
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 March 2019
Sec. Viral Immunology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00566
This article is part of the Research Topic Current Insights into Host Immune Responses to Human Respiratory Syncytial Virus (RSV) and Challenges Towards Efficient Treatments and Vaccines against RSV View all 11 articles
 Diego R. Hijano1†
Diego R. Hijano1† Luan D. Vu2,3†
Luan D. Vu2,3† Lawrence M. Kauvar4
Lawrence M. Kauvar4 Ralph A. Tripp5
Ralph A. Tripp5 Fernando P. Polack6
Fernando P. Polack6 Stephania A. Cormier2,3*
Stephania A. Cormier2,3*Respiratory syncytial virus (RSV) is the most common cause of lower respiratory tract disease in children <2 years of age. Increased morbidity and mortality have been reported in high-risk patients, such as premature infants, patients with cardiac disease, and severely immune compromised patients. Severe disease is associated with the virulence of the virus as well as host factors specifically including the innate immune response. The role of type I interferons (IFNs) in the response to RSV infection is important in regulating the rate of virus clearance and in directing the character of the immune response, which is normally associated with protection and less severe disease. Two RSV non-structural proteins, NS1 and NS2, as well as the envelope G glycoprotein are known to suppress type I IFN production and a robust type I IFN response to RSV does not occur in human infants or neonatal mouse models of RSV infection. Additionally, presence of type I IFNs are associated with mild symptoms in infants and administration of IFN-α prior to infection of neonatal mice with RSV reduces immunopathology. This evidence has driven RSV prophylaxis and therapeutic efforts to consider strategies for enhancing type I IFN production.
Respiratory syncytial virus (RSV) is a common cause of lower respiratory tract disease in infants and young children (1–3). Although 30–70% of infants develop bronchiolitis upon primary RSV infection, only 1–3% are hospitalized (4). Despite this heterogeneous course of disease, the global burden of RSV disease is estimated at 64 million cases and 160,000 deaths annually (5, 6). Increased morbidity and mortality have been reported in high-risk patients, such as premature infants, infants with cardiac disease, and severely immuno-compromised patients (7–9). Moreover, the consequences of severe RSV infection are long lasting and constitute a risk factor for childhood asthma and bronchiolitis (10–14). The elderly and immune compromised also suffer from RSV, particularly those with prior pulmonary problems (15). Notwithstanding the advances in our understanding of the immune response to RSV and the recently determined high resolution structures of the two major immunogenic viral proteins, the RSV F and G proteins, we still lack adequate therapeutics as well as a safe, robust, and effective vaccine (16).
Both viral and host immune factors have been implicated in severe infections (17–20). RSV is an orthopneumovirus in the Paramyxoviridae family (21, 22). The RNA genome contains 10 genes encoding 11 proteins. The envelope of the virus is formed by the matrix (M) protein, the small hydrophobic (SH) protein, and two abundant, glycosylated surface proteins: the fusion (F) and attachment (G) proteins. The G and F proteins control the initial phases of infection (23, 24). The G protein is composed of three epitope regions identified by murine monoclonal antibodies: mostly invariant epitopes in the central conserved domain (CCD); group-specific epitopes (subtype A or B); and strain-specific epitopes in the C-terminal hypervariable region of the G protein ectodomain (25, 26). The two antigenically distinct subtypes, A and B, can co-circulate during the same epidemic season (27–29). The clinical impact of different subtypes likely contributes to different disease severity. While the F protein has historically been the major target for antiviral and vaccine development, both G and F proteins are naturally targeted by neutralizing antibodies induced by infection (23, 24, 30–33). The two non-structural proteins, NS1 and NS2, suppress IFN production (34–36), with NS1 known to bind RIG-I within the cytoplasm of host cells thereby abrogating the signal transmitted via MAVS (2). Further, the G protein also impedes IFN-α expression through the interaction of the CX3C chemokine-like motif in G, which interacts with CX3CR1 and impairs the immune response to RSV. Infection with an RSV strain that lacks the CX3C motif (mimic of the human chemokine called fractalkine or CX3CL1) or treatment with an anti-G monoclonal antibody (MAb) that blocks binding to CX3CR1 result in increased levels of type I/III IFN (37).
The fractalkine receptor, CX3CR1, is expressed on human plasmacytoid dendritic cells (pDCs) and epithelial cells (37–39). The former are specialized immune cells that infiltrate the lung to produce large amounts of type I IFN in response to viral infection (40, 41).
The link between RSV G protein and type I IFN expression is well established (42–44) with details elucidated that include TLR4 signaling and SOCS3 regulation of type I IFN (45–50). For example, the RSV G protein contributes to immune evasion by modifying host cytokine and chemokine responses whose expression is negatively regulated by suppressor of cytokine signaling (SOCS) proteins (48). SOCS1 and SOCS3 are closely related and well characterized members of the family acting through the JAK/STAT pathway to regulate cytokine expression via a kinase inhibitory region (51). SOCS1 and SOCS3 are downstream from toll like receptors (TLR) and can indirectly regulate them (52). Specifically, SOCS3 induction by TLR is dependent on Myd88 (52). SOCS1 and SOCS3 strongly suppress TLR7-mediated type I IFN production by binding IFN regulatory factor 7 (53). In addition, SOCS1 modulates TIRAP which is downstream of TLR1/2, TLR2/6 and TLR4 but not TLR9 (51). It has been shown that SOCS1 and SOCS3 regulate type I IFN in normal fully-differentiated human bronchial epithelial (NHBE) cells, with the pathway including interferon-regulatory factor (IRF)-3 activation and nuclear translocation (48). Further, interferon-stimulated gene (ISG)-15 expression is altered very early after infection and RSV infection has been shown to upregulate SOCS 1 and SOCS 3 in epithelial cells (46). NHBE cells infected with an RSV mutant virus lacking the G gene have distinct responses as compared to wild-type RSV (30). Notably, RSV mutant strains without secreted G induced less CCL2 and CCL5 with no apparent lung disease in mice. Interestingly, mice developed good antibody responses despite the attenuated infection (54). These findings suggest that RSV surface proteins signal through multiple pathways, and this may be an important means of reducing anti-viral type I IFN expression, thereby promoting virus replication.
Of interest, RSV does not induce robust, long term immunity and people may be repeatedly infected with the same and different strains of RSV (55, 56). These finding are particularly relevant to the multiple failed RSV vaccine trails to date, including the original formalin inactivated RSV (FI-RSV) vaccine as well as more recent subunit and live attenuated vaccines. The deficient response to both natural and artificial exposure to RSV antigens in human represents a barrier to the development of novel therapeutic or preventive strategies (57–64). Further, the immune response to both primary and repeat infections with RSV needs further study to better understand short- and long-term immunity. More detailed characterization of the response of healthy adults as compared to the elderly and to infants is also needed. The importance of elucidating the host response to RSV infection is underscored by recent clinical evaluation of prophylaxis with the anti-F protein monoclonal antibody (mAb) palivizumab in healthy preterm infants. In this single-blind, randomized, placebo-controlled trial, suppression of RSV replication did not have a major effect on reducing the RSV-associated asthma incidence at age 6 years, suggesting that other factors besides viral load contribute to the clinical severity (11, 65).
Type I IFNs are a group of related proteins that help regulate the activity of the immune system. The mammalian types are named IFN-α (alpha), IFN-β (beta), IFN-κ (kappa), IFN-δ (delta), IFN-ε (epsilon), IFN-τ (tau), IFN-ω (omega), and IFN-ζ (zeta) (66, 67). IFN-α has 13 different subtypes in humans (α1/13; α2; α4; α5; α6; α7; α8; α10; α14; α16; α17; α21) (68) and is primarily produced by pDCs, while IFN-β is produced largely by fibroblasts; both have antiviral activity that is an important component of the innate immune response. Quantitative and qualitative differences in gene expression have been observed, with type I IFN being notably absent in the RSV infected cells (69). This result is consistent with results from the INFANT study, conducted by Argentine doctors to investigate the causes of respiratory diseases that seriously affect children such as RSV associated asthma and bronchiolitis, and pneumonia and influenza virus infection. In the INFANT study, RSV infection failed to induce a robust type I IFN response in the nasal mucosa of infants even when co-infected with influenza, which normally induces a robust response (70). Intriguingly, neonatal mouse models of RSV infection recapitulate these data from humans. Specifically, neonatal mice infected with RSV fail to induce a type I IFN response to RSV in contrast to adult mice infected with RSV (71). Furthermore, as compared to non-treated controls, administration of IFN-α during infection of the neonate enhances the immune response to RSV infection 5 weeks later and prevents Th2 biased immune responses (including perivascular inflammation and mucus production) and airway hyperreactivity (71). Notably, studies examining human cord blood-derived pDCs exposed to RSV showed reduced type I IFN production when compared to vehicle control or left unstimulated (40). These recent correlations between type I IFN responses and RSV disease severity in infants merit further investigation. Here, we review the mechanism surrounding RSV and type I IFN production in humans and mouse models and discuss its implications for development of therapeutics and vaccines.
Human IFNs are classified as type I (IFN-I), type II (IFN-II), or type III (IFN-III) with each class binding to specific receptors. All type I IFNs bind to a specific cell surface receptor complex known as the IFN-α receptor (IFNAR) that consists of IFNAR1 and IFNAR2 chains (72). The ability to produce and respond to IFN-I is distributed in a wide variety of cells. This confers several autocrine and paracrine effects that have been extensively characterized, mainly in viral infections. IFN-I signaling is mediated through a common cell surface receptor, the IFN-I receptor (IFNAR), signaling through the JAK-STAT cascade leading to transcriptional upregulation of the IFN-ISGs. The IFN-II family is represented by a single gene product, IFN-γ, and is mainly produced by T lymphocytes and natural killer (NK) cells. The associated receptor (IFNGR) regulates several cell functions related to host defense to intracellular pathogens. IFN-λ comprises four subtypes: IFN-λ1, IFN-λ2, IFN-λ3, and IFN-λ4. The members of this IFN-III family interact through a unique receptor, the IFN-λ receptor (IFN-λR). It has been shown that IFN systems differ in terms of tissue distribution of their receptors (73, 74). While IFN-α/β systems are more prominent on endothelial cells, they are expressed on all cells. On the other hand, IFN-λ expression is more restricted occurring predominantly on epithelial cells of the intestines and lungs (73). RSV infection induces high expression levels of IFN-λ 1–3 in the lungs, and these have been associated with more severe disease in children (75).
Type I and III IFNs are induced in virtually all cell types upon recognition of viral proteins by cytoplasmic and endosomal receptors (67, 68). IFN induction by RSV involves the recognition of RSV by TLRs which activate innate and acquired immunity (47, 49, 76–78). Leukocytes express several TLRs, including TLR2, TLR6, TLR3, TLR4, and TLR7 (79). Using knockout mice, TLR2 and TLR6 signaling in leukocytes has been shown to activate innate immunity against RSV by promoting TNF-α (tumor necrosis factor), IL-6 (interleukin-6), CCL2 (monocyte chemoattractant protein 1), and CCL5 (RANTES) (80). TLR4 was shown to also contribute to cytokine activation, and TLR2 and TLR6 activation was shown to be important for controlling viral replication in vivo in mice (81). TLR2 interactions with RSV promoted neutrophil migration and dendritic cell activation within the lung. TLR3 has been associated with more severe disease in mice models (82).
