 Ruth Brignall1,2
Ruth Brignall1,2 Amy T. Moody
Amy T. Moody Shibin Mathew
Shibin Mathew Suzanne Gaudet
Suzanne Gaudet- 1Center for Cancer Systems Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA, United States
- 2Department of Genetics, Harvard Medical School, Blavatnik Institute, Boston, MA, United States
- 3Laboratory for Systems Pharmacology, Harvard Medical School, Blavatnik Institute, Boston, MA, United States
- 4Department of Microbiology, Tufts University School of Medicine, Boston, MA, United States
The NF-κB transcription regulation system governs a diverse set of responses to various cytokine stimuli. With tools from in vitro biochemical characterizations, to omics-based whole genome investigations, great strides have been made in understanding how NF-κB transcription factors control the expression of specific sets of genes. Nonetheless, these efforts have also revealed a very large number of potential binding sites for NF-κB in the human genome, and a puzzle emerges when trying to explain how NF-κB selects from these many binding sites to direct cell-type- and stimulus-specific gene expression patterns. In this review, we surmise that target gene transcription can broadly be thought of as a function of the nuclear abundance of the various NF-κB dimers, the affinity of NF-κB dimers for the regulatory sequence and the availability of this regulatory site. We use this framework to place quantitative information that has been gathered about the NF-κB transcription regulation system into context and thus consider questions it answers, and questions it raises. We end with a brief discussion of some of the future prospects that new approaches could bring to our understanding of how NF-κB transcription factors orchestrate diverse responses in different biological contexts.
Introduction
The nuclear factor-κB (NF-κB) family of transcription factors regulate the expression of genes that are crucial to a wide variety of biological processes, ranging from immune, stress, and inflammatory responses, to cell apoptosis. The NF-κB family is made up of five proteins, p105/p50 (encoded by NFKB1), p100/p52 (encoded by NFKB2), RelA (also known as p65), RelB, and c-Rel, which can form a range of homo- and hetero-dimeric complexes [Figures 1A,B; (6)]. When partnered with inhibitory IκB proteins, NF-κB dimers are preferentially shuttled to the cytoplasm where they are held inactive. In response to stimuli, IκB is phosphorylated and subsequently degraded, thus releasing NF-κB and allowing it to accumulate in the nucleus (Figure 1C). Once in the nucleus an NF-κB dimer can bind to κB sites to activate or repress the transcription of its target genes. The best-studied κB sites fit the consensus κB site pattern, 5′-GGGRNWYYCC-3′ (where R, W, Y, and N, respectively denote purine, adenine or thymine, pyrimidine, and any nucleotide) (7–9). In the human genome encompassing 3 × 109 base pairs, there are undoubtedly myriads of sequences matching the consensus κB site. Indeed, early on, ChIP-chip (chromatin immunoprecipitation to microarray) experiments interrogating the sequence of human chromosome 22 suggested that there are more than 1.4 × 104 of these consensus sites contacted by NF-κB dimers during a response to stimulus (2, 10). More recent ChIP-seq experiments have identified 20,000–50,000 RelA-bound peaks, although it is unclear whether RelA directly contacts the DNA at all of these sites (3, 4). Just considering the RelA subunit, 1 × 105-1.5 × 105 molecules enter the nucleus following stimulation (11), a portion of these molecules bind to DNA and this regulates the expression of just ~600 genes [a curated list of known NF-κB target genes can be found at the Boston University NF-κB Transcription Factors website; (12)]. The large number of RelA molecules in comparison to the relatively small number of regulated transcripts suggests a complex relationship between the amount of NF-κB in the nucleus and the subsequent expression of target genes.
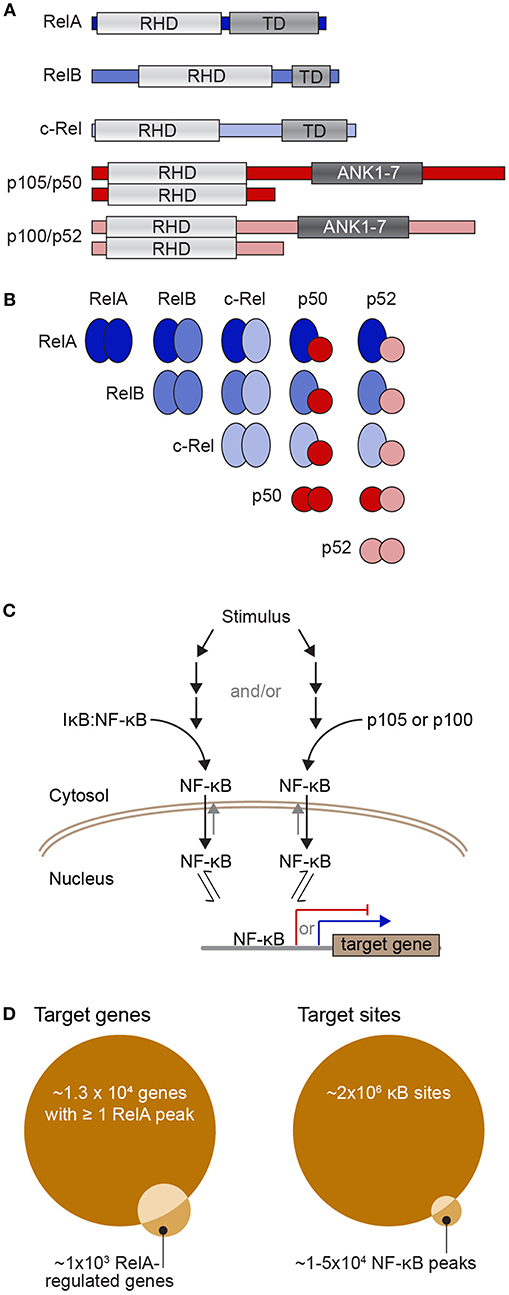
Figure 1. The NF-κB transcription factor family and its dimerization and DNA interactions. (A) Diagrams of the five NF-κB subunits showing their Rel homology domain (RHD), which encompasses both their DNA-binding domain and dimerization region, the transactivation domains (TD) of RelA, RelB, and c-Rel, as well as the ankyrin-rich region of p105 and p100 (repeats 1-7; ANK1-7), which is cleaved to yield p50 and p52. (B) Diagram of the ways in which the NF-κB subunits can partner to form dimers that contain zero (all red), one (blue/red), or two (all blue) transactivation domains. (C) Simple schematic of the process of activation of NF-κB dimers. Upon stimulation, a series of events leads to phosphorylation and proteasome-mediated degradation of IκB to release NF-κB dimers (left) and/or cleavage of p105 or p100 to remove their IκB-like ankyrin-rich domain and again release NF-κB dimers (right). Free NF-κB dimers are preferentially shuttled into the nucleus where they have access to the regulatory sequences of NF-κB target genes. TD-containing NF-κB dimers can activate transcription of target genes (blue arrow), while TD-lacking NF-κB dimers can act as transcriptional repressors (red). (D) Venn diagrams representing potential target sites (right) and potential target genes (left). There may be up to 2 × 106 consensus κB sites or half-sites in the human genome (1) although various ChIP-seq studies have reported that there may be between 1 and 5 × 104 NF-κB-bound peaks in a mammalian genome across a population of stimulated cells [e.g., (2–4)], of which 30–50% contain a consensus κB site. Because many gene regulatory sequences have multiple NF-κB-bound peaks, one estimate is that around 1.3 × 104 genes have at least one RelA peak in their regulatory region (5). However, the same study found only ~1,000 genes were detectably regulated by RelA-containing NF-κB with ~60% of these having a RelA ChIP-seq peak in gene-proximal regulatory regions (5).
Numerous ChIP-seq and whole genome sequencing experiments have shown that the recruitment of many transcription factors, including NF-κB, to chromatinized DNA is dependent on the cellular context and therefore must be highly regulated [reviewed in (10)]. However, despite the wealth of genomic data now available, the mechanisms by which NF-κB-DNA interactions generate specific gene expression profiles following stimulation remain largely unknown. For example, by allocating ChIP-Seq peaks to their nearest gene, RelA-containing NF-κB was found to bind ~13,600 genes in TNF-stimulated HeLa cells, yet only ~1,000 genes were up or down-regulated in response to RelA perturbation, and only ~600 of these were directly bound by NF-κB [Figure 1D, left; (5)]. Thus, a vast majority of the genes that are bound by NF-κB in response to stimulus are not regulated. This raises the questions: how do NF-κB dimers select their binding sites and why are only some of the bound genes transcriptionally regulated? Seeking to answer these questions, we will focus herein on three key sets of factors that regulate NF-κB recruitment to DNA: abundance of NF-κB dimers and κB binding sites, binding affinity, and the availability of the κB sites at any given time.
Abundance
κB Binding Sites
If, as Martone et al. (2) estimated, there are ~104 consensus κB sites in the genome that are bound by RelA and ~1 × 105 RelA-containing dimers enter the nucleus upon cell stimulation [estimated by Hottiger et al. (11)], a simple view of the system would predict rapid saturation of these consensus κB sites (see Box 1). However, experiments demonstrate that many consensus κB sites are not bound and, in fact, this lack of saturation of the system is necessary to generate stimulus- and cell-type-specific gene expression profiles (16–18). One explanation for this apparent dichotomy is that, in addition to consensus κB sites, NF-κB can bind to degenerate κB sites. Structural, biochemical, and in vivo assays have demonstrated that NF-κB dimers can bind to κB half sites, sites whose sequences deviate from the consensus sequence, and even unrelated sites (3, 19–24). With these additional non-consensus binding sites, the total number of potential NF-κB sites in the human genome could easily climb to 2 × 106 (1). This flips the NF-κB protein vs. NF-κB binding site calculus (Figure 1D, right), and our first question becomes: how do the relatively sparse NF-κB dimers decide which of the numerous potential κB binding sites to interact with?
Box 1. Computing fraction of binding sites occupied by transcription factor.
