2102033
258. Paruchuri S, Tashimo H, Feng C, Maekawa A, Xing W, Jiang Y, et al. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. (2009) 206:2543–55. doi: 10.1084/jem.20091240
259. Lussana F, Di Marco F, Terraneo S, Parati M, Razzari C, Scavone M, et al. Effect of prasugrel in patients with asthma: results of PRINA, a randomized, double-blind, placebo-controlled, cross-over study. J Thromb Haemost. (2015) 13:136–41. doi: 10.1111/jth.12779
260. Maekawa A, Balestrieri B, Austen KF, Kanaoka Y. GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc Natl Acad Sci USA. (2009) 106:11685–90. doi: 10.1073/pnas.0905364106
261. Marucci G, Dal Ben D, Lambertucci C, Santinelli C, Spinaci A, Thomas A, et al. The G protein-coupled receptor GPR17: overview and update. Chem. Med. Chem. (2016) 11:2567–74. doi: 10.1002/cmdc.201600453
262. Kanaoka Y, Maekawa A, Austen KF. Identification of GPR99 protein as a potential third cysteinyl leukotriene receptor with a preference for leukotriene E4 ligand. J Biol Chem. (2013) 288:10967–72. doi: 10.1074/jbc.C113.453704
263. Bankova LG, Lai J, Yoshimoto E, Boyce JA, Austen KF, Kanaoka Y, et al. Leukotriene E4 elicits respiratory epithelial cell mucin release through the G-protein-coupled receptor, GPR99. Proc Natl Acad Sci USA. (2016) 113:6242–7. doi: 10.1073/pnas.1605957113
264. Lazarinis N, Bood J, Gomez C, Kolmert J, Lantz AS, Gyllfors P, et al. Leukotriene E4 induces airflow obstruction and mast cell activation through the cysteinyl leukotriene type 1 receptor. J Allergy Clin Immunol. (2018) 142:1080–1089. doi: 10.1016/j.jaci.2018.02.024
265. Vachier I, Bonnans C, Chavis C, Farce M, Godard P, Bousquet J, et al. Severe asthma is associated with a loss of LX4, an endogenous anti-inflammatory compound. J Allergy Clin Immunol. (2005) 115:55–60. doi: 10.1016/j.jaci.2004.09.038
266. Higham A, Cadden P, Southworth T, Rossall M, Kolsum U, Lea S, et al. Leukotriene B4 levels in sputum from asthma patients. ERJ Open Res. (2016) 2:00088-2015. doi: 10.1183/23120541.00088-2015
267. Basyigit I, Yildiz F, Ozkara SK, Boyaci H, Ilgazli A. Inhaled corticosteroid effects both eosinophilic and non-eosinophilic inflammation in asthmatic patients. Mediators Inflamm. (2004) 13:285–91. doi: 10.1080/09629350400003118
268. Peters SP, MacGlashan DW Jr, Schulman ES, Schleimer RP, Hayes EC, Rokach J, Lichtenstein LM, et al. Arachidonic acid metabolism in purified human lung mast cells. J Immunol. (1984) 132:1972–9.
269. Weller CL, Collington SJ, Brown JK, Miller HR, Al-Kashi A, Clark P, et al. Leukotriene B4, an activation product of mast cells, is a chemoattractant for their progenitors. J Exp Med. (2005) 201:1961–71. doi: 10.1084/jem.20042407
270. Ott VL, Cambier JC, Kappler J, Marrack P, Swanson BJ. Mast cell-dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat Immunol. (2003) 4:974–81. doi: 10.1038/ni971
271. Tager AM, Bromley SK, Medoff BD, Islam SA, Bercury SD, Friedrich EB, et al. Leukotriene B4 receptor BLT1 mediates early effector T cell recruitment. Nat Immunol. (2003) 4:982–90. doi: 10.1038/ni970
272. Terawaki K, Yokomizo T, Nagase T, Toda A, Taniguchi M, Hashizume K, et al. Absence of leukotriene B4 receptor 1 confers resistance to airway hyperresponsiveness and Th2-type immune responses. J Immunol. (2005) 175:4217–25. doi: 10.4049/jimmunol.175.7.4217
273. Miyahara N, Ohnishi H, Miyahara S, Takeda K, Matsubara S, Matsuda H, et al. Leukotriene B4 release from mast cells in IgE-mediated airway hyperresponsiveness and inflammation. Am J Respir Cell Mol Biol. (2009) 40:672–82. doi: 10.1165/rcmb.2008-0095OC
274. Matsunaga Y, Fukuyama S, Okuno T, Sasaki F, Matsunobu T, Asai Y, et al. Leukotriene B4 receptor BLT2 negatively regulates allergic airway eosinophilia. FASEB J. (2013) 27:3306–14. doi: 10.1096/fj.12-217000
