 Renata Curciarello
Renata Curciarello Karina Eva Canziani
Karina Eva Canziani Guillermo Horacio Docena
Guillermo Horacio Docena Cecilia Isabel Muglia
Cecilia Isabel Muglia- 1Instituto de Estudios Inmunológicos y Fisiopatológicos, CONICET, Facultad de Ciencias Exactas, Universidad Nacional de La Plata, La Plata, Argentina
- 2Departamento de Ciencias Biológicas, Facultad de Ciencias Exactas, Universidad Nacional de La Plata, La Plata, Argentina
The mucosal immune system constitutes a physical and dynamic barrier against foreign antigens and pathogens and exerts control mechanisms to maintain intestinal tolerance to the microbiota and food antigens. Chronic alterations of the intestinal homeostasis predispose to inflammatory diseases of the gastrointestinal tract, such as Inflammatory Bowel Diseases (IBD). There is growing evidence that the frequency and severity of these diseases are increasing worldwide, which may be probably due to changes in environmental factors. Several stromal and immune cells are involved in this delicate equilibrium that dictates homeostasis. In this review we aimed to summarize the role of epithelial cells and fibroblasts in the induction of mucosal inflammation in the context of IBD. It has been extensively described that environmental factors are key players in this process, and the microbiome of the gastrointestinal tract is currently being intensively investigated due to its profound impact the immune response. Recent findings have demonstrated the interplay between dietary and environmental components, the gut microbiome, and immune cells. “Western” dietary patterns, such as high caloric diets, and pollution can induce alterations in the gut microbiome that in turn affect the intestinal and systemic homeostasis. Here we summarize current knowledge on the influence of dietary components and air particulate matters on gut microbiome composition, and the impact on stromal and immune cells, with a particular focus on promoting local inflammation.
Gut Inflammation
Inflammation is a central component of innate immunity, comprising the physiopathological response to infection or tissue damage. As a local response to cellular injury, it is initiated when tissue-resident cells of the innate immune system detect the damaging insult and alarm resident cells and circulating neutrophils. These cells migrate to the inflamed tissue, promote recruitment of inflammatory monocytes and potentiate the pro-inflammatory environment, allowing to deal with the harmful agent (1). Hence, the acute inflammatory response is marked by increased blood flow, capillary dilatation, leukocyte infiltration, and the localized production of chemical mediators, which serves to initiate the elimination of toxic agents and the repair of the damaged tissue. Hence, the acute inflammatory response is a physiological process committed to control an offending stimulus.
The intestinal mucosa has evolved as a well-structured barrier against physical, chemical, and microbial insults. The epithelial layer, mucus, antimicrobial peptides, secreted immunoglobulin A, and innate and adaptive immune cells, together help to establish a beneficial environment to tolerate the diverse community of microbes of the microbiota and food antigens. The intestinal mucosal surface constitutes the major interface between the internal tissues and a potentially hostile outer environment. To deal with this universe of antigenic components, intestinal homeostasis has been evolutionary developed through a constant crosstalk between metabolites and microbes of the microbiota, intestinal stromal cells, and the mucosal immune system. Perturbations of the homeostatic state can result in severe inflammatory conditions in the gut that may lead to tissue damage. Therefore, intestinal inflammation is a double-edge sword that should be tightly regulated. Although it is an essential component for immunosurveillance and host defense, chronic inflammatory processes may promote pathology such as inflammatory bowel diseases (IBD) (2), irritable bowel syndrome (IBS) (3), diverticular disease (4), food allergy (5), celiac disease (4), etc.
In this homeostatic scenario, the controlled and physiological inflammation of the gut promotes a barrier permeability that allows the penetration of luminal antigens to the underlying mucosal tissue. In chronic inflammatory disorders, such as IBD, microbial components of the microbiota are translocated through the damaged mucosal barrier, and trigger and maintain a sustained inflammatory response, as it is represented in Figure 1. Epithelial cells, dendritic cells (DCs), macrophages, and innate lymphocytes (ILCs), which sense the presence of microbes or an altered tissue environment, are activated and promote the induction of the adaptive immune response. As it is summarized in Figure 1, the pro-inflammatory cytokines secreted by innate cells and activated T cells are key to amplify and perpetuate mucosal inflammation. Therefore, the mucosal immune system is responsible for the induction of the inflammatory process, while tissue damage results from continuous activation and differentiation of local cells, such as myofibroblasts, that release noxious mediators (2, 6).
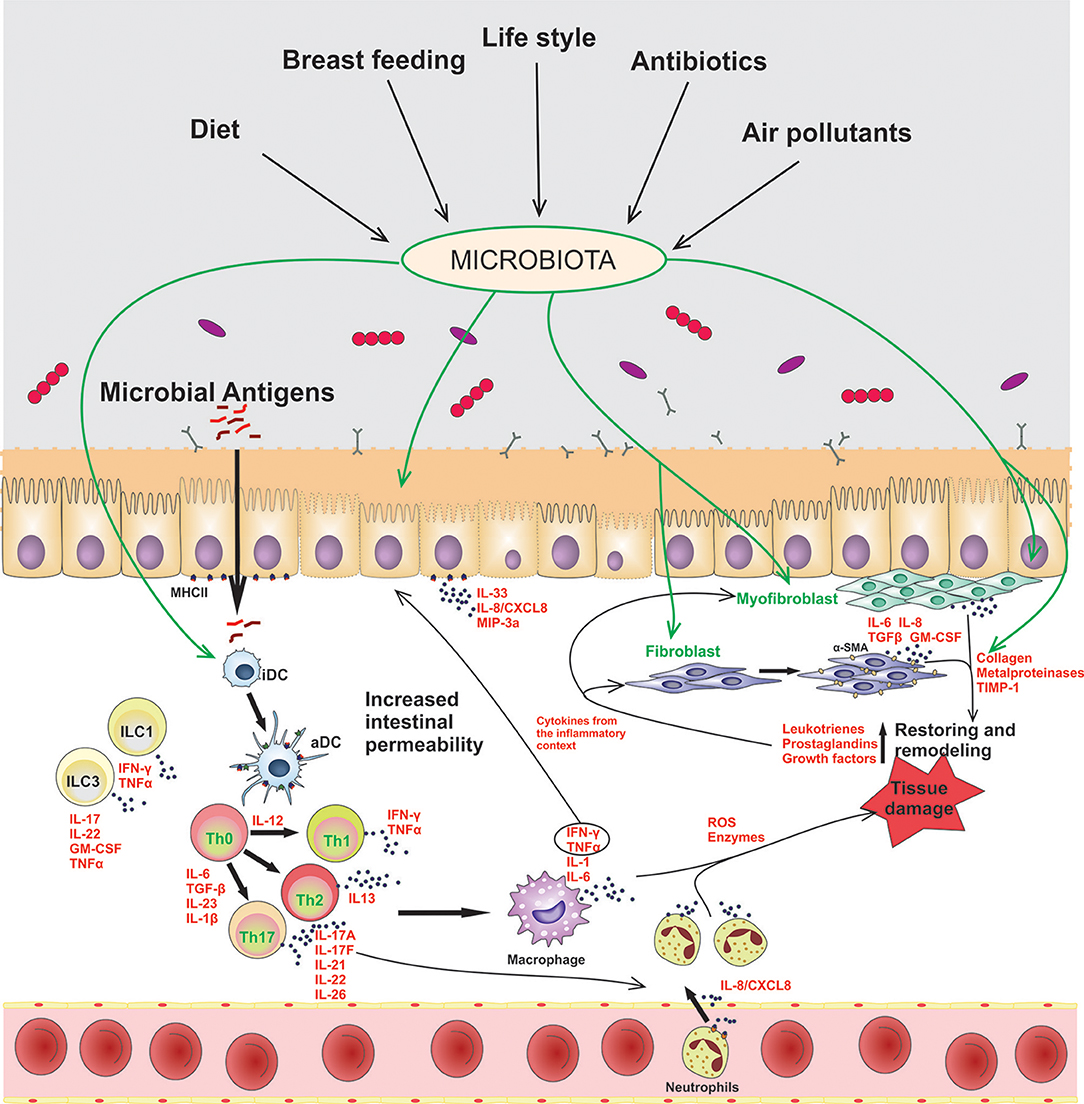
Figure 1. Contribution of epithelial cells and intestinal fibroblasts to the induction and modulation of mucosal inflammation in IBD. In this disorder the intestinal barrier permeability is impaired, allowing the passage of luminal antigens into the lamina propria. The exposure of stromal and immune cells to the luminal content induces cell activation, inflammatory soluble-mediators release (IFN-γ, TNF-α, IL-1β, IL-6, IL-17A), cell-crosstalk (represented with arrows) and neutrophil recruitment to the inflamed tissue. Several environmental factors can modulate the microbiota composition and the activation of stromal and immune cells in the gut. iDC, innactivated dendritic cells; aDC, activated dendritic cells; ILCs, innate lymphoid cells; Th, T helper cells.