TLR4 is upregulated by RSV F protein interaction with TLR4 (76, 77). RSV G protein reduced TLR4 activity to baseline levels even in the presence of LPS (lipopolysaccharide), a strong stimulus, as assayed using a luciferase reporter construct for TLR4 signaling (76). As previously noted, RSV infection of normal human bronchoepithelial cells has been shown to modulate expression of SOCS, an effect mediated by G protein, leading to inhibition of type I IFN and ISG15 expression (48). These findings suggest that RSV surface proteins signal through multiple TLRs, and that enhanced expression and activation of type I IFNs may promote viral replication. Accordingly, IFN-α has been considered as an adjuvant for RSV vaccines as it is known to promote the activation and survival of virus-specific T cells (83).
The role of type I IFN in RSV infection, shedding, and disease severity in humans has been a subject of interest for decades (84, 85). While early studies struggled to identify a role for type I IFN in RSV disease (84–88), novel findings in recent years implicate type I IFN as determinants of RSV pathogenesis and immune responses (40, 41, 89, 90). RSV is a poor inducer of IFN and as a consequence, these IFNs and related cytokines have been speculated to have a limited role in the host defense against viral infection (84, 85, 87, 88). In fact, most hypotheses for RSV disease susceptibility in infants have been based on unique structural respiratory factors such as smaller airway size, lack of interalveolar pores and channels and different innervation patterns, inflammatory responses, and Th2 polarization of the adaptive immune response (78, 91, 92). Reconsideration of this bias is needed. Unlike the case in infants and children infected with influenza virus, IFN levels were undetectable or low in nasal secretions of infants and young children with RSV lower respiratory tract illness and did not correlate with resolution of clinical signs (84, 85). In a more recent study of infants in Argentina, type I IFN was detected more frequently in those infected with influenza A virus than in those infected with RSV or hMPV (93). RSV infected infants hospitalized with bronchiolitis displayed low, intermittent concentrations of IFN-α in respiratory secretions (87). No significant correlation was seen between these low respiratory IFN levels and RSV shedding (88). In human macrophages and peripheral blood mononuclear cells, RSV infection also induced minimal IFN activity and elicited no detectable transcription of IFN-α or IFN-β gene products (86), which is consistent with low IFN-α production in monocyte cultures from young infants (40).
Intriguingly, RSV-induced IFN-α expression by primary pDC collected from older children (from 1 to 5-year-olds) was notably higher than that of healthy full-term infant counterparts suggesting expression may be linked to age of the patient. Likewise, higher IFN-α expression was detected in primary pDCs obtained from healthy adults (40). Age at the time of initial infection is an important predictive factor for disease severity (94, 95). Cohort studies demonstrated that young infants (<6 months of age at initial infection) are at greater risk for severe disease than older infants (96, 97). Furthermore, long-term consequences of RSV infection, such as development of asthma, are closely associated with severity of infection (10, 13). Extrapolation of response to vaccines or therapeutics in adults to those in young infants is thus highly problematic.
While clear linkage between IFN expression and RSV infection in humans has been elusive, a factor that needs further study is the prolonged incubation period of RSV disease in infants for whom the mean time from infection to symptoms is 4–6 days (87) in sharp contrast to the considerably shorter incubation period for influenza virus (average of 2 days). Type I IFN levels peak early after infection, and therefore sampling of respiratory secretions after symptoms appear may be too late to detect its antiviral effects for infants infected with RSV (84, 85, 93). Support for a function of type I IFNs in RSV pathogenesis is also growing from analysis of developmental innate immune mechanisms associated with poor type I IFN responses in newborn and young infants. For instance, and as mentioned above, RSV-induced IFN-α production appears to be primarily mediated by pDC, (40, 41). Indeed, compared to adult pDC production of type I IFN during RSV infection is substantially impaired in infants when disease is particularly severe (40, 90). Impairment in infants is explained by deficits either in MAVS or RIG-I at the post-translational level or by signaling events downstream of MAVS (40).
Additional evidence supporting a role for type I IFN in RSV infection and illness is the strong inhibition of IFN induction and signaling mediated by the two earliest genes transcribed among the 11 RSV gene products, NS1 and NS2 (89). NS1 and NS2 have been postulated to have various roles in RSV pathogenesis, generally linked to their anti-IFN activity. In addition to antagonizing type I IFN, NS1, and NS2 may negatively modulate dendritic cell maturation, affect Th17 lymphocyte proliferation, and promote Th2 polarization (35, 98–105). Deletion of anti-IFN proteins NS1 and NS2 in RSV live vaccines is responsible for attenuated phenotypes (89).
In the era prior to availability of antibodies against RSV, topical administration of recombinant IFN-α-2a accelerated control of upper respiratory tract symptoms during RSV infection in a randomized, double-blinded trial while not affecting duration or magnitude of viral shedding (106). This early result is of interest in the context of a more recent study of nasal epithelial cells from children with wheeze and/or atopy that showed reduced IFN-β in the nasal swabs in response to RSV infection, which was associated with increased viral shedding (107). However, consistent with other successful immunotherapies, this regimen elicited adverse effects and severity of those effects were dose-dependent (108). Common side effects due to IFN-α include flu-like symptoms, pulmonary toxicity (109), gastrointestinal symptoms (110), and neurotoxicity (111). Lethal toxicities associated with IFN-α regimen are rare and severe toxicities due to IFN-α are manageable if recognized expeditiously (112, 113). Importantly, IFN-α therapy in children (114) and infants with RSV-induced bronchiolitis (115) is generally safe and well tolerated. However, caution is still warranted in use of recombinant IFN-α in the context of an RSV infection, due to the side effects mentioned above.
It is also possible that antiviral agents may benefit from restoring natural type I IFN responses, which may lead to faster clearance of the virus. Two studies using healthy adult volunteers experimentally infected with RSV and treated with antivirals showed that rapid RSV clearance was related to reduced disease (116, 117). Similarly, a higher RSV load was linked to an increased risk for severe bronchiolitis in a large multicenter trial in the United States (28). None of these studies have attempted to define the mechanism by which higher viral load contributes to disease severity. In that regard, a study in infants with RSV bronchiolitis that described an association between viral load and disease severity (length of hospital stay) is of interest since a correlation was also noted with relative expression of ISG-56 (118). Finally, additional evidence for the role of type I IFN in disease severity comes from two studies of rare loss-of-function variants in IFIH1 (which encodes a RIG-I-like receptor involved in the sensing of viral RNA); the variants result in defective innate recognition of RNA viruses preventing the activation of an efficient antiviral IFN response. These rare but serious immunodeficiencies lead to extreme susceptibility to RSV and other respiratory viruses (119, 120).
Mice provide a semi-permissive model for human RSV and while attempts to adapt a strain to this model have repeatedly failed (121) data from numerous laboratories demonstrate similarities in age related immune responses between humans and neonatal mice. Since, our current understanding of the features that contribute to severe RSV disease in infants is tied to our understanding of developmental immunity during the first year of life, the neonatal mouse model of RSV infection is a helpful tool (122–124). Numerous studies utilizing mouse models of RSV infection have revealed a bias toward a T helper type 2 (Th2) cytokine response when mice are initially infected as neonates as compared to adults (71, 125–128). Upon reinfection, mice initially infected as neonates mount significantly greater Th2 responses as compared to mice initially infected as adults (126). This skewed Th2 response upon reinfection is associated with lung dysfunction (lung eosinophilia, increased mucus production, and air hyperresponsiveness) (126, 127, 129). Such responses mirror observations made in infants with severe RSV disease (130–132). Production of type I IFN by pDC during RSV infection of the neonate mouse, as in humans, is considerably impaired. However, both pDC number and production of type I IFN in response to RSV increase with age; adult mice recruit substantially higher numbers of pDCs to the lungs after RSV infection when compared to those of same age that are not infected and to neonatal mice infected with RSV (71). A single dose of IFN-α or adoptive transfer of adult-derived pDCs (capable of mounting a type I IFN response), prior to a primary RSV infection, substantially impedes the Th2-biased immunopathology observed during reinfection (71). A related strategy to revert poor outcomes associated with RSV infection in neonatal mice has been administering Flt3 ligand to neonates before RSV infection (133). Ftl3 ligand is a growth factor that stimulates the proliferation of hematopoietic cells that triggers expansion of cDCs and pDCs in human cord blood and strongly promotes IFN-α production by pDCs in response to viral exposure (134, 135). This treatment has led to increased lung DC numbers and reconditioning of the type I IFN pathway toward Th1-mediated immunity. In addition, these mice were protected from exacerbated airway disease upon adult re-exposure to RSV (133).
Treating mice with neutralizing mAbs against the RSV G protein reduced G protein-mediated lung inflammation. Specifically, TRL3D3, a human mAb against the G protein CCD, enhanced IFN responses, decreased airway inflammation, and improved lung function upon secondary infection, whereas mice treated with an anti-F mAb (palivizumab) had less IFN than mock infected mice (30, 33). Since RSV infection is inhibited by IFN-induced transmembrane proteins (71, 117), the impact of counteracting the G protein's suppressive effect on IFN production likely also contributes to the antiviral effect of such mAbs. Consistent with these results, intranasal IFN-α administration in neonatal mice prior to RSV infection appreciably reduced RSV viral load in both nasal associated lymphoid tissue and lungs when compared to age-matched controls (136).
Interestingly, while the IFN-α response to RSV progressively increases with age (40, 136); another cytokine IL33, an alarmin cytokine, decreases with age (126). Recent work has demonstrated that IL-33 is significantly greater in neonatal compared to adult mice during RSV infection. IL-33 signaling in the neonatal mouse model of RSV has been shown to induce RSV immunopathogenesis including Th2 bias (126). Elevations in IL-33 are inversely correlated with age at RSV infection (126) and severity of RSV infantile disease has been associated with elevated levels of respiratory IL-33 and polymorphisms within ST2, the receptor for IL-33, (137). IL-33 promotes Th2 responses via multiple signaling pathways that are summarized in Figure 1. Similarly, intranasal instillation of IL-33 significantly impaired the production of IFN-α/λ in the BALF and reduced the expression of IFN-stimulated genes in the lung following PVM infection (138). Table 1 summarizes the significant advances in the role of age-dependent differences in various immune and non-immune cells related to the immune pathogenesis of RSV infection in infants. Figure 1 highlights age-dependent differences in RSV-mediated immune pathogenesis.
