 Eduardo I. Tognarelli
Eduardo I. Tognarelli Susan M. Bueno
Susan M. Bueno Pablo A. González
Pablo A. González
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 15 April 2019
Sec. Viral Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00810
This article is part of the Research Topic Current Insights into Host Immune Responses to Human Respiratory Syncytial Virus (RSV) and Challenges Towards Efficient Treatments and Vaccines against RSV View all 11 articles
The human respiratory syncytial virus (hRSV) is the leading cause of pneumonia in infants and produces a significant burden in the elderly. It can also infect and produce disease in otherwise healthy adults and recurrently infect those previously exposed to the virus. Importantly, recurrent infections are not necessarily a consequence of antigenic variability, as described for other respiratory viruses, but most likely due to the capacity of this virus to interfere with the host's immune response and the establishment of a protective and long-lasting immunity. Although some genes encoded by hRSV are known to have a direct participation in immune evasion, it seems that repeated infection is mainly given by its capacity to modulate immune components in such a way to promote non-optimal antiviral responses in the host. Importantly, hRSV is known to interfere with dendritic cell (DC) function, which are key cells involved in establishing and regulating protective virus-specific immunity. Notably, hRSV infects DCs, alters their maturation, migration to lymph nodes and their capacity to activate virus-specific T cells, which likely impacts the host antiviral response against this virus. Here, we review and discuss the most important and recent findings related to DC modulation by hRSV, which might be at the basis of recurrent infections in previously infected individuals and hRSV-induced disease. A focus on the interaction between DCs and hRSV will likely contribute to the development of effective prophylactic and antiviral strategies against this virus.
The human respiratory syncytial virus (hRSV) is the leading cause of infant pneumonia worldwide and also elicits significant morbidity in the elderly and children (1–3). Importantly, infants with partial airway development due to premature birth, airway hyperreactivity, pulmonary hypertension, cystic fibrosis, Down syndrome, neurologic conditions, congenital heart disease, and those that are immunosuppressed are at increased risk of developing severe complications due to hRSV infection, which may even lead to death (4, 5). Nevertheless, individuals that are otherwise healthy, such as infants 2 months old or older can also be infected with hRSV and suffer respiratory illness leading to significant morbidity and eventually life-threatening disease, mainly because of complicated pneumonia (3, 6, 7). Noteworthy, at present there are no vaccines available against hRSV, yet many are under development and being assessed clinically, although few would be suitable for direct application on to newborns (8–11).
An important feature of hRSV is that it is capable of re-infecting healthy children and adults that have been previously infected with this virus (12, 13). hRSV can be classified into two groups (A or B) that mainly differentiate from each other based on nucleotide variability in the attachment glycoprotein G (14). Thanks to affordable sequencing costs and high-throughput sequencing techniques, at present circulating hRSV isolates, can be further sub-classified into at least 14 A genotype groups and 23 B genotypes groups, based on similarities of the G protein gene (14). Furthermore, improved access to whole genome sequencing has opened the possibility for molecular classification and molecular epidemiology studies on hRSV (15). However, despite nucleotide and antigenic variability in the attachment G protein of hRSV, recurrent infections with the same virus can occur at a high frequency within healthy individuals. For instance, adults that had a previous natural infection with hRSV and were then exposed to a virus belonging to the same strain group subsequently manifested several reinfections. At 26 months, 73% of individuals were shown to have two or more re-infections, and 47% had three or more infections (16). Thus, other immune-evasion mechanisms distinct from antigenic variation are likely at the base of host reinfections with hRSV. Because relatively few hRSV-encoded genes are known to directly interfere with the host's antiviral response in a somewhat direct manner, one could suggest that the capacity of hRSV to repeatedly infect the host may derive from its ability to elicit an adaptive antiviral immune response that is non-optimal for the host. Indeed, hRSV has been exhaustively described to skew the host's antiviral immune response toward phenotypes that promote exacerbated lung inflammation in response to lung infection which favor the virus (17–20). Importantly, hRSV lung infection can induce macrophages and monocyte-derived macrophages in this tissue to upregulate the surface expression of PD-L1, which will likely have adverse effects over the function of T cells (21). Furthermore, hRSV can elicit human neonatal regulatory B cells to secrete IL-10, which may also result in non-optimal antiviral T cell responses (22). However, an immune cell of choice targeted by hRSV seems to be dendritic cells (DCs), critical immune cells that initiate and regulate antigen-specific adaptive antiviral immunes responses. Indeed, the phenotype and function of these cells have been broadly reported to be modulated during hRSV infection, both in vitro and in vivo. Here, we review the latest studies that describe the interaction between hRSV and DCs and how the outcome affects relevant functions of these cells, which will ultimately impact the establishment of an effective antiviral response against hRSV in the host.
hRSV is an enveloped, negative-sense, and single-stranded RNA virus that encodes 10 genes that are translated into 11 proteins (Figure 1) (23). Its replication and gene transcription occur in the cytoplasm, thanks to the aid of an RNA-dependent RNA-polymerase that is encoded within the viral genome by the L gene (24, 25). For its adequate function, the L protein requires the viral phosphoprotein P, which associates to this RNA polymerase (26, 27). Importantly, the replication of the viral genome and the transcription of its genes are modulated by the hRSV-encoded factors M2-1 and M2-2, which are generated from a common mRNA transcript by a ribosome shift that occurs on the mRNA after producing the M2-1 protein; initiation of M2-2 takes place at a start codon that overlaps with the M2-1 open reading frame (ORF) (23, 28). Noteworthy, in the virion and infected cells, the viral genome is covered by the nucleoprotein N, which is highly expressed within infected cells (29–31). Covering the nucleocapsid, yet beneath the envelope is the matrix protein M, which has been reported to travel to the nucleus of infected cells during the replication cycle of hRSV to inhibit the transcription of host genes and was recently described to interact with actin within infected cells, likely contributing to the transport of virion components into budding virions (30, 32, 33). Importantly, the virion envelope is covered on its surface by the attachment glycoprotein G, which may be dispensable for infection in some cells (34–36), the fusion F glycoprotein which is a type-I integral membrane protein that binds nucleolin for cell infection (37, 38), and the transmembrane protein SH, which forms a viroporin that transports cationic ions (39, 40). Importantly, to date, there is accumulating data that describes the molecular interactions between hRSV structural components, which has allowed establishing an overall comprehensive scenario of how the virus' components are coordinately assembled within infected cells to favor its replication and exit (30). Finally, the non-structural (NS) genes NS1 and NS2 that are at the foremost 3′ of the viral genome, are solely expressed within infected cells (not contained within the virion), and are known to negatively modulate the cellular antiviral interferon type-I response early after infection (Figure 1) (41, 42). Importantly, several host factors that modulate the replication cycle of hRSV, such as factors involved in the regulation of host transcription, innate immune responses, regulation of the cytoskeleton, membrane remodeling, and cellular trafficking have been identified and confirmed. These factors could eventually be overexpressed or silenced in host cells to reduce infection or hamper virus replication during infection to avoid pathology (43–45). Although many of the abovementioned hRSV proteins have been studied individually in vitro, only few of them have been assessed within immune cells or more specifically DCs, which if performed could eventually reveal relevant immune-evasion or immune-modulation properties for hRSV-encoded viral factors and help identify key viral and host factors that modulate the virus' replication cycle in these cells.
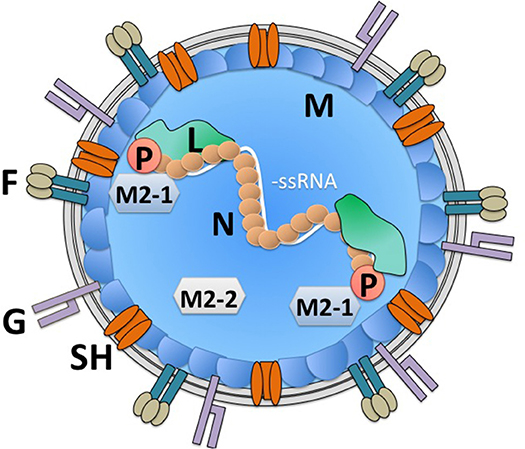
Figure 1. hRSV virion structure. The hRSV genome is a negative-sense single-stranded RNA virus. Its genome is wrapped by the nucleoprotein (N). An RNA-dependent RNA polymerase (L) is associated to the viral genome in the virion. Also, within the virion is the viral phosphoprotein (P), which is required by the L polymerase for its function. Additionally, within the virion are the viral proteins M2-1 and M2-2, derived from a single mRNA, which modulate transcription of viral genes and genome replication by the L polymerase. Beneath the virus envelope is the matrix protein (M), which has been described to inhibit host gene translation in the nucleus of infected cells. Three proteins are immersed in the virus envelope: the small hydrophobic (SH) protein, which forms a viroporin, that transports cations, the attachment glycoprotein (G) and the fusion protein, which arranges as a trimer on the virion surface (F). The viral proteins NS1 and NS2 are non-structural and hence, are not found within the virion.
Dendritic cells (DCs) are immune cells that play vital roles in initiating and regulating antigen-specific immune responses against foreign and self-antigens in the organism (46–48). DCs are strategically located both, at peripheral sites and internal organs in such a way to sense and capture both, foreign and self-proteins. If the captured protein is immunogenic or associated with activating molecules, DCs undergo phenotypic transformations, and migrate to lymph nodes (LNs) to present protein-derived peptides to antigen-specific CD8+ and CD4+ T cells in MHC-I and MHC-II molecules, respectively (49–52). Importantly, DCs express a battery of molecular sensors that detect pathogen-associated molecular patterns (PAMPs), which leads in most cases to transcriptional and phenotypical changes in these cells in a process known as DC maturation (46, 53, 54). In turn, this process will lead to the activation and modulation of other immune cells that can help resolve infection (49, 50, 55, 56). If DCs capture virus components DCs, these cells will ideally encounter, activate and differentiate virus-specific CD4+ T cells into helper cells (Th) that support the production of antiviral antibodies by B cells, as well as promote the generation of cytotoxic T cells (CTLs) that eliminate virus-infected cells (57–59). Given the crucial role of DCs in initiating antigen-specific adaptive antiviral immune responses, mainly through the activation and differentiation of T cells, such as CD4+ T helper cells, numerous viruses and other pathogens have evolved molecular determinants and mechanisms to interfere with the function of DCs, in such a way to impair the establishment of an effective antiviral immune response (60–67).