Non-immune Cells Involved in Gut Inflammation
Epithelial Cells
The alteration of the intestinal epithelial barrier permeability leads to local inflammation. The fact that mice expressing dominant negative N-cadherin adherent junction protein suffer from spontaneous inflammatory bowel disease, clearly highlights the fundamental role of barrier integrity in the development of colitis and, probably, IBD (7). Many cytokines that are increased in IBD favor gut permeability, such as TNF-α. Indeed, impaired barrier function has been described for Crohn's disease (CD) and ulcerative colitis (UC) patients and it is a marker for predicting the course of these relapsing diseases (8, 9). Intestinal pro-inflammatory cytokines have been documented to modify the expression of different tight junction proteins. Claudins showed deregulation and differential expression in human active IBD (8, 9). However, studies in mice null for claudins and other tight junction proteins were controversial, demonstrating that other factors may contribute to gut permeability (10). Overall, these reported evidences highlight the relevance of epithelial cells in intestinal inflammation. Amongst these, Paneth cells secrete antimicrobial peptides that modulate the gut microbial composition (11), and it has been recently described that metabolites produced by luminal microbes control the secretion of these peptides (12). In other words, there is a mutual monitoring of the microbiota composition and the mucosal immune system. Defects in defensin production have been found in patients with CD and NOD-2 mutations (13). Taking into account the role of the microbiome in intestinal homeostasis, these findings should not be underestimated. Recently, a study involving Paneth cell deficient mice showed that they develop dysbiosis and visceral hypersensitivity (14). Other relevant epithelial cell type are Goblet cells, responsible for secretion of mucus and sampling of luminal antigens (15, 16). These cells showed a protective role in gut inflammation since MUC2-null mice developed spontaneous colitis (17), and patients with UC showed polymorphism in MUC2 in the Dutch population (18). Goblet cell loss and decreased mucous levels are commonly observed in UC patients, with endoplasmic reticulum stress and accumulation of MUC2 precursors (17, 19). In addition, micro-fold cells, or M cells, found in the follicle-associated epithelium of the gut, have a key function in the immunosurveillance of the luminal content (16, 20, 21). There is no report showing M cell dysfunction in colitis or any other inflammatory condition. Enteroendocrine cells, which are also sensors of gut luminal content, were found to be altered in mouse models of colitis and in patients with active IBD (22).
The most abundant cells in the epithelial compartment are the absorptive cells, which not only constitute a physical barrier against luminal antigens, but mediate the crosstalk between intestinal microbiome and the host immune system, mainly through innate immune receptors. Conserved molecular patterns are mainly recognized by Toll-like receptors (TLRs) and Nod-like receptors (NLR), among others, which are expressed along the intestinal tract (23–26). Healthy human small intestine expresses TLR-2 and−4 proteins, whereas high levels of TLR-5 are found in the colon. These receptors show a polarized distribution, being located in the basolateral membranes, ensuring that commensal bacteria do not trigger an inflammatory response in homeostasis (27). These particular expression patterns of innate receptors are modified in an inflammatory setting. IBD patients have increased levels of TLR4 expression, and lower level of TLR2 and TLR5 in epithelial cells, while TLR4 was shown to be expressed in the apical surface of epithelial cells (28). Current studies have demonstrated the importance of TLR1 in the prevention of gut inflammation (29). In addition to NOD-2 mutations, abnormal mucosal NLRP3 activity has been reported in IBD and in experimental colitis; GWAS studies revealed polymorphisms in these receptors (30–33).
Intestinal epithelial cells (IECs) also contribute to intestinal homeostasis through interaction with microbiota and secretion of TGF-β and IL-10 (34, 35). Although IECs crosstalk with T cells through cell-cell interactions (36), the role of these cells as antigen presenting cells in vivo is controversial (37). They express class 2 MHC, but no co-stimulatory molecules, though they do express the ICOS-L; of note, variants in this gene have been related to the IBD early onset clinical entity (38). Colonic epithelial cell isolated from active IBD patients have shown to secrete the neutrophil-attracting IL-8/CXCL8 (39, 40) and IL-33 (41–44), which contribute to inflammation (Figure 1). IL-1β, produced through activation of the inflammasome, and IL-17 are also secreted (45, 46), contributing to Th1 and Th17 responses in IBD. IL-21R expression is up-regulated in IECs of IBD patients, which leads to increased CCL20 synthesis, a T cell and DC chemo attractant (47, 48). IL-22, IL-31, and IL-33 have been also described to be augmented in IBD, which leads to deregulation of IEC proliferation and migration functions, whereas they stimulate IL-1, TNF-α, IL-6, and IL-8 secretion (49–51). Cytokines generated by innate and adaptive responses, on the other hand, interact with IECs, activating them and altering barrier permeability. It is well known that TNF-α induces IECs apoptosis (52) and that IFN-γ alters epithelial permeability by affecting tight junctions and bacterial translocation (53), as represented in Figure 1. All these factors contribute to immune cell activation and cytokine secretion which, in the context of IBD, favor an inflammation and damage perpetuation.
In conclusion, IECs are relevant cell sources of pro-inflammatory and regulatory mediators, which should be tightly controlled to achieve intestinal homeostasis.
Fibroblasts
Intestinal myofibroblasts are localized beneath the epithelial compartment, particularly around the crypts of the small intestine and the colon, and participate in the repair process. Fibroblasts, which are involved in formation of the extracellular matrix (ECM), differentiate into contractile myofibroblasts, which are also involved in the inflammatory response to injury through the secretion of cytokines (54–56). Furthermore, the origin of myofibroblasts in chronic inflammation is not clear, and they may have an epithelial origin through an epithelial-mesenchymal transition (57).
Myofibroblast activation is associated with tissue injury and inflammation (Figure 1). They have the ability to migrate to the injured sites, where they contract the wound area and produce extracellular matrix components that restore and remodel the damaged mucosal tissue. However, persistent myofibroblast activation in an inflammatory environment may promote an irreversible damage of the affected tissue with fibrosis and cell proliferation in the submucosa (58). Cells can secrete a number of soluble cytokines (IL-6), chemokines (IL-8, MCP-1), growth factors (TGF-β, GM-CSF), collagen, metalloproteinases (MMP-1, MMP-3, MMP-9, MMP-12), and MMPs' inhibitor (TIMP-1), that attract other cells to perpetuate the inflammation. Depending on the balance of MMPs and TIMP-1, and collagen deposition, it will be the consequence of tissue remodeling in IBD, thus resulting in fibrosis, stricture formation or ulceration (59–62). It is unknown why in some cases the intestinal inflammation induces penetrating damage with perforation and fistulae, and with increased risk of colorectal cancer in UC patients, whereas in CD patients the long-term complications include abscesses, granuloma, strictures, obstruction, fibrosis, and stenosis. Overall, the control of myofibroblast differentiation is critical to prevent or reverse complications in CD and UC patients.
Inflammation Is Modulated by Different Factors
It has been previously described that the immune system is regulated at different points for homeostasis, while aberrant or inappropriate regulation may result in failure to protect the body from pathogens or any injury.