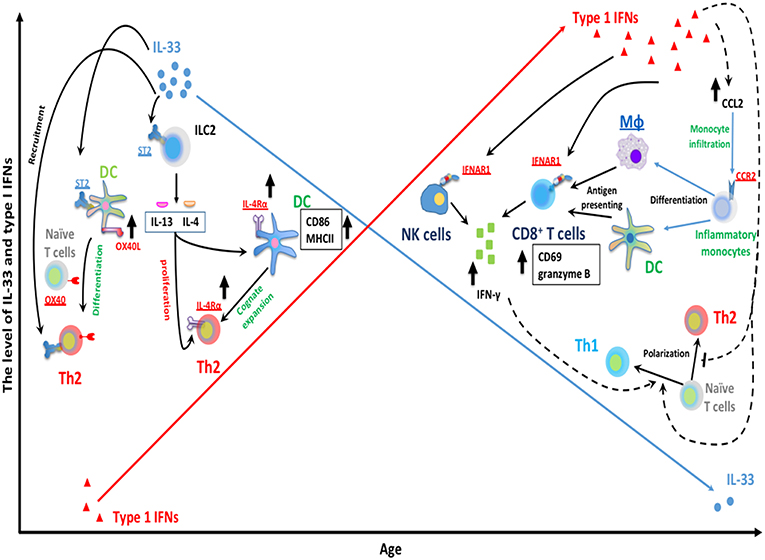
Figure 1. Age-dependent differences RSV-mediated immune pathogenesis. The expression of RSV-induced IFNα is limited during infantile RSV infection and progressively increases with age. Type I IFNs are capable of suppressing Th2 development and promoting type 1 immunity. Type I IFNs have been implicated in the regulation of NK and CD8+ T cells functionality. Type I IFNs can elicit the activation of cytotoxic IFN-γ+ CD8+ T cells by enhancing the recruitment of inflammatory myeloid cells into infected lungs. These infiltrating myeloid cells then differentiate into macrophage and DC, and acquire antigen presenting ability, subsequently activate CD8+ T cells and trigger CD8+ T cells IFN-γ production. In addition, type I IFNs can also act directly on CD8+ T cells and NK cells by targeting its receptor IFNAR1 on membrane of CD8+ T cells and NK cells. This results in the production of IFNγ, a key mediator for type 1 immunity, which presumably favors Th1 polarization from naïve CD4+ T cells. In contrast with IFN-α, IL-33 has been implicated in the induction of Th2-biased immune pathogenesis during neonatal RSV infection. A large amount of IL-33 is rapidly released following RSV infection in neonatal but not adult mice. IL-33 can elicit ILC2-mediated IL-13/IL-4 production through its cellular receptor ST2 on ILC2. ILC2-derived IL13/IL-4 then can facilitate cognate expansion of Th2 by upregulating the expression of Th2 costimulatory molecules (CD86 and MHCII) on DC. ILC2-derived IL-4 also promotes the proliferation of Th2 cells. It involves the upregulation of IL4Rα on both DC and Th2 cells. Similarly, IL-33 can promote Th2 polarization from naïve CD4+ T cells by targeting DC via ST2 receptors on DC and then enhance the expression of OX40L on DC (ligand for cellular receptor OX40 on naïve T cells).
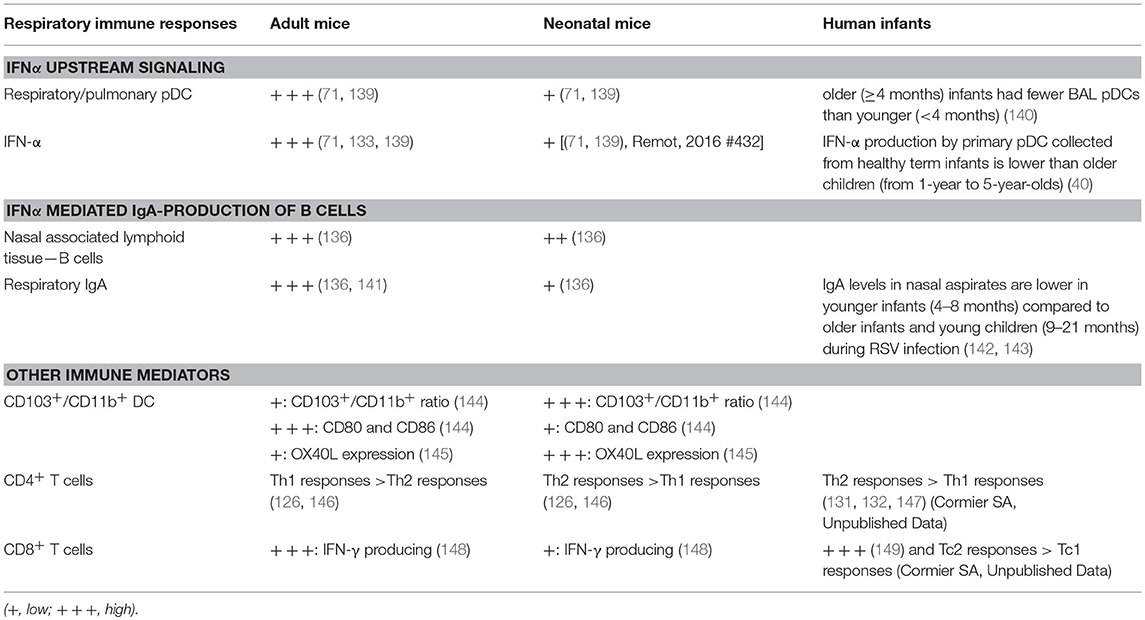
Table 1. Differences in immunological responses toward RSV in the respiratory tract.
Current RSV vaccine candidates seek to induce high levels of RSV-specific serum neutralizing antibodies, which are associated with reduced RSV-related hospitalization rates. However, serum neutralizing antibodies may not be sufficient to prevent infection and/or induce protective responses. This feature of RSV biology was exemplified by the antibody responses induced to the FI-RSV vaccine in the 1960's, which elicited lower avidity, non-protective antibodies as compared to those that develop after natural RSV infection (150). Furthermore, mucosal antibodies have been shown to correlate better with RSV protection than serum antibodies in both infants and adults (151–153).
The majority of vaccine efforts to date have focused on the RSV F protein, based on the assumption that reducing RSV load will reduce or eliminate disease. While mAbs against RSV F protein (palivizumab) given to premature infants (at or before 35 weeks) do help to protect children with certain lung or heart conditions who are at high risk for severe RSV disease, such treatment does not fully protect from disease. Further, in a recent study of viral burden in healthy full-term infants (<70 days old), nearly a third experienced a multi-log rebound in viral load at around 2 weeks after onset of symptoms (154). Since viral load had declined by several orders of magnitude by that point, the most likely cause was mutational escape which is a well characterized response to anti-F protein mAbs (155).
In short, the role of RSV viral load as a driver for severity of infection remains controversial. On the one hand, quantitative RT-PCR correlation with disease severity in patients showed that viral load was associated with disease severity in younger patients although not in older patients (63). For patients intubated due to respiratory distress, RSV infection resulted in higher viral load than those not intubated, and higher viral loads were associated with longer hospitalization (156). In the adult human RSV challenge model, virus replication is inversely correlated with the level of nasal secretory neutralizing antibody prior to infection (157). Higher nasal immunoglobulin (Ig) A predicts lower infectivity and lower measures of viral replication (151) and low RSV-specific nasal IgA is an independent significant risk factor for RSV infection (158). On the other hand, several groups have failed to find an association of higher viral load in nasopharyngeal lavage (159) or nasal aspirates with either length of hospitalization, duration of oxygen supplement or severe bronchiolitis in either infants (160) or children (161).
The picture that is emerging is that primary reduction in viral load is useful, but not sufficient, to reduce the clinically relevant pathology. Accordingly, a combination of an anti-viral agent with an agent that reduces the RSV induced alteration in the innate immune response is the most likely route to improved outcomes. Targeting the F protein addresses the first issue. Targeting the G protein addresses the second issue; since anti-G protein mAbs also have potent antiviral activity, targeting the G protein alone may be sufficient to achieve both goals.
The optimal type of RSV vaccine employed, i.e., RSV F and/or G protein, will likely be dependent on the host target population (162, 163), with four groups being of interest: (1) infants and young children, (2) adults, (3) the elderly, and (4) pregnant woman. Immunization schedule (prime/boost) and the specific platform for delivery of the vaccine are also likely to be important (162, 164, 165). Consequently, there are a spectrum of RSV vaccines being tested that include live-attenuated and chimeric virus, purified F protein (including variants engineered to present predominantly the pre-fusion conformation), particle and vector-based presentations of the antigen(s) (165). For example, RSV F protein particle-based (Novovax) (166, 167) and RSV F subunit (GSK; NIH) vaccines are being evaluated for use in pregnant mothers, while RSV F protein particle-based (Novovax; Mucosis) and live-attenuated vaccines such as RSV deletion mutant vaccines, e.g., ΔM2-2 and ΔNS2 constructs (Sanofi; NIH) are being targeted for the pediatric population with potential extension to older children and young adults (168). An important caveat for using live vaccines is the need to prevent transmission to the immune compromised, or those with reduced or waning immunity. An additional issue for vaccinating infants and young children is that the vaccine needs to balance safety (higher attenuation) and efficacy (lower attenuation). A promising recent study of an RSV vaccine candidate having a deletion of the M2-2 coding sequence showed downregulation of viral replication and upregulation of transcription and antigen synthesis (169). For healthy older adults, several RSV vaccine candidates are being considered, including vector-based platforms such as VXA-RSV F oral (Vaxart) and Ad26.RSV.preF (Janssen) (168). Given the high transmissibility of RSV, even a safe and effective vaccine will likely leave gaps in protection for high-risk, very young infants. Vaccinating pregnant women has become an area of high interest to induce passive protection in the infant by generating high maternal antibody titers.
Antibodies directed to dominant antigenic sites on the F protein have variable neutralization capacities with the most potent neutralization epitopes associated with the pre-F conformation (170–174). Stabilized F protein antigen in both pre- and post-fusion morphology are being explored (31, 172, 175, 176). The typical benchmark is achieving a protective titer at a defined time point, but the time course of increase in antibody titer is also an important parameter, which will likely differ according to vaccine type and composition. The RSV G protein is also an antigenic target for neutralizing antibodies, but despite this fact, the G protein has not usually been considered as a RSV vaccine candidate because of its variability across RSV strains (175–177). However, with the recent discovery of the G protein structure (29, 32), and the known role of the G protein oligomer on the virus surface vs. its monomeric secreted form (54, 178), there has been reinvigorated interest in its potential as a RSV vaccine candidate.
Passive transfer of antibodies is protective against severe RSV infection using polyclonal or monoclonal antibodies (mAbs; RSV-IVIG, palivizumab) (179) The ratio of antibody transfer and decay kinetics is considered a principal parameter to measure protection. More recent versions of mAbs have become available with improved antibody transfer and decay kinetics such as MEDI-8897 which is optimized from the human antibody D25 that targets RSV pre-F protein (24, 170, 172, 180, 181). This type of treatment potentially offers novel immunotherapeutic strategies to bridge gaps with RSV vaccine candidates.
Many studies indicate that certain cytokines can mediate strong vaccine responses associated with a good outcome. For example, IFN-α2b is an FDA-approved therapy for adjuvant treatment of patients with certain cancers (182) and hepatitis C (183). Of particular interest is the recent demonstration that administration to neonatal mice of IFN-α prior to RSV infection increased RSV specific IgA production in nasal washes when compared to age matched controls (136). Furthermore, IgA levels became comparable to those of adult mice infected with RSV (136). In addition, IFN-α induced expression of B cell activating factor (BAFF) in nasal associated lymphoid tissue (NALT) (136). BAFF, a B cell survival factor and mediator of B cell activation and class switching, and APRIL, a TNF ligand family member that shares receptors with BAFF, regulate B cell survival, proliferation and differentiation. Gene expression analysis from NALT and lung homogenates further support a role for IFN-α in regulating granulocyte migration and neutrophil-mediated immunity (136).
