 Shafaqat Ali
Shafaqat Ali Ritu Mann-Nüttel
Ritu Mann-Nüttel Anja Schulze1
Anja Schulze1 Lisa Richter
Lisa Richter Judith Alferink
Judith Alferink Stefanie Scheu
Stefanie Scheu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 April 2019
Sec. Antigen Presenting Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00778
This article is part of the Research Topic Harnessing the Participation of Dendritic Cells in Immunity and Tolerance View all 40 articles
Type I Interferons (IFNs) are hallmark cytokines produced in immune responses to all classes of pathogens. Type I IFNs can influence dendritic cell (DC) activation, maturation, migration, and survival, but also directly enhance natural killer (NK) and T/B cell activity, thus orchestrating various innate and adaptive immune effector functions. Therefore, type I IFNs have long been considered essential in the host defense against virus infections. More recently, it has become clear that depending on the type of virus and the course of infection, production of type I IFN can also lead to immunopathology or immunosuppression. Similarly, in bacterial infections type I IFN production is often associated with detrimental effects for the host. Although most cells in the body are thought to be able to produce type I IFN, plasmacytoid DCs (pDCs) have been termed the natural “IFN producing cells” due to their unique molecular adaptations to nucleic acid sensing and ability to produce high amounts of type I IFN. Findings from mouse reporter strains and depletion experiments in in vivo infection models have brought new insights and established that the role of pDCs in type I IFN production in vivo is less important than assumed. Production of type I IFN, especially the early synthesized IFNβ, is rather realized by a variety of cell types and cannot be mainly attributed to pDCs. Indeed, the cell populations responsible for type I IFN production vary with the type of pathogen, its tissue tropism, and the route of infection. In this review, we summarize recent findings from in vivo models on the cellular source of type I IFN in different infectious settings, ranging from virus, bacteria, and fungi to eukaryotic parasites. The implications from these findings for the development of new vaccination and therapeutic designs targeting the respectively defined cell types are discussed.
The cytokine family of type I IFNs fulfills key functions in anti-viral immunity but is also produced in the immune responses to other classes of pathogens covering viruses, bacteria, parasites, and fungi (1). Additionally, these cytokines are functionally involved in the pathogenesis of inflammatory autoimmune diseases (2).
Together with IFNβ, type I IFNs comprise multiple IFNα subtypes (11 in mice and 13 in humans), IFNε, IFNκ, and IFNω in most mammals. In addition, IFNδ, IFNζ (limitin), and IFNτ have been detected exclusively in pigs, mice, and ruminants, respectively (3–6). Type I IFNs are encoded by intronless genes clustered in mice on chromosome 4 and in humans on chromosome 9 (3–6). Induction of type I IFN expression is facilitated after activation of a diverse set of pathogen sensing pattern recognition receptor (PRR) pathways by binding of IFN regulatory factors (IRFs) and NF-κB to acute response elements in the promoters of type I IFN gene loci (7). All type I IFNs bind to a common heterodimeric IFNα receptor (IFNAR), which is composed of the IFNAR1 and IFNAR2 subunits and is expressed by virtually all nucleated cells of the body. Following IFNAR engagement by its ligands, canonical type I IFN signaling activates the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway, leading to transcription of IFN-stimulated genes (ISGs) (5, 8). ISG-encoded proteins mediate induction of cell-intrinsic antimicrobial states in infected and neighboring cells that limit the spread of infectious agents, particularly viral pathogens. Additionally, ISGs influence innate and adaptive immune responses by promoting antigen presentation and NK cell functions, modulating inflammatory cytokine production, and activating high-affinity antigen-specific T and B cell responses and immunological memory (9). Type I IFN production, however, can also have deleterious roles in chronic viral and bacterial infections, and can lead to immunopathologies such as inflammatory disorders and autoimmunity (1, 2, 10, 11).
IFNβ was originally defined as the antiviral factor produced by fibroblasts after viral infections (12) and has been thought to be produced by virtually all cells of the body. Later pDCs specialized in the rapid secretion of high amounts of type I IFN have been termed the natural “IFN producing cells” (IPCs). Recent findings, however, indicate that production of type I IFN, especially the early synthesized IFNβ, in anti-infectious immune responses can occur independently of pDCs and that the cell type responsible for type I IFN production rather depends on the specific infectious setting. In this review we summarize the recent findings on the identity and function of type I IFN producing cells in infection by focusing on insights gained from in vivo mouse models covering type I IFN reporter mice and models of cell type specific ablation.
To devise novel anti-infectious treatment regimens targeting a specific cellular subtype, it is crucial to know the identity of the cells responsible for the production of type I IFN in the course of an infection. Early on, pDCs were considered primary producers of IFNα during virus infections (13, 14). For human pDCs it has been reported that IFNα/β transcripts account for an astounding 50% of all mRNAs in the cell after viral activation (15). More than 40 years ago, pDCs were first described in humans as natural IPCs that activate NK cells upon exposure to viruses (16, 17). The murine equivalent was described in 2001 as type I IFN producing cells with plasmacytoid morphology (18–20). These cells detect RNA and DNA viruses through two endosomal sensors, TLR7 and TLR9, respectively, which induce secretion of type I IFN through the MyD88-IRF7 signaling pathway (21–24). Specifically, TLR7/9-ligand interactions in early endosomes result in type I IFN production while ligand recognition in late endosomes or lysosomes rather leads to inflammatory cytokine production and pDC maturation (25, 26). At least in the mouse, TLR7 and 9 are also expressed by monocytes, conventional DCs (cDCs), and B cells (27, 28). Therefore, the contribution of those cell types to type I IFN production triggered via the TLR7/9-MyD88-IRF7 pathway has to be considered. B cells, for instance, have recently been shown to produce type I IFN in vivo after optimized stimulation conditions using the TLR9 ligand CpG-A (29). A specific feature of pDCs is that they can produce type I IFN independently of IFNAR mediated feedback signaling (30). However, they do respond to type I IFN by generating an autocrine circuit through IFNAR, which augments type I IFN secretion and induces their activation and migration (31, 32).
In humans, pDCs, monocytes, and other myeloid cells also produce type I IFN after stimulation of the TLR8-MyD88-IRF7 pathway by viral single-stranded RNA (ssRNA) (33, 34). The mouse TLR8 was initially considered non-functional (33, 34). More recently it has been shown that mouse TLR8 can be stimulated by a combination of oligodeoxynucleotides (ODNs) and human TLR8 ligands. Further, mouse pDCs produce type I IFN after stimulation with vaccinia virus (VV) in a TLR8 dependent way (35, 36). Two additional TLRs, TLR3 and 4, are able to induce type I IFN expression independently of the MyD88 pathway via recruiting the TIR domain-containing adaptor protein inducing interferon beta (TRIF; also known as TIR domain-containing adapter molecule 1, TICAM-1). This activates the transcription factor IRF3 thus initiating type I IFN, in particular IFNβ expression (37, 38). TLR3 is absent in mouse pDCs but highly expressed in endosomes of murine CD8α+ and CD103+ and human CD141+ cDCs of the DC1 subtype that are efficient in cross-presenting (39, 40). It recognizes double-stranded RNA (dsRNA) as viral replication intermediates as well as ssRNA containing stem loops (41). In addition to DCs, TLR3 activation can lead to type I IFN expression in macrophages, fibroblasts, and epithelial cells (42). While TLR3 exclusively signals via the TRIF pathway, TLR4 utilizes MyD88 as well as TRIF signaling routes after recognizing its cognate ligand bacterial lipopolysaccharide (LPS). Analogous to TLR3 activation, LPS binding to TLR4 induces type I IFN expression via TRIF-IRF3 (43). The majority of hematopoietic cells of the myeloid and lymphoid lineage, with the exception of human pDCs, and few other cell types such as pancreatic β-cells express TLR4 (44).
In contrast to pDCs, cDCs, and macrophages mainly produce type I IFN in response to virus challenge by utilizing retinoic acid-inducible gene I (RIG-I)-like helicases (RLHs) (43, 45–47). RLHs, including RIG-I and melanoma differentiation-associated gene 5 (MDA5), are cytoplasmic dsRNA receptors that transmit their signal through the mitochondrial antiviral-signaling protein, virus-induced signaling adapter (MAVS, aka IFNb promoter stimulator (IPS)-1 or Cardif). This activates IRF3 and IRF7 to induce the transcription of type I IFN and other antiviral genes (48–50).
Finally, soluble sensors in the cytoplasm detect dsDNA in a sequence-independent manner, exhibit a broad expression spectrum including pDCs, cDCs, macrophages, and mouse embryonic fibroblasts (MEFs), and activate signaling pathways leading to type I IFN expression (47, 51). These sensors include the cyclic GMP-AMP synthase (cGAS)/STING pathway, the RNA polymerase III/RIG-I/MAVS pathway, DNA-dependent activator of IRFs (DAI), IFNγ-inducible protein 16 (IFI16), and the DDX family (47, 51–58).
Several models of cytokine reporter mice have been developed for the detection of type I IFN production in vivo, as intracellular staining is not sensitive in most cases (Table 1 and Figure 1). Earlier published IFNβ knock-out mouse lines already contained reporter elements to detect Ifnb promoter-driven gene transcription. For example, coding sequences for the mouse immunoglobulin λ2 chain, a green fluorescent protein (GFP), or the human CD2 had been inserted immediately downstream of the Ifnb promoter to visualize IFNβ expression on a cellular level (73–75). However, the reporter features in these mouse strains have not been used in vivo so far.
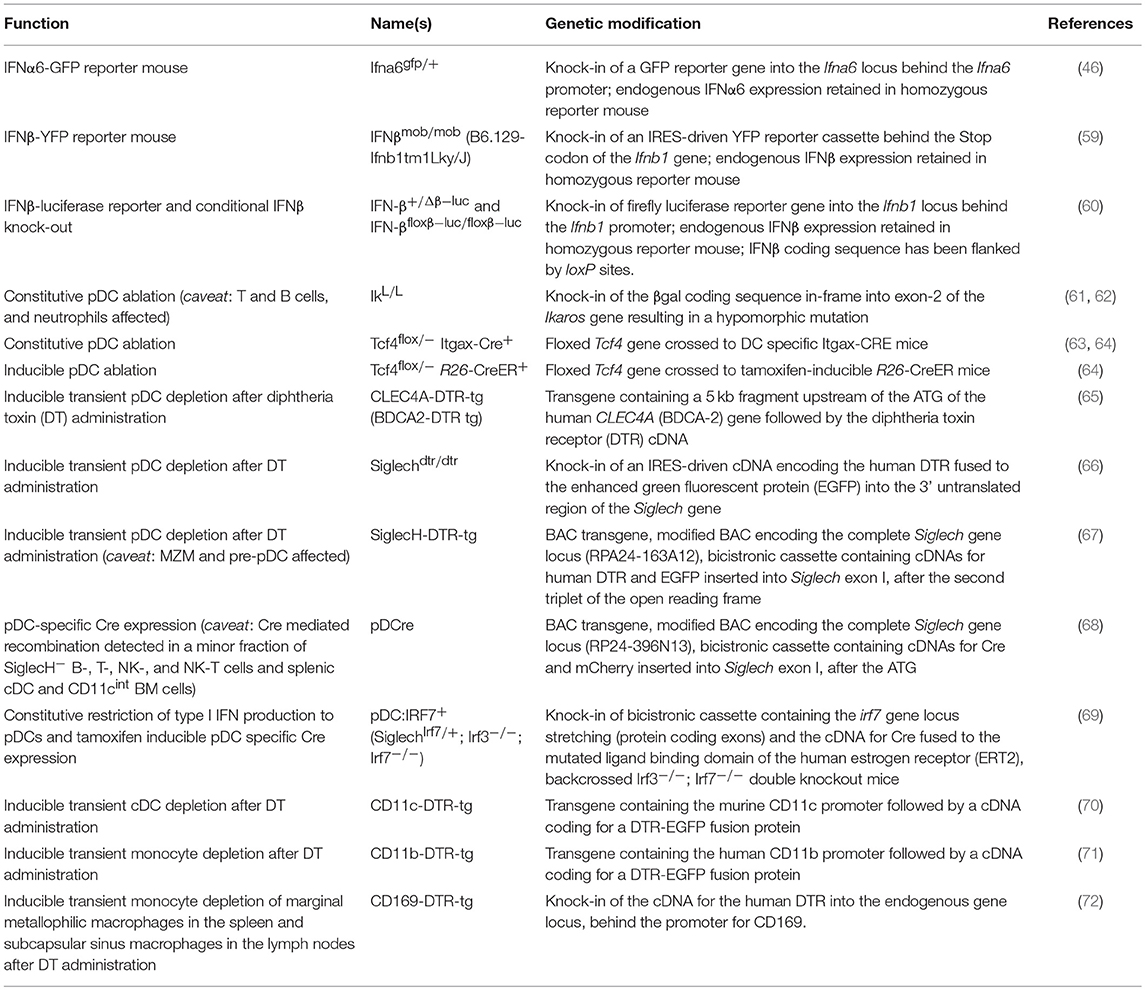
Table 1. Genetically modified mouse models to visualize or define the function of type I IFN producing cells.