Computational models provide a powerful means to examine, interrogate, and ultimately better understand the relationships between inputs and outputs of complicated biological processes. Here, we use a simple mass-action kinetics model to illustrate how (i) binding affinity, (ii) abundance of transcription factors and their binding sites, and (iii) the availability of these binding sites due to the presence or absence of a binding competitor species affect the fraction of sites bound by the transcription factor. Although in reality, binding by a transcription factor is only a rough correlate of gene transcription in response to stimuli, this toy model shows us how the interplay between quantitative aspects of protein-DNA interactions potentially affects transcription regulation. Previous studies have used similar kinetics models to calculate fraction of binding sites (13).
In the simple scenario that we depict (Figure Box 1A), we model the binding of transcription factors to their cognate sites on the genome as a simple adsorption process—where molecules bind to sites, unchanged. This model therefore gives us a theoretical limit on the fraction of bound sites when the process is activation energy-limited (i.e., within-nucleus transport is much faster than DNA binding) and the process of a transcription factor finding a binding site is random. We also make additional simplifying assumptions: (1) the contents of the nucleus are well mixed and both genomic and non-genomic compartments are homogeneous; (2) all binding sites are equivalent with identical affinities for the transcription factor and competitor species; (3) the total nuclear concentrations of transcription factor and competitor species are fixed, under the assumption that any change occurs on a time scale slower than that of the binding process (and therefore, in this very simplistic model, we assume that the steady state is reached faster than changes in nuclear abundance and post-translational modifications of transcription factors). Given the stated assumptions, we will let X be free nuclear transcription factor, Xcomp be free nuclear competitor species, and Y represent the transcription factor binding site. Y can be bound by X or Xcomp creating the complexes Y:X and Y:Xcomp, respectively. Binding of transcription factor and competitor species to DNA can then be modeled by two reaction equations:
As we assumed identical affinities to DNA binding sites for the transcription factor and its competitor, we will also assume the same association rate parameter α and dissociation rate parameter γ. Using mass action kinetics and mass balance equations, our reaction system can be fully described using two ordinary differential equations (ODEs):
Here, XT, and YT are the total number of molecules or sites for a given nucleus and, under our assumption of time scale separation (#3), they are assumed constant while solving the ODEs. We also define , the dissociation constant (which is, as usual, the inverse of the binding affinity constant). Solving the system of ODEs gives the concentration of each species over time and at steady state. Solving the ODEs for different sets of parameter values and initial concentrations for XT, and YT allows us to illustrate the relationships between these parameters and initial concentrations and the steady state (ss) fraction of sites bound by the transcription factor, calculated as .
To survey a biologically relevant range of concentration values, we considered a typical HeLa cell, with a total cell volume of 2,700 μm3, a cytoplasmic to nuclear volume ratio of 3.3 (14), and ratios of transcription factors and available binding sites from 103:106 to 106:103 molecules/site. In the absence of the competitor (Figure Box 1B; continuous lines) and for KD > 10 nM, we need a large amount of transcription factor () to effectively saturate most of the binding sites at steady state. If we consider the RelA:p50 heterodimer, which has been reported to bind to the consensus κB site with a KD of 12.8 ± 2.2 nM (15), and ~1.5 × 105 heterodimers in a nucleus (11) with ~1.4 × 104 binding sites (2), we obtain , and the simple model finds that >90% of the DNA binding sites would be occupied. In the presence of the competitor species (in abundance equal to that of the dimers) the achievable occupancy is reduced to half (Figure Box 1B, dashed lines). Even a simplistic illustrative model such as the one we used here clearly shows how the interplay between abundance and affinity changes the fraction of bound sites in a nonlinear fashion, and how one mechanism for regulating availability of the sites—competition—can substantially reduce the number of sites occupied by a transcription factor. As we discuss in this review, there are many other nuances to abundance, affinity, and availability which will require more complex models to fully capture.
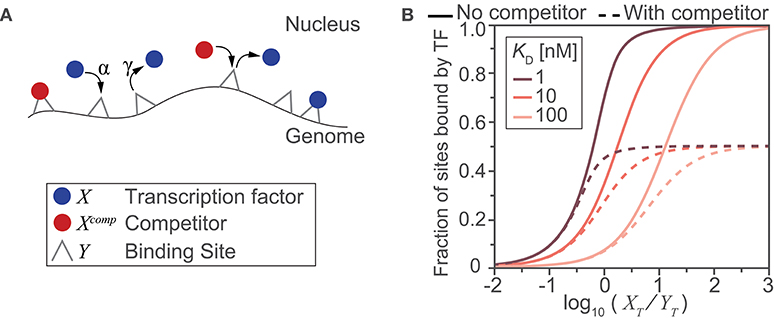
Figure Box 1. A toy model illustrates the impact of abundance, affinity and availability on the fraction of potential sites bound. (A) Schematic diagram of the reactions and molecular species included in the model. DNA binding sites are present on the genome and can be bound by a transcription factor (blue) or by a competitor protein (red) with an on-rate of α and off-rate of γ. (B) Model-derived input-output relationships between the ratio of total transcription factor to potential binding sites () and the steady state fraction of sites that are bound by a transcription factor. The relationship was derived for three different transcription factor binding affinities for the DNA sites (expressed using the dissociation constant, ), in the presence (dashed lines) or absence (solid lines) of a competitor species (where, ).
In recent years, innovative live-cell imaging techniques based on fluorescence recovery after photobleaching (FRAP), along with kinetic modeling of the collected data, have started to shed light on the dynamic nature of the transcription factor-DNA interaction process. Broadly speaking, this work indicates that most transcription factors may rapidly diffuse through the nucleus (with diffusion coefficients of ~0.5–5 μm2s−1 depending on transcription factor size) while “scanning” the genome for high-specificity sites (25, 26). Of note, the use of the term “scanning” should not necessarily evoke the image of a transcription factor gliding along chromatin, although such one-dimensional sliding models have been posited following single-molecule imaging studies of the p53 transcription factor (27, 28). Instead, many transcription factors, including NF-κB dimers, may “scan” by visiting multiple sites in a trial-and-error series of short-duration binding events (29). Therefore, transcription factors undergo thousands of these transient encounters with chromatin that ultimately will have no direct consequence on gene expression.
Interestingly, it is now thought that most functional NF-κB interactions with chromatin—interactions that lead to a change in transcription—are fleeting. Early, in vitro, bulk biochemical measurements of NF-κB interactions with κB sites indicated the formation of very stable complexes with a half-life of up to 45 min (30); using bulk, ChIP-based assays, similarly long interaction half-lives have been measured for other transcription factors (31) and shown to be regulated by ubiquitylation [reviewed in (32, 33)]. However, a subsequent study using FRAP in live cells expressing RelA-GFP showed that most RelA-DNA interactions are actually quite dynamic, with half-lives of a few seconds (16). Using stopped-flow kinetics and surface plasmon resonance, which can both directly measure association and dissociation kinetics, in vitro measurements made in physiological salt and pH conditions recapitulated these faster kinetics [yielding half-lives of 1.5 and 40 s, respectively for RelA:p50-DNA (34) and a range of a few seconds to a few minutes for a variety of NF-κB dimers and binding sites (22)]. Strikingly, IκBα can further accelerate the RelA:p50-DNA dissociation by up to ~40-fold, “stripping” dimers from DNA in a process that has now been extensively characterized (34–39). Indeed, a recent study used single-molecule tracking of individual Halo-tagged RelA molecules in live cells to show that the majority (~96%) of RelA undergoes short-lived interactions lasting on average ~0.5 s, while just ~4% of RelA molecules form more stable complexes with a lifetime of ~4 s (40). Because the ability of the RelA fusion proteins to induce transcription of target genes was verified in both the FRAP and single-molecule in vivo studies, these results suggest that long-lasting NF-κB binding may not be required for preinitiation complex assembly or for the activation of transcription. However, the mechanisms that distinguish NF-κB-DNA binding events that change transcription of a target gene from those that do not remain unclear.
Recent studies have found that while individual interactions are very brief, the integrated target site occupancy of Sox2 and Oct4 transcription factors can be highly sensitive to the nuclear concentration of the transcription factor (41). This implies that even when transcription factor occupancy at target sites is short-lived, high nuclear concentrations facilitate rapid turnover and, overall, increase the frequency of these short interactions. In this way, high rates of transcription factor sampling at binding sites may generate enough cumulative site occupancy to affect transcription (29). Having many binding sites across the human genome, NF-κB may also use this mechanism to tune the spatiotemporal patterns of gene expression it generates in response to stimulation by, for example, having a larger effect on sites that have the highest cumulative occupancy.
Intriguingly, high frequencies of transcription factor sampling have also been observed at non-consensus sites, yet these interactions were shown to have no direct effect on transcription (42). This observation has revived ideas first proposed years ago, whereby a key part of the target search process is transcription factors making non-specific contacts with DNA and then proceeding to slide or hop around the local chromatin environment until either a specific contact is formed, or contact and access to DNA is lost (43, 44). This model could partially explain the large number of sites detected by static, end-point biochemical binding assays including ChIP-Seq studies, which appear to be non-functional. Although these sites are “visited” in the search process, their cumulative occupancy may not be sufficiently long, or their interaction qualitatively suitable, to affect transcription.