Comparative studies of genetic background of mice has shown diverse influences on Th cell differentiation by controlling the capacity for IL-2-induced IL-4 production by naive CD4+ T cells. BALB/c mice are Th2-prone, while C57BL/6 mice are Th1-prone (28, 184–186). Notably, type I IFN pathways are reconditioned in neonatal BALB/c mice after RSV infection as lung dendritic cells (DC) numbers increase; the associated shift toward a Th1 response protected the mice from exacerbated airway disease (187). Adult mice produce considerably higher levels of type I IFNs in response to RSV than do neonatal mice. Finally, recent studies have implicated the type III IFN-λ as being significant for mucosal antiviral immune responses to RSV infection (41, 65).
Since SOCS-1 and SOCS-3 negatively regulate the IFN-induced signal cascade, and NS1, NS2, and G protein inhibit the type I IFN response, any of these viral proteins may prove to be useful targets to induce a more effective innate immune response (45, 50). Understanding how these viral proteins modify host immune responses is thus crucial to the development of effective countermeasures. Although no animal model perfectly mimics the human response, the mouse offers a far greater set of tools for analyzing the immune system than other popular models, such as the cotton rat, and the mouse has for that reason become the nearly exclusive model for studies on RSV and the host immune response.
Over the past decade, targeting the F protein has repeatedly produced disappointing clinical results. In particular, agents targeting the F protein have not been proven effective post-infection. This is not only problematic for the multiple populations in need of treatment but also for vaccines since healthy full term infants (<70 days old) experienced a significant rebound in viral load at around 2 weeks after onset of symptoms in nearly a third of the study population (154). Moreover, palivizumab is only approved for prophylaxis in premature infants and those at high risk for severe RSV disease. Retrospective analysis of samples from the clinical trials leading to approval of this drug revealed a striking skewing of TLR4 polymorphisms (188). Mutations that interfere with function of this key innate immune system receptor have an incidence in the general population of ~10%, but 90% of the high-risk premature birth infants had a TLR4 mutation. This striking result has been replicated (78). As described above, the RSV F protein stimulates TLR4, while the G protein suppresses this signaling pathway (48). In the premature birth population, antibody mediated removal of the TLR4 stimulus should not impact the overall response since the pathway is already suppressed genetically. In the broader population, however, removal of that beneficial stimulus may contribute to the lower observed efficacy compared to what was expected.
In light of these empirical failures and the improved understanding of RSV disease mechanisms, interest has increased in the role of the other major viral envelope protein, the RSV G protein, on viral entry, on viral neutralization, and most critically on RSV-mediated pathology (33). In mouse pDCs, mutating the G protein CCD prevented suppression of IFN-α attributable to the G protein; the Fab of a murine mAb against this region of the G protein was nearly as effective as the mutation (39).
Human mAbs targeting the CCD of RSV G protein (189) have recently been compared to anti-F mAbs, as both prophylactic and therapeutic treatment in BALB/c mice. The results showed that targeting the G protein was more effective for reducing viral load, leukocyte infiltration, and pro-inflammatory cytokine expression in cell-free bronchial alveolar lavage (BAL) supernatants (190). These results are consistent with in vitro studies on the type I IFN response of normal human bronchial epithelial cells to RSV in conjunction with mAbs to either the F or G protein which showed clear superiority for targeting the G protein (48).
TLR3D3 is a native human mAb that binds the G protein CCD with low pM affinity; it has strong activities as both an antiviral and for immune response normalization (189). It is currently in IND-enabling preclinical development. In light of the accumulated results summarized here on the mechanisms underlying RSV disease, it is appropriate to test this agent as a post-infection treatment. If proven effective, design of a vaccine to induce comparable mAbs will benefit from recently published structural analysis of the binding of TRL3D3 to the G protein CCD (32).
RSV infections continue to be a major cause of morbidity and mortality around the world affecting a wide variety of patients. Infants, the elderly, and those with comorbidities are at particularly high risk of hospitalization and death. Mainstream therapy remains restricted to supportive care. Despite successful antigen presentation leading to high titer of neutralizing antibodies by several approaches, we still do not have a licensed vaccine. Although the single licensed monoclonal antibody, palivizumab, is effective, it protects only a minor fraction of the population at high risk. Advances in therapeutic and vaccine development for RSV has mainly been hampered by the lack of understanding of the immune response to the virus both in the setting of primary infection as well as recurrent reinfections. Diverse approaches have converged over the last few years on identification of Type I IFN as a key actor and a readily measured biomarker of the broader innate immune response. Clinical studies in human infants have shown that RSV is a poor inducer of type I IFN responses, and there is accumulating literature reporting an inverse correlation between type I IFN responses and disease severity.
As our understanding improves of how viral proteins modify host immune responses, and the age dependence of those responses, research efforts can focus on development of effective countermeasures to overcome the virus's sophisticated sabotage of the host immune system. Animal models, complemented by studies on human cells in vitro, continue to be essential in the discovery and/or confirmation of the key features surrounding the host-virus interaction. Mouse models have proven to be particularly informative, including demonstrations that neonatal mice fail to produce IFN-α in the setting of RSV infection due to poor pDC recruitment, and that administration of IFNα decreases Th2-biased immunopathology and viral load. In addition, and importantly, administration of IFNα enhances mucosal RSV specific IgA production, which is critical given the clinical evidence that suggests that mucosal antibodies correlate better than systemic antibodies with protection. Although the known toxicities of recombinant IFN precludes use in this setting, a variety of approaches to restoring the normal IFN response have been identified, offering new opportunities for both therapeutic and vaccine discovery.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
LK is an employee of Trellis Bioscience, and holds an equity interest in the company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by grants from NIH to SC (R01AI090059, R01ES015050, and P42ES013648), BMGF (OPP1157162) and NIEHS Director's Challenge Award to FP, the Georgia Research Alliance to RT, SBIR grant from NIAID to LK (2R44AI122360-03).
1. Hall CB. Respiratory syncytial virus and parainfluenza virus. N Engl J Med. (2001) 344:1917–28. doi: 10.1056/NEJM200106213442507
2. Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, et al. The burden of respiratory syncytial virus infection in young children. N Engl J Med. (2009) 360:588–98. doi: 10.1056/NEJMoa0804877
3. Hall CB, Simoes EA, Anderson LJ. Clinical and epidemiologic features of respiratory syncytial virus. Curr Top Microbiol Immunol. (2013) 372:39–57. doi: 10.1007/978-3-642-38919-1_2
4. Blanco JCG, Boukhvalova MS, Morrison TG, Vogel SN. A multifaceted approach to RSV vaccination. Hum Vaccin Immunother. (2018) 14:1734–45. doi: 10.1080/21645515.2018.1472183
5. Shi T, McAllister DA, O'Brien KL, Simoes EAF, Madhi SA, Gessner BD, et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. (2017) 390:946–58. doi: 10.1016/S0140-6736(17)30938-8
6. Stein RT, Bont LJ, Zar H, Polack FP, Park C, Claxton A, et al. Respiratory syncytial virus hospitalization and mortality: Systematic review and meta-analysis. Pediatr Pulmonol. (2017) 52:556–69. doi: 10.1002/ppul.23570
7. Higgins D, Trujillo C, Keech C. Advances in RSV vaccine research and development - A global agenda. Vaccine. (2016) 34:2870–5. doi: 10.1016/j.vaccine.2016.03.109
8. Paes B, Fauroux B, Figueras-Aloy J, Bont L, Checchia PA, Simoes EA, et al. Defining the risk and associated morbidity and mortality of severe respiratory syncytial virus infection among infants with chronic lung disease. Infect Dis Ther. (2016) 5:453–71. doi: 10.1007/s40121-016-0137-7
9. Checchia PA, Paes B, Bont L, Manzoni P, Simoes EA, Fauroux B, et al. Defining the risk and associated morbidity and mortality of severe respiratory syncytial virus infection among infants with congenital heart disease. Infect Dis Ther. (2017) 6:37–56. doi: 10.1007/s40121-016-0142-x
10. Carroll KN, Wu P, Gebretsadik T, Griffin MR, Dupont WD, Mitchel EF, et al. Season of infant bronchiolitis and estimates of subsequent risk and burden of early childhood asthma. J Allergy Clin Immunol. (2009) 123:964–6. doi: 10.1016/j.jaci.2008.12.011
11. Gelfand EW. Development of asthma is determined by the age-dependent host response to respiratory virus infection: therapeutic implications. Curr Opin Immunol. (2012) 24:713–9. doi: 10.1016/j.coi.2012.08.011
12. Caballero MT, Jones MH, Karron RA, Hartert TV, Simoes EA, Stein RT, et al. The impact of respiratory syncytial virus disease prevention on pediatric asthma. Pediatr Infect Dis J. (2016) 35:820–2. doi: 10.1097/INF.0000000000001167
13. Lu S, Hartert TV, Everard ML, Giezek H, Nelsen L, Mehta A, et al. Predictors of asthma following severe respiratory syncytial virus (RSV) bronchiolitis in early childhood. Pediatr Pulmonol. (2016) 51:1382–92. doi: 10.1002/ppul.23461
14. Dupont W, Wu P, Gebretsadik T, Hartert T. RSV prevention in infancy and asthma in later life. Lancet Respir Med. (2018) 6:e32. doi: 10.1016/S2213-2600(18)30231-5
15. Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. (2005) 352:1749–59. doi: 10.1056/NEJMoa043951
16. Jorquera PA, Tripp RA. Respiratory syncytial virus: prospects for new and emerging therapeutics. Expert Rev Respir Med. (2017) 11:609–15. doi: 10.1080/17476348.2017.1338567
17. Walsh EE, McConnochie KM, Long CE, Hall CB. Severity of respiratory syncytial virus infection is related to virus strain. J Infect Dis. (1997) 175:814–20. doi: 10.1086/513976
18. DeVincenzo JP, Wilkinson T, Vaishnaw A, Cehelsky J, Meyers R, Nochur S, et al. Viral load drives disease in humans experimentally infected with respiratory syncytial virus. Am J Respir Crit Care Med. (2010) 182:1305–14. doi: 10.1164/rccm.201002-0221OC
19. Caserta MT, Qiu X, Tesini B, Wang L, Murphy A, Corbett A, et al. Development of a Global Respiratory Severity Score for Respiratory Syncytial Virus Infection in Infants. J Infect Dis. (2017) 215:750–6.