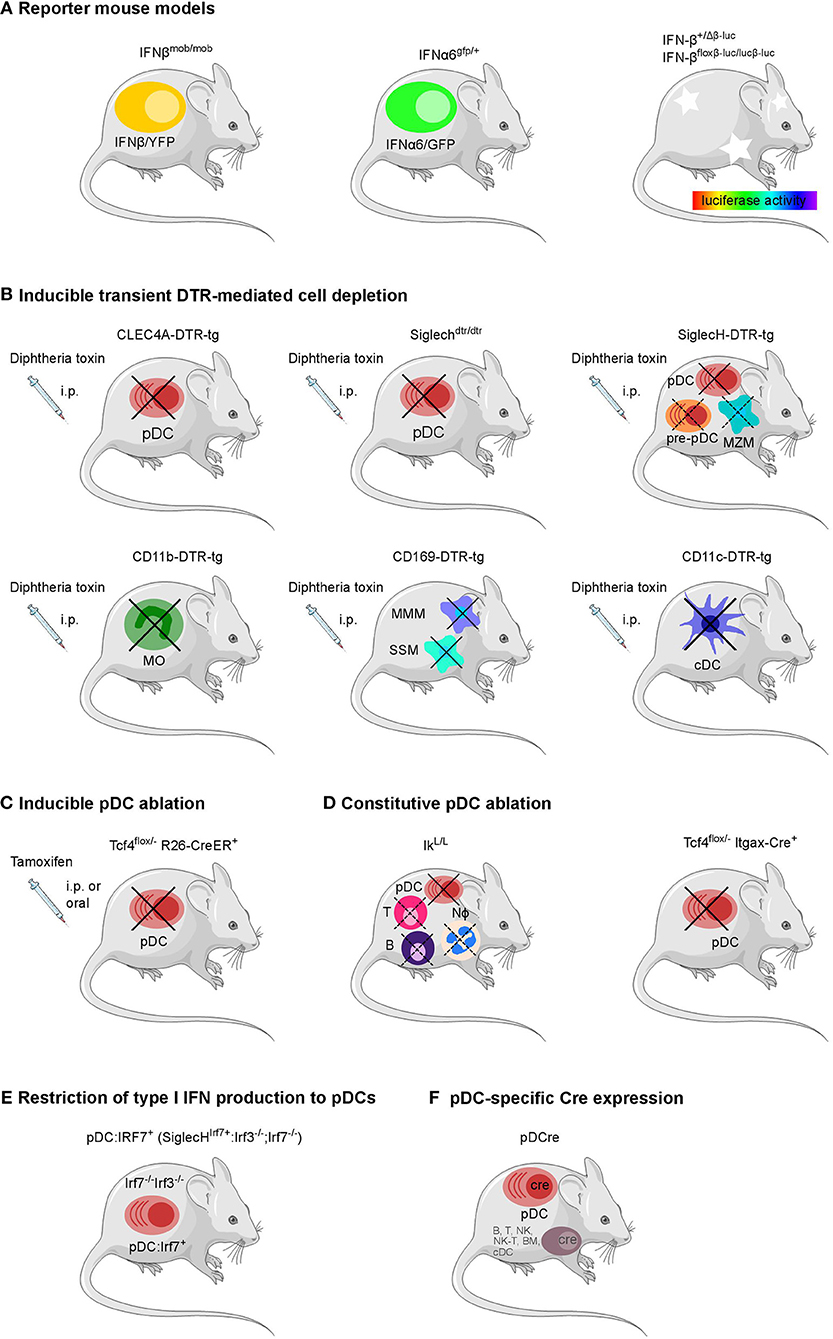
Figure 1. Overview of genetically modified mouse models available to define the cellular source and impact of type I IFNs. (A) Reporter mouse models for the detection of type I IFN producing cells, (B) mouse strains for the transient DTR-mediated cell depletion and (C) inducible and (D) constitutive ablation of pDCs, (E) pDC-specific Cre expression, and (F) a mouse line with a restriction of the type I IFN production to pDCs have been employed in various infection settings in vivo.Each model system harbors specific advantages and caveats as further described in Table 1. B, B cell; BM, bone marrow cell; cDC, conventional dendritic cell; MMM, marginal metallophilic macrophage; MZM, marginal zone macrophage; MO, monocyte; NK, natural killer cell; Nφ, neutrophil; SSM, subcapsular sinus macrophage; T, T cell; pDC, plasmacytoid dendritic cell. The figure was created using Servier Medical Art according to Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/). Changes were made to the original cartoons.
More recently, a mouse line expressing GFP under the control of the Ifna6 promoter (Ifna6gfp/+) recapitulates the expression of various IFNα genes and has been employed to define the cellular source of IFNα in virus infection models (32, 46, 76). Also, for IFNβ a fluorescence reporter-knock-in mouse model (IFNβmob/mob) has been generated. Here, yellow fluorescent protein (YFP) is expressed from a bicistronic mRNA linked by an internal ribosomal entry site (IRES) to the endogenous IFNβ mRNA (59). Ifna6gfp/+ as well as IFNβmob/mob reporter mice have each been shown to report for the majority of type I IFNs. However, in vitro analyses on BM-derived DCs from the double reporter mouse line generated by intercrossing the Ifna6gfp/+ and IFNβmob/mob reporter strains revealed that specific type I IFN subtypes can be produced by distinct cell subpopulations (77).
In an alternative reporter mouse system, a firefly luciferase reporter gene has been placed under the control of the Ifnb promoter (IFN-β+/Δβ−luc). Rather than IFNβ expression on a single cell level, this model detects in vivo kinetics of IFNβ expression in the mouse paralleling the spread of pathogens through the organism under infectious conditions (60). Additionally, in this mouse line the IFNβ coding sequence is flanked by loxP sites (IFN-βfloxβ−luc/floxβ−luc) providing the possibility to characterize the impact of IFNβ production by a given cell type on the pathophysiology of various infections via tissue- or cell-specific Cre-mediated deletion of IFNβ (60).
Several experimental strategies have been developed to determine the in vivo contribution of a specific cell type to the type I IFN response during infections (78). Initially, antibody mediated depletion has been utilized frequently to ablate pDCs and monocytes (79). Antibodies against Ly6G/C (also known as Gr1) and CD317 (also known as BST-2) have been used to deplete pDCs in vivo and in vitro (18, 79–87). However, these antibodies generally target multiple cell types in addition to pDCs: The antibody RB6-8C5 directed against Ly6G/C reacts strongly with neutrophil-specific Ly6G antigen, but cross-reacts also with the Ly6C Ag (88) expressed on pDCs as well as on monocytes/macrophages, activated T cells, NK cells, plasma cells, and endothelial cells (89–92). Likewise, CD317 is recognized by the three different antibody clones 120G8.04, JF05-1C2.4.1 (also known as PDCA-1), and eBio927, and is expressed in naïve mice by pDCs, but also plasma cells. Following stimulation with type I IFNs and IFNγ CD317 is upregulated, additionally, on several other myeloid and lymphoid cells (79, 93). Finally, in vivo treatment with clodronate-containing liposomes depletes phagocytes in mice, but also disturbs the microarchitecture of secondary lymphoid organs (94, 95).
In the past years, several genetically modified mouse lines with a constitutive or inducible lack of specific cell types attributed to produce type I IFN have become available (Table 1 and Figure 1) (78). For pDCs, already several mouse models exist for constitutive or inducible ablation. Mice carrying a hypomorphic mutation at the Ikaros locus express low levels of the transcription factor Ikaros (IkL/L) and therefore lack peripheral pDCs, but no other DC subsets (61). When using this line as a “pDC-less” model, one has to take into account that other hematopoietic lineages including T and B cells and neutrophils are also affected by the IkL/L mutation, and that IkL/L mice start to develop thymic lymphomas by 10 weeks of age (62, 96, 97). Constitutive deletion of E2-2, the basic helix-loop-helix transcription factor, also known as TCF4, that controls development and maintenance of pDCs, results in perinatal lethality in mice (98). To overcome this lethality and to specifically ablate the pDC lineage, mice harboring a constitutively deleted and a floxed Tfc4 allele (Tcf4flox/−) have been crossed to Itgax-Cre (CD11c-Cre) or Rosa26-CreER mice in which Cre is expressed in DCs or can be induced ubiquitously after tamoxifen administration, respectively (63, 64, 99, 100). Another strategy uses Diphtheria toxin receptor (DTR)-mediated conditional and targeted cell depletion. CLEC4A-DTR-tg mice express DTR under the human pDC specific promotor of the C-type lectin domain family 4 member A (CLEC4A; also known as blood dendritic cell antigen 2, BDCA2). Administration of diphtheria toxin (DT) in these CLEC4A-DTR-tg mice results in transient but specific depletion of pDCs (65). In an alternative approach, a cDNA encoding the human DTR fused to the enhanced green fluorescent protein (EGFP) and preceded by an IRES was inserted into the 3′ untranslated region of the Siglech gene. This Siglechdtr/dtr mouse model allows specific elimination of pDCs in vivo via injection of DT (66). An analogous mouse line termed SiglecH-DTR-tg was generated using bacterial artificial chromosome (BAC) transgenic technology (67). SiglecH represents a sialic acid–binding Ig-like lectin that exerts immunomodulatory roles in antiviral immune responses. In SiglecHeGFP/+mice, heterozygous for the reporter gene, it was shown that in addition to pDCs, SiglecH was expressed in specialized macrophage subsets, such as marginal zone macrophages (MZM), lymph node medullary macrophages, and microglia. SiglecH was also found in immediate precursors of pDCs (pre-pDCs) in the BM, which have the plasticity to differentiate into pDCs and cDCs (67, 101). Despite of SiglecH expression on above described other cell types Loschko et al. showed pDC specific antigen delivery in mice by using SiglecH as a target structure (102) suggesting the usability of SiglecH as a lead molecule for the generation of pDC specific transgenic animals. A side by side comparison showed a higher susceptibility to Listeria monocytogenes infection in DT-treated SiglecH-DTR-tg vs. CLEC4A-DTR-tg mice. This finding was attributed to the additional lack of MZM in SiglecH-DTR-tg mice after DT treatment which was not observed in CLEC4A-DTR-tg mice (67).
With the aim to specifically express the Cre recombinase in pDCs a BAC-tg “pDCre” mouse line was generated which expresses Cre under the control of the Siglech promoter (68). By crossing these mice with a reporter mouse line that indicates Cre activity via red fluorescent protein (RFP) expression the authors found ~30% of SiglecH+ pDCs terminally labeled with RFP. Additionally, RFP expression was observed in a minor fraction of SiglecH− B-, T-, NK-, and NK-T cells and splenic cDCs and CD11cint BM cells suggesting that a small fraction of early lymphoid progenitors actively transcribes the SiglecH locus. Thus, the broader expression pattern of SiglecH should be considered when using SiglecH-DTR-tg mice to evaluate pDC functions in vivo.
Recently, a novel mouse model has been described in which type I IFN production is restricted to pDCs. In this knock-in model Irf7 expression is driven by the Siglech promoter (SiglechIrf7/+). The SiglechIrf7/+ mice were then backcrossed onto Irf3−/−/Irf7−/− double knock-out mice which are deficient in type I IFN production. This yielded animals (referred to as “pDC:Irf7+” mice) in which IRF7 signaling required for type I IFN expression is functional exclusively in pDCs (69). Additionally, in these mice an IRES site followed by coding sequences for Cre fused to the mutated ligand binding domain of the human estrogen receptor (ERT2) (103) was inserted behind the Irf7 gene into the Siglech gene locus (69). Therefore, this mouse line can potentially be used in the future for tamoxifen inducible pDC specific Cre expression and thus pDC specific gene deletion when crossed to the respective floxed mouse lines.
Also, for other cell types than pDCs, the DTR-mediated depletion approach has been employed. In recent years, mice expressing the DTR under the control of the CD11b-, CD11c-, and CD169-promoters have been generated and successfully used for depletion of monocytes, cDCs, and CD169+ macrophage subpopulations such as MZMs and subcapsular sinus macrophages in the spleen and lymph nodes (70–72, 104–109).
In the following sections we will discuss approaches designed to define the type I IFN producing cell types in infection using the in vivo mouse models described above.
In this chapter we will focus on more recent findings from in vivo models aimed at visualizing IFNα/β producing cell types and defining their contribution to the overall type I IFN production and their impact on the course of viral infections (Table 2). For a more generalized overview of the cellular sources of type I IFN in viral infections we kindly refer to an expert review by Swiecki et al. (127).
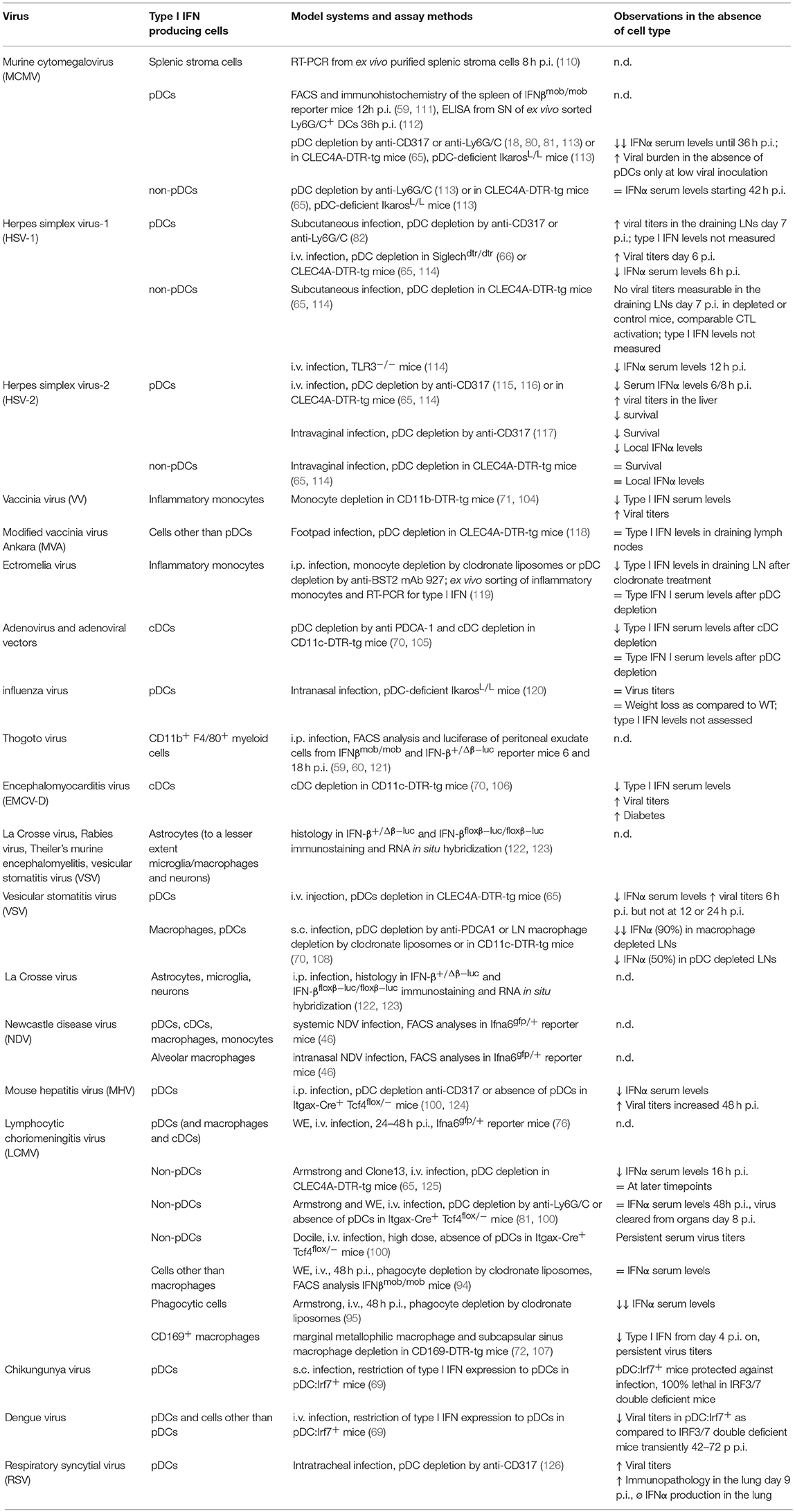
Table 2. Cellular sources of type I IFN production in viral infections in vivo.
