
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 29 March 2019
Sec. NK and Innate Lymphoid Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00661
This article is part of the Research Topic Emerging Concepts on the NKG2D Receptor- Ligand Axis in Health and Diseases View all 19 articles
 Alexandra Frazao1Louise Rethacker1Meriem Messaoudene1,2Marie-Françoise Avril3,4
Alexandra Frazao1Louise Rethacker1Meriem Messaoudene1,2Marie-Françoise Avril3,4 Antoine Toubert1,4,5
Antoine Toubert1,4,5 Nicolas Dulphy1,4,5
Nicolas Dulphy1,4,5 Anne Caignard1*
Anne Caignard1*The antitumor functions of NK cells are regulated by the integration of positive and negative signals triggered by numerous membrane receptors present on the NK cells themselves. Among the main activating receptors, NKG2D binds several stress-induced molecules on tumor targets. Engagement of NKG2D by its ligands (NKG2D-Ls) induces NK cell activation leading to production of cytokines and target cell lysis. These effects have therapeutic potential as NKG2D-Ls are widely expressed by solid tumors, whereas their expression in healthy cells is limited. Here, we describe the genetic and environmental factors regulating the NKG2D/NKG2D-L pathway in tumors. NKG2D-L expression is linked to cellular stress and cell proliferation, and has been associated with oncogenic mutations. Tumors have been found to alter their to NKG2D-L expression as they progress, which interferes with the antitumor function of the pathway. Nevertheless, this pathway could be advantageously exploited for cancer therapy. Various cancer treatments, including chemotherapy and targeted therapies, indirectly interfere with the cellular and soluble forms of NKG2D-Ls. In addition, NKG2D introduced into chimeric antigen receptors in T- and NK cells is a promising tumor immunotherapy approach.
The immunosurveillance theory described by Robert Schreiber in 2002 (1) suggested that NK cells are involved in the early control of tumor development, before the successive equilibrium and escape phases when tumor-induced immunosuppression results in the emergence of immune-resistant tumor variants. During the elimination phase, NK cells detect and kill emerging transformed cells. NK cells naturally express receptors detecting stress-induced molecules and altered expression of Major Histocompatibility Complex (MHC) class-I molecules on transformed targets (2). NK cells also potentiate the adaptive immune response through cytokine secretion and by stimulating dendritic cells (DC), notably within lymph nodes (3–8).
Several groups have used samples obtained after curative resection to investigate the role played by NK cells in primary solid tumors. Their results indicated that NK cell infiltrates may correlate with clinical outcome (9–14). In most reports, NK cells were present at low numbers within tumors, as reviewed in (15). In contrast, in colorectal carcinoma (16), lung cancers (14), and gastrointestinal stromal tumors (GIST) (17) numerous NK cells were present in peritumoral areas. NK cells are generally underrepresented among tumor-infiltrating lymphocytes compared to their circulating proportions, and their effector functions are also altered within the tumor microenvironment, as shown in breast and lung malignancies (18, 19). NK cell dysfunction, based on reduced cytotoxicity and cytokine release, correlates with downregulation of NK-activating receptors (18–21). However, this is a tumor-specific phenomenon, described in renal cell carcinoma (RCC) (22), GIST (17), neuroblastoma (23), melanoma (24, 25), and acute myeloid leukemia (26, 27). Altogether, these data revealed that, more than NK cell numbers, expression of NK cell receptors (including activating NK receptors or inhibitory KIRs) strongly influence prognosis and disease outcome. The only notable exception to this conclusion is chronic myeloid leukemia (CML) patients for whom treatment with imatinib was interrupted; in these patients, NK cell numbers were a significant predictive parameter for relapse (28, 29).
In addition, NK cells are thought to play a role in the emergence of metastases, as high numbers of circulating or tumor-infiltrating NK cells inversely correlated with metastatic disease (30, 31). The mechanisms deployed to limit cancer dissemination could involve activating NK receptors, including NKG2D or NKp46. In murine models of melanoma and prostate cancer, IFN-γ production by NK cells triggered by NKp46 activation was found to induce expression of the extracellular matrix protein fibronectin 1 in tumor cells, altering tumor architecture and controlling metastatic invasion (32).
NK cell activation is regulated by signals from activating receptors and inhibitory NK receptors, which bind to HLA-class-I molecules. In addition to natural cytotoxicity receptors, or Ig-type family receptors—the ligands for which have not yet been clearly identified—NKG2D (Natural Killer Group 2, member D)—a C-type lectin receptor—is a major activating receptor for NK cells. In humans, the gene encoding NKG2D (KLRK1) lies amid a cluster of genes referred to as the “NK-complex” (NKC) that includes several genes expressed by NK cells [KLRD1 (CD94), KLRC4 (NKG2F), KLRC3 (NKG2E), KLRC2 (NKG2C), and KLRC1 (NKG2A)]. NKG2D was first identified on the surface of NK cells as an immunosurveillance receptor. It is also expressed by most CD8+ and a small subset of CD4+ cytotoxic αβ T cells, as well as innate-like immune cells, such as some iNKT cells and γδ T cells (33, 34). NKG2D is a type II transmembrane protein and, in humans, it associates with the transmembrane domain of the adaptor protein DAP-10 (DNAX-Activating Protein 10). Ligand binding causes dimerization of two NKG2D monomers to form an active receptor which phosphorylates DAP10 and triggers NK cell activation signaling pathways which promote Ca2+ influx, actin-based cytoskeleton reorganization, and microtubule polarization (35). This signaling cascade leads to the release of the contents of cytolytic granules, and in some cases elicits the production of cytokines by NK cells (36). NKG2D provides co-stimulatory signals in activated T cells (37). NKG2D expression in NK cells and CD8+ T cells can be upregulated, in particular in response to cytokines, such as interleukin (IL)-2, and IL-15, while transforming growth factor (TGF)-β can decrease NKG2D expression.
Our current understanding of the role played by NKG2D in controlling tumor development through NK cell and cytotoxic T-lymphocyte (CTL) activity was aided by the early characterization of its ligands (38). NKG2D binds to eight molecules, members of the following two families: MHC class-I-related chains A or B (MICA/B), and UL16-binding proteins (ULBP1-6). Seven MIC genes (MICA to MICGI) have been identified, located on chromosome 6p21.33. Only MICA and MICB genes are translated into proteins. As to ULBP, six protein-coding genes (ULBP1 to ULBP6I) have been described on chromosome 6q24.2–25.3. Among all these proteins, MICA, MICB, ULBP4, and ULBP5 are transmembrane-anchored glycoproteins, whereas ULBP1, ULBP2, ULBP3, and ULBP6 are bound to the cell surface by a glycophosphatidylinositol (GPI) motif (39). All NKG2D-Ls are composed of one alpha 1 and one alpha 2 extracellular immunoglobulin (Ig)-like domain, which share a strong homology to the corresponding domains in classical HLA-class-I molecules (40). MICA and MICB contain a third, additional extracellular Ig-like domain (alpha 3). NKG2D-Ls are not associated with β2-microglobulin and bind no antigenic peptide.
