 Patrick Neubert
Patrick Neubert Agnes Schröder2
Agnes Schröder2 Dominik N. Müller
Dominik N. Müller Jonathan Jantsch
Jonathan Jantsch- 1Institute of Clinical Microbiology and Hygiene, University Hospital Regensburg, University of Regensburg, Regensburg, Germany
- 2Department of Orthodontics, University Hospital Regensburg, University of Regensburg, Regensburg, Germany
- 3Experimental and Clinical Research Center, A Joint Cooperation of Max-Delbrück Center for Molecular Medicine and Charité-Universitätsmedizin Berlin, Berlin, Germany
- 4Max-Delbrück Center for Molecular Medicine in the Helmholtz Association, Berlin, Germany
Local Na+ balance emerges as an important factor of tissue microenvironment. On the one hand, immune cells impact on local Na+ levels. On the other hand, Na+ availability is able to influence immune responses. In contrast to macrophages, our knowledge of dendritic cells (DCs) in this state of affair is rather limited. Current evidence suggests that the impact of increased Na+ on DCs is context dependent. Moreover, it is conceivable that DC immunobiology might also be influenced by Na+-rich-diet-induced changes of the gut microbiome.
Dendritic cells (DCs) represent important sentinel cells that continuously scan their microenvironment and play a key role in inducing immune responses and maintaining immunogenic tolerance [reviewed in (1–3)]. It is accepted that DCs are able to respond to a plethora of proteinaceous, lipid or carbohydrate molecules as well as nucleic acids via specialized receptors and signaling pathways [reviewed in (4–6)]. Recently, however, it emerged that the local Na+ electrolyte abundance impacts on innate and adaptive immune cell function and vice versa [reviewed in (7, 8)].
Extrarenal Na+ Storage
In general, body Na+ and fluid homeostasis are known to be regulated in very narrow limits. Disturbing this balance by excessive dietary salt intake is linked to various diseases including hypertension and autoimmunity, which ultimately results in increased morbidity and mortality [reviewed in (9, 10)]. Traditionally, the kidney was seen as the sole organ that controls body salt content and fluid regulation. For that purpose, Na+ concentrations of about 400 mM can be reached at the renal loop bend accompanied by osmolalities of up to about 1,200 mOsm/kg in the renal medulla (11). The remaining extracellular body fluids are thought to readily equilibrate with plasma. Therefore, extra-renal regulation of total body and certain tissue Na+ content and concentration was largely ignored [reviewed in (7, 12–15)], even though evidence of interstitial salt storage was provided already in 1909, when chloride storage was found in the skin during pre-clinical studies (16, 17). Within the last twenty years, however, the interstitium of the skin has emerged as important organ involved in maintaining body Na+ balance. For instance chemical analysis in rodents revealed that the effective osmolyte concentration in skin tissue (i.e., skin (Na++K+)/skin water) can reach levels of about 190 mM which is substantially higher than the effective osmolyte concentration in plasma of about 145 mM (18). Recent evidence using 23Na MRI and mathematical modeling demonstrate that very high Na+ concentrations are present at the epidermal and dermal junction zone (19, 20). Of note, chemical analysis of skin biopsies confirmed that the skin may serve as a Na+ buffer also in humans (21). This Na+-storage is reversible by dialysis (22, 23) and is able to strengthen the innate immune barrier by invigorating macrophage-dependent responses against intruding pathogens (24).
Elevated Na+ deposition is paralleled by changes in the gel-like cutaneous collagen matrix (25–27). Upon Na+-rich diets, there is an increased sulfation of glycosaminoglycan (GAGs) which might enable cutaneous Na+ storage [reviewed in (15)]. In addition to high Na+ containing diets (18, 27–29), it emerged that superficial skin infections (24) and chronic inflammatory processes (30) are able to trigger local Na+ accumulation. The mechanisms underlying both, the diet-dependent and diet-independent Na+ accumulation in the skin are, however, unknown. It is tempting to speculate that soluble or cell-bound mediators are able to modulate the GAG network's ability to serve as a negative charge capacitor facilitating local Na+ accumulation (27). Moreover, aldosterone and glucocorticoids may play an important role in this state of affair (31).
