 Dylan A. Galloway
Dylan A. Galloway Alexandra E. M. Phillips
Alexandra E. M. Phillips David R. J. Owen2
David R. J. Owen2 Craig S. Moore
Craig S. Moore- 1Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, St. John's, NL, Canada
- 2Division of Brain Sciences, Department of Medicine Hammersmith Hospital, Imperial College London, London, United Kingdom
Microglia are resident macrophages of the central nervous system and significantly contribute to overall brain function by participating in phagocytosis during development, homeostasis, and diseased states. Phagocytosis is a highly complex process that is specialized for the uptake and removal of opsonized and non-opsonized targets, such as pathogens, apoptotic cells, and cellular debris. While the role of phagocytosis in mediating classical innate and adaptive immune responses has been known for decades, it is now appreciated that phagocytosis is also critical throughout early neural development, homeostasis, and initiating repair mechanisms. As such, modulating phagocytic processes has provided unexplored avenues with the intent of developing novel therapeutics that promote repair and regeneration in the CNS. Here, we review the functional consequences that phagocytosis plays in both the healthy and diseased CNS, and summarize how phagocytosis contributes to overall pathophysiological mechanisms involved in brain injury and repair.
Introduction
Phagocytosis is the process through which cells recognize, engulf, and digest large particles (>0.5 microns), including, but not limited to, bacteria, apoptotic cells, and cell debris. Phagocytosis is a receptor-mediated process involving three major steps: “find me,” “eat me,” and “digest me,” with each of these steps being regulated by multiple receptors, unique molecules, and signaling pathways. Specific receptors involved in phagocytosis can be either opsonic (i.e., Fc receptors, complement receptors) or non-opsonic (i.e., C-type lectin receptors, phosphatidylserine receptors). Following recognition by phagocytic receptors, the plasma membrane extends around the phagocytic target in an actin-dependent manner, with particles ultimately being enclosed within a vesicular phagosome. Following formation, this nascent phagosome proceeds through a series of maturation steps, culminating in fusion with lysosomes (phagolysosome) for the eventual destruction of the phagocytosed particles. Importantly, following destruction, byproducts must be effectively dealt with by the phagocytic cell, either through storage, recycling or efflux mechanisms. The basic cell biology of phagocytosis has been extensively reviewed elsewhere (1).
Adding additional complexity to phagocytosis is the requirement for specific outcomes in the context of different phagocytic targets. For example, while recognition and phagocytosis of bacteria requires rapid induction of proinflammatory responses, a similar reaction to apoptotic cells induces detrimental autoinflammation (2). As such, specific immune responses to phagocytic targets are tailored to by a variety of context-dependent signals, including the engagement of phagocytic receptors that utilize distinct inflammatory signaling pathways (pro vs. anti-inflammatory) and microenvironment-derived signals that promote quiescence or inflammation (3, 4).
Within the CNS, phagocytosis is a critical process required for proper neural circuit development and maintaining homeostasis. To assist in maintaining homeostasis in the CNS, synapses, apoptotic cells, and debris must be continuously removed to maintain optimal neural function. While phagocytosis is primarily attributed to microglia (the professional phagocytes in the CNS), non-professional phagocytes (e.g., astrocytes or oligodendrocytes) may also participate (5). Arising from discrete pathologies, specific phagocytic targets, such as insoluble protein aggregates or myelin debris add further burden to the phagocytic machinery within the CNS. It has been hypothesized that failures during phagocytic processes may actually promote inflammation and/or neurodegenerative processes. Herein, we review how phagocytosis contributes to both the maintenance of homeostasis and disease within the CNS (Figure 1).
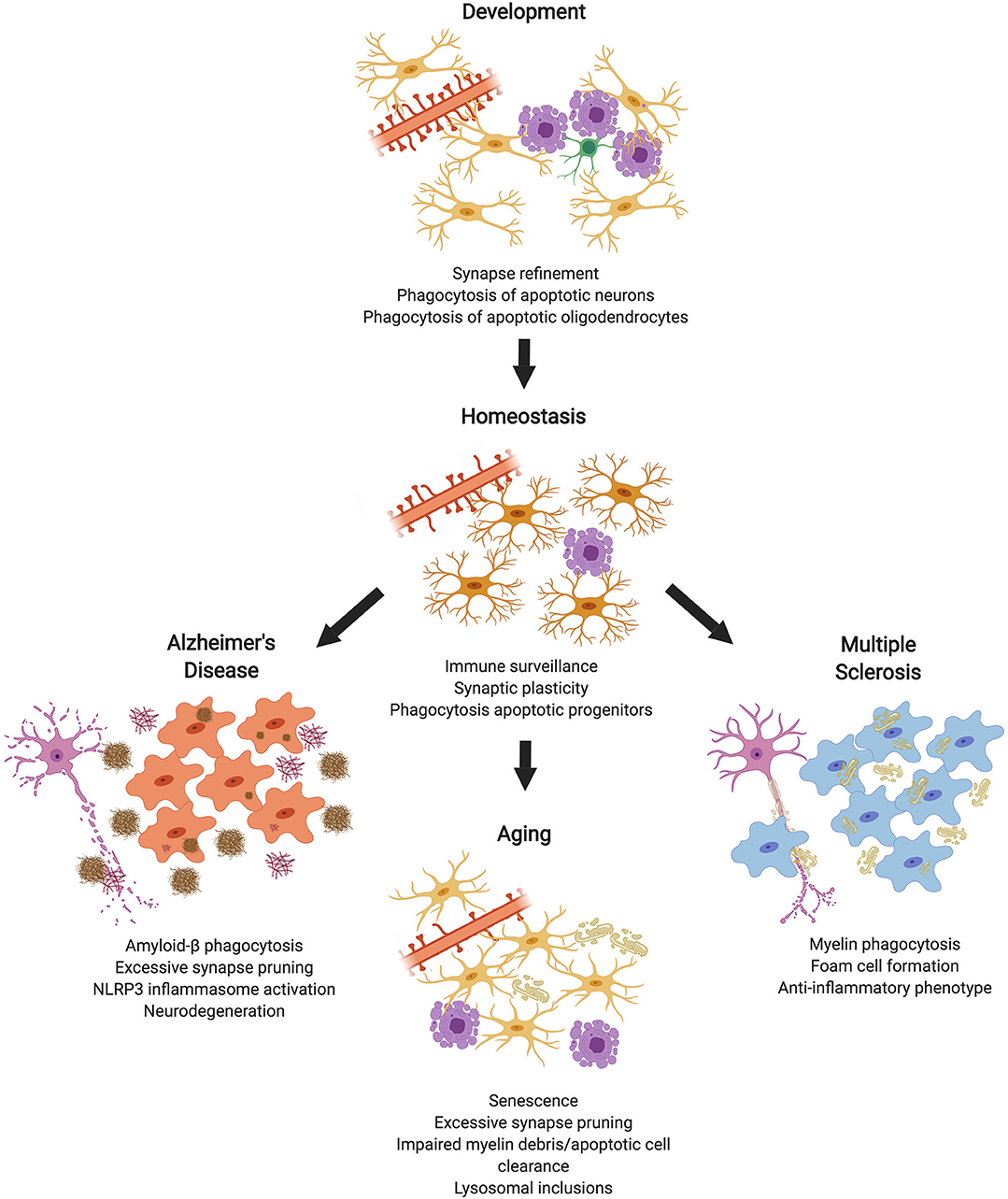
Figure 1. Microglial Phagocytosis in the CNS. During development, microglial phagocytosis is essential for the refinement of excessive synapses, as well as the removal of apoptotic neurons and oligodendrocytes that are overproduced during development. Homeostatic microglia in the adult brain constantly survey the brain's parenchyma, contributing to synaptic plasticity and phagocytosing apoptotic progenitor cells. With advanced age, microglia undergo senescence, display impaired debris clearance, and excessively prune synapses. In diseases, such as Alzheimer's or multiple sclerosis, microglia act as key contributors to pathology, which is partially mediated by phagocytosis of substrates, such as amyloid-β or myelin debris (made in ©BioRender - biorender.com).