If transcription factors rapidly sample many sites, would a cluster of non-functional binding sites near a target gene promoter or enhancer increases, or decreases, the local concentration of transcription factor? An increase in local concentration could occur if the brief interactions with clustered sites keep more transcription factor molecules nearby, increasing the probability that one binds to the functional target site [e.g., via an avidity effect as theoretically considered by (45)]. Alternatively, if the non-functional sites sequester transcription factors away from the functional target sites, acting as “natural decoys” (46), they would effectively decrease the local concentration of transcription factors. An early study showed that transfection of double-stranded oligonucleotides with κB sites inhibits the NF-κB-induced production of inflammatory cytokines in a rat model of myocardial infarction (47) showing a decoy-site effect. However, in that scenario, the transfected oligonucleotides likely reduce the global, not local, concentration of available NF-κB dimers and thus globally suppress transcription NF-κB-driven transcription. A more direct test of the effects of clusters of sites would be to manipulate the sequences near an NF-κB target gene promoter. In budding yeast, synthetic promoters were used to show that adding clustered sites for a transcriptional activator reduce the transcriptional output, as expected for decoys (48). In addition, the clustered sites could qualitatively change a transcriptional response from a graded response, correlated to transcription factor abundance, to a threshold-based, non-linear response (48). A combination of mathematical models and synthetic LacI-based constructs in E. coli showed that varying the number and chromosomal context of repressor binding sites can also quantitatively and qualitatively change the response (49). With so many possible nuances driving up or down the probability of transcription factor-DNA interactions, it may well be that the effect of additional sites on the transcription of an NF-κB target gene, whether enhancing or dampening, is highly context-dependent. The relative affinity, number, and two- or three-dimensional clustering of the sites could all modulate their effects and diversify the response of target genes to one NF-κB signal.
In one more layer of complexity, many NF-κB target genes have multiple κB sites within their regulatory regions [a common feature of many transcription factor binding motifs; (50)]. In fact, 95% of up-regulated and 91% of down-regulated NF-κB target genes have been shown to contain more than three κB sites in their regulatory regions [e.g., (2, 5)]. For many years, the predominant model for transcriptional regulation was that having multiple sites within gene promoters would drive cooperativity in DNA binding by the cognate transcription factors (51). This cooperative binding was then thought to lead to rapid, binary switching between fully unoccupied inactive promoters and fully occupied active promoters, yielding a largely all-or-none transcriptional activation (or repression) response. However, more recent studies have shown that NF-κB (via RelA) does not generally show cooperative binding to DNA, and instead NF-κB-dependent transcriptional activity scales gradually with NF-κB nuclear concentration (52). Therefore, Giorgetti and colleagues propose that the presence of multiple κB sites in one regulatory region increases the dynamic range of transcriptional outputs, with promoters with more consensus κB sites driving higher transcription at the same nuclear concentration of NF-κB, thus providing yet another means to quantitatively modulate NF-κB-dependent gene expression.
In summary, these observations indicate that the distribution of κB sites in the genome is non-uniform and clustering of the consensus and non-consensus sites in combination with highly frequent interactions of NF-κB with these sites can influence the transcriptional logic as well as shape the dynamic range of transcription. As, in fact, most human transcription factors are generally observed to bind to only a fraction of their consensus sites in any given cell type (53); this site selection process may be a generalized mechanism to achieve specific transcriptional responses.
Nucleus-Localized NF-κB Dimers
One challenge for anyone surveying the NF-κB literature with a quantitative mindset is that the terms “NF-κB” and “RelA” are often used interchangeably, and most studies reporting on the abundance of “NF-κB” in the nucleus focus exclusively on the RelA subunit. By ignoring other dimer species, these numbers muddle the relationship between signal, nuclear “NF-κB,” and DNA binding or transcription output. Different stimuli can lead to the nuclear accumulation of specific NF-κB dimers, indicating the importance of considering more than just the RelA subunit [e.g., (54)]. As other reviews have considered stimulus-specific activation of particular NF-κB dimers (17, 55); here, we specifically consider how the nuclear abundance of different dimer species can modulate NF-κB-driven transcriptional responses (Figure 2).
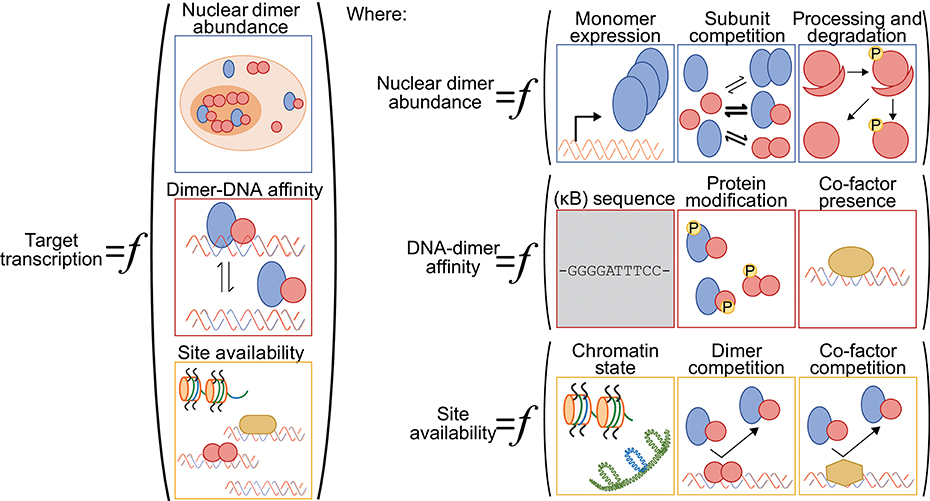
Figure 2. A generalized function for NF-κB-driven gene regulation. Schematic diagram of quantitative and qualitative factors that can differentially modulate NF-κB-driven gene regulation gene-by-gene and across various cellular contexts. Broadly speaking, target gene transcription is set by the nuclear abundance of NF-κB dimers, the NF-κB-DNA binding affinity and the availability of the DNA binding sites (left). The nuclear abundance of NF-κB dimers is itself a function of NF-κB subunit monomer expression, of NF-κB subunit competition in the various dimerization reactions, and of processing and degradation of inhibitory domains (ankyrin-rich domains of p105 and p100) and inhibitory proteins (IκBs) (top right panel). The NF-κB-DNA binding affinity is influenced by the DNA sequence (for both consensus κB sites and non-consensus sites), by NF-κB dimer identity and their post-translation modifications, and by the presence of regulatory co-factors that may help recruit NF-κB dimers to DNA or stabilize the interactions (center right panel). We note here that the DNA sequence is, arguably, the only factor that is not cell-type specific (gray shading). Finally, the availability of DNA binding sites for interaction with an NF-κB dimer is a function of the chromatin state, including the presence of histones and histone modifications, of competition from other NF-κB dimers and their relative affinities for the same site, and of competition with other regulatory factors that may bind to and occlude the potential binding site (bottom right panel).
The five NF-κB subunits can dimerize in almost every combination, each with unique but overlapping DNA and protein binding affinities [Figure 1B; (3, 22, 23, 56)]. RelA, RelB, and c-Rel each contain a transactivation domain (TD), capable of recruiting the transcription machinery, and thus NF-κB dimers including at least one of these subunits can activate transcription. In contrast, p50 and p52 do not have a TD and homodimers or heterodimers made up of only p50 and p52 are not capable of inducing transcription without recruiting an additional TD-containing transcription factor. Bound to the same κB site, a TD-containing NF-κB dimer will likely act as a transcriptional activator while a TD-lacking NF-κB dimer can act as a transcriptional repressor.
Although RelA:p50 is frequently cited as the most abundant NF-κB dimer, this may be dependent on cellular context. Other dimer species can also be highly expressed, and some are more likely to be found in the nucleus prior to stimulation. For example, p50 homodimers localize to the nucleus in resting mouse bone marrow derived macrophages (BMDMs) at a concentration of ~200 nM, which is similar to the maximum nuclear RelA:p50 concentration following stimulation in these cells (57). Thus, a quantitative framework that seeks to explain or predict NF-κB-DNA interactions and NF-κB-driven transcription at target genes but considers only RelA:p50 dimers is greatly oversimplifying the system. The 1.5 × 105 molecule per cell figure that we have considered overlooks contributions from other dimer species, the nuclear concentration of which is not necessarily correlated with that of RelA:p50. Therefore, the simple assumption that nuclear RelA:p50 is the major contributor to NF-κB-driven transcription not only underestimates total nuclear NF-κB abundance, but may also obscure the true relationship between “NF-κB” abundance and DNA-binding and transcription activation (see also Box 1).
Finally, the abundance of the different NF-κB dimers is not a static quantity. Some stimuli induce the production of specific dimer species, for example via processing of the p100 subunit to p52, which leads to an increase in p52-containing dimers [Figures 1C, 2; (55)]. There is also competition between the various NF-κB subunits for dimerization, due to the similar affinity of multiple subunits for a given subunit dimerization partner [Figure 2; (58)]. Therefore, if, for example, p52 abundance increases, not only could this induce an increase in repressive p52:p52 dimers, competition for NF-κB dimerization will reduce the abundance of lower dimerization affinity subunit pairs, which could lead to splitting of dimers containing two TD domains to generate p52-containing heterodimers, and effectively increase the abundance of transcription activating NF-κB dimers. Overall, although many studies consider only one protein, the RelA subunit, the total nuclear abundance of NF-κB factors could be substantially higher and the relative abundances of various dimers dynamically modulated. In the section ‘Competition between NF-κB dimers' below, we come back to this and discuss how different dimer abundances can impinge on κB binding site availability.
As we add resolution to quantitative understanding and models of NF-κB-driven transcription in various cellular contexts, we will need to reevaluate simplifying assumptions about the abundance of NF-κB dimers and consider the contributions of the combinatorial possibilities of the “NF-κB dimer network” (17). Because of dimer-specific activities, transcription is certainly impacted by subunit abundance and competition for partnering with TD-containing subunits.