Findings on the cellular sources of type I IFN during relevant infection models for DNA viruses and the respective in vivo experimental strategies are highlighted in the following sections.
Infection with the human cytomegalovirus (HCMV) causes mostly asymptomatic, latent infections in the immunocompetent host. In immunosuppressed individuals or newborns infected in utero, an infection with this virus can lead to severe illness and permanent organ damage. The murine cytomegalovirus (MCMV) exhibits high structural and biological similarity to HCMV and is thus widely used as a model system for antiviral immune responses (128). MCMV induces a biphasic type I IFN response, with peak expressions occurring at 8 h and 36–72 h p.i. which are triggered by the initial virus contact and viral particles entering the system after completion of the first viral replication cycle, respectively (110). Early type I IFN expression is independent of TLR signaling and predominantly generated by stromal cells infected by the virus (110). Using IFNβmob/mob reporter mice, IFNβ production was detected in splenic pDCs as early as 6–12 h p.i. (59, 111). After in vivo depletion of pDCs by anti-CD317 or anti-Ly6G/C treatment IFNα serum levels were severely reduced 36 h after MCMV infection (18, 80, 81, 113). Under these conditions, however, other cell types secrete IL-12 and ensure sufficient IFNγ and NK cell responses leading to control of MCMV infection (18, 80). Of note, 44 h after MCMV infection IFNα serum levels in pDC depleted mice were no longer reduced as compared to untreated mice (113). Similar observations were made in IkL/L mice that lack pDCs (61) or CLEC4A-DTR-tg mice that have been transiently depleted of pDCs (65, 113). Thus, transient type I IFN production at the first day of MCMV infection was pDC-dependent, while cells other than pDCs are responsible for the type I IFN levels measured at later timepoints, at least when relatively high inocula of MCMV are used. In contrast, at lower doses of MCMV which are presumably closer to a natural infection setting, pDCs can limit viral burden in the spleen and liver. Here, pDCs have been shown to promote NK cell activation and cytotoxicity in the early phase of MCMV infection (65). While it is well-established that pDCs sense the MCMV via the TLR9 and TLR7 mediated pathways (18, 80, 113, 129, 130), also the TLR3 and TLR2 pathways which are functionally used by other cells than pDCs have been shown to be involved in the induction of type I IFN production (104, 131, 132). These findings are in accordance with multiple observations that defects in MyD88 signaling have a more severe impact on anti-MCMV immune responses than TLR9 deficiency or pDC depletion (113, 129). So far, the identity of the non-pDC cell types involved in anti-MCMV type I IFN response remain incompletely defined.
One report indicated that vaccinia virus (VV) and to a lesser extend MCMV induce type I IFN in CD11c− CD11b+ Ly6C+ inflammatory monocytes, but not macrophages or other types of DCs, in a TLR2 dependent way using IFNβmob/mob reporter mice. Further, CD11b-DTR-tg mice depleted of monocytes exhibited increased viral titers in the liver and decreased serum levels of type I IFN after VV infection (104). This is similar to other studies using footpad infection of modified vaccinia virus Ankara (MVA) and pDC depletion in the CLEC4A-DTR-tg mouse model, where type I IFN levels in the draining lymph nodes were comparable to control mice indicating that pDCs are not required for mounting an intact type I IFN response after local infection with this dsDNA virus (118).
The dsDNA adenovirus is used as a vector for the development of gene therapy applications but can also cause severe disease in immunocompromised individuals. By using CD11c-DTR-tg mice and anti-CD317 treatment to ablate cDCs vs. pDCs in vivo it has been shown that wildtype (WT) adenovirus as well as adenoviral vectors induce rapid IFNα/β production almost exclusively in splenic cDCs rather than in pDCs (105).
For Herpes Simplex Virus (HSV) local (subcutaneous or genital) as well as systemic (i.v.) infection models have been analyzed. After subcutaneous HSV-1 infection, pDCs were shown to provide type I IFN necessary for licensing of cDCs which in turn induce effective cytotoxic T cell responses. Here, mice depleted for pDCs by anti-Ly6G/C treatment displayed increased viral titers in the draining lymph nodes at day 7 p.i. as compared to controls (82). Similarly, in a genital HSV-2 herpes model, mice depleted for pDCs using anti-CD317 antibodies succumbed earlier to the infection and exhibited reduced local IFNα levels, while the Th1 response in draining lymph nodes developed normally (117). In contrast to findings from antibody-mediated depletion, in pDC depleted CLEC4A-DTR-tg mice neither differences in viral burden nor survival after vaginal HSV-2 infection was found nor were pDCs found to contribute significantly to antiviral CD8 T cell responses after subcutaneous HSV-1 inoculation (114). These contradicting findings have been explained by the antibody-mediated depletion of additional cell types other than pDCs in contrast to the more restricted depletion in the CLEC4A-DTR-tg genetic mouse model. On the other hand, it cannot be excluded that DTR mediated depletion is less effective and therefore a residual pDC activity retained after DT administration. Slight differences in the respective experimental settings might contribute as well as e.g. after antibody-mediated pDC depletion IFNα levels were measured in vaginal washes while in the genetic depletion model total protein amount was assessed in the vaginal and cervical tissue itself. As for MCMV, TLR3-expressing cells, such as CD8+ DCs or other hematopoietic and non-hematopoietic cells, are essential for type I IFN production in local HSV infection at later timepoints rather than pDCs (114).
After systemic challenge with UV-irradiated HSV in an early study immunohistological stainings for IFNα/β indicated that the majority of type I IFN producing cells in the spleen represent marginal metallophilic macrophages and to a lesser extend MZMs (133). However, IFNα levels were markedly reduced in pDC depleted Siglechdtr/dtr mice 6 h after i.v. infection with HSV-1 and viral titers were found increased in the spleen as compared to control animals pointing toward pDCs as the major type I IFN producers in this situation (66). Similar results were obtained in pDC depleted CLEC4A-DTR-tg mice, with the exception that no viral replication was detectable in the spleens of either DT-treated CLEC4A-DTR-tg or control mice (114). This discrepancy may reflect differences in the strains of HSV-1 used or differences in the promoters used to drive DTR expression (CLEC4A vs. SiglecH) with a slightly divergent expression pattern as discussed above. For systemic HSV-2 infections, results from antibody depletion and pDC ablation in CLEC4A-DTR-tg mice corresponded well since in both cases a reduction of IFNα serum levels were observed together with increased viral titers and reduced survival (114–116). Thus, similar to vaccinia virus the cell type responsible for the production of type I IFN in HSV infection may depend on the route of pathogen entry with pDCs controlling the infection once the virus has spread systemically.
Ectromelia virus (ECTV), a large DNA orthopoxvirus, is the causative agent of mousepox, the mouse homolog of human smallpox. ECTV causes systemic disease after s.c. infection of the footpad. In vivo it was shown by clodronate and anti-CD317 mediated depletion of monocytes vs. pDCs and ex vivo sorting and RT-PCR analyses that infected inflammatory monocytes are the major producers of type I IFN in the draining lymph nodes (119).
In summary, the cellular source for type I IFN production during DNA virus infection depends on the virus type itself, the dosage, timepoint as well as route of infection. Early after infection with MCMV pDCs are the primary source of type I IFN production capable of reducing virus titers at low concentrations of the virus. However, at later timepoints of infection CD8+ DCs rather than pDCs become the key source of type I IFN production. In addition to pDCs, other cell types such as cDCs in adenovirus infection, metallophilic macrophages and MZMs during HSV exposure, stromal cells in MCMV infection and inflammatory monocytes in response to ECTV are an essential source of type I IFN.
A recent meta-transcriptomics survey defined 196 vertebrate-specific RNA virus species the majority of which is able to infect humans and cause diseases of varying severity (134, 135). At the moment only few mouse models are available to elucidate the host immune response to these viruses. In this chapter we summarize the in vivo model studies aimed at visualizing type I IFN producing cell types and defining their contribution to the type I IFN production and RNA virus control.
For systemic infections with RNA viruses, such as after i.v. inoculation with the paramyxovirus Newcastle disease virus (NDV), it has been shown that pDCs and also cDCs, macrophages, and monocytes, produced IFNα (46). Here, pDCs mount an antiviral type I IFN response in a viral replication-independent manner through virus recognition by TLR7 and the activation of the type I IFN positive feedback loop. Only in the absence of this type I IFN positive feedback, the virus infects and also replicates in pDCs. In this case, type I IFN induction occurs in pDCs via cytoplasmic RLHs (32). However, other ssRNA viruses have been reported to induce type I IFN expression in pDCs in a replication dependent manner (136, 137). Especially for vesicular stomatitis virus (VSV), the capture of the replicating virus in the autophagosome is required for its transfer to the TLR7 containing endosomal compartment (137). After local infection with NDV, here after intranasal infection, the IFNα-producing cells shifted from pDCs to alveolar macrophages and cDCs that utilize the RLH system for type I IFN induction (46).
Upon s.c. VSV infection, draining LNs contained ~90% less IFNα when depleted of macrophages by clodronate liposomes. However, when pDCs were depleted by anti-CD317 treatment, IFNα levels induced by VSV were reduced only by half as compared to controls. It was concluded that infected CD169+ subcapsular sinus macrophages produce IFNα, yet half of the type I IFN is produced by pDCs stimulated directly or indirectly by the infected macrophages (108). Later it was shown that CD169+ macrophages in the spleen represent a compartment of enhanced viral replication (138). Thus, it is conceivable that CD169+ macrophages potentiate the type I IFN response indeed indirectly via activating pDCs. When VSV was inoculated i.v. in CLEC4A-DTR-tg mice transiently depleted for pDCs, IFNα was found reduced and viral titers increased only at very early timepoints, again pointing to a rather transient role of pDCs in anti-viral immunity (65).
For the distantly related arboviruses Dengue (DENV) and Chikungunya (CHIKV) virus it was recently shown that pDCs are sufficient to control these viruses via IRF7-regulated type I IFN responses in both systemic as well as local infection settings. In this report novel pDC:Irf7+ mice were introduced in which IRF7-driven type I IFN production is restricted to pDCs and were compared to IRF3/7 double deficient mice that are completely devoid of type I IFN expression (69). After i.v. infection with DENV pDC:Irf7+ mice exhibited a lower viral load than Irf3/7 double deficient mice. However, as compared to WT mice higher viral tiers were detected in pDC:Irf7+ mice (69). After s.c. infection with CHIKV Irf3/7 double deficient mice succumb to the virus while 100% of pDC:Irf7+ mice survive the infection exhibiting no overt clinical symptoms similar to WT mice. Early control of viremia in pDC:Irf7+ mice was reduced as compared to WT but still improved as compared to Irf3/7 double deficient mice. Thus, analogous to findings from other virus infection models also for these RNA viruses, antiviral response mounted by pDCs controls infection once the virus spreads systemically.
In infection models with RNA viruses exhibiting a specific tropism, pDCs play only a minor role. In the brain of mice infected with the ssRNA La Crosse virus, IFNβ production was assessed by the IFN-β+/Δβ−luc luciferase reporter mouse model (60) and detected in astrocytes, microglia, and to a lesser extend also in infected neurons (122). This confirmed earlier findings where IFNα/β expression in these cell types after La Crosse virus infection was visualized by immunostaining and RNA in situ hybridization (123). Utilizing the conditional reporter activity of the IFN-βfloxβ−luc/floxβ−luc mice it was shown for several neurotropic viruses such as rabies virus (RABV), Theiler's murine encephalomyelitis virus (TMEV), and VSV that astrocytes are the main producers of IFNβ after infection of the brain (139).
Another example for type I IFN expression by non-myeloid cells represent β-islet cells. The encephalomyocarditis virus (EMCV) strain D, an ssRNA picornavirus with tropism for the insulin-producing β cells of the pancreas, can induce diabetes and myocarditis in certain mouse strains. CD11c+ cells in this model have been shown to be protective as DT treated CD11c-DTR-tg mice developed diabetes and exhibited increased viral titers in the pancreas, spleen, and heart associated with reduced type I IFN levels as compared to non-depleted controls (106).