Dendritic Cells as Potential Regulators of Cutaneous Na+ Stores
While the mechanisms that allow for local cutaneous Na+ accumulation remain elusive, depletion of mononuclear phagocytes using clodronate liposomes unraveled that these cells play an important role in regulating cutaneous Na+ stores (18, 28). In addition, targeting the osmoprotective transcription factor nuclear factor of activated T cells 5 (Nfat5) in myeloid cells using Lyz2 (Lysozym2)/ LysM-Cre deleter mice revealed that this transcription factor plays an important role in sensing Na+-rich diet-induced local hypertonic environments (29). This myeloid cell specific osmoprotective response included the upregulation of the Nfat5 target gene vascular endothelial cell growth factor C (Vegfc) which ultimately leads to lymphcapillary hyperplasia facilitating removal of Na+ from the skin (18, 29). Recent evidence also suggests that local Na+ storage additionally increases lymph flow in muscle and skin (32).
However, clodronate liposomes are known to deplete various mononuclear phagocytes in the skin including monocytes, macrophages and DCs (33). Moreover, although Lyz2 Cre primarily induces recombination in granulocytes, monocytes and macrophages, there is some recombination occurring in DCs (34, 35). In the Immgen Database (www.immgen.org), DCs, for instance, from skin draining lymph nodes (LN) (CD11c+, MHCIIhi, Langerin−, CD11b− CD103− CD8a− CD4−; CD11c+, MHCIIhi, Langerin−, CD11b+ CD103− CD8a− CD4−; CD11c+, MHCIIhi, Langerin+, CD11blow, CD103+, CD8a−, CD4−; CD11c+, MHCIIhi, Langerin+, CD11b+ CD103− CD8a− CD4−) express very high Nfat5 levels, suggesting that these cells might be involved in organization and regulation of cutaneous Na+ balance. To the best of our knowledge, the relative contribution of different mononuclear phagocyte subtypes including various DC subtypes in this state of affair is, however, unexplored. The use of novel DC- and macrophage-specific (transcriptional) reporter mouse strains and ablation strategies might be useful to uncover the relative contribution of distinct mononuclear phagocyte subtypes [reviewed in (36–38)]. It is likely that, in addition to macrophages, DCs might fulfill distinct tasks in regulating cutaneous Na+ balance. Recently, Randolph and colleagues demonstrated that lymphatic vessel permeability is controlled by DCs in a G protein-coupled homing receptor CCR7-dependent manner. Further analysis revealed that this task is fulfilled by IFN regulatory factor 4-positive DC subset (39). Taking these observations and the data from the Immgen database into account it is possible that DC-mediated regulation of the lymphatic vessels might be involved in facilitating the drainage of excess Na+ from cutaneous interstitial space (Figure 1).
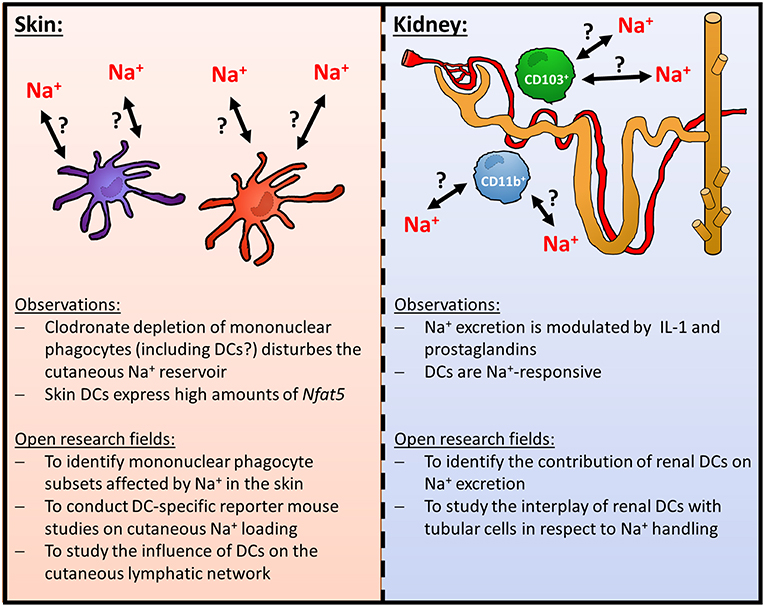
Figure 1. Role of DCs in homeostatic cutaneous and renal Na+-handling.