Affinity of NF-κB Dimers for κB Binding Sequences
Biochemical DNA binding studies of a wide variety of 9–12 base-pair sequences have revealed that different NF-κB dimers bind far more sequences than previously thought, with different dimer species exhibiting specific but overlapping affinities for consensus and non-consensus κB site sequences (3, 22, 23). Although specific NF-κB dimer-DNA affinity values are hard to pin down because they are strongly condition-dependent (15), a constant is that for a given sequence and assay, the affinities of different dimers are consistent with more than one dimer being able to bind this sequence in cells [e.g.,(15, 22, 34)]. Many sequences that contain only a single consensus half-site also show substantial dimer binding (22). Furthermore, structural studies showed that in certain conformations, only one subunit of NF-κB dimers is involved in sequence-specific DNA interactions (24). Taken together, these studies indicate that κB half sites are sufficient for functional NF-κB dimer binding and that the state of the dimer may direct its binding toward certain sequences. Importantly, just as dimers exhibit preferences for different DNA sequences (Figure 2), the corollary must be true, that different DNA sequences may recruit one specific dimer combination over another.
Interestingly, once bound to DNA, each NF-κB dimer has been shown to induce different amounts of transcriptional activity from target genes [reviewed in (56)]. The clearest example, as mentioned above, is that because neither p50 or p52 possesses a TD, dimers containing just these subunits are unable to activate transcription alone. More subtle differences have also been reported, for example, the decreased recruitment of RNA polymerase II (RNAPII) as the IL12B promoter switches from binding RelA-containing dimers to RelB-containing dimers (59). The combination of dimer specificity with dimer switching during a response can thus provide a mechanism to generate temporally diverse NF-κB-dependent transcription responses. On the one hand, a response could be abbreviated when TD-containing dimers driving transcription are replaced with TD-lacking repressing dimers, to switch off gene transcription. In a specific example, the stabilization of p50 homodimers during the response of macrophages to LPS stimulation leads to curtailing of the pro-inflammatory transcription of TNFA (60), likely via a switch from transcriptionally active dimers to inactive p50 homodimers at the promoter region. By contrast, a switch to the p52/RelB heterodimer, which is insensitive to inhibitory IκB proteins, was found to facilitate the sustained activation of target genes such as NFKBIA and NFKB2 [encoding IκBα and p100/p52, respectively; (59)]. Therefore, the intricacies of sequence-specific affinities of NF-κB dimers and dimer-specific RNAPII-recruiting activities can enable not only tuning of the strength but also the duration and temporal patterns of transcriptional responses at target gene promoters.
How might different consensus κB sites modulate the activity of the NF-κB dimers? Structure-function studies have shown that binding to different consensus κB sites can alter the conformation of the bound NF-κB dimers, thus dictating dimer function [(61, 62), reviewed in (10, 63)]. When an NF-κB dimer interacts with a DNA sequence, side chains of the amino acids located in the DNA-binding domains of dimers contact the bases exposed in the groove of the DNA. For different consensus κB site sequences different bases are exposed in this groove, and NF-κB seems to alter its conformation to maximize interactions with the DNA and maintain high binding affinity (61). Changes in conformation may in turn impact NF-κB binding to co-regulators of transcription, whether these are activating or inhibitory, to specify the strength and dynamics of the transcriptional response (64). These findings again highlight how the huge array of κB binding site sequences must play a key role in modulating the transcription of target genes.
Finally, as an additional layer of dimer and sequence-specific regulation, each of the subunits can be phosphorylated at multiple sites with, depending on the site, effects on nearly every step of NF-κB activation [reviewed in (55)]. While the function of each phosphorylation site is still emerging, there are clear examples of phosphorylation events that have κB-sequence-specific effects on DNA binding and transcription (Figure 2). One of these is the phosphorylation of serine 329 (Ser329) of p50. This phosphorylation attenuates the affinity of p50 for consensus κB sites with a cytosine (C) vs. adenosine (A) at position −1, leading to differential transcriptional activation at A- vs. C-containing sites (65). In addition to effects on NF-κB dimer affinity for DNA, we note that phosphorylation at other sites on the NF-κB subunits has also been shown to affect dimer abundances, via effects on dimerization, monomer and dimer stability, affinity of IκBs, and nuclear translocation rates [reviewed in (55)].
Overall, when considering the various ways in which NF-κB dimer abundances and their affinity for DNA can be modulated, it becomes clear that with these multiple cascading effects, small differences in consensus κB site sequences and small a priori differences in interaction affinities can ultimately have a large impact on the transcriptional response to NF-κB pathway activation.
Availability of High Affinity κB Binding Sequences
Chromatin State
So far, in our discussion of the large numbers of κB sites on DNA and the high nuclear abundance of NF-κB dimers upon stimulation, we made a strong implicit assumption that all the consensus κB sites and half sites are available for binding. Given their high abundance, nuclear NF-κB dimers should be able to locate and bind to many consensus κB sites and half sites within minutes of an initial cell stimulation. However, ChIP-PCR studies in the murine monocytic cell line Raw 264.7, have shown that while recruitment of NF-κB occurs rapidly after LPS addition for a subset of genes (e.g., NFKBIA and CXCL2), other gene promoters containing high affinity κB sites remain unbound by NF-κB dimers for over an hour (e.g., CCL5 and IL6) despite the continued presence of nuclear NF-κB dimers (66). This kinetic complexity of the recruitment of NF-κB dimers to DNA during a stimulus-induced response has been largely attributed to variable, chromatin-state-dependent accessibility following stimulation.
The promoter regions of early response genes have abundant histone acetylation or trimethylation prior to stimulation [e.g., H3K27ac, (67) and H4K20me3, (66)], a chromatin state “poised” for immediate activation. This chromatin state may lead to a more open chromatin structure, constitutively accessible to transcription factor binding (66, 67). In contrast, promoters of late genes often have hypo-acetylated histones, requiring conformational changes to the chromatin to become accessible. They are therefore unable to recruit NF-κB for up to several hours after stimulation (68), due to the slow process of chromatin remodeling. Of note, we recently observed that recruitment of RelA-containing dimers displayed similar rapid binding kinetics at highly and poorly acetylated H3 HIV LTR insertions, but recruitment of RNAPII was different, with highly acetylated H3 correlating with more efficient transcription (69). Others have also reported early appearance of nascent transcripts of late genes, again hinting that, at least in some contexts, recruitment of NF-κB dimers may take place early, but that differential stability or processing of the transcript leads them to accumulating only later (70–72). Nevertheless, taken together, these different studies of chromatin state and NF-κB dimer binding suggest that despite the large repertoire of potential binding sites, only a fraction of these sites are available for binding, or for active recruitment of RNAPII, at any given time. This accessible repertoire can change upon stimulation and is dictated by the epigenetic state of the cell.
Indeed, another aspect of the NF-κB DNA-binding response that has been revealed by ChIP-seq experiments is its cell-type- and stimulus-specific nature, with different NF-κB subunits binding to diverse sites under different experimental conditions. For example, Xing et al. (5) compared the genes that were bound and regulated by RelA in TNF-treated HeLa human cervical carcinoma cells, to the direct, transcriptionally regulated target genes identified in LPS-treated U937 and THP-1 human monocytic cells. They found a strikingly small overlap between the sets of genes directly regulated by NF-κB in all three of these scenarios. Although deeper and less stringent analyses of these data may reveal a greater overlap, it is clear that cell type and stimulus combine to regulate chromatin accessibility and focus NF-κB dimer-DNA interactions at a subset of all consensus κB binding sites. Therefore, one role of the very large number of potential κB binding sites may be to allow context-specific and diverse use of the NF-κB pathway in response to a variety of stimuli and across different cell types and states.
Beyond the binding events monitored by ChIP-seq and other protein-DNA interaction assays, the “function” of a binding event is generally assessed by determining the transcriptional outcome of the nearest gene. However, this simple view may need to be revisited. Indeed, until recently, it was assumed that the regulatory elements of a gene must be located within several kilobases of its locus, and situated on the same chromosome (73). Contrary to this, there is mounting evidence of functional long-range interactions occurring between genomic regions that are situated megabases apart, and even located on different chromosomes (74, 75). Moreover, chromosome organization studies have implicated RelA-containing NF-κB dimers in the initiation or maintenance of higher-order intra- and inter-chromosomal complexes (76, 77). In particular, Apostolou and Thanos (77) found that RelA-containing NF-κB binding to specialized Alu repeats plays an important role in initiating interchromosomal interactions, and in the initiation of the IFNB1 enhanceosome assembly during the early stages of Sendai virus infection (77, 78). Alu repeats are ubiquitous repetitive DNA transposable elements that had been shown to contain putative κB-binding sites; they were later shown to represent 11% of p52-, RelB-, and RelA-bound sites in HeLa cells (1). What becomes clear is that NF-κB dimers, at least RelA-containing dimers, can use long-range intra- and inter-chromosomal interactions to regulate gene expression, meaning that the “nearest gene” method of assessing impact of consensus κB sites likely misestimates the number of functional sites.
Taken together, chromatin state and chromatin organization strongly influence the selection of DNA binding sites by NF-κB dimers and, most likely, the selection of the target genes that are regulated by these protein-DNA interaction events. Analyses that consider binding events in the context of three-dimensional nuclear organization and chromatin composition will be required to generate a more accurate view of the ways in which NF-κB-DNA binding affects gene transcription.
Competition Between NF-κB Dimers
In addition to cell-state specific chromatin modifications and chromatin conformation, NF-κB-driven transcriptional responses can also be modulated by competition between different dimer species for response element binding (Figure 2). Indeed, while global NF-κB dimer abundance may set the global number of sites that are occupied, which dimers are present pre- and post-stimulus will modulate which sites are transcriptionally activated or repressed, based on relative abundances and affinities. In particular, competition between dimers is consequential when dimers lacking a TD occupy consensus κB sites and limit site availability for newly translocated TD-containing NF-κB dimer binding. As mentioned above, this mechanism of transcription repression has been studied most extensively for the p50 homodimer, which has been shown to play a critical role dampening the inflammatory response [reviewed in (79, 80)]. Specifically, NFKB1 (p50-encoding) knockout mice have been shown to be more susceptible to several types of infection or infection models [e.g., (81–83)], and some of these responses have been linked to disruption of the transcriptional regulation of inflammatory signals (83, 84). By contrast, perturbations that increase nuclear p50:p50 lead to increased promoter binding by p50:p50 and reduced transcription in response to stimulation of many inflammatory genes (60, 85). Those two examples represent relative extremes of dimer concentrations modulation. Yet, given that, as we discussed above, the nuclear abundances of TD-containing NF-κB dimers appear far from saturating conditions, even moderate changes in nuclear concentration of TD-lacking dimers should affect consensus κB site availability to TD-containing, transcription activating dimer binding (see also Box 1). Taken together, differences in dimer abundances, along with competition for κB sites, help explain why different cell types or states exhibit varied responses to NF-κB-activating stimuli.