The pioneering work by Imai et al. attributed a major role to NKG2D polymorphisms in cancer immunosurveillance (41), and in the prevention of cancer formation (42). An 11-years follow-up survey of a cohort including >3,500 members indicated that medium and high natural cytotoxic activity of peripheral-blood lymphocytes was associated with a reduced cancer risk, whereas low natural cytotoxicity correlated with a higher incidence of cancer. These findings suggest a role for NK cell-mediated immunity in controlling cancer. Analysis of 25 SNPs reported with an allele frequency of >10% in the NKC gene cluster identified eight SNPs in the NKG2D locus (KLRK1) that form two haplotype blocks (NKG2Dhb1 and hb2). Each of these blocks can generate two major alleles linked to low (LNK) or high (HNK) cytotoxic activity. Patients with the HNK1/HNK1 NK2GDhb1 haplotype had a lower incidence of cancer compared to those with the LNK1/LNK1 haplotype (41). In a Japanese population the HNK1/HNK1 genotype was associated with decreased colorectal (43) and aerodigestive tract cancer (44). A recent report indicated that NKG2D gene polymorphisms also correlated with control of CML by dasatinib (45). Thus, patients with the NKG2D HNK1/HNK1 haplotype achieved deep molecular response (MR4.5) more quickly than those with other haplotypes. Interestingly, phosphorylation of VAV1 on Tyr174, which was proposed as a major mechanism by which dasatinib intensifies NK cell activity (46), could also be enhanced by expression of the NKG2D HNK1 allele (45). These data suggest that the NKG2D HNK1/HNK1 haplotype may influence cancer development and modulate treatment response (Figure 1).
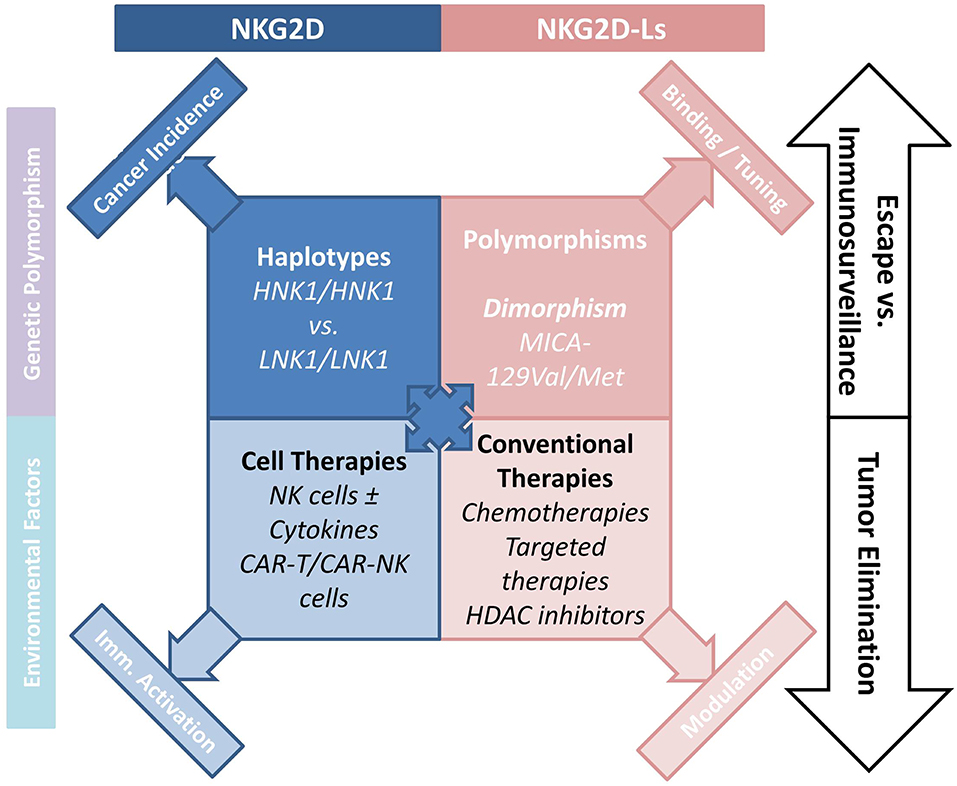
Figure 1. How the NKG2D/NKG2D-L axis is involved NK-cell mediated cancer immunosurveillance. The axis interferes with tumor development and progression, and regulates the antitumor function of NK cells. Various cancer treatments including chemotherapy and targeted therapies indirectly interfere with the cellular and soluble forms of NKG2D-Ls, emphasizing the interest to which this pathway is involved in cancer therapy. Novel CAR-T- and CAR-NK-cell-based therapies hold considerable promise.
Importantly, MICA and MICB as well as ULBP molecules are highly polymorphic and allelic variation can alter their expression levels or their affinity for NKG2D. As a result, NKG2D-L polymorphisms may strongly influence NKG2D-mediated NK cell triggering by tumor cells or other stressed targets (47). To date, 107 MICA and 47 MICB alleles have been described (updated allele numbers can be found at http://hla.alleles.org/nomenclature/index.html). SNPs are located within regions encoding the α1 and α2 extracellular domains. These alleles are transcribed to produce a total of 82 MICA and 30 MICB proteins. The impact of MICA polymorphisms on protein expression and function remains only partly characterized, and MICA-129 is the only SNP described so far that affects NKG2D receptor affinity (48). MICA-129 (rs1051792) dimorphism—the substitution of a methionine (Met) for a valine (Val) at position 129—alters MICA affinity for the NKG2D receptor: MICA-129Met has an affinity 10- to 50-fold higher than MICA-129Val. Significantly, expression levels for MICA-129Met isoforms are reduced compared to the MICA-129Val molecule (49), but they nevertheless have a higher capacity to trigger the NKG2D pathway, leading to enhanced NK cell activity (50).
