 Joseph W. Saelens
Joseph W. Saelens Gopinath Viswanathan
Gopinath Viswanathan David M. Tobin
David M. Tobin- 1Department of Molecular Genetics and Microbiology, Duke University School of Medicine, Durham, NC, United States
- 2Department of Immunology, Duke University School of Medicine, Durham, NC, United States
Over the past 200 years, tuberculosis (TB) has caused more deaths than any other infectious disease, likely infecting more people than it has at any other time in human history. Mycobacterium tuberculosis (Mtb), the etiologic agent of TB, is an obligate human pathogen that has evolved through the millennia to become an archetypal human-adapted pathogen. This review focuses on the evolutionary framework by which Mtb emerged as a specialized human pathogen and applies this perspective to the emergence of specific lineages that drive global TB burden. We consider how evolutionary pressures, including transmission dynamics, host tolerance, and human population patterns, may have shaped the evolution of diverse mycobacterial genomes.
Introduction
Tuberculosis (TB) is a critical health crisis in our modern world. TB is one of the top ten causes of death worldwide, killing an estimated 1.7 million people in 2017 (1). Despite years of coordinated global efforts to reduce the burden of TB, it is estimated that around 10 million new infections developed around the world in 2017 (1).
Mycobacterium tuberculosis (Mtb), the etiologic agent of TB, has evolved through the millennia to become a highly specialized obligate human pathogen. Indeed, some consider Mtb as the archetypal human-adapted pathogen (2). Unlike the non-pathogenic soil-dwellers and the opportunistically pathogenic species of mycobacteria, Mtb has no known environmental reservoir and does not survive outside of its human host. For its survival, Mtb has evolved to subvert and co-opt the very mechanisms the human immune system deploys to clear bacterial infections for its own advantage. However, the host is capable of limiting mycobacterial growth and, in some cases, inducing latency (3, 4), or sterilizing the infection (5, 6). Latent or subclinical disease provides mechanisms whereby Mtb can remain in the host and reactivate following immune suppression, transmitting to new hosts (7), although our previous understanding of the nature and significance of latent disease is now being rethought (8, 9). Nonetheless, this balance between host and pathogen is central to the evolutionary survival strategy of Mtb as an obligate human pathogen. Indeed, it is estimated that 90% of people that are infected by Mtb either contain or clear the infection (10). Yet the 10% of patients who develop active disease transmit Mtb to such a degree that one quarter of the world's population is estimated to have mounted an immune response to the pathogen (11). TB has caused over 1 billion deaths in the past 200 years, surpassing all other infectious diseases (12). In this review, we discuss the features of Mtb that were central to its emergence as a human pathogen and how genetic diversity among strains contributes to phenotypic diversity in disease presentation, with a focus on the evolutionary interplay between pathogen and host. Bacterial factors that engage the host promote bacterial growth, survival, and transmission in human populations. Yet, overall, an evolutionary balance has been reached in which host mechanisms of containment and tolerance counteract many of these bacterial features.
The Origins of Mtb
The timing of events that contributed to Mtb's specialized adaptation to human hosts remains a matter of debate. Some point to an early origin of Mtb ~70,000 years ago (13, 14), while others have more conservative estimates of 35,000 years ago (15). Other studies suggest a more recent emergence of ~6,000 years ago is most likely (16). These estimates are based on different assumptions and study materials, and have therefore led to a wide dispersion of estimates.
Most studies have employed inference methods based on DNA sequence among extant strains of Mtb. This method relies on the calibration of a molecular clock, which uses genetic distance as a measure of time since divergence (17). Mtb demonstrates a clonal population structure that can be divided into seven major lineages (Figure 1), and the divergence between these lineages and the other members of the Mycobacterium tuberculosis complex (MTBC) has been used by some to calibrate the molecular clock for Mtb (25).
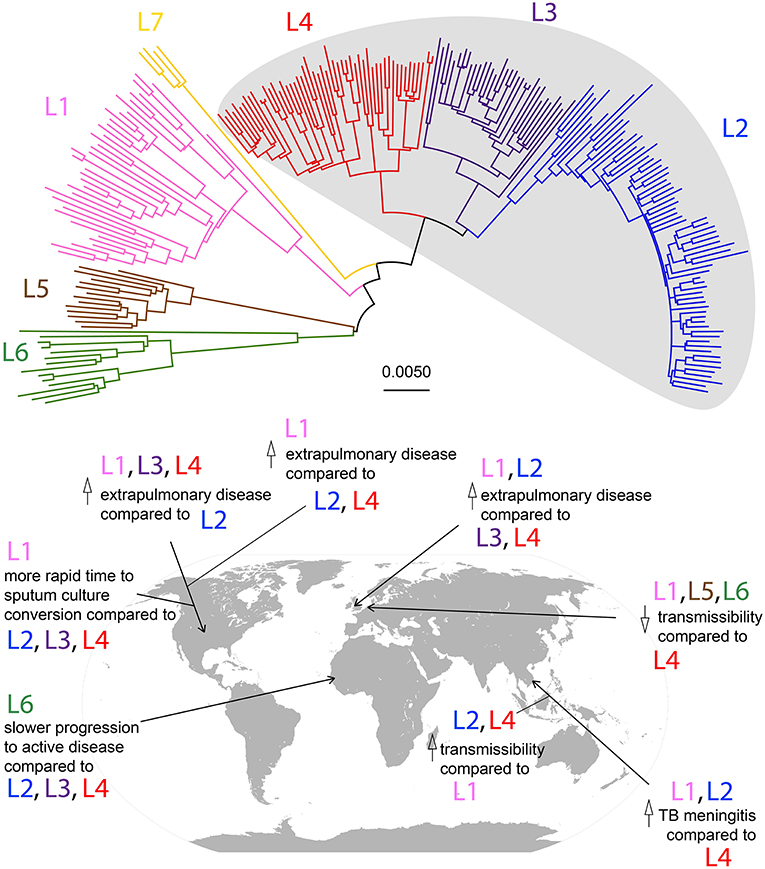
Figure 1. Phylogeny of Mtb lineages and geographic associations of disease characteristics. Neighbor-joining phylogeny based on 35,787 SNPs among 225 strains from Comas et al. (13). Lineages are color-coded according to the scheme described in Bos et al. (16), and modern lineages are shaded in gray. Scale bar represents relative number of substitutions per known variant. Disease characteristic associations with Mtb lineages in geographic locations by studies described in Table 1 are marked on a world map.
Prior to the advent of widespread accessibility to whole genome sequencing, Mtb's molecular clock was estimated using variable numbers of tandem-repeats (VNTR) in microsatellite-like loci (26). This method proposed an origin of the MTBC approximately 40,000 years ago, and highlighted the likelihood of Mtb dispersing throughout Africa and Eurasia via human migration (27). However, the use of VNTR in constructing phylogenies can lead to phylogenetic arrangements incongruent with known genetic relationships due to convergent evolution at these loci (28). Therefore, the current gold standard for calibrating a molecular clock is genome sequencing. However, as demonstrated below, the method by which Mtb's molecular clock is calibrated will have a significant impact on the resulting estimates.