Co-regulators of Transcription
Because DNA binding by NF-κB may not necessarily require high affinity and high specificity [e.g., (3, 22)] and non-NF-κB transcription factors can also bind consensus κB sites due to degeneracies in recognized sequences (86) or as they search for their targets (43, 44), it follows that other transcription factors could act as co-regulators of transcription by competing with or helping recruit NF-κB dimers (Figure 2). In addition, TD-containing NF-κB subunits are also known to interact via their TD with a variety of transcriptional co-factors that modify the chromatin landscape to facilitate NF-κB recruitment and initiate transcription [e.g., (87, 88), and reviewed in (89, 90)]. Here, there are two potential scenarios: (1) the partner transcription regulators pre-exist at the κB sites and activation is rapid, or (2) the partner transcriptional regulators must be activated by the same stimulus that activates NF-κB. In the latter scenario, the time scale of NF-κB's ability to affect transcription will be dependent on the time scale of activation of its co-regulator. If this partner is required for releasing a binding site from a competitor or other barrier to site accessibility, then delayed partner activation is another factor that could explain the delayed NF-κB occupancy at high affinity sites. With these different possibilities, co-regulators could effectively modulate the abundance and identity of available κB sites over time.
How do NF-κB dimers interact with the chromatin modifying machinery? One way is via post-translationally modified NF-κB dimers that are known to recruit the histone acetyltransferases (HATs) p300 and CBP, which promote chromatin accessibility and transcription factor binding. For example, RelA interacts with the CBP/p300 complex once RelA Ser276 has been phosphorylated, following the degradation of IκBα (87, 91). When Ser276 is mutated to alanine to prevent phosphorylation, RelA cannot recruit CBP/p300 and fails to induce transcription at a subset of NF-κB-dependent genes (92). Intriguingly, which genes are affected does not seem directly related to whether they normally show delayed expression or not; some genes whose response normally peaks early show reduced transcription when RelA cannot recruit HATs (92), so there may be additional factors that come into play to determine site accessibility and its timing.
In contrast to RelA, the p50 and p52 NF-κB subunits lack a TD and as such bind to histone deacetylases (HDACs) instead of HATs [e.g., (93)]. HDACs are associated with formation of closed chromatin and transcriptional repression (94). In the context of κB sites found in the HIV genome, the recruitment of HDACs by the p50:p50 homodimer acts to maintain transcriptional repression and latency (93). Upon stimulation with cytokines that activate the NF-κB pathway, RelA-containing NF-κB dimers can displace p50:p50 homodimers and HDACs, leading to restoration of an acetylated histone state and transcriptional reactivation of the HIV genome (93). A similar mechanism has been suggested for the transcriptional regulation of pro-inflammatory cytokine genes in hepatic cells (84, 95). Taken together, these studies show that the interactions of NF-κB dimers with different modifiers of chromatin can result in the dynamic regulation of the chromatin state and of the availability of consensus κB sites for transcriptionally repressive or activating interactions with these dimers.
NF-κB has also been reported to bind cooperatively to many general transcription factors in vitro. For example, the in vitro assembly of the interferon-β (IFNB1) enhanceosome was shown to be dependent upon interactions with the AP-1 family member c-Jun, interferon regulatory factors (IRFs) and activating transcription factor 2 (ATF2) [reviewed in (10) and in (96)]. However, in vivo these factors are recruited to the enhanceosome in a stepwise manner, without any dependence upon cooperative interactions (77, 78). As such, there is currently little in vivo evidence that NF-κB dimers binding to consensus κB site is enhanced by association with partner transcription factors. Nevertheless, ChIP-seq experiments have detected many instances where NF-κB dimers may be brought to enhancer or promoter sequences lacking κB sites via interactions with another transcription regulator such as PU.1 or ZNF143 (3). Overall, with promoter and enhancer sequences replete with transcription factor binding sites, NF-κB dimers likely partner with other transcription factors to modulate target genes transcription, but how these interactions are coordinated and how they impact transcription is still unclear for most of these targets.
Although here we have only discussed a few specific examples, there are several other points of cross-talk between NF-κB dimers and other families of transcription factors [e.g., nuclear hormone receptors (97) and (33, 98), as well as STATs and IRFs, recently reviewed in (99)]. Overall, it is clear that the differing abilities of NF-κB dimers to recruit other transcriptional regulators via protein-protein interactions and the specific sets of DNA-protein interactions that can take place at each gene's regulatory region could potentially be combined to produce a vast diversity of context-specific, temporally distinct NF-κB-dependent gene expression patterns.
The Future of NF-κB Research
Understanding the mechanisms that regulate NF-κB-DNA interactions is critical to elucidating the intricacies of NF-κB-specific gene expression profiles. In this review we have discussed the relationships between NF-κB and κB binding sites, and some of the many known complexities of these relationships that affect the regulation of target genes (Table 1). However, despite the wealth of information that has already been uncovered by studies of the NF-κB transcription factors, a comprehensive understanding of the mechanisms underpinning NF-κB-DNA interactions that explain stimulus- and cell type-specific responses remains elusive as numerous questions are yet unanswered.
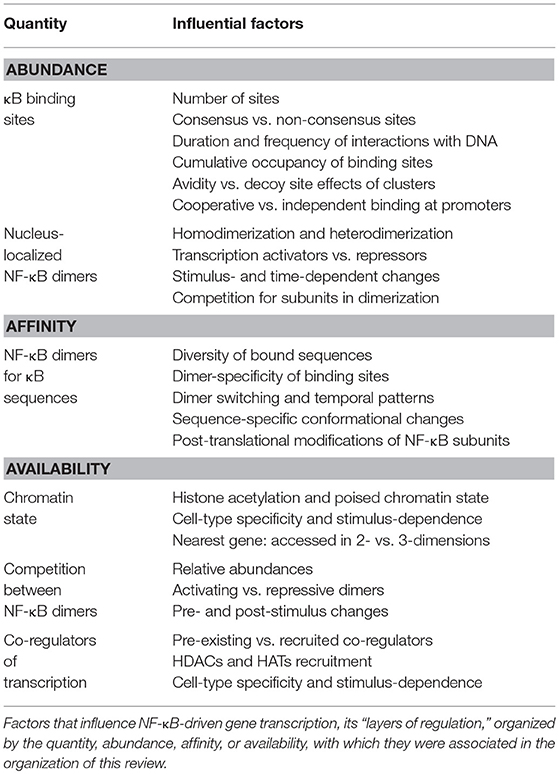
Table 1. Summary of layers of regulations influencing NF-κB-driven gene transcription.
In recent years, our ability to probe chromatin and visualize transcription has considerably advanced, and these advances will be key to developing a better understanding of the complex regulatory processes in the NF-κB system. Chromosome conformation capture (3C) and its subsequent iterations, most recently Hi-C (100), have facilitated the observation of chromatin folding and identification of long-range interactions on a genome-wide scale [reviewed in (101)]. Of particular interest for the study of the interactions of promoters of NF-κB target genes with other regions of the genome, the Promoter Capture Hi-C assay takes promoter-containing fragments from Hi-C libraries and performs paired-end sequencing to identify long-range promoter interactions with distal regulatory elements (102). However, it is important to keep in mind that such methods inform us on the enrichment of particular interactions in bulk populations of cells, an average readout of chromosomal interactions and conformation. Other approaches will be required to understand how different instances of the system vary and how this variability translates into different NF-κB-driven gene expression programs.
Simultaneous developments in the fields of biomolecular labeling and imaging technology have facilitated the visualization of transcription factor dynamics in living cells [reviewed in (29)]. These approaches offer unparalleled insights into the interactions occurring between transcription factors and DNA at the single-cell, single-molecule level. Early studies of transcription factor diffusion and DNA-binding dynamics often used fluorescent proteins and fluorescence recovery after photobleaching (FRAP) assays (29, 103). In FRAP assays, the rate of fluorescence recovery after bleaching then provides information regarding the diffusion and binding kinetics (kon and koff) of a large population of fluorescently labeled molecules (104, 105). However, FRAP measurements fail to accurately capture the heterogeneity in binding dynamics (26, 106).
Single-molecule tracking approaches promise a more complete picture of the different types of dynamic interactions, slow and fast, between NF-κB dimers and DNA. However, two difficult challenges from the use of fluorescent protein tags in these approaches are that the low photostability of fluorescent proteins can severely limit the duration of tracking and the generally high number of expressed fusion proteins yields densely packed, difficult to resolve, transcription factors. The advent of high-brightness, photostable, self-labeling dye tags, relying on fusion with the enzymatic HaloTag and SNAPTag (107, 108) is facilitating long-term imaging of single-molecules at high signal-to-noise ratios. In addition, the development of genome-editing techniques has enabled the tagging of endogenous proteins instead of relying on high-expression exogenous promoters for fusion proteins, thus generating more sparsely labeled populations of molecules to track. Combining these approaches with super-resolution imaging modalities such as photoactivated localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM) should open the door to the development of a clearer picture of the NF-κB-DNA interactions and subsequent gene expression.