Pneumonia virus of mice (PVM) infection led to a marked infiltration of pDCs and increased expression of type I IFN in WT but not TLR7- or MyD88-deficient mice. Transfer of TLR7-competent, but not TLR7-deficient pDCs led to a significantly diminished virus recovery in TLR7−/− animals on day 7 after infection with PVM indicating that TLR7-mediated signaling by pDC is required for appropriate innate responses to acute PVM infection (140).
For intratracheal infection with respiratory syncytial virus (RSV) it has been shown that anti-CD317 mediated depletion of pDCs completely abolished IFNα expression and protein levels in the lungs. This correlated with increased viral titers and exacerbated immunopathology of the lungs of pDC depleted mice (126). Thus, pDCs fulfill a substantial protective role during local RSV infection.
Initial in vitro studies showed that spleen cells from mice that were depleted for pDCs by anti-Ly6G/C injection did not produce IFNα in response to stimulation with inactivated influenza virus in contrast to splenocytes from untreated animals (18). IFNα production in vitro could be attributed to the CD317+ CD11c+ pDC population of sorted mouse spleen cells (86). However, in vivo intranasal infection with sublethal doses of influenza virus in pDC-deficient IkarosL/L and WT mice revealed a similar course of disease, as determined e.g. by weight loss and viral titers (120). Thus, pDCs are able to produce type I IFN after stimulation by influenza but are dispensable for a successful antiviral immune response. Albeit, type I IFN levels in vivo were not assessed for this infection model.
For the influenza virus-like orthomyxovirus Thogoto virus (THOV) type I IFN production in the peritoneal cavity was mainly attributed to CD11b+ F4/80+ myeloid cells that was independent of the type I IFN receptor mediated feedback loop and coincided with the tropism of this virus (121).
After i.p. infection with Mouse hepatitis virus (MHV), pDC depletion by anti-CD317 was accompanied by severely diminished IFNα serum levels (124). The transient pDC depletion did not lead to lethality following the low-dose MHV infection used in this study. Nevertheless, initial viral titers in spleens were found increased more than 1,000-fold in pDC-depleted compared to control mice (124). Very similar observations were made in Itgax-Cre+ Tcf4flox/− mice lacking pDCs. These mice show reduced serum IFNα levels and elevated viral loads in the liver and spleen (100). Thus, pDCs appear to be essential for type IFN I mediated protection against systemic infection with the prototypical acute cytopathic coronavirus MHV.
Lymphocytic choriomeningitis virus (LCMV) infection is widely used to study acute as well as chronic infections. In an acute infection setting in Ifna6gfp/+ reporter mice pDCs were found to be the major type I IFN producers after infection with the WE strain of LCMV. Additionally, few cDCs and macrophages specifically in the spleen exhibited GFP-reporter activity (76). Also, in IFNβmob/mob reporter mice macrophages could be excluded as major type I IFN producers and depletion of phagocytic cells by clodronate liposomes did not affect type I IFN serum levels (94). In contrast, another study using the Armstrong strain of LCMV reported severely reduced IFNα/β serum levels after clodronate treatment (95). Specifically, a small population of CD169+ macrophages in the spleen and lymph nodes has recently been shown to release high amounts of type I IFN after LCMV infection. Selective depletion of these cells in CD169-DTR-tg mice resulted in reduced type I IFN levels from day 4 p.i. onward and persistent viral titers. As a consequence, CD169 depleted mice exhibited severe immunopathology and died quickly after infection (107). In line with this, production of serum type I IFN was not reduced in LCMV infected mice treated with the pDC depleting anti-Ly6G/C antibody as compared to those injected with control antibody (81). Also, in congenitally pDC-deficient Itgax-Cre+ Tcf4flox/− mice, virus titers early after infection were comparable to WT controls confirming that pDCs are dispensable for the control of acute LCMV infection (100). Still, pDCs have been shown to be a transient source of type I IFN as pDC depletion in CLEC4A-DTR-tg mice led to reduced serum IFNα levels at 16 h p.i. with LCMV Armstrong or clone 13, but not at later timepoints (125). Contrasting the observations in acute LCMV infection, in a chronic infection setup using LCMV Docile the virus persisted until day 53 in the blood of Itgax-Cre+ Tcf4flox/− mice while the virus was cleared between day 21 and 28 in WT mice. This was attributed to a failure of sufficient CD4+ and CD8+ T cell activation in the absence of pDCs and indicated that pDCs are essential for generating a functional adaptive immunity to chronic viral infections (100).
Taken together, pDCs are a major source of type I IFN and are required for type I IFN mediated protection against systemic infection in most of the RNA virus infections such as NDV, VSV, DENV, CHIKV, PVM, RSV, MHV, and LCMV. However, contribution of pDCs in type I IFN release and type I IFN mediated protection depends on the titer of the virus, time after infection, and the route of the infection. In addition to pDCs, other cell types such as cDCs, macrophages, and monocytes in NDV, macrophages in VSV, astrocytes, microglia, and neurons in La Crosse virus, astrocytes in RABV, TMEV, and VSV, β-islet cells and cDCs in EMCV, and cDCs and macrophages in LCMV infection significantly contribute to type I IFN production. Thus, similar to infection with DNA viruses, also after infection with RNA viruses pDCs are functionally involved in type I IFN production mostly early during infection but are dispensable for virus control during later stages of infection. In chronic infection, however, pDCs provide type I IFN to support and preserve T cell functions.
HIV activates pDCs to produce high levels of IFNα most likely via activation of TLR7 (141). Also, it is assumed that type I IFNs are produced during HIV infection predominantly by pDCs as decreased IFNα production in HIV-infected patients correlates with numerical and functional deficiencies in circulating pDCs (142). A direct assessment of the contribution of pDCs to type I IFN levels in HIV-infection, however, has not been performed. Although type I IFNs are known to mediate antiviral immunity, there has always been caution toward a detrimental role of type I IFNs during HIV/AIDS because of their proinflammatory nature (11, 143, 144). Thus, many studies have shown that pDCs are a source of type I IFN in retroviral and other virus infections in vivo. However, additional cellular sources of type I IFN are required to fully control viral infections. In summary, pDCs are a known source of type I IFN in retroviral infection. However, the relative contribution of pDCs vs. other type I IFN producers to the overall type I IFN response and immune control or pathology after retrovirus infection, is not fully understood.
While a considerable number of studies have been undertaken to define the cellular source of type I IFN and the functions of these cell types in viral infections, fewer data exist for non-viral infections. In bacterial infections, type I IFNs can act as activators of protective immune responses or mediate immunosuppressive functions leading to exacerbation of the infection. This ambivalent role of type I IFN has been reviewed recently (1, 11, 145). In this chapter we will focus on the efforts to clarify the identity and impact of type I IFN producing cells as knowledge on these has increased significantly paralleling the availability of newly developed mouse models.
It has been well established that CD4 T cells as well as secreted effector cytokines TNF, IL-12, and IFNγ exert protective functions in host resistance to the intracellular bacterial pathogen Mycobacterium tuberculosis (Mtb) (146). In contrast, the role of type I IFN during Mtb infection appears to promote infection instead of controlling infection. Type I IFNs downregulate IFNGR1 expression and thereby suppress IFNγ signaling (147, 148) and IFNAR-deficient mice displayed increased bacterial clearance to infection with Mtb, although bacterial growth in the lung was unaffected (149). In in vitro studies, BM-derived macrophages and DCs have been identified as a possible source of type I IFN in response to Mtb (149, 150). Also, human peripheral blood mononuclear cell (PBMC)-derived macrophages and especially DCs were shown to produce type I IFN after in vitro infection with Mtb (151, 152).
Similar to Mtb, IFNγ promotes antimicrobial activity against Mycobacterium leprae whereas type I IFNs contribute to pathogenesis (153). Here, PBMC-derived monocytes expressed IFNβ and IFN-stimulated genes including the immunosuppressive cytokine IL-10 during M. leprae infection in vitro (153). So far the cell type expressing type I IFN in the context of mycobacterial infections in vivo as well as definition of the functional impact of these cells await clarification.
Type I IFN not only inhibits antibacterial signaling pathways and promotes infection in the case of mycobacteria. Also, L. monocytogenes has evolved mechanisms to activate the type I IFN pathway for the benefit of this intracellular pathogen. Mice deficient in IFNAR signaling are more resistant to systemic L. monocytogenes infection as compared to WT controls. Mechanistically, type I IFNs enhance susceptibility to systemic Listeria infection by reducing responsiveness to IFNγ, decreasing the number of pro-inflammatory myeloid cells, promoting the expression of proapoptotic genes, and enhancing T cell sensitivity to apoptosis (148, 154–156). Of note, in intragastric or foodborne infection with L. monocytogenes type I IFN receptor mediated signaling contributed positively to survival of infected mice or did not have an impact at all, respectively (157, 158). This emphasizes again, that the route of infection contributes significantly to differences in the impact of type I IFN in infection.
Four distinct cell types have been reported as sources for type I IFN production during systemic L. monocytogenes infection in vivo (Table 3). For one, a FACS-purified splenic cell population from infected mice that displays surface antigens typical of macrophages and not pDCs was identified as the main producer of type I IFN (159). Also, the apathogenic Listeria mutant lacking listeriolysin O which is unable to escape from the phagolysosome into the cytoplasm of the infected cell, does not stimulate IFNβ synthesis (164, 165). Later, Tip-DCs, an effector subtype of Mac-3hi inflammatory monocytes, which produce TNF and iNOS were identified as the major IFNβ-producing cells in vivo in systemic L. monocytogenes infections using IFNβmob/mob and IFN-β+/Δβ−luc reporter mice (160, 161). IFNβ-producing TiP-DCs harbored high bacterial loads and were located within the foci of infection in the splenic white pulp ideally positioned to activate T cells as well as NK cells via type I IFN (160). Bacterial loads in the spleen were severely increased in mice deficient in CCR2 and thus lacking TiP-DCs (162). Thus, this subtype of inflammatory monocytes has been attributed an important role in early containment of L. monocytogenes infection (162, 166). The overall role of TiP-DCs in this infection may therefore be ambiguous, having a regulatory function in controlling the balance between containment of infection and at the same time mediating detrimental effects of type I IFN on the host. Interestingly, in the spleens of Listeria-infected CCR2−/− mice increased levels of type I IFN were observed indicating that alternative cell types produce type I IFN in the absence of TiP-DCs which are triggered additionally by increased bacterial load. Along this line, a detrimental role for pDCs in controlling L. monocytogenes infection was demonstrated in Siglechdtr/dtr mice where ablation of pDCs caused significantly increased survival and decreased bacterial burden at day 3 p.i., while type I IFN levels themselves were not analyzed under these conditions (66). At earlier timepoints, however, anti-PDCA-1 mediated depletion of pDCs did not lead to a difference in bacterial load or levels of type I IFN in the spleen as compared to control animals (161). In one report, CD317+ SiglecH− CD19+ B cells have been found to be able to induce IFNα after stimulation with heat-killed L. monocytogenes (163). Ex vivo isolated CD317+ SiglecH− CD19+ B cells activated cytotoxic function of NK cells in an IFNα-dependent manner. In vivo, this B cell subset contributed positively to resistance to L. monocytogenes infection as Btk−/− mice deficient for B-cells and unable to generate CD317+ CD19+ B cells displayed increased susceptibility to L. monocytogenes infection, while adoptive transfer of CD317+ CD19+ B cells to Btk−/− mice normalized their resistance to L. monocytogenes infection (163).
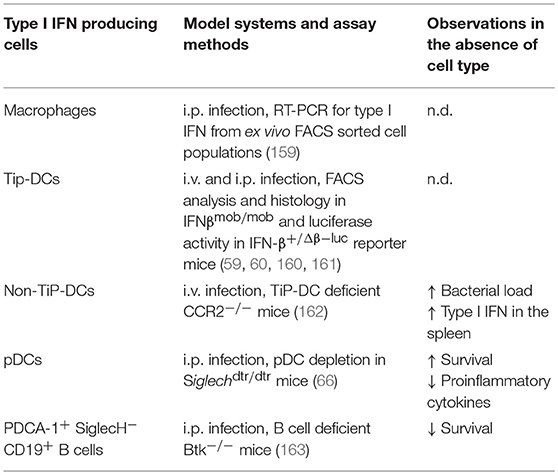
Table 3. Cellular sources of type I IFN production in Listeria monocytogenes infection in vivo.
As for intracellular bacteria also for extracellularly replicating bacterial pathogens type I IFN can either be detrimental or essential for host defense (145). Group B streptococci (GBS) are important neonatal pathogens and type I IFN receptor signaling is reported to contribute to host resistance against this pathogen (167). Mice i.p. infected with GBS express elevated levels of IFNβ and IFNα4 mRNA in the spleen. In vitro, GBS activated type I IFN expression in peritoneal macrophages, BM-derived cDCs and to a lesser extent also in macrophages, while pDCs were completely unable to produce type I IFN after GBS stimulation (167, 168).
In contrast to GBS, type I IFN induction in the mixed bacterial sepsis model of colon ascendens stent peritonitis (CASP) has been shown to have a detrimental effect on the host. Septic peritonitis induced in IFNAR−/− mice showed improved survival and bacterial clearance as compared to WT controls. Splenic CD11b+ CD11c− macrophage-like cells could be identified as major producers of IFNβ ex vivo by RT-PCR analyses from sorted cells, while no IFNα subtypes were detected (169).