Noteworthy, hRSV infects DCs in vitro and is known to interfere with their functions, even though DCs seem not to be an optimal viral substrate for this virus. Indeed, many in vitro studies report relatively low virus yields from hRSV-infected DCs, even at multiplicity of infection (MOI) values that generally lead to complete infection of epithelial cell cultures (MOI >3) (66, 68–71). This phenomenon is suggestive of abortive hRSV infection in a significant proportion of DCs (66, 68, 69, 71, 72). Thus, it seems that hRSV likely infects DCs as a strategy to target a pivotal immune component to indirectly favor its infectious process in the host, namely the infection of epithelial lung cells that yield high amounts of infective virions, which will expand the magnitude of the infection within the individual and promote its dissemination onto others. Interestingly, hRSV may reach other tissues besides the airways during infection, such as the central nervous system (CNS) (73, 74).
Although cell surface receptors that lead to hRSV cell infection have been identified, such as cellular heparan sulfate glycosaminoglycans that act as attachment factors for the hRSV G glycoprotein (75, 76), as well as nucleolin (37) and ICAM1 (77) as ligands for the F fusion protein, the exact mechanism by which hRSV enters DCs has not been corroborated and could eventually be different compared to that observed in other cells, such as epithelial cells (78). Noteworthy, opsonized hRSV particles (hRSV covered with virus-specific antibodies), which is known to hamper virus-infection of epithelial cells, were recently shown to be nevertheless capable of infecting DCs and interfere with their function, such as activating T cells (Figure 2). Importantly, this process was shown to be mediated by Fcγ receptors (FcγRs) expressed on the surface of DCs (79). Because opsonized hRSV particles retained the same ability as free hRSV to interfere with DC activation of T cells, this process would favor impaired DC function in time despite the individual having anti-hRSV antibodies. Thus, hindered DC function by hRSV would ensue during each exposure to the virus, likely hampering the capacity of the host to mount an effective response against this virus.
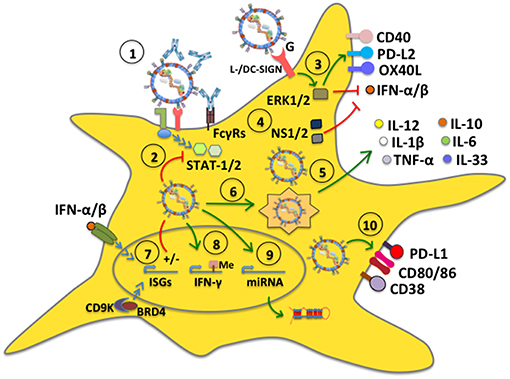
Figure 2. hRSV modulates dendritic cell function. (1) DC infection with hRSV can occur even in the presence of antibodies bound to the virus (opsonized virus), which enter DCs through Fcγ receptors (FcγRs). (2) hRSV is capable of inhibiting antiviral signaling pathways mediated by STAT-1 and STAT-2, likely through its NS proteins. (3) The G glycoprotein signals through L-/DC-SIGN and phosphorylates ERK1/2, which translates into the upregulation of surface expression of CD40, OX40L, and PD-L2, whereas it downregulates IFN-α secretion. (4) The hRSV NS1 and NS2 proteins interfere with type-I interferon secretion. (5) hRSV induces the secretion of proinflammatory cytokines by DCs. Some mDC subsets (BDCA-1+ and BDCA-3+) secrete IL-10. (6) hRSV induces autophagy and is processed by the autophagosome leading to cytokine release and lung inflammation. (7) hRSV differentially modulates the expression of interferon-stimulated genes (ISGs), through IFN-dependent and independent pathways. (8) hRSV induces the activity of demethylases to modulate gene expression, such as IFN-γ, preventing an antiviral response. (9) hRSV upregulates the expression of specific host microRNAs. (10) hRSV stimulates the expression of CD80 and CD86. Additionally, the virus upregulates PD-L1 and CD38 expression on the DC surface to modulate inflammation in the lungs.
Growing amounts of stuDies have focused on the relationship between hRSV and DCs increasing our knowledge on the outcome of this interaction. While some reports indicate that DCs infected with hRSV can sense viral components, which can lead to somewhat activating signaling pathways within these cells, other reports indicate that hRSV determinants interfere with antiviral signaling pathways within DCs, such as those related to interferon type-I responses, which is mediated by STAT-1 and STAT-2 (80). Importantly, the activation or inhibition of distinct intracellular signaling pathways within DCs by hRSV generally leads to DC outcomes that are overall poorly activating for T cells, with hRSV-infected DCs displaying poor- or only partial-maturation phenotypes both, in human and murine DCs (58, 59, 66, 68, 69, 72, 81).
NS1 and NS2 have been reported as two hRSV factors that are directly involved in inhibiting the maturation of human DCs and impairing the secretion of type-I IFNs by myeloid DCs (mDCs), which can enhance the differentiation of CD4+ T cells into Th2-phenotypes and promote the generation of Th2-polarized anti-hRSV immune responses in the host. Consequently, these immune responses can be detrimental to the host, as they promote exacerbated inflammation in the lungs (80, 82). Another hRSV-encoded determinant that has been reported to alter the maturation of DCs directly is the surface glycoprotein G, which was described to trigger ERK1 and ERK2 phosphorylation within these cells, mainly through DC- and L-SIGN molecules on the DC surface. Neutralization of DC- and L-SIGN induced significant secretion of IFN-α, MIP-1α, and MIP-1β in plasmacytoid DCs (pDCs) inoculated with hRSV, suggesting that this virus alters DC maturation through this signaling pathway thanks to this glycoprotein (Figure 2) (83). Such intracellular signaling events induced by the hRSV G glycoprotein in these cells may explain why mice immunized with a recombinant vaccinia virus (rVV) expressing hRSV G and subsequently challenged with hRSV displayed lung inflammatory DCs that expressed increased levels of the programmed cell death 1 ligand 2 (PD-L2), as well as low CD40 and OX40 ligand (OX40L), when compared to mice inoculated with a rVV expressing the hRSV F fusion protein, which were also challenged with hRSV. Noteworthy, the expression or not of these co-stimulatory molecules on the DC surface was shown to have profound effects over T cell activation, suggesting that the hRSV G glycoprotein has some important immune-modulatory properties, possibly mediated through DCs (84).
Other studies have found that hRSV infection promotes DC maturation and the secretion of pro-inflammatory cytokines by these cells, either directly or through the infection of other cells. For instance, primary human DCs characterized as mDC1, mDC2 or pDC were found to upregulate phenotypic markers associated to maturation after hRSV inoculation, which was dependent on divalent cations suggesting the participation of C-type lectin receptors in this process (71). Other human DC subsets studied with hRSV have been BDCA-1+ and BDCA-3+ mDCs obtained from peripheral blood. Similar to other DC subsets, these cells were susceptible to infection with hRSV, and while they expressed increased amounts of CD80 and CD86 in response to this virus as compared to non-infected cells, they also expressed the inhibitory costimulatory receptor PD-L1 and secreted IL-10. Furthermore, hRSV-infected BDCA-1+ mDCs produced pro-inflammatory cytokines and chemokines, namely IL-1β, IL-6, IL-12, MIP-1α, and TNF-α and displayed a reduced capacity to stimulate T cells (85). Hence, hRSV can produce significant changes in DCs once infected, namely by modulating the expression of T cell-activating molecules on their surface. etSuch modulation was accompanied by the expression of inhibitory receptors and the secretion of numerous immune-modulatory cytokines, mostly inflammatory.
Another study reported that depending on the hRSV strain used; human DCs can respond differentially to this virus by secreting different kinds of type-I and type-III IFNs, and transcribe distinct interferon-stimulated genes (ISGs). Although both serotypes of hRSV A and B induced the expressing of IFN-β, IFN-α1, IFN-α8, and IFN-λ1-3, only the serotype A2 induced IFN-α2, -α14, and -α21 (86). Type-I IFN-dependent activation of ISGs during an hRSV infection was shown to be modulated by the virus' ability to downregulate suppressor of cytokine signaling (SOCS1 and SOCS3) through its RSV G protein, in turn affecting IFN-β and ISG15 expression (87). Moreover, during hRSV infection, airway epithelial cells activate cyclin-dependent kinase 9 (CDK9) and associates with bromodomain 4 (BRD4) to activate IRF3-dependent IFN-stimulated genes, independent of IFN-signaling. Altogether, these processes contribute to increased RSV-induced airway inflammation and disease (88, 89). Although these findings may have important implications over disease severity and the outcome of the host's immune response, as well as the modulation of immunity, the implications of different hRSV serotypes in clinical infections and in vitro studies are somewhat seldom assessed.