Dendritic cells as Potential Regulators of Renal Na+ handling
In addition to regulating local Na+ balance in the skin, it is conceivable that DCs play a key role in orchestrating renal electrolyte handling. It is well-established that there is a dense network of mononuclear phagocytes including macrophages and DCs throughout the kidney. These cells play an important role in various inflammatory and fibrotic kidney injury models [reviewed in (40–43)]. Furthermore, they are able to change their shape and motility upon tissue damage (44, 45) and are involved in curtailing and/ or promoting inflammatory responses after various insults (46–52). Under steady state, the mononuclear phagocyte compartment of the mouse kidney mainly consists of CD103+ and CD11b+ renal mononuclear phagocyte subsets [reviewed in (41, 43, 53)]. The CD103+ mononuclear phagocytes are derived from bona fide DC precursors and these renal DCs play an important anti-inflammatory role upon renal damage (52, 54). The CD11b+ renal mononuclear phagocytes represent over 90% of the renal mononuclear phagocyte population and comprise DCs and macrophages [reviewed in (41, 43, 53)]. In contrast to the CD103+ renal mononuclear phagocytes/ DCs, the DC subset of these CD11b+ mononuclear phagocytes exerts proinflammatory functions (54). Of note, recent evidence using a transcriptional reporter mouse for DCs (zinc finger and BTB domain containing 46 [Zbtb42]-GFP; visualizing both CD103+ and CD11b+ DCs) demonstrates that CD103+ and CD11b+ renal DC subsets are round-shaped and located around blood vessels while in contrast counterintuitively most of the other renal mononuclear phagocytes (i.e., macrophages) are dendritically shaped (54).
While there is substantial evidence that these DCs are involved in inflammatory responses in the kidney it is currently unclear whether DCs contribute to the regulation of renal Na+ excretion. Recent data indicates that renal mononuclear phagocytes play an important role as accessory cells in regulating Na+ transport of renal tubular cells. Crowley and co-workers uncovered that IL-1-signaling modulates tubular Na+ excretion via mononuclear phagocytes in mice (55). Moreover, using a CD11b-Cre deleter mouse strain, Zhang et al. reported that prostaglandins derived from renal mononuclear phagocytes modulate the activity of renal Na+-Cl− cotransporters (56). As the CD11b-Cre deleter mouse strain recombines in DCs as well (35), it is tempting to speculate that renal DCs are involved in this state of affair. This idea is further supported by the fact that murine DCs express specific molecules that facilitate the transport of Na+ and thus sensing of increased extracellular Na+ levels such as the sodium-potassium chloride cotransporter-1 (NKCC1), chloride cotransporter (NCC), the sodium-calcium exchanger (NCX) and the α and γ subunits of the epithelial sodium channel (ENaC) (57). Murine DCs are able to express gap junction proteins such as Connexin 43 (58), which are able to facilitate Na+ entry in addition to other molecules (59). It is tempting to speculate that DCs are able to form functional syncytial cell aggregates with tubular cells and thereby regulate renal Na+ handling. However, the contribution of these molecules in electrolyte physiology is unexplored and warrants further studies.
Impact of Na+ on Dendritic Cell Immunobiology
DCs might not only be important regulators of local Na+ balance. For instance there is robust evidence that increases in Na+ levels limits the anti-inflammatory capacity of macrophages while promoting their proinflammatory status (24, 60–65). Enhanced induction of proinflammatory macrophage activation required the activity of the osmoprotective transcription factor Nfat5 (24, 64). Recently, Buxade et al. reported that Nfat5 regulates the expression of MHCII molecules under standard cell culture conditions (i.e., normal salt conditions) and thereby regulates CD4+ T cell responses (66). This regulatory circuit only operates in macrophages but not in DCs (66). Surprisingly, the impact of increased Na+ levels on DC immunobiology has been studied in less detail and the data available are controversial (Figure 2).
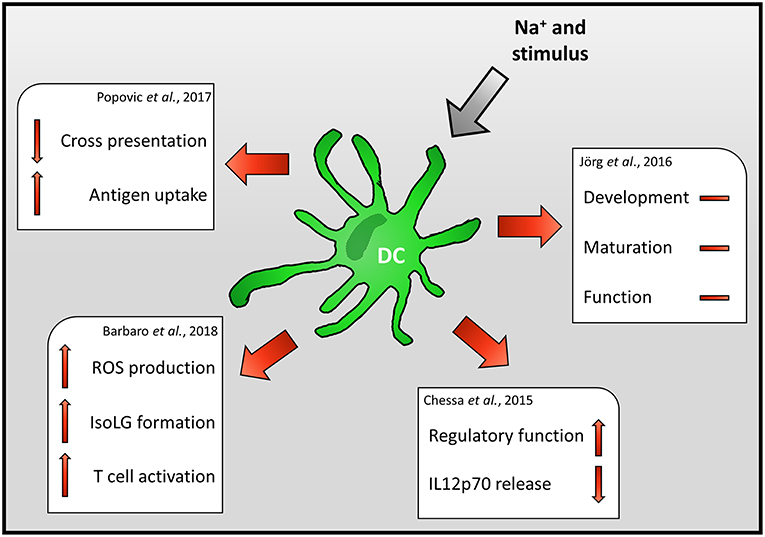
Figure 2. Impact of Na+ on DC immunobiology.