Multiple studies have employed genome sequencing to determine the molecular clock of Mtb and have arrived at vastly different estimates for the age of Mtb. The calibration of the molecular clock underlies these differences. Comas et al. estimate Mtb's origins as far back as 70,000 years ago (13). This estimate is based on the parallels of mitochondrial DNA (mtDNA) haplogroups and the lineages of Mtb that are most commonly found among the corresponding human populations, and then calibrating the molecular clock using key events in human evolution reflected by mtDNA. This generated an estimated mutation rate in Mtb of 2.58 × 10−9 substitutions/site/year, which is low compared to estimates derived from contemporary outbreaks (1.1 × 10−7 substitutions/site/year) (29). However, their estimates produced multiple time points for Mtb's emergence, and 70,000 years was chosen as the most likely. The researchers who put forth this hypothesis on the origin of Mtb had previously published work proposing the dispersal of Mtb via human migration out of Africa (14). While the phylogeographic distribution of the major lineages of Mtb coincide with concordant patterns in human migration (25), calibrating Mtb's molecular clock based on these patterns to determine when Mtb emerged presupposes its own hypothesis that Mtb emerged with modern humans.
Others have challenged this hypothesis and proposed a much later time frame for Mtb's emergence (30). Instead of mtDNA, Pepperell et al. based their estimates on historical samples of MTBC strains and determined that the emergence of the most basal species of Mtb, M. africanum, occurred approximately 2,200 years ago. The most recently evolved strains of Mtb, those among the so-called “modern” lineages, are estimated to have arisen ~1,300 years ago. The estimated mutation rate of Mtb from this study (1.3 × 10−7 substitutions/site/year) was significantly higher than that of Comas et al. Furthermore, based on this early estimate for the origin of Mtb, Pepperell et al. propose the estimates for human population divergence do not correlate with the divergence of the Mtb lineages, and therefore did not disperse concurrently (30). Another study has put forth an origin estimate similar to that of Pepperell et al. The mummified remains of human samples from Peru dated between AD 1028 and AD 1280 demonstrated skeletal lesions indicative of TB (31–33). Sequenced ancient DNA (aDNA) from these samples revealed disease was caused by M. pinipedii, a member of the Mycobacterium tuberculosis complex (MTBC) that primarily infects seals (16). Comparing the aDNA against a current strain of M. pinipedii generated an estimate of MTBC's emergence occurring 6,000 years ago, with a mutation rate intermediate to the estimates of Comas et al. and Pepperell et al. (4.6 × 10−8 substitutions/site/year). However, the reliance on aDNA comes with the caveat that post-mortem DNA decays due to physical and chemical damage, leading to strand breakage and the hydrolytic deamination of cytosine to uracil (34). Therefore, additional bioinformatic corrections must be implemented to sort out decay artifacts, leading to the possibility of erroneous or missed variant calls in aDNA samples.
The variety of conclusions from these studies demonstrates that the calibration of the molecular clock is critical to the resulting estimates, and raises the question as to how well-suited Mtb is for molecular clock estimations. The application of molecular clocks relies on satisfying certain assumptions that could be problematic when applied to Mtb: namely, a constant mutation rate through time and the broad applicability of this rate across lineages (17). It is not at all clear that the mutation rate of Mtb is stable over evolutionary time, as no study has been able to collect longitudinal data from historical samples. Additionally, the health status of human hosts across space and time is highly variable, creating different pressures on the infecting strains. Furthermore, even among the extant lineages of Mtb, which are much more closely related to each other than they are to other members of the MTBC, variable mutation rates have been observed (35). A recent analysis highlights the complexities, uncertainties, and limitations of different methods used to calibrate an Mtb molecular clock (36).
The earliest claim of mycobacterial disease comes from a 500,000 year old fossil of Homo erectus, which demonstrated lesions characteristic of mycobacterial infection (37). As no ancient DNA (aDNA) was recovered from this sample, it is impossible to determine what species of mycobacteria might have caused the lesions. Using lipid profiles unique to pathogenic mycobacteria and the IS6110 insertion element (38), a feature found only in members of the MTBC (39), the oldest confirmed sample of mycobacterial disease was found in bovid fossils in North America, dating back approximately 17,000 years (40, 41). The earliest known association of the MTBC with humans comes from Atlit-Yam, Israel, dating back 9,000 years (42). Interestingly, this sample bears the TbD1 marker, a genomic deletion found exclusively in the evolutionarily “modern” lineages of Mtb (43). Linking definitive archaeological findings with aDNA sequencing will provide the most compelling evidence to settle the divergent estimates. As the techniques for collecting and sequencing aDNA continue to advance, our insight into Mtb's origins will similarly improve, and we may better understand the evolutionary forces and constraints leading to modern Mtb and the nature of its interactions with its hosts.
The Evolution of Mycobacterium tuberculosis as a Specialized Pathogen
Mycobacteria range from environmental, non-pathogenic species, to opportunistic pathogens that infect immune-compromised hosts, to professional pathogens. The vast majority of Mycobacteria are non-pathogenic in nature. Comparative genomic studies have revealed the evolutionary trajectory to pathogenicity, in which environmental mycobacteria acquired virulence loci and became opportunists, and opportunists adapted to their host environments to become professional pathogens. The pathogenic species include but are not limited to: Mycobacterium ulcerans (the agent of Buruli ulcer), Mycobacterium leprae (leprosy), Mycobacterium marinum, Mycobacterium canetti, and the range of species that make up the MTBC. The MTBC contains the closely related species of pathogenic mycobacteria that, together, cause the vast majority of TB. Several of these species are animal-adapted strains that cause disease across a range of mammalian species. These include Mycobacterium bovis (infecting cows), Mycobacterium caprae (goats and sheep), Mycobacterium pinipedii (seals and sea lions), Mycobacterium microti (voles), and Mycobacterium orygis (oryxes) (44, 45). Mtb and Mycobacterium africanum cause the majority of disease in humans. Among all of these pathogenic mycobacteria, M. tuberculosis sensu stricto has emerged as the most prevalent mycobacterial species and one of the most historically successful human pathogens. The key features and events that underlie the adaptation of mycobacteria into a specialized pathogen are discussed below and have also been highlighted in previous reviews [e.g., (2)].
From the Environment to New Hosts
The soil-dwelling mycobacteria Mycobacterium kansasii is an environmental, opportunistic mycobacterial pathogen closely related to the MTBC. This genetic relationship provides insight into the late-stage events conferring Mtb's specialized adaptation that allowed it to expand and persist as an obligate human pathogen. Unlike the nonpathogenic mycobacterial species, M. kansasii contains an array of virulence determinants for host adaptation. There are five ESX loci in Mtb, and all five are present in M. kansasii (46). Furthermore, M. kansasii has expanded its set of PE/PPE proteins and, in fact, encodes a greater number of PE/PPE proteins than Mtb and other members of the MTBC. Despite these similarities M. kansasii is only rarely found in patients, whereas Mtb infection in humans is prevalent (47, 48). Therefore, the ESX secretion systems and its effectors are not sufficient to explain the pathogenicity of Mtb. Given the shared virulence features of M. kansasii with Mtb but their vastly different impact on global health, what other features separate Mtb from M. kansasii?