IL-33 is a key cytokine involved in Th2 immune responses and inflammatory airway diseases and is usually secreted in high amounts by epithelial cells in this tissue (90, 91). Interestingly, hRSV-infected DCs within the lungs of hRSV-infected animals have been reported to have elevated levels of IL-33 mRNA and were suggested to be a relevant source of IL-33 in the lungs of hRSV-infected mice (92). Noteworthy, blocking TLR3 or TLR7 signaling with antagonists significantly reduced the levels of IL-33 mRNA produced by DCs, suggesting that IL-33 expression in these cells upon hRSV infection is TLR-dependent (92). Interestingly, a study on the identification of enzymes that alter the methylation status of the host DNA suggests that the profile of cytokines secreted by DCs in response to hRSV may be driven by specific demethylases induced by infection with this virus (Figure 2). In a study by Ptaschinski and colleagues, it was shown that hRSV upregulates the expression of Kdm5b/Jarid1b H3K4 demethylase in response to in vitro hRSV infection of DCs and that inhibiting this factor with siRNA led to a 10-fold increase in IFN-β production, as well as other cytokines. Furthermore, mice that had Kdm5b specifically deleted in DCs showed higher production of IFN-γ and reduced IL-4 and IL-5 secretion after hRSV infection, as well as lesser lung inflammatory mucus production in this tissue. Some of these findings were mirrored in human DCs treated with an inhibitor of KDM5B suggesting that this factor, which is induced by hRSV can directly inhibit the expression of type-I IFN and other cytokines within infected DCs, likely favoring hRSV replication and virus-induced lung disease (93). This finding calls for further studies assessing the roles of such DNA-modification enzymes in host cells and how they are modulated by hRSV infection, potentially unveiling novel antiviral strategies.
Studies that have assessed the role of autophagy in hRSV-infected DCs have found that this process is involved in driving the production of cytokines that lead to lung inflammation (Figure 2). Indeed, inhibition of autophagy with inhibitors such as siRNA, or experiments with Beclin+/− mouse-derived DCs, or exposing Beclin+/− mice to hRSV significantly reduced the production of cytokines by CD4+ T cells. In these cases, hRSV-infected lungs displayed increased amounts of mucus secretion, and cellular infiltrates, unveiling important roles for autophagy in DCs in response to hRSV infection (94, 95). Additionally, Beclin-1+/− DCs were shown to express reduced amounts of MHC class II molecules on their surface and were less effective at stimulating the production of IFN-γ and IL-17 in co-cultures with CD4+ T cells, as compared to controls; furthermore, they promoted the secretion of Th2-cytokines by these T cells. On the other hand, transfer of hRSV-infected Beclin-1+/− DCs into the airways of wild-type mice elicited lung disease accompanied with the production of significant amounts of Th2 cytokines upon later challenge with hRSV (94, 95). Notably, a recent study found that hRSV induces Sirtuin-1 (SIRT1) expression in DCs, which is a NAD(+)-dependent deacetylase that is associated with the induction of autophagy. In this study, it was found that SIRT1 exerts antiviral effects against hRSV in vitro and that using an inhibitor of this enzyme, siRNA o analyzing the specific effect of SIRT1 knockout in DCs not only attenuated autophagy in these cells, but these animals manifested exacerbated hRSV-pathology (96).
When searching for particular markers induced by hRSV infection in DCs or cytokines elicited by hRSV-infected DCs, a recent study found that this virus induces CD38 expression in these cells, which is an ectoenzyme that catalyzes the synthesis of cyclic ADPR (cADPR). The expression of this enzyme was found to be dependent on hRSV-induced type-I IFN and inhibitors of CD38 significantly reduced the expression of type-I/III IFNs, suggesting that CD38 is regulated by- and influences IFNs in DCs and thus, modulating this enzyme may be an intriguing target for improving the host's response to hRSV infection and pathology (97).
Despite poor, or relatively low expression of surface markers associated with the potential capacity of DCs to activate or promote the activation of T cells, a common feature that has been repeatedly observed in hRSV-infected DCs is the secretion of cytokines that may promote the differentiation of T cells into phenotypes that are not favorable for the effective resolution of infection, such as IL-6 and IL-10, which lead to Th2 CD4+ T cell responses (66, 81). Concomitantly, cytokines such as IL-12 that tend to elicit T cells with phenotypes that are commonly associated with efficient viral clearance, such as Th1 are usually not secreted by hRSV-infected DCs, (66, 68, 69, 98–101). Interestingly, a study reported that the secretion of different cytokine profiles by hRSV-infected human DCs depends on whether these cells originate from neonates or adults. For instance, DCs derived from blood cord samples secrete more TGF-β1 than DCs obtained from adult blood in response to hRSV, suggesting the existence of age-related phenotypes in DCs that may translate into differential responses to hRSV (further discussed below) (102).
Interestingly, a somewhat novel approach that is being undertaken to study the relationship between hRSV and DCs is analyzing the profile of miRNA expression in these cells (Figure 2). A recent study found that DC infection with hRSV elicited the upregulation of a specific miRNA, namely let-7b (103). This study is complemented by another report that found that hRSV infection induces significant expression of three miRNAs, namely hsa-miR-4448, hsa-miR-30a-5p, and hsa-miR-4634 in human DCs (104). Interestingly, this latter study also performed comparative analyses of miRNA profiles between DCs infected with hRSV and a related virus, namely the human metapneumovirus, and found that both viruses induced the expression of elevated levels of hsa-miR-4634. Elucidating the contribution of these miRNAs in DCs in response to hRSV remains to be determined.
Although the study of DC infection with hRSV in vitro has provided valuable insights on the consequences that hRSV infection has over these cells, studying the effects of hRSV over DCs at the site of infection is likely key for understanding the contribution of this interaction to airway disease. They are also important as they help determine if the results obtained in vitro mirror what occurs in the respiratory tissue. Interestingly, several studies have addressed the question of how DCs respond to lung infection with hRSV, yet only a few have directly assessed whether the analyzed DCs are actually infected with hRSV, or if the observed effects are driven by viral antigen or other factors in the virus-infected environment. Evidence for the participation of hRSV-infected DCs in the exacerbated inflammatory response to hRSV has been reported by the instillation of hRSV-infected DCs directly into the airways, which produced a pathological Th2-type response in mice (105). Regarding how DCs are infected by hRSV in vivo, a study by Ugonna et al. explored in an in vitro setting whether cells present in the respiratory tissues may contribute to hRSV access to DCs. Interestingly, by analyzing the interrelationship between DCs and epithelial cells, and their reciprocal infection in co-culture transwell assays they found that macrophages on the apical surface of differentiated epithelia helped hRSV infect DCs in the basal chamber, suggesting that lung macrophages may have a potentially relevant, and previously unknown role in DC infection with hRSV (106). However, this remains to be assessed and demonstrated in in vivo settings. Furthermore, other reports have analyzed whether cells that are usually adjacent to DCs in the infected tissues may influence the outcome of DCs. Interestingly, one study found that hRSV-infected rat airway epithelial cells elicited DC activation, increasing MHC-II and CD86 surface expression, as well as enhancing T cell proliferation in mixed lymphocyte reactions. Noteworthy, this activation was dependent on thymic stromal lymphopoietin (TSLP), a pleiotropic cytokine implicated in inflammatory diseases, which was secreted by hRSV-infected airway epithelial cells (Figure 3) (107, 108). On the other hand, airway DCs incubated with inflammatory mediators secreted by hRSV-infected lung epithelial cells was shown to induce their differentiation into functional DCs capable of activating T cells characterized by a type-I IFN antiviral response. Nevertheless, these DCs only had a partial mature phenotype, as they were unable to up-regulate CD80, CD83, CD86, and CCR7, and were unresponsive to TLR triggering, suggesting that the airway epithelium elicits DCs with a somewhat suppressive phenotype, even under inflammatory conditions induced in the lungs after infection with hRSV (109).
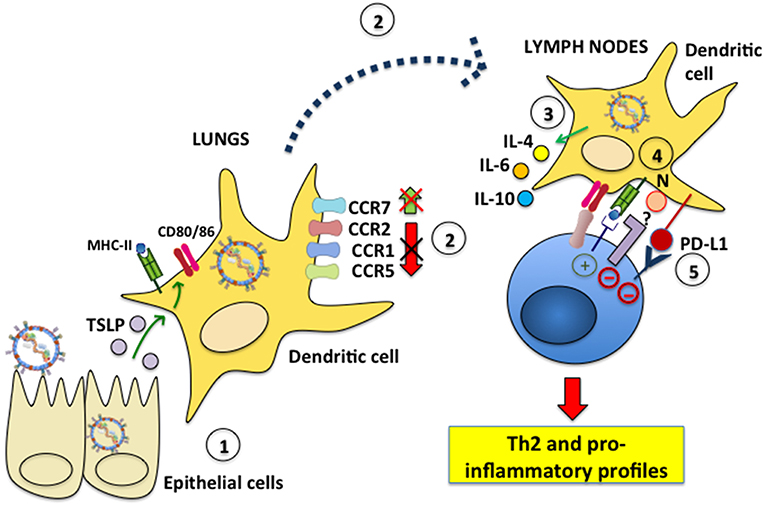
Figure 3. hRSV modulates dendritic cell migration and induces inflammatory profiles in T cells. (1) Epithelial cells infected with hRSV produce TSLP, which elicits MHC-II, CD80, and CD86 expression in lung DCs. (2) hRSV-infected DCs fail to downmodulate the surface expression of the chemokine receptors CCR1, CCR2 and CCR5, which is needed for effective DC migration to lymph nodes. Additionally, CCR7, which favors DC migration to the LNs, is not upregulated on the surface of hRSV-infected DCs. (3) Nevertheless, during infection hRSV-infected DCs migrate to the LNs to interact with T cells. hRSV-infected DCs secrete IL-4, IL-6, and IL-10 and promote the differentiation of CD4+ helper T cells toward a Th2 phenotype. (4) The hRSV N protein expressed on the DC surface mediates impaired immunological synapse assembly. The signaling events led by host proteins that interact with N are unknown. (5) hRSV induces PD-L1 expression on the DC surface which signals negatively within inflammatory T cells.
Given the existence of diverse types of DCs in the lung tissue, different studies have focused on analyzing the effects of hRSV over distinct subtypes of DCs in the respiratory tissue and their contribution to hRSV-associated lung pathology (110). Early studies on the dynamics of DCs in vivo in the lungs of hRSV-infected animals showed that pDCs accumulate in this tissue and secrete type-I IFNs, thus contributing to limit viral replication and the extension of pathology induced by hRSV infection (111, 112). Interestingly, other subsets besides pDCs, such as conventional DCs (cDCs) also accumulate in the lungs of hRSV-infected animals (113, 114). Noteworthy, together with increased accumulation of DCs in the lungs, an increase in the amount of these cells in the associated LNs also occurs, with DCs exhibiting varying phenotypes at this site (114–118). However, in most cases, it is unclear whether the analyzed DCs are infected by hRSV, or if their migration is influenced by other factors within the infected tissue, such as hRSV antigens or cytokines elicited in the infected tissue.