Jörg et al., for instance, reported that high Na+ levels do not impact the generation, maturation or function of mouse DCs but rather directly impact on T cells (67). In contrast to these findings, Chessa et al. demonstrate that increasing extracellular Na+ levels, found in the renal medulla during DC development, skews murine DCs to a macrophage-like regulatory phenotype and suppresses the release of the Th1 priming cytokine IL-12p70 (68).
In line with this, Popovic et al. reported that the ability of mouse DCs to cross-present the model antigen ovalbumin is severely impaired (69). Decreased cross-presentation was recorded despite enhanced antigen uptake, processing, and presentation. Of note, increased Na+ levels resulted in enhanced expression of co-inhibitor and co-stimulatory molecules. Using knock out strategies and blocking antibodies the authors exclude that enhanced expression of co-inhibitory/ -stimulatory molecules or reduced production of IL-12 underlies this phenotype. The authors provide evidence that the suppressive effect of high salt conditions (HS) on cross-presentation is dependent on TIR-domain-containing adapter-inducing interferon-β (TRIF) regulated process. However, the TRIF-dependent mechanism that ultimately results in impaired cross-presentation requires further investigation (69). Recently, Zhang et al. reported that exposure of virally infected mouse macrophages to increased Na+ levels boosts the release of Type 1 interferon (65). Since TRIF and type 1 interferon production are intertwined [reviewed in (70)] and type 1 interferon signaling has the potential to inhibit antigen-presentation (71), it is conceivable that exposure to increased Na+ levels triggers an overshooting type 1 interferon response which ultimately inhibits cross-presentation by DCs.
In line with enhanced degradative activity of DCs upon HS exposure (69), Barbaro et al. found that increasing extracellular Na+ levels result in enhanced ROS production and formation of isolevuglandin (IsoLG)-protein adducts in mouse DCs. However, in contrast to the study using the model antigen ovalbumin, Barbaro et al. reported increased frequencies of IFN-γ and IL-17 producing T cells after co-incubation of DCs with T cells. Moreover, transfer of DCs exposed to high Na+ environments, increased the blood pressure of mice subjected to low levels of angiotensin II (57). These findings suggest that increased local Na+ levels enhance the inflammatory potential of DCs and, thus might propagate inflammatory circuits that ultimately result in arterial hypertension and cardiovascular death.
Of note, increases in dietary Na+ might not only directly influence the immunobiology of dendritic cells. Recently, Wilck et al. demonstrated that dietary high salt conditions change the composition of the microbiome by removal of Lactobacillus murinus (72). Depletion of Lactobacillus was accompanied by reduction of the tryptophan metabolites such as indole 3-lactic acid (ILA) and indole 3-acetic acid. Increased levels of ILA directly inhibit the proliferation of TH17 cells in vitro (72). In addition, it is possible that these tryptophan degradation products are impacting on gut dendritic cells, which in turn orchestrate e.g., regulatory T cell, TH22 and TH17 effector cell balance (73, 74). In line with this, there are several reports that Na+-rich diets increases the production of cytokines that are key players in screwing the induction of TH1 and TH17 cells in inflamed gut tissue such as Il12b and IL-23 (75, 76).
Conclusion
Na+ availability emerges as a new factor of tissue microenvironment which on the one hand is regulated by immune cells and on the other hand is able to impact on their immunological function. In contrast to macrophages, our knowledge regarding DCs is rather limited. Current evidence suggests that the impact of increased Na+ levels on DCs is context dependent. However, the role of DCs in regulating local Na+ stores is unexplored and warrants further studies.
Data Availability
Publicly available datasets were analyzed in this study. This data can be found here: “www.immgen.org.”
Author Contributions
PN and JJ: conception and writing of the manuscript. AS and DM: contributed to the writing.
Funding
This work was supported by grants from the Deutsche Forschungsgemeinschaft (JA1993/4-1) and faculty grants from the University Regensburg (Reform C) to JJ.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Alexander Steinkasserer for critically reading the manuscript.