The enhanced virulence of Mtb may have been the result of acquiring pathogenicity islands via horizontal gene transfer (HGT) (49–52). Comparative genomics reveals the presence of 55 genes in Mtb absent from M. kansasii (51). The majority of these genes contain an unusual GC content for mycobacteria and appear in clusters flanked by the vehicles that provide mechanisms for HGT (mycobacteriophage genes, transposons, and toxin-antitoxin systems). Notably, some of these HGT-acquired genes, encoding factors responsible for cell adhesion (53), arresting phagosome maturation (54, 55), the production of PGLs that function in oxidative stress resistance (56) and modulation of the host immune system (57) have been implicated in Mtb's adaptation to survival within a host (55, 58).
Mycobacterial species comprising the “smooth tubercle bacilli” (STB) are thought to be an evolutionary bridge between the environmental opportunistic species, such as M. kansasii, to the pathogenic MTBC (46). Unlike the MTBC, genome sequencing reveals that M. canetti demonstrates a non-clonal population structure with >60,000 SNPs separating some strains (50). While the environmental reservoir of M. canetti remains unknown, cases are highly geographically restricted and arise predominantly in patients who have some form of contact with East-Africa (59). Like M. kansasii, M. canetti harbors compelling signatures of HGT in its genome (60, 61). Boritsch et al. offered conclusive experimental evidence that HGT occurs in M. canetti, finding the transfer of DNA fragments as large as 117.6 kilobase pairs (kbp) (62). Like M. canetti, the most basal lineages in the MTBC, including L5, L6, and L7, are also strongly geographically restricted to Africa (14, 63, 64). These observations and experiments support a scenario in which an M. canetti-like species of mycobacteria in Africa acquired virulence loci via HGT, thus giving rise to the pathogenic progenitor of the MTBC.
The role of ongoing HGT in Mtb, however, remains controversial. Most evidence suggests that Mtb demonstrates clonal evolution without ongoing recombination events. In the same experiments in which HGT was detected in M. canetti, HGT could not be detected among MTBC species (62). The lack of ongoing HGT in the MTBC is supported by the congruence of phylogenetic trees based on a variety of molecular markers (65–67), stable G+C content across the majority of the genome (68), a low frequency of homoplasic mutations (14, 28), and that all known drug-resistance factors arise via de novo mutation (69). The mechanism by which Mtb lost capacity for ongoing genetic recombination, however, remains unknown. Together, this evidence provides strong support for the role of HGT as a critical component in the emergence of Mtb, and that subsequently Mtb appears to have lost significant capacity for genetic recombination and evolved in a clonal fashion.
Genetic and Phenotypic Diversity in Mtb
Mtb is an obligate human pathogen and has no known environmental reservoir. As such, its population structure is largely isomorphic to its human host population. Despite the clonal evolution of Mtb, significant genetic variation exists and based on this it is divided into seven major lineages. These lineages can be grouped into evolutionarily “ancient” and “modern” lineages, with the TbD1 deletion serving as a genetic marker separating the two groups (43). The ancient lineages (L1, L5, L6, L7) demonstrate a high degree of geographic constraint (14, 63, 64), whereas the more recently evolved modern lineages (L2, L3, L4) are found more broadly throughout the world (70). L1 predominantly circulates in Southeast Asia, L5 and L6 in West Africa, and L7 in the Horn of Africa. L2 is strongly associated with an East Asian origin (71), but also causes significant disease burden in Eurasia, South Africa, and Peru. Over the past 200 years, the population size of L2 strains has dramatically increased, and can be found in most countries throughout the world (72). L3 strains circulate mostly in India and Central Asia. L4 strains cause the most global disease and are the most widely distributed among the Mtb lineages (73). Interestingly, discrete sublineages within L4 differ in their geographic distribution, suggesting that some L4 strains are more capable of spreading to new host populations (74).
The genetic lineages of Mtb were first defined by lineage-specific deletions, referred to as large sequence polymorphisms (LSPs) (25). Due to the extreme rarity of ongoing horizontal gene transfer (HGT) among species of the MTBC, these markers are thought to be largely irreversible and well-suited to lineage classification (73). Single nucleotide polymorphisms (SNPs) are also phylogenetically informative in Mtb due to the lack of ongoing HGT, and help to increase the resolution of relationships among strains within a lineage (75–77). From the application of these markers in constructing the phylogenetic relationships among Mtb lineages, it has become clear that the ancestral lineages separate into distinct phylogenetic groups, and are thus paraphyletic in nature. The modern lineages, conversely, are more closely related and share a more recent common ancestor (i.e., monophyletic) than the ancient lineages are with one another. These lineages have evolved independently in separate human populations, resulting in distinct induction of inflammatory phenotypes (78, 79) and differential modulation of innate immune signaling (80). Furthermore, the variable geographic distribution and disease burden of the different lineages raises the question as to how the existing variation among Mtb strains contributes to disease phenotype, and whether this variation explains the uneven distribution of Mtb's lineages.
Phenotypic Diversity Among Mtb Lineages
Strain variation in disease severity, transmission potential, and resistance to drug therapy is of significant interest to global health. Identifying virulent and/or drug-resistant clones informs current and future treatment. Numerous studies have investigated the phenotypes associated with the different lineages and strains of Mtb. By the mid-20th century, TB research had begun to investigate virulence traits among clinical and reference strains of Mtb (81, 82). The first attempts to correlate virulence with strain background via typing techniques, however, did not occur until 1978 (83). In a landmark study, Valway et al. utilized IS6110 typing patterns to identify a strain associated with a particularly virulent outbreak (84). The outbreak was characterized by extensive transmission among patients, and the researchers correlated a significant increase in in vivo replication as a potential underlying cause using a mouse infection model. Following the adoption of the restriction fragment length polymorphism (RFLP) typing technique (85) to describe the population structure of Mtb, strains originating in China and Mongolia, the so-called “Beijing” strains (now known as L2), demonstrated increased replication in cell culture and mouse models in addition to increased mortality in vivo (86, 87). In a rabbit infection model, L2 strains rapidly disseminated to extrapulmonary sites resulting in severe meningeal disease presentation (88). However, we should exercise caution when applying strain-specific characteristics broadly across its genetic lineage, as infection phenotypes can vary widely among strains from the same lineage (79, 89). Correspondingly, L2 strains demonstrate variable virulence patterns. The most recently evolved L2 strains, those comprising the so-called “modern Beijing” sublineage, exhibit increased virulence compared to the ancestral strains (90). These and earlier studies (91, 92) focused attention on the apparent increased virulence of the L2 strains, and their impact on the immune response was identified as an avenue of future research.