Importantly, differences in the phenotype of DCs obtained from the lungs of hRSV-infected animals have also been assessed based on the age of the individual, by analyzing these cells in the lungs of neonates and adults. One such study found that while CD103+ DCs dominated the response to hRSV in neonates, CD11b+ DCs were underrepresented in this group both, in number and function as compared to adult animals. For instance, pDCs from neonate animals display limited type-I IFN responses during hRSV infection, as compared to adult pDCs (119). Noteworthy, the transfer of adult pDCs into neonate animals reduced the Th2-biased immunopathology produced by hRSV which was elicited after a subsequent challenge with hRSV, further evidencing significant differences between DCs obtained from these different age-groups in response to hRSV infection (120). In line, with this notion, another study found that neonatal CD11b+ mDCs expressed increased levels of the IL-4 receptor IL-4Rα, as compared to adult DCs and that specifically deleting this cytokine receptor from CD11b+ mDCs significantly decreased hRSV-induced immunopathophysiology. Concomitantly, overexpression of IL-4Rα on the surface of CD11b+ DCs of adult animals and transferring them into adult mice elicited hRSV-induced immunopathology. Finally, an important finding in this study was also the fact that increased IL-4Rα expression in DCs was associated with reduced maturation of DCs during hRSV infection (121). Interestingly, another study found that age-dependent DC responses against hRSV could be modified through the use of TLR agonists, such as agonists for TLR4 or TLR9 at the time of infection. By using such agonists, a significant change in the response of hRSV-specific CD8+ T cells could be observed, evidenced as a shift in the immunodominance of the antigens to which these T cells responded when activated by neonate DCs. The shifted response found resembled more that was observed in adults, which is associated with less severe disease (122). Overall, the findings outlined above suggest particular and distinctive features between lung DCs from neonates and adults after hRSV infection, at least in the mouse model, and could be considered in the future for potential therapeutic and prophylaxis approaches in neonates and adults.
Another area of intense research regarding the interaction between hRSV and DCs is the migration of these cells, as hRSV lung infection may result in alterations on the of migration pattern of different subsets of DCs from the lungs to LNs. Interestingly, an in vitro study found that human monocyte-derived DCs infected with hRSV failed to downregulate CCR1, CCR2, and CCR5 from their surface, which is required for DCs to effectively migration to LNs. Indeed, these infected DCs migrated significantly less in chemokine gradients in in vitro assays. Furthermore, hRSV-infected DCs failed to upregulate CCR7 on their surface, which is known to promote the migration of antigen-exposed DCs to LNs for presenting antigens to T cells (123). Nevertheless, these findings need to be corroborated in hRSV-infected individuals. Even though cDCs accumulate in the lungs of hRSV-infected animals as mentioned above (113, 114), it has been reported that monocyte DC precursors are depleted during infection from this tissue. Importantly, this phenomenon has been suggested to favor opportunistic lung infections by pathogens, such as bacteria (116). Notably, two major subsets of lung tissue cDCs have been shown to transport hRSV RNA to the LNs, namely CD103+/CD11blow/CD11c+ and CD103−/CD11bhigh/CD11c+ cDCs and present hRSV antigens to T cells on MHC-I and MHC-II molecules (118). Interestingly, a study that was mentioned in the section above which analyzed the effects of TLR agonists over DCs infected with hRSV showed that the treatment with these molecules increased the numbers of CD11b+ and CD103+ DCs migrating from the lungs to draining LNs in neonates, likely supporting an improved antiviral response thanks to DCs with adult-like phenotypes migrating to this site for optimal T cell activation (122).
Although some studies suggest a positive role for lung cDCs during hRSV infection, other reports indicate that these cells may play detrimental functions for the host during hRSV infection (111, 113, 115). These effects have been evidenced, for example by blocking the chemokine CCL20 in hRSV-infected animals or knocking-out its associated receptor (CCR6), which significantly reduced the presence of cDCs in the airway tissue without affecting pDCs. These scenarios overall translated into improved outcomes of hRSV infection, suggesting that a balance between pDCs and cDCs in the lungs is likely associated with hRSV-induced pathology (114, 124). The finding supports this notion that depletion of pDCs from the lungs of animals significantly increases pulmonary disease after a challenge with hRSV (112). Concomitantly, activation of pDCs in the lungs of hRSV-infected animals was shown to limit the replication of hRSV in the airways and decrease hRSV-associated pathology (114). Thus, pDCs, as well as cDCs in the airways, are considered to interplay limiting hRSV replication and regulating inflammation (111, 112, 114). However, whether the findings described above in the mouse model hold in humans remains to be determined. Interestingly, some observations performed in animal models have been mirrored in patients, such as individuals experiencing hRSV bronchiolitis having significantly higher numbers of cDCs than pDCs in the blood, suggesting an imbalance in the proportion of DC subtypes in children with bronchiolitis, as compared to healthy individuals after hRSV infection (125).
A substantial effect of hRSV over DC function is its ability to reduce the capacity of hRSV-infected DCs to effectively activate CD4+ and CD8+ T cells (Figure 3). Although this phenomenon has been reported in vitro and is not necessarily echoed in vivo in humans or animal models, a relationship between the in vitro observations and potential in vivo effects likely exists in terms of non-optimal T cell activation taking place, as a result of DC infection with hRSV (66, 68, 72, 126). Given that the reported phenotype of hRSV-infected DCs is generally associated to weak or incomplete maturation, it is somewhat expected that the activation of T cells by infected DCs will not be optimal and may lead to less potent, or inadequately differentiated or polarized hRSV-specific T cells. Interestingly, some studies have identified particular hRSV factors that are involved in hampering the capacity of DCs to activate T cells. For instance, a report published in 2008 that studied the interaction between DCs and hRSV suggested that hRSV factors driving poor T cell activation by hRSV-infected DCs were membrane-bound and interfered with the establishment of the immunological synapse (IS) between T cells and hRSV-infected DCs in vitro, which is essential for productive T cell activation (49, 66). This notion was reinforced by the fact that supernatants from hRSV-infected DCs enhanced the activation of T cells in the presence of plate-bound activating anti-CD3 and anti-CD28 antibodies (66). However, another study suggested that soluble factors secreted by hRSV-infected DCs were involved in impaired T cell activation by hRSV-infected DCs (68, 99). It is possible thus that both, membrane-bound and soluble factors secreted on to the extracellular media, or at the DC-T cell immunological synapse negatively modulate the activation of T cells, given that the is highly susceptible to modulation by factors of both natures (49).
A membrane-bound hRSV factor that has been reported to mediate negative effects over T cells is the hRSV F fusion protein, which expressed on the surface of epithelial cells was shown to inhibit T cell activation in vitro (127). However, the effect of the hRSV F protein has not been assessed in the context of DC-T cell interactions. Still, a study that assessed the role of the hRSV N nucleoprotein in mediating detrimental effects over the DC-T cell interaction found that this protein was present on the surface of hRSV-infected DCs and could directly mediate the inhibition of T cell activation (128). An interesting finding in this study was the fact that the hRSV N protein was shown to be able to interfere with the establishment of productive immunological synapses between T cells and cognate ligand mounted on lipid bilayers (128). Importantly, the identification of the hRSV N protein on the surface of infected cells had not been previously reported for this virus. Another hRSV factor known to hamper the capacity of DCs to activate T cells is NS1, which has been found to negatively modulate the capacity of human DCs to activate both, CD4+ and CD8+ T cells (129). Additionally, NS1 has also been reported to favor the differentiation of DCs toward phenotypes that promote the activation of CD4+ T cells that secrete IL-4, yet by a mechanism that is independent of its capacity to modulate IFN-I signaling (129).
Again, depending on whether DCs inoculated with hRSV originate from neonate or adult animals, a study by Thornburg et al. reported differences in the capacity of such hRSV-infected DCs to activate autologous T cells, with DCs from adult mice eliciting IFN-γ, TNF-α, and IL-12 secretion in co-cultures and neonate DCs (from blood cords) eliciting IL-1β, IL-4, IL-6, and IL-17 release (102). Furthermore, neonatal CD103+ DCs have been shown to promote the proliferation of T cells differently, as compared to adult CD103+ DCs, namely by eliciting the expansion of T cells against distinct antigens, defining different hRSV antigenic hierarchal profiles. The differences observed with DCs from animals of different ages in this and other studies, suggest that neonatal DCs overall display limited T cell co-stimulatory properties when compared to adult DCs, which could eventually relate to infants being more susceptible to severe disease than adults after hRSV infection (119). Another report found two phenotypically and functionally distinct populations of CD103+ DCs in the lungs of neonatal mice following hRSV infection, and that those that were CD103lo were functionally limited at activating hRSV-specific T cells, while those that were CD103hi were capable of potently activating T cells (130). Whether such differences mirror the adaptive immune responses to hRSV in vivo in both adults and infant humans, remains to be determined.
Although several studies have reported impaired T cell activation by hRSV-infected DCs in vitro, in vivo studies reveal that hRSV-specific T cells are expanded in the organism after infection, yet they generally display pro-inflammatory phenotypes that likely contribute, or are the root of exacerbated lung damage during hRSV infection (131–133). Thus, reduced activation of T cells in vitro seems to translate in vivo as the activation of virus-specific T cells with detrimental phenotypes that respond to an hRSV lung infection. Yet, a role for hRSV-infected DCs has also been described in regulating or controlling pathogenic T cells during infection. Analyses of human and murine lung DCs report that these cells express PD-L1 and that this molecule is critical for suppressing the activity of inflammatory T cells (Figure 3). This finding suggests a vital role for the PD-L1/PD-1 axis in DC-T cell interactions for limiting the inflammatory response of T cells to hRSV (134). Nevertheless, the results of this study contrast with those of another report that found that hRSV inhibited the capacity of pDCs to produce a regulatory T cell response to inhaled antigens, eliciting an alteration in their immunotolerogenic potential (135).