Environmental factors influence the incidence and development of IBD in many ways that are not fully understood, with a higher incidence in developed countries and urban populations, compared to rural areas and underdeveloped regions of the world. It has been shown that young migrants from low incidence countries have a similar incidence of IBD compared to non-immigrants, highlighting the importance of the environment in these diseases (63). Several studies discuss associations of different dietary components in gut inflammation and IBD. High meat/fat (64) consumption has been linked to a higher risk of IBD onset, and studies in animal models correlate this finding with the high heme intake (65). Also, trans- and poly-unsaturated long-chain fatty acids have been related to the disease (66, 67), while unsaturated fatty acids seem to exert a preventive role (67). Emulsifiers, which are commonly found in processed foods, have a pro-inflammatory effect in the gut (68). On the other hand, dietary fiber, especially from fruits, has been associated to a lower IBD incidence (69). Also, vitamin consumption is thought to be beneficial (70). These results should now be reevaluated after the recently published study in which the effect of different dietary ingredients was addressed in the context of IBD, using experimental colitis models in pathogen-free and germ-free mice in order to identify specific triggers (71).
More recently, the microbiota has been targeted as a critical player in establishing and sustaining the tight equilibrium of the immune system that is constantly exposed to a myriad of antigens in the mucosal surfaces. The intestinal microbiota is key for maintaining intestinal homeostasis, and it may be involved in inflammatory disorders when the composition and diversity is modified, which is called dysbiosis. In all mucosa an associated microbiota has been described and it is known that the gut microbiota might be the most complex and dynamic one. It is established during the intrauterine life and modified after birth. Several factors have been described as contributing to condition the composition of the microbiota throughout life. There is a link between diet, gut composition and gut metabolism which undoubtedly impact on the gastrointestinal health. There is growing evidence that a dysbiotic intestinal microbiota is associated with immune and non-immune disorders. However, it is debatable whether dysbiosis is a cause or a consequence of the inflammatory process (72, 73). Commonly, dysbiosis implies a change from a diverse anaerobe community, rich in Firmicutes and Bacteroidetes to a lower diversity community with enrichment of facultative anaerobes including Proteobacteria and Bacilli, although it depends on the pathology (74). There is broad consensus that pathology-associated dysbiosis is accompanied with a restriction in the diversity of species (75).
Microbiota composition may be affected by different external factors: diet (fiber, calories, etc.), urbanization, use of antibiotics, age, mode of birth, exposure to air pollutants, etc. (76–78). A westernized diet with high red meat-high fat and processed carbohydrates content, is associated with a loss in gut microbial diversity, with an increase of pathogenic adherent-invasive E. coli (AIEC), as it has been reported in IBD patients (79). It has been demonstrated that a high-fat/high-sugar diet leads to dysbiosis with increased Bacteroides spp and Ruminococcus torques in mice (79). On the other hand, digestible fibers, which are fermented by bacteria in the gut, generate beneficial, anti-inflammatory short-chain fatty acids (SCFA). Modern diet components such as artificial sweeteners and emulsifiers are being subjected to investigation as they are suspected to induce dysbiosis (80–82). Human and murine studies have demonstrated that the use of antibiotics generates variations in gut microbiota mostly according to the type and the period of time used (78). Antibiotics directed to anaerobes, such as vancomicin, seem to have a more severe impact on gut microbiota composition (76). The contamination of food with particulate matter, as occurs in a contaminated environment, has shown to impair intestinal permeability and to generate inflammation, altering microbial gut composition (77, 83).
There are several evidences that highlight the relevance of the microbiota in IBD: germ-free animals do not develop experimental colitis (84), and antibiotic therapy has been successful in the treatment of CD, while it has given promising results in some forms of UC (84), etc. Dysbiosis has been well characterized in CD, with a decrease in Clostridiales and an increase in Enterobacteriales (85), while a diminished, but less defined, microbial diversity has been described for UC (85). Emerging insights on intestinal dysbiosis during immune and non-immune disorders has attracted the attention to target the microbiota composition as a novel therapeutic approach to control intestinal and extra-intestinal inflammation. However, it should be considered that the microbiota-derived metabolites are the true messengers that control the development, differentiation, and activity of the immune system associated to the local, and also, distant mucosa. Microbiota transfer strategies, or fecal microbiota transplant (FMT), have been used in Chinese ancestral medicine for centuries and have proven to be useful for Clostridium difficile recurrent infections (86). Several clinical trials have demonstrated that FMT restores gut microbial diversity, diminishing disbiosis and controlling mucosal inflammation. More recently, FMT has been explored in IBD (87). Most clinical trials and randomized controlled studies have been made in UC, with promising results when they were performed early in the disease course (88). Results in CD have been variable and rigorous randomized controlled trials are needed (89). More studies are mandatory to confirm the beneficial effect of this therapy.
Another therapeutic approach aimed to modify the gut microbiome has been the use of probiotics. Numerous studies using animal models (90–93) and in vitro approaches (94–96) provide evidence on the beneficial use of probiotics in colitis. Results depend on the specific strain of probiotics used, and vary with the experimental model used. In particular, Bifidobacterium and Lactobacillus are the most widely used probiotic bacteria (97), although strains of E. coli (98–101), Propionibacterium (102–105), Bacillus (106–108), and Saccharomyces (109, 110) are amongst those mostly studied.
Different mechanisms of action have been described for probiotics, including binding to IECs and thus preventing the binding of pathogenic microorganisms (109, 110), acidification of the lumen of the colon by nutrient fermentation, production of SCFA like acetate, propionate, and butyric acid as a source of energy, but also with immune modulating properties and anti-inflammatory effects (109, 110), as enhancement of epithelial barrier integrity (97), etc. Mack et al. (111) described an increased production of MUC2 and MUC3 secretion by epithelial cells stimulated with Lactobacillus plantarum and Lactobacillus rhamnosus in HT-29 intestinal cell line, while Anderson et al. described that L. plantarum MB452 increased the trans-epithelial electrical resistance of Caco-2 cells monolayer through the induction of tight junction proteins (112). Recently, components of L. amylovorus DSM 16698 cell wall demonstrated to have protective effects toward E. coli induced damage on Caco-2/TC7 cells, protecting membrane leakage and reducing the phosphorylation of the p65 component of the NF-κB intracellular signaling pathway (113). The inhibition of NF-κB activation has also been described for other Lactobacilli strains and for Bifidobacterium (114–116). The increased expression of tight junction proteins is linked to an activation of the ERK and p38 MAPK signaling cascades, via Toll like receptors (114). Probiotics have also been described for preventing IECs apoptosis by reducing oxidative stress, as in the case of L. amylovorous DSM16698 (117), and to inhibit apoptosis through the activation of the epidermal growth factor receptor by the release of a soluble protein known as p40 by L. rhamnosus GG (114, 118). In addition, probiotics act by diminishing pro-inflammatory responses and contributing to tolerogenic responses, modulating TLR-2 and TLR-4 signaling (114), and driving DCs to a suppressive phenotype, which further promote the generation of Tregs (119, 120).
It has been demonstrated in the inflamed gut, that the mucosal damage not only affects the epithelial compartment, but also colonic myofibroblasts located beneath the epithelium. It has been reported that the exposure to colonic microbiota products promotes cell activation through TLR (121). Beswick and col. demonstrated that isolated myofibroblasts from normal human colonic mucosa respond to TLR4 stimulation by LPS with the induction of PD-L1, which mediates the suppression of activated CD4+ PD-1+ T cell response and inhibition of IFN-γ secretion in vitro. Authors also showed that in vivo up-regulation of PD-L1 in colonic myofibroblast is MyD88-dependent in colitis (122). This provides evidence that colonic myofibroblasts might help to maintain the equilibrium between tolerance and immunity to protect the colonic mucosa against inflammatory responses toward the microbiota. In agreement with these findings, Scheibe and col. reported that IL-36R ligands released upon mucosal damage activate IL-36R+ colonic fibroblasts via MyD88, thereby inducing expression of chemokines, granulocyte-macrophage colony-stimulating factor and IL-6. These mediators induce the migration and recruitment of leukocytes and neutrophils to the inflamed colon and contribute to control mucosal healing (123).