Mtb Lineages and Disease Presentation
Transmission of Mtb depends on disease within pulmonary tissue in human hosts. Given its status as an obligate human pathogen, there are no environmental reservoirs for Mtb to transmit from, and extrapulmonary sites do not afford transmission. This leads to the question: Do particular Mtb lineages demonstrate variable disease presentations? Are more transmissible strains less often be associated with non-transmissible disease sites, i.e., extrapulmonary tissues? In a marmoset model of infection, a strain from the ancient L6 group was found to develop lower bacterial load in pulmonary tissue compared to modern strains from L2 and L4, but disseminated to extrapulmonary sites more compared to L4 (93). Interestingly, the L2 strain demonstrated the highest burden in all organs assayed, effectively replicating within the lung and disseminating to extrapulmonary sites. This study suggests the modern strains are more capable of transmitting by establishing pulmonary disease, but L2 also spreads effectively to extrapulmonary sites. Based on the characteristics of infection, it is possible that the L2 and L6 strains disseminated to other tissues by different mechanisms, where L2's dissemination was a byproduct of increased overall virulence as described in the preceding sections. While this study offers novel visualization methods to assess disease progression of diverse tuberculosis lineages in a primate infection model, it is not clear how generalizable these phenotypes are across these lineages.
There are few studies that have compared patterns of disease presentation among a diverse range of strains from more than two lineages in a large sample population (summarized in Figure 1 and Table 1). Even in these, associations between lineage and disease presentation have been variable, and the comparisons differ. In the United States, L1, L3, and L4 strains were more likely to cause extrapulmonary disease compared to strains from L2 (20). In Vietnam, L1 and L2 strains were associated with TB meningitis compared to L4 strains (18). In the UK, L1 and L2 were associated with increased likelihood of exclusively extrapulmonary disease compared to L3 and L4 (22). Aside from site of disease, characteristics such as time to sputum culture conversion and transmissibility differ between lineages as well. In the United States, L1 strains demonstrate a more rapid time to sputum culture conversion compared to strains from the modern lineages (L2, L3, and L4) (21). Additionally, in Gambia, L6 strains progressed to active disease at a significantly lower rate compared to strains from the modern lineages, but displayed no differences in transmissibility (19). However, in the Netherlands, ancient strains (L1, L5, and L6) demonstrated reduced transmissibility compared to L4 strains (23). In Florida, L1 strains were associated with higher rates of extrapulmonary disease compared to L2 and L4 strains (24). Together, these studies indicate that significant differences exist in disease presentation among the different lineages of Mtb (particularly between ancient and modern strains), and these patterns can be observed experimentally and in human populations.
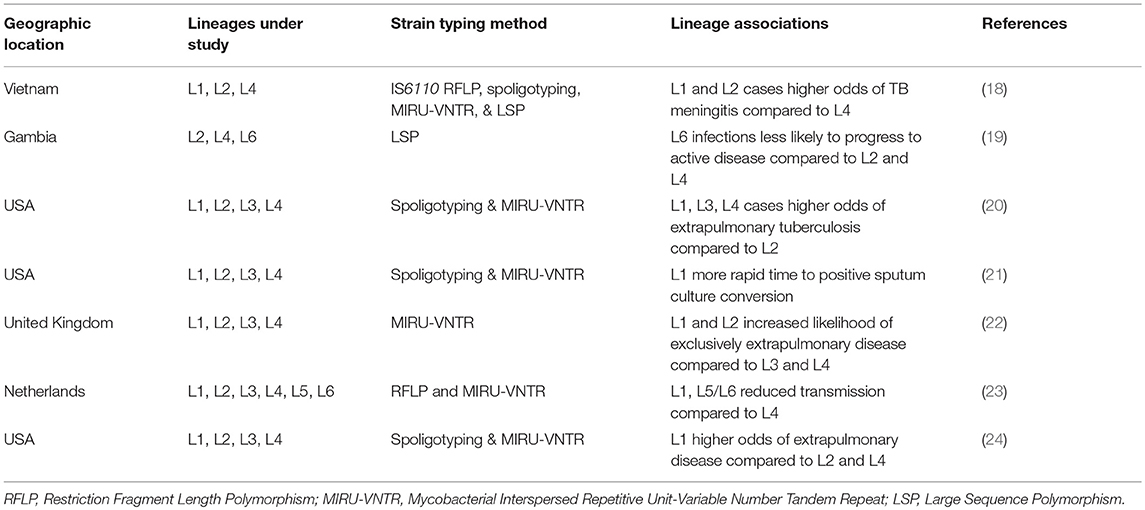
Table 1. Studies investigating multiple Mtb lineages and their associations with disease characteristics.
Bacterial Determinants of Virulence
Mtb lineages show varying geographic distribution patterns with ancient lineages being geographically restricted in comparison to the modern lineages. Several factors like population density, migration pattern, economic and health conditions, and more recently the HIV/AIDS pandemic and emergence of MDR strains could influence this distribution (94). However, bacterial genetic variation within and between each lineage may reflect evolutionary history and pressures.
Cell Envelope-Associated Lipids
As a pathogen, Mtb must interface with its host, and the mycobacterial cell envelope makes first contact. The mycobacterial cell envelope is a complex multi-layered structure containing the plasma membrane, cell wall skeleton, mycomembrane and a capsule (95–97). It contains several lipids unique to pathogenic mycobacteria which contributes to their in vivo survival by modulating the host immune response, and have been the subject of more comprehensive reviews [e.g. (98)]. These include mannose capped lipoarabinomannan (ManLAM), phenolic glycolipid (PGL) and phthiocerol dimycocerosate (PDIM) (99–101). These features are highlighted in Figure 2.
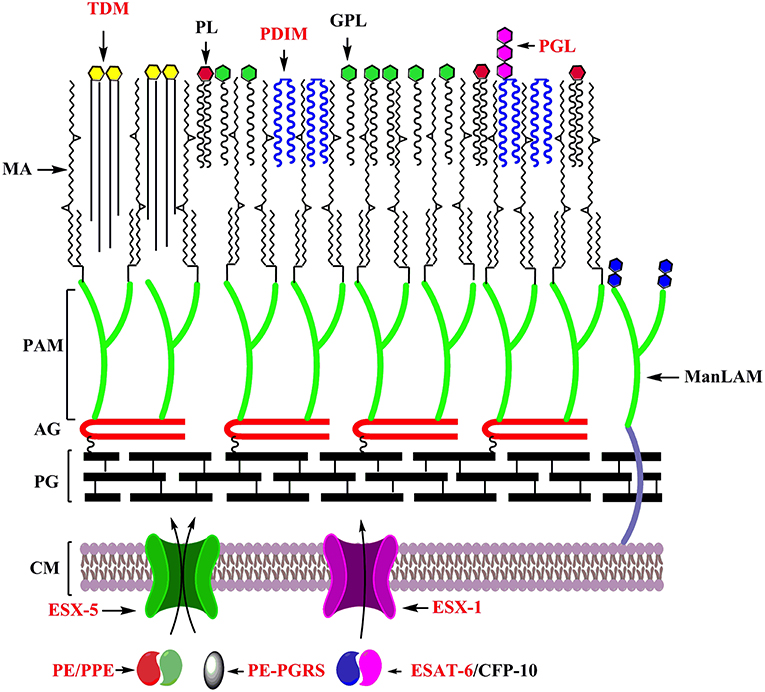
Figure 2. Key features underlying the adaptation of mycobacteria as specialized pathogens: Cell envelope of mycobacteria with factors playing distinct roles in its adaptation as a specialized pathogens labeled in red. CM, Cell membrane; PG, Peptidoglycan; AG, Arabinogalactan; PAM, Penta arabinosyl motif; MA, Mycolic acids; TDM, Trehalose dimycolate; PL, Phospholipids; PDIM, Pthiocerol dimycocerosate; GPL, Glycopeptidolipids; PGL, Phenolic glycolipids; ManLAM, Mannose capped lipoarabinomannan.