References
1. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. (1998) 392:245–52. doi: 10.1038/32588
2. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, et al. Immunobiology of dendritic cells. Annu Rev Immunol. (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
3. Reis E, Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. (2006) 6:476–83. doi: 10.1038/nri1845
4. Satoh T, Akira S. Toll-like receptor signaling and its inducible proteins. Microbiol Spectr. (2016) 4:e00402016. doi: 10.1128/microbiolspec.mchd-0040-2016
5. Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by antibodies: recent progress. Annu Rev Immunol. (2017) 35:285–311. doi: 10.1146/annurev-immunol-051116-052433
6. Brown GD, Willment JA, Whitehead L. C-type lectins in immunity and homeostasis. Nat Rev Immunol. (2018) 18:374–89. doi: 10.1038/s41577-018-0004-8
7. Schatz V, Neubert P, Schroder A, Binger K, Gebhard M, Muller DN, et al. Elementary immunology: Na+ as a regulator of immunity. Pediatr Nephrol. (2017) 32:201–10. doi: 10.1007/s00467-016-3349-x
8. Muller DN, Wilck N, Haase S, Kleinewietfeld M, Linker RA. Sodium in the microenvironment regulates immune responses and tissue homeostasis. Nat Rev Immunol. (2019). doi: 10.1038/s41577-018-0113-4. [Epub ahead of print].
9. Manzel A, Muller DN, Hafler DA, Erdman SE, Linker RA, Kleinewietfeld M. Role of “Western diet” in inflammatory autoimmune diseases. Curr Allergy Asthma Rep. (2014) 14:404. doi: 10.1007/s11882-013-0404-6
10. Selvarajah V, Connolly K, Mceniery C, Wilkinson I. Skin sodium and hypertension: a paradigm shift? Curr Hypertens Rep. (2018) 20:94. doi: 10.1007/s11906-018-0892-9
11. Dantzler WH, Layton AT, Layton HE, Pannabecker TL. Urine-concentrating mechanism in the inner medulla: function of the thin limbs of the loops of Henle. Clin J Am Soc Nephrol. (2014) 9:1781–9. doi: 10.2215/CJN.08750812
12. Titze J, Dahlmann A, Lerchl K, Kopp C, Rakova N, Schroder A, et al. Spooky sodium balance. Kidney Int. (2013) 85:759–67. doi: 10.1038/ki.2013.367
13. Jantsch J, Binger KJ, Muller DN, Titze J. Macrophages in homeostatic immune function. Front Physiol. (2014) 5:146. doi: 10.3389/fphys.2014.00146
14. Titze J, Rakova N, Kopp C, Dahlmann A, Jantsch J, Luft FC. Balancing wobbles in the body sodium. Nephrol Dial Transplant. (2015) 31:1078–81. doi: 10.1093/ndt/gfv343
15. Wiig H, Luft FC, Titze JM. The interstitium conducts extrarenal storage of sodium and represents a third compartment essential for extracellular volume and blood pressure homeostasis. Acta Physiol. (2018) 222:e13006. doi: 10.1111/apha.13006
17. Wahlgren V. Über die bedeutung der gewebe als chlordepots. Arch exp Pathol Pharm. (1909) 61:97–112. doi: 10.1007/BF01841955
18. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. (2009) 15:545–52. doi: 10.1038/nm.1960
19. Hofmeister LH, Perisic S, Titze J. Tissue sodium storage: evidence for kidney-like extrarenal countercurrent systems? Pflugers Arch. (2015) 467:551–8. doi: 10.1007/s00424-014-1685-x
20. Linz P, Santoro D, Renz W, Rieger J, Ruehle A, Ruff J, et al. Skin sodium measured with 23Na MRI at 7.0 T. NMR Biomed. (2015) 28:54–62. doi: 10.1002/nbm.3224
21. Selvarajah V, Maki-Petaja KM, Pedro L, Bruggraber SFA, Burling K, Goodhart AK, et al. Novel mechanism for buffering dietary salt in humans: effects of salt loading on skin sodium, vascular endothelial growth factor C, and blood pressure. Hypertension. (2017) 70:930–7. doi: 10.1161/HYPERTENSIONAHA.117.10003
22. Dahlmann A, Dorfelt K, Eicher F, Linz P, Kopp C, Mossinger I, et al. Magnetic resonance-determined sodium removal from tissue stores in hemodialysis patients. Kidney Int. (2015) 87:434–41. doi: 10.1038/ki.2014.269
23. Kopp C, Linz P, Maier C, Wabel P, Hammon M, Nagel AM, et al. Elevated tissue sodium deposition in patients with type 2 diabetes on hemodialysis detected by 23Na magnetic resonance imaging. Kidney Int. (2018) 93:1191–7. doi: 10.1016/j.kint.2017.11.021
24. Jantsch J, Schatz V, Friedrich D, Schroder A, Kopp C, Siegert I, et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. (2015) 21:493–501. doi: 10.1016/j.cmet.2015.02.003
25. Ivanova LN, Archibasova VK, Shterental I. [Sodium-depositing function of the skin in white rats]. Fiziol Zh SSSR Im I M Sechenova. (1978) 64:358–63.