On the other hand, dysbiosis and specific bacterial taxa correlated with fibrostenosis in a CD cohort study (32), although there are no studies showing the contribution of the microbiome to fibrosis and fibroblasts modulation in the gut. It has recently been published the first evidence that fibroblast activation and the intestinal fibrosis require specific microbial cues provided by the mouse microbiota. Authors reported that intestinal fibrosis is microbiota-dependent, by giving gavages of feces from germ-free mice, pathogen-free feces or healthy human donor feces, in TL1A-overexpressing mice. In addition, they found that the microbial composition affects fibroblast differentiation into myofibroblasts. Furthermore, they identified several candidates that correlated directly with the fibrosis degree in mice (123, 124). Although the key role of TL1A in inducing fibrostenosis is known (125, 126), the interplay between microbiota, TL1A and fibroblasts is novel (124).
Given that there are a number of chronic inflammatory disorders with fibrotic evolution, such as rheumatoid arthritis, cirrhosis, IBD, and pulmonary fibrosis, there is great effort to develop therapies that could control or reverse fibrosis. Particularly, intestinal fibrosis has been exclusively associated to IBD, mostly CD. The use of probiotics is becoming increasingly important for the prevention and treatment of gastrointestinal conditions. In a mouse model of DSS-induced colitis, administration of Lactobacillus acidophilus ameliorated collagen mucosal deposition and improved intestinal fibrosis. Indeed, L. acidophilus treated mice showed decreased α-SMA and collagen I expression levels, compared to untreated mice (127). Alternatively to probiotics, a role for Vitamin D in modulating the immune system and the integrity of intestinal epithelium and gut microbioma has also been proposed. In this sense, vitamin D supplementation improves IBD patient's condition (128) and regarding intestinal fibrosis, it was demonstrated that vitamin D exerted protective effects on colonic fibrosis caused by TNBS-induced chronic colitis, through direct inhibition of TGFβ-1/Smad3 pathway and up/regulation of vitamin D receptor in sub-epithelial myofibroblasts (129). More recently, the overexpression of fibroblasts activation protein (FAP), an inducible surface glycoprotein, has been associated with fibrosis in strictured CD patients. In vitro assays with anti-FAP treatment have shown promising results in controlling ECM deposition (130). Overall, despite the widely described contribution of fibroblasts to intestinal inflammation, the control of fibrosis in chronic intestinal inflammatory disorders still remains a big challenge for therapeutic purposes.
Conclusions
As the intestinal mucosa surface constitutes the major surface of the body which is in direct contact with the outer environment, intestinal immune homeostasis must be accurately regulated. The interplay between commensal microbiota, intestinal stromal cells, and the mucosal immune system components should guarantee the intestinal homeostasis to avoid a sustained inflammation that could induce tissue damage. However, several factors can lead to inflammation through homeostasis breakdown. Figure 1 summarizes the main points that have been reviewed here. We have described what it is known so far about the role of epithelial cells and intestinal fibroblasts in the induction and modulation of mucosal inflammation in IBD. In this chronic inflammatory disorders the intestinal barrier permeability is compromised and the selective passage of luminal antigens into the lamina propria is altered, triggering cell activation, and inflammation. A plethora of evidences demonstrate the impact of dietary and environmental factors on the gut microbioma and on the modulation of the intestinal immunity. Notwithstanding the efforts made to find alternatives to conventional anti-inflammatory treatments (steroids, antibiotics, immunosuppressive drugs and biologics) by modulation of non-immune cells response, no current evidences arise that support the replacement of conventional therapies. Regarding probiotics, for instance, it has recently been demonstrated that the use of probiotics to achieve effective mucosal protection should be personalized according to the individual affection (131–133).
In conclusion, in this review we summarized the most recent findings in animal models and cohort studies, that show the contribution of epithelial cells and fibroblasts to gut inflammation with the influence of different environmental and dietary factors. Considering that the frequency and severity of IBD are increasing worldwide, changes on environmental factors and dietary habits should not be underestimated. Based on these observations, and those regarding the modulation of the intestinal microbioma and mucosal immune cells, it has been prompted to develop novel therapeutic interventions to prevent, control or reverse gut inflammation.
Author Contributions
RC, CIM, KEC, and GHD conceptualized the review. RC and CIM provided an initial draft of the manuscript and KEC provided the figure. RC, CIM, and GHD performed the final editions.
Funding
RC, CIM, and GHD are researchers from CONICET. Fundings from Agencia Nacional de Promoción Científica were received for the research projects conducted by GHD (PICT2015-1648), CIM (PICT2016-0648), and RC (PICT2016-2387). KEC is Ph.D. fellow from CONICET.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Mantovani A, Cassatella MA, Costantini C, Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat Rev Immunol. (2011) 11:519–31. doi: 10.1038/nri3024
2. Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. (2007) 369:1627–40. doi: 10.1016/S0140-6736(07)60750-8
3. Choung RS, Locke GR. Epidemiology of IBS. Gastroenterol Clin North Am. (2011) 40:1–10. doi: 10.1016/j.gtc.2010.12.006
4. Strate LL, Modi R, Cohen E, Spiegel BMR. Diverticular disease as a chronic illness: evolving epidemiologic and clinical insights. Am J Gastroenterol. (2012) 107:1486–93. doi: 10.1038/ajg.2012.194
5. Tordesillas L, Berin MC, Sampson HA. Immunology of food allergy. Immunity. (2017) 47:32–50. doi: 10.1016/j.immuni.2017.07.004
6. MacDonald TT, Monteleone I, Fantini MC, Monteleone G. Regulation of homeostasis and inflammation in the intestine. Gastroenterology. (2011) 140:1768–75. doi: 10.1053/j.gastro.2011.02.047
7. Hermiston ML, Gordon JI. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science. (1995) 270:1203–7. doi: 10.1126/science.270.5239.1203