Variations in these components among different strains and lineages may correspond to discrete evolutionary trajectories. For example, variation in ManLAM has been observed in clinical strains leading to altered virulence (102, 103). A subset of lineage 2 strains with truncated and more branched forms of ManLAM exhibited defects in phagocytosis by primary human macrophages when compared to lineage 4 reference strains (103).
Variations in PDIM, PGL and other lipids may also contribute to disease progression. PDIM can neutralize oxidative and nitrosative free radicals and has been proposed to play a role in protecting Mtb from these stress causing agents (104, 105). Further, PDIM may also have a role in immune evasion by masking cell wall pathogen-associated molecular patterns (PAMPs) (57), and also is required for proper secretion of ESX-1 substrates (106). Among the modern lineages, L2 strains but not L4 strains produce the phenolic glycolipid PGL, which may play an important role in promoting their virulence and transmission (107). In mycobacterium-infected macrophages, PGL induces the production of chemokine CCL2 which recruits monocytes to the site of infection. This facilitates mycobacterial escape from bactericidal macrophages to permissive monocytes (108). A point mutation in Rv2952 encoding the S-adenosylmethionine-dependent methyltransferase in Beijing strains resulted in structural variations in PDIM and PGL compared to other lineage strains (109). As noted above, a deletion in the pks1/15 locus encoding a polyketide synthase in L4 strains leads to defective production of PGL (110). These lipids can also inhibit the production or secretion of proinflammatory cytokines by the host leading to the establishment of infection (105, 107, 111, 112).
The abundant cell wall lipid trehalose dimycolate (TDM) plays multiple roles in pathogenesis (113–118). Specific cyclopropane modifications to the mycolic acids that comprise TDM are associated with pathogenic mycobacteria, but not with non-pathogenic species; PcaA-mediated modification of TDM modulates the host immune response to mycobacterial infection (119, 120). This cyclopropanated TDM plays an important role in inducing or accelerating host angiogenesis around the mycobacterial granuloma, a response that helps to support bacterial growth during early infection (121–123). Thus, intricately structured and complex lipid species provide important host modulatory activities and may be important substrates for evolution. Notably, lineage-specific differences in cytokine induction upon exposure of macrophages to lipid extracts from different lineages have been reported (78).
Type VII Secretion Systems
The ESAT-6 secretion (ESX/Type VII) systems and their secretion substrates are key features that contribute to the pathogenicity of Mtb (124). The ESX secretion systems were discovered after genomic analysis of the M. bovis BCG vaccine strain revealed a large deletion [Region of Difference (RD) 1] that interrupted the ESX-1 system (125). This system was lost in M. bovis following 11-year serial culture by Calmette and Guerin in the pursuit of a TB vaccine. The absence of this system was subsequently shown to account for a significant share of BCG's attenuation, and much attention has been paid to the role of this and other ESX systems and their secreted substrates on Mtb's virulence (126, 127).
ESX secretion systems are encoded in clusters throughout mycobacterial genomes. Mtb contains five ESX loci, which have expanded through gene duplication, diversification, and insertions of the ancestral ESX-4 locus (128). These clusters share six core genes encoding: three ESX conserved-components (EccB, EccC, EccD), a mycosin (MycP), and two small, secreted Esx proteins. Besides the most ancestral ESX-4 locus, the ESX clusters also encode genes for PE, PPE, EccA, EccE, and ESX-1-specific component (Esp) proteins. The esp genes are not specific to ESX-1, but they are most abundant in that system. Orthologs of ESX-4 can be found among mycobacterial and non-mycobacterial species in the phylum Actinobacteria (128, 129). ESX-4 is the simplest gene cluster among the ESX secretion systems, containing only seven genes. ESX-4 encodes the FtsK/SpoIIIE protein EccC4, the WXG proteins EsxU and EsxT, the conserved ESX core components EccB4 and EccD4, the mycosin protease MycP4, and the hypothetical valine and alanine rich protein Rv3446c.
The components of the ESX systems can be divided into cytosolic, membrane bound, and secreted proteins. EspG and EccA function in the cytosol. EspG is found in all ESX clusters besides ESX-4, and is thought to function as a specific chaperone for PE and PPE proteins (130–132). EccA is an AAA+ family (ATPase associated with various cellular activities) protein that is thought to form a hexamer and functions in the secretion of Esx and PE-PPE proteins (133–136). The conserved membrane components of ESX secretion systems (EccB, EccC, EccD, EccE, and MycP) are essential for secretion in all of the studied loci (137–141). These proteins contain large hydrophilic domains in either the N- or C-terminus and a range of transmembrane domains. EccB, EccC, EccD, and EccE are thought to form the transport channel through which the ESX substrates are transported across the inner membrane. EccB, EccC, EccD, and EccE form a stable membrane complex of ~1,500 kDa that can be co-immunoprecipitated (139). MycP, a mycosin, is a subtilisin-like protease containing a C-terminal transmembrane domain that tethers the protein on the cell membrane (142, 143). Its role in secretion remains unknown.
The components described thus far have been localized to the inner membrane. The inner membrane, however, is surrounded by a thick, lipid-rich cell wall (also referred to as the outer membrane or mycomembrane) in addition to another thick capsular layer [reviewed in (144)]. How ESX substrates are exported beyond these structural boundaries has been a mystery. Recently, Lou et al. discovered that EspC forms a long filamentous structure that localizes to the cell membrane, and its expression is required for secretion of EsxA (145).
The conserved secreted effectors of ESX systems are comprised of Esx and PE/PPE proteins (the latter is described in more detail in the following section). The Esx proteins are also referred to as WxG100 proteins due to a conserved tryptophan-X-glycine motif that causes a turn between two helical domains in the ~100 amino acid proteins (146). The most well-studied Esx proteins are EsxA and EsxB, encoded within the ESX-1 locus. ESX-1, the prototypical ESX secretion system in tuberculosis research, has been demonstrated to be essential for the intracellular survival of Mtb due to its critical role in host-pathogen interaction during Mtb infection via secretion of its substrates, many of which are secreted in a codependent manner (147). EsxA and EsxB are secreted as antiparallel heterodimers (148, 149) via recognition of an ESX secretion signal on the C-terminus of EsxB (150). EsxA has long-been associated as a cytolytic virulence factor of Mtb (126, 135, 151, 152). Experiments demonstrating recombinant EsxA could induce its cytolytic effect in the absence of infection led to the notion that EsxA was primarily responsible for ESX-1's pathogenicity (152, 153). However, recent work has definitively demonstrated that the cytolytic effect of recombinant EsxA was due to a residual detergent in the extract (154). Therefore, the cytolytic effect is dependent on other factors dependent on Mtb's ESX-1 secretion system.