275. Okuno T, Iizuka Y, Okazaki H, Yokomizo T, Taguchi R, Shimizu T. 12(S)-Hydroxyheptadeca−5Z, 8E, 10E-trienoic acid is a natural ligand for leukotriene B4 receptor 2. J Exp Med. (2008) 205:759–66. doi: 10.1084/jem.20072329
276. Claar D, Hartert TV, Peebles RS Jr. The role of prostaglandins in allergic lung inflammation and asthma. Expert Rev Respir Med. (2015) 9:55–72. doi: 10.1586/17476348.2015.992783
277. Luna-Gomes T, Magalhaes KG, Mesquita-Santos FP, Bakker-Abreu I, Samico RF, Molinaro R, et al. Eosinophils as a novel cell source of prostaglandin D2: autocrine role in allergic inflammation. J Immunol. (2011) 187:6518–26. doi: 10.4049/jimmunol.1101806
278. Fajt ML, Gelhaus SL, Freeman B, Uvalle CE, Trudeau JB, Holguin F, et al. Prostaglandin D(2) pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol. (2013) 131:1504–12. doi: 10.1016/j.jaci.2013.01.035
279. Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. (2001) 193:255–61. doi: 10.1084/jem.193.2.255
280. Boie Y, Sawyer N, Slipetz DM, Metters KM, Abramovitz M. Molecular cloning and characterization of the human prostanoid DP receptor. J Biol Chem. (1995) 270:18910–6. doi: 10.1074/jbc.270.32.18910
281. Taketomi Y, Ueno N, Kojima T, Sato H, Murase R, Yamamoto K, et al. Mast cell maturation is driven via a group III phospholipase A2-prostaglandin D2-DP1 receptor paracrine axis. Nat Immunol. (2013) 14:554–63. doi: 10.1038/ni.2586
282. Matsuoka T, Hirata M, Tanaka H, Takahashi Y, Murata T, Kabashima K, et al. Prostaglandin D2 as a mediator of allergic asthma. Science. (2000) 287:2013–7. doi: 10.1126/science.287.5460.2013
283. Honda K, Arima M, Cheng G, Taki S, Hirata H, Eda F, et al. Prostaglandin D2 reinforces Th2 type inflammatory responses of airways to low-dose antigen through bronchial expression of macrophage-derived chemokine. J Exp Med. (2003) 198:533–43. doi: 10.1084/jem.20022218
284. Hammad H, de Heer HJ, Soullie T, Hoogsteden HC, Trottein F, Lambrecht BN. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J Immunol. (2003) 171:3936–40. doi: 10.4049/jimmunol.171.8.3936
285. Hammad H, Kool M, Soullie T, Narumiya S, Trottein F, Hoogsteden HC, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J Exp Med. (2007) 204:357–67. doi: 10.1084/jem.20061196
286. Nagata K, Hirai H, Tanaka K, Ogawa K, Aso T, Sugamura K, et al. CRTH2, an orphan receptor of T-helper−2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. (1999) 459:195–9. doi: 10.1016/S0014-5793(99)01251-X
287. Mjosberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, et al. Human IL−25- and IL−33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. (2011) 12:1055–62. doi: 10.1038/ni.2104
288. Boehme SA, Franz-Bacon K, Chen EP, Ly TW, Kawakami Y, Bacon KB. Murine bone marrow-derived mast cells express chemoattractant receptor-homologous molecule expressed on T-helper class 2 cells (CRTh2). Int Immunol. (2009) 21:621–32. doi: 10.1093/intimm/dxp031
289. Shirasaki H, Kikuchi M, Kanaizumi E, Himi T. Accumulation of CRTH2-positive leukocytes in human allergic nasal mucosa. Ann Allergy Asthma Immunol. (2009) 102:110–5. doi: 10.1016/S1081-1206(10)60239-6
290. Moon TC, Campos-Alberto E, Yoshimura T, Bredo G, Rieger AM, Puttagunta L, et al. Expression of DP2 (CRTh2), a prostaglandin D(2) receptor, in human mast cells. PLoS ONE. (2014) 9:0108595. doi: 10.1371/journal.pone.0108595
291. Spik I, Brenuchon C, Angeli V, Staumont D, Fleury S, Capron M, et al. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. (2005) 174:3703–8. doi: 10.4049/jimmunol.174.6.3703
292. Chevalier E, Stock J, Fisher T, Dupont M, Fric M, Fargeau H, et al. Cutting edge: chemoattractant receptor-homologous molecule expressed on Th2 cells plays a restricting role on IL−5 production and eosinophil recruitment. J Immunol. (2005) 175:2056–60. doi: 10.4049/jimmunol.175.4.2056