8. Zeissig S, Burgel N, Gunzel D, Richter J, Mankertz J, Wahnschaffe U, et al. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn's disease. Gut. (2007) 56:61–72. doi: 10.1136/gut.2006.094375
9. Martini E, Krug SM, Siegmund B, Neurath MF, Becker C. Mend your fences. Cell Mol Gastroenterol Hepatol. (2017) 4:33–46. doi: 10.1016/j.jcmgh.2017.03.007
10. Ahmad R, Sorrell MF, Batra SK, Dhawan P, Singh AB. Gut permeability and mucosal inflammation: bad, good or context dependent. Mucosal Immunol. (2017) 10:307–17. doi: 10.1038/mi.2016.128
11. Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjöberg J, Amir E, et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat Immunol. (2010) 11:76–83. doi: 10.1038/ni.1825
12. Schoenborn AA, von Furstenberg RJ, Valsaraj S, Hussain FS, Stein M, Shanahan MT, et al. The enteric microbiota regulates jejunal Paneth cell number and function without impacting intestinal stem cells. Gut Microbes. (2018) 8:1–32. doi: 10.1080/19490976.2018.1474321
13. Gassler N. Paneth cells in intestinal physiology and pathophysiology. World J Gastrointest Pathophysiol. (2017) 8:150–60. doi: 10.4291/wjgp.v8.i4.150
14. Riba A, Olier M, Lacroix-Lamandé S, Lencina C, Bacquié V, Harkat C, et al. Paneth cell defects induce microbiota dysbiosis in mice and promote visceral hypersensitivity. Gastroenterology. (2017) 153:1594–1606.e2. doi: 10.1053/j.gastro.2017.08.044
15. McGuckin MA, Lindén SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol. (2011) 9:265–78. doi: 10.1038/nrmicro2538
16. Knoop KA, McDonald KG, McCrate S, McDole JR, Newberry RD. Microbial sensing by goblet cells controls immune surveillance of luminal antigens in the colon. Mucosal Immunol. (2015) 8:198–210. doi: 10.1038/mi.2014.58
17. Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. Schreiber S, editor. PLoS Med. (2008) 5:e54. doi: 10.1371/journal.pmed.0050054
18. Visschedijk MC, Alberts R, Mucha S, Deelen P, de Jong DJ, Pierik M, et al. Pooled resequencing of 122 ulcerative colitis genes in a large dutch cohort suggests population-specific associations of rare variants in MUC2. Prokunina-Olsson L, editor. PLoS ONE. (2016) 11:e0159609. doi: 10.1371/journal.pone.0159609
19. Gersemann M, Becker S, Kübler I, Koslowski M, Wang G, Herrlinger KR, et al. Differences in goblet cell differentiation between Crohn's disease and ulcerative colitis. Differentiation. (2009) 77:84–94. doi: 10.1016/j.diff.2008.09.008
20. Tahoun A, Mahajan S, Paxton E, Malterer G, Donaldson DS, Wang D, et al. Salmonella transforms follicle-associated epithelial cells into M cells to promote intestinal invasion. Cell Host Microbe. (2012) 12:645–56. doi: 10.1016/j.chom.2012.10.009
21. Ohno H, Kanaya T, Williams IR. M cell differentiation: distinct lineage or phenotypic transition? Salmonella provides answers. Cell Host Microbe. (2012) 12:607–9. doi: 10.1016/j.chom.2012.11.003
22. Harrison E, Lal S, McLaughlin JT. Enteroendocrine cells in gastrointestinal pathophysiology.Curr Opin Pharmacol. (2013) 13(6):941–5. doi: 10.1016/j.coph.2013.09.012
23. Claes A-K, Zhou JY, Philpott DJ. NOD-like receptors: guardians of intestinal mucosal barriers. Physiology. (2015) 30:241–50. doi: 10.1152/physiol.00025.2014
24. Hug H, Mohajeri MH, La Fata G. Toll-like receptors: regulators of the immune response in the human gut. Nutrients. (2018) 10:203. doi: 10.3390/nu10020203
25. Latorre E, Layunta E, Grasa L, Pardo J, García S, Alcalde AI, et al. Toll-like receptors 2 and 4 modulate intestinal IL-10 differently in ileum and colon. United Eur Gastroenterol J. (2018) 6:446–53. doi: 10.1177/2050640617727180
26. Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat Rev Immunol. (2010) 10:131–44. doi: 10.1038/nri2707
27. Yu S, Gao N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell Mol Life Sci. (2015) 72:3343–53. doi: 10.1007/s00018-015-1931-1
28. Cario E. Toll-like receptors in inflammatory bowel diseases: a decade later. Inflamm Bowel Dis. (2010) 16:1583–97. doi: 10.1002/ibd.21282
29. Kamdar K, Johnson AMF, Chac D, Myers K, Kulur V, Truevillian K, et al. Innate recognition of the microbiota by TLR1 promotes epithelial homeostasis and prevents chronic inflammation. J Immunol. (2018) 201:230–42. doi: 10.4049/jimmunol.1701216
30. Hugot JP. CARD15/NOD2 mutations in Crohn's disease. Ann N Y Acad Sci. (2006) 1072:9–18. doi: 10.1196/annals.1326.011
31. Cummings JRF, Cooney RM, Clarke G, Beckly J, Geremia A, Pathan S, et al. The genetics of NOD-like receptors in Crohn's disease. Tissue Antigens. (2010) 76:48–56. doi: 10.1111/j.1399-0039.2010.01470.x
32. Kugathasan S, Denson LA, Walters TD, Kim M-O, Marigorta UM, Schirmer M, et al. Prediction of complicated disease course for children newly diagnosed with Crohn's disease: a multicentre inception cohort study. Lancet. (2017) 389:1710–8. doi: 10.1016/S0140-6736(17)30317-3
33. Hanaei S, Sadr M, Rezaei A, Shahkarami S, Ebrahimi Daryani N, Bidoki AZ, et al. Association of NLRP3 single nucleotide polymorphisms with ulcerative colitis: a case-control study. Clin Res Hepatol Gastroenterol. (2018) 42:269–75. doi: 10.1016/j.clinre.2017.09.003
34. Martin-Gallausiaux C, Béguet-Crespel F, Marinelli L, Jamet A, Ledue F, Blottière HM, et al. Butyrate produced by gut commensal bacteria activates TGF-beta1 expression through the transcription factor SP1 in human intestinal epithelial cells. Sci Rep. (2018) 8:9742. doi: 10.1038/s41598-018-28048-y
35. Fonseca-Camarillo G, Yamamoto-Furusho JK. Immunoregulatory pathways involved in inflammatory bowel disease. Inflamm Bowel Dis. (2015) 21:2188–93. doi: 10.1097/MIB.0000000000000477
36. Cruickshank SM, McVay LD, Baumgart DC, Felsburg PJ, Carding SR. Colonic epithelial cell mediated suppression of CD4 T cell activation. Gut. (2004) 53:678–84. doi: 10.1136/gut.2003.029967
37. Nakazawa A, Dotan I, Brimnes J, Allez M, Shao L, Tsushima F, et al. The expression and function of costimulatory molecules B7H and B7-H1 on colonic epithelial cells. Gastroenterology. (2004) 126:1347–57. doi: 10.1053/j.gastro.2004.02.004
38. Imielinski M, Baldassano RN, Griffiths A, Russell RK, Annese V, Dubinsky M, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. (2009) 41:1335–40. doi: 10.1038/ng.489
39. Izutani R, Loh EY, Reinecker H-C, Ohno Y, Fusunyan RD, Lichtenstein GR, et al. Increased expression of interleukin-8 mRNA in ulcerative colitis and Crohn's disease mucosa and epithelial cells. Inflamm Bowel Dis. (1995) 1:37–47.