ESX-1 has been ascribed numerous roles in Mtb's pathogenesis. As previously mentioned, ESX-1 is required for membrane disruptions in its host cell, allowing Mtb to escape from the phagosome and enter the cytosol whereupon necrosis-like cell death is induced (155–157). While EsxA has been shown to be insufficient to induce membrane disruptions, this process is dependent on its presence and secretion (154). EspB, which is encoded outside of the ESX-1 locus and depends on secretion of EsxA and EsxB for its own secretion, forms a ring-shaped heptamer with a hydrophobic domain, suggesting the possibility that it could be involved in membrane disruption via EsxA and EsxB (158). EsxA has been shown to induce expression of matrix metalloproteinase-9 (MMP9), which recruits additional phagocytes to the site of infection and facilitates its spread to new cells (156). The recurrent recruitment of additional leukocytes to take up the apoptotic debris of the former round of infected macrophages amplifies the bacterial population in successive waves and leads to the formation of the tuberculosis granuloma (159).
The regulation of ESX-1 differs among MTBC species, perhaps contributing to distinct infection phenotypes among lineages. The PhoPR regulon, a two-component regulation system, regulates the production and secretion of, among other things, EsxA and EsxB (160), and is central to the pathogenesis of Mtb (161). Strains from L5, L6 and the animal-adapted species all contain a missense mutation in phoR that downregulates the PhoPR system when genetically transferred into L2 and L4 strains (162). Intriguingly, Gonzalo-Asensio et al. noted that there were no significant differences in the production of proteins induced by PhoPR in the L5, L6 and animal-adapted species compared to L2 and L4, and that a deletion found only in the former rescued the defect. The authors went on to show that an outbreak of an unusually virulent strain of M. bovis that was transmitting among humans was associated with the insertion of an IS6110 sequence upstream of phoP, serving as a promoter to increase the expression of the PhoPR regulon (162).
The pathogenic species of mycobacteria possess two additional ESX secretion systems, ESX-2 and ESX-5, that are not found in the rapid-growing, non-pathogenic mycobacteria (128, 163). The duplication of these systems in pathogenic mycobacteria is linked to the expansion of the PE and PPE gene families (163). The PE and PPE proteins, the other core substrates of ESX secretion systems, and their role in pathogenesis are discussed below.
PE/PPE Family Proteins
Initial sequencing of the Mtb genome led to a surprise finding that 10% of its genes code for a unique family of proteins with signature proline-glutamate and proline-proline-glutamate residues conserved at their N-termini, linked to a variable C-terminus. Due to their variable C-termini, initially they were thought to be a source of antigenic variation to evade host immune system (68). The pe/ppe genes have greatly expanded in the pathogenic species of mycobacteria and have been critical for host adaptation (164, 165). This family of proteins are thought to help in Mtb survival and dissemination through diverse modes. This includes upregulation of anti-inflammatory cytokine levels (166), induction of apoptosis in macrophages (167) and increased secretion of chemokine MCP-1 (168). They also interact with TLR-2, leading to macrophage activation, promote apoptosis and necrosis in host cells (164). PE-PGRS a subfamily of PE family is unique to MTBC and related species (165). Mutations in their corresponding genes have been associated with impaired replication and decreased persistence in the host indicating a direct role for this class of genes in virulence (169). The “modern” Beijing strains from L2 have been demonstrated to harbor a deletion affecting ppe38, a consequential mutation that increases the virulence of affected strains (170). The authors found that the absence of ppe38 inhibits the secretion of a large number of PPE_PGRS and PPE_MPTR (major polymorphic tandem repeats) substrates through ESX-5, and postulate that this mutation played a significant role in the global spread of the “modern” Beijing L2 strains. Thus, variation in these gene classes may contribute to the degree of virulence, transmissibility, and evolutionary success for mycobacterial species and strains within discrete hosts and genetic backgrounds.
Mycobacterial Genetic Diversity and Its Intersection With Host Tolerance
Variation in mycobacterial lipids, ESX secretion systems and their effectors among the genetic lineages and sublineages of Mtb intersect with the nature of the host response to mycobacterial infection. Evidence from experimental infection models suggests that different Mtb lineages exhibit diverse growth phenotypes and elicit variable host immune responses. Hence in addition to these factors, the role of variable host tolerance among these lineages in shaping their diversity may be important. Some of the first evidence supporting this argument came from aerosol infections in mice with Mtb strains CDC1551, HN878, and HN60. CDC1551 belongs to lineage 4 whereas HN878 and HN60 belong to lineage 2. Mice infected with HN878 and HN60 succumbed earlier. This observation correlated with the cytokine profiles of CDC1551 infected mice which showed increased production of pro inflammatory cytokines TNF-α, IL-12, and IFN-γ in comparison to HN878 and HN60 infected mice (91). Moreover, strains from the modern lineages 2, 3, and 4 induced significantly lower levels of pro inflammatory cytokines than ancient lineages in a human monocyte-derived macrophages infection model (79).
Mtb sublineages too exhibit significant differences in virulence and immune modulatory functions. The M—Strain, a highly prevalent strain in Argentina belonging to the Haarlem family of Lineage 4 failed to induce PMN apoptosis and ROS production as opposed to the LAM family of the same lineage (171). Collectively these findings may help explain the emergence and evolutionary success of the modern lineages.
Recent work on tolerance in animal models of TB suggests that specific host factors can contribute differentially to bacterial restriction and host tolerance. For example, Phox-deficient mice are not compromised for resistance to infection but do display tolerance defects (172–174). Similarly, previous work in the zebrafish model of mycobacterial infection suggested that, in addition to overall bacterial load, inflammatory state influences disease outcome (175, 176). Thus, the degree of host tolerance to infection has important consequences to host survival, bacterial burden, and presumably transmission; indeed the majority of humans who do not manifest active disease upon exposure to Mtb suggests a high level of tolerance to infection (177). Reciprocally, how variation within distinct bacterial lineages and strains influences inflammation, tolerance, pathogenesis, and ultimately successful transmission, may determine the evolutionary trajectories of both pathogen and host.