Several studies implicated MICA polymorphisms in viral infections and autoimmunity [reviewed in (51)], but few have investigated the impact of MICA polymorphism in cancers. Initial studies focused on cervical cancer and found no association between MICA polymorphism and disease susceptibility (52). In contrast, MICA polymorphism was found to be a significant risk factor for other tumors. For example, MICA-129Val is associated with poor prognosis in nasopharyngeal carcinoma (53) or breast cancer (54) in a Tunisian population. In melanoma, Isernhagen et al. (49) showed that MICA-129Val-homozygous melanoma cell lines expressed higher surface levels of MICA than cells with the Met/Met genotype, which released more soluble MICA. A group of frequent MICA alleles, named MICA-A5.1 (prototype MICA*008), produce a truncated protein that acquires a GPI anchor allowing it to be recruited to exosomes, from where it can downmodulate NKG2D expression (55). A GWAS study in cervical cancer patients linked one MICA-adjacent region to the disease and identified a SNP (rs2516448) linked to the MICA-A5.1 frame-shift mutation, suggesting that this allele may cause impaired immune activation resulting in cancer development (56). In colorectal liver metastases, in contrast, the MICA-A5.1 polymorphism was associated with better tumor control and response to treatment (57).
Few studies have investigated ULBP polymorphisms, probably because of the limited number of SNP identified within these genes. However, one ULBP6 dimorphism (two SNP at positions 106 and 147) plays a significant role in determining affinity of the protein for the NKG2D receptor. The ULBP0602 molecule (which contains Leu106 and Thr147 in contrast to ULPB0601's Arg106 and Ile147) binds to NKG2D with a 10- to 1,000-fold higher affinity than other ULBPs. This difference in binding could result in decreased interaction of NKG2D with other ligands; it could thus have a negative effect on NK cell function (58).
NKG2D-Ls are rarely expressed by healthy cells, but are induced at the cell surface when the cell is stressed as a result of viral infection or malignant transformation; they are therefore called “induced-self” ligands. NKG2D-L-positive cells are detected and eliminated, mostly by NK cells. Expression of NKG2D-Ls is regulated by several mechanisms, which may be transcriptional, translational or post-translational. Expression of NKG2D-Ls is induced by DNA damage, a characteristic of tumor transformation, which leads to the activation of the ATM-ATR DNA repair pathways (59). In mouse models, hyper-proliferation can also induce NKG2D-L expression through activation of the E2F transcription factor (60). Indeed, following HER2/HER3 or BCR-ABL activation, proliferative signals can induce MICA/B and ULBP expression (61). Cellular stress, such as heat shock, has also been reported to induce heat shock factor 1-mediated MICA/B expression (62, 63).
The NKG2D/NKG2D-L pathway is triggered early in cancer development and participates in the elimination of tumor cells. However, during tumor progression, profound changes occur and the NKG2D and/or NKG2D-Ls are targeted by a range of tumor escape mechanisms. Cancer can sculpt the immune environment by selecting immune-ligand-negative variants (1, 64). Tumor cells expressing high levels of NKG2D-Ls can thus be eliminated as part of the tumor immunoediting process, which involves NK cells and NKG2D, and progressively results in the emergence of NKG2D-resistant variants (65). Persistent NKG2D-L expression by tumor cells may cause systemic immunosuppression as a result of NK exhaustion and perturbation of the immune synapse (26). Epigenetic and transcriptional regulation mechanisms are often perturbed in tumor cells, and NKG2D-L expression may be altered. In melanoma, endoplasmic reticulum stress can reduce MICA transcription by modulating activation of the transcription factor E2F1 (66). Indeed, immature isoforms of MICA are retained in the endoplasmic reticulum, resulting in limited membrane-display of MICA (67). Tumors can also inhibit NKG2D-L expression by altering cell surface glycosylation (68), notably in a hypoxic environment (69). In CML, BCR/ABL controls MICA expression through post-transcriptional mechanisms (70), including MICA glycosylation (71). Finally, histone deacetylases (HDAC) may also regulate NKG2D-L expression (72, 73).
Soluble factors secreted by tumor cells and cells from their microenvironment can also alter NKG2D-L expression levels. Thus, TGF-β and IL-10 secreted by regulatory T cells (Tregs) and myeloid-derived suppressor cells downmodulate the expression of NKG2D-Ls (74, 75). Some tumors were demonstrated to secrete TGF-β, resulting in reduced NK cell-mediated lysis (10). ADAM metallopeptidases can catalyze shedding of NKG2D-Ls from the cell's surface, and the released soluble forms can hamper NKG2D signaling (76). Upregulation of ADAM10 or ADAM17 expression in tumors has been linked to the release of solMICA/B, decreased membrane MICA/B expression, and reduced NKG2D expression on NK cells or CD8 T cells (77, 78). Importantly, high serum levels of soluble MIC associated with poor clinical prognosis and the emergence of metastases in RCC (79) and prostate cancer (80). Most leukemia patients present high levels of at least one solNKG2D-L, associated with reduced NKG2D expression by NK cells and impaired anti-leukemic function. In patients who entered complete remission following treatment, solNKG2D-Ls were no longer detected, and NK function was restored (81). Following publication of these data, a meta-analysis of 19 studies, comprising 2,588 patients with 10 different types of tumor, showed that serum concentrations of solMICA/B represent a potential prognostic marker in human cancer (82). In metastatic melanoma, levels of solULBPs are associated with reduced survival in patients treated with immune checkpoint blockers (83); solNKG2D-Ls could thus be a relevant biomarker to select melanoma patients for immunotherapy.
The frequent and high expression of NKG2D-Ls by tumor cells in various human cancers, and the potent antitumor function of the NKG2D/NKG2D-L pathway are now established (Figure 1). Cancer therapies aiming to improve or restore NK- and T-cell responses through NKG2D activation have attracted considerable interest. Among the options proposed, conventional cancer treatments can be used to increase NKGD2 expression and signaling. Indeed, a number of cancer treatments increase NKG2D-L expression on tumor cells. Thus, HDAC inhibitors (Valporic acid) acting on epigenetic regulation of NKG2D-Ls can upregulate their membrane expression (84, 85). Cisplatin, Gemcitabine, Oxiplatin, or 5-fluorouracil chemotherapies have all been shown to increase MICA/B expression on tumor cells through modulation of the ATM-ATR pathway. Other chemotherapies directly kill tumor cells while also inducing NKG2D-L expression, which stimulates tumor elimination by activating immune processes (86–89). Proteasome inhibitors inducing ULBP2 expression and decreasing HLA-I molecules may promote NK cell immunosurveillance of hematologic malignancies (90). ADAM10 or MMP inhibitors could be used to inhibit NKG2D-L-shedding and promote NK cell activity, this approach has been shown to restore NKG2D-Ls in some cancers (91–93). Similarly, antibodies targeting the proteolytic site can prevent shedding of MICA/B proteins, and was shown to limit tumor growth and reduce the formation of metastases in a humanized murine melanoma model (94).