40. Struyf S, Gouwy M, Dillen C, Proost P, Opdenakker G, Van Damme J. Chemokines synergize in the recruitment of circulating neutrophils into inflamed tissue. Eur J Immunol. (2005) 35:1583–91. doi: 10.1002/eji.200425753
41. Beltrán CJ, Núñez LE, Díaz-Jiménez D, Farfan N, Candia E, Heine C, et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflamm Bowel Dis. (2010) 16:1097–107. doi: 10.1002/ibd.21175
42. Di Salvo E, Ventura-Spagnolo E, Casciaro M, Navarra M, Gangemi S. IL-33/IL-31 axis: a potential inflammatory pathway. Mediators Inflamm. (2018) 2018:3858032. doi: 10.1155/2018/3858032
43. Travers J, Rochman M, Caldwell JM, Besse JA, Miracle CE, Rothenberg ME. IL-33 is induced in undifferentiated, non-dividing esophageal epithelial cells in eosinophilic esophagitis. Sci Rep. (2017) 7:17563. doi: 10.1038/s41598-017-17541-5
44. De la Fuente M, MacDonald TT, Hermoso MA. The IL-33/ST2 axis: role in health and disease. Cytokine Growth Factor Rev. (2015) 26:615–23. doi: 10.1016/j.cytogfr.2015.07.017
45. Lazaridis L-D, Pistiki A, Giamarellos-Bourboulis EJ, Georgitsi M, Damoraki G, Polymeros D, et al. Activation of NLRP3 inflammasome in inflammatory bowel disease: differences between Crohn's disease and ulcerative colitis. Dig Dis Sci. (2017) 62:2348–56. doi: 10.1007/s10620-017-4609-8
46. Friedrich M, Diegelmann J, Schauber J, Auernhammer CJ, Brand S. Intestinal neuroendocrine cells and goblet cells are mediators of IL-17A-amplified epithelial IL-17C production in human inflammatory bowel disease. Mucosal Immunol. (2015) 8:943–58. doi: 10.1038/mi.2014.124
47. Henderson P, van Limbergen JE, Schwarze J, Wilson DC. Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm Bowel Dis. (2011) 17:382–95. doi: 10.1002/ibd.21379
48. Caruso R, Fina D, Peluso I, Stolfi C, Fantini MC, Gioia V, et al. A functional role for interleukin-21 in promoting the synthesis of the T-cell chemoattractant, MIP-3alpha, by gut epithelial cells. Gastroenterology. (2007) 132:166–75. doi: 10.1053/j.gastro.2006.09.053
49. Mizoguchi A, Yano A, Himuro H, Ezaki Y, Sadanaga T, Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J Gastroenterol. (2018) 53:465–74. doi: 10.1007/s00535-017-1401-7
50. Andoh A, Yagi Y, Shioya M, Nishida A, Tsujikawa T, Fujiyama Y. Mucosal cytokine network in inflammatory bowel disease. World J Gastroenterol. (2008) 14:5154–61. doi: 10.3748/wjg.14.5154
51. Netea MG, Azam T, Ferwerda G, Girardin SE, Walsh M, Park J-S, et al. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 ligands for IL-1beta and IL-6 production through a caspase 1-dependent mechanism. Proc Natl Acad Sci USA. (2005) 102:16309–14. doi: 10.1073/pnas.0508237102
52. Watson AJM, Hughes KR. TNF-α-induced intestinal epithelial cell shedding: implications for intestinal barrier function. Ann N Y Acad Sci. (2012) 1258:1–8. doi: 10.1111/j.1749-6632.2012.06523.x
53. Smyth D, McKay CM, Gulbransen BD, Phan VC, Wang A, McKay DM. Interferon-gamma signals via an ERK1/2-ARF6 pathway to promote bacterial internalization by gut epithelia. Cell Microbiol. (2012) 14:1257–70. doi: 10.1111/j.1462-5822.2012.01796.x
54. Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Liver Physiol. (2011) 300:G684–96. doi: 10.1152/ajpgi.00474.2010
55. Pinchuk IV, Mifflin RC, Saada JI, Powell DW. Intestinal mesenchymal cells. Curr Gastroenterol Rep. (2010) 12:310–8. doi: 10.1007/s11894-010-0135-y
56. Valentich JD, Powell DW. Intestinal subepithelial myofibroblasts and mucosal immunophysiology. Curr Opin Gastroenterol. (1994) 10:645–51. doi: 10.1097/00001574-199411000-00013
57. Willis BC, duBois RM, Borok Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc Am Thorac Soc. (2006) 3:377–82. doi: 10.1513/pats.200601-004TK
58. Chen W, Lu C, Hirota C, Iacucci M, Ghosh S, Gui X. Smooth muscle hyperplasia/hypertrophy is the most prominent histological change in Crohn's fibrostenosing bowel strictures: a semiquantitative analysis by using a novel histological grading scheme. J Crohn's Colitis. (2017) 11:92–104. doi: 10.1093/ecco-jcc/jjw126
59. Lawrance IC, Maxwell L, Doe W. Altered response of intestinal mucosal fibroblasts to profibrogenic cytokines in inflammatory bowel disease. Inflamm Bowel Dis. (2001) 7:226–36. doi: 10.1097/00054725-200108000-00008
60. Bamba S, Andoh A, Yasui H, Araki Y, Bamba T, Fujiyama Y. Matrix metalloproteinase-3 secretion from human colonic subepithelial myofibroblasts: role of interleukin-17. J Gastroenterol. (2003) 38:548–54. doi: 10.1007/s00535-002-1101-8
61. Di Sabatino A, Jackson CL, Pickard KM, Buckley M, Rovedatti L, Leakey NAB, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn's disease strictures. Gut. (2009) 58:777–89. doi: 10.1136/gut.2008.149096
62. Biancheri P, Pender SL, Ammoscato F, Giuffrida P, Sampietro G, Ardizzone S, et al. The role of interleukin 17 in Crohn's disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair. (2013) 6:13. doi: 10.1186/1755-1536-6-13
63. Benchimol EI, Mack DR, Guttmann A, Nguyen GC, To T, Mojaverian N, et al. Inflammatory bowel disease in immigrants to Canada and their children: a population-based cohort study. Am J Gastroenterol. (2015) 110:553–63. doi: 10.1038/ajg.2015.52
64. Niewiadomski O, Studd C, Wilson J, Williams J, Hair C, Knight R, et al. Influence of food and lifestyle on the risk of developing inflammatory bowel disease. Intern Med J. (2016) 46:669–76. doi: 10.1111/imj.13094
65. Constante M, Fragoso G, Calvé A, Samba-Mondonga M, Santos MM. Dietary heme induces gut dysbiosis, aggravates colitis, and potentiates the development of adenomas in mice. Front Microbiol. (2017) 8:1809. doi: 10.3389/fmicb.2017.01809
66. Ananthakrishnan AN, Khalili H, Konijeti GG, Higuchi LM, de Silva P, Fuchs CS, et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn's disease. Gut. (2014) 63:776–84. doi: 10.1136/gutjnl-2013-305304
67. IBD in EPIC Study Investigators. Tjonneland A, Overvad K, Bergmann MM, Nagel G, Linseisen J, et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut. (2009) 58:1606–11. doi: 10.1136/gut.2008.169078
68. Viennois E, Merlin D, Gewirtz AT, Chassaing B. Dietary emulsifier–induced low-grade inflammation promotes colon carcinogenesis. Cancer Res. (2017) 77:27–40. doi: 10.1158/0008-5472.CAN-16-1359
69. Amre DK, D'Souza S, Morgan K, Seidman G, Lambrette P, Grimard G, et al. Imbalances in dietary consumption of fatty acids, vegetables, and fruits are associated with risk for Crohn's disease in children. Am J Gastroenterol. (2007) 102:2016–25. doi: 10.1111/j.1572-0241.2007.01411.x
70. Reif S, Klein I, Lubin F, Farbstein M, Hallak A, Gilat T. Pre-illness dietary factors in inflammatory bowel disease. Gut. (1997) 40:754–60. doi: 10.1136/gut.40.6.754
71. Llewellyn SR, Britton GJ, Contijoch EJ, Vennaro OH, Mortha A, Colombel J-F, et al. Interactions between diet and the intestinal microbiota alter intestinal permeability and colitis severity in mice. Gastroenterology. (2018) 154:1037–1046.e2
72. Lee N, Kim W-U. Microbiota in T-cell homeostasis and inflammatory diseases. Exp Mol Med. (2017) 49:e340. doi: 10.1038/emm.2017.36
73. Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. Phimister EG, editor. N Engl J Med. (2016) 375:2369–79. doi: 10.1056/NEJMra1600266
74. Kriss M, Hazleton KZ, Nusbacher NM, Martin CG, Lozupone CA. Low diversity gut microbiota dysbiosis: drivers, functional implications and recovery. Curr Opin Microbiol. (2018) 44:34–40. doi: 10.1016/j.mib.2018.07.003
75. Brown K, DeCoffe D, Molcan E, Gibson DL. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients. (2012) 4:1095–119. doi: 10.3390/nu4081095
76. Vrieze A, Out C, Fuentes S, Jonker L, Reuling I, Kootte RS, et al. Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol. (2014) 60:824–31. doi: 10.1016/j.jhep.2013.11.034
77. Salim SY, Kaplan GG, Madsen KL. Air pollution effects on the gut microbiota. Gut Microbes. (2014) 5:215–9. doi: 10.4161/gmic.27251