A number of examples exist in which bacterial-host interactions appear to be specific to lineage. For example Lineage 2 mediated TB has been shown to be associated with C allele of TLR-2—T597C, and NRAMP1—D543N polymorphisms (18, 178). The−261TT variant in the Immunity-related GTPase Family M (IRGM) confers defense against pathogens including Lineage 4 Mtb which lacks pks1/15, but is not associated with M. africanum mediated TB. This gene is associated with PGL biosynthesis highlighting a potential role of the lipid in inhibiting IRGM mediated autophagy (179). Lineage 4 contains both ubiquitous (presumed to be generalist) and specialized (geographically restricted) sublineages, suggesting that at least some Mtb strains may have specialized to specific host populations (74). More recently, a large study in a Vietnamese population identified increased transmission of Lineage 2 Beijing strains between individuals than endemic strains, consistent with previous studies of transmission of Beijing strains in other regions (180–182). These studies underscore the need for further research that integrates data on Mtb strains and lineages with human genotypes to understand how this intersection contributes to the clinical outcome of Mtb infection. Ongoing studies with larger cohorts and deeper descriptions of clinical phenotypes should provide additional insight into these interactions.
Mtb genetic diversity and evolution may reflect the genetic arms race between successful pathogen and its host, leading to reciprocal genetic changes. There is newfound appreciation that host tolerance to mycobacterial infection is an important component of this interplay, contributing to disease trajectory and transmission patterns. Thus, genetic variation in aspects of host tolerance—generated through both bacterial and host mechanisms—is another important consideration in understanding the complex interactions between host and pathogen that have evolved during the long association between Mtb and its human hosts.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a National Science Foundation GRFP Award (JS), a Clinical and Translational Science Award UL1TR002553 (JS), and National Institutes of Health grants AI130236, AI125517, and AI127115 (DT).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to J. Stout and members of the Tobin laboratory for helpful discussions.
References
2. Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. (2014) 159:1497–509. doi: 10.1016/j.cell.2014.11.024
3. Chao MC, Rubin EJ. Letting sleeping dos lie: does dormancy play a role in tuberculosis? Ann Rev Microbiol. (2010) 64:293–311. doi: 10.1146/annurev.micro.112408.134043
4. Rittershaus ES, Baek SH, Sassetti CM. The normalcy of dormancy: common themes in microbial quiescence. Cell Host Microbe. (2013) 13:643–51. doi: 10.1016/j.chom.2013.05.012
5. Cosma CL, Sherman DR, Ramakrishnan L. The secret lives of the pathogenic mycobacteria. Ann Rev Microbiol. (2003) 57:641–76. doi: 10.1146/annurev.micro.57.030502.091033
6. Feldman WH, Baggenstoss AH. The residual infectivity of the primary complex of tuberculosis. Am J Pathol. (1938) 14:473–90 473.
7. Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. (2012) 12:581–91. doi: 10.1038/nri3259
8. Behr MA, Edelstein PH, Ramakrishnan L. Revisiting the timetable of tuberculosis. BMJ. (2018) 362:k2738. doi: 10.1136/bmj.k2738
9. Lin PL, Flynn JL. The end of the binary Era: revisiting the spectrum of tuberculosis. J Immunol. (2018) 201:2541–8. doi: 10.4049/jimmunol.1800993
10. Zumla A, Raviglione M, Hafner R, von Reyn CF. Tuberculosis. N Engl J Med. (2013) 368:745–55. doi: 10.1056/NEJMra1200894
11. Houben RM, Dodd PJ. The global burden of latent tuberculosis infection: a re-estimation using mathematical modelling. PLoS Med. (2016) 13:e1002152. doi: 10.1371/journal.pmed.1002152
13. Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M, et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. (2013) 45:1176–82. doi: 10.1038/ng.2744
14. Hershberg R, Lipatov M, Small PM, Sheffer H, Niemann S, Homolka S, et al. High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLoS Biol. (2008) 6:e311. doi: 10.1371/journal.pbio.0060311
15. Hughes AL, Friedman R, Murray M. Genomewide pattern of synonymous nucleotide substitution in two complete genomes of Mycobacterium tuberculosis. Emerg Infect Dis. (2002) 8:1342–6. doi: 10.3201/eid0811.020064
16. Bos KI, Harkins KM, Herbig A, Coscolla M, Weber N, Comas I, et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature. (2014) 514:494–7. doi: 10.1038/nature13591
17. Ho SY, Chen AX, Lins LS, Duchene DA, Lo N. The Genome as an Evolutionary Timepiece. Genome Biol Evol. (2016) 8:3006–10. doi: 10.1093/gbe/evw220
18. Caws M, Thwaites G, Dunstan S, Hawn TR, Lan NT, Thuong NT, et al. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog. (2008) 4:e1000034. doi: 10.1371/journal.ppat.1000034
19. de Jong BC, Hill PC, Aiken A, Awine T, Antonio M, Adetifa IM, et al. Progression to active tuberculosis, but not transmission, varies by Mycobacterium tuberculosis lineage in The Gambia. J Infect Dis. (2008) 198:1037–43. doi: 10.1086/591504
20. Click ES, Moonan PK, Winston CA, Cowan LS, Oeltmann JE. Relationship between Mycobacterium tuberculosis phylogenetic lineage and clinical site of tuberculosis. Clin Infect Dis. (2012) 54:211–9. doi: 10.1093/cid/cir788
21. Click ES, Winston CA, Oeltmann JE, Moonan PK, Mac Kenzie WR. Association between Mycobacterium tuberculosis lineage and time to sputum culture conversion. Int J Tubercul Lung Dis. (2013) 17:878–84. doi: 10.5588/ijtld.12.0732
22. Pareek M, Evans J, Innes J, Smith G, Hingley-Wilson S, Lougheed KE, et al. Ethnicity and mycobacterial lineage as determinants of tuberculosis disease phenotype. Thorax. (2013) 68:221–9. doi: 10.1136/thoraxjnl-2012-201824
23. Nebenzahl-Guimaraes H, Verhagen LM, Borgdorff MW, van Soolingen D. Transmission and Progression to Disease of Mycobacterium tuberculosis phylogenetic lineages in The Netherlands. J Clin Microbiol. (2015) 53:3264–71. doi: 10.1128/JCM.01370-15
24. Seraphin MN, Doggett R, Johnston L, Zabala J, Gerace AM, Lauzardo M. Association between Mycobacterium tuberculosis lineage and site of disease in Florida, 2009-2015. Infect Genet Evol. (2017) 55:366–71. doi: 10.1016/j.meegid.2017.10.004
25. Gagneux S, Small PM. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. (2007) 7:328–37. doi: 10.1016/S1473-3099(07)70108-1
26. Wirth T, Hildebrand F, Allix-Beguec C, Wolbeling F, Kubica T, Kremer K, et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. (2008) 4:e1000160. doi: 10.1371/journal.ppat.1000160
27. Mellars P. Going east: new genetic and archaeological perspectives on the modern human colonization of Eurasia. Science. (2006) 313:796–800. doi: 10.1126/science.1128402
28. Comas I, Homolka S, Niemann S, Gagneux S. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS ONE. (2009) 4:e7815. doi: 10.1371/journal.pone.0007815
29. Walker TM, Ip CL, Harrell RH, Evans JT, Kapatai G, Dedicoat MJ, et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect Dis. (2013) 13:137–46. doi: 10.1016/S1473-3099(12)70277-3
30. Pepperell CS, Casto AM, Kitchen A, Granka JM, Cornejo OE, Holmes EC, et al. The role of selection in shaping diversity of natural M. tuberculosis populations PLoS Pathog. (2013) 9:e1003543. doi: 10.1371/journal.ppat.1003543
31. Allison MJ, Mendoza D, Pezzia A. Documentation of a case of tuberculosis in Pre-Columbian America. Am Rev Respir Dis. (1973) 107:985–91.