Because of the key role of DCs in mounting and regulating immune responses to viruses such as hRSV, novel vaccines are needed to strategically seek and target these cells in a specific manner. A recently described approach that directly involves DCs consists on a DNA vaccine encoding the ectodomain of the hRSV F protein fused to a single-chain variable fragment F (scFv) that directly targets the viral antigen to DEC205 on the DC surface. This viral protein is then phagocytosis by DCs through this receptor and processed for antigen presentation to T cells. Interestingly, this strategy has been reported to elicit high levels of anti-hRSV antibodies with neutralizing capacity and induce F-specific CD8+ T cells that elicit a Th1 response in mice (136).
Over the last years, new studies have revealed novel features of the DC-hRSV interaction, providing unanticipated outcomes in DCs after infection with this virus and helping identify different host and viral factors that participate in these processes. Because DCs play pivotal roles in initiating and regulating antigen-specific immune responses to infections, it seems relevant that particular focus should be given to these cells both, before and after interacting with hRSV. Indeed, these cells are needed for establishing an effective antiviral immune response in the lungs to promote viral clearance, while altogether avoid exacerbated inflammation of the airways. The fact that hRSV can repeatedly reinfect the host without the need of varying its antigens calls for special attention to the steps that determine the founding events of the host antiviral response, in such a way to train the immune system to withstand the negative immune-modulatory properties of this virus or counteract its potent Th-skewing effects. In both cases, a protective immune response elicited against hRSV, such as one that could be induced by a vaccine, should be strong enough to bear subsequent viral reinfections that will push to revert this outcome and elicit scenarios that are favorable for the virus. Overall, significant efforts should be invested in identifying viral and host factors that hamper hRSV-infected DCs, or bystander DCs in the infected tissue from promoting effective antiviral immune responses against this virus. Importantly, promoting positive hRSV-DC interactions during re-infections, after virus-specific immune components have already been polarized toward detrimental phenotypes by hRSV may be more complicated than promoting an effective immune response before primary infection. Indeed, shifting a pre-existing antigen-specific immune profile has proven somewhat challenging in the context of other diseases, such as cancer and autoimmunity, although the antigens involved in these pathologies are seldom known, which is not the case for hRSV.
Finally, some questions that remain open regarding the roles of DCs in hRSV infection are: How can we enable hRSV-infected DCs to elicit effective antiviral immune responses during primary infection and re-infections against this virus? Are there hRSV factors, or hRSV-induced factors elicited during reinfections that revert, through their effects over DCs, otherwise effective primary anti-hRSV immune responses? Do the different circulating hRSV A and B genotypes affect the outcomes of hRSV-infected DCs equally? What are the roles of hRSV-infected and non-infected DCs in the lungs of hRSV-infected individuals? Do the findings reported in the mouse model hold in humans? Hopefully, answers to these and many other questions regarding DCs and their interaction with hRSV will provide novel insights that will help limit the burden and mortality associated with the epidemiology of this important respiratory virus.
PG and SB wrote, revised, and edited the article and figures. ET wrote and revised the article and drew the figures.
Authors are supported by FONDECYT (Fondo Nacional de Desarrollo Científico y Tecnológico) grants #1190864 and #1170964 from the Comisión Nacional de Investigación Científica y Tecnológica (CONICYT), INNOVA-CORFO grant 13CTI-21526 and the Millennium Institute on Immunology and Immunotherapy (MIII), grant #P09-016-F.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
hRSV, Human respiratory syncytial virus; DCs, dendritic cells; IL, interleukin, LNs, lymph nodes.
1. Shi T, Mcallister DA, O'brien KL, Simoes EAF, Madhi SA, Gessner BD, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. (2017) 390:946–58. doi: 10.1016/S0140-6736(17)30938-8
2. Ackerson B, Tseng HF, Sy LS, Solano Z, Slezak J, Luo Y, et al. Severe morbidity and mortality associated with respiratory syncytial virus versus influenza infection in hospitalized older adults. Clin Infect Dis. (2018). doi: 10.1093/cid/ciy991
3. Haber N. Respiratory syncytial virus infection in elderly adults. Med Mal Infect. (2018) 48:377–82. doi: 10.1016/j.medmal.2018.01.008
4. Welliver RC. Review of epidemiology and clinical risk factors for severe respiratory syncytial virus (RSV) infection. J Pediatr. (2003) 143:S112–7. doi: 10.1067/S0022-3476(03)00508-0
5. Manzoni P, Figueras-Aloy J, Simoes EAF, Checchia PA, Fauroux B, Carbonell-Estrany X, et al. Defining the incidence and associated morbidity and mortality of severe respiratory syncytial virus infection among children with chronic diseases. Infect Dis Ther. (2017) 6:383–411. doi: 10.1007/s40121-017-0160-3
6. Walsh EE. Respiratory syncytial virus infection in adults. Semin Respir Crit Care Med. (2011) 32:423–32. doi: 10.1055/s-0031-1283282
7. Alimi Y, Lim WS, Lansbury L, Leonardi-Bee J, Nguyen-Van-Tam JS. Systematic review of respiratory viral pathogens identified in adults with community-acquired pneumonia in Europe. J Clin Virol. (2017) 95:26–35. doi: 10.1016/j.jcv.2017.07.019
8. Bueno SM, Gonzalez PA, Cautivo KM, Mora JE, Leiva ED, Tobar HE, et al. Protective T cell immunity against respiratory syncytial virus is efficiently induced by recombinant BCG. Proc Natl Acad Sci USA. (2008) 105:20822–7. doi: 10.1073/pnas.0806244105
9. Cespedes PF, Rey-Jurado E, Espinoza JA, Rivera CA, Canedo-Marroquin G, Bueno SM, et al. A single, low dose of a cGMP recombinant BCG vaccine elicits protective T cell immunity against the human respiratory syncytial virus infection and prevents lung pathology in mice. Vaccine. (2017) 35:757–66. doi: 10.1016/j.vaccine.2016.12.048
10. Rey-Jurado E, Soto J, Galvez N, Kalergis AM. A safe and efficient BCG vectored vaccine to prevent the disease caused by the human respiratory syncytial virus. Hum Vaccin Immunother. (2017) 13:2092–7. doi: 10.1080/21645515.2017.1334026
11. Mazur NI, Higgins D, Nunes MC, Melero JA, Langedijk AC, Horsley N, et al. The respiratory syncytial virus vaccine landscape: lessons from the graveyard and promising candidates. Lancet Infect Dis. (2018) 18:e295–311. doi: 10.1016/S1473-3099(18)30292-5
12. Cane PA. Molecular epidemiology of respiratory syncytial virus. Rev Med Virol. (2001) 11:103–16. doi: 10.1002/rmv.305
13. O'shea MK, Ryan MA, Hawksworth AW, Alsip BJ, Gray GC. Symptomatic respiratory syncytial virus infection in previously healthy young adults living in a crowded military environment. Clin Infect Dis. (2005) 41:311–7. doi: 10.1086/431591
14. Obodai E, Odoom JK, Adiku T, Goka B, Wolff T, Biere B, et al. The significance of human respiratory syncytial virus (HRSV) in children from Ghana with acute lower respiratory tract infection: a molecular epidemiological analysis, 2006 and 2013-2014. PLoS ONE. (2018) 13:e0203788. doi: 10.1371/journal.pone.0203788
15. Pangesti KNA, Abd El Ghany M, Walsh MG, Kesson AM, Hill-Cawthorne GA. Molecular epidemiology of respiratory syncytial virus. Rev Med Virol. (2018) 28:e1968. doi: 10.1002/rmv.1968
16. Hall CB, Walsh EE, Long CE, Schnabel KC. Immunity to and frequency of reinfection with respiratory syncytial virus. J Infect Dis. (1991) 163:693–8. doi: 10.1093/infdis/163.4.693
17. Gonzalez PA, Bueno SM, Carreno LJ, Riedel CA, Kalergis AM. Respiratory syncytial virus infection and immunity. Rev Med Virol. (2012) 22:230–44. doi: 10.1002/rmv.1704
18. Acosta PL, Caballero MT, Polack FP. Brief history and characterization of enhanced respiratory syncytial virus disease. Clin Vaccine Immunol. (2015) 23:189–95. doi: 10.1128/CVI.00609-15
19. Bohmwald K, Espinoza JA, Rey-Jurado E, Gomez RS, Gonzalez PA, Bueno SM, et al. Human respiratory syncytial virus: infection and pathology. Semin Respir Crit Care Med. (2016) 37:522–37. doi: 10.1055/s-0036-1584799
20. Restori KH, Srinivasa BT, Ward BJ, Fixman ED. Neonatal immunity, respiratory virus infections, and the development of asthma. Front Immunol. (2018) 9:1249. doi: 10.3389/fimmu.2018.01249
21. Staples KJ, Nicholas B, Mckendry RT, Spalluto CM, Wallington JC, Bragg CW, et al. Viral infection of human lung macrophages increases PDL1 expression via IFNβ. PLoS ONE. (2015) 10:e0121527. doi: 10.1371/journal.pone.0121527
22. Zhivaki D, Lemoine S, Lim A, Morva A, Vidalain PO, Schandene L, et al. Respiratory Syncytial virus infects regulatory B cells in human neonates via chemokine receptor CX3CR1 and promotes lung disease severity. Immunity. (2017) 46:301–14. doi: 10.1016/j.immuni.2017.01.010
23. Lay MK, Gonzalez PA, Leon MA, Cespedes PF, Bueno SM, Riedel CA, et al. Advances in understanding respiratory syncytial virus infection in airway epithelial cells and consequential effects on the immune response. Microbes Infect. (2013) 15:230–42. doi: 10.1016/j.micinf.2012.11.012
24. Braun MR, Deflube LR, Noton SL, Mawhorter ME, Tremaglio CZ, Fearns R. RNA elongation by respiratory syncytial virus polymerase is calibrated by conserved region V. PLoS Pathog. (2017) 13:e1006803. doi: 10.1371/journal.ppat.1006803
25. Cox RM, Toots M, Yoon JJ, Sourimant J, Ludeke B, Fearns R, et al. Development of an allosteric inhibitor class blocking RNA elongation by the respiratory syncytial virus polymerase complex. J Biol Chem. (2018) 293:16761–77. doi: 10.1074/jbc.RA118.004862
26. Asenjo A, Villanueva N. Phosphorylation of the human respiratory syncytial virus P protein mediates M2-2 regulation of viral RNA synthesis, a process that involves two P proteins. Virus Res. (2016) 211:117–25. doi: 10.1016/j.virusres.2015.10.011