In summary, type I IFN production has a detrimental effect for the host after infection with intracellular bacteria such as mycobacteria and L. monocytogenes. BMDCs and PBMC-derived DCs and macrophages are the responsible cell types for type I IFN production during mycobacteria infection. For L. monocytogenes, four cell types have been identified as type I IFN producers, namely macrophages, Tip-DCs, inflammatory monocytes, and B cells. In the case of extracellular bacteria, the cell types identified as type I IFN producers include macrophages and BMDCs. However, with the exception of the intracellular model organism L. monocytogenes, the knowledge on the cellular source of type I IFN in bacterial infection is rather scarce.
As for bacterial infections, the effect of type I IFN in mouse models for infections with pathogenic fungi has been reported as beneficial or detrimental for the host depending on the fungal species and the route of infection. Additionally, controversial results obtained from very similar infection settings have been explained by the possible impact of differences in the microbiome in the respective mouse colonies (1, 11). The cell type responsible for type I IFN production in fungal infections in vivo, however, awaits clarification. To our knowledge only for the important opportunistic fungal pathogen Aspergillus fumigatus in vivo studies in this direction have been undertaken. The type I IFN response triggered by A. fumigatus was analyzed initially in human pDCs isolated from PBMCs. When these cells were stimulated in vitro with A. fumigatus hyphae IFNα was detected in the supernatant (170). IFNAR−/− mice or mice depleted of pDCs by anti-CD317 treatment exhibited an increased susceptibility to pulmonary or i.v. infection with A. fumigatus conidia. A direct impact of pDC depletion on type I IFN levels in vivo after infection, however, has not been analyzed in this study (170). Therefore, the hypothesis that pDCs mediate their protective function in this fungal infection directly via type I IFN remains to be tested.
Infection with a wide variety of protozoan parasites can trigger type I IFN expression in mammalian hosts as reviewed recently (171, 172). For Plasmodium, Leishmania, and Trypanosoma in vivo infection models several studies have been carried out in the last few years which allowed the identification of cellular sources of type I IFN in response to intracellular parasite infections. This will be the focus of the following chapter and summarized in Table 4.
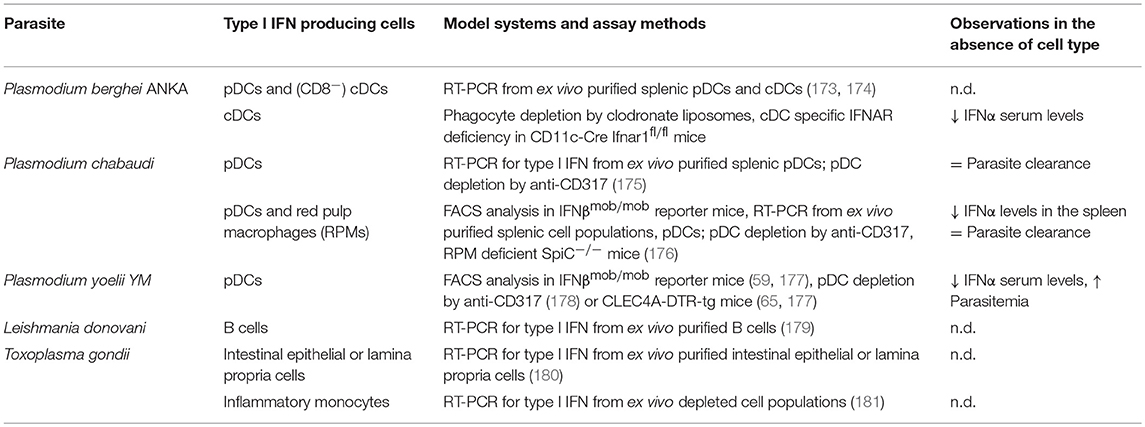
Table 4. Cellular sources of type I IFN production in intracellular parasite infections in vivo.
Malaria is an important parasitic disease predominantly in tropical and subtropical African regions. It is caused by the protozoan parasite Plasmodium with P. falciparum being responsible for its most severe forms. In humans, malarial parasites are transmitted at sporozoite-stage by infected mosquitoes (182). The transmitted sporozoites rapidly travel to the liver, where they infect hepatocytes and initiate clinically silent but immunologically active liver-stage infection (171). Well-established in vivo mouse models include the lethal Plasmodium yoelii YM and P. berghei ANKA leading to high parasitemia and cerebral malaria (CM), respectively, after inoculation with Plasmodium-infected erythrocytes. Further, P. chabaudi is used as a chronic infection model. Various cellular sources for type I IFNs have been proposed after Plasmodium infection in vitro (182–184).
After inoculation with P. berghei ANKA infected erythrocytes in vivo, isolated splenic pDCs as well as CD8− cDCs expressed type I IFN (173, 174). Using anti-CD317 mediated pDC depletion and cDC depletion in CD11c-DTR-tg mice it was shown that cDCs but not pDCs are required for the induction of CM (173). Additionally, cDCs require IFNAR dependent signaling for systemic IFNα production in this model as indicated by substantially lower levels of serum IFNα in CD11c-Cre Ifnar1fl/fl mice, compared to those in infected Ifnar1fl/fl littermate controls (174).
In contrast to the P. berghei ANKA model, P. chabaudi infection did not induce IFNα in splenic cDCs but rather in pDCs via the TLR9 sensing pathway (175). However, pDCs were not essential for parasite clearance in P. chabaudi infection (175). Direct in vivo analysis performed in IFNβmob/mob reporter mice (59) revealed that in P. chabaudi infection about 75% of IFN producing cells are pDCs (176). In addition to pDCs, splenic red pulp macrophages (RPMs) can generate significant quantities of IFNβ in response to P. chabaudi infection. Contribution of both cell types to the type I IFN response in this system was defined by pDC depletion via anti-CD317 treatment and in RPM deficient SpiC−/− mice (176).
In the lethal malaria mouse model of P. yoelii YM infection, type I IFN enhances inflammatory blood leukocyte activation and lethal outcome (177). IFNβmob/mob reporter mice indicated here that type I IFN is produced in high amounts by BM and blood pDCs and to lesser extent by tissue resident pDCs (177). Depletion of pDCs by anti-CD317 or using pDC specific CLEC4A-DTR-tg mice confirmed pDCs as the major cellular source of type I IFN in this severe malaria model (177, 178). However, depletion of pDCs also resulted in a slight but significant increase of parasitemia (178). Further, priming of pDCs by plasmodium activated CD169+ macrophages was essential (177). It was proposed that in in vivo settings the low levels of secreted type I IFN produced by monocytes and macrophages prime pDCs for systemic production of type I IFN in malaria.
From data available so far, pDCs as well as cDCs and macrophage subtypes are the cell types responsible for the generation of the type I IFN response, depending on the Plasmodium species. Similar to LCMV, Mycobacteria, or Listeria infections (153, 156, 185), it is thought that early robust production of type I IFN in the first 24 h is essential to induce protective innate and adaptive immunity against Plasmodium, while late production of type I IFN impairs host anti-malaria immune responses by induction of negative immune regulators such as PD-L1 and IL-10 (178).
Leishmania spp. are transmitted to mammalian organisms by the bite of infected sand flies (186). The parasites preferentially infect macrophages, but can also be found in other cell types, such as fibroblasts, neutrophils, and DCs (172). Depending on the parasite species and strain Leishmania causes a mild to severe cutaneous, mucocutaneous or visceral leishmaniasis (171, 187). Increased production of type I IFN has been observed in local tissues and in the draining lymph nodes of L. major infected mice (187, 188). There are diverging reports about the role of type I IFN production in the control of parasite burden and development of disease pathology. Depending on the time course of infection and type I IFN induction it can exert detrimental or protective effects for the host in Leishmaniasis (189). Most of the studies addressing the cellular source of type I IFN in Leishmania infection were performed in vitro. For example, infection of murine macrophages with L. major or L. amazonensis lead to type I IFN production (188, 190). In vitro exposure of BM-derived as well as splenic pDCs to L. major, L. infantum, or L. braziliensis promastigotes induces release of IFNα and IFNβ in a TLR9-dependent manner (191). Intriguingly, the amounts of type I IFN produced in response to Leishmania spp. are comparable to the type I IFN levels produced in response to stimulation with CpG ODNs in these experiments (191). Recently in vitro exposure to the parasite L. donovani was reported to trigger IFNβ production in splenic B cells. Here, also high levels of type I IFN mRNA were detected in splenic B cells purified from in vivo L. donovani infected mice (179). Taken together, depending on the Leishmania subtypes, pDCs and B cells are the source of type I IFN when the cells are directly exposed to the pathogen in vitro. Information on the type I IFN producers in vivo remain scarce so far for this important protozoan parasite model but could be increased significantly making use of the now available mouse models.
Toxoplasma gondii is an intracellular protozoan parasite that has infected at least 50% of the human population. It causes severe toxoplasmosis in immune-suppressed patients. T. gondii can infect a wide range of warm-blooded animals, is able to invade any nucleated cell but survives outside of the mammalian host as well (171, 172). The gut epithelium is a strategic barrier to prevent or limit parasite dissemination upon oral infection with T. gondii. In the initial phase of oral T. gondii infection elevated IFNβ mRNA levels were observed in the small intestine. Intestinal epithelial cells (IECs) and cells from the lamina propria are the source of local IFNβ production in early infection as assessed by real-time PCR performed on cells isolated from infected mice (180). In in vitro infection, T. gondii has been reported to induce or suppress type I IFN induction depending on the host species, the cell type, and the parasite strain analyzed. One publication showed that BM-derived murine pDCs produce IFNα after infection with T. gondii (192). Murine pDCs recognized T. gondii profilin via TLR11 and TLR12 and produce type I IFN in a MyD88 dependent fashion (192, 193). In contrast to murine pDCs, human pDCs lack TLR11 and TLR12 and are unable to produce type I IFN despite of direct infection with T. gondii. Active infection with T. gondii in vitro rather functionally inactivates human pDCs (194). In particular macrophages and DCs serve as reservoirs of T. gondii infection and facilitate early dissemination (195). Most of the Toxoplasma strains tested are unable to induce type I IFN production in murine BM-derived macrophages after in vitro infection (196, 197). T. gondii mediated suppression of type I IFN expression has been reported also for monocytes, macrophages, and several DC subsets in vitro (181, 195, 196). On the other hand, few atypical Toxoplasma strains such as COUGAR and RUB can induce IFNβ production in murine BM-derived macrophages as well as in human skin fibroblasts in in vitro infection systems (196). In a physiological oral infection mouse model ex vivo isolated inflammatory monocytes in the gut-draining mesenteric lymph nodes were identified the major producers of IFNβ. The expression of IFNβ by inflammatory monocytes required phagocytic uptake of T. gondii, while active invasion did not trigger IFNβ induction (181). Thus, depending on the host species, the cell type, and the parasite strain, T. gondii may induce or suppress type I IFN production. Epithelial, skin fibroblasts, pDCs, macrophages and inflammatory monocytes here are known cellular sources of type I IFN. In T. gondii infection most of the knowledge about the cellular sources of type I IFN is deduced from in vitro experiments. Analysis of type I IFN reporter mouse models with and without ablation of different cell types is missing, so far.
Taken together, the major type I IFN producing cell types and their contribution to immunity against many protozoan parasites remain to be defined. To our knowledge, no direct study to elucidate the cellular sources of type I IFN in multicellular parasite such as helminth infections in vivo has been published. In order to understand the cellular sources of type I IFN and their relevance with regard to disease elimination in multicellular parasites such as helminths, type I IFN reporter mouse models and cell specific depletion models remain to be analyzed. As for the other pathogen types reviewed above, parasite numbers and the site of infection might influence the sensing pathway and cell type activated to produce type I IFN.
In recent years the generation of novel animal models has remarkably advanced our understanding of the mode of action of IFNs and the cell type responsible for its production in the context of an infection. The existing knowledge does not allow to depict any cell type as a single cell type responsible for the entire type I IFN production in the course of any infection. Rather, depending on the type of infection a wide variety of cells have exhibited the capacity to produce type I IFN. Decisive factors for the type of cell initiating type I IFN production are the type and amount of pathogen and the site and stage of the infection. Additionally, the genetic background of the mouse model and its microbiome status contribute as well and need to be further analyzed.
Even though pDCs are more specialized than other cell types in type I IFN production, it is getting increasingly clear that in vivo their contribution to antiviral immunity and also to immune responses to bacterial, fungal, and parasitic infection exhibits restricted patterns in time of induction and duration. The importance of pDCs as the source of type I IFN early in virus infections does not hold true at later timepoints when other host cells take over as dominant producers of type I IFNs. The impact of pDCs also depends on the route of infection. While pDCs provide an important source of type I IFN in systemic infections, their requirement for I IFN-mediated antiviral immune responses in local tissues seems to be necessary only if other lines of defense are broken. However, there are exceptions to the rule as shown for local infections with MHV and HSV-2 where pDC-derived type I IFN in mice is critical for viral control and survival. Indeed, the limitation of pDC responses is caused by an upregulation of pro-apoptotic molecules and apoptosis induction in pDCs in a type I IFN-dependent manner during systemic viral infections (198). This has been suggested as a mechanism to prevent immunopathology due to sustained pDC-mediated type I IFN production. Besides pDCs, mainly macrophages, inflammatory monocytes and cDCs are able to mount significant anti-infectious type I IFN responses in vivo. Instead of a single specialized cell type, it is rather the orchestrated type I IFN expression by multiple cellular sources that ensures protective anti-infectious immune responses mediated by type I IFN. To elucidate synergisms and redundancies between the different type I IFN producing cells will be a topic of future studies.
Advanced single cell functional profiling and systems biology approaches will contribute significantly in the near future to identify the exact functions of specific cell types, even cell subtypes, in the different stages of an infection. The spatio-temporal interaction of the type I IFN producing cell with the pathogen and the immune cells that are activated by type I IFN could help to better dissect the diverse functions of type I IFN in the immune response at different stages of infection. Importantly, due to the severe side effects of type I IFN treatment, there is a dire need to better control its activity and thereby increase its beneficial net effect. Strategies such as modifying the affinity of type I IFNs or modulating its time of availability have been reviewed recently (199). These new approaches to develop and improve vaccination strategies and to define novel therapeutic leads for infectious diseases are urgently called for in a time where antibiotic resistances are projected to increase rapidly.