Inhibitors of oncogene-driven mutations are actively being developed to treat various tumor types. Such inhibitors decrease the constitutive activation of kinases involved in tumor cell proliferation and also affect NKG2D-L expression (61, 95). Treatment with imatinib controls the expression of NKG2D-Ls and membrane ganglioside (GM1), and was shown to interfere with NK cell recognition and cytolysis of BCR/ABL cells (70, 71). Treatment of melanoma cells with BRAF and MEK inhibitors modulates the expression of MICA/B and ULBP2, attenuating their recognition by NK cells (96). These effects can be overcome by the simultaneous application of HDAC inhibitors, restoring NKG2D-L expression and stimulating NK cell recognition and function (97). Erk activation was shown to increase NKG2D-L expression (98, 99), but, by acting on MMP, Erk/MEK activation can disrupt the equilibrium between membrane and soluble isoforms of NKG2D-Ls and alter NKG2D function (100, 101). It would therefore be relevant to control membrane and solNKG2D-Ls when performing immunotherapy trials.
One of the most promising approaches to cancer immunotherapy relies on revisited immune-cell-based therapies, T cells engineered to express chimeric antigen receptors (CARs) can be highly tumor-specific and have a high killing potential (102). CAR-T cells present a major clinical benefit for patients with malignant hemopathies. For instance, CAR-T cells were shown to be potent against CD19-expressing hematologic tumors in several trials (103, 104). However, the safety of CAR-T cells remains a major obstacle, and CARs must be optimized to increase efficacy and limit treatment-related morbidities due to cytokine-release syndrome (105). Up to now, the use of CAR-T cells for treating solid tumors has been limited by the lack of appropriate tumor antigens (106). Production of modified NK cells expressing CARs could represent an alternative for treatment of solid tumors as they recognize numerous tumor cell types. In addition, infusion of large numbers of NK cells is known not to induce autoreactivity. CAR-NK cells could thus be used in complement or as an alternative to CAR-T cells (Figure 1). NK cells have recently been engineered with CARs to enhance their killing activity, and trials of these cells for treatment of refractory solid tumors have been initiated (107). NK-92 cells were engineered with tumor antigen-specific CARs (EGFR, EpCAM) and successfully used in xenograft models (108, 109). NK cells have a limited lifespan and do not produce memory cells, as a result the excessive activation observed with CAR-T cells should be avoided. In addition, engineered CAR-NK cell lines could be mass-produced, circumventing the need to generate autologous products for each patient, a process that remains challenging and expensive (110). Another promising option would be to express CARs in human iPSC-derived NK cells. This approach might present several advantages, as it could provide a universal cell therapy product (111).
When considering CAR constructs the NKG2D receptor is worthy of attention. Indeed, as we have seen, NKG2D-Ls are widely expressed by a number of solid human tumors, and their relatively selective expression by transformed cells compared to healthy cells makes them an attractive receptor for CARs constructs in T cells. The extracellular domain of NKG2D is used in different CARs constructs with a view to promoting tumor-reactive T reactions (112–114). However, investigations must be performed with care as NKG2D-ligands can also be expressed by healthy cells (115), potentially leading to significant toxicity (116).
The expression of NKG2D-Ls by myeloid cells, Tregs and endothelial cells in the tumor microenvironment suggests that CAR NKG2D cells could also be used to control in situ immunosuppression (117). Thus, CAR-NK cells transduced with NKG2D fused to the TCR CD3z chain could be used to target suppressive myeloid cells and improve infiltration and function of subsequent infusions of tumor-specific CAR-T cells (118).
NKG2D-based CARs with full-length NKG2D or NKG2D-ligand binding domains represent a novel strategy to target several types of solid tumors, and would have the capacity to induce potent antitumor immunity in patients. NKG2D CARs could not only target tumors but also myeloid immunosuppressive cells and Tregs, as well as others cells in the microenvironment that promote tumor progression.
AF, ND, and AC wrote the manuscript. AT, M-FA, MM, and LR read and corrected the manuscript.
This work was supported by grants from Institut National du Cancer PAIR Melanome (2013-0662013), la Ligue Nationale contre le Cancer (PhD grant for MM).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
2. Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
3. Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat Med. (2018) 24:1178–91. doi: 10.1038/s41591-018-0085-8
4. Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. (2018) 172:1022–37 e1014. doi: 10.1016/j.cell.2018.01.004
5. Ferlazzo G, Munz C. NK cell compartments and their activation by dendritic cells. J Immunol. (2004) 172:1333–9. doi: 10.4049/jimmunol.172.3.1333
6. Gustafsson K, Ingelsten M, Bergqvist L, Nystrom J, Andersson B, Karlsson-Parra A. Recruitment and activation of natural killer cells in vitro by a human dendritic cell vaccine. Cancer Res. (2008) 68:5965–71. doi: 10.1158/0008-5472.CAN-07-6494
7. Mailliard RB, Son YI, Redlinger R, Coates PT, Giermasz A, Morel PA, et al. Dendritic cells mediate NK cell help for Th1 and CTL responses: two-signal requirement for the induction of NK cell helper function. J Immunol. (2003) 171:2366–73. doi: 10.4049/jimmunol.171.5.2366
8. Saito T, Takayama T, Osaki T, Nagai S, Suzuki T, Sato M, et al. Combined mobilization and stimulation of tumor-infiltrating dendritic cells and natural killer cells with Flt3 ligand and IL-18 in vivo induces systemic antitumor immunity. Cancer Sci. (2008) 99:2028–36. doi: 10.1111/j.1349-7006.2008.00907.x