27. Pereira N, Cardone C, Lassoued S, Galloux M, Fix J, Assrir N, et al. New insights into structural disorder in human respiratory syncytial virus phosphoprotein and implications for binding of protein partners. J Biol Chem. (2017) 292:2120–31. doi: 10.1074/jbc.M116.765958
28. Selvaraj M, Yegambaram K, Todd E, Richard CA, Dods RL, Pangratiou GM, et al. The structure of the human respiratory syncytial virus M2-1 protein bound to the interaction domain of the phosphoprotein P defines the orientation of the complex. MBio. (2018) 9: e01554–18. doi: 10.1128/mBio.01554-18
29. Bakker SE, Duquerroy S, Galloux M, Loney C, Conner E, Eleouet JF, et al. The respiratory syncytial virus nucleoprotein-RNA complex forms a left-handed helical nucleocapsid. J Gen Virol. (2013) 94:1734–8. doi: 10.1099/vir.0.053025-0
30. Gonzalez PA, Carreno LJ, Bueno SM, Riedel CA, Kalergis AM. Understanding respiratory syncytial virus infection to improve treatment and immunity. Curr Mol Med. (2013) 13:1122–39. doi: 10.2174/1566524011313070007
31. Galloux M, Gabiane G, Sourimant J, Richard CA, England P, Moudjou M, et al. Identification and characterization of the binding site of the respiratory syncytial virus phosphoprotein to RNA-free nucleoprotein. J Virol. (2015) 89:3484–96. doi: 10.1128/JVI.03666-14
32. Ward C, Maselko M, Lupfer C, Prescott M, Pastey MK. Interaction of the human respiratory syncytial virus matrix protein with cellular adaptor protein complex 3 plays a critical role in trafficking. PLoS ONE. (2017) 12:e0184629. doi: 10.1371/journal.pone.0184629
33. Shahriari S, Wei KJ, Ghildyal R. Respiratory syncytial virus matrix (M) protein interacts with actin in vitro and in cell culture. Viruses. (2018) 10:E535. doi: 10.3390/v10100535
34. Karron RA, Buonagurio DA, Georgiu AF, Whitehead SS, Adamus JE, Clements-Mann ML, et al. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B mutant. Proc Natl Acad Sci USA. (1997) 94:13961–6. doi: 10.1073/pnas.94.25.13961
35. Techaarpornkul S, Barretto N, Peeples ME. Functional analysis of recombinant respiratory syncytial virus deletion mutants lacking the small hydrophobic and/or attachment glycoprotein gene. J Virol. (2001) 75:6825–34. doi: 10.1128/JVI.75.15.6825-6834.2001
36. Fedechkin SO, George NL, Wolff JT, Kauvar LM, Dubois RM. Structures of respiratory syncytial virus G antigen bound to broadly neutralizing antibodies. Sci Immunol. (2018) 3:eaar3534. doi: 10.1126/sciimmunol.aar3534
37. Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat Med. (2011) 17:1132–5. doi: 10.1038/nm.2444
38. Bilawchuk LM, Griffiths CD, Jensen LD, Elawar F, Marchant DJ. The susceptibilities of respiratory syncytial virus to nucleolin receptor blocking and antibody neutralization are dependent upon the method of virus purification. Viruses. (2017) 9:207. doi: 10.3390/v9080207
39. Gan SW, Ng L, Lin X, Gong X, Torres J. Structure and ion channel activity of the human respiratory syncytial virus (hRSV) small hydrophobic protein transmembrane domain. Protein Sci. (2008) 17:813–20. doi: 10.1110/ps.073366208
40. Araujo GC, Silva RH, Scott LP, Araujo AS, Souza FP, De Oliveira RJ. Structure and functional dynamics characterization of the ion channel of the human respiratory syncytial virus (hRSV) small hydrophobic protein (SH) transmembrane domain by combining molecular dynamics with excited normal modes. J Mol Model. (2016) 22:286. doi: 10.1007/s00894-016-3150-6
41. Spann KM, Tran KC, Collins PL. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J Virol. (2005) 79:5353–62. doi: 10.1128/JVI.79.9.5353-5362.2005
42. Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, et al. Respiratory syncytial virus NS1 protein degrades STAT2 by using the elongin-Cullin E3 ligase. J Virol. (2007) 81:3428–36. doi: 10.1128/JVI.02303-06
43. Kipper S, Hamad S, Caly L, Avrahami D, Bacharach E, Jans DA, et al. New host factors important for respiratory syncytial virus (RSV) replication revealed by a novel microfluidics screen for interactors of matrix (M) protein. Mol Cell Proteom. (2015) 14:532–43. doi: 10.1074/mcp.M114.044107
44. Pokharel SM, Shil NK, Bose S. Autophagy, TGF-β, and SMAD-2/3 signaling regulates interferon-β response in respiratory syncytial virus infected macrophages. Front Cell Infect Microbiol. (2016) 6:174. doi: 10.3389/fcimb.2016.00174
45. Mehedi M, Collins PL, Buchholz UJ. A novel host factor for human respiratory syncytial virus. Commun Integr Biol. (2017) 10:e1319025. doi: 10.1080/19420889.2017.1319025
46. Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. (2013) 31:563–604. doi: 10.1146/annurev-immunol-020711-074950
47. Said A, Weindl G. Regulation of dendritic cell function in inflammation. J Immunol Res. (2015) 2015:743169. doi: 10.1155/2015/743169
48. Audiger C, Rahman MJ, Yun TJ, Tarbell KV, Lesage S. The importance of dendritic cells in maintaining immune tolerance. J Immunol. (2017) 198:2223–31. doi: 10.4049/jimmunol.1601629
49. Gonzalez PA, Carreno LJ, Figueroa CA, Kalergis AM. Modulation of immunological synapse by membrane-bound and soluble ligands. Cytokine Growth Factor Rev. (2007) 18:19–31. doi: 10.1016/j.cytogfr.2007.01.003
50. Carreno LJ, Gonzalez PA, Bueno SM, Riedel CA, Kalergis AM. Modulation of the dendritic cell-T-cell synapse to promote pathogen immunity and prevent autoimmunity. Immunotherapy. (2011) 3:6–11. doi: 10.2217/imt.11.38
51. Alloatti A, Kotsias F, Magalhaes JG, Amigorena S. Dendritic cell maturation and cross-presentation: timing matters! Immunol Rev. (2016) 272:97–108. doi: 10.1111/imr.12432
52. Worbs T, Hammerschmidt SI, Forster R. Dendritic cell migration in health and disease. Nat Rev Immunol. (2017) 17:30–48. doi: 10.1038/nri.2016.116
53. Arnold-Schrauf C, Berod L, Sparwasser T. Dendritic cell specific targeting of MyD88 signalling pathways in vivo. Eur J Immunol. (2015) 45:32–9. doi: 10.1002/eji.201444747
54. Macri C, Pang ES, Patton T, O'keeffe M. Dendritic cell subsets. Semin Cell Dev Biol. (2018) 84:11–21. doi: 10.1016/j.semcdb.2017.12.009
55. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
56. Peron G, De Lima Thomaz L, Camargo Da Rosa L, Thome R, Cardoso Verinaud LM. Modulation of dendritic cell by pathogen antigens: where do we stand? Immunol Lett. (2018) 196:91–102. doi: 10.1016/j.imlet.2018.02.001
57. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
58. Steinman RM, Hemmi H. Dendritic cells: translating innate to adaptive immunity. Curr Top Microbiol Immunol. (2006) 311:17–58. doi: 10.1007/3-540-32636-7_2
59. Kawai T, Akira S. Antiviral signaling through pattern recognition receptors. J Biochem. (2007) 141:137–45. doi: 10.1093/jb/mvm032
60. Morrow G, Slobedman B, Cunningham AL, Abendroth A. Varicella-zoster virus productively infects mature dendritic cells and alters their immune function. J Virol. (2003) 77:4950–9. doi: 10.1128/JVI.77.8.4950-4959.2003
61. Tobar JA, Gonzalez PA, Kalergis AM. Salmonella escape from antigen presentation can be overcome by targeting bacteria to Fc gamma receptors on dendritic cells. J Immunol. (2004) 173:4058–65. doi: 10.4049/jimmunol.173.6.4058
62. Majumder B, Janket ML, Schafer EA, Schaubert K, Huang XL, Kan-Mitchell J, et al. Human immunodeficiency virus type 1 Vpr impairs dendritic cell maturation and T-cell activation: implications for viral immune escape. J Virol. (2005) 79:7990–8003. doi: 10.1128/JVI.79.13.7990-8003.2005
63. Raftery MJ, Winau F, Kaufmann SH, Schaible UE, Schonrich G. CD1 antigen presentation by human dendritic cells as a target for herpes simplex virus immune evasion. J Immunol. (2006) 177:6207–14. doi: 10.4049/jimmunol.177.9.6207
64. Tobar JA, Carreno LJ, Bueno SM, Gonzalez PA, Mora JE, Quezada SA, et al. Virulent Salmonella enterica serovar typhimurium evades adaptive immunity by preventing dendritic cells from activating T cells. Infect Immun. (2006) 74:6438–48. doi: 10.1128/IAI.00063-06
65. Bueno SM, Gonzalez PA, Carreno LJ, Tobar JA, Mora GC, Pereda CJ, et al. The capacity of Salmonella to survive inside dendritic cells and prevent antigen presentation to T cells is host specific. Immunology. (2008) 124:522–33. doi: 10.1111/j.1365-2567.2008.02805.x
66. Gonzalez PA, Prado CE, Leiva ED, Carreno LJ, Bueno SM, Riedel CA, et al. Respiratory syncytial virus impairs T cell activation by preventing synapse assembly with dendritic cells. Proc Natl Acad Sci USA. (2008) 105:14999–5004. doi: 10.1073/pnas.0802555105
67. Retamal-Diaz A, Weiss KA, Tognarelli EI, Freire M, Bueno SM, Herold BC, et al. US6 gene deletion in herpes simplex virus Type 2 enhances dendritic cell function and T cell activation. Front Immunol. (2017) 8:1523. doi: 10.3389/fimmu.2017.01523
68. De Graaff PM, De Jong EC, Van Capel TM, Van Dijk ME, Roholl PJ, Boes J, et al. Respiratory syncytial virus infection of monocyte-derived dendritic cells decreases their capacity to activate CD4 T cells. J Immunol. (2005) 175:5904–11. doi: 10.4049/jimmunol.175.9.5904
69. Guerrero-Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME, et al. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol. (2006) 34:320–9. doi: 10.1165/rcmb.2005-0287OC
70. Jones A, Morton I, Hobson L, Evans GS, Everard ML. Differentiation and immune function of human dendritic cells following infection by respiratory syncytial virus. Clin Exp Immunol. (2006) 143:513–22. doi: 10.1111/j.1365-2249.2005.03004.x
71. Johnson TR, Johnson CN, Corbett KS, Edwards GC, Graham BS. Primary human mDC1, mDC2, and pDC dendritic cells are differentially infected and activated by respiratory syncytial virus. PLoS ONE. (2011) 6:e16458. doi: 10.1371/journal.pone.0016458
72. Le Nouen C, Munir S, Losq S, Winter CC, Mccarty T, Stephany DA, et al. Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology. (2009) 385:169–82. doi: 10.1016/j.virol.2008.11.043
73. Espinoza JA, Bohmwald K, Cespedes PF, Gomez RS, Riquelme SA, Cortes CM, et al. Impaired learning resulting from respiratory syncytial virus infection. Proc Natl Acad Sci USA. (2013) 110:9112–7. doi: 10.1073/pnas.1217508110
74. Bohmwald K, Espinoza JA, Gonzalez PA, Bueno SM, Riedel CA, Kalergis AM. Central nervous system alterations caused by infection with the human respiratory syncytial virus. Rev Med Virol. (2014) 24:407–19. doi: 10.1002/rmv.1813