Another quantitative aspect of NF-κB biology that merits a revisit in the future is how the landscape of dimer abundances changes across different cellular contexts and across time. Our current understanding of this landscapes relies mostly on bulk, population-based, endpoint biochemical assays, as well as inferences made from lots of accumulated knowledge from in vitro affinity measurements and disruptions of cellular contents with knockout of specific NF-κB subunits [reviewed in (56)]. From many single-cell studies of the dynamics of RelA translocation to the nucleus in different cell types and under different stimuli, we have learned that these dynamics are quite variable [e.g., (109–113)] and, importantly, that the observed variability is absolutely consequential for target gene expression and cellular outcomes [e.g., (14, 69, 72, 111, 114–121)]. However, in all these studies, we are left to assume which RelA-containing NF-κB dimers are actually present in each cell, and each nucleus, and we still lack a similar body of knowledge on NF-κB dimers that do not contain RelA. Capturing live-cell dynamics of the nuclear abundance of other NF-κB subunits and how these dynamics affect transcriptional output of target genes should help us figure out whether what we learned for RelA also applies to these other subunits. Finally, the application of fluorescence correlation spectroscopy (FCS) and fluorescence-lifetime imaging microscopy (FLIM) has begun to reveal aspects of protein dimerization and multimerization in other systems [e.g., p53 tetramerization in irradiated human cells (122) and cell type-determining transcription factors in the Arabidopsis root (123)]. In the future, similar approaches should help us broaden our understanding of how NF-κB transcription factors interact with each other, interact with other transcription regulators and interact with DNA in a complex, tunable system that regulates gene expression in many cellular decision processes.
Conclusions
Over the years, studies have dissected the NF-κB pathway, uncovering many factors and nuances that influence the outcome of DNA binding in this complex system. With hundreds of thousands of DNA binding proteins, and millions of potential DNA binding sites, the recruitment of NF-κB to DNA is regulated in complex ways. This regulation generates gene-, stimulus- and cell type-specific NF-κB responses, allowing NF-κB to respond to numerous different inputs, with a diverse array of outputs. However, a complete, mechanistic understanding of these processes remains unresolved. As we collect better measurements from single-molecule to genome-wide scales, systems biology models may now help us reassemble this dissected system into a framework that can predict ensembles of transcriptional responses.
Author Contributions
RB, AM, SM, and SG all contributed to the conceptualization, writing and editing of this review. SM performed the simulations presented in the Box 1.
Funding
This work was funded by NIH grant R01-GM104247 and funding from the Louis B. Mayer Foundation to SG and NIH award F32-AI136422 to AM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the reviewers for their helpful comments and suggestions. As part of the NF-κB community, we are fortunate to stand on the shoulders of giants, although it means that it is not possible to include all the seminal studies in one review and we are forced to choose illustrative examples. We apologize to all whose work we were not able to cite.
References
1. Antonaki A, Demetriades C, Polyzos A, Banos A, Vatsellas G, Lavigne MD, et al. Genomic analysis reveals a novel nuclear factor-kappaB (NF-kappaB)-binding site in Alu-repetitive elements. J Biol Chem. (2011) 286:38768–82. doi: 10.1074/jbc.M111.234161
2. Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci USA. (2003) 100:12247–52. doi: 10.1073/pnas.2135255100
3. Zhao B, Barrera LA, Ersing I, Willox B, Schmidt SC, Greenfeld H, et al. The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. (2014) 8:1595–606. doi: 10.1016/j.celrep.2014.07.037
4. Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, et al. Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell. (2018) 173:1796–1809 e1717. doi: 10.1016/j.cell.2018.04.018
5. Xing Y, Yang Y, Zhou F, Wang J. Characterization of genome-wide binding of NF-kappaB in TNFalpha-stimulated HeLa cells. Gene. (2013) 526:142–9. doi: 10.1016/j.gene.2013.05.001
6. Wan F, Lenardo MJ. Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb Perspect Biol. (2009) 1:a000067. doi: 10.1101/cshperspect.a000067
7. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. (1986) 46:705–16. doi: 10.1016/0092-8674(86)90346-6
8. Lenardo M, Pierce JW, Baltimore D. Protein-binding sites in Ig gene enhancers determine transcriptional activity and inducibility. Science. (1987) 236:1573–7. doi: 10.1126/science.3109035
9. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: a blossoming of relevance to human pathobiology. Cell. (2017) 168:37–57. doi: 10.1016/j.cell.2016.12.012
10. Natoli G, Saccani S, Bosisio D, Marazzi I. Interactions of NF-kappaB with chromatin: the art of being at the right place at the right time. Nat Immunol. (2005) 6:439–45. doi: 10.1038/ni1196
11. Hottiger MO, Felzien LK, Nabel GJ. Modulation of cytokine-induced HIV gene expression by competitive binding of transcription factors to the coactivator p300. EMBO J. (1998) 17:3124–34. doi: 10.1093/emboj/17.11.3124
12. Gilmore T. D. NF-kB Target Genes (online). Available online at: http://www.bu.edu/nf-kb/gene-resources/target-genes/ (Accessed 2018).
13. Stewart-Ornstein J, Nelson C, DeRisi J, Weissman JS, El-Samad H. Msn2 coordinates a stoichiometric gene expression program. Curr Biol. (2013) 23:2336–45. doi: 10.1016/j.cub.2013.09.043
14. Lee RE, Walker SR, Savery K, Frank DA, Gaudet S. Fold change of nuclear NF-kappaB determines TNF-induced transcription in single cells. Mol Cell. (2014) 53:867–79. doi: 10.1016/j.molcel.2014.01.026
15. Phelps CB, Sengchanthalangsy LL, Malek S, Ghosh G. Mechanism of kappa B DNA binding by Rel/NF-kappa B dimers. J Biol Chem. (2000) 275:24392–9. doi: 10.1074/jbc.M003784200
16. Bosisio D, Marazzi I, Agresti A, Shimizu N, Bianchi ME, Natoli G. A hyper-dynamic equilibrium between promoter-bound and nucleoplasmic dimers controls NF-kappaB-dependent gene activity. EMBO J. (2006) 25:798–810. doi: 10.1038/sj.emboj.7600977
17. Basak S, Behar M, Hoffmann A. Lessons from mathematically modeling the NF-kappaB pathway. Immunol Rev. (2012) 246:221–38. doi: 10.1111/j.1600-065X.2011.01092.x
18. Wang Z, Potoyan DA, Wolynes PG. Modeling the therapeutic efficacy of NFkappaB synthetic decoy oligodeoxynucleotides (ODNs). BMC Syst Biol. (2018) 12:4. doi: 10.1186/s12918-018-0525-6
19. Zabel U, Schreck R, Baeuerle PA. DNA binding of purified transcription factor NF-kappa B. Affinity, specificity, Zn2+ dependence, and differential half-site recognition. J Biol Chem. (1991) 266:252–60.
20. Chen FE, Ghosh G. Regulation of DNA binding by Rel/NF-kappaB transcription factors: structural views. Oncogene. (1999) 18:6845–52. doi: 10.1038/sj.onc.1203224
21. Huang DB, Chen YQ, Ruetsche M, Phelps CB, Ghosh G. X-ray crystal structure of proto-oncogene product c-Rel bound to the CD28 response element of IL-2. Structure. (2001) 9:669–78. doi: 10.1016/S0969-2126(01)00635-9
22. Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, et al. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-kappaB family DNA binding. Nat Immunol. (2011) 13:95–102. doi: 10.1038/ni.2151
23. Wong D, Teixeira A, Oikonomopoulos S, Humburg P, Lone IN, Saliba D, et al. Extensive characterization of NF-kappaB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. (2011) 12:R70. doi: 10.1186/gb-2011-12-7-r70
24. Mulero MC, Huang DB, Nguyen HT, Wang VY, Li Y, Biswas T, et al. DNA-binding affinity and transcriptional activity of the RelA homodimer of nuclear factor kappaB are not correlated. J Biol Chem. (2017) 292:18821–30. doi: 10.1074/jbc.M117.813980
25. Mueller F, Wach P, McNally JG. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys J. (2008) 94:3323–39. doi: 10.1529/biophysj.107.123182
26. Mueller F, Mazza D, Stasevich TJ, McNally JG. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: what do we really know? Curr Opin Cell Biol. (2010) 22:403–11. doi: 10.1016/j.ceb.2010.03.002
27. Leith JS, Tafvizi A, Huang F, Uspal WE, Doyle PS, Fersht AR, et al. Sequence-dependent sliding kinetics of p53. Proc Natl Acad Sci USA. (2012) 109:16552–7. doi: 10.1073/pnas.1120452109
28. Murata A, Itoh Y, Mano E, Kanbayashi S, Igarashi C, Takahashi H, et al. One-dimensional search dynamics of tumor suppressor p53 regulated by a disordered C-terminal domain. Biophys J. (2017) 112:2301–14. doi: 10.1016/j.bpj.2017.04.038
29. Liu Z, Tjian R. Visualizing transcription factor dynamics in living cells. J Cell Biol. (2018) 217:1181–91. doi: 10.1083/jcb.201710038