Phagocytosis in CNS Development and Homeostasis
Synapse Elimination
Within the developing CNS, phagocytosis is necessary for the refinement of synaptic connectivity, as the developing CNS overproduces both neurons and synapses (6). Furthermore, the removal of unwanted synapses refines neural networks, thus contributing to learning and memory (7). Microglia are important cells that execute the pruning of synaptic connections, utilizing immune signaling pathways, such as the complement pathway, although other signaling pathways also contribute (8). Microglia continuously survey the brain's parenchyma (9) and make frequent physical contacts with synapses that are mediated in part by sensing neuronal activity via nucleotides, such as ATP or ADP, and suggest that nucleotides may act as “find me” signals that guide microglial processes toward active synapses (10, 11). Studies investigating synapse pruning have relied extensively on the developing visual system, which is a well-defined sensory system that allows for easy manipulation. Modulation of neuronal activity within the visual system using visual deprivation (to reduce the frequency of action potentials) has demonstrated that neuronal activity is essential for synaptic pruning by microglia (12). Consistent with the role of nucleotides serving as a “find me” signal within the CNS, mice lacking the ADP receptor P2Y12, or pharmacological blockade of P2Y12 signaling, result in impaired synapse pruning within the developing visual cortex (13). Additionally, CX3CR1 knockout mice demonstrate increased hippocampal spine density during development (14), suggesting CX3CL1 may also act as a “find me” signal regulating synapse pruning. However, CX3CL1 is dispensable for synapse pruning within the developing visual system (15). In regards to recognition and engulfment of synapses, the classical complement system has been extensively studied. Briefly, the classical complement system functions via tagging of targets with C1q, which catalyzes the production of C3 convertase. C3 convertase subsequently cleaves C3 producing both C3a (a pro-inflammatory mediator) and C3b, an opsonin that triggers phagocytosis via complement receptors on phagocytes. Early reports demonstrated that the classical complement components C1q and C3 tag synapses for phagocytosis and are required for proper refinement of the developing lateral geniculate (16). Additional reports observed that microglial complement receptor 3 (CR3) is required for the clearance of complement tagged synapses (17). Highlighting the relevance of complement in synapse pruning during development, mice lacking C1q exhibit spontaneous seizure activity as a result of impaired synapse removal (18). The importance of C1q in tagging synapse (19) for elimination is supported by the finding of local apoptotic mechanisms within presynaptic elements that results in C1q accumulation (20). Recent work leveraging the power of correlative light and electron microscopy (CLEM) and live-cell imaging demonstrated that microglia frequently contact synapses within the healthy brain and can be seen engulfing presynaptic, but not post-synaptic, elements via a specialized form of phagocytosis termed trogocytosis, which results in the partial removal of cell constituents (21). While supporting the notion that microglia remove synaptic elements, this study fails to demonstrate that microglia actively phagocytose entire synapses, and further, the authors observed no role for the receptor CR3 in this process. In addition to the classical complement system, both TREM2 and CD47-SIRPα signaling contribute to synapse pruning by microglia (22, 23), although these pathways have been less extensively studied.
In addition to microglia, astrocytes also participate in refining synaptic connectivity (24, 25). It was initially reported that astrocytes participate in the phagocytosis of synapses utilizing the receptors MerTK and MEGF10, whereby astrocyte-specific deletion of these receptors results in a failure to refine synapses in the developing visual system (26). Furthermore, the ability of human astrocytes to phagocytose synapses in dissociated cultures and cerebral organoids has been demonstrated (27, 28), suggesting astrocytes actively prune synapses in the human CNS. Additionally, the Alzheimer-associated gene ApoE regulates the phagocytic capacity of astrocytes and C1q accumulation on synapses during aging (29). It has also been reported that sleep deprivation causes increased synaptic pruning by astrocytes, likely mediated by MerTK (30).
Removal of Apoptotic Cells
Apoptotic cells are constantly generated and phagocytosed throughout the nervous system during both development and homeostasis. Within the subgranular zone (SGZ) and subventricular zone (SVZ), the major regions containing neural progenitor cells (NPCs), microglia are required to constantly phagocytose apoptotic NPCs throughout the lifespan of the organism. Despite the majority of newborn cells in neurogenic niches undergoing apoptosis, identification of apoptotic cells is difficult due to their rapid clearance by microglia. During inflammatory insult, increased apoptosis of NPCs is coupled to increased phagocytosis by SGZ microglia, suggesting that microglia continue to remove apoptotic progenitors regardless of inflammatory status (31). Mechanistically, the phagocytosis of apoptotic NPCs appears to depend on the TAM family receptors MerTK and AXL, as evidenced by a buildup of apoptotic within the SVZ when these receptors are genetically deleted (32). Interestingly, NPCs may also participate in the phagocytosis of neighboring apoptotic cells and may be required for efficient neurogenesis (33). Apoptotic neurons and oligodendrocytes are also generated throughout development, and phagocytosis is required to clear these cells (34–36). The phagocytosis of apoptotic neurons depends on receptors including TREM2, CD11b, BAI1 and TIM-4, as well as the v-ATPase transporter that is required for the degradation of apoptotic corpses (36–39). Importantly, recent work has identified a novel microglia subset associated with developmental white matter that is specialized for the phagocytosis and removal of apoptotic oligodendrocytes (40). Thus, it appears that microglia may acquire distinct phenotypes that are required for region-specific phagocytic functions which may not be extrapolated from one region to another.
Phagocytosis During Aging
Within the aging CNS, there is abundant synapse loss and myelin degeneration which is believed to contribute to age-related cognitive decline (41, 42). Microglia- and complement-mediated synapse elimination has been suggested to underlie excessive synapse elimination during normal aging. Increases in complement protein C1q are observed throughout the aged brain, and knockout of C1q prevents age-related cognitive decline, suggesting that excessive synapse pruning during aging potentiates cognitive decline (19). Genetic deletion of the complement component C3 reduces synapse loss and cognitive decline in aged mice, further implicating excessive complement-dependent synapse elimination as a key contributor to age-related cognitive impairment (43). In regards to myelin degeneration, ultrastructural analysis and in vivo imaging has demonstrated that large amounts of myelin debris are generated during normal aging, and this debris is continuously phagocytosed by microglia (44, 45). With advanced age, clearance of myelin debris becomes impaired, resulting in insoluble intracellular aggregates (lipofuscin granules) within microglia and microglial senescence (46). This microglial dysfunction is exacerbated by increased production of myelin debris or impairment of lysosomal processing; indicating pathways downstream of engulfment are essential to effectively deal with myelin debris in the healthy CNS. While microglial phagocytosis appears to be an important contributor to the aging brain, further investigations are needed to confirm and expand on these findings.
Phagocytosis in Disease States
As we begin to understand the roles of microglia in the development and homeostasis of the CNS, it has become increasingly evident that changes in the key functions of these highly active cells can exert significant effects on the progression of multiple CNS diseases (47) (Table 1).

Table 1. Current evidence of phagocytosis alterations resulting from variants in disease-associated genes expressed in microglia.
Acute Injury
The cellular response to acute CNS injury, such as traumatic brain injury (TBI), spinal cord injury (SCI), and stroke is multiphasic and has been studied in a range of models. The initial phase involves rapid activation of CNS-resident microglia (9, 84, 85) resulting in pro-inflammatory cytokine release and recruitment of peripheral immune cells, including neutrophils, monocytes, and monocyte-derived macrophages (MDMs), to the lesion site (86, 87). This early response by microglia limits the spread of the lesion (85, 88, 89) but also generates inflammatory cytokines and reactive oxygen species (ROS), which may be detrimental to recovery and contribute to secondary injury if not resolved (90).
Phagocytosis, initially performed by microglia and subsequently by recruited MDMs, acts to restore homeostasis and minimize chronic activation (91). Phagocytosis of apoptotic cells prevents the release of cytotoxic and immunogenic intracellular contents (92, 93) and the removal of damaged myelin has been shown to be important for axon regeneration and remyelination (94–96). Additionally, live neurons may be phagocytosed by microglia during injury (97), inducing a form of cell death termed phagoptosis, which contributes to neuronal cell death during pathological states (98, 99). Importantly, the presence of “don't eat me” signals, primarily CD47-SIRPα or CD200-CD200L signaling, from neurons to microglia suppresses aberrant phagocytosis and is critical for maintaining microglia in a quiescent state [reviewed in (100)].
Recruited MDMs play an important role in post-injury clearance. In a model of cerebral ischemia in mice, MDMs, once localized to the site of injury, have been shown to have a higher phagocytic capacity than microglia (101). However, as demonstrated in a SCI model, recruited MDMs are less capable of processing phagocytic material intracellularly and are also more susceptible to apoptotic and necrotic cell death (102). Therefore, although peripherally-derived MDMs take up more debris post-injury, the inefficient processing of phagocytosed material results in cell stress, which ultimately contributes to the inflammatory milieu.
Whether the activity of infiltrating MDMs following CNS injury is beneficial for repair or a detrimental contributor to inflammation is still disputed (103–106). Following disease onset in models of stroke and SCI, infiltrating MDMs have been found to alter microglial gene expression, downregulating both beneficial (phagocytosis) and neurotoxic (pro-inflammatory) functions (102, 107, 108). Indeed, blockade of MDM infiltration or MDM ablation has been shown to be beneficial in SCI (109–111) and TBI (112, 113). Conversely, other models suggest that preventing the infiltration and functioning of certain MDMs populations in CNS injury results in worsened outcomes (107, 114–116). Contradictory data may be due to inconsistent spatial and temporal assessment of inflammation, phagocytic activity and outcomes in the various models (117) as well as difficulties in differentiating CNS-resident from infiltrating myeloid cells. The use of double transgenic models, such as the Cx3cr1GFP/+Ccr2RFP/+ mouse, would enable improved distinction between phagocytosis performed by resident microglia and infiltrating monocytes, as used to differentiate these myeloid cell types in studies of stroke (118, 119), SCI (105) and inflammation (120).