26. Titze J, Lang R, Ilies C, Schwind KH, Kirsch KA, Dietsch P, et al. Osmotically inactive skin Na+ storage in rats. Am J Physiol Renal Physiol. (2003) 285:F1108–17. doi: 10.1152/ajprenal.00200.2003
27. Titze J, Shakibaei M, Schafflhuber M, Schulze-Tanzil G, Porst M, Schwind KH, et al. Glycosaminoglycan polymerization may enable osmotically inactive Na+ storage in the skin. Am J Physiol Heart Circ Physiol. (2004) 287:H203–8. doi: 10.1152/ajpheart.01237.2003
28. Machnik A, Dahlmann A, Kopp C, Goss J, Wagner H, Van Rooijen N, et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension. (2010) 55:755–61. doi: 10.1161/HYPERTENSIONAHA.109.143339
29. Wiig H, Schroder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. (2013) 123:2803–15. doi: 10.1172/JCI60113
30. Kopp C, Beyer C, Linz P, Dahlmann A, Hammon M, Jantsch J, et al. Na+ deposition in the fibrotic skin of systemic sclerosis patients detected by 23Na-magnetic resonance imaging. Rheumatology. (2017) 56:674. doi: 10.1093/rheumatology/kex149
31. Rakova N, Juttner K, Dahlmann A, Schroder A, Linz P, Kopp C, et al. Long-term space flight simulation reveals infradian rhythmicity in human Na+ balance. Cell Metab. (2013) 17:125–31. doi: 10.1016/j.cmet.2012.11.013
32. Karlsen TV, Nikpey E, Han J, Reikvam T, Rakova N, Castorena-Gonzalez JA, et al. High-salt diet causes expansion of the lymphatic network and increased lymph flow in skin and muscle of rats. Arterioscler Thromb Vasc Biol. (2018) 38:2054–64. doi: 10.1161/ATVBAHA.118.311149
33. Ward NL, Loyd CM, Wolfram JA, Diaconu D, Michaels CM, Mccormick TS. Depletion of antigen-presenting cells by clodronate liposomes reverses the psoriatic skin phenotype in KC-Tie2 mice. Br J Dermatol. (2011) 164:750–8. doi: 10.1111/j.1365-2133.2010.10129.x
34. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. (1999) 8:265–77. doi: 10.1023/A:1008942828960
35. Abram CL, Roberge GL, Hu Y, Lowell CA. Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods. (2014) 408:89–100. doi: 10.1016/j.jim.2014.05.009
36. Guilliams M, van De Laar L. A hitchhiker's guide to myeloid cell subsets: practical implementation of a novel mononuclear phagocyte classification system. Front Immunol. (2015) 6:406. doi: 10.3389/fimmu.2015.00406
37. Schraml BU, Reis E Sousa C. Defining dendritic cells. Curr Opin Immunol. (2015) 32:13–20. doi: 10.1016/j.coi.2014.11.001
38. Durai V, Murphy KM. Functions of murine dendritic cells. Immunity. (2016) 45:719–36. doi: 10.1016/j.immuni.2016.10.010
39. Ivanov S, Scallan JP, Kim KW, Werth K, Johnson MW, Saunders BT, et al. CCR7 and IRF4-dependent dendritic cells regulate lymphatic collecting vessel permeability. J Clin Invest. (2016) 126:1581–91. doi: 10.1172/JCI84518
40. Kurts C, Panzer U, Anders HJ, Rees AJ. The immune system and kidney disease: basic concepts and clinical implications. Nat Rev Immunol. (2013) 13:738–53. doi: 10.1038/nri3523
41. Gottschalk C, Kurts C. The debate about dendritic cells and macrophages in the kidney. Front Immunol. (2015) 6:435. doi: 10.3389/fimmu.2015.00435
42. Weisheit CK, Engel DR, Kurts C. Dendritic cells and macrophages: sentinels in the kidney. Clin J Am Soc Nephrol. (2015) 10:1841–51. doi: 10.2215/CJN.07100714
43. Viehmann SF, Bohner AMC, Kurts C, Brahler S. The multifaceted role of the renal mononuclear phagocyte system. Cell Immunol. (2018) 330:97–104. doi: 10.1016/j.cellimm.2018.04.009
44. Snelgrove SL, Kausman JY, Lo C, Ooi JD, Coates PT, Hickey MJ, et al. Renal dendritic cells adopt a pro-inflammatory phenotype in obstructive uropathy to activate T cells but do not directly contribute to fibrosis. Am J Pathol. (2012) 180:91–103. doi: 10.1016/j.ajpath.2011.09.039