78. Dudek-Wicher RK, Junka A, Bartoszewicz M. The influence of antibiotics and dietary components on gut microbiota. Gastroenterol Rev. (2018) 13:85–92. doi: 10.5114/pg.2018.76005
79. Agus A, Denizot J, Thévenot J, Martinez-Medina M, Massier S, Sauvanet P, et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci Rep. (2016) 6:19032. doi: 10.1038/srep19032
80. Pearlman M, Obert J, Casey L. The association between artificial sweeteners and obesity. Curr Gastroenterol Rep. (2017) 19:64. doi: 10.1007/s11894-017-0602-9
81. Chi L, Bian X, Gao B, Tu P, Lai Y, Ru H, et al. Effects of the artificial sweetener neotame on the gut microbiome and fecal metabolites in mice. Molecules. (2018) 23:367. doi: 10.3390/molecules23020367
82. Nickerson KP, McDonald C. Crohn's disease-associated adherent-invasive Escherichia coli adhesion is enhanced by exposure to the ubiquitous dietary polysaccharide maltodextrin. Mizoguchi E, editor. PLoS ONE. (2012) 7:e52132. doi: 10.1371/journal.pone.0052132
83. Kish L, Hotte N, Kaplan GG, Vincent R, Tso R, Gänzle M, et al. Environmental particulate matter induces murine intestinal inflammatory responses and alters the gut microbiome. PLoS ONE. (2013) 8:e62220. doi: 10.1371/journal.pone.0062220
84. Onderdonk AB, Hermos JA, Bartlett JG. The role of the intestinal microflora in experimental colitis. Am J Clin Nutr. (1977) 30:1819–25. doi: 10.1093/ajcn/30.11.1819
85. Pascal V, Pozuelo M, Borruel N, Casellas F, Campos D, Santiago A, et al. A microbial signature for Crohn's disease. Gut. (2017) 66:813–22. doi: 10.1136/gutjnl-2016-313235
86. Kim P, Gadani A, Abdul-Baki H, Mitre R, Mitre M. Fecal microbiota transplantation in recurrent Clostridium difficile infection: a retrospective single-center chart review. JGH Open. (2018) 3:4–9. doi: 10.1002/jgh3.12093
87. Chen T, Zhou Q, Zhang D, Jiang F, Wu J, Zhou J-Y, et al. Effect of faecal microbiota transplantation for treatment of clostridium difficile infection in patients with inflammatory bowel disease: a systematic review and meta-analysis of cohort studies. J Crohn's Colitis. (2018) 12:710–7. doi: 10.1093/ecco-jcc/jjy031
88. Allegretti J, Eysenbach LM, El-Nachef N, Fischer M, Kelly C, Kassam Z. The current landscape and lessons from fecal microbiota transplantation for inflammatory bowel disease: past, present, and future. Inflamm Bowel Dis. (2017) 23:1710–7. doi: 10.1097/MIB.0000000000001247
89. Baktash A, Terveer EM, Zwittink RD, Hornung BVH, Corver J, Kuijper EJ, et al. Mechanistic insights in the success of fecal microbiota transplants for the treatment of clostridium difficile infections. Front Microbiol. (2018) 9:1242. doi: 10.3389/fmicb.2018.01242
90. Madsen KL, Doyle JS, Jewell LD, Tavernini MM, Fedorak RN. Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology. (1999) 116:1107–14. doi: 10.1016/S0016-5085(99)70013-2
91. Hvas CL, Bendix M, Dige A, Dahlerup JF, Agnholt J. Current, experimental, and future treatments in inflammatory bowel disease: a clinical review. Immunopharmacol Immunotoxicol. (2018) 10:1–15. doi: 10.1080/08923973.2018.1469144
92. Kim DH, Kim S, Lee JH, Kim JH, Che X, Ma HW, et al. Lactobacillus acidophilus suppresses intestinal inflammation by inhibiting endoplasmic reticulum stress. J Gastroenterol Hepatol. (2018) 34:178–85. doi: 10.1111/jgh.14362
93. Je I-G, Lee D-G, Jeong D-G, Hong D, Yoon J-M, Moon JS, et al. The probiotic, ID-JPL934, attenuates dextran sulfate sodium-induced colitis in mice through inhibition of proinflammatory cytokines expression. J Med Food. (2018) 23:jmf.2017.4152. doi: 10.1089/jmf.2017.4152
94. Nishitani Y, Tanoue T, Yamada K, Ishida T, Yoshida M, Azuma T, et al. Lactococcus lactis subsp. cremoris FC alleviates symptoms of colitis induced by dextran sulfate sodium in mice. Int Immunopharmacol. (2009) 9:1444–51. doi: 10.1016/j.intimp.2009.08.018
95. Borthakur A, Bhattacharyya S, Kumar A, Anbazhagan AN, Tobacman JK, Dudeja PK. Lactobacillus acidophilus alleviates platelet-activating factor-induced inflammatory responses in human intestinal epithelial cells. Dahiya R, editor. PLoS ONE. (2013) 8:e75664. doi: 10.1371/journal.pone.0075664
96. Mattar AF, Drongowski RA, Coran AG, Harmon CM. Effect of probiotics on enterocyte bacterial translocation in vitro. Pediatr Surg Int. (2001) 17:265–8. doi: 10.1007/s003830100591
97. Plaza-Diaz J, Gomez-Llorente C, Fontana L, Gil A. Modulation of immunity and inflammatory gene expression in the gut, in inflammatory diseases of the gut and in the liver by probiotics. World J Gastroenterol. (2014) 20:15632. doi: 10.3748/wjg.v20.i42.15632
98. Rodríguez-Nogales A, Algieri F, Garrido-Mesa J, Vezza T, Utrilla MP, Chueca N, et al. The administration of Escherichia coli nissle 1917 ameliorates development of DSS-induced colitis in mice. Front Pharmacol. (2018) 9:468. doi: 10.3389/fphar.2018.00468
99. Lodinová-Žádniková R, Sonnenborn U. Effect of preventive administration of a nonpathogenic Escherichia coli strain on the colonization of the intestine with microbial pathogens in newborn infants. Neonatology. (1997) 71:224–32.
100. Barbaro MR, Fuschi D, Cremon C, Carapelle M, Dino P, Marcellini MM, et al. Escherichia coli Nissle 1917 restores epithelial permeability alterations induced by irritable bowel syndrome mediators. Neurogastroenterol Motil. (2018) 30:e13388. doi: 10.1111/nmo.13388
101. Secher T, Maillet I, Mackowiak C, Le Bérichel J, Philippeau A, Panek C, et al. The probiotic strain Escherichia coli Nissle 1917 prevents papain-induced respiratory barrier injury and severe allergic inflammation in mice. Sci Rep. (2018) 8:11245. doi: 10.1038/s41598-018-29689-9
102. Fukumoto S, Toshimitsu T, Matsuoka S, Maruyama A, Oh-oka K, Takamura T, et al. Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunol Cell Biol. (2014) 92:460–5. doi: 10.1038/icb.2014.2
103. Le Maréchal C, Peton V, Plé C, Vroland C, Jardin J, Briard-Bion V, et al. Surface proteins of Propionibacterium freudenreichii are involved in its anti-inflammatory properties. J Proteomics. (2015) 113:447–61. doi: 10.1016/j.jprot.2014.07.018
104. Barouei J, Moussavi M, Hodgson DM. Effect of maternal probiotic intervention on HPA axis, immunity and gut microbiota in a rat model of irritable bowel syndrome. Heimesaat MM, editor. PLoS ONE. (2012) 7:e46051. doi: 10.1371/journal.pone.0046051
105. Okada Y, Tsuzuki Y, Narimatsu K, Sato H, Ueda T, Hozumi H, et al. 1,4-Dihydroxy-2-naphthoic acid from Propionibacterium freudenreichii reduces inflammation in interleukin-10-deficient mice with colitis by suppressing macrophage-derived proinflammatory cytokines. J Leukoc Biol. (2013) 94:473–80. doi: 10.1189/jlb.0212104
106. De Cupere F, Deprez P, Demeulenaere D, Muylle E. Evaluation of the effect of 3 probiotics on experimental Escherichia coli enterotoxaemia in weaned piglets. Zentralbl Veterinarmed B. (1992) 39:277–84. doi: 10.1111/j.1439-0450.1992.tb01169.x
107. Sudha MR, Jayanthi N, Aasin M, Dhanashri RD, Anirudh T. Efficacy of Bacillus coagulans unique IS2 in treatment of irritable bowel syndrome in children: a double blind, randomised placebo controlled study. Benef Microbes. (2018) 9:563–72. doi: 10.3920/BM2017.0129
108. Yang Y, Jing Y, Yang J, Yang Q. Effects of intranasal administration with Bacillus�subtilis on immune cells in the nasal mucosa and tonsils of piglets. Exp Ther Med. (2018) 15:5189–98. doi: 10.3892/etm.2018.6093
109. Sivananthan K, Petersen AM. Review of Saccharomyces boulardii as a treatment option in IBD. Immunopharmacol Immunotoxicol. (2018) 17:1–11. doi: 10.1080/08923973.2018.1469143
110. Roussel C, Sivignon A, de Vallée A, Garrait G, Denis S, Tsilia V, et al. Anti-infectious properties of the probiotic Saccharomyces cerevisiae CNCM I-3856 on enterotoxigenic E. coli (ETEC) strain H10407. Appl Microbiol Biotechnol. (2018) 102:6175–89. doi: 10.1007/s00253-018-9053-y
111. Mack DR, Michail S, Wei S, McDougall L, Hollingsworth MA. Probiotics inhibit enteropathogenic E. coli adherence in vitro by inducing intestinal mucin gene expression. Am J Physiol. (1999) 276(4 Pt 1):G941–50.