SA, RM-N, AS, LR, JA, and SS wrote the manuscript. SA, RM-N, and SS designed and generated tables. RM-N designed and generated the figure. All authors read and approved the final manuscript.
This work was supported by the Deutsche Forschungsgemeinschaft (RTG 2158 Natural products and natural product analogs against therapy-resistant tumors and microorganisms: new lead structures and modes of action), the Manchot Graduate School Molecules of Infection II and III, the Research Commission of the Medical Faculty of the University of Düsseldorf, Germany (30/2016) (to SS) and by the Deutsche Forschungsgemeinschaft (FOR 2107, AL 1145/5–2), the IZKF (Alf3/018/16), the DFG EXC 1003, Grant FF-2014-01 Cells in Motion–Cluster of Excellence, Münster, Germany, and the Alzheimer Forschung Initiative e.V. (14835) (to JA).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. Type I interferons in infectious disease. Nat Rev Immunol. (2015) 15:87–103. doi: 10.1038/nri3787
2. Kretschmer S, Lee-Kirsch MA. Type I interferon-mediated autoinflammation and autoimmunity. Curr Opin Immunol. (2017) 49:96–102. doi: 10.1016/j.coi.2017.09.003
3. Decker T, Muller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. (2005) 5:675–87. doi: 10.1038/nri1684
4. Hardy MP, Owczarek CM, Jermiin LS, Ejdeback M, Hertzog PJ. Characterization of the type I interferon locus and identification of novel genes. Genomics. (2004) 84:331–45. doi: 10.1016/j.ygeno.2004.03.003
5. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. (2005) 5:375–86. doi: 10.1038/nri1604
6. van Pesch V, Lanaya H, Renauld JC, Michiels T. Characterization of the murine alpha interferon gene family. J Virol. (2004) 78:8219–28. doi: 10.1128/JVI.78.15.8219-8228.2004
7. Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. (2006) 25:349–60. doi: 10.1016/j.immuni.2006.08.009
8. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. (2014) 14:36–49. doi: 10.1038/nri3581
9. Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. (2014) 32:513–45. doi: 10.1146/annurev-immunol-032713-120231
10. Trinchieri G. Type I interferon: friend or foe? J Exp Med. (2010) 207:2053–63. doi: 10.1084/jem.20101664
11. Stifter SA, Feng CG. Interfering with immunity: detrimental role of type I IFNs during infection. J Immunol. (2015) 194:2455–65. doi: 10.4049/jimmunol.1402794
12. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B Biol Sci. (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
13. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. (2005) 23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843
14. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. (1999) 5:919–23. doi: 10.1038/11360
15. Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. (2006) 107:2423–31. doi: 10.1182/blood-2005-07-2709
16. Trinchieri G, Santoli D. Anti-viral activity induced by culturing lymphocytes with tumor-derived or virus-transformed cells. Enhancement of human natural killer cell activity by interferon and antagonistic inhibition of susceptibility of target cells to lysis. J Exp Med. (1978) 147:1314–33. doi: 10.1084/jem.147.5.1314
17. Trinchieri G, Santoli D, Dee RR, Knowles BB. Anti-viral activity induced by culturing lymphocytes with tumor-derived or virus-transformed cells. Identification of the anti-viral activity as interferon and characterization of the human effector lymphocyte subpopulation. J Exp Med. (1978) 147:1299–313. doi: 10.1084/jem.147.5.1299
18. Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. (2001) 2:1144–50. doi: 10.1038/ni736
19. Bjorck P. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. (2001) 98:3520–6. doi: 10.1182/blood.V98.13.3520
20. Nakano H, Yanagita M, Gunn MD. CD11c(+)B220(+)Gr-1(+) cells in mouse lymph nodes and spleen display characteristics of plasmacytoid dendritic cells. J Exp Med. (2001) 194:1171–8. doi: 10.1084/jem.194.8.1171
21. Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. (2010) 32:305–15. doi: 10.1016/j.immuni.2010.03.012
22. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. (2010) 11:373–84. doi: 10.1038/ni.1863
23. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. (2011) 29:163–83. doi: 10.1146/annurev-immunol-031210-101345
24. Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, et al. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. (2004) 5:1061–8. doi: 10.1038/ni1118
25. Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. (2006) 203:1999–2008. doi: 10.1084/jem.20060401
26. Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. (2005) 434:1035–40. doi: 10.1038/nature03547
27. Gururajan M, Jacob J, Pulendran B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS ONE. (2007) 2:e863. doi: 10.1371/journal.pone.0000863
28. Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. (2004) 5:987–95. doi: 10.1038/ni1112
29. Akkaya M, Akkaya B, Miozzo P, Rawat M, Pena M, Sheehan PW, et al. B cells produce Type 1 IFNs in response to the TLR9 agonist CpG-A conjugated to cationic lipids. J Immunol. (2017) 199:931–40. doi: 10.4049/jimmunol.1700348
30. Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med. (2002) 195:507–16. doi: 10.1084/jem.20011666
31. Asselin-Paturel C, Brizard G, Chemin K, Boonstra A, O'Garra A, Vicari A, et al. Type I interferon dependence of plasmacytoid dendritic cell activation and migration. J Exp Med. (2005) 201:1157–67. doi: 10.1084/jem.20041930
32. Kumagai Y, Kumar H, Koyama S, Kawai T, Takeuchi O, Akira S. Cutting Edge: TLR-dependent viral recognition along with type I IFN positive feedback signaling masks the requirement of viral replication for IFN-{alpha} production in plasmacytoid dendritic cells. J Immunol. (2009) 182:3960–4. doi: 10.4049/jimmunol.0804315
33. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. (2004) 303:1526–9. doi: 10.1126/science.1093620
34. Jurk M, Heil F, Vollmer J, Schetter C, Krieg AM, Wagner H, et al. Human TLR7 or TLR8 independently confer responsiveness to the antiviral compound R-848. Nat Immunol. (2002) 3:499. doi: 10.1038/ni0602-499
35. Gorden KK, Qiu XX, Binsfeld CC, Vasilakos JP, Alkan SS. Cutting edge: activation of murine TLR8 by a combination of imidazoquinoline immune response modifiers and polyT oligodeoxynucleotides. J Immunol. (2006) 177:6584–7. doi: 10.4049/jimmunol.177.10.6584
36. Martinez J, Huang X, Yang Y. Toll-like receptor 8-mediated activation of murine plasmacytoid dendritic cells by vaccinia viral DNA. Proc Natl Acad Sci USA. (2010) 107:6442–7. doi: 10.1073/pnas.0913291107
37. Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol. (2003) 4:161–7. doi: 10.1038/ni886
38. Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. (2003) 301:640–3. doi: 10.1126/science.1087262
39. Jelinek I, Leonard JN, Price GE, Brown KN, Meyer-Manlapat A, Goldsmith PK, et al. TLR3-specific double-stranded RNA oligonucleotide adjuvants induce dendritic cell cross-presentation, CTL responses, and antiviral protection. J Immunol. (2011) 186:2422–9. doi: 10.4049/jimmunol.1002845
40. Azuma M, Ebihara T, Oshiumi H, Matsumoto M, Seya T. Cross-priming for antitumor CTL induced by soluble Ag + polyI:C depends on the TICAM-1 pathway in mouse CD11c(+)/CD8alpha(+) dendritic cells. Oncoimmunology. (2012) 1:581–92. doi: 10.4161/onci.19893
41. Tatematsu M, Nishikawa F, Seya T, Matsumoto M. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat Commun. (2013) 4:1833. doi: 10.1038/ncomms2857
42. Matsumoto M, Funami K, Tanabe M, Oshiumi H, Shingai M, Seto Y, et al. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J Immunol. (2003) 171:3154–62. doi: 10.4049/jimmunol.171.6.3154
43. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
44. Vaure C, Liu Y. A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol. (2014) 5:316. doi: 10.3389/fimmu.2014.00316
45. Barchet W, Krug A, Cella M, Newby C, Fischer JA, Dzionek A, et al. Dendritic cells respond to influenza virus through TLR7- and PKR-independent pathways. Eur J Immunol. (2005) 35:236–42. doi: 10.1002/eji.200425583
46. Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, et al. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. (2007) 27:240–52. doi: 10.1016/j.immuni.2007.07.013
47. Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol. (2014) 32:461–88. doi: 10.1146/annurev-immunol-032713-120156
48. Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. (2006) 203:1795–803. doi: 10.1084/jem.20060792
49. Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. (2008) 20:17–22. doi: 10.1016/j.coi.2008.01.002
50. Reikine S, Nguyen JB, Modis Y. Pattern recognition and signaling mechanisms of RIG-I and MDA5. Front Immunol. (2014) 5:342. doi: 10.3389/fimmu.2014.00342
51. Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. (2006) 24:93–103. doi: 10.1016/j.immuni.2005.12.003
52. Holm CK, Paludan SR, Fitzgerald KA. DNA recognition in immunity and disease. Curr Opin Immunol. (2013) 25:13–8. doi: 10.1016/j.coi.2012.12.006
53. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
54. Paludan SR, Bowie AG. Immune sensing of DNA. Immunity. (2013) 38:870–80. doi: 10.1016/j.immuni.2013.05.004
55. Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. (2007) 448:501–5. doi: 10.1038/nature06013
56. Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, et al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. (2010) 11:997–1004. doi: 10.1038/ni.1932
57. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, et al. cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–4. doi: 10.1038/nature12306
58. Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular nucleic acid detection in autoimmunity. Annu Rev Immunol. (2017) 35:313–36. doi: 10.1146/annurev-immunol-051116-052331
59. Scheu S, Dresing P, Locksley RM. Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo. Proc Natl Acad Sci USA. (2008) 105:20416–21. doi: 10.1073/pnas.0808537105
60. Lienenklaus S, Cornitescu M, Zietara N, Lyszkiewicz M, Gekara N, Jablonska J, et al. Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol. (2009) 183:3229–36. doi: 10.4049/jimmunol.0804277
61. Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, et al. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood. (2006) 108:4025–34. doi: 10.1182/blood-2006-03-007757
62. Kirstetter P, Thomas M, Dierich A, Kastner P, Chan S. Ikaros is critical for B cell differentiation and function. Eur J Immunol. (2002) 32:720–30. doi: 10.1002/1521-4141(200203)32:3<720::AID-IMMU720>3.0.CO;2-P
63. Caton ML, Smith-Raska MR, Reizis B. Notch-RBP-J signaling controls the homeostasis of CD8- dendritic cells in the spleen. J Exp Med. (2007) 204:1653–64. doi: 10.1084/jem.20062648
64. Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. (2008) 135:37–48. doi: 10.1016/j.cell.2008.09.016
65. Swiecki M, Gilfillan S, Vermi W, Wang Y, Colonna M. Plasmacytoid dendritic cell ablation impacts early interferon responses and antiviral NK and CD8(+) T cell accrual. Immunity. (2010) 33:955–66. doi: 10.1016/j.immuni.2010.11.020
66. Takagi H, Fukaya T, Eizumi K, Sato Y, Sato K, Shibazaki A, et al. Plasmacytoid dendritic cells are crucial for the initiation of inflammation and T cell immunity in vivo. Immunity. (2011) 35:958–71. doi: 10.1016/j.immuni.2011.10.014
67. Swiecki M, Wang Y, Riboldi E, Kim AH, Dzutsev A, Gilfillan S, et al. Cell depletion in mice that express diphtheria toxin receptor under the control of SiglecH encompasses more than plasmacytoid dendritic cells. J Immunol. (2014) 192:4409–16. doi: 10.4049/jimmunol.1303135
68. Puttur F, Arnold-Schrauf C, Lahl K, Solmaz G, Lindenberg M, Mayer CT, et al. Absence of Siglec-H in MCMV infection elevates interferon alpha production but does not enhance viral clearance. PLoS Pathog. (2013) 9:e1003648. doi: 10.1371/journal.ppat.1003648
69. Webster B, Werneke SW, Zafirova B, This S, Coleon S, Decembre E, et al. Plasmacytoid dendritic cells control dengue and Chikungunya virus infections via IRF7-regulated interferon responses. Elife. (2018) 7:e34273. doi: 10.7554/eLife.34273
70. Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. (2002) 17:211–20. doi: 10.1016/S1074-7613(02)00365-5
71. Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S, et al. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J Immunol. (2005) 174:2336–42. doi: 10.4049/jimmunol.174.4.2336
72. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest. (2007) 117:2268–78. doi: 10.1172/JCI31990
73. Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, et al. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr Biol. (1998) 8:223–6. doi: 10.1016/S0960-9822(98)70086-7
74. Deonarain R, Alcami A, Alexiou M, Dallman MJ, Gewert DR, Porter AC. Impaired antiviral response and alpha/beta interferon induction in mice lacking beta interferon. J Virol. (2000) 74:3404–9. doi: 10.1128/JVI.74.7.3404-3409.2000
75. Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. (2000) 288:2357–60. doi: 10.1126/science.288.5475.2357
76. Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, et al. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J Virol. (2008) 82:196–206. doi: 10.1128/JVI.01640-07
77. Frenz T, Graalmann L, Detje CN, Doring M, Grabski E, Scheu S, et al. Independent of plasmacytoid dendritic cell (pDC) infection, pDC triggered by virus-infected cells mount enhanced type I IFN responses of different composition as opposed to pDC stimulated with free virus. J Immunol. (2014) 193:2496–503. doi: 10.4049/jimmunol.1400215
78. Reizis B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity. (2019) 50:37–50. doi: 10.1016/j.immuni.2018.12.027
79. Swiecki M, Colonna M. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev. (2010) 234:142–62. doi: 10.1111/j.0105-2896.2009.00881.x
80. Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, et al. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. (2004) 21:107–19. doi: 10.1016/j.immuni.2004.06.007
81. Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, et al. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. (2002) 195:517–28. doi: 10.1084/jem.20011672
82. Yoneyama H, Matsuno K, Toda E, Nishiwaki T, Matsuo N, Nakano A, et al. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med. (2005) 202:425–35. doi: 10.1084/jem.20041961
83. Kuwajima S, Sato T, Ishida K, Tada H, Tezuka H, Ohteki T. Interleukin 15-dependent crosstalk between conventional and plasmacytoid dendritic cells is essential for CpG-induced immune activation. Nat Immunol. (2006) 7:740–6. doi: 10.1038/ni1348
84. Goubier A, Dubois B, Gheit H, Joubert G, Villard-Truc F, Asselin-Paturel C, et al. Plasmacytoid dendritic cells mediate oral tolerance. Immunity. (2008) 29:464–75. doi: 10.1016/j.immuni.2008.06.017
85. Shen H, Iwasaki A. A crucial role for plasmacytoid dendritic cells in antiviral protection by CpG ODN-based vaginal microbicide. J Clin Invest. (2006) 116:2237–43. doi: 10.1172/JCI28681
86. Asselin-Paturel C, Brizard G, Pin JJ, Briere F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. (2003) 171:6466–77. doi: 10.4049/jimmunol.171.12.6466
87. Loschko J, Schlitzer A, Dudziak D, Drexler I, Sandholzer N, Bourquin C, et al. Antigen delivery to plasmacytoid dendritic cells via BST2 induces protective T cell-mediated immunity. J Immunol. (2011) 186:6718–25. doi: 10.4049/jimmunol.1004029
88. Fleming TJ, Fleming ML, Malek TR. Selective expression of Ly-6G on myeloid lineage cells in mouse bone marrow. RB6-8C5 mAb to granulocyte-differentiation antigen (Gr-1) detects members of the Ly-6 family. J Immunol. (1993) 151:2399–408.