45. Engel DR, Krause TA, Snelgrove SL, Thiebes S, Hickey MJ, Boor P, et al. CX3CR1 reduces kidney fibrosis by inhibiting local proliferation of profibrotic macrophages. J Immunol. (2015) 194:1628–38. doi: 10.4049/jimmunol.1402149
46. Kitagawa K, Wada T, Furuichi K, Hashimoto H, Ishiwata Y, Asano M, et al. Blockade of CCR2 ameliorates progressive fibrosis in kidney. Am J Pathol. (2004) 165:237–46. doi: 10.1016/S0002-9440(10)63292-0
47. Scholz J, Lukacs-Kornek V, Engel DR, Specht S, Kiss E, Eitner F, et al. Renal dendritic cells stimulate IL-10 production and attenuate nephrotoxic nephritis. J Am Soc Nephrol. (2008) 19:527–37. doi: 10.1681/ASN.2007060684
48. Heymann F, Meyer-Schwesinger C, Hamilton-Williams EE, Hammerich L, Panzer U, Kaden S, et al. Kidney dendritic cell activation is required for progression of renal disease in a mouse model of glomerular injury. J Clin Invest. (2009) 119:1286–97. doi: 10.1172/JCI38399
49. Tadagavadi RK, Reeves WB. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J Am Soc Nephrol. (2010) 21:53–63. doi: 10.1681/ASN.2009040407
50. Hochheiser K, Engel DR, Hammerich L, Heymann F, Knolle PA, Panzer U, et al. Kidney dendritic cells become pathogenic during crescentic glomerulonephritis with proteinuria. J Am Soc Nephrol. (2011) 22:306–16. doi: 10.1681/ASN.2010050548
51. Peng X, Zhang J, Xiao Z, Dong Y, Du J. CX3CL1-CX3CR1 interaction increases the population of Ly6C(-)CX3CR1(hi) macrophages contributing to unilateral ureteral obstruction-induced fibrosis. J Immunol. (2015) 195:2797–805. doi: 10.4049/jimmunol.1403209
52. Evers BD, Engel DR, Bohner AM, Tittel AP, Krause TA, Heuser C, et al. CD103+ kidney dendritic cells protect against crescentic GN by maintaining IL-10-producing regulatory T cells. J Am Soc Nephrol. (2016) 27:3368–82. doi: 10.1681/ASN.2015080873
53. Nelson PJ, Rees AJ, Griffin MD, Hughes J, Kurts C, Duffield J. The renal mononuclear phagocytic system. J Am Soc Nephrol. (2012) 23:194–203. doi: 10.1681/ASN.2011070680
54. Brahler S, Zinselmeyer BH, Raju S, Nitschke M, Suleiman H, Saunders BT, et al. Opposing roles of dendritic cell subsets in experimental GN. J Am Soc Nephrol. (2018) 29:138–54. doi: 10.1681/ASN.2017030270
55. Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, Mcdonough AA, et al. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin II-induced hypertension via the NKCC2 co-transporter in the nephron. Cell Metab. (2015) 23:360–8. doi: 10.1016/j.cmet.2015.11.013
56. Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, et al. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest. (2015) 125:4281–94. doi: 10.1172/JCI81550
57. Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. (2017) 21:1009–20. doi: 10.1016/j.celrep.2017.10.002
58. Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity. (2014) 40:248–61. doi: 10.1016/j.immuni.2013.12.012
59. Li F, Sugishita K, Su Z, Ueda I, Barry WH. Activation of connexin-43 hemichannels can elevate [Ca2+]i and [Na+]i in rabbit ventricular myocytes during metabolic inhibition. J Mol Cell Cardiol. (2001) 33:2145–55. doi: 10.1006/jmcc.2001.1477
60. Binger KJ, Gebhardt M, Heinig M, Rintisch C, Schroeder A, Neuhofer W, et al. High salt reduces the activation of IL-4- and IL-13-stimulated macrophages. J Clin Invest. (2015) 125:4223–38. doi: 10.1172/JCI80919
61. Ip WK, Medzhitov R. Macrophages monitor tissue osmolarity and induce inflammatory response through NLRP3 and NLRC4 inflammasome activation. Nat Commun. (2015) 6:6931. doi: 10.1038/ncomms7931
62. Zhang WC, Zheng XJ, Du LJ, Sun JY, Shen ZX, Shi C, et al. High salt primes a specific activation state of macrophages, M(Na). Cell Res. (2015) 25:893–910. doi: 10.1038/cr.2015.87