20. Walsh EE, Wang L, Falsey AR, Qiu X, Corbett A, Holden-Wiltse J, et al. Virus-specific antibody, viral load, and disease severity in respiratory syncytial virus infection. J Infect Dis. (2018) 218:208–17. doi: 10.1093/infdis/jiy106
21. Tregoning JS, Schwarze J. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev. (2010) 23:74–98. doi: 10.1128/CMR.00032-09
22. Vandini S, Biagi C, Lanari M. Respiratory syncytial virus: the influence of serotype and genotype variability on clinical course of infection. Int J Mol Sci. (2017) 18:E1717. doi: 10.3390/ijms18081717
23. McLellan JS, Chen M, Joyce MG, Sastry M, Stewart-Jones GB, Yang Y, et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science. (2013) 342:592–8. doi: 10.1126/science.1243283
24. McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science. (2013) 340:1113–7. doi: 10.1126/science.1234914
25. Johnson PR, Spriggs MK, Olmsted RA, Collins PL. The G glycoprotein of human respiratory syncytial viruses of subgroups A and B: extensive sequence divergence between antigenically related proteins. Proc Natl Acad Sci USA. (1987) 84:5625–9. doi: 10.1073/pnas.84.16.5625
26. Cortjens B, Yasuda E, Yu X, Wagner K, Claassen YB, Bakker AQ, et al. Broadly reactive anti-respiratory syncytial virus G antibodies from exposed individuals effectively inhibit infection of primary airway epithelial cells. J Virol. (2017) 91:e02357–16. doi: 10.1128/JVI.02357-16
27. Borchers AT, Chang C, Gershwin ME, Gershwin LJ. Respiratory syncytial virus–a comprehensive review. Clin Rev Allergy Immunol. (2013) 45:331–79. doi: 10.1007/s12016-013-8368-9
28. Rodriguez-Fernandez R, Tapia LI, Yang CF, Torres JP, Chavez-Bueno S, Garcia C, et al. Respiratory syncytial virus genotypes, host immune profiles, and disease severity in young children hospitalized with bronchiolitis. J Infect Dis. (2017) 217:24–34. doi: 10.1093/infdis/jix543
29. Jones HG, Ritschel T, Pascual G, Brakenhoff JPJ, Keogh E, Furmanova-Hollenstein P, et al. Structural basis for recognition of the central conserved region of RSV G by neutralizing human antibodies. PLoS Pathog. (2018) 14:e1006935. doi: 10.1371/journal.ppat.1006935
30. Kauvar LM, Harcourt JL, Haynes LM, Tripp RA. Therapeutic targeting of respiratory syncytial virus G-protein. Immunotherapy. (2010) 2:655–61. doi: 10.2217/imt.10.53
31. Capella C, Chaiwatpongsakorn S, Gorrell E, Risch ZA, Ye F, Mertz SE, et al. Prefusion F, postfusion F, G antibodies, and disease severity in infants and young children with acute respiratory syncytial virus infection. J Infect Dis. (2017) 216:1398–406. doi: 10.1093/infdis/jix489
32. Fedechkin SO, George NL, Wolff JT, Kauvar LM, DuBois RM. Structures of respiratory syncytial virus G antigen bound to broadly neutralizing antibodies. Sci Immunol. (2018) 3:eaar3534. doi: 10.1126/sciimmunol.aar3534
33. Tripp RA, Power UF, Openshaw PJM, Kauvar LM. Respiratory syncytial virus: targeting the G protein provides a new approach for an old problem. J Virol. (2018) 92:e01302–17. doi: 10.1128/JVI.01302-17
34. Bossert B, Marozin S, Conzelmann KK. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol. (2003) 77:8661–8. doi: 10.1128/JVI.77.16.8661-8668.2003
35. Spann KM, Tran KC, Chi B, Rabin RL, Collins PL. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J Virol. (2004) 78:4363–9. doi: 10.1128/JVI.78.8.4363-4369.2004
36. Appiah GD, Blanton L, D'Mello T, Kniss K, Smith S, Mustaquim D, et al. Influenza activity - United States, 2014-15 season and composition of the 2015-16 influenza vaccine. MMWR Morb Mortal Wkly Rep. (2015) 64:583–90.
37. Chirkova T, Boyoglu-Barnum S, Gaston KA, Malik FM, Trau SP, Oomens AG, et al. Respiratory syncytial virus G protein CX3C motif impairs human airway epithelial and immune cell responses. J Virol. (2013) 87:13466–79. doi: 10.1128/JVI.01741-13
38. Chirkova T, Lin S, Oomens AG, Gaston KA, Boyoglu-Barnum S, Meng J, et al. CX3CR1 is an important surface molecule for respiratory syncytial virus infection in human airway epithelial cells. J Gen Virol. (2015) 96:2543–56. doi: 10.1099/vir.0.000218
39. Boyoglu-Barnum S, Todd SO, Meng J, Barnum TR, Chirkova T, Haynes LM, et al. Mutating the CX3C motif in the G protein should make a live respiratory syncytial virus vaccine safer and more effective. J Virol. (2017) 91:e02059–16. doi: 10.1128/JVI.02059-16
40. Marr N, Wang TI, Kam SH, Hu YS, Sharma AA, Lam A, et al. Attenuation of respiratory syncytial virus-induced and RIG-I-dependent type I IFN responses in human neonates and very young children. J Immunol. (2014) 192:948–57. doi: 10.4049/jimmunol.1302007
41. Hillyer P, Mane VP, Chen A, Dos Santos MB, Schramm LM, Shepard RE, et al. Respiratory syncytial virus infection induces a subset of types I and III interferons in human dendritic cells. Virology. (2017) 504:63–72. doi: 10.1016/j.virol.2017.01.017
42. Barik S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr Top Microbiol Immunol. (2013) 372:173–91. doi: 10.1007/978-3-642-38919-1_9
43. Russell CD, Unger SA, Walton M, Schwarze J. The human immune response to respiratory syncytial virus infection. Clin Microbiol Rev. (2017) 30:481–502. doi: 10.1128/CMR.00090-16
44. Hillyer P, Shepard R, Uehling M, Krenz M, Sheikh F, Thayer KR, et al. Differential responses by human respiratory epithelial cell lines to respiratory syncytial virus reflect distinct patterns of infection control. J Virol. (2018) 92:e02202–17. doi: 10.1128/JVI.02202-17
45. Moore EC, Barber J, Tripp RA. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol J. (2008) 5:116. doi: 10.1186/1743-422X-5-116
46. Hashimoto K, Ishibashi K, Ishioka K, Zhao D, Sato M, Ohara S, et al. RSV replication is attenuated by counteracting expression of the suppressor of cytokine signaling (SOCS) molecules. Virology. (2009) 391:162–70. doi: 10.1016/j.virol.2009.06.026
47. Klein Klouwenberg P, Tan L, Werkman W, van Bleek GM, Coenjaerts F. The role of Toll-like receptors in regulating the immune response against respiratory syncytial virus. Crit Rev Immunol. (2009) 29:531–50. doi: 10.1615/CritRevImmunol.v29.i6.40
48. Oshansky CM, Krunkosky TM, Barber J, Jones LP, Tripp RA. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a toll-like receptor pathway. Viral Immunol. (2009) 22:147–61. doi: 10.1089/vim.2008.0098
49. Mukherjee S, Lukacs NW. Innate immune responses to respiratory syncytial virus infection. Curr Top Microbiol Immunol. (2013) 372:139–54. doi: 10.1007/978-3-642-38919-1_7
50. Wang S, Zheng G, Zhao L, Xu F, Qian J. Shp-2 contributes to anti-RSV activity in human pulmonary alveolar epithelial cells by interfering with the IFN-alpha-induced Jak/Stat1 pathway. J Cell Mol Med. (2015) 19:2432–40. doi: 10.1111/jcmm.12629
51. Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol. (2008) 19:414–22. doi: 10.1016/j.semcdb.2008.07.010
52. Baetz A, Frey M, Heeg K, Dalpke AH. Suppressor of cytokine signaling (SOCS) proteins indirectly regulate toll-like receptor signaling in innate immune cells. J Biol Chem. (2004) 279:54708–15. doi: 10.1074/jbc.M410992200
53. Yu CF, Peng WM, Schlee M, Barchet W, Eis-Hubinger AM, Kolanus W, et al. SOCS1 and SOCS3 target IRF7 degradation to suppress TLR7-mediated type I IFN production of human plasmacytoid dendritic cells. J Immunol. (2018) 200:4024–35. doi: 10.4049/jimmunol.1700510
54. Maher CF, Hussell T, Blair E, Ring CJ, Openshaw PJ. Recombinant respiratory syncytial virus lacking secreted glycoprotein G is attenuated, non-pathogenic but induces protective immunity. Microbes Infect. (2004) 6:1049–55. doi: 10.1016/j.micinf.2004.07.001
55. Scott PD, Ochola R, Ngama M, Okiro EA, James Nokes D, Medley GF, et al. Molecular analysis of respiratory syncytial virus reinfections in infants from coastal Kenya. J Infect Dis. (2006) 193:59–67. doi: 10.1086/498246
56. Zhang W, Lockey RF, Mohapatra SS. Respiratory syncytial virus: immunopathology and control. Expert Rev Clin Immunol. (2006) 2:169–79. doi: 10.1586/1744666X.2.1.169
57. Woolums AR, Singer RS, Boyle GA, Gershwin LJ. Interferon gamma production during bovine respiratory syncytial virus (BRSV) infection is diminished in calves vaccinated with formalin-inactivated BRSV. Vaccine. (1999) 17:1293–7. doi: 10.1016/S0264-410X(98)00379-X
58. Haynes LM, Jones LP, Barskey A, Anderson LJ, Tripp RA. Enhanced disease and pulmonary eosinophilia associated with formalin-inactivated respiratory syncytial virus vaccination are linked to G glycoprotein CX3C-CX3CR1 interaction and expression of substance P. J Virol. (2003) 77:9831–44. doi: 10.1128/JVI.77.18.9831-9844.2003
59. Plotnicky H, Siegrist CA, Aubry JP, Bonnefoy JY, Corvaia N, Nguyen TN, et al. Enhanced pulmonary immunopathology following neonatal priming with formalin-inactivated respiratory syncytial virus but not with the BBG2NA vaccine candidate. Vaccine. (2003) 21:2651–60. doi: 10.1016/S0264-410X(03)00055-0
60. Johnson TR, Graham BS. Contribution of respiratory syncytial virus G antigenicity to vaccine-enhanced illness and the implications for severe disease during primary respiratory syncytial virus infection. Pediatr Infect Dis J. (2004) 23(Suppl. 1):S46–57. doi: 10.1097/01.inf.0000108192.94692.d2
61. Johnson TR, Teng MN, Collins PL, Graham BS. Respiratory syncytial virus (RSV) G glycoprotein is not necessary for vaccine-enhanced disease induced by immunization with formalin-inactivated RSV. J Virol. (2004) 78:6024–32. doi: 10.1128/JVI.78.11.6024-6032.2004
62. Castilow EM, Olson MR, Varga SM. Understanding respiratory syncytial virus (RSV) vaccine-enhanced disease. Immunol Res. (2007) 39:225–39. doi: 10.1007/s12026-007-0071-6
63. Fuller JA, Njenga MK, Bigogo G, Aura B, Ope MO, Nderitu L, et al. Association of the CT values of real-time PCR of viral upper respiratory tract infection with clinical severity, Kenya. J Med Virol. (2013) 85:924–32. doi: 10.1002/jmv.23455
64. Loebbermann J, Durant L, Thornton H, Johansson C, Openshaw PJ. Defective immunoregulation in RSV vaccine-augmented viral lung disease restored by selective chemoattraction of regulatory T cells. Proc Natl Acad Sci USA. (2013) 110:2987–92. doi: 10.1073/pnas.1217580110
65. Scheltema NM, Nibbelke EE, Pouw J, Blanken MO, Rovers MM, Naaktgeboren CA, et al. Respiratory syncytial virus prevention and asthma in healthy preterm infants: a randomised controlled trial. Lancet Respir Med. (2018) 6:257–64. doi: 10.1016/S2213-2600(18)30055-9
67. Durbin RK, Kotenko SV, Durbin JE. Interferon induction and function at the mucosal surface. Immunol Rev. (2013) 255:25–39. doi: 10.1111/imr.12101
68. Gibbert K, Schlaak JF, Yang D, Dittmer U. IFN-α subtypes: distinct biological activities in anti-viral therapy. Br J Pharmacol. (2013) 168:1048–58. doi: 10.1111/bph.12010
69. Ioannidis I, McNally B, Willette M, Peeples ME, Chaussabel D, Durbin JE, et al. Plasticity and virus specificity of the airway epithelial cell immune response during respiratory virus infection. J Virol. (2012) 86:5422–36. doi: 10.1128/JVI.06757-11
70. Scotta MC, Greff D, Goecks Oliveira S, de Moura A, Rhoden Estorgato G, Duarte de Souza AP, et al. Evaluation of nasal levels of interferon and clinical severity of influenza in children. J Clin Virol. (2019) doi: 10.1016/j.jcv.2019.02.003. [Epub ahead of print].