Multiple Sclerosis
Destruction of myelin sheaths within the CNS, as occurs in multiple sclerosis (MS), produces degenerating myelin at sites of injury and inflammation. This degenerating myelin, termed myelin debris, must be cleared from sites of injury to promote timely repair. CNS (but not PNS) myelin acts as a potent inhibitor of oligodendrocyte differentiation (121), and the introduction of exogenous myelin into demyelinated lesions halts oligodendrocyte differentiation at the pre-myelinating stage (122). The removal of myelin debris within MS lesions and experimental animal models, such as experimental autoimmune encephalomyelitis (EAE) or cuprizone-induced demyelination, is primarily mediated by microglia and macrophages. Resident microglia and peripheral macrophages are capable of phagocytosing and degrading large quantities of myelin as highlighted by rapid clearance of myelin debris in animal models, although myelin debris can persist in MS patient lesions (123). Microglia and macrophages differ in their ability to uptake myelin. Specifically, resident microglia demonstrate a greater ability to engulf myelin then peripheral macrophages (124–126), and are more resistant to apoptosis following myelin phagocytosis (102), indicating microglia are more efficient at both engulfing and degrading myelin debris. The mechanism underlying this difference is unknown, although both ontogeny and exposure to the CNS microenvironment likely contribute. For example, astrocyte-conditioned media has been shown to promote myelin phagocytosis by macrophages and microglia in vitro (127, 128), suggesting the CNS microenvironment programs myeloid cells for efficient myelin phagocytosis. The phenotype of myeloid cells (pro-inflammatory vs. reparative) also has a large influence on the phagocytic ability of myeloid cells, as inflammatory myeloid cells (e.g., LPS stimulated) display reduced myelin phagocytosis in comparison to reparative, anti-inflammatory myeloid cells (e.g., IL-4 stimulated) (126).
Early investigations into myelin phagocytosis examined the effects of opsonization, demonstrating that both immunoglobulins and complement proteins promote the phagocytosis of myelin, and blocking Fc or complement receptors reduced myelin phagocytosis in vitro (125, 129, 130). In addition, evidence from MS lesions suggests that Fc receptors and complement play active roles in myelin phagocytosis (131). Interestingly, myelin debris is capable of activating complement in the absence of myelin reactive antibodies (132). Furthermore, myelin phagocytosis in vitro relies on scavenger and C-type lectin receptors for recognition and internalization of myelin debris (125, 133). More recently, the TAM family receptors MerTK and AXL, which bind phosphatidylserine via the bridging molecules Protein S and Gas6, respectively, have been identified as essential regulators of myelin phagocytosis. Within the EAE animal model, deletion of AXL results in increased clinical severity and impaired myelin clearance, while delivery of exogenous Gas6 is protective (134). Loss of AXL/Gas6 during cuprizone-induced demyelination results in increased neuroinflammation and impaired remyelination, indicating that signaling via the apoptotic cell receptor AXL is required to promote the resolution of inflammation following demyelination (135). Studies utilizing human macrophages and microglia have demonstrated that MerTK is an essential phagocytic receptor for myelin, expression of MerTK correlates with myelin phagocytosis in vitro, and MerTK levels are reduced within MS patient macrophages (82, 83). Polymorphisms within the MerTK gene are associated with MS susceptibility, suggesting MerTK plays an important role in the development of MS (80, 81). Finally, TREM2 has also been implicated in myelin phagocytosis. TREM2 binds myelin lipids directly to facilitate internalization, and studies using the EAE animal model observed that blockade of TREM2 increases EAE severity, while TREM2 overexpression is protective, in part mediated by effects on clearance of myelin debris (136–138). Moreover, TREM2 KO mice display faulty myelin debris clearance and remyelination in the cuprizone model of toxic demyelination, with TREM2 knockout microglia failing to upregulation genes associated with phagocytosis and lipid metabolism (138, 139).
In addition to myeloid cells, astrocytes have been observed to engulf myelin debris. Within MS lesions, hypertrophic astrocytes contain myelin inclusions (140), and astrocytes have been demonstrated to uptake myelin debris in vitro (141). Transcriptomic analysis of astrocytes reveals expression of several complete phagocytic pathways and apoptotic cell receptors, such as MerTK, AXL and LRP1 (142). This is supported by the description of neurotoxic “A1” astrocytes, which downregulate phagocytic receptors including MerTK and show impaired myelin phagocytosis in vitro (143). Astrocytes phagocytose myelin at quantities several-fold lower than myeloid cells (144), questioning the functional significance of astrocyte-mediated myelin phagocytosis in vivo. Recently, phagocytosis of myelin by astrocytes has been shown to induce the expression of multiple chemokines both in vitro and in vivo (145), suggesting that astrocytes sense myelin debris and respond by recruiting professional phagocytes to sites of injury. These results are in line with the demonstration that astrocyte ablation impairs myeloid cell recruitment and phagocytosis in the cuprizone model, partly due to lack of CXCL10 expression (146).
Alzheimer's Disease and CNS Phagocytosis During Aging
Alzheimer's disease (AD) is characterized by the accumulation of extracellular plaques of toxic amyloid-beta (Aβ) and intracellular neurofibrillary tangles. The amyloid cascade hypothesis posits that an imbalance between the production and the clearance of Aβ initiates the pathological cascade of synapse loss, neuron death, and brain atrophy found in AD (147). The contribution of microglia to AD pathogenesis is becoming increasingly recognized as genome wide association studies (GWAS) and transcriptomic analyses highlight links between microglial genes and AD risk, as well as between microglial signaling pathways and disease progression (47).
In the case of the more common, late onset AD (LOAD), it has been argued that impairment in the clearance of Aβ has a greater impact on disease progression than its overproduction (148). As the primary resident phagocytes of the CNS, microglia play an important role in preventing the accumulation of this toxic protein through both phagocytosis and the production of degrading intra- and extracellular enzymes (149). In vitro, Aβ initiates cell stress responses, synapse loss, mitochondrial dysfunction, and neuronal apoptosis. In the early stages of AD, microglial function is neuroprotective, acting to clear apoptotic cells and pathological protein aggregates (150) as well as forming a barrier around plaques to restrict their growth and diffusion of synaptotoxic Aβ oligomers (151, 152). However, long term exposure to Aβ induces chronic microglial activation-associated dysfunction known as reactive microgliosis (153), in which phenotypic changes result in the adoption of a continuous pro-inflammatory status and compromised phagocytosis (154–156).
It is important to note that many in vitro and in vivo phagocytosis assays rely heavily on determining the uptake, but not measuring the subsequent degradation, of pathogenic proteins. In order to prevent intracellular accumulation of Aβ, it must be appropriately degraded and cleared through the endosome-lysosomal pathway (157, 158). The uptake of fibrillar and soluble Aβ has been reported in multiple models of microglia in vitro and in vivo, however, whether complete degradation of this protein, in particular the fibrillar (f) form, occurs remains disputed. Following in vitro culture of control microglia with fAβ, phagosomes have been found to contain incompletely degraded Aβ for up to 20 days (159–161). It has been reported that acidification of microglial lysosomes, for example by treatment with MCSF, can improve the efficiency of intracellular fAβ breakdown (162, 163).
The presence of complement activation in AD pathology has been observed for several decades (164, 165) however, it is only recently that genetic analyses have identified complement components as playing a role in AD pathogenesis (166–169). As discussed previously, complement-mediated pruning of synapses is a key microglial function during development and, whilst complement factors aid plaque clearance (170, 171), phagocytosis of synapses appears to become dysregulated during AD. Synapse loss has been identified early on in AD and correlates strongly with cognitive decline (172, 173). Inhibition or knockout of the complement components C1q, C3, and CR3, required for microglial synapse refinement during development, reduced the synapse loss found in mouse AD models (174–176).
GWAS studies have identified the rare variant R47H in the gene encoding the phagocytic receptor TREM2 as a risk factor for the development of LOAD (48, 49). TREM2 expression is necessary for the phagocytosis of a range of particles (36, 53, 54); TREM2 knockout microglia are less efficient at phagocytosing Aβ than WT microglia (53, 55) and mutations in TREM2 affect the detection of damage-associated lipids by microglia (56), which may explain their reduced ability to take up apoptotic cells (60, 61, 177). It has recently been suggested that TREM2 drives the expression of the scavenger receptor CD36, via the upregulation of C/EBPα (52), augmenting phagocytosis. TREM2 knockout AD models have produced contrasting results regarding amyloid burden (56–59), however it can be argued that this is an indirect measure of microglial phagocytosis of Aβ, as amyloid burden can be altered by other reported TREM2-mediated effects including microglial migration and plaque barrier formation (55, 178, 179).