9. McKay K, Moore PC, Smoller BR, Hiatt KM. Association between natural killer cells and regression in melanocytic lesions. Hum Pathol. (2011) 42:1960–4. doi: 10.1016/j.humpath.2011.02.019
10. Pasero C, Gravis G, Guerin M, Granjeaud S, Thomassin-Piana J, Rocchi P, et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. (2016) 76:2153–65. doi: 10.1158/0008-5472.CAN-15-1965
11. Schleypen JS, Baur N, Kammerer R, Nelson PJ, Rohrmann K, Grone EF, et al. Cytotoxic markers and frequency predict functional capacity of natural killer cells infiltrating renal cell carcinoma. Clin Cancer Res. (2006) 12:718–25. doi: 10.1158/1078-0432.CCR-05-0857
12. Takanami I, Takeuchi K, Giga M. The prognostic value of natural killer cell infiltration in resected pulmonary adenocarcinoma. J Thorac Cardiovasc Surg. (2001) 121:1058–63. doi: 10.1067/mtc.2001.113026
13. Villegas FR, Coca S, Villarrubia VG, Jimenez R, Chillon MJ, Jareno J, et al. Prognostic significance of tumor infiltrating natural killer cells subset CD57 in patients with squamous cell lung cancer. Lung Cancer. (2002) 35:23–8. doi: 10.1016/S0169-5002(01)00292-6
14. Jin S, Deng Y, Hao JW, Li Y, Liu B, Yu Y, et al. NK cell phenotypic modulation in lung cancer environment. PLoS ONE. (2014) 9:e109976. doi: 10.1371/journal.pone.0109976
15. Desbois M, Rusakiewicz S, Locher C, Zitvogel L, Chaput N. Natural killer cells in non-hematopoietic malignancies. Front Immunol. (2012) 3:395. doi: 10.3389/fimmu.2012.00395
16. Sconocchia G, Zlobec I, Lugli A, Calabrese D, Iezzi G, Karamitopoulou E, et al. Tumor infiltration by FcgammaRIII (CD16)+ myeloid cells is associated with improved survival in patients with colorectal carcinoma. Int J Cancer. (2011) 128:2663–72. doi: 10.1002/ijc.25609
17. Rusakiewicz S, Semeraro M, Sarabi M, Desbois M, Locher C, Mendez R, et al. Immune infiltrates are prognostic factors in localized gastrointestinal stromal tumors. Cancer Res. (2013) 73:3499–510. doi: 10.1158/0008-5472.CAN-13-0371
18. Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. (2011) 121:3609–22. doi: 10.1172/JCI45816
19. Platonova S, Cherfils-Vicini J, Damotte D, Crozet L, Vieillard V, Validire P, et al. Profound coordinated alterations of intratumoral NK cell phenotype and function in lung carcinoma. Cancer Res. (2011) 71:5412–22. doi: 10.1158/0008-5472.CAN-10-4179
20. Carrega P, Morandi B, Costa R, Frumento G, Forte G, Altavilla G, et al. Natural killer cells infiltrating human nonsmall-cell lung cancer are enriched in CD56 bright CD16(-) cells and display an impaired capability to kill tumor cells. Cancer. (2008) 112:863–75. doi: 10.1002/cncr.23239
21. Chretien AS, Le Roy A, Vey N, Prebet T, Blaise D, Fauriat C, et al. Cancer-induced alterations of NK-mediated target recognition: current and investigational pharmacological strategies aiming at restoring NK-mediated anti-tumor activity. Front Immunol. (2014) 5:122. doi: 10.3389/fimmu.2014.00122
22. Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res. (2013) 19:4079–91. doi: 10.1158/1078-0432.CCR-12-3847
23. Semeraro M, Rusakiewicz S, Minard-Colin V, Delahaye NF, Enot D, Vely F, et al. Clinical impact of the NKp30/B7-H6 axis in high-risk neuroblastoma patients. Sci Transl Med. (2015) 7:283ra255. doi: 10.1126/scitranslmed.aaa2327
24. Fregni G, Messaoudene M, Fourmentraux-Neves E, Mazouz-Dorval S, Chanal J, Maubec E, et al. Phenotypic and functional characteristics of blood natural killer cells from melanoma patients at different clinical stages. PLoS ONE. (2013) 8:e76928. doi: 10.1371/journal.pone.0076928
25. Fregni G, Perier A, Pittari G, Jacobelli S, Sastre X, Gervois N, et al. Unique functional status of natural killer cells in metastatic stage IV melanoma patients and its modulation by chemotherapy. Clin Cancer Res. (2011) 17:2628–37. doi: 10.1158/1078-0432.CCR-10-2084
26. Khaznadar Z, Henry G, Setterblad N, Agaugue S, Raffoux E, Boissel N, et al. Acute myeloid leukemia impairs natural killer cells through the formation of a deficient cytotoxic immunological synapse. Eur J Immunol. (2014) 44:3068–80. doi: 10.1002/eji.201444500
27. Ruggeri L, Capanni M, Urbani E, Perrucio K, Shlomchik W, Tosti A, et al. Effectiveness of donor natral killer cell alloreactivity in mismatched hematopoietic transplants. Science. (2002) 295:2375–82. doi: 10.1126/science.1068440
28. Ilander M, Olsson-Stromberg U, Schlums H, Guilhot J, Bruck O, Lahteenmaki H, et al. Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. (2017) 31:1108–16. doi: 10.1038/leu.2016.360
29. Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. (2017) 102:1368–77. doi: 10.3324/haematol.2017.165001
30. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
31. Smyth MJ, Swann J, Kelly JM, Cretney E, Yokoyama WM, Diefenbach A, et al. NKG2D recognition and perforin effector function mediate effective cytokine immunotherapy of cancer. J Exp Med. (2004) 200:1325–35. doi: 10.1084/jem.20041522
32. Glasner A, Levi A, Enk J, Isaacson B, Viukov S, Orlanski S, et al. NKp46 receptor-mediated interferon-gamma production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity. (2018) 48:396–8. doi: 10.1016/j.immuni.2018.01.010
33. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. (1999) 285:727–9. doi: 10.1126/science.285.5428.727
34. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. (2003) 3:781–90. doi: 10.1038/nri1199
35. Graham DB, Cella M, Giurisato E, Fujikawa K, Miletic AV, Kloeppel T, et al. Vav1 controls DAP10-mediated natural cytotoxicity by regulating actin and microtubule dynamics. J Immunol. (2006) 177:2349–55. doi: 10.4049/jimmunol.177.4.2349
36. Wu J, Song Y, Bakker AB, Bauer S, Spies T, Lanier LL, et al. An activating immunoreceptor complex formed by NKG2D and DAP10. Science. (1999) 285:730–2. doi: 10.1126/science.285.5428.730
37. Diefenbach A, Tomasello E, Lucas M, Jamieson AM, Hsia JK, Vivier E, et al. Selective associations with signaling proteins determine stimulatory versus costimulatory activity of NKG2D. Nat Immunol. (2002) 3:1142–9. doi: 10.1038/ni858