89. Jutila MA, Kroese FG, Jutila KL, Stall AM, Fiering S, Herzenberg LA, et al. Ly-6C is a monocyte/macrophage and endothelial cell differentiation antigen regulated by interferon-gamma. Eur J Immunol. (1988) 18:1819–26. doi: 10.1002/eji.1830181125
90. Sato N, Yahata T, Santa K, Ohta A, Ohmi Y, Habu S, et al. Functional characterization of NK1.1 + Ly-6C+ cells. Immunol Lett. (1996) 54:5–9. doi: 10.1016/S0165-2478(96)02632-6
91. Takahama Y, Sharrow SO, Singer A. Expression of an unusual T cell receptor (TCR)-V beta repertoire by Ly-6C+ subpopulations of CD4+ and/or CD8+ thymocytes. Evidence for a developmental relationship between Ly-6C+ thymocytes and CD4-CD8-TCR-alpha beta+ thymocytes. J Immunol. (1991) 147:2883–91.
92. Wrammert J, Kallberg E, Agace WW, Leanderson T. Ly6C expression differentiates plasma cells from other B cell subsets in mice. Eur J Immunol. (2002) 32:97–103. doi: 10.1002/1521-4141(200201)32:1<97::AID-IMMU97>3.0.CO;2-Y
93. Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. (2006) 177:3260–5. doi: 10.4049/jimmunol.177.5.3260
94. Lang PA, Recher M, Honke N, Scheu S, Borkens S, Gailus N, et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology. (2010) 52:25–32. doi: 10.1002/hep.23640
95. Louten J, van Rooijen N, Biron CA. Type 1 IFN deficiency in the absence of normal splenic architecture during lymphocytic choriomeningitis virus infection. J Immunol. (2006) 177:3266–72. doi: 10.4049/jimmunol.177.5.3266
96. Dumortier A, Jeannet R, Kirstetter P, Kleinmann E, Sellars M, dos Santos NR, et al. Notch activation is an early and critical event during T-Cell leukemogenesis in Ikaros-deficient mice. Mol Cell Biol. (2006) 26:209–20. doi: 10.1128/MCB.26.1.209-220.2006
97. Dumortier A, Kirstetter P, Kastner P, Chan S. Ikaros regulates neutrophil differentiation. Blood. (2003) 101:2219–26. doi: 10.1182/blood-2002-05-1336
98. Zhuang Y, Cheng P, Weintraub H. B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2-2, and HEB. Mol Cell Biol. (1996) 16:2898–905. doi: 10.1128/MCB.16.6.2898
99. Bergqvist I, Eriksson M, Saarikettu J, Eriksson B, Corneliussen B, Grundstrom T, et al. The basic helix-loop-helix transcription factor E2-2 is involved in T lymphocyte development. Eur J Immunol. (2000) 30:2857–63. doi: 10.1002/1521-4141(200010)30:10<2857::AID-IMMU2857>3.0.CO;2-G
100. Cervantes-Barragan L, Lewis KL, Firner S, Thiel V, Hugues S, Reith W, et al. Plasmacytoid dendritic cells control T-cell response to chronic viral infection. Proc Natl Acad Sci USA. (2012) 109:3012–7. doi: 10.1073/pnas.1117359109
101. Schlitzer A, Heiseke AF, Einwachter H, Reindl W, Schiemann M, Manta CP, et al. Tissue-specific differentiation of a circulating CCR9- pDC-like common dendritic cell precursor. Blood. (2012) 119:6063–71. doi: 10.1182/blood-2012-03-418400
102. Loschko J, Heink S, Hackl D, Dudziak D, Reindl W, Korn T, et al. Antigen targeting to plasmacytoid dendritic cells via Siglec-H inhibits Th cell-dependent autoimmunity. J Immunol. (2011) 187:6346–56. doi: 10.4049/jimmunol.1102307
103. Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. (1997) 237:752–7. doi: 10.1006/bbrc.1997.7124
104. Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. (2009) 10:1200–7. doi: 10.1038/ni.1792
105. Fejer G, Drechsel L, Liese J, Schleicher U, Ruzsics Z, Imelli N, et al. Key role of splenic myeloid DCs in the IFN-alphabeta response to adenoviruses in vivo. PLoS Pathog. (2008) 4:e1000208. doi: 10.1371/journal.ppat.1000208
106. McCartney SA, Vermi W, Lonardi S, Rossini C, Otero K, Calderon B, et al. RNA sensor-induced type I IFN prevents diabetes caused by a beta cell-tropic virus in mice. J Clin Invest. (2011) 121:1497–507. doi: 10.1172/JCI44005
107. Shaabani N, Duhan V, Khairnar V, Gassa A, Ferrer-Tur R, Haussinger D, et al. CD169(+) macrophages regulate PD-L1 expression via type I interferon and thereby prevent severe immunopathology after LCMV infection. Cell Death Dis. (2016) 7:e2446. doi: 10.1038/cddis.2016.350
108. Iannacone M, Moseman EA, Tonti E, Bosurgi L, Junt T, Henrickson SE, et al. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. (2010) 465:1079–83. doi: 10.1038/nature09118
109. Sapoznikov A, Fischer JA, Zaft T, Krauthgamer R, Dzionek A, Jung S. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J Exp Med. (2007) 204:1923–33. doi: 10.1084/jem.20062373
110. Schneider K, Loewendorf A, De Trez C, Fulton J, Rhode A, Shumway H, et al. Lymphotoxin-mediated crosstalk between B cells and splenic stroma promotes the initial type I interferon response to cytomegalovirus. Cell Host Microbe. (2008) 3:67–76. doi: 10.1016/j.chom.2007.12.008
111. Bauer J, Dress RJ, Schulze A, Dresing P, Ali S, Deenen R, et al. Cutting edge: IFN-beta expression in the spleen is restricted to a subpopulation of plasmacytoid dendritic cells exhibiting a specific immune modulatory transcriptome signature. J Immunol. (2016) 196:4447–51. doi: 10.4049/jimmunol.1500383
112. Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, et al. Dendritic cell responses to early murine cytomegalovirus infection: subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. (2003) 197:885–98. doi: 10.1084/jem.20021522
113. Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. (2005) 175:6723–32. doi: 10.4049/jimmunol.175.10.6723
114. Swiecki M, Wang Y, Gilfillan S, Colonna M. Plasmacytoid dendritic cells contribute to systemic but not local antiviral responses to HSV infections. PLoS Pathog. (2013) 9:e1003728. doi: 10.1371/journal.ppat.1003728
115. Stout-Delgado HW, Du W, Shirali AC, Booth CJ, Goldstein DR. Aging promotes neutrophil-induced mortality by augmenting IL-17 production during viral infection. Cell Host Microbe. (2009) 6:446–56. doi: 10.1016/j.chom.2009.09.011
116. Stout-Delgado HW, Yang X, Walker WE, Tesar BM, Goldstein DR. Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol. (2008) 181:6747–56. doi: 10.4049/jimmunol.181.10.6747
117. Lund JM, Linehan MM, Iijima N, Iwasaki A. Cutting Edge: Plasmacytoid dendritic cells provide innate immune protection against mucosal viral infection in situ. J Immunol. (2006) 177:7510–4. doi: 10.4049/jimmunol.177.11.7510
118. Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, et al. CD8(+) T cells orchestrate pDC-XCR1(+) dendritic cell spatial and functional cooperativity to optimize priming. Immunity. (2017) 46:205–19. doi: 10.1016/j.immuni.2017.01.003
119. Xu RH, Wong EB, Rubio D, Roscoe F, Ma X, Nair S, et al. Sequential activation of two pathogen-sensing pathways required for type I interferon expression and resistance to an acute DNA virus infection. Immunity. (2015) 43:1148–59. doi: 10.1016/j.immuni.2015.11.015
120. Wolf AI, Buehler D, Hensley SE, Cavanagh LL, Wherry EJ, Kastner P, et al. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J Immunol. (2009) 182:871–9. doi: 10.4049/jimmunol.182.2.871
121. Kochs G, Anzaghe M, Kronhart S, Wagner V, Gogesch P, Scheu S, et al. In vivo conditions enable IFNAR-independent type I interferon production by peritoneal CD11b+ cells upon thogoto virus infection. J Virol. (2016) 90:9330–7. doi: 10.1128/JVI.00744-16
122. Kallfass C, Ackerman A, Lienenklaus S, Weiss S, Heimrich B, Staeheli P. Visualizing production of beta interferon by astrocytes and microglia in brain of La Crosse virus-infected mice. J Virol. (2012) 86:11223–30. doi: 10.1128/JVI.01093-12
123. Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, et al. Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA. (2006) 103:7835–40. doi: 10.1073/pnas.0602460103
124. Cervantes-Barragan L, Zust R, Weber F, Spiegel M, Lang KS, Akira S, et al. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. (2007) 109:1131–7. doi: 10.1182/blood-2006-05-023770
125. Wang Y, Swiecki M, Cella M, Alber G, Schreiber RD, Gilfillan S, et al. Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe. (2012) 11:631–42. doi: 10.1016/j.chom.2012.05.003
126. Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med. (2006) 203:1153–9. doi: 10.1084/jem.20052359
127. Swiecki M, Colonna M. Type I interferons: diversity of sources, production pathways and effects on immune responses. Curr Opin Virol. (2011) 1:463–75. doi: 10.1016/j.coviro.2011.10.026
128. Alcami A, Koszinowski UH. Viral mechanisms of immune evasion. Mol Med Today. (2000) 6:365–72. doi: 10.1016/S1357-4310(00)01775-5
129. Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, et al. Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. (2008) 180:5799–803. doi: 10.4049/jimmunol.180.9.5799
130. Doring M, Lessin I, Frenz T, Spanier J, Kessler A, Tegtmeyer P, et al. M27 expressed by cytomegalovirus counteracts effective type I interferon induction of myeloid cells but not of plasmacytoid dendritic cells. J Virol. (2014) 88:13638–50. doi: 10.1128/JVI.00216-14
131. Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. (2006) 80:4286–91. doi: 10.1128/JVI.80.9.4286-4291.2006
132. Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. (2004) 101:3516–21. doi: 10.1073/pnas.0400525101
133. Eloranta ML, Alm GV. Splenic marginal metallophilic macrophages and marginal zone macrophages are the major interferon-alpha/beta producers in mice upon intravenous challenge with herpes simplex virus. Scand J Immunol. (1999) 49:391–4. doi: 10.1046/j.1365-3083.1999.00514.x
134. ME JW, Adair K, Brierley L. RNA viruses: a case study of the biology of emerging infectious diseases. Microbiol Spectr. (2013) 1:1–11. doi: 10.1128/microbiolspec.OH-0001-2012
135. Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, et al. The evolutionary history of vertebrate RNA viruses. Nature. (2018) 556:197–202. doi: 10.1038/s41586-018-0012-7
136. Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann KK, et al. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol. (2004) 173:5935–43. doi: 10.4049/jimmunol.173.10.5935
137. Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. (2007) 315:1398–401. doi: 10.1126/science.1136880
138. Honke N, Shaabani N, Cadeddu G, Sorg UR, Zhang DE, Trilling M, et al. Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat Immunol. (2011) 13:51–7. doi: 10.1038/ni.2169
139. Pfefferkorn C, Kallfass C, Lienenklaus S, Spanier J, Kalinke U, Rieder M, et al. Abortively infected astrocytes appear to represent the main source of interferon beta in the virus-infected brain. J Virol. (2016) 90:2031–8. doi: 10.1128/JVI.02979-15
140. Davidson S, Kaiko G, Loh Z, Lalwani A, Zhang V, Spann K, et al. Plasmacytoid dendritic cells promote host defense against acute pneumovirus infection via the TLR7-MyD88-dependent signaling pathway. J Immunol. (2011) 186:5938–48. doi: 10.4049/jimmunol.1002635
141. O'Brien M, Manches O, Bhardwaj N. Plasmacytoid dendritic cells in HIV infection. Adv Exp Med Biol. (2013) 762:71–107. doi: 10.1007/978-1-4614-4433-6_3
142. Feldman S, Stein D, Amrute S, Denny T, Garcia Z, Kloser P, et al. Decreased interferon-alpha production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin Immunol. (2001) 101:201–10. doi: 10.1006/clim.2001.5111
143. Herbeuval JP, Shearer GM. HIV-1 immunopathogenesis: how good interferon turns bad. Clin Immunol. (2007) 123:121–8. doi: 10.1016/j.clim.2006.09.016
144. Hosmalin A, Lebon P. Type I interferon production in HIV-infected patients. J Leukoc Biol. (2006) 80:984–93. doi: 10.1189/jlb.0306154
145. Kovarik P, Castiglia V, Ivin M, Ebner F. Type I interferons in bacterial infections: a Balancing Act. Front Immunol. (2016) 7:652. doi: 10.3389/fimmu.2016.00652
146. O'Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annu Rev Immunol. (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939