The ApolipoproteinE–ε4 allele is the greatest genetic risk factor for the development of LOAD (180). APOE has been found to be an endogenous TREM2 ligand (53, 181), suggesting an interaction between the two most significant AD genetic risk factors on the surface of microglia. APOE binds to both Aβ (181, 182) and apoptotic cells (181) and therefore may facilitate the detection and phagocytosis of Aβ and apoptotic cells by TREM2-expressing microglia. The TREM2-APOE signaling pathway has been reported to suppress the homeostatic signature of microglia in several CNS disease models, inducing a shift to a neurodegenerative phenotype (183). Therefore, this signaling axis may exert effects on both beneficial and detrimental microglial functions.
Single-cell RNA-sequencing has identified a new subset of highly phagocytic, AD-associated microglia (DAM) surrounding the plaques in an AD mouse model (184). Interestingly, this subset of microglia was also found in models of aging and ALS so may represent a generalized response to age-related neurodegeneration, or loss of homeostasis, rather than to a specific disease-associated protein. Whether this adoption of a highly phagocytic phenotype is beneficial or deleterious for AD progression has not yet been established.
It has been argued that aging and the expression of genetic risk factors, either independently or in combination, limit the ability of microglia to prevent or slow the pathogenesis of neurodegenerative diseases (185). Age is the single biggest risk factor for the development of LOAD and the recent identification of a unique phenotype of aged human microglia, in which susceptibility genes for both AD and MS were found to be enriched (186), suggests that age could significantly impact microglial function. In the aged brain, microglia exhibit marked changes in their morphology and activity; compared to young cells there is an increase in soma size, a loss of the characteristic tiled tissue distribution, and shorter, less dynamic processes (187–189). Primary mouse microglia demonstrate age-dependent, substrate-specific decreases in phagocytosis of fibrillary Aβ (190) and α-synuclein oligomers (191). Age-related phagocytic activation of microglia, which correlated with cognitive impairment, was reported in aged rhesus monkey brain (192). In this study, immunohistochemical analysis of white matter regions indicated that with age, increasing numbers of microglia simultaneously expressed galectin-3, a phagocytic marker, and HLA-DR MHC II, a marker of microglial activation. However, in another study, the presence of large, heterogeneous intracellular inclusions suggested that increased uptake, but inefficient lysosomal digestion, of particles may be associated with aged microglia (193). Poor Aβ protein degradation is also found in aged mouse microglia (188, 194). Therefore, age-related changes may increase the susceptibility to abnormally folded proteins and accumulating debris, resulting in a loss of homeostasis and the persistence of cytotoxic conditions.
Other Neurodegenerative Diseases
Microglial phagocytosis has been implicated in a range of other neurodegenerative diseases (195), in particular proteopathic diseases in which the balance of protein production, clearance and degradation becomes dysregulated. In prion disease, the pathogenic form of prion protein (PrPsc) is not taken up by microglia (196), and alters the uptake of other particles (197), resulting in the accumulation of pathology. Multiple studies have demonstrated the ability of α-synuclein (α-SYN), the pathogenic protein found in Lewy bodies in Parkinson's disease (PD), to alter microglial phagocytosis, although this is conformation and expression-level dependent (198). Mutations in LRRK2 are the most commonly found variants in familial PD and recent work has demonstrated that LRRK2 influences myeloid cell phagocytosis via interactions with the actin remodeling protein WAVE2 (79). LRRK2 deficiency in mouse microglia attenuated phagocytosis of beads, whereas expression of PD-associated LRRK2–G2019S augmented phagocytosis in mouse microglia and patient-derived macrophages, which may result in neuronal damage due to overactive phagocytosis during disease (79).
The TREM2 R47H variant associated with AD has also been found to be a risk factor for PD and frontotemporal dementia (FTD), suggesting a common role for TREM2 dysfunction in multiple neurodegenerative diseases (50). Nasu-Hakola disease (NHD) is a progressive, presenile dementia in which phagocytic alterations are a primary cause of pathology. Rare but lethal, NHD is caused by a loss of function of TREM2 or its signaling partner DAP12 (51). Significant demyelination is found in patient brains (199) and in mouse models (200, 201), however signs of Aβ and tau pathology are limited, despite the role of TREM2 in AD (202). Overactive microglial phagocytosis is also a driver of the pathology found in FTD; loss of expression of functional progranulin results in increased C1q production and complement-mediated synapse loss during aging (203).
Modeling Phagocytosis in the CNS
CNS phagocytosis can be modeled and studied using a range of in vitro and in vivo techniques. Flow cytometry and microscopy are frequently and easily utilized to assess the uptake by cell lines or primary cells in vitro of fluorescently-labeled synthetic or physiological particles, including latex, Aβ, myelin, zymosan, and dextran (204). Flow cytometry allows the rapid assessment of large numbers of cells, whereas microscopy provides additional information on the motility and morphology of the cells as they perform this actin-associated function. Time-lapse microscopy allows the monitoring of the clearance or, in the case of some pathogenic proteins, the persistence of phagocytosed material within the cells. The ability to fluorescently label a range of particles enables disease- or context-specific analysis of phagocytosis, however, in order to ensure the identity of the phagocytic cell, these experiments are frequently performed in monoculture. Monocultures of primary cells are valuable for understanding the morphological changes and cellular pathways of specific substrate uptake during phagocytosis but do not provide information on the interactions between different cell types during this process. It has also been shown that the isolation and culture of microglia rapidly alters their transcriptomic signature (205), so in vitro assays may not accurately recapitulate CNS phagocytosis.
In vivo, phagocytosis can be inferred in tissue by the expression of phagocytic markers, such as CD206 and CD68, or live cell imaging either transcranially or organotypic cultures. Using acutely prepared and organotypic slices, microglial phagocytosis of apoptotic neurons has been observed (206, 207). These techniques preserve the structure and physiological conditions within the tissue, and, when combined with fluorescent labels, such as lectin, and two-photon or confocal time lapse imaging, allow the study of interactions between different cell types (208). Transgenic mouse lines in which enhanced green fluorescent protein (EGFP) is expressed under the control of myeloid cell-specific gene promoters, including CX3CR1 (209), Iba1 (210), and Cfs1r (211), allow live fluorescent imaging of microglial migration and phagocytosis (12).
Species-specific differences between mouse and human cells have been found with respect to phagocytosis (212) so, in the case of investigating human diseases, human studies are necessary. Primary human microglia can be obtained from fetal or post-mortem samples, however these resources can be difficult to obtain particularly within acceptable post-mortem delay conditions. The recent development of methods for the generation of induced pluripotent stem cell-derived microglia-like cells (iMGLs) allow the study of human microglia function in vitro (70, 213–215). Mimicking scarce primary human microglia, iMGLs are capable of phagocytosing synaptosomes, as found during development, and also disease-associated Aβ and tau (213). This model system is valuable for investigating interactions between genetic risk factors and pathogenesis, as recently demonstrated by iMGLs carrying patient-derived TREM2 variants phagocytosing less apoptotic cell material than controls (60), whilst avoiding the caveats associated with mouse models of human disease. It should be noted that these cells have never been exposed to cues arising from the CNS microenvironment, which may alter the differentiation and function of the iMGLs.
Until recently, markers for distinguishing microglia from monocytes were lacking, making it difficult to determine whether phagocytosis was being performed by CNS resident microglia or infiltrating myeloid cells. The discovery of microglia-specific proteins, such as TMEM119 (216), will allow more accurate investigations of the microglial-specific contributions to homeostasis and disease. These tools may also allow the resolution of current discrepancies, including those regarding phagocytosis, found between different disease models (56, 57, 117).
Conclusions and Perspectives
In this review we have summarized the critical role phagocytosis plays in both CNS homeostasis and disease. While much progress has been made in recent years, many unanswered questions remain. How phagocytosis in the CNS is influenced by numerous factors, such as microenvironment or phagocytic target, have yet to be fully resolved. Additionally, the utilization of novel technologies, including in vivo imaging techniques (217), iPSC-derived microglia (213) and high-throughput screens (66), will likely contribute to further identification of phagocytic pathways and consequences of phagocytosis within the CNS. As targeting myeloid cells in neuroinflammatory and neurodegenerative diseases is receiving increased interest (218), drugs modulating phagocytic pathways may emerge as novel therapeutics for brain disease.
Author Contributions
DG and AP performed the literature search and wrote the manuscript. DO and CM oversaw preparation of the manuscript, and contributed to writing and editing of the manuscript.