38. Carapito R, Bahram S. Genetics, genomics, and evolutionary biology of NKG2D ligands. Immunol Rev. (2015) 267:88–116. doi: 10.1111/imr.12328
39. Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol. (2001) 2:443–51. doi: 10.1038/87757
40. Spreu J, Stehle T, Steinle A. Human cytomegalovirus-encoded UL16 discriminates MIC molecules by their alpha2 domains. J Immunol. (2006) 177:3143–9. doi: 10.4049/jimmunol.177.5.3143
41. Hayashi T, Imai K, Morishita Y, Hayashi I, Kusunoki Y, Nakachi K. Identification of the NKG2D haplotypes associated with natural cytotoxic activity of peripheral blood lymphocytes and cancer immunosurveillance. Cancer Res. (2006) 66:563–70. doi: 10.1158/0008-5472.CAN-05-2776
42. Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. (2000) 356:1795–9. doi: 10.1016/S0140-6736(00)03231-1
43. Furue H, Matsuo K, Kumimoto H, Hiraki A, Suzuki T, Yatabe Y, et al. Decreased risk of colorectal cancer with the high natural killer cell activity NKG2D genotype in Japanese. Carcinogenesis. (2008) 29:316–20. doi: 10.1093/carcin/bgm260
44. Furue H, Kumimoto H, Matsuo K, Suzuki T, Hasegawa Y, Shinoda M, et al. Opposite impact of NKG2D genotype by lifestyle exposure to risk of aerodigestive tract cancer among Japanese. Int J Cancer. (2008) 123:181–6. doi: 10.1002/ijc.23456
45. Hara R, Onizuka M, Matsusita E, Kikkawa E, Nakamura Y, Matsushita H, et al. NKG2D gene polymorphisms are associated with disease control of chronic myeloid leukemia by dasatinib. Int J Hematol. (2017) 106:666–74. doi: 10.1007/s12185-017-2294-1
46. Hassold N, Seystahl K, Kempf K, Urlaub D, Zekl M, Einsele H, et al. Enhancement of natural killer cell effector functions against selected lymphoma and leukemia cell lines by dasatinib. Int J Cancer. (2012) 131:E916–27. doi: 10.1002/ijc.27537
47. Zuo J, Mohammed F, Moss P. The biological influence and clinical relevance of polymorphism within the NKG2D ligands. Front Immunol. (2018) 9:1820. doi: 10.3389/fimmu.2018.01820
48. Steinle A, Li P, Morris DL, Groh V, Lanier LL, Strong RK, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics. (2001) 53:279–87. doi: 10.1007/s002510100325
49. Isernhagen A, Schilling D, Monecke S, Shah P, Elsner L, Walter L, et al. The MICA-129Met/Val dimorphism affects plasma membrane expression and shedding of the NKG2D ligand MICA. Immunogenetics. (2016) 68:109–23. doi: 10.1007/s00251-015-0884-8
50. Isernhagen A, Malzahn D, Viktorova E, Elsner L, Monecke S, von Bonin F, et al. The MICA-129 dimorphism affects NKG2D signaling and outcome of hematopoietic stem cell transplantation. EMBO Mol Med. (2015) 7:1480–502. doi: 10.15252/emmm.201505246
51. Isernhagen A, Malzahn D, Bickeboller H, Dressel R. Impact of the MICA-129Met/Val dimorphism on NKG2D-mediated biological functions and disease risks. Front Immunol. (2016) 7:588. doi: 10.3389/fimmu.2016.00588
52. Ghaderi M, Hjelmstrom P, Hallmans G, Wiklund F, Lenner P, Dillner J, et al. MICA gene polymorphism and the risk to develop cervical intraepithelial neoplasia. Hum Immunol. (1999) 60:970–3. doi: 10.1016/S0198-8859(99)00087-7
53. Douik H, Ben Chaaben A, Attia Romdhane N, Romdhane HB, Mamoghli T, Fortier C, et al. Association of MICA-129 polymorphism with nasopharyngeal cancer risk in a Tunisian population. Hum Immunol. (2009) 70:45–8. doi: 10.1016/j.humimm.2008.10.008
54. Ouni N, Ben Chaaben A, Kablouti G, Lajnef M, Ayari F, Abaza H, et al. MICA-129Met/Val polymorphism is associated with early-onset breast cancer risk. Immunol Invest. (2017) 46:603–14. doi: 10.1080/08820139.2017.1336175
55. Ashiru O, Lopez-Cobo S, Fernandez-Messina L, Pontes-Quero S, Pandolfi R, Reyburn HT, et al. A GPI anchor explains the unique biological features of the common NKG2D-ligand allele MICA*008. Biochem J. (2013) 454:295–302. doi: 10.1042/BJ20130194
56. Chen D, Juko-Pecirep I, Hammer J, Ivansson E, Enroth S, Gustavsson I, et al. Genome-wide association study of susceptibility loci for cervical cancer. J Natl Cancer Inst. (2013) 105:624–33. doi: 10.1093/jnci/djt051
57. Nishioka Y, Shindoh J, Inagaki Y, Gonoi W, Mitsui J, Abe H, et al. Host MICA polymorphism as a potential predictive marker in response to chemotherapy for colorectal liver metastases. Digest Dis. (2018) 36:437–45. doi: 10.1159/000490411
58. Zuo J, Willcox CR, Mohammed F, Davey M, Hunter S, Khan K, et al. A disease-linked ULBP6 polymorphism inhibits NKG2D-mediated target cell killing by enhancing the stability of NKG2D ligand binding. Sci Signal. (2017) 10:eaai8904. doi: 10.1126/scisignal.aai8904
59. Gasser S, Orsulic S, Brown EJ, Raulet DH. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature. (2005) 436:1186–90. doi: 10.1038/nature03884
60. Jung H, Hsiung B, Pestal K, Procyk E, Raulet DH. RAE-1 ligands for the NKG2D receptor are regulated by E2F transcription factors, which control cell cycle entry. J Exp Med. (2012) 209:2409–22. doi: 10.1084/jem.20120565
61. Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, et al. HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol. (2012) 188:2136–45. doi: 10.4049/jimmunol.1102237
62. Groh V, Bahram S, Bauer S, Herman A, Beauchamp M, Spies T. Cell stress-regulated human major histocompatibility complex class I gene expressed in gastrointestinal epithelium. Proc Natl Acad Sci USA. (1996) 93:12445–50. doi: 10.1073/pnas.93.22.12445
63. Venkataraman GM, Suciu D, Groh V, Boss JM, Spies T. Promoter region architecture and transcriptional regulation of the genes for the MHC class I-related chain A and B ligands of NKG2D. J Immunol. (2007) 178:961–9. doi: 10.4049/jimmunol.178.2.961
64. Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. (2004) 21:137–48. doi: 10.1016/j.immuni.2004.07.017
65. O'Sullivan T, Dunn GP, Lacoursiere DY, Schreiber RD, Bui JD. Cancer immunoediting of the NK group 2D ligand H60a. J Immunol. (2011) 187:3538–45. doi: 10.4049/jimmunol.1100413