30. Zabel U, Baeuerle PA. Purified human I kappa B can rapidly dissociate the complex of the NF-kappa B transcription factor with its cognate DNA. Cell. (1990) 61:255–65. doi: 10.1016/0092-8674(90)90806-P
31. Nalley K, Johnston SA, Kodadek T. Proteolytic turnover of the Gal4 transcription factor is not required for function in vivo. Nature. (2006) 442:1054–7. doi: 10.1038/nature05067
32. Kodadek T, Sikder D, Nalley K. Keeping transcriptional activators under control. Cell. (2006) 127:261–4. doi: 10.1016/j.cell.2006.10.002
33. Pascual G, Glass CK. Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab. (2006) 17:321–7. doi: 10.1016/j.tem.2006.08.005
34. Bergqvist S, Alverdi V, Mengel B, Hoffmann A, Ghosh G, Komives EA. Kinetic enhancement of NF-kappaBxDNA dissociation by IkappaBalpha. Proc Natl Acad Sci USA. (2009) 106:19328–33. doi: 10.1073/pnas.0908797106
35. Sue SC, Alverdi V, Komives EA, Dyson HJ. Detection of a ternary complex of NF-kappaB and IkappaBalpha with DNA provides insights into how IkappaBalpha removes NF-kappaB from transcription sites. Proc Natl Acad Sci USA. (2011) 108:1367–72. doi: 10.1073/pnas.1014323108
36. Alverdi V, Hetrick B, Joseph S, Komives EA. Direct observation of a transient ternary complex during IkappaBalpha-mediated dissociation of NF-kappaB from DNA. Proc Natl Acad Sci USA. (2014) 111:225–30. doi: 10.1073/pnas.1318115111
37. Potoyan DA, Zheng W, Komives EA, Wolynes PG. Molecular stripping in the NF-kappaB/IkappaB/DNA genetic regulatory network. Proc Natl Acad Sci USA. (2016) 113:110–5. doi: 10.1073/pnas.1520483112
38. Dembinski HE, Wismer K, Vargas JD, Suryawanshi GW, Kern N, Kroon G, et al. Functional importance of stripping in NFkappaB signaling revealed by a stripping-impaired IkappaBalpha mutant. Proc Natl Acad Sci USA. (2017) 114:1916–21. doi: 10.1073/pnas.1610192114
39. Potoyan DA, Bueno C, Zheng W, Komives EA, Wolynes PG. Resolving the NFkappaB heterodimer binding paradox: strain and frustration guide the binding of dimeric transcription factors. J Am Chem Soc. (2017) 139:18558–66. doi: 10.1021/jacs.7b08741
40. Callegari A, Sieben C, Benke A, Suter DM, Fierz B, Mazza D, et al. Single-molecule dynamics and genome-wide transcriptomics reveal that NF-kB (p65)-DNA binding times can be decoupled from transcriptional activation. PLoS Genet. (2019) 15:e1007891. doi: 10.1371/journal.pgen.1007891
41. Chen J, Zhang Z, Li L, Chen BC, Revyakin A, Hajj B, et al. Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell. (2014) 156:1274–85. doi: 10.1016/j.cell.2014.01.062
42. Fisher WW, Li JJ, Hammonds AS, Brown JB, Pfeiffer BD, Weiszmann R, et al. DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila. Proc Natl Acad Sci USA. (2012) 109:21330–5. doi: 10.1073/pnas.1209589110
43. Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. (1981) 20:6929–48. doi: 10.1021/bi00527a028
44. Gowers DM, Wilson GG, Halford SE. Measurement of the contributions of 1D and 3D pathways to the translocation of a protein along DNA. Proc Natl Acad Sci USA. (2005) 102:15883–8. doi: 10.1073/pnas.0505378102
45. Kitov PI, Bundle DR. On the nature of the multivalency effect: a thermodynamic model. J Am Chem Soc. (2003) 125:16271–84. doi: 10.1021/ja038223n
46. Kemme CA, Nguyen D, Chattopadhyay A, Iwahara J. Regulation of transcription factors via natural decoys in genomic DNA. Transcription. (2016) 7:115–20. doi: 10.1080/21541264.2016.1188873
47. Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, et al. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. (1997) 3:894–9. doi: 10.1038/nm0897-894
48. Lee TH, Maheshri N. A regulatory role for repeated decoy transcription factor binding sites in target gene expression. Mol Syst Biol. (2012) 8:576. doi: 10.1038/msb.2012.7
49. Brewster RC, Weinert FM, Garcia HG, Song D, Rydenfelt M, Phillips R. The transcription factor titration effect dictates level of gene expression. Cell. (2014) 156:1312–23. doi: 10.1016/j.cell.2014.02.022
50. Wunderlich Z, Mirny LA. Different gene regulation strategies revealed by analysis of binding motifs. Trends Genet. (2009) 25:434–40. doi: 10.1016/j.tig.2009.08.003
51. Walters MC, Fiering S, Eidemiller J, Magis W, Groudine M, Martin DI. Enhancers increase the probability but not the level of gene expression. Proc Natl Acad Sci USA. (1995) 92:7125–9. doi: 10.1073/pnas.92.15.7125
52. Giorgetti L, Siggers T, Tiana G, Caprara G, Notarbartolo S, Corona T, et al. Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol Cell. (2010) 37:418–28. doi: 10.1016/j.molcel.2010.01.016
53. Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, et al. The human transcription factors. Cell. (2018) 172:650–65. doi: 10.1016/j.cell.2018.01.029
54. Banoth B, Chatterjee B, Vijayaragavan B, Prasad MV, Roy P, Basak S. Stimulus-selective crosstalk via the NF-kappaB signaling system reinforces innate immune response to alleviate gut infection. Elife. (2015) 4:e05648. doi: 10.7554/eLife.05648
55. Christian F, Smith EL, Carmody RJ. The regulation of NF-kappaB subunits by phosphorylation. Cells. (2016) 5:E12. doi: 10.3390/cells5010012
56. Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med. (2016) 8:227–41. doi: 10.1002/wsbm.1331
57. Cheng CS, Feldman KE, Lee J, Verma S, Huang DB, Huynh K, et al. The specificity of innate immune responses is enforced by repression of interferon response elements by NF-kappaB p50. Sci Signal. (2011) 4:ra11. doi: 10.1126/scisignal.2001501
58. Tsui R, Kearns JD, Lynch C, Vu D, Ngo KA, Basak S, et al. IkappaBbeta enhances the generation of the low-affinity NFkappaB/RelA homodimer. Nat Commun. (2015) 6:7068. doi: 10.1038/ncomms8068
59. Saccani S, Pantano S, Natoli G. Modulation of NF-kappaB activity by exchange of dimers. Mol Cell. (2003) 11:1563–74. doi: 10.1016/S1097-2765(03)00227-2
60. Wessells J, Baer M, Young HA, Claudio E, Brown K, Siebenlist U, et al. BCL-3 and NF-kappaB p50 attenuate lipopolysaccharide-induced inflammatory responses in macrophages. J Biol Chem. (2004) 279:49995–50003. doi: 10.1074/jbc.M404246200
61. Leung TH, Hoffmann A, Baltimore D. One nucleotide in a kappaB site can determine cofactor specificity for NF-kappaB dimers. Cell. (2004) 118:453–64. doi: 10.1016/j.cell.2004.08.007
62. Natoli G. Little things that count in transcriptional regulation. Cell. (2004) 118:406–8. doi: 10.1016/j.cell.2004.08.003
63. Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. (2006) 25:6706–16. doi: 10.1038/sj.onc.1209933
64. Wang VY, Huang W, Asagiri M, Spann N, Hoffmann A, Glass C, et al. The transcriptional specificity of NF-kappaB dimers is coded within the kappaB DNA response elements. Cell Rep. (2012) 2:824–39. doi: 10.1016/j.celrep.2012.08.042
65. Crawley CD, Raleigh DR, Kang S, Voce DJ, Schmitt AM, Weichselbaum RR, et al. DNA damage-induced cytotoxicity is mediated by the cooperative interaction of phospho-NF-kappaB p50 and a single nucleotide in the kappaB-site. Nucleic Acids Res. (2013) 41:764–74. doi: 10.1093/nar/gks1120
66. Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med. (2001) 193:1351–9. doi: 10.1084/jem.193.12.1351
67. Ramirez-Carrozzi VR, Nazarian AA, Li CC, Gore SL, Sridharan R, Imbalzano AN, et al. Selective and antagonistic functions of SWI/SNF and Mi-2beta nucleosome remodeling complexes during an inflammatory response. Genes Dev. (2006) 20:282–96. doi: 10.1101/gad.1383206
68. Kayama H, Ramirez-Carrozzi VR, Yamamoto M, Mizutani T, Kuwata H, Iba H, et al. Class-specific regulation of pro-inflammatory genes by MyD88 pathways and IkappaBzeta. J Biol Chem. (2008) 283:12468–77. doi: 10.1074/jbc.M709965200
69. Wong VC, Bass VL, Bullock ME, Chavali AK, Lee REC, Mothes W, et al. NF-kappaB-chromatin interactions drive diverse phenotypes by modulating transcriptional noise. Cell Rep. (2018) 22:585–99. doi: 10.1016/j.celrep.2017.12.080
70. Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol. (2009) 10:281–8. doi: 10.1038/ni.1699
71. Hao S, Baltimore D. RNA splicing regulates the temporal order of TNF-induced gene expression. Proc Natl Acad Sci USA. (2013) 110:11934–9. doi: 10.1073/pnas.1309990110
72. Zambrano S, De Toma I, Piffer A, Bianchi ME, Agresti A. NF-kappaB oscillations translate into functionally related patterns of gene expression. Elife. (2016) 5:e09100. doi: 10.7554/eLife.09100
73. Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. (2013) 93:779–97. doi: 10.1016/j.ajhg.2013.10.012
74. Fraser P, Bickmore W. Nuclear organization of the genome and the potential for gene regulation. Nature. (2007) 447:413–7. doi: 10.1038/nature05916
75. Miele A, Dekker J. Long-range chromosomal interactions and gene regulation. Mol Biosyst. (2008) 4:1046–57. doi: 10.1039/b803580f
76. Liu Z, Garrard WT. Long-range interactions between three transcriptional enhancers, active Vkappa gene promoters, and a 3' boundary sequence spanning 46 kilobases. Mol Cell Biol. (2005) 25:3220–31. doi: 10.1128/MCB.25.8.3220-3231.2005