147. Kearney SJ, Delgado C, Eshleman EM, Hill KK, O'Connor BP, Lenz LL. Type I IFNs downregulate myeloid cell IFN-gamma receptor by inducing recruitment of an early growth response 3/NGFI-A binding protein 1 complex that silences ifngr1 transcription. J Immunol. (2013) 191:3384–92. doi: 10.4049/jimmunol.1203510
148. Rayamajhi M, Humann J, Penheiter K, Andreasen K, Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med. (2010) 207:327–37. doi: 10.1084/jem.20091746
149. Stanley SA, Johndrow JE, Manzanillo P, Cox JS. The Type I IFN response to infection with Mycobacterium tuberculosis requires ESX-1-mediated secretion and contributes to pathogenesis. J Immunol. (2007) 178:3143–52. doi: 10.4049/jimmunol.178.5.3143
150. Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, et al. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity. (2011) 35:1023–34. doi: 10.1016/j.immuni.2011.12.002
151. Giacomini E, Iona E, Ferroni L, Miettinen M, Fattorini L, Orefici G, et al. Infection of human macrophages and dendritic cells with Mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T cell response. J Immunol. (2001) 166:7033–41. doi: 10.4049/jimmunol.166.12.7033
152. Remoli ME, Giacomini E, Lutfalla G, Dondi E, Orefici G, Battistini A, et al. Selective expression of type I IFN genes in human dendritic cells infected with Mycobacterium tuberculosis. J Immunol. (2002) 169:366–74. doi: 10.4049/jimmunol.169.1.366
153. Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. (2013) 339:1448–53. doi: 10.1126/science.1233665
154. Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. (2004) 200:527–33. doi: 10.1084/jem.20040976
155. Carrero JA, Calderon B, Unanue ER. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J Immunol. (2004) 172:4866–74. doi: 10.4049/jimmunol.172.8.4866
156. O'Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. (2004) 200:437–45. doi: 10.1084/jem.20040712
157. Kernbauer E, Maier V, Rauch I, Muller M, Decker T. Route of infection determines the impact of type I interferons on innate immunity to listeria monocytogenes. PLoS ONE. (2013) 8:e65007. doi: 10.1371/journal.pone.0065007
158. Pitts MG, Myers-Morales T, D'Orazio SE. Type I IFN does not promote susceptibility to foodborne listeria monocytogenes. J Immunol. (2016) 196:3109–16. doi: 10.4049/jimmunol.1502192
159. Stockinger S, Kastner R, Kernbauer E, Pilz A, Westermayer S, Reutterer B, et al. Characterization of the interferon-producing cell in mice infected with Listeria monocytogenes. PLoS Pathog. (2009) 5:e1000355. doi: 10.1371/journal.ppat.1000355
160. Dresing P, Borkens S, Kocur M, Kropp S, Scheu S. A fluorescence reporter model defines “Tip-DCs” as the cellular source of interferon beta in murine listeriosis. PLoS ONE. (2010) 5:e15567. doi: 10.1371/journal.pone.0015567
161. Solodova E, Jablonska J, Weiss S, Lienenklaus S. Production of IFN-beta during Listeria monocytogenes infection is restricted to monocyte/macrophage lineage. PLoS ONE. (2011) 6:e18543. doi: 10.1371/journal.pone.0018543
162. Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, Pamer EG. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. (2003) 19:59–70. doi: 10.1016/S1074-7613(03)00171-7
163. Bao Y, Han Y, Chen Z, Xu S, Cao X. IFN-alpha-producing PDCA-1+ Siglec-H- B cells mediate innate immune defense by activating NK cells. Eur J Immunol. (2011) 41:657–68. doi: 10.1002/eji.201040840
164. O'Riordan M, Yi CH, Gonzales R, Lee KD, Portnoy DA. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc Natl Acad Sci USA. (2002) 99:13861–6. doi: 10.1073/pnas.202476699
165. Stockinger S, Materna T, Stoiber D, Bayr L, Steinborn R, Kolbe T, et al. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J Immunol. (2002) 169:6522–9. doi: 10.4049/jimmunol.169.11.6522
166. Serbina NV, Shi C, Pamer EG. Monocyte-mediated immune defense against murine Listeria monocytogenes infection. Adv Immunol. (2012) 113:119–34. doi: 10.1016/B978-0-12-394590-7.00003-8
167. Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. (2007) 178:3126–33. doi: 10.4049/jimmunol.178.5.3126
168. Mancuso G, Gambuzza M, Midiri A, Biondo C, Papasergi S, Akira S, et al. Bacterial recognition by TLR7 in the lysosomes of conventional dendritic cells. Nat Immunol. (2009) 10:587–94. doi: 10.1038/ni.1733
169. Weighardt H, Kaiser-Moore S, Schlautkotter S, Rossmann-Bloeck T, Schleicher U, Bogdan C, et al. Type I IFN modulates host defense and late hyperinflammation in septic peritonitis. J Immunol. (2006) 177:5623–30. doi: 10.4049/jimmunol.177.8.5623
170. Ramirez-Ortiz ZG, Lee CK, Wang JP, Boon L, Specht CA, Levitz SM. A nonredundant role for plasmacytoid dendritic cells in host defense against the human fungal pathogen Aspergillus fumigatus. Cell Host Microbe. (2011) 9:415–24. doi: 10.1016/j.chom.2011.04.007
171. Beiting DP. Protozoan parasites and type I interferons: a cold case reopened. Trends Parasitol. (2014) 30:491–8. doi: 10.1016/j.pt.2014.07.007
172. Silva-Barrios S, Stager S. Protozoan parasites and type I IFNs. Front Immunol. (2017) 8:14. doi: 10.3389/fimmu.2017.00014
173. deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, et al. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol. (2007) 178:6033–7. doi: 10.4049/jimmunol.178.10.6033
174. Haque A, Best SE, Montes de Oca M, James KR, Ammerdorffer A, Edwards CL, et al. Type I IFN signaling in CD8- DCs impairs Th1-dependent malaria immunity. J Clin Invest. (2014) 124:2483–96. doi: 10.1172/JCI70698
175. Voisine C, Mastelic B, Sponaas AM, Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. Int J Parasitol. (2010) 40:711–9. doi: 10.1016/j.ijpara.2009.11.005
176. Kim CC, Nelson CS, Wilson EB, Hou B, DeFranco AL, DeRisi JL. Splenic red pulp macrophages produce type I interferons as early sentinels of malaria infection but are dispensable for control. PLoS ONE. (2012) 7:e48126. doi: 10.1371/journal.pone.0048126
177. Spaulding E, Fooksman D, Moore JM, Saidi A, Feintuch CM, Reizis B, et al. STING-licensed macrophages prime type I IFN production by plasmacytoid dendritic cells in the bone marrow during severe Plasmodium yoelii malaria. PLoS Pathog. (2016) 12:e1005975. doi: 10.1371/journal.ppat.1005975
178. Yu X, Cai B, Wang M, Tan P, Ding X, Wu J, et al. Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity. (2016) 45:1093–107. doi: 10.1016/j.immuni.2016.10.001
179. Silva-Barrios S, Smans M, Duerr CU, Qureshi ST, Fritz JH, Descoteaux A, et al. Innate immune B cell activation by Leishmania donovani exacerbates disease and mediates hypergammaglobulinemia. Cell Rep. (2016) 15:2427–37. doi: 10.1016/j.celrep.2016.05.028
180. Minns LA, Menard LC, Foureau DM, Darche S, Ronet C, Mielcarz DW, et al. TLR9 is required for the gut-associated lymphoid tissue response following oral infection of Toxoplasma gondii. J Immunol. (2006) 176:7589–97. doi: 10.4049/jimmunol.176.12.7589
181. Han SJ, Melichar HJ, Coombes JL, Chan SW, Koshy AA, Boothroyd JC, et al. Internalization and TLR-dependent type I interferon production by monocytes in response to Toxoplasma gondii. Immunol Cell Biol. (2014) 92:872–81. doi: 10.1038/icb.2014.70
182. Sebina I, Haque A. Effects of type I interferons in malaria. Immunology. (2018) 155:176–85. doi: 10.1111/imm.12971
183. Pichyangkul S, Yongvanitchit K, Kum-arb U, Hemmi H, Akira S, Krieg AM, et al. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J Immunol. (2004) 172:4926–33. doi: 10.4049/jimmunol.172.8.4926
184. Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med. (2014) 20:47–53. doi: 10.1038/nm.3424
185. Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science. (2013) 340:202–7. doi: 10.1126/science.1235208
186. Beignon AS, McKenna K, Skoberne M, Manches O, DaSilva I, Kavanagh DG, et al. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J Clin Invest. (2005) 115:3265–75. doi: 10.1172/JCI26032
187. Schleicher U, Liese J, Justies N, Mischke T, Haeberlein S, Sebald H, et al. Type I interferon signaling is required for CpG-Oligodesoxynucleotide-induced control of Leishmania major, but not for spontaneous cure of subcutaneous primary or secondary L. major infection. Front Immunol. (2018) 9:79. doi: 10.3389/fimmu.2018.00079
188. Diefenbach A, Schindler H, Donhauser N, Lorenz E, Laskay T, MacMicking J, et al. Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity. (1998) 8:77–87. doi: 10.1016/S1074-7613(00)80460-4
189. Bogdan C, Mattner J, Schleicher U. The role of type I interferons in non-viral infections. Immunol Rev. (2004) 202:33–48. doi: 10.1111/j.0105-2896.2004.00207.x
190. Vivarini Ade C, Pereira Rde M, Teixeira KL, Calegari-Silva TC, Bellio M, Laurenti MD, et al. Human cutaneous leishmaniasis: interferon-dependent expression of double-stranded RNA-dependent protein kinase (PKR) via TLR2. FASEB J. (2011) 25:4162–73. doi: 10.1096/fj.11-185165
191. Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, et al. NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J Exp Med. (2007) 204:893–906. doi: 10.1084/jem.20061293
192. Pepper M, Dzierszinski F, Wilson E, Tait E, Fang Q, Yarovinsky F, et al. Plasmacytoid dendritic cells are activated by Toxoplasma gondii to present antigen and produce cytokines. J Immunol. (2008) 180:6229–36. doi: 10.4049/jimmunol.180.9.6229
193. Koblansky AA, Jankovic D, Oh H, Hieny S, Sungnak W, Mathur R, et al. Recognition of profilin by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity. (2013) 38:119–30. doi: 10.1016/j.immuni.2012.09.016
194. Pierog PL, Zhao Y, Singh S, Dai J, Yap GS, Fitzgerald-Bocarsly P. Toxoplasma gondii inactivates human plasmacytoid dendritic cells by functional Mimicry of IL-10. J Immunol. (2018) 200:186–95. doi: 10.4049/jimmunol.1701045
195. Leng J, Butcher BA, Denkers EY. Dysregulation of macrophage signal transduction by Toxoplasma gondii: past progress and recent advances. Parasite Immunol. (2009) 31:717–28. doi: 10.1111/j.1365-3024.2009.01122.x
196. Melo MB, Nguyen QP, Cordeiro C, Hassan MA, Yang N, McKell R, et al. Transcriptional analysis of murine macrophages infected with different Toxoplasma strains identifies novel regulation of host signaling pathways. PLoS Pathog. (2013) 9:e1003779.
197. Beiting DP, Peixoto L, Akopyants NS, Beverley SM, Wherry EJ, Christian DA, et al. Differential induction of TLR3-dependent innate immune signaling by closely related parasite species. PLoS ONE. (2014) 9:e88398. doi: 10.1371/journal.pone.0088398
198. Swiecki M, Wang Y, Vermi W, Gilfillan S, Schreiber RD, Colonna M. Type I interferon negatively controls plasmacytoid dendritic cell numbers in vivo. J Exp Med. (2011) 208:2367–74. doi: 10.1084/jem.20110654
Keywords: type I interferon, plasmacytoid dendritic cells, interferon producing cells, infection, pathogen, virus, immunopathology, immune activation
Citation: Ali S, Mann-Nüttel R, Schulze A, Richter L, Alferink J and Scheu S (2019) Sources of Type I Interferons in Infectious Immunity: Plasmacytoid Dendritic Cells Not Always in the Driver's Seat. Front. Immunol. 10:778. doi: 10.3389/fimmu.2019.00778
Received: 31 August 2018; Accepted: 25 March 2019;
Published: 12 April 2019.
Edited by:
Diana Dudziak, Universitätsklinikum Erlangen, GermanyReviewed by:
Susan Kovats, Oklahoma Medical Research Foundation, United StatesCopyright © 2019 Ali, Mann-Nüttel, Schulze, Richter, Alferink and Scheu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefanie Scheu, c3RlZmFuaWUuc2NoZXVAdW5pLWR1ZXNzZWxkb3JmLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.