66. Obiedat A, Seidel E, Mahameed M, Berhani O, Tsukerman P, Voutetakis K, et al. Transcription of the NKG2D ligand MICA is suppressed by the IRE1/XBP1 pathway of the unfolded protein response through the regulation of E2F1. FASEB J. (2019) 33:3481–95. doi: 10.1096/fj.201801350RR
67. Fuertes MB, Girart MV, Molinero LL, Domaica CI, Rossi LE, Barrio MM, et al. Intracellular retention of the NKG2D ligand MHC class I chain-related gene A in human melanomas confers immune privilege and prevents NK cell-mediated cytotoxicity. J Immunol. (2008) 180:4606–14. doi: 10.4049/jimmunol.180.7.4606
68. Mellergaard M, Skovbakke SL, Schneider CL, Lauridsen F, Andresen L, Jensen H, et al. N-glycosylation of asparagine 8 regulates surface expression of major histocompatibility complex class I chain-related protein A (MICA) alleles dependent on threonine 24. J Biol Chem. (2014) 289:20078–91. doi: 10.1074/jbc.M114.573238
69. Yamada N, Kato-Kogoe N, Yamanegi K, Nishiura H, Fujihara Y, Fukunishi S, et al. Inhibition of asparagine-linked glycosylation participates in hypoxia-induced down-regulation of cell-surface MICA expression. Anticancer Res. (2018) 38:1353–9. doi: 10.21873/anticanres.12358
70. Boissel N, Rea D, Tieng V, Dulphy N, Brun M, Cayuela JM, et al. BCR/ABL oncogene directly controls MHC class I chain-related molecule A expression in chronic myelogenous leukemia. J Immunol. (2006) 176:5108–16. doi: 10.4049/jimmunol.176.8.5108
71. Cebo C, Da Rocha S, Wittnebel S, Turhan AG, Abdelali J, Caillat-Zucman S, et al. The decreased susceptibility of Bcr/Abl targets to NK cell-mediated lysis in response to imatinib mesylate involves modulation of NKG2D ligands, GM1 expression, and synapse formation. J Immunol. (2006) 176:864–72. doi: 10.4049/jimmunol.176.2.864
72. Kato N, Tanaka J, Sugita J, Toubai T, Miura Y, Ibata M, et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia. (2007) 21:2103–8. doi: 10.1038/sj.leu.2404862
73. Lopez-Soto A, Folgueras AR, Seto E, Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. (2009) 28:2370–82. doi: 10.1038/onc.2009.117
74. Eisele G, Wischhusen J, Mittelbronn M, Meyermann R, Waldhauer I, Steinle A, et al. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain. (2006) 129:2416–25. doi: 10.1093/brain/awl205
75. Serrano AE, Menares-Castillo E, Garrido-Tapia M, Ribeiro CH, Hernandez CJ, Mendoza-Naranjo A, et al. Interleukin 10 decreases MICA expression on melanoma cell surface. Immunol Cell Biol. (2011) 89:447–57. doi: 10.1038/icb.2010.100
76. Schmiedel D, Mandelboim O. NKG2D ligands-critical targets for cancer immune escape and therapy. Front Immunol. (2018) 9:2040. doi: 10.3389/fimmu.2018.02040
77. Chitadze G, Bhat J, Lettau M, Janssen O, Kabelitz D. Generation of soluble NKG2D ligands: proteolytic cleavage, exosome secretion and functional implications. Scand J Immunol. (2013) 78:120–9. doi: 10.1111/sji.12072
78. Chitadze G, Lettau M, Bhat J, Wesch D, Steinle A, Furst D, et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: heterogeneous involvement of the “a disintegrin and metalloproteases” 10 and 17. Int J Cancer. (2013) 133:1557–66. doi: 10.1002/ijc.28174
79. Zhao YK, Jia CM, Yuan GJ, Liu W, Qiu Y, Zhu QG. Expression and clinical value of the soluble major histocompatibility complex class I-related chain A molecule in the serum of patients with renal tumors. Genet Mol Res. (2015) 14:7233–40. doi: 10.4238/2015.June.29.16
80. Wu JD, Higgins LM, Steinle A, Cosman D, Haugk K, Plymate SR. Prevalent expression of the immunostimulatory MHC class I chain-related molecule is counteracted by shedding in prostate cancer. J Clin Invest. (2004) 114:560–8. doi: 10.1172/JCI200422206
81. Hilpert J, Grosse-Hovest L, Grunebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol. (2012) 189:1360–71. doi: 10.4049/jimmunol.1200796
82. Zhao Y, Chen N, Yu Y, Zhou L, Niu C, Liu Y, et al. Prognostic value of MICA/B in cancers: a systematic review and meta-analysis. Oncotarget. (2017) 8:96384–95. doi: 10.18632/oncotarget.21466
83. Maccalli C, Giannarelli D, Chiarucci C, Cutaia O, Giacobini G, Hendrickx W, et al. Soluble NKG2D ligands are biomarkers associated with the clinical outcome to immune checkpoint blockade therapy of metastatic melanoma patients. Oncoimmunology. (2017) 6:e1323618. doi: 10.1080/2162402X.2017.1323618
84. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, et al. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. (2005) 65:6321–9. doi: 10.1158/0008-5472.CAN-04-4252
85. Diermayr S, Himmelreich H, Durovic B, Mathys-Schneeberger A, Siegler U, Langenkamp U, et al. NKG2D ligand expression in AML increases in response to HDAC inhibitor valproic acid and contributes to allorecognition by NK-cell lines with single KIR-HLA class I specificities. Blood. (2008) 111:1428–36. doi: 10.1182/blood-2007-07-101311
86. Buddingh EP, Schilham MW, Ruslan SE, Berghuis D, Szuhai K, Suurmond J, et al. Chemotherapy-resistant osteosarcoma is highly susceptible to IL-15-activated allogeneic and autologous NK cells. Cancer Immunol Immunother. (2011) 60:575–86. doi: 10.1007/s00262-010-0965-3
87. Fine JH, Chen P, Mesci A, Allan DS, Gasser S, Raulet DH, et al. Chemotherapy-induced genotoxic stress promotes sensitivity to natural killer cell cytotoxicity by enabling missing-self recognition. Cancer Res. (2010) 70:7102–13. doi: 10.1158/0008-5472.CAN-10-1316
88. Hervieu A, Rebe C, Vegran F, Chalmin F, Bruchard M, Vabres P, et al. Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth. J Invest Dermatol. (2013) 133:499–508. doi: 10.1038/jid.2012.273
89. Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, et al. ATM-ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood. (2009) 113:3503–11. doi: 10.1182/blood-2008-08-173914
90. Vales-Gomez M, Chisholm SE, Cassady-Cain RL, Roda-Navarro P, Reyburn HT. Selective induction of expression of a ligand for the NKG2D receptor by proteasome inhibitors. Cancer Res. (2008) 68:1546–54. doi: 10.1158/0008-5472.CAN-07-2973