77. Apostolou E, Thanos D. Virus Infection Induces NF-kappaB-dependent interchromosomal associations mediating monoallelic IFN-beta gene expression. Cell. (2008) 134:85–96. doi: 10.1016/j.cell.2008.05.052
78. Schoenfelder S, Fraser P. Interchromosomal huddle kickstarts antiviral defense. Cell. (2008) 134:14–6. doi: 10.1016/j.cell.2008.06.041
79. Perkins ND. The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer. (2012) 12:121–32. doi: 10.1038/nrc3204
80. Cartwright T, Perkins ND, L Wilson C. NFKB1: a suppressor of inflammation, ageing and cancer. FEBS J. (2016) 283:1812–22. doi: 10.1111/febs.13627
81. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. (1995) 80:321–30. doi: 10.1016/0092-8674(95)90415-8
82. Artis D, Speirs K, Joyce K, Goldschmidt M, Caamano J, Hunter CA, et al. NF-kappa B1 is required for optimal CD4+ Th1 cell development and resistance to Leishmania major. J Immunol. (2003) 170:1995–2003. doi: 10.4049/jimmunol.170.4.1995
83. Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. (2006) 281:26041–50. doi: 10.1074/jbc.M602222200
84. Elsharkawy AM, Oakley F, Lin F, Packham G, Mann DA, Mann J. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol. (2010) 53:519–27. doi: 10.1016/j.jhep.2010.03.025
85. Calegari-Silva TC, Vivarini AC, Miqueline M, Dos Santos GR, Teixeira KL, Saliba AM, et al. The human parasite Leishmania amazonensis downregulates iNOS expression via NF-kappaB p50/p50 homodimer: role of the PI3K/Akt pathway. Open Biol. (2015) 5:150118. doi: 10.1098/rsob.150118
86. Inukai S, Kock KH, Bulyk ML. Transcription factor-DNA binding: beyond binding site motifs. Curr Opin Genet Dev. (2017) 43:110–9. doi: 10.1016/j.gde.2017.02.007
87. Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. (2002) 9:625–36. doi: 10.1016/S1097-2765(02)00477-X
88. Lecoq L, Raiola L, Chabot PR, Cyr N, Arseneault G, Legault P, et al. Structural characterization of interactions between transactivation domain 1 of the p65 subunit of NF-kappaB and transcription regulatory factors. Nucleic Acids Res. (2017) 45:5564–76. doi: 10.1093/nar/gkx146
89. O'Shea JM, Perkins ND. Regulation of the RelA (p65) transactivation domain. Biochem Soc Trans. (2008) 36(Pt 4):603–8. doi: 10.1042/BST0360603
90. Bhatt D, Ghosh S. Regulation of the NF-kappaB-mediated transcription of inflammatory genes. Front Immunol. (2014) 5:71. doi: 10.3389/fimmu.2014.00071
91. Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. (1998) 1:661–71. doi: 10.1016/S1097-2765(00)80066-0
92. Dong J, Jimi E, Zhong H, Hayden MS, Ghosh S. Repression of gene expression by unphosphorylated NF-kappaB p65 through epigenetic mechanisms. Genes Dev. (2008) 22:1159–73. doi: 10.1101/gad.1657408
93. Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. (2006) 25:139–49. doi: 10.1038/sj.emboj.7600900
94. Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. (1998) 20:615–26. doi: 10.1002/(SICI)1521-1878(199808)20:8andlt;615::AID-BIES4andgt;3.0.CO;2-H
95. Wilson CL, Jurk D, Fullard N, Banks P, Page A, Luli S, et al. NFkappaB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat Commun. (2015) 6:6818. doi: 10.1038/ncomms7818
96. Hoffmann A, Baltimore D. Circuitry of nuclear factor kappaB signaling. Immunol Rev. (2006) 210:171–86. doi: 10.1111/j.0105-2896.2006.00375.x
97. Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. (2005) 122:707–21. doi: 10.1016/j.cell.2005.06.029
98. Sacta MA, Tharmalingam B, Coppo M, Rollins DA, Deochand DK, Benjamin B, et al. Gene-specific mechanisms direct glucocorticoid-receptor-driven repression of inflammatory response genes in macrophages. Elife. (2018) 7:e34864. doi: 10.7554/eLife.34864
99. Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFkappaB in inflammation. Front Immunol. (2018) 9:2542. doi: 10.3389/fimmu.2018.02542
100. Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat Genet. (2015) 47:598–606. doi: 10.1038/ng.3286
101. Yu M, Ren B. The three-dimensional organization of mammalian genomes. Annu Rev Cell Dev Biol. (2017) 33:265–89. doi: 10.1146/annurev-cellbio-100616-060531
102. Schoenfelder S, Javierre BM, Furlan-Magaril M, Wingett SW, Fraser P. Promoter capture Hi-C: high-resolution, genome-wide profiling of promoter interactions. J Vis Exp. (2018). doi: 10.3791/57320
103. Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J. (1976) 16:1055–69. doi: 10.1016/S0006-3495(76)85755-4
104. Voss TC, Hager GL. Visualizing chromatin dynamics in intact cells. Biochim Biophys Acta. (2008) 1783:2044–51. doi: 10.1016/j.bbamcr.2008.06.022
105. Nissim-Rafinia M, Meshorer E. Photobleaching assays (FRAP and FLIP) to measure chromatin protein dynamics in living embryonic stem cells. J Vis Exp. (2011) 2696. doi: 10.3791/2696
106. Mazza D, Abernathy A, Golob N, Morisaki T, McNally JG. A benchmark for chromatin binding measurements in live cells. Nucleic Acids Res. (2012) 40:e119. doi: 10.1093/nar/gks701
107. Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods. (2015) 12, 244–50, 243 p following 250. doi: 10.1038/nmeth.3256
108. Grimm JB, Brown TA, English BP, Lionnet T, Lavis LD. Synthesis of Janelia Fluor HaloTag and SNAP-Tag ligands and their use in cellular imaging experiments. Methods Mol Biol. (2017) 1663:179–88. doi: 10.1007/978-1-4939-7265-4_15
109. Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, et al. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science. (2004) 306:704–8. doi: 10.1126/science.1099962
110. Tay S, Hughey JJ, Lee TK, Lipniacki T, Quake SR, Covert MW. Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature. (2010) 466:267–71. doi: 10.1038/nature09145
111. Cheng Z, Taylor B, Ourthiague DR, Hoffmann A. Distinct single-cell signaling characteristics are conferred by the MyD88 and TRIF pathways during TLR4 activation. Sci Signal. (2015) 8:ra69. doi: 10.1126/scisignal.aaa5208
112. Ankers JM, Awais R, Jones NA, Boyd J, Ryan S, Adamson AD, et al. Dynamic NF-kappaB and E2F interactions control the priority and timing of inflammatory signalling and cell proliferation. Elife. (2016) 5:e10473. doi: 10.7554/eLife.10473
113. Harper CV, Woodcock DJ, Lam C, Garcia-Albornoz M, Adamson A, Ashall L, et al. Temperature regulates NF-kappaB dynamics and function through timing of A20 transcription. Proc Natl Acad Sci USA. (2018) 115:E5243–9. doi: 10.1073/pnas.1803609115
114. Ashall L, Horton CA, Nelson DE, Paszek P, Harper CV, Sillitoe K, et al. Pulsatile stimulation determines timing and specificity of NF-kappaB-dependent transcription. Science. (2009) 324:242–6. doi: 10.1126/science.1164860
115. Sung MH, Salvatore L, De Lorenzi R, Indrawan A, Pasparakis M, Hager GL, et al. Sustained oscillations of NF-kappaB produce distinct genome scanning and gene expression profiles. PLoS ONE. (2009) 4:e7163. doi: 10.1371/journal.pone.0007163
116. Sung MH, Li N, Lao Q, Gottschalk RA, Hager GL, Fraser ID. Switching of the relative dominance between feedback mechanisms in lipopolysaccharide-induced NF-kappaB signaling. Sci Signal. (2014) 7:ra6. doi: 10.1126/scisignal.2004764
117. Adamson A, Boddington C, Downton P, Rowe W, Bagnall J, Lam C, et al. Signal transduction controls heterogeneous NF-kappaB dynamics and target gene expression through cytokine-specific refractory states. Nat Commun. (2016) 7:12057. doi: 10.1038/ncomms12057
118. Junkin M, Kaestli AJ, Cheng Z, Jordi C, Albayrak C, Hoffmann A, et al. High-content quantification of single-cell immune dynamics. Cell Rep. (2016) 15:411–22. doi: 10.1016/j.celrep.2016.03.033
119. Lee RE, Qasaimeh MA, Xia X, Juncker D, Gaudet S. NF-kappaB signalling and cell fate decisions in response to a short pulse of tumour necrosis factor. Sci Rep. (2016) 6:39519. doi: 10.1038/srep39519
120. Zhang Q, Gupta S, Schipper DL, Kowalczyk GJ, Mancini AE, Faeder JR, et al. NF-kappaB dynamics discriminate between TNF doses in single cells. Cell Syst. (2017) 5:638–45 e635. doi: 10.1016/j.cels.2017.10.011
121. Bagnall J, Boddington C, England H, Brignall R, Downton P, Alsoufi Z, et al. Quantitative analysis of competitive cytokine signaling predicts tissue thresholds for the propagation of macrophage activation. Sci. Signal. (2018) 11:eaaf3998. doi: 10.1126/scisignal.aaf3998
122. Gaglia G, Guan Y, Shah JV, Lahav G. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci USA. (2013) 110:15497–501. doi: 10.1073/pnas.1311126110
Keywords: NF-κB, transcription regulation, specificity, accessibility, competition
Citation: Brignall R, Moody AT, Mathew S and Gaudet S (2019) Considering Abundance, Affinity, and Binding Site Availability in the NF-κB Target Selection Puzzle. Front. Immunol. 10:609. doi: 10.3389/fimmu.2019.00609
Received: 13 November 2018; Accepted: 07 March 2019;
Published: 29 March 2019.
Edited by:
Myong-Hee Sung, National Institutes of Health (NIH), United StatesReviewed by:
Alexander Hoffmann, University of California, Los Angeles, United StatesTrevor Siggers, Boston University, United States
Copyright © 2019 Brignall, Moody, Mathew and Gaudet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suzanne Gaudet, c3V6YW5uZV9nYXVkZXRAZGZjaS5oYXJ2YXJkLmVkdQ==