293. Uller L, Mathiesen JM, Alenmyr L, Korsgren M, Ulven T, Hogberg T, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res. (2007) 8:16. doi: 10.1186/1465-9921-8-16
294. Lukacs NW, Berlin AA, Franz-Bacon K, Sasik R, Sprague LJ, Ly TW, et al. CRTH2 antagonism significantly ameliorates airway hyperreactivity and downregulates inflammation-induced genes in a mouse model of airway inflammation. Am J Physiol Lung Cell Mol Physiol. (2008) 295:L767–79. doi: 10.1152/ajplung.90351.2008
295. Liu W, Min J, Jiang H, Mao B. Chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2) antagonists in asthma: a systematic review and meta-analysis protocol. BMJ Open. (2018) 8:e020882. doi: 10.1136/bmjopen-2017-020882
296. Barnes N, Pavord I, Chuchalin A, Bell J, Hunter M, Lewis T, et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. (2012) 42:38–48. doi: 10.1111/j.1365-2222.2011.03813.x
297. Bateman ED, Guerreros AG, Brockhaus F, Holzhauer B, Pethe A, Kay RA, et al. Fevipiprant, an oral prostaglandin DP2 receptor (CRTh2) antagonist, in allergic asthma uncontrolled on low-dose inhaled corticosteroids. Eur Respir J. (2017) 50:1700670. doi: 10.1183/13993003.00670-2017
298. Hall IP, Fowler AV, Gupta A, Tetzlaff K, Nivens MC, Sarno M, et al. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm Pharmacol Ther. (2015) 32:37–44. doi: 10.1016/j.pupt.2015.03.003
299. Saluja R, Kumar A, Jain M, Goel SK, Jain A. Role of sphingosine−1-phosphate in mast cell functions and asthma and its regulation by non-coding RNA. Front Immunol. (2017) 8:587. doi: 10.3389/fimmu.2017.00587
300. Rivera J, Proia RL, Olivera A. The alliance of sphingosine−1-phosphate and its receptors in immunity. Nat Rev Immunol. (2008) 8:753. doi: 10.1038/nri2400
301. Ammit AJ, Hastie AT, Edsall LC, Hoffman RK, Amrani Y, Krymskaya VP, et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. (2001) 15:1212–4. doi: 10.1096/fj.00-0742fje
302. Rosenfeldt HM, Amrani Y, Watterson KR, Murthy KS, Panettieri RA Jr, Spiegel S. Sphingosine−1-phosphate stimulates contraction of human airway smooth muscle cells. FASEB J. (2003) 17:1789–99. doi: 10.1096/fj.02-0836com
303. Lai WQ, Goh HH, Bao Z, Wong WS, Melendez AJ, Leung BP. The role of sphingosine kinase in a murine model of allergic asthma. J Immunol. (2008) 180:4323–9. doi: 10.4049/jimmunol.180.6.4323
304. Roviezzo F, Di Lorenzo A, Bucci M, Brancaleone V, Vellecco V, De Nardo M, et al. Sphingosine−1-phosphate/sphingosine kinase pathway is involved in mouse airway hyperresponsiveness. Am J Respir Cell Mol Biol. (2007) 36:757–62. doi: 10.1165/rcmb.2006-0383OC
305. Roviezzo F, D'Agostino B, Brancaleone V, De Gruttola L, Bucci M, De Dominicis G, et al. Systemic administration of sphingosine−1-phosphate increases bronchial hyperresponsiveness in the mouse. Am J Respir Cell Mol Biol. (2010) 42:572–7. doi: 10.1165/rcmb.2009-0108OC
306. Roviezzo FEA. Toll-like receptor 4 is essential for the expression of sphingosine−1-phosphate-dependent asthma-like disease in mice. Front Immuol. (2018) 8:1336. doi: 10.3389/fimmu.2017.01336
307. Oskeritzian CA, Hait NC, Wedman P, Chumanevich A, Kolawole EM, Price MM, et al. The sphingosine−1-phosphate/sphingosine−1-phosphate receptor 2 axis regulates early airway T-cell infiltration in murine mast cell-dependent acute allergic responses. J Allergy Clin Immunol. (2015) 135:1008–18.e1. doi: 10.1016/j.jaci.2014.10.044
308. Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. (2010) 9:883–97. doi: 10.1038/nrd3248
309. Boulton C, David OJ, Meiser K, Schmouder R. Tolerability and pulmonary pharmacodynamic effects during treatment initiation of once-daily oral fingolimod in subjects with moderate asthma. Clin Pharmacol Drug Dev. (2013) 2:2–10. doi: 10.1002/cpdd.4
310. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. (2018) 18:454–66. doi: 10.1038/s41577-018-0006-6