71. Cormier SA, Shrestha B, Saravia J, Lee GI, Shen L, DeVincenzo JP, et al. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J Virol. (2014) 88:9350–60. doi: 10.1128/JVI.00818-14
72. Crisler WJ, Lenz LL. Crosstalk between type I and II interferons in regulation of myeloid cell responses during bacterial infection. Curr Opin Immunol. (2018) 54:35–41. doi: 10.1016/j.coi.2018.05.014
73. Sommereyns C, Paul S, Staeheli P, Michiels T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. (2008) 4:e1000017. doi: 10.1371/journal.ppat.1000017
74. Blumer T, Coto-Llerena M, Duong FHT, Heim MH. SOCS1 is an inducible negative regulator of interferon lambda (IFN-lambda)-induced gene expression in vivo. J Biol Chem. (2017) 292:17928–38. doi: 10.1074/jbc.M117.788877
75. Selvaggi C, Pierangeli A, Fabiani M, Spano L, Nicolai A, Papoff P, et al. Interferon lambda 1-3 expression in infants hospitalized for RSV or HRV associated bronchiolitis. J Infect. (2014) 68:467–77. doi: 10.1016/j.jinf.2013.12.010
76. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. (2000) 1:398–401. doi: 10.1038/80833
77. Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. (2001) 75:10730–7. doi: 10.1128/JVI.75.22.10730-10737.2001
78. Caballero MT, Serra ME, Acosta PL, Marzec J, Gibbons L, Salim M, et al. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. J Clin Invest. (2015) 125:571–82. doi: 10.1172/JCI75183
79. Said EA, Tremblay N, Al-Balushi MS, Al-Jabri AA, Lamarre D. Viruses seen by our cells: the role of viral RNA sensors. J Immunol Res. (2018) 2018:9480497. doi: 10.1155/2018/9480497
80. Murawski MR, Bowen GN, Cerny AM, Anderson LJ, Haynes LM, Tripp RA, et al. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J Virol. (2009) 83:1492–500. doi: 10.1128/JVI.00671-08
81. Kim TH, Lee HK. Innate immune recognition of respiratory syncytial virus infection. BMB Rep. (2014) 47:184–91. doi: 10.5483/BMBRep.2014.47.4.050
82. Dou Y, Zhao Y, Zhang Z-Y, Mao H-W, Tu W-W, Zhao X-D. Respiratory syncytial virus infection induces higher Toll-like receptor-3 expression and TNF-α production than human metapneumovirus infection. PLoS ONE. (2013) 8:e73488. doi: 10.1371/journal.pone.0073488
83. Pletneva LM, Haller O, Porter DD, Prince GA, Blanco JC. Induction of type I interferons and interferon-inducible Mx genes during respiratory syncytial virus infection and reinfection in cotton rats. J Gen Virol. (2008) 89(Pt 1):261–70. doi: 10.1099/vir.0.83294-0
84. Hall CB, Douglas RG Jr, Simons RL, Geiman JM. Interferon production in children with respiratory syncytial, influenza, and parainfluenza virus infections. J Pediatr. (1978) 93:28–32. doi: 10.1016/S0022-3476(78)80594-0
85. McIntosh K. Interferon in nasal secretions from infants with viral respiratory tract infections. J Pediatr. (1978) 93:33–6. doi: 10.1016/S0022-3476(78)80595-2
86. Krilov LR, Hendry RM, Godfrey E, McIntosh K. Respiratory virus infection of peripheral blood monocytes: correlation with ageing of cells and interferon production in vitro. J Gen Virol. (1987) 68:1749–53. doi: 10.1099/0022-1317-68-6-1749
87. Isaacs D. Production of interferon in respiratory syncytial virus bronchiolitis. Arch Dis Child. (1989) 64:92–5. doi: 10.1136/adc.64.1.92
88. Taylor CE, Webb MS, Milner AD, Milner PD, Morgan LA, Scott R, et al. Interferon alfa, infectious virus, and virus antigen secretion in respiratory syncytial virus infections of graded severity. Arch Dis Child. (1989) 64:1656–60. doi: 10.1136/adc.64.12.1656
89. Teng MN, Whitehead SS, Bermingham A, St Claire M, Elkins WR, Murphy BR, et al. Recombinant respiratory syncytial virus that does not express the NS1 or M2-2 protein is highly attenuated and immunogenic in chimpanzees. J Virol. (2000) 74:9317–21. doi: 10.1128/JVI.74.19.9317-9321.2000
90. Hornung V, Schlender J, Guenthner-Biller M, Rothenfusser S, Endres S, Conzelmann KK, et al. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J Immunol. (2004) 173:5935–43. doi: 10.4049/jimmunol.173.10.5935
91. Martinez FD, Morgan WJ, Wright AL, Holberg CJ, Taussig LM. Diminished lung function as a predisposing factor for wheezing respiratory illness in infants. N Engl J Med. (1988) 319:1112–7. doi: 10.1056/NEJM198810273191702
92. Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax. (2000) 55:1023–7. doi: 10.1136/thorax.55.12.1023
93. Melendi GA, Coviello S, Bhat N, Zea-Hernandez J, Ferolla FM, Polack FP. Breastfeeding is associated with the production of type I interferon in infants infected with influenza virus. Acta Paediatr. (2010) 99:1517–21. doi: 10.1111/j.1651-2227.2010.01862.x
94. DeVincenzo JP, El Saleeby CM, Bush AJ. Respiratory syncytial virus load predicts disease severity in previously healthy infants. J Infect Dis. (2005) 191:1861–8. doi: 10.1086/430008
95. El Saleeby CM, Somes GW, DeVincenzo JP, Gaur AH. Risk factors for severe respiratory syncytial virus disease in children with cancer: the importance of lymphopenia and young age. Pediatrics. (2008) 121:235–43. doi: 10.1542/peds.2007-1102
96. Sommer C, Resch B, Simões EA. Risk factors for severe respiratory syncytial virus lower respiratory tract infection. Open Microbiol J. (2011) 5:144–54. doi: 10.2174/1874285801105010144
97. Rodríguez DA, Rodríguez-Martínez CE, Cárdenas AC, Quilaguy IE, Mayorga LY, Falla LM, et al. Predictors of severity and mortality in children hospitalized with respiratory syncytial virus infection in a tropical region. Pediatr Pulmonol. (2014) 49:269–76. doi: 10.1002/ppul.22781
98. Lo MS, Brazas RM, Holtzman MJ. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J Virol. (2005) 79:9315–9. doi: 10.1128/JVI.79.14.9315-9319.2005
99. Becker Y. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy–a review. Virus Genes. (2006) 33:235–52. doi: 10.1007/s11262-006-0064-x
100. Bitko V, Shulyayeva O, Mazumder B, Musiyenko A, Ramaswamy M, Look DC, et al. Nonstructural proteins of respiratory syncytial virus suppress premature apoptosis by an NF-kappaB-dependent, interferon-independent mechanism and facilitate virus growth. J Virol. (2007) 81:1786–95. doi: 10.1128/JVI.01420-06
101. Gonzalez PA, Prado CE, Leiva ED, Carreno LJ, Bueno SM, Riedel CA, et al. Respiratory syncytial virus impairs T cell activation by preventing synapse assembly with dendritic cells. Proc Natl Acad Sci USA. (2008) 105:14999–5004. doi: 10.1073/pnas.0802555105
102. Munir S, Le Nouen C, Luongo C, Buchholz UJ, Collins PL, Bukreyev A. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J Virol. (2008) 82:8780–96. doi: 10.1128/JVI.00630-08
103. Munir S, Hillyer P, Le Nouen C, Buchholz UJ, Rabin RL, Collins PL, et al. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. (2011) 7:e1001336. doi: 10.1371/journal.ppat.1001336
104. Okabayashi T, Kojima T, Masaki T, Yokota S, Imaizumi T, Tsutsumi H, et al. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. (2011) 160:360–6. doi: 10.1016/j.virusres.2011.07.011
105. Chatterjee S, Luthra P, Esaulova E, Agapov E, Yen BC, Borek DM, et al. Structural basis for human respiratory syncytial virus NS1-mediated modulation of host responses. Nat Microbiol. (2017) 2:17101. doi: 10.1038/nmicrobiol.2017.101
106. Sung RY, Yin J, Oppenheimer SJ, Tam JS, Lau J. Treatment of respiratory syncytial virus infection with recombinant interferon alfa-2a. Arch Dis Child. (1993) 69:440–2. doi: 10.1136/adc.69.4.440
107. Spann KM, Baturcam E, Schagen J, Jones C, Straub CP, Preston FM, et al. Viral and host factors determine innate immune responses in airway epithelial cells from children with wheeze and atopy. Thorax. (2014) 69:918–25. doi: 10.1136/thoraxjnl-2013-204908
108. Sriskandan K, Garner P, Watkinson J, Pettingale KW, Brinkley D, Calman FMB, et al. A toxicity study of recombinant interferon-gamma given by intravenous infusion to patients with advanced cancer. Cancer Chemother Pharmacol. (1986) 18:63–8. doi: 10.1007/BF00253067
109. Savale L, Sattler C, Gunther S, Montani D, Chaumais MC, Perrin S, et al. Pulmonary arterial hypertension in patients treated with interferon. Eur Respir J. (2014) 44:1627–34. doi: 10.1183/09031936.00057914
110. Schmidinger M. Understanding and managing toxicities of vascular endothelial growth factor (VEGF) inhibitors. EJC Suppl. (2013) 11:172–91. doi: 10.1016/j.ejcsup.2013.07.016
111. Caraceni A, Gangeri L, Martini C, Belli F, Brunelli C, Baldini M, et al. Neurotoxicity of interferon-alpha in melanoma therapy: results from a randomized controlled trial. Cancer. (1998) 83:482–9. doi: 10.1002/(SICI)1097-0142(19980801)83:3<482::AID-CNCR17>3.0.CO;2-S
112. Gogas H, Bafaloukos D, Ioannovich J, Skarlos D, Polyzos A, Fountzilas G, et al. Tolerability of adjuvant high-dose interferon alfa-2b: 1 month versus 1 year–a Hellenic Cooperative Oncology Group study. Anticancer Res. (2004) 24:1947–52.