91. Arai J, Goto K, Tanoue Y, Ito S, Muroyama R, Matsubara Y, et al. Enzymatic inhibition of MICA sheddase ADAM17 by lomofungin in hepatocellular carcinoma cells. Int J Cancer. (2018) 143:2575–83. doi: 10.1002/ijc.31615
92. Huang B, Sikorski R, Sampath P, Thorne SH. Modulation of NKG2D-ligand cell surface expression enhances immune cell therapy of cancer. J Immunother. (2011) 34:289–96. doi: 10.1097/CJI.0b013e31820e1b0d
93. Shiraishi K, Mimura K, Kua LF, Koh V, Siang LK, Nakajima S, et al. Inhibition of MMP activity can restore NKG2D ligand expression in gastric cancer, leading to improved NK cell susceptibility. J Gastroenterol. (2016) 51:1101–11. doi: 10.1007/s00535-016-1197-x
94. Ferrari de Andrade L, Tay RE, Pan D, Luoma AM, Ito Y, Badrinath S, et al. Antibody-mediated inhibition of MICA and MICB shedding promotes NK cell-driven tumor immunity. Science. (2018) 359:1537–42. doi: 10.1126/science.aao0505
95. He S, Yin T, Li D, Gao X, Wan Y, Ma X, et al. Enhanced interaction between natural killer cells and lung cancer cells: involvement in gefitinib-mediated immunoregulation. J Transl Med. (2013) 11:186. doi: 10.1186/1479-5876-11-186
96. Frazao A, Colombo M, Fourmentraux-Neves E, Messaoudene M, Rusakiewicz S, Zitvogel L, et al. Shifting the balance of activating and inhibitory natural killer receptor ligands on BRAF(V600E) melanoma lines with vemurafenib. Cancer Immunol Res. (2017) 5:582–93. doi: 10.1158/2326-6066.CIR-16-0380
97. Lopez-Cobo S, Pieper N, Campos-Silva C, Garcia-Cuesta EM, Reyburn HT, Paschen A, et al. Impaired NK cell recognition of vemurafenib-treated melanoma cells is overcome by simultaneous application of histone deacetylase inhibitors. Oncoimmunology. (2018) 7:e1392426. doi: 10.1080/2162402X.2017.1392426
98. Borchers MT, Harris NL, Wesselkamper SC, Vitucci M, Cosman D. NKG2D ligands are expressed on stressed human airway epithelial cells. Am J Physiol. Lung Cell Mol Physiol. (2006) 291:L222–31. doi: 10.1152/ajplung.00327.2005
99. Ogbomo H, Michaelis M, Klassert D, Doerr HW, Cinatl J Jr. Resistance to cytarabine induces the up-regulation of NKG2D ligands and enhances natural killer cell lysis of leukemic cells. Neoplasia. (2008) 10:1402–10. doi: 10.1593/neo.08972
100. Huntington JT, Shields JM, Der CJ, Wyatt CA, Benbow U, Slingluff CL Jr, et al. Overexpression of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive melanoma cells: role of BRAF mutation and fibroblast growth factor signaling. J Biol Chem. (2004) 279:33168–76. doi: 10.1074/jbc.M405102200
101. Lu H, Liu S, Zhang G, Kwong LN, Zhu Y, Miller JP, et al. Oncogenic BRAF-mediated melanoma cell invasion. Cell Rep. (2016) 15:2012–24. doi: 10.1016/j.celrep.2016.04.073
102. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. (2013) 3:388–98. doi: 10.1158/2159-8290.CD-12-0548
103. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. (2013) 5:177ra138. doi: 10.1126/scitranslmed.3005930
104. Salter AI, Pont MJ, Riddell SR. Chimeric antigen receptor-modified T cells: CD19 and the road beyond. Blood. (2018) 131:2621–9. doi: 10.1182/blood-2018-01-785840
105. Daniyan AF, Brentjens RJ. At the bench: chimeric antigen receptor (CAR) T cell therapy for the treatment of B cell malignancies. J Leukoc Biol. (2016) 100:1255–64. doi: 10.1189/jlb.5BT1215-556RR
106. Watanabe K, Kuramitsu S, Posey AD Jr, June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol. (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
107. Bollino D, Webb TJ. Chimeric antigen receptor-engineered natural killer and natural killer T cells for cancer immunotherapy. Transl Res. (2017) 187:32–43. doi: 10.1016/j.trsl.2017.06.003
108. Zhang Q, Tian K, Xu J, Zhang H, Li L, Fu Q, et al. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res. (2017) 2017:6915912. doi: 10.1155/2017/6915912
109. Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res. (2018) 2018:4263520. doi: 10.1155/2018/4263520
110. Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. (2018) 51:146–53. doi: 10.1016/j.coi.2018.03.013
111. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. (2018) 23:181–92 e185. doi: 10.1016/j.stem.2018.06.002
112. Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. (2006) 66:5927–33. doi: 10.1158/0008-5472.CAN-06-0130
113. Zhang T, Barber A, Sentman CL. Chimeric NKG2D modified T cells inhibit systemic T-cell lymphoma growth in a manner involving multiple cytokines and cytotoxic pathways. Cancer Res. (2007) 67:11029–36. doi: 10.1158/0008-5472.CAN-07-2251
114. Zhang T, Sentman CL. Cancer immunotherapy using a bispecific NK receptor fusion protein that engages both T cells and tumor cells. Cancer Res. (2011) 71:2066–76. doi: 10.1158/0008-5472.CAN-10-3200
115. Eagle RA, Jafferji I, Barrow AD. Beyond stressed self: evidence for NKG2D ligand expression on healthy cells. Curr Immunol Rev. (2009) 5:22–34. doi: 10.2174/157339509787314369
116. Sentman ML, Murad JM, Cook WJ, Wu MR, Reder J, Baumeister SH, et al. Mechanisms of acute toxicity in NKG2D chimeric antigen receptor T cell-treated mice. J Immunol. (2016) 197:4674–85. doi: 10.4049/jimmunol.1600769
117. Stojanovic A, Correia MP, Cerwenka A. The NKG2D/NKG2DL axis in the crosstalk between lymphoid and myeloid cells in health and disease. Front Immunol. (2018) 9:827. doi: 10.3389/fimmu.2018.00827
Keywords: NKG2D, natural killer cells, NKG2D ligands, tumor immunosurveillance, NKG2D CARs
Citation: Frazao A, Rethacker L, Messaoudene M, Avril M-F, Toubert A, Dulphy N and Caignard A (2019) NKG2D/NKG2-Ligand Pathway Offers New Opportunities in Cancer Treatment. Front. Immunol. 10:661. doi: 10.3389/fimmu.2019.00661
Received: 07 December 2018; Accepted: 11 March 2019;
Published: 29 March 2019.
Edited by:
Nadia Guerra, Imperial College London, United KingdomReviewed by:
Alessandra Zingoni, Sapienza University of Rome, ItalyCopyright © 2019 Frazao, Rethacker, Messaoudene, Avril, Toubert, Dulphy and Caignard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne Caignard, YW5uZS5jYWlnbmFyZEBpbnNlcm0uZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.