75. Feldman SA, Hendry RM, Beeler JA. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J Virol. (1999) 73:6610–7.
76. Hallak LK, Collins PL, Knudson W, Peeples ME. Iduronic acid-containing glycosaminoglycans on target cells are required for efficient respiratory syncytial virus infection. Virology. (2000) 271:264–75. doi: 10.1006/viro.2000.0293
77. Behera AK, Matsuse H, Kumar M, Kong X, Lockey RF, Mohapatra SS. Blocking intercellular adhesion molecule-1 on human epithelial cells decreases respiratory syncytial virus infection. Biochem Biophys Res Commun. (2001) 280:188–95. doi: 10.1006/bbrc.2000.4093
78. Mclellan JS, Ray WC, Peeples ME. Structure and function of respiratory syncytial virus surface glycoproteins. Curr Top Microbiol Immunol. (2013) 372:83–104. doi: 10.1007/978-3-642-38919-1_4
79. Gomez RS, Ramirez BA, Cespedes PF, Cautivo KM, Riquelme SA, Prado CE, et al. Contribution of Fcγ receptors to human respiratory syncytial virus pathogenesis and the impairment of T-cell activation by dendritic cells. Immunology. (2016) 147:55–72. doi: 10.1111/imm.12541
80. Jie Z, Dinwiddie DL, Senft AP, Harrod KS. Regulation of STAT signaling in mouse bone marrow derived dendritic cells by respiratory syncytial virus. Virus Res. (2011) 156:127–33. doi: 10.1016/j.virusres.2011.01.007
81. Boogaard I, Van Oosten M, Van Rijt LS, Muskens F, Kimman TG, Lambrecht BN, et al. Respiratory syncytial virus differentially activates murine myeloid and plasmacytoid dendritic cells. Immunology. (2007) 122:65–72. doi: 10.1111/j.1365-2567.2007.02613.x
82. Munir S, Le Nouen C, Luongo C, Buchholz UJ, Collins PL, Bukreyev A. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J Virol. (2008) 82:8780–96. doi: 10.1128/JVI.00630-08
83. Johnson TR, Mclellan JS, Graham BS. Respiratory syncytial virus glycoprotein G interacts with DC-SIGN and L-SIGN to activate ERK1 and ERK2. J Virol. (2012) 86:1339–47. doi: 10.1128/JVI.06096-11
84. Wythe SE, Dodd JS, Openshaw PJ, Schwarze J. OX40 ligand and programmed cell death 1 ligand 2 expression on inflammatory dendritic cells regulates CD4 T cell cytokine production in the lung during viral disease. J Immunol. (2012) 188:1647–55. doi: 10.4049/jimmunol.1103001
85. Gupta MR, Kolli D, Garofalo RP. Differential response of BDCA-1+ and BDCA-3+ myeloid dendritic cells to respiratory syncytial virus infection. Respir Res. (2013) 14:71. doi: 10.1186/1465-9921-14-71
86. Hillyer P, Mane VP, Chen A, Dos Santos MB, Schramm LM, Shepard RE, et al. Respiratory syncytial virus infection induces a subset of types I and III interferons in human dendritic cells. Virology. (2017) 504:63–72. doi: 10.1016/j.virol.2017.01.017
87. Moore EC, Barber J, Tripp RA. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol J. (2008) 5:116. doi: 10.1186/1743-422X-5-116
88. Tian B, Zhao Y, Kalita M, Edeh CB, Paessler S, Casola A, et al. CDK9-dependent transcriptional elongation in the innate interferon-stimulated gene response to respiratory syncytial virus infection in airway epithelial cells. J Virol. (2013) 87:7075–92. doi: 10.1128/JVI.03399-12
89. Tian B, Yang J, Zhao Y, Ivanciuc T, Sun H, Garofalo RP, et al. BRD4 Couples NF-kappaB/RelA with airway inflammation and the IRF-RIG-I amplification loop in respiratory syncytial virus infection. J Virol. (2017) 91:e00007–17. doi: 10.1128/JVI.00007-17
90. Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, et al. Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. (2013) 123:3967–82. doi: 10.1172/JCI65570
91. Hardman CS, Panova V, Mckenzie AN. IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur J Immunol. (2013) 43:488–98. doi: 10.1002/eji.201242863
92. Qi F, Wang D, Liu J, Zeng S, Xu L, Hu H, et al. Respiratory macrophages and dendritic cells mediate respiratory syncytial virus-induced IL-33 production in TLR3- or TLR7-dependent manner. Int Immunopharmacol. (2015) 29:408–15. doi: 10.1016/j.intimp.2015.10.022
93. Ptaschinski C, Mukherjee S, Moore ML, Albert M, Helin K, Kunkel SL, et al. RSV-induced H3K4 demethylase KDM5B leads to regulation of dendritic cell-derived innate cytokines and exacerbates pathogenesis in vivo. PLoS Pathog. (2015) 11:e1004978. doi: 10.1371/journal.ppat.1004978
94. Morris S, Swanson MS, Lieberman A, Reed M, Yue Z, Lindell DM, et al. Autophagy-mediated dendritic cell activation is essential for innate cytokine production and APC function with respiratory syncytial virus responses. J Immunol. (2011) 187:3953–61. doi: 10.4049/jimmunol.1100524
95. Reed M, Morris SH, Jang S, Mukherjee S, Yue Z, Lukacs NW. Autophagy-inducing protein beclin-1 in dendritic cells regulates CD4 T cell responses and disease severity during respiratory syncytial virus infection. J Immunol. (2013) 191:2526–37. doi: 10.4049/jimmunol.1300477
96. Owczarczyk AB, Schaller MA, Reed M, Rasky AJ, Lombard DB, Lukacs NW. Sirtuin 1 regulates dendritic cell activation and autophagy during respiratory syncytial virus-induced immune responses. J Immunol. (2015) 195:1637–46. doi: 10.4049/jimmunol.1500326
97. Schiavoni I, Scagnolari C, Horenstein AL, Leone P, Pierangeli A, Malavasi F, et al. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology. (2018) 154:122–31. doi: 10.1111/imm.12873
98. Bartz H, Turkel O, Hoffjan S, Rothoeft T, Gonschorek A, Schauer U. Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-gamma in naive T cells. Immunology. (2003) 109:49–57. doi: 10.1046/j.1365-2567.2003.01629.x
99. Chi B, Dickensheets HL, Spann KM, Alston MA, Luongo C, Dumoutier L, et al. Alpha and lambda interferon together mediate suppression of CD4 T cells induced by respiratory syncytial virus. J Virol. (2006) 80:5032–40. doi: 10.1128/JVI.80.10.5032-5040.2006
100. Vallbracht S, Unsold H, Ehl S. Functional impairment of cytotoxic T cells in the lung airways following respiratory virus infections. Eur J Immunol. (2006) 36:1434–42. doi: 10.1002/eji.200535642
101. Bueno SM, Gonzalez PA, Pacheco R, Leiva ED, Cautivo KM, Tobar HE, et al. Host immunity during RSV pathogenesis. Int Immunopharmacol. (2008) 8:1320–9. doi: 10.1016/j.intimp.2008.03.012
102. Thornburg NJ, Shepherd B, Crowe JEJr. Transforming growth factor beta is a major regulator of human neonatal immune responses following respiratory syncytial virus infection. J Virol. (2010) 84:12895–902. doi: 10.1128/JVI.01273-10
103. Thornburg NJ, Hayward SL, Crowe JEJr. Respiratory syncytial virus regulates human microRNAs by using mechanisms involving beta interferon and NF-kappaB. MBio. (2012) 3:e00220–12. doi: 10.1128/mBio.00220-12
104. Banos-Lara MDR, Zabaleta J, Garai J, Baddoo M, Guerrero-Plata A. Comparative analysis of miRNA profile in human dendritic cells infected with respiratory syncytial virus and human metapneumovirus. BMC Res Notes. (2018) 11:432. doi: 10.1186/s13104-018-3541-0
105. Jang S, Smit J, Kallal LE, Lukacs NW. Respiratory syncytial virus infection modifies and accelerates pulmonary disease via DC activation and migration. J Leukoc Biol. (2013) 94:5–15. doi: 10.1189/jlb.0412195
106. Ugonna K, Bingle CD, Plant K, Wilson K, Everard ML. Macrophages are required for dendritic cell uptake of respiratory syncytial virus from an infected epithelium. PLoS ONE. (2014) 9:e91855. doi: 10.1371/journal.pone.0091855
107. Soumelis V, Reche PA, Kanzler H, Yuan W, Edward G, Homey B, et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat Immunol. (2002) 3:673–80. doi: 10.1038/ni805