112. Anderson RC, Cookson AL, McNabb WC, Park Z, McCann MJ, Kelly WJ, et al. Lactobacillus plantarum MB452 enhances the function of the intestinal barrier by increasing the expression levels of genes involved in tight junction formation. BMC Microbiol. (2010) 10:316. doi: 10.1186/1471-2180-10-316
113. Dai C, Zhao DH, Jiang M. VSL#3 probiotics regulate the intestinal epithelial barrier in vivo and in vitro via the p38 and ERK signaling pathways. Int J Mol Med. (2012) 29:202–8. . doi: 10.3892/ijmm.2011.839
114. Llewellyn A, Foey A. Probiotic modulation of innate cell pathogen sensing and signaling events. Nutrients. (2017) 9:E1156. doi: 10.3390/nu9101156
115. Kim SW, Kim HM, Yang KM, Kim S-A, Kim S-K, An MJ, et al. Bifidobacterium lactis inhibits NF-kappaB in intestinal epithelial cells and prevents acute colitis and colitis-associated colon cancer in mice. Inflamm Bowel Dis. (2010) 16:1514–25. doi: 10.1002/ibd.21262
116. Tomosada Y, Villena J, Murata K, Chiba E, Shimazu T, Aso H, et al. Immunoregulatory effect of bifidobacteria strains in porcine intestinal epithelial cells through modulation of ubiquitin-editing enzyme A20 expression. PLoS ONE. (2013) 8:e59259. doi: 10.1371/journal.pone.0059259
117. Finamore A, Ambra R, Nobili F, Garaguso I, Raguzzini A, Serafini M. Redox role of Lactobacillus casei shirota against the cellular damage induced by 2,2′-Azobis (2-Amidinopropane) dihydrochloride-induced oxidative and inflammatory stress in enterocytes-like epithelial cells. Front Immunol. (2018) 9:1131. doi: 10.3389/fimmu.2018.01131
118. Yan F, Liu L, Dempsey PJ, Tsai Y-H, Raines EW, Wilson CL, et al. A Lactobacillus rhamnosus GG-derived soluble protein, p40, stimulates ligand release from intestinal epithelial cells to transactivate epidermal growth factor receptor. J Biol Chem. (2013) 288:30742–51. doi: 10.1074/jbc.M113.492397
119. Coombes JL, Siddiqui KRR, Arancibia-Cárcamo CV, Hall J, Sun C-M, Belkaid Y, et al. A functionally specialized population of mucosal CD103 + DCs induces Foxp3 + regulatory T cells via a TGF-β- and retinoic acid–dependent mechanism. J Exp Med. (2007) 204:1757–64. doi: 10.1084/jem.20070590
120. Iliev ID, Spadoni I, Mileti E, Matteoli G, Sonzogni A, Sampietro GM, et al. Human intestinal epithelial cells promote the differentiation of tolerogenic dendritic cells. Gut. (2009) 58:1481–9. doi: 10.1136/gut.2008.175166
121. Otte J-M, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. (2003) 124:1866–78. doi: 10.1016/S0016-5085(03)00403-7
122. Beswick EJ, Johnson JR, Saada JI, Humen M, House J, Dann S, et al. TLR4 activation enhances the PD-L1-mediated tolerogenic capacity of colonic CD90+ stromal cells. J Immunol. (2014) 193:2218–29. doi: 10.4049/jimmunol.1203441
123. Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, et al. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut. (2016) 66:823–38. doi: 10.1136/gutjnl-2015-310374
124. Jacob N, Jacobs JP, Kumagai K, Ha CWY, Kanazawa Y, Lagishetty V, et al. Inflammation-independent TL1A-mediated intestinal fibrosis is dependent on the gut microbiome. Mucosal Immunol. (2018) 11:1466–76. doi: 10.1038/s41385-018-0055-y
125. Shih DQ, Zheng L, Zhang X, Zhang H, Kanazawa Y, Ichikawa R, et al. Inhibition of a novel fibrogenic factor Tl1a reverses established colonic fibrosis. Mucosal Immunol. (2014) 7:1492–503. doi: 10.1038/mi.2014.37
126. Barrett R, Zhang X, Koon HW, Vu M, Chang J-Y, Yeager N, et al. Constitutive TL1A expression under colitogenic conditions modulates the severity and location of gut mucosal inflammation and induces fibrostenosis. Am J Pathol. (2012) 180:636–49. doi: 10.1016/j.ajpath.2011.10.026
127. Park JS, Choi JW, Jhun J, Kwon JY, Lee BI, Yang CW, et al. Lactobacillus acidophilus improves intestinal inflammation in an acute colitis mouse model by regulation of th17 and treg cell balance and fibrosis development. J Med Food. (2018) 21:215–24. doi: 10.1089/jmf.2017.3990
128. Gubatan J, Moss AC. Vitamin D in inflammatory bowel disease: more than just a supplement. Curr Opin Gastroenterol. (2018) 34:217–25. doi: 10.1097/MOG.0000000000000449
129. Tao Q, Wang B, Zheng Y, Jiang X, Pan Z, Ren J. Vitamin D prevents the intestinal fibrosis via induction of vitamin D receptor and inhibition of transforming growth factor-beta1/Smad3 pathway. Dig Dis Sci. (2015) 60:868–75. doi: 10.1007/s10620-014-3398-6
130. Truffi M, Sorrentino L, Monieri M, Fociani P, Mazzucchelli S, Bonzini M, et al. Inhibition of fibroblast activation protein restores a balanced extracellular matrix and reduces fibrosis in Crohn's disease strictures ex vivo. Inflamm Bowel Dis. (2018) 24:332–45. doi: 10.1093/ibd/izx008
131. Suez J, Zmora N, Zilberman-Schapira G, Mor U, Dori-Bachash M, Bashiardes S, et al. Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell. (2018) 174:1406–23.e16. doi: 10.1016/j.cell.2018.08.047
132. Zmora N, Zilberman-Schapira G, Suez J, Mor U, Dori-Bachash M, Bashiardes S, et al. Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell. (2018) 174:1388–405.e21. doi: 10.1016/j.cell.2018.08.041
Keywords: gut inflammation, inflammatory bowel disease, intestinal epithelial cells, intestinal fibroblasts, immune cell activation/modulation, intestinal microbiota
Citation: Curciarello R, Canziani KE, Docena GH and Muglia CI (2019) Contribution of Non-immune Cells to Activation and Modulation of the Intestinal Inflammation. Front. Immunol. 10:647. doi: 10.3389/fimmu.2019.00647
Received: 14 September 2018; Accepted: 11 March 2019;
Published: 10 April 2019.
Edited by:
Marcela A. Hermoso, Universidad de Chile, ChileReviewed by:
Rita Carsetti, Bambino Gesù Children Hospital (IRCCS), ItalyMarkus Bosmann, Boston University, United States
Copyright © 2019 Curciarello, Canziani, Docena and Muglia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Isabel Muglia, Y211Z2xpYWVuYXRvckBnbWFpbC5jb20=