113. Minami T, Kishikawa T, Sato M, Tateishi R, Yoshida H, Koike K. Meta-analysis: mortality and serious adverse events of peginterferon plus ribavirin therapy for chronic hepatitis C. J Gastroenterol. (2013) 48:254–68. doi: 10.1007/s00535-012-0631-y
114. Dubois J, Hershon L, Carmant L, Bélanger S, Leclerc J-M, David M. Toxicity profile of interferon alfa-2b in children: a prospective evaluation. J Pediatrics. (1999) 135:782–5. doi: 10.1016/S0022-3476(99)70104-6
115. Portnoy J, Hicks R, Pacheco F, Olson L. Pilot study of recombinant interferon alpha-2a for treatment of infants with bronchiolitis induced by respiratory syncytial virus. Antimicrob Agents Chemother. (1988) 32:589–91. doi: 10.1128/AAC.32.4.589
116. DeVincenzo JP, Whitley RJ, Mackman RL, Scaglioni-Weinlich C, Harrison L, Farrell E, et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N Engl J Med. (2014) 371:711–22. doi: 10.1056/NEJMoa1401184
117. DeVincenzo JP, McClure MW, Symons JA, Fathi H, Westland C, Chanda S, et al. Activity of oral ALS-008176 in a respiratory syncytial virus challenge study. N Engl J Med. (2015) 373:2048–58. doi: 10.1056/NEJMoa1413275
118. Scagnolari C, Midulla F, Selvaggi C, Monteleone K, Bonci E, Papoff P, et al. Evaluation of viral load in infants hospitalized with bronchiolitis caused by respiratory syncytial virus. Med Microbiol Immunol. (2012) 201:311–7. doi: 10.1007/s00430-012-0233-6
119. Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci USA. (2017) 114:8342–7. doi: 10.1073/pnas.1704259114
120. Lamborn IT, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, et al. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med. (2017) 214:1949–72. doi: 10.1084/jem.20161759
121. Taylor G. Animal models of respiratory syncytial virus infection. Vaccine. (2017) 35:469–80. doi: 10.1016/j.vaccine.2016.11.054
122. Cormier SA, You D, Honnegowda S. The use of a neonatal mouse model to study respiratory syncytial virus infections. Expert Rev Anti Infect Ther. (2010) 8:1371–80. doi: 10.1586/eri.10.125
123. Ruckwardt TJ, Malloy AM, Gostick E, Price DA, Dash P, McClaren JL, et al. Neonatal CD8 T-cell hierarchy is distinct from adults and is influenced by intrinsic T cell properties in respiratory syncytial virus infected mice. PLoS Pathog. (2011) 7:e1002377. doi: 10.1371/journal.ppat.1002377
124. Battles MB, Langedijk JP, Furmanova-Hollenstein P, Chaiwatpongsakorn S, Costello HM, Kwanten L, et al. Molecular mechanism of respiratory syncytial virus fusion inhibitors. Nat Chem Biol. (2016) 12:87–93. doi: 10.1038/nchembio.1982
125. Adderson E, Branum K, Sealy RE, Jones BG, Surman SL, Penkert R, et al. Safety and immunogenicity of an intranasal Sendai virus-based human parainfluenza virus type 1 vaccine in 3- to 6-year-old children. Clin Vaccine Immunol. (2015) 22:298–303. doi: 10.1128/CVI.00618-14
126. Saravia J, You D, Shrestha B, Jaligama S, Siefker D, Lee GI, et al. Respiratory syncytial virus disease is mediated by age-variable IL-33. PLoS Pathog. (2015) 11:e1005217. doi: 10.1371/journal.ppat.1005217
127. Chu HY, Chin J, Pollard J, Zerr DM, Englund JA. Clinical outcomes in outpatient respiratory syncytial virus infection in immunocompromised children. Influenza Other Respir Viruses. (2016) 10:205–10. doi: 10.1111/irv.12375
128. Shrestha B, You D, Saravia J, Siefker DT, Jaligama S, Lee GI, et al. IL-4Ralpha on dendritic cells in neonates and Th2 immunopathology in respiratory syncytial virus infection. J Leukoc Biol. (2017) 102:153–61. doi: 10.1189/jlb.4A1216-536R
129. Ripple MJ, You D, Honnegowda S, Giaimo JD, Sewell AB, Becnel DM, et al. Immunomodulation with IL-4R alpha antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J Immunol. (2010) 185:4804–11. doi: 10.4049/jimmunol.1000484
130. Roman M, Calhoun WJ, Hinton KL, Avendano LF, Simon V, Escobar AM, et al. Respiratory syncytial virus infection in infants is associated with predominant Th-2-like response. Am J Respir Crit Care Med. (1997) 156:190–5. doi: 10.1164/ajrccm.156.1.9611050
131. Pinto RA, Arredondo SM, Bono MR, Gaggero AA, Diaz PV. T helper 1/T helper 2 cytokine imbalance in respiratory syncytial virus infection is associated with increased endogenous plasma cortisol. Pediatrics. (2006) 117:e878–886. doi: 10.1542/peds.2005-2119
132. Bermejo-Martin JF, Garcia-Arevalo MC, De Lejarazu RO, Ardura J, Eiros JM, Alonso A, et al. Predominance of Th2 cytokines, CXC chemokines and innate immunity mediators at the mucosal level during severe respiratory syncytial virus infection in children. Eur Cytokine Netw. (2007) 18:162–7. doi: 10.1684/ecn.2007.0096
133. Remot A, Descamps D, Jouneau L, Laubreton D, Dubuquoy C, Bouet S, et al. Flt3 ligand improves the innate response to respiratory syncytial virus and limits lung disease upon RSV reexposure in neonate mice. Eur J Immunol. (2016) 46:874–84. doi: 10.1002/eji.201545929
134. Pulendran B, Banchereau J, Burkeholder S, Kraus E, Guinet E, Chalouni C, et al. Flt3-ligand and granulocyte colony-stimulating factor mobilize distinct human dendritic cell subsets in vivo. J Immunol. (2000) 165:566–72. doi: 10.4049/jimmunol.165.1.566
135. Willems F, Vollstedt S, Suter M. Phenotype and function of neonatal DC. Eur J Immunol. (2009) 39:26–35. doi: 10.1002/eji.200838391
136. Hijano DR, Siefker DT, Shrestha B, Jaligama S, Vu LD, Tillman H, et al. Type I interferon potentiates IgA immunity to respiratory syncytial virus infection during infancy. Sci Rep. (2018) 8:11034. doi: 10.1038/s41598-018-29456-w
137. Faber TE, Schuurhof A, Vonk A, Koppelman GH, Hennus MP, Kimpen JL, et al. IL1RL1 gene variants and nasopharyngeal IL1RL-a levels are associated with severe RSV bronchiolitis: a multicenter cohort study. PLoS ONE. (2012) 7:e34364. doi: 10.1371/journal.pone.0034364
138. Lynch JP, Werder RB, Simpson J, Loh Z, Zhang V, Haque A, et al. Aeroallergen-induced IL-33 predisposes to respiratory virus-induced asthma by dampening antiviral immunity. J Allergy Clin Immunol. (2016) 138:1326–37. doi: 10.1016/j.jaci.2016.02.039
139. Lynch JP, Werder RB, Loh Z, Sikder MAA, Curren B, Zhang V, et al. Plasmacytoid dendritic cells protect from viral bronchiolitis and asthma through semaphorin 4a–mediated T reg expansion. J Exp Med. (2018) 215:537–57. doi: 10.1084/jem.20170298
140. Kerrin A, Fitch P, Errington C, Kerr D, Waxman L, Riding K, et al. Differential lower airway dendritic cell patterns may reveal distinct endotypes of RSV bronchiolitis. Thorax. (2017) 72:620–7. doi: 10.1136/thoraxjnl-2015-207358
141. Singleton R, Etchart N, Hou S, Hyland L. Inability to evoke a long-lasting protective immune response to respiratory syncytial virus infection in mice correlates with ineffective nasal antibody responses. J Virol. (2003) 77:11303–11. doi: 10.1128/JVI.77.21.11303-11311.2003
142. Murphy BR, Graham BS, Prince GA, Walsh EE, Chanock RM, Karzon DT, et al. Serum and nasal-wash immunoglobulin G and A antibody response of infants and children to respiratory syncytial virus F and G glycoproteins following primary infection. J Clin Microbiol. (1986) 23:1009–14.
143. Tsutsumi H, Matsuda K, Yamazaki H, Ogra PL, Chiba S. Different kinetics of antibody responses between IgA and IgG classes in nasopharyngeal secretion in infants and children during primary respiratory syncytial virus infection. Acta Paediatr Jpn. (1995) 37:464–8. doi: 10.1111/j.1442-200X.1995.tb03356.x
144. Ruckwardt TJ, Malloy AMW, Morabito KM, Graham BS. Quantitative and qualitative deficits in neonatal lung-migratory dendritic cells impact the generation of the CD8+ T cell response. PLoS Pathog. (2014) 10:e1003934. doi: 10.1371/journal.ppat.1003934
145. Han J, Dakhama A, Jia Y, Wang M, Zeng W, Takeda K, et al. Responsiveness to respiratory syncytial virus in neonates is mediated through thymic stromal lymphopoietin and OX40 ligand. J Allergy Clin Immunol. (2012) 130:1175–86.e79. doi: 10.1016/j.jaci.2012.08.033
146. You D, Marr N, Saravia J, Shrestha B, Lee GI, Turvey SE, et al. IL-4Rα on CD4+ T cells plays a pathogenic role in respiratory syncytial virus reinfection in mice infected initially as neonates. J Leukocyte Biol. (2013) 93:933–42. doi: 10.1189/jlb.1012498
147. Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am J Respir Crit Care Med. (2003) 168:633–9. doi: 10.1164/rccm.200210-1148OC
148. Tregoning JS, Yamaguchi Y, Harker J, Wang B, Openshaw PJM. The role of T cells in the enhancement of respiratory syncytial virus infection severity during adult reinfection of neonatally sensitized mice. J Virol. (2008) 82:4115–24. doi: 10.1128/JVI.02313-07
149. Heidema J, Lukens MV, van Maren WW, van Dijk ME, Otten HG, van Vught AJ, et al. CD8+ T cell responses in bronchoalveolar lavage fluid and peripheral blood mononuclear cells of infants with severe primary respiratory syncytial virus infections. J Immunol. (2007) 179:8410–7. doi: 10.4049/jimmunol.179.12.8410
150. Delgado MF, Coviello S, Monsalvo AC, Melendi GA, Hernandez JZ, Batalle JP, et al. Lack of antibody affinity maturation due to poor Toll-like receptor stimulation leads to enhanced respiratory syncytial virus disease. Nat Med. (2009) 15:34–41. doi: 10.1038/nm.1894
151. Bagga B, Cehelsky JE, Vaishnaw A, Wilkinson T, Meyers R, Harrison LM, et al. Effect of preexisting serum and mucosal antibody on experimental Respiratory Syncytial Virus (RSV) challenge and infection of adults. J Infect Dis. (2015) 212:1719–25. doi: 10.1093/infdis/jiv281
152. Habibi MS, Jozwik A, Makris S, Dunning J, Paras A, DeVincenzo JP, et al. Impaired antibody-mediated protection and defective IgA B-cell memory in experimental infection of adults with respiratory syncytial virus. Am J Respir Crit Care Med. (2015) 191:1040–9. doi: 10.1164/rccm.201412-2256OC
153. Vissers M, Ahout IM, de Jonge MI, Ferwerda G. Mucosal IgG levels correlate better with respiratory syncytial virus load and inflammation than plasma IgG levels. Clin Vaccine Immunol. (2015) 23:243–5. doi: 10.1128/CVI.00590-15
154. Brint ME, Hughes JM, Shah A, Miller CR, Harrison LG, Meals EA, et al. Prolonged viral replication and longitudinal viral dynamic differences among respiratory syncytial virus infected infants. Pediatr Res. (2017) 82:872–80. doi: 10.1038/pr.2017.173
155. Bates JT, Keefer CJ, Slaughter JC, Kulp DW, Schief WR, Crowe JE Jr. Escape from neutralization by the respiratory syncytial virus-specific neutralizing monoclonal antibody palivizumab is driven by changes in on-rate of binding to the fusion protein. Virology. (2014) 454-455:139–44. doi: 10.1016/j.virol.2014.02.010
156. DeVincenzo JP, Buckingham SC. Relationship between respiratory syncytial virus load and illness severity in children. J Infect Dis. (2002) 186:1376–7; author reply 1377. doi: 10.1086/344331
157. Mills JT, Van Kirk JE, Wright PF, Chanock RM. Experimental respiratory syncytial virus infection of adults. Possible mechanisms of resistance to infection and illness. J Immunol. (1971) 107:123–30.