Funding
CM is supported by grants from the Canada Research Chair program, Canadian Institutes of Health Research and the MS Society of Canada. DG is supported by a studentship from the MS Society of Canada. AP is supported by a grant from the Medical Research Council.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Flannagan RS, Jaumouille V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol. (2012) 7:61–98. doi: 10.1146/annurev-pathol-011811-132445
2. Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. (2010) 140:619–30. doi: 10.1016/j.cell.2010.02.014
3. Gonzalez A-N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
4. Roberts AW, Lee BL, Deguine J, John S, Shlomchik MJ, Barton GM. Tissue-resident macrophages are locally programmed for silent clearance of apoptotic cells. Immunity. (2017) 47:913–27 e916. doi: 10.1016/j.immuni.2017.10.006
5. Watabe K, Osborne D, Kim SU. Phagocytic activity of human adult astrocytes and oligodendrocytes in culture. J Neuropathol Exp Neurol. (1989) 48:499–506. doi: 10.1097/00005072-198909000-00001
6. Schafer DP, Stevens B. Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Curr Opin Neurobiol. (2013) 23:1034–40. doi: 10.1016/j.conb.2013.09.012
7. Lichtman JW, Colman H. Synapse elimination and indelible memory. Neuron. (2000) 25:269–78. doi: 10.1016/S0896-6273(00)80893-4
8. Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. (2012) 35:369–89. doi: 10.1146/annurev-neuro-061010-113810
9. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
10. Li Y, Du XF, Liu CS, Wen ZL, Du JL. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Dev Cell. (2012) 23:1189–202. doi: 10.1016/j.devcel.2012.10.027
11. Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB, MacVicar BA. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci. (2014) 34:10511–27. doi: 10.1523/JNEUROSCI.0405-14.2014
12. Tremblay ME, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. (2010) 8:e1000527. doi: 10.1371/journal.pbio.1000527
13. Sipe GO, Lowery RL, Tremblay ME, Kelly EA, Lamantia CE, Majewska AK. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat Commun. 7:10905. doi: 10.1038/ncomms10905
14. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. (2011) 333:1456–8. doi: 10.1126/science.1202529
15. Lowery RL, Tremblay ME, Hopkins BE, Majewska AK. The microglial fractalkine receptor is not required for activity-dependent plasticity in the mouse visual system. Glia. (2017) 65:1744–61. doi: 10.1002/glia.23192
16. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. (2007) 131:1164–78. doi: 10.1016/j.cell.2007.10.036
17. Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. (2012) 74:691–705. doi: 10.1016/j.neuron.2012.03.026
18. Chu Y, Jin X, Parada I, Pesic A, Stevens B, Barres B, et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci USA. (2010) 107:7975–80. doi: 10.1073/pnas.0913449107
19. Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. (2013) 33:13460–74. doi: 10.1523/JNEUROSCI.1333-13.2013
20. Gyorffy BA, Kun J, Torok G, Bulyaki E, Borhegyi Z, Gulyassy P, et al. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc Natl Acad Sci USA. (2018) 115:6303–8. doi: 10.1073/pnas.1722613115
21. Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun. (2018) 9:1228. doi: 10.1038/s41467-018-03566-5
22. Filipello F, Morini R, Corradini I, Zerbi V, Canzi A, Michalski B, et al. The microglial innate immune receptor TREM2 is required for synapse elimination and normal brain connectivity. Immunity. (2018) 48:979–91 e978. doi: 10.1016/j.immuni.2018.04.016
23. Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, et al. CD47 protects synapses from excess microglia-mediated pruning during development. Neuron. (2018) 100:120–34 e126. doi: 10.1016/j.neuron.2018.09.017
24. Chung WS, Allen NJ, Eroglu C. Astrocytes control synapse formation, function, and elimination. Cold Spring Harb Perspect Biol. (2015) 7:a020370. doi: 10.1101/cshperspect.a020370
25. Allen NJ, Eroglu C. Cell biology of astrocyte-synapse interactions. Neuron. (2017) 96:697–708. doi: 10.1016/j.neuron.2017.09.056
26. Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. (2013) 504:394–400. doi: 10.1038/nature12776
27. Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. (2016) 89:37–53. doi: 10.1016/j.neuron.2015.11.013
28. Sloan SA, Darmanis S, Huber N, Khan TA, Birey F, Caneda C, et al. Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron. (2017) 95:779–90 e776. doi: 10.1016/j.neuron.2017.07.035
29. Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci USA. (2016) 113:10186–91. doi: 10.1073/pnas.1609896113
30. Bellesi M, de Vivo L, Chini M, Gilli F, Tononi G, Cirelli C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J Neurosci. (2017) 37:5263–73. doi: 10.1523/JNEUROSCI.3981-16.2017
31. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. (2010) 7:483–95. doi: 10.1016/j.stem.2010.08.014
32. Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, et al. TAM receptors regulate multiple features of microglial physiology. Nature. (2016) 532:240–4. doi: 10.1038/nature17630
33. Lu Z, Elliott MR, Chen Y, Walsh JT, Klibanov AL, Ravichandran KS, et al. Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat Cell Biol. (2011) 13:1076–83. doi: 10.1038/ncb2299
34. Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. (1997) 137:459–68. doi: 10.1083/jcb.137.2.459
35. Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cells. Neuron. (2004) 41:535–47. doi: 10.1016/S0896-6273(04)00069-8
36. Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. (2005) 201:647–57. doi: 10.1084/jem.20041611
37. Peri F, Nusslein-Volhard C. Live imaging of neuronal degradation by microglia reveals a role for v0-ATPase a1 in phagosomal fusion in vivo. Cell. (2008) 133:916–27. doi: 10.1016/j.cell.2008.04.037
38. Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. (2008) 28:8138–43. doi: 10.1523/JNEUROSCI.1006-08.2008
39. Mazaheri F, Breus O, Durdu S, Haas P, Wittbrodt J, Gilmour D, et al. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat Commun. 5:4046. doi: 10.1038/ncomms5046
40. Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. (2019) 101:207–23 e210. doi: 10.1016/j.neuron.2018.12.006
41. Peters A, Moss MB, Sethares C. Effects of aging on myelinated nerve fibers in monkey primary visual cortex. J Comp Neurol. (2000) 419:364–76. doi: 10.1002/(SICI)1096-9861(20000410)419:3%3C364::AID-CNE8%3E3.0.CO;2-R
42. Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. (2012) 13:240–50. doi: 10.1038/nrn3200
43. Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, et al. Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci. (2015) 35:13029–42. doi: 10.1523/JNEUROSCI.1698-15.2015
44. Peters A. The effects of normal aging on myelin and nerve fibers: a review. J Neurocytol. (2002) 31:581–93. doi: 10.1023/A:1025731309829
45. Hill RA, Li AM, Grutzendler J. Lifelong cortical myelin plasticity and age-related degeneration in the live mammalian brain. Nat Neurosci. (2018) 21:683–95. doi: 10.1038/s41593-018-0120-6
46. Safaiyan S, Kannaiyan N, Snaidero N, Brioschi S, Biber K, Yona S, et al. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat Neurosci. (2016) 19:995–8. doi: 10.1038/nn.4325
47. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. (2017) 23:1018–27. doi: 10.1038/nm.4397
48. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al., Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. N Engl J Med. (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
49. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med. (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
50. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener. (2013) 8:19. doi: 10.1186/1750-1326-8-19
51. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. (2002) 71:656–62. doi: 10.1086/342259
52. Kim SM, Mun BR, Lee SJ, Joh Y, Lee HY, Ji KY, et al. TREM2 promotes Abeta phagocytosis by upregulating C/EBPalpha-dependent CD36 expression in microglia. Sci Rep. (2017) 7:11118. doi: 10.1038/s41598-017-11634-x
53. Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. (2016) 91:328–40. doi: 10.1016/j.neuron.2016.06.015
54. Kleinberger G, Yamanishi Y, Suarez-Calvet M, Czirr E, Lohmann E, Cuyvers E, et al. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. (2014) 6:243ra286. doi: 10.1126/scitranslmed.3009093
55. Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. (2016) 90:724–39. doi: 10.1016/j.neuron.2016.05.003
56. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. (2015) 160:1061–71. doi: 10.1016/j.cell.2015.01.049
57. Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med. (2015) 212:287–95. doi: 10.1084/jem.20142322
58. Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer's disease. J Neurosci. (2017) 37:637–47. doi: 10.1523/JNEUROSCI.2110-16.2016
59. Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. (2014) 9:20. doi: 10.1186/1750-1326-9-20