108. Qiao J, Li A, Jin X. TSLP from RSV-stimulated rat airway epithelial cells activates myeloid dendritic cells. Immunol Cell Biol. (2011) 89:231–8. doi: 10.1038/icb.2010.85
109. Sluijs KF, Obregon C, Geiser TK, Muhlemann K, Nicod LP. Monocyte differentiation toward regulatory dendritic cells is not affected by respiratory syncytial virus-induced inflammatory mediators. Am J Respir Cell Mol Biol. (2011) 44:655–64. doi: 10.1165/rcmb.2010-0136OC
110. Kim TH, Lee HK. Differential roles of lung dendritic cell subsets against respiratory virus infection. Immune Netw. (2014) 14:128–37. doi: 10.4110/in.2014.14.3.128
111. Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med. (2006) 203:1153–9. doi: 10.1084/jem.20052359
112. Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol. (2006) 177:6263–70. doi: 10.4049/jimmunol.177.9.6263
113. Beyer M, Bartz H, Horner K, Doths S, Koerner-Rettberg C, Schwarze J. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J Allergy Clin Immunol. (2004) 113:127–33. doi: 10.1016/j.jaci.2003.10.057
114. Smit JJ, Lindell DM, Boon L, Kool M, Lambrecht BN, Lukacs NW. The balance between plasmacytoid DC versus conventional DC determines pulmonary immunity to virus infections. PLoS ONE. (2008) 3:e1720. doi: 10.1371/journal.pone.0001720
115. Gill MA, Palucka AK, Barton T, Ghaffar F, Jafri H, Banchereau J, et al. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J Infect Dis. (2005) 1991:1105–15 doi: 10.1086/428589
116. Wang H, Peters N, Laza-Stanca V, Nawroly N, Johnston SL, Schwarze J. Local CD11c+ MHC class II- precursors generate lung dendritic cells during respiratory viral infection, but are depleted in the process. J Immunol. (2006) 177:2536–42. doi: 10.4049/jimmunol.177.4.2536
117. Guerrero-Plata A, Kolli D, Hong C, Casola A, Garofalo RP. Subversion of pulmonary dendritic cell function by paramyxovirus infections. J Immunol. (2009) 182:3072–83. doi: 10.4049/jimmunol.0802262
118. Lukens MV, Kruijsen D, Coenjaerts FE, Kimpen JL, Van Bleek GM. Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J Virol. (2009) 83:7235–43. doi: 10.1128/JVI.00452-09
119. Ruckwardt TJ, Malloy AM, Morabito KM, Graham BS. Quantitative and qualitative deficits in neonatal lung-migratory dendritic cells impact the generation of the CD8+ T cell response. PLoS Pathog. (2014) 10:e1003934. doi: 10.1371/journal.ppat.1003934
120. Cormier SA, Shrestha B, Saravia J, Lee GI, Shen L, Devincenzo JP, et al. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J Virol. (2014) 88:9350–60. doi: 10.1128/JVI.00818-14
121. Shrestha B, You D, Saravia J, Siefker DT, Jaligama S, Lee GI, et al. IL-4Rα on dendritic cells in neonates and Th2 immunopathology in respiratory syncytial virus infection. J Leukoc Biol. (2017) 102:153–61. doi: 10.1189/jlb.4A1216-536R
122. Malloy AM, Ruckwardt TJ, Morabito KM, Lau-Kilby AW, Graham BS. Pulmonary dendritic cell subsets shape the respiratory syncytial virus-specific CD8+ T cell immunodominance hierarchy in neonates. J Immunol. (2017) 198:394–403. doi: 10.4049/jimmunol.1600486
123. Le Nouen C, Hillyer P, Winter CC, Mccarty T, Rabin RL, Collins PL, et al. Low CCR7-mediated migration of human monocyte derived dendritic cells in response to human respiratory syncytial virus and human metapneumovirus. PLoS Pathog. (2011) 7:e1002105. doi: 10.1371/journal.ppat.1002105
124. Kallal LE, Schaller MA, Lindell DM, Lira SA, Lukacs NW. CCL20/CCR6 blockade enhances immunity to RSV by impairing recruitment of DC. Eur J Immunol. (2010) 40:1042–52. doi: 10.1002/eji.200939778
125. Weng K, Zhang J, Mei X, Wu A, Zhang B, Cai M, et al. Lower number of plasmacytoid dendritic cells in peripheral blood of children with bronchiolitis following respiratory syncytial virus infection. Influen Other Respir Virus. (2014) 8:469–73. doi: 10.1111/irv.12242
126. Rothoeft T, Fischer K, Zawatzki S, Schulz V, Schauer U, Korner Rettberg C. Differential response of human naive and memory/effector T cells to dendritic cells infected by respiratory syncytial virus. Clin Exp Immunol. (2007) 150:263–73. doi: 10.1111/j.1365-2249.2007.03497.x
127. Schlender J, Walliser G, Fricke J, Conzelmann K-K. Respiratory syncytial virus fusion protein mediates inhibition of mitogen-induced T-cell proliferation by contact. J Virol. (2002) 76:1163–70. doi: 10.1128/JVI.76.3.1163-1170.2002
128. Cespedes PF, Bueno SM, Ramirez BA, Gomez RS, Riquelme SA, Palavecino CE, et al. Surface expression of the hRSV nucleoprotein impairs immunological synapse formation with T cells. Proc Natl Acad Sci USA. (2014) 111:E3214–3223. doi: 10.1073/pnas.1400760111
129. Munir S, Hillyer P, Le Nouen C, Buchholz UJ, Rabin RL, Collins PL, et al. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. (2011) 7:e1001336. doi: 10.1371/journal.ppat.1001336
130. Ruckwardt TJ, Morabito KM, Bar-Haim E, Nair D, Graham BS. Neonatal mice possess two phenotypically and functionally distinct lung-migratory CD103(+) dendritic cell populations following respiratory infection. Mucosal Immunol. (2018) 11:186–98. doi: 10.1038/mi.2017.28
131. Christiaansen AF, Knudson CJ, Weiss KA, Varga SM. The CD4 T cell response to respiratory syncytial virus infection. Immunol Res. (2014) 59:109–17. doi: 10.1007/s12026-014-8540-1
132. Wang J, Kong L, Luo Q, Li B, Wu J, Liu B, et al. Dual effects of respiratory syncytial virus infections on airway inflammation by regulation of Th17/Treg responses in ovalbumin-challenged mice. Inflammation. (2014) 37:1984–2005. doi: 10.1007/s10753-014-9931-0
133. Miyauchi K. Helper T cell responses to respiratory viruses in the lung: development, virus suppression, and pathogenesis. Viral Immunol. (2017) 30:421–30. doi: 10.1089/vim.2017.0018
134. Yao S, Jiang L, Moser EK, Jewett LB, Wright J, Du J, et al. Control of pathogenic effector T-cell activities in situ by PD-L1 expression on respiratory inflammatory dendritic cells during respiratory syncytial virus infection. Mucosal Immunol. (2015) 8:746–59. doi: 10.1038/mi.2014.106
135. Tsuchida T, Matsuse H, Fukahori S, Kawano T, Tomari S, Fukushima C, et al. Effect of respiratory syncytial virus infection on plasmacytoid dendritic cell regulation of allergic airway inflammation. Int Arch Allergy Immunol. (2012) 157:21–30. doi: 10.1159/000324676
136. Hua Y, Jiao YY, Ma Y, Peng XL, Fu YH, Zhang XJ, et al. Enhanced humoral and CD8+ T cell immunity in mice vaccinated by DNA vaccine against human respiratory syncytial virus through targeting the encoded F protein to dendritic cells. Int Immunopharmacol. (2017) 46:62–9. doi: 10.1016/j.intimp.2017.02.023
Keywords: dendritic cells (DCs), DC maturation, antigen presentation, T cell activation, inflammation, recurrent infection, immune evasion, immunity
Citation: Tognarelli EI, Bueno SM and González PA (2019) Immune-Modulation by the Human Respiratory Syncytial Virus: Focus on Dendritic Cells. Front. Immunol. 10:810. doi: 10.3389/fimmu.2019.00810
Received: 14 December 2018; Accepted: 26 March 2019;
Published: 15 April 2019.
Edited by:
Steven Varga, The University of Iowa, United StatesReviewed by:
Jürgen Schwarze, University of Edinburgh, United KingdomCopyright © 2019 Tognarelli, Bueno and González. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pablo A. González, cGFnb256YWxlekBiaW8ucHVjLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.