158. Walsh EE, Falsey AR. Humoral and mucosal immunity in protection from natural respiratory syncytial virus infection in adults. J Infect Dis. (2004) 190:373–8. doi: 10.1086/421524
159. de Souza AP, de Freitas DN, Antuntes Fernandes KE, D'Avila da Cunha M, Antunes Fernandes JL, Benetti Gassen R, et al. Respiratory syncytial virus induces phosphorylation of mTOR at ser2448 in CD8 T cells from nasal washes of infected infants. Clin Exp Immunol. (2016) 183:248–57. doi: 10.1111/cei.12720
160. Wright PF, Gruber WC, Peters M, Reed G, Zhu Y, Robinson F, et al. Illness severity, viral shedding, and antibody responses in infants hospitalized with bronchiolitis caused by respiratory syncytial virus. J Infect Dis. (2002) 185:1011–8. doi: 10.1086/339822
161. Franz A, Adams O, Willems R, Bonzel L, Neuhausen N, Schweizer-Krantz S, et al. Correlation of viral load of respiratory pathogens and co-infections with disease severity in children hospitalized for lower respiratory tract infection. J Clin Virol. (2010) 48:239–45. doi: 10.1016/j.jcv.2010.05.007
162. Giersing BK, Karron RA, Vekemans J, Kaslow DC, Moorthy VS. Meeting report: WHO consultation on Respiratory Syncytial Virus (RSV) vaccine development, Geneva, 25-26 April 2016. Vaccine. (2017). doi: 10.1016/j.vaccine.2017.02.068. [Epub ahead of print].
163. Villafana T, Falloon J, Griffin MP, Zhu Q, Esser MT. Passive and active immunization against respiratory syncytial virus for the young and old. Expert Rev Vaccines. (2017) 16:1–13. doi: 10.1080/14760584.2017.1333425
164. Giersing BK, Vekemans J, Nava S, Kaslow DC, Moorthy V. Report from the World Health Organization's third Product Development for Vaccines Advisory Committee (PDVAC) meeting, Geneva, 8-10th June 2016. Vaccine. (2017). doi: 10.1016/j.vaccine.2016.10.090. [Epub ahead of print].
165. Noor A, Krilov LR. Respiratory syncytial virus vaccine: where are we now and what comes next? Expert Opin Biol Ther. (2018) 18:1247–56. doi: 10.1080/14712598.2018.1544239
166. Esposito S, Pietro GD. Respiratory syncytial virus vaccines: an update on those in the immediate pipeline. Future Microbiol. (2016) 11:1479–90. doi: 10.2217/fmb-2016-0106
167. Roberts JN, Graham BS, Karron RA, Munoz FM, Falsey AR, Anderson LJ, et al. Challenges and opportunities in RSV vaccine development: meeting report from FDA/NIH workshop. Vaccine. (2016) 34:4843–9. doi: 10.1016/j.vaccine.2016.07.057
168. Mazur NI, Higgins D, Nunes MC, Melero JA, Langedijk AC, Horsley N, et al. The respiratory syncytial virus vaccine landscape: lessons from the graveyard and promising candidates. Lancet Infect Dis. (2018) 18:e295–311. doi: 10.1016/S1473-3099(18)30292-5
169. Groppo R, DiNapoli J, Il Jeong K, Kishko M, Jackson N, Kleanthous H, et al. Effect of genetic background and delivery route on the preclinical properties of a live attenuated RSV vaccine. PLoS ONE. (2018) 13:e0199452. doi: 10.1371/journal.pone.0199452
170. Ngwuta JO, Chen M, Modjarrad K, Joyce MG, Kanekiyo M, Kumar A, et al. Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med. (2015) 7:309ra162. doi: 10.1126/scitranslmed.aac4241
171. Boyington JC, Joyce MG, Sastry M, Stewart-Jones GB, Chen M, Kong WP, et al. Structure-based design of head-only fusion glycoprotein immunogens for respiratory syncytial virus. PLoS ONE. (2016) 11:e0159709. doi: 10.1371/journal.pone.0159709
172. Tian D, Battles MB, Moin SM, Chen M, Modjarrad K, Kumar A, et al. Structural basis of respiratory syncytial virus subtype-dependent neutralization by an antibody targeting the fusion glycoprotein. Nat Commun. (2017) 8:1877. doi: 10.1038/s41467-017-01858-w
173. Mousa JJ, Binshtein E, Human S, Fong RH, Alvarado G, Doranz BJ, et al. Human antibody recognition of antigenic site IV on Pneumovirus fusion proteins. PLoS Pathog. (2018) 14:e1006837. doi: 10.1371/journal.ppat.1006837
174. Zhu Q, Lu B, McTamney P, Palaszynski S, Diallo S, Ren K, et al. Prevalence and significance of substitutions in the fusion protein of respiratory syncytial virus resulting in neutralization escape from antibody MEDI8897. J Infect Dis. (2018) 218:572–80. doi: 10.1093/infdis/jiy189
175. Graham BS. Vaccine development for respiratory syncytial virus. Curr Opin Virol. (2017) 23:107–12. doi: 10.1016/j.coviro.2017.03.012
176. Taleb SA, Al Thani AA, Al Ansari K, Yassine HM. Human respiratory syncytial virus: pathogenesis, immune responses, and current vaccine approaches. Eur J Clin Microbiol Infect Dis. (2018) 37, 1817–27. doi: 10.1007/s10096-018-3289-4
177. Anderson R, Huang Y, Langley JM. Prospects for defined epitope vaccines for respiratory syncytial virus. Future Microbiol. (2010) 5:585–602. doi: 10.2217/fmb.10.22
178. Teng MN, Whitehead SS, Collins PL. Contribution of the respiratory syncytial virus G glycoprotein and its secreted and membrane-bound forms to virus replication in vitro and in vivo. Virology. (2001) 289:283–96. doi: 10.1006/viro.2001.1138
179. Collins PL, Melero JA. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res. (2011) 162:80–99. doi: 10.1016/j.virusres.2011.09.020
180. Zhu Q, McLellan JS, Kallewaard NL, Ulbrandt ND, Palaszynski S, Zhang J, et al. A highly potent extended half-life antibody as a potential RSV vaccine surrogate for all infants. Sci Transl Med. (2017) 9:eaaj1928. doi: 10.1126/scitranslmed.aaj1928
181. Rossey I, McLellan JS, Saelens X, Schepens B. Clinical potential of prefusion RSV F-specific antibodies. Trends Microbiol. (2018) 26:209–19. doi: 10.1016/j.tim.2017.09.009
182. Carillo MC, Alvarez Mde L, Quiroga AD. Interferon alfa-2b triggers transforming growth factor-beta-induced apoptosis on preneoplasticliver. Ann Hepatol. (2006) 5:244–50.
183. Jadoon SM, Jadoon S, Muhammad I. Response to standard interferon A2b and ribavirin combination therapy in chronic hepatitis C treatment naive patients. J Ayub Med Coll Abbottabad. (2010) 22:164–6.
184. Biopharma A. Ansun BioPharma Announces Breakthrough Designation for its Experimental Drug DAS181 [Online]. (2017) Available: http://www.ansunbiopharma.com/news/ (Accessed September 3, 2018).
185. Stanisavljevic S, Dedovic N, Vujicic M, Saksida T, Jevtic B, Milovanovic B, et al. Strain-specific helper T cell profile in the gut-associated lymphoid tissue. Immunol Lett. (2017) 190:282–8. doi: 10.1016/j.imlet.2017.08.017
186. Petrovic R, Bufan B, Arsenovic-Ranin N, Zivkovic I, Minic R, Radojevic K, et al. Mouse strain and sex as determinants of immune response to trivalent influenza vaccine. Life Sci. (2018) 207:117–26. doi: 10.1016/j.lfs.2018.05.056
187. Harker JA, Yamaguchi Y, Culley FJ, Tregoning JS, Openshaw PJ. Delayed sequelae of neonatal respiratory syncytial virus infection are dependent on cells of the innate immune system. J Virol. (2014) 88:604–11. doi: 10.1128/JVI.02620-13
188. Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, et al. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J Immunol. (2007) 179:3171–7. doi: 10.4049/jimmunol.179.5.3171
189. Collarini EJ, Lee FE, Foord O, Park M, Sperinde G, Wu H, et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from B cells of infected patients. J Immunol. (2009) 183:6338–45. doi: 10.4049/jimmunol.0901373
190. Caidi H, Miao C, Thornburg NJ, Tripp RA, Anderson LJ, Haynes LM. Anti-respiratory syncytial virus (RSV) G monoclonal antibodies reduce lung inflammation and viral lung titers when delivered therapeutically in a BALB/c mouse model. Antiviral Res. (2018) 154:149–57. doi: 10.1016/j.antiviral.2018.04.014
Keywords: infant immunity, respiratory syncytial virus, type I interferons, human, mouse, vaccine
Citation: Hijano DR, Vu LD, Kauvar LM, Tripp RA, Polack FP and Cormier SA (2019) Role of Type I Interferon (IFN) in the Respiratory Syncytial Virus (RSV) Immune Response and Disease Severity. Front. Immunol. 10:566. doi: 10.3389/fimmu.2019.00566
Received: 15 January 2019; Accepted: 04 March 2019;
Published: 26 March 2019.
Edited by:
Alexis M. Kalergis, Pontificia Universidad Católica de Chile, ChileReviewed by:
Jan Rehwinkel, University of Oxford, United KingdomCopyright © 2019 Hijano, Vu, Kauvar, Tripp, Polack and Cormier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephania A. Cormier, c3RlcGhhbmlhY29ybWllckBsc3UuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.