311. Mukai K, Tsai M, Saito H, Galli SJ. Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev. (2018) 282:121–50. doi: 10.1111/imr.12634
312. Bradding P, Feather IH, Howarth PH, Mueller R, Roberts JA, Britten K, et al. Interleukin 4 is localized to and released by human mast cells. J Exp Med. (1992) 176:1381–6. doi: 10.1084/jem.176.5.1381
313. Liu AY, Dwyer DF, Jones TG, Bankova LG, Shen S, Katz HR, et al. Mast cells recruited to mesenteric lymph nodes during helminth infection remain hypogranular and produce IL−4 and IL−6. J Immunol. (2013) 190:1758–66. doi: 10.4049/jimmunol.1202567
314. Bradding P, Roberts JA, Britten KM, Montefort S, Djukanovic R, Mueller R, et al. Interleukin−4, −5, and −6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. (1994) 10:471–80. doi: 10.1165/ajrcmb.10.5.8179909
315. Kobayashi H, Okayama Y, Ishizuka T, Pawankar R, Ra C, Mori M. Production of IL−13 by human lung mast cells in response to Fcepsilon receptor cross-linkage. Clin Exp Allergy. (1998) 28:1219–27. doi: 10.1046/j.1365-2222.1998.00377.x
316. Brightling CE, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID, Bradding P. Interleukin−4 and −13 expression is co-localized to mast cells within the airway smooth muscle in asthma. Clin Exp Allergy. (2003) 33:1711–6. doi: 10.1111/j.1365-2222.2003.01827.x
317. Ohkawara Y, Yamauchi K, Tanno Y, Tamura G, Ohtani H, Nagura H, et al. Human lung mast cells and pulmonary macrophages produce tumor necrosis factor-alpha in sensitized lung tissue after IgE receptor triggering. Am J Respir Cell Mol Biol. (1992) 7:385–92. doi: 10.1165/ajrcmb/7.4.385
318. Stassen M, Muller C, Arnold M, Hultner L, Klein-Hessling S, Neudorfl C, et al. IL−9 and IL−13 production by activated mast cells is strongly enhanced in the presence of lipopolysaccharide: NF-kappa B is decisively involved in the expression of IL−9. J Immunol. (2001) 166:4391–8. doi: 10.4049/jimmunol.166.7.4391
319. Holroyd KJ, Martinati LC, Trabetti E, Scherpbier T, Eleff SM, Boner AL, et al. Asthma and bronchial hyperresponsiveness linked to the XY long arm pseudoautosomal region. Genomics. (1998) 52:233–5. doi: 10.1006/geno.1998.5445
320. Kauppi P, Laitinen T, Ollikainen V, Mannila H, Laitinen LA, Kere J. The IL9R region contribution in asthma is supported by genetic association in an isolated population. Eur J Hum Genet. (2000) 8:788–92. doi: 10.1038/sj.ejhg.5200541
321. Shimbara A, Christodoulopoulos P, Soussi-Gounni A, Olivenstein R, Nakamura Y, Levitt RC, et al. IL−9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J Allergy Clin Immunol. (2000) 105(1 Pt 1):108–15. doi: 10.1016/S0091-6749(00)90185-4
322. Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. (1998) 188:1307–20. doi: 10.1084/jem.188.7.1307
323. Gong F, Pan YH, Huang X, Zhu HY, Jiang DL. From bench to bedside: therapeutic potential of interleukin−9 in the treatment of asthma. Exp Ther Med. (2017) 13:389–94. doi: 10.3892/etm.2017.4024
324. Parker JM, Oh CK, LaForce C, Miller SD, Pearlman DS, Le C, et al. Safety profile and clinical activity of multiple subcutaneous doses of MEDI−528, a humanized anti-interleukin−9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm Med. (2011) 11:14. doi: 10.1186/1471-2466-11-14
Keywords: mast cell, mast cell progenitors, allergic asthma, mast cell development, mast cell activation
Citation: Méndez-Enríquez E and Hallgren J (2019) Mast Cells and Their Progenitors in Allergic Asthma. Front. Immunol. 10:821. doi: 10.3389/fimmu.2019.00821
Received: 10 December 2018; Accepted: 28 March 2019;
Published: 29 May 2019.
Edited by:
Clinton Mathias, Western New England University, United StatesReviewed by:
Lennart K. A. Lundblad, Meakins-Christie Laboratories, CanadaHydar Ali, University of Pennsylvania, United States
Copyright © 2019 Méndez-Enríquez and Hallgren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny Hallgren, amVubnkuaGFsbGdyZW5AaW1iaW0udXUuc2U=