63. Hucke S, Eschborn M, Liebmann M, Herold M, Freise N, Engbers A, et al. Sodium chloride promotes pro-inflammatory macrophage polarization thereby aggravating CNS autoimmunity. J Autoimmun. (2016) 67:90–101. doi: 10.1016/j.jaut.2015.11.001
64. Berry MR, Mathews RJ, Ferdinand JR, Jing C, Loudon KW, Wlodek E, et al. Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell. (2017) 170:860–74.e819. doi: 10.1016/j.cell.2017.07.022
65. Zhang WC, Du LJ, Zheng XJ, Chen XQ, Shi C, Chen BY, et al. Elevated sodium chloride drives type I interferon signaling in macrophages and increases antiviral resistance. J Biol Chem. (2018) 293:1030–9. doi: 10.1074/jbc.M117.805093
66. Buxade M, Huerga Encabo H, Riera-Borrull M, Quintana-Gallardo L, Lopez-Cotarelo P, Tellechea M, et al. Macrophage-specific MHCII expression is regulated by a remote Ciita enhancer controlled by NFAT5. J Exp Med. (2018) 215:2901–18. doi: 10.1084/jem.20180314
67. Jörg S, Kissel J, Manzel A, Kleinewietfeld M, Haghikia A, Gold R, et al. High salt drives Th17 responses in experimental autoimmune encephalomyelitis without impacting myeloid dendritic cells. Exp Neurol. (2016) 279:212–22. doi: 10.1016/j.expneurol.2016.03.010
68. Chessa F, Mathow D, Wang S, Hielscher T, Atzberger A, Porubsky S, et al. The renal microenvironment modifies dendritic cell phenotype. Kidney Int. (2015) 89:82–94. doi: 10.1038/ki.2015.292
69. Popovic ZV, Embgenbroich M, Chessa F, Nordstrom V, Bonrouhi M, Hielscher T, et al. Hyperosmolarity impedes the cross-priming competence of dendritic cells in a TRIF-dependent manner. Sci Rep. (2017) 7:311. doi: 10.1038/s41598-017-00434-y
70. Pfaender S, Grabski E, Detje CN, Riebesehl N, Lienenklaus S, Steinmann E, et al. Hepatitis C virus stimulates murine CD8alpha-like dendritic cells to produce type I interferon in a TRIF-dependent manner. PLoS Pathog. (2016) 12:e1005736. doi: 10.1371/journal.ppat.1005736
71. Schwandt T, Schumak B, Gielen GH, Jungerkes F, Schmidbauer P, Klocke K, et al. Expression of type I interferon by splenic macrophages suppresses adaptive immunity during sepsis. EMBO J. (2012) 31:201–13. doi: 10.1038/emboj.2011.380
72. Wilck N, Matus MG, Kearney SM, Olesen SW, Forslund K, Bartolomaeus H, et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature. (2017) 551:585–9. doi: 10.1038/nature24628
73. Bekiaris V, Persson EK, Agace WW. Intestinal dendritic cells in the regulation of mucosal immunity. Immunol Rev. (2014) 260:86–101. doi: 10.1111/imr.12194
74. Sun M, He C, Cong Y, Liu Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. (2015) 8:969–78. doi: 10.1038/mi.2015.49
75. Aguiar SLF, Miranda MCG, Guimaraes MAF, Santiago HC, Queiroz CP, Cunha PDS, et al. High-salt diet induces IL-17-dependent gut inflammation and exacerbates colitis in mice. Front Immunol. (2017) 8:1969. doi: 10.3389/fimmu.2017.01969
Keywords: dendritic cells, Na+ balance, antigen-presentation, Nfat5, kidney, skin microenvironment
Citation: Neubert P, Schröder A, Müller DN and Jantsch J (2019) Interplay of Na+ Balance and Immunobiology of Dendritic Cells. Front. Immunol. 10:599. doi: 10.3389/fimmu.2019.00599
Received: 30 October 2018; Accepted: 06 March 2019;
Published: 29 March 2019.
Edited by:
Bart Everts, Leiden University Medical Center, NetherlandsReviewed by:
Cristina Lopez-Rodriguez, Universidad Pompeu Fabra, SpainNicole C. Kaneider, University of Cambridge, United Kingdom
Copyright © 2019 Neubert, Schröder, Müller and Jantsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Jantsch, amFudHNjaEB1a3IuZGU=">jonathanamFudHNjaEB1a3IuZGU=