60. Garcia-Reitboeck P, Phillips A, Piers TM, Villegas-Llerena C, Butler M, Mallach A, et al. Human induced pluripotent stem cell-derived microglia-like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep. (2018) 24:2300–11. doi: 10.1016/j.celrep.2018.07.094
61. Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, et al. A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem. (2009) 109:1144–56. doi: 10.1111/j.1471-4159.2009.06042.x
62. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet. (2011) 43:429–35. doi: 10.1038/ng.803
63. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet. (2011) 43:436–41. doi: 10.1038/ng.801
64. Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. (2013) 78:631–43. doi: 10.1016/j.neuron.2013.04.014
65. Jakobsdottir J, van der Lee SJ, Bis JC, Chouraki V, Li-Kroeger D, Yamamoto S, et al. Rare functional variant in TM2D3 is associated with late-onset Alzheimer's disease. PLoS Genet. (2016) 12:e1006327. doi: 10.1371/journal.pgen.1006327
66. Haney MS, Bohlen CJ, Morgens DW, Ousey JA, Barkal AA, Tsui CK, et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat Genet. (2018) 50:1716–27. doi: 10.1038/s41588-018-0254-1
67. Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci. (2017) 20:1052–61. doi: 10.1038/nn.4587
68. Smith AM, Gibbons HM, Oldfield RL, Bergin PM, Mee EW, Faull RL, et al. The transcription factor PU.1 is critical for viability and function of human brain microglia. Glia. (2013) 61:929–42. doi: 10.1002/glia.22486
69. Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. (2003) 302:841. doi: 10.1126/science.1090278
70. Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Rep. (2017) 8:1727–42. doi: 10.1016/j.stemcr.2017.05.017
71. Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. (2006) 442:916–9. doi: 10.1038/nature05016
72. Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. (2006) 442:920–4. doi: 10.1038/nature05017
73. Brouwers N, Sleegers K, Engelborghs S, Maurer-Stroh S, Gijselinck I, van der Zee J, et al. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology. (2008) 71:656–64. doi: 10.1212/01.wnl.0000319688.89790.7a
74. Cortini F, Fenoglio C, Guidi I, Venturelli E, Pomati S, Marcone A, et al. Novel exon 1 progranulin gene variant in Alzheimer's disease. Eur J Neurol. (2008) 15:1111–7. doi: 10.1111/j.1468-1331.2008.02266.x
75. Minami SS, Min SW, Krabbe G, Wang C, Zhou Y, Asgarov R, et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer's disease mouse models. Nat Med. (2014) 20:1157–64. doi: 10.1038/nm.3672
76. N'Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C, et al. TREM-2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol. (2009) 184:215–23. doi: 10.1083/jcb.200808080
77. Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. (2005) 365:415–6. doi: 10.1016/S0140-6736(05)17830-1
78. Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. (2004) 44:601–7. doi: 10.1016/j.neuron.2004.11.005
79. Kim KS, Marcogliese PC, Yang J, Callaghan SM, Resende V, Abdel-Messih E, et al. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson's disease. Proc Natl Acad Sci USA. (2018) 115:E5164–73. doi: 10.1073/pnas.1718946115
80. International Multiple Sclerosis Genetics Consortium Wellcome Trust Case Control Consortium, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. (2011) 476:214–9. doi: 10.1038/nature10251
81. Ma GZ, Stankovich J, Australia New Zealand Multiple Sclerosis Genetics Consortium, Kilpatrick TJ, Binder MD, Field J. Polymorphisms in the receptor tyrosine kinase MERTK gene are associated with multiple sclerosis susceptibility. PLoS ONE. (2011) 6:e16964. doi: 10.1371/journal.pone.0016964
82. Healy LM, Perron G, Won SY, Michell-Robinson MA, Rezk A, Ludwin SK, et al. MerTK is a functional regulator of myelin phagocytosis by human myeloid cells. J Immunol. (2016) 196:3375–84. doi: 10.4049/jimmunol.1502562
83. Healy LM, Jang JH, Won SY, Lin YH, Touil H, Aljarallah S, et al. MerTK-mediated regulation of myelin phagocytosis by macrophages generated from patients with MS. Neurol Neuroimmunol Neuroinflamm. (2017) 4:e402. doi: 10.1212/NXI.0000000000000402
84. Morioka T, Kalehua AN, Streit WJ. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J Comp Neurol. (1993) 327:123–32. doi: 10.1002/cne.903270110
85. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. (2005) 8:752–8. doi: 10.1038/nn1472
86. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. (2009) 40:1849–57. doi: 10.1161/STROKEAHA.108.534503
87. Mawhinney LA, Thawer SG, Lu WY, Rooijen N, Weaver LC, Brown A, et al. Differential detection and distribution of microglial and hematogenous macrophage populations in the injured spinal cord of lys-EGFP-ki transgenic mice. J Neuropathol Exp Neurol. (2012) 71:180–97. doi: 10.1097/NEN.0b013e3182479b41
88. Hines DJ, Hines RM, Mulligan SJ, Macvicar BA. Microglia processes block the spread of damage in the brain and require functional chloride channels. Glia. (2009) 57:1610–8. doi: 10.1002/glia.20874
89. Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, et al. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun. 7:11499. doi: 10.1038/ncomms11499
90. David S, Kroner A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat Rev Neurosci. (2011) 12:388–99. doi: 10.1038/nrn3053
91. Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. (2015) 16:907–17. doi: 10.1038/ni.3253
92. Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. (2010) 189:1059–70. doi: 10.1083/jcb.201004096
93. Green DR, Oguin TH, Martinez J. The clearance of dying cells: table for two. Cell Death Differ. (2016) 23:915–26. doi: 10.1038/cdd.2015.172
94. Kotter MR, Zhao C, van Rooijen N, Franklin RJ. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol Dis. (2005) 18:166–75. doi: 10.1016/j.nbd.2004.09.019
95. Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. (2009) 132(Pt 2):288–95. doi: 10.1093/brain/awn109
96. Schwab ME, Strittmatter SM. Nogo limits neural plasticity and recovery from injury. Curr Opin Neurobiol. (2014) 27:53–60. doi: 10.1016/j.conb.2014.02.011
97. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
98. Fricker M, Neher JJ, Zhao JW, Thery C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. (2012) 32:2657–66. doi: 10.1523/JNEUROSCI.4837-11.2012
99. Neher JJ, Emmrich JV, Fricker M, Mander PK, Thery C, Brown GC. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci USA. (2013) 110:E4098–4107. doi: 10.1073/pnas.1308679110
100. Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, et al. Roles of microglia in brain development, tissue maintenance and repair. Brain. (2015) 138(Pt 5):1138–59. doi: 10.1093/brain/awv066
101. Ritzel RM, Patel AR, Grenier JM, Crapser J, Verma R, Jellison ER, et al. Functional differences between microglia and monocytes after ischemic stroke. J Neuroinflammation 12:106. doi: 10.1186/s12974-015-0329-1
102. Greenhalgh AD, David S. Differences in the phagocytic response of microglia and peripheral macrophages after spinal cord injury and its effects on cell death. J Neurosci. (2014) 34:6316–22. doi: 10.1523/JNEUROSCI.4912-13.2014
103. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. (2009) 29:13435–44. doi: 10.1523/JNEUROSCI.3257-09.2009
104. Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity. (2013) 38:555–69. doi: 10.1016/j.immuni.2013.02.012
105. Evans TA, Barkauskas DS, Myers JT, Hare EG, You JQ, Ransohoff RM, et al. High-resolution intravital imaging reveals that blood-derived macrophages but not resident microglia facilitate secondary axonal dieback in traumatic spinal cord injury. Exp Neurol. (2014) 254:109–20. doi: 10.1016/j.expneurol.2014.01.013
106. David S, Greenhalgh AD, Kroner A. Macrophage and microglial plasticity in the injured spinal cord. Neuroscience. (2015) 307:311–8. doi: 10.1016/j.neuroscience.2015.08.064
107. Greenhalgh AD, Zarruk JG, Healy LM, Baskar Jesudasan SJ, Jhelum P, Salmon CK, et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol. (2018) 16:e2005264. doi: 10.1371/journal.pbio.2005264
108. Zarruk JG, Greenhalgh AD, David S. Microglia and macrophages differ in their inflammatory profile after permanent brain ischemia. Exp Neurol. (2018) 301(Pt B):120–32. doi: 10.1016/j.expneurol.2017.08.011
109. Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. (1999) 158:351–65. doi: 10.1006/exnr.1999.7118
110. Nishio Y, Koda M, Hashimoto M, Kamada T, Koshizuka S, Yoshinaga K, et al. Deletion of macrophage migration inhibitory factor attenuates neuronal death and promotes functional recovery after compression-induced spinal cord injury in mice. Acta Neuropathol. (2009) 117:321–8. doi: 10.1007/s00401-008-0476-x
111. Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, et al. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. (2011) 31:9910–22. doi: 10.1523/JNEUROSCI.2114-11.2011
112. Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci. (2015) 35:748–60. doi: 10.1523/JNEUROSCI.2405-14.2015
113. Makinde HM, Cuda CM, Just TB, Perlman HR, Schwulst SJ. Nonclassical monocytes mediate secondary injury, neurocognitive outcome, and neutrophil infiltration after traumatic brain injury. J Immunol. (2017) 199:3583–91. doi: 10.4049/jimmunol.1700896
114. Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med. (2009) 6:e1000113. doi: 10.1371/journal.pmed.1000113
115. Francos-Quijorna I, Amo-Aparicio J, Martinez-Muriana A, Lopez-Vales R. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia. (2016) 64:2079–92. doi: 10.1002/glia.23041
116. Russo MV, McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. (2016) 353:783–5. doi: 10.1126/science.aaf6260
117. Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. (2008) 209:378–88. doi: 10.1016/j.expneurol.2007.06.009
118. Umekawa T, Osman AM, Han W, Ikeda T, Blomgren K. Resident microglia, rather than blood-derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia-ischemia. Glia. (2015) 63:2220–30. doi: 10.1002/glia.22887
119. Garcia-Bonilla L, Faraco G, Moore J, Murphy M, Racchumi G, Srinivasan J, et al. Spatio-temporal profile, phenotypic diversity, and fate of recruited monocytes into the post-ischemic brain. J Neuroinflammation. (2016) 13:285. doi: 10.1186/s12974-016-0750-0
120. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med. (2014) 211:1533–49. doi: 10.1084/jem.20132477
121. Robinson S, Miller RH. Contact with central nervous system myelin inhibits oligodendrocyte progenitor maturation. Dev Biol. (1999) 216:359–68. doi: 10.1006/dbio.1999.9466
122. Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. (2006) 26:328–32. doi: 10.1523/JNEUROSCI.2615-05.2006
123. Kotter MR, Stadelmann C, Hartung HP. Enhancing remyelination in disease–can we wrap it up? Brain. (2011) 134(Pt 7):1882–900. doi: 10.1093/brain/awr014
124. Smith ME. Phagocytosis of myelin by microglia in vitro. J Neurosci Res. (1993) 35:480–7. doi: 10.1002/jnr.490350504
125. Mosley K, Cuzner ML. Receptor-mediated phagocytosis of myelin by macrophages and microglia: effect of opsonization and receptor blocking agents. Neurochem Res. (1996) 21:481–7. doi: 10.1007/BF02527713
126. Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, et al. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. (2012) 60:717–27. doi: 10.1002/glia.22298
127. Smith ME, Hoerner MT. Astrocytes modulate macrophage phagocytosis of myelin in vitro. J Neuroimmunol. (2000) 102:154–62. doi: 10.1016/S0165-5728(99)00218-0
128. Tcw J, Wang M, Pimenova AA, Bowles KR, Hartley BJ, Lacin E, et al. An efficient platform for astrocyte differentiation from human induced pluripotent stem cells. Stem Cell Rep. (2017) 9:600–14. doi: 10.1016/j.stemcr.2017.06.018
129. Bruck W, Friede RL. Anti-macrophage CR3 antibody blocks myelin phagocytosis by macrophages in vitro. Acta Neuropathol. (1990) 80:415–8. doi: 10.1007/BF00307696
130. DeJong BA, Smith ME. A role for complement in phagocytosis of myelin. Neurochem Res. (1997) 22:491–8. doi: 10.1023/A:1027372129989
131. Prineas JW, Graham JS. Multiple sclerosis: capping of surface immunoglobulin G on macrophages engaged in myelin breakdown. Ann Neurol. (1981) 10:149–58. doi: 10.1002/ana.410100205
132. Vanguri P, Koski CL, Silverman B, Shin ML. Complement activation by isolated myelin: activation of the classical pathway in the absence of myelin-specific antibodies. Proc Natl Acad Sci USA. (1982) 79:3290–4. doi: 10.1073/pnas.79.10.3290
133. da Costa CC, van der Laan LJ, Dijkstra CD, Bruck W. The role of the mouse macrophage scavenger receptor in myelin phagocytosis. Eur J Neurosci. (1997) 9:2650–7. doi: 10.1111/j.1460-9568.1997.tb01694.x
134. Weinger JG, Brosnan CF, Loudig O, Goldberg MF, Macian F, Arnett HA, et al. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J Neuroinflammation 8:49. doi: 10.1186/1742-2094-8-49
135. Ray AK, DuBois JC, Gruber RC, Guzik HM, Gulinello ME, Perumal G, et al. Loss of Gas6 and Axl signaling results in extensive axonal damage, motor deficits, prolonged neuroinflammation, and less remyelination following cuprizone exposure. Glia. (2017) 65:2051–69. doi: 10.1002/glia.23214
136. Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH, et al. Blockade of TREM-2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. (2007) 37:1290–301. doi: 10.1002/eji.200636837
137. Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. (2007) 4:e124. doi: 10.1371/journal.pmed.0040124
138. Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest. (2015) 125:2161–70. doi: 10.1172/JCI77983
139. Cantoni C, Bollman B, Licastro D, Xie M, Mikesell R, Schmidt R, et al. TREM2 regulates microglial cell activation in response to demyelination in vivo. Acta Neuropathol. (2015) 129:429–47. doi: 10.1007/s00401-015-1388-1
140. Ludwin SK, Rao V, Moore CS, Antel JP. Astrocytes in multiple sclerosis. Mult Scler. (2016) 22:1114–24. doi: 10.1177/1352458516643396
141. Vinores SA, Herman MM. Phagocytosis of myelin by astrocytes in explants of adult rabbit cerebral white matter maintained on Gelfoam matrix. J Neuroimmunol. (1993) 43:169–76. doi: 10.1016/0165-5728(93)90088-G
142. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. (2008) 28:264–78. doi: 10.1523/JNEUROSCI.4178-07.2008
143. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. (2017) 541:481–7. doi: 10.1038/nature21029
144. Williams K, Ulvestad E, Waage A, Antel JP, McLaurin J. Activation of adult human derived microglia by myelin phagocytosis in vitro. J Neurosci Res. (1994) 38:433–43. doi: 10.1002/jnr.490380409
145. Ponath G, Ramanan S, Mubarak M, Housley W, Lee S, Sahinkaya FR, et al. Myelin phagocytosis by astrocytes after myelin damage promotes lesion pathology. Brain. (2017) 140:399–413. doi: 10.1093/brain/aww298
146. Skripuletz T, Hackstette D, Bauer K, Gudi V, Pul R, Voss E, et al. Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain. (2013) 136(Pt 1):147–67. doi: 10.1093/brain/aws262
147. Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. (1992) 256:184–5. doi: 10.1126/science.1566067
148. Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. (2010) 330:1774. doi: 10.1126/science.1197623
149. Ries M, Sastre M. Mechanisms of abeta clearance and degradation by glial cells. Front Aging Neurosci. 8:160. doi: 10.3389/fnagi.2016.00160
150. Lee CY, Landreth GE. The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna). (2010) 117:949–60. doi: 10.1007/s00702-010-0433-4
151. Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. (2008) 28:4283–92. doi: 10.1523/JNEUROSCI.4814-07.2008
152. Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Abeta42 hotspots around plaques. Nat Commun. 6:6176. doi: 10.1038/ncomms7176
153. Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. (1999) 57:563–81. doi: 10.1016/S0301-0082(98)00069-0
154. Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, et al. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer's disease. Mol Neurodegener. (2011) 6:45. doi: 10.1186/1750-1326-6-45
155. Hellwig S, Masuch A, Nestel S, Katzmarski N, Meyer-Luehmann M, Biber K. Forebrain microglia from wild-type but not adult 5xFAD mice prevent amyloid-beta plaque formation in organotypic hippocampal slice cultures. Sci Rep. (2015) 5:14624. doi: 10.1038/srep14624
156. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. (2015) 14:388–405. doi: 10.1016/S1474-4422(15)70016-5
157. Majumdar A, Chung H, Dolios G, Wang R, Asamoah N, Lobel P, et al. Degradation of fibrillar forms of Alzheimer's amyloid beta-peptide by macrophages. Neurobiol Aging. (2008) 29:707–15. doi: 10.1016/j.neurobiolaging.2006.12.001
158. Zuroff L, Daley D, Black KL, Koronyo-Hamaoui M. Clearance of cerebral Abeta in Alzheimer's disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci. (2017) 74:2167–201. doi: 10.1007/s00018-017-2463-7
159. Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol. (1992) 84:225–33. doi: 10.1007/BF00227813
160. Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid beta-protein by microglial cells. J Biol Chem. (1997) 272:29390–7. doi: 10.1074/jbc.272.46.29390
161. Chung H, Brazil MI, Soe TT, Maxfield FR. Uptake, degradation, and release of fibrillar and soluble forms of Alzheimer's amyloid beta-peptide by microglial cells. J Biol Chem. (1999) 274:32301–8. doi: 10.1074/jbc.274.45.32301
162. Majumdar A, Cruz D, Asamoah N, Buxbaum A, Sohar I, Lobel P, et al. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. (2007) 18:1490–6. doi: 10.1091/mbc.e06-10-0975
163. Majumdar A, Capetillo-Zarate E, Cruz D, Gouras GK, Maxfield FR. Degradation of Alzheimer's amyloid fibrils by microglia requires delivery of ClC-7 to lysosomes. Mol Biol Cell. (2011) 22:1664–76. doi: 10.1091/mbc.e10-09-0745
164. Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ. Localization and cell association of C1q in Alzheimer's disease brain. Exp Neurol. (1996) 138:22–32. doi: 10.1006/exnr.1996.0043
165. Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer's disease brain. Am J Pathol. (1999) 154:927–36. doi: 10.1016/S0002-9440(10)65340-0
166. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. (2009) 41:1088–93. doi: 10.1038/ng.440
167. Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. (2009) 41:1094–9. doi: 10.1038/ng.439
168. Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet. (2013) 45:1452–8. doi: 10.1038/ng.2802
169. Morgan BP. Complement in the pathogenesis of Alzheimer's disease. Semin Immunopathol. (2018) 40:113–24. doi: 10.1007/s00281-017-0662-9
170. Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. (2000) 161:127–38. doi: 10.1006/exnr.1999.7260
171. Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, et al. Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia. (2012) 60:993–1003. doi: 10.1002/glia.22331
172. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. (1991) 30:572–80. doi: 10.1002/ana.410300410
173. Jack Jr CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. (2010) 9:119–28. doi: 10.1016/S1474-4422(09)70299-6
174. Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer's disease. J Neurosci. (2004) 24:6457–65. doi: 10.1523/JNEUROSCI.0901-04.2004
175. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. (2016) 352:712–6. doi: 10.1126/science.aad8373
176. Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. (2017) 9:eaaf6295. doi: 10.1126/scitranslmed.aaf6295
177. Kawabori M, Kacimi R, Kauppinen T, Calosing C, Kim JY, Hsieh CL, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) deficiency attenuates phagocytic activities of microglia and exacerbates ischemic damage in experimental stroke. J Neurosci. (2015) 35:3384–96. doi: 10.1523/JNEUROSCI.2620-14.2015
178. Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. (2016) 213:667–75. doi: 10.1084/jem.20151948
179. Condello C, Yuan P, Grutzendler J. Microglia-mediated neuroprotection, TREM2, and Alzheimer's disease: evidence from optical imaging. Biol Psychiatry. (2018) 83:377–87. doi: 10.1016/j.biopsych.2017.10.007
180. Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer's disease. Neurology. (1993) 43:1467–72. doi: 10.1212/WNL.43.8.1467
181. Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem. (2015) 290:26043–50. doi: 10.1074/jbc.M115.679043
182. Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. (2009) 63:287–303. doi: 10.1016/j.neuron.2009.06.026
183. Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. (2017) 47:566–81 e569. doi: 10.1016/j.immuni.2017.08.008
184. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. (2017) 169:1276–90 e1217. doi: 10.1016/j.cell.2017.05.018
185. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer's disease. J Cell Biol. (2018) 217:459–72. doi: 10.1083/jcb.201709069
186. Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ, et al. A transcriptomic atlas of aged human microglia. Nat Commun. (2018) 9:539. doi: 10.1038/s41467-018-02926-5
187. Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K. Microglia derived from aging mice exhibit an altered inflammatory profile. Glia. (2007) 55:412–24. doi: 10.1002/glia.20468
188. Tremblay ME, Zettel ML, Ison JR, Allen PD, Majewska AK. Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia. (2012) 60:541–58. doi: 10.1002/glia.22287
189. Hefendehl JK, Neher JJ, Suhs RB, Kohsaka S, Skodras A, Jucker M. Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell. (2014) 13:60–9. doi: 10.1111/acel.12149
190. Floden AM, Combs CK. Microglia demonstrate age-dependent interaction with amyloid-beta fibrils. J Alzheimers Dis. (2011) 25:279–93. doi: 10.3233/JAD-2011-101014
191. Bliederhaeuser C, Grozdanov V, Speidel A, Zondler L, Ruf WP, Bayer H, et al. Age-dependent defects of alpha-synuclein oligomer uptake in microglia and monocytes. Acta Neuropathol. (2016) 131:379–91. doi: 10.1007/s00401-015-1504-2
192. Shobin E, Bowley MP, Estrada LI, Heyworth NC, Orczykowski ME, Eldridge SA, et al. Microglia activation and phagocytosis: relationship with aging and cognitive impairment in the rhesus monkey. Geroscience. (2017) 39:199–220. doi: 10.1007/s11357-017-9965-y
193. Peters A, Josephson K, Vincent SL. Effects of aging on the neuroglial cells and pericytes within area 17 of the rhesus monkey cerebral cortex. Anat Rec. (1991) 229:384–98. doi: 10.1002/ar.1092290311
194. Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. (2012) 33:195 e191-112. doi: 10.1016/j.neurobiolaging.2010.05.008
195. Clayton KA, Van Enoo AA, Ikezu T. Alzheimer's disease: the role of microglia in brain homeostasis and proteopathy. Front Neurosci. (2017) 11:680. doi: 10.3389/fnins.2017.00680
196. Hughes MM, Field RH, Perry VH, Murray CL, Cunningham C. Microglia in the degenerating brain are capable of phagocytosis of beads and of apoptotic cells, but do not efficiently remove PrPSc, even upon LPS stimulation. Glia. (2010) 58:2017–30. doi: 10.1002/glia.21070
197. Ciesielski-Treska J, Grant NJ, Ulrich G, Corrotte M, Bailly Y, Haeberle AM, et al. Fibrillar prion peptide (106–126) and scrapie prion protein hamper phagocytosis in microglia. Glia. (2004) 46:101–15. doi: 10.1002/glia.10363
198. Janda E, Boi L, Carta AR. Microglial phagocytosis and its regulation: a therapeutic target in Parkinson's disease? Front Mol Neurosci. (2018) 11:144. doi: 10.3389/fnmol.2018.00144
199. Satoh J, Tabunoki H, Ishida T, Yagishita S, Jinnai K, Futamura N, et al. Immunohistochemical characterization of microglia in Nasu-Hakola disease brains. Neuropathology. (2011) 31:363–75. doi: 10.1111/j.1440-1789.2010.01174.x
200. Kaifu T, Nakahara J, Inui M, Mishima K, Momiyama T, Kaji M, et al. Osteopetrosis and thalamic hypomyelinosis with synaptic degeneration in DAP12-deficient mice. J Clin Invest. (2003) 111:323–32. doi: 10.1172/JCI16923
201. Roumier A, Bechade C, Poncer JC, Smalla KH, Tomasello E, Vivier E, et al. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J Neurosci. (2004) 24:11421–8. doi: 10.1523/JNEUROSCI.2251-04.2004
202. Satoh JI, Kino Y, Yanaizu M, Saito Y. Alzheimer's disease pathology in Nasu-Hakola disease brains. Intractable Rare Dis Res. (2018) 7:32–6. doi: 10.5582/irdr.2017.01088
203. Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin deficiency promotes circuit-specific synaptic pruning by microglia via complement activation. Cell. (2016) 165:921–35. doi: 10.1016/j.cell.2016.04.001
204. Lian H, Roy E, Zheng H. Microglial phagocytosis assay. Bio Protoc. (2016) 6:e1988. doi: 10.21769/BioProtoc.1988
205. Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JCM, Sajti E, et al. An environment-dependent transcriptional network specifies human microglia identity. Science. (2017) 356:eaa13222. doi: 10.1126/science.aal3222
206. Petersen MA, Dailey ME. Diverse microglial motility behaviors during clearance of dead cells in hippocampal slices. Glia. (2004) 46:195–206. doi: 10.1002/glia.10362
207. Dailey ME, Eyo U, Fuller L, Hass J, Kurpius D. Imaging microglia in brain slices and slice cultures. Cold Spring Harb Protoc. (2013) 2013:1142–8. doi: 10.1101/pdb.prot079483
208. Beccari S, Diaz-Aparicio I, Sierra A. Quantifying microglial phagocytosis of apoptotic cells in the brain in health and disease. Curr Protoc Immunol. (2018) 24:e49. doi: 10.1002/cpim.49
209. Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, et al. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. (2000) 20:4106–14. doi: 10.1128/MCB.20.11.4106-4114.2000
210. Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, et al. Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J Neurosci Res. (2005) 81:357–62. doi: 10.1002/jnr.20480
211. Sasmono RT, Williams E. Generation and characterization of MacGreen mice, the Cfs1r-EGFP transgenic mice. Methods Mol Biol. (2012) 844:157–76. doi: 10.1007/978-1-61779-527-5_11
212. Smith AM, Dragunow M. The human side of microglia. Trends Neurosci. (2014) 37:125–35. doi: 10.1016/j.tins.2013.12.001
213. Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA, et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron. (2017) 94:278–93 e279. doi: 10.1016/j.neuron.2017.03.042
214. Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, et al. Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. (2017) 8:1516–24. doi: 10.1016/j.stemcr.2017.04.023
215. Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci. (2017) 20:753–9. doi: 10.1038/nn.4534
216. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA. (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
217. Nimmerjahn A. Two-photon imaging of microglia in the mouse cortex in vivo. Cold Spring Harb. Protoc. (2012) 2012:pdb.prot069294. doi: 10.1101/pdb.prot069294
Keywords: phagocytosis, microglia, macrophage, neurodegeneration, neuroinflammation
Citation: Galloway DA, Phillips AEM, Owen DRJ and Moore CS (2019) Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 10:790. doi: 10.3389/fimmu.2019.00790
Received: 15 January 2019; Accepted: 26 March 2019;
Published: 16 April 2019.
Edited by:
Bieke Broux, University of Hasselt, BelgiumReviewed by:
Amanda Sierra, Achucarro Basque Center for Neuroscience, SpainAndre Ortlieb Guerreiro Cacais, Karolinska Institute (KI), Sweden
Copyright © 2019 Galloway, Phillips, Owen and Moore. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig S. Moore, Y3JhaWcubW9vcmVAbXVuLmNh
†These authors have contributed equally to this work