32. Arriaza BT, Salo W, Aufderheide AC, Holcomb TA. Pre-Columbian tuberculosis in northern Chile: molecular and skeletal evidence. Am J Phys Anthropol. (1995) 98:37–45. doi: 10.1002/ajpa.1330980104
33. Salo WL, Aufderheide AC, Buikstra J, Holcomb TA. Identification of Mycobacterium tuberculosis DNA in a pre-Columbian Peruvian mummy. Proc Natl Acad Sci USA. (1994) 91:2091–4. doi: 10.1073/pnas.91.6.2091
34. Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Paabo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. (2001) 29:4793–9. doi: 10.1093/nar/29.23.4793
35. Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat Genet. (2013) 45:784–90. doi: 10.1038/ng.2656
36. Menardo F, Duchene S, Brites D, Gagneux S. The molecular clock of Mycobacterium tuberculosis. bioRxiv. (2019) 532390. doi: 10.1101/532390
37. Roberts CA, Pfister LA, Mays S. Letter to the editor: was tuberculosis present in Homo erectus in Turkey? Am J Phys Anthropol. (2009) 139:442–4. doi: 10.1002/ajpa.21056
38. Gernaey AM, Minnikin DE, Copley MS, Dixon RA, Middleton JC, Roberts CA. Mycolic acids and ancient DNA confirm an osteological diagnosis of tuberculosis. Tuberculosis. (2001) 81:259–65. doi: 10.1054/tube.2001.0295
39. Thierry D, Cave MD, Eisenach KD, Crawford JT, Bates JH, Gicquel B, et al. IS6110, an IS-like element of Mycobacterium tuberculosis complex. Nucleic Acids Res. (1990) 18:188. doi: 10.1093/nar/18.1.188
40. Lee OY, Wu HH, Besra GS, Rothschild BM, Spigelman M, Hershkovitz I, et al. Lipid biomarkers provide evolutionary signposts for the oldest known cases of tuberculosis. Tuberculosis. (2015) 95(Suppl. 1):S127–32. doi: 10.1016/j.tube.2015.02.013
41. Lee OY, Wu HH, Donoghue HD, Spigelman M, Greenblatt CL, Bull ID, et al. Mycobacterium tuberculosis complex lipid virulence factors preserved in the 17,000-year-old skeleton of an extinct bison, Bison antiquus. PLoS ONE. (2012) 7:e41923. doi: 10.1371/journal.pone.0041923
42. Hershkovitz I, Donoghue HD, Minnikin DE, Besra GS, Lee OY, Gernaey AM, et al. Detection and molecular characterization of 9,000-year-old Mycobacterium tuberculosis from a Neolithic settlement in the Eastern Mediterranean. PLoS ONE. (2008) 3:e3426. doi: 10.1371/journal.pone.0003426
43. Brosch R, Gordon SV, Marmiesse M, Brodin P, Buchrieser C, Eiglmeier K, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. (2002) 99:3684–9. doi: 10.1073/pnas.052548299
44. Smith NH, Kremer K, Inwald J, Dale J, Driscoll JR, Gordon SV, et al. Ecotypes of the Mycobacterium tuberculosis complex. J Theor Biol. (2006) 239:220–5. doi: 10.1016/j.jtbi.2005.08.036
45. van Ingen J, Rahim Z, Mulder A, Boeree MJ, Simeone R, Brosch R, et al. Characterization of Mycobacterium orygis as M. tuberculosis complex subspecies Emerg Infect Dis. (2012) 18:653–5. doi: 10.3201/eid1804.110888
46. Wang J, McIntosh F, Radomski N, Dewar K, Simeone R, Enninga J, et al. Insights on the emergence of Mycobacterium tuberculosis from the analysis of Mycobacterium kansasii. Genome Biol Evol. (2015) 7:856–70. doi: 10.1093/gbe/evv035
47. Canueto-Quintero J, Caballero-Granado FJ, Herrero-Romero M, Dominguez-Castellano A, Martin-Rico P, Verdu EV, et al. Epidemiological, clinical, and prognostic differences between the diseases caused by Mycobacterium kansasii and Mycobacterium tuberculosis in patients infected with human immunodeficiency virus: a multicenter study. Clin Infect Dis. (2003) 37:584–90. doi: 10.1086/376987
48. Lillo M, Orengo S, Cernoch P, Harris RL. Pulmonary and disseminated infection due to Mycobacterium kansasii: a decade of experience. Rev Infect Dis. (1990) 12:760–7. doi: 10.1093/clinids/12.5.760
49. Becq J, Gutierrez MC, Rosas-Magallanes V, Rauzier J, Gicquel B, Neyrolles O, et al. Contribution of horizontally acquired genomic islands to the evolution of the tubercle bacilli. Mol Biol Evol. (2007) 24:1861–71. doi: 10.1093/molbev/msm111
50. Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, Khanna V, et al. Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet. (2013) 45:172–9. doi: 10.1038/ng.2517
51. Veyrier F, Pletzer D, Turenne C, Behr MA. Phylogenetic detection of horizontal gene transfer during the step-wise genesis of Mycobacterium tuberculosis. BMC Evol Biol. (2009) 9:196. doi: 10.1186/1471-2148-9-196
52. Wang J, Behr MA. Building a better bacillus: the emergence of Mycobacterium tuberculosis. Front Microbiol. (2014) 5:139. doi: 10.3389/fmicb.2014.00139
53. Rosas-Magallanes V, Stadthagen-Gomez G, Rauzier J, Barreiro LB, Tailleux L, Boudou F, et al. Signature-tagged transposon mutagenesis identifies novel Mycobacterium tuberculosis genes involved in the parasitism of human macrophages. Infect Immunity. (2007) 75:504–7. doi: 10.1128/IAI.00058-06
54. Brodin P, Poquet Y, Levillain F, Peguillet I, Larrouy-Maumus G, Gilleron M, et al. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog. (2010) 6:e1001100. doi: 10.1371/journal.ppat.1001100
55. Pethe K, Swenson DL, Alonso S, Anderson J, Wang C, Russell DG. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci USA. (2004) 101:13642–7. doi: 10.1073/pnas.0401657101
56. Chan J, Fujiwara T, Brennan P, McNeil M, Turco SJ, Sibille JC, et al. Microbial glycolipids: possible virulence factors that scavenge oxygen radicals. Proc Natl Acad Sci USA. (1989) 86:2453–7. doi: 10.1073/pnas.86.7.2453
57. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. (2014) 505:218–22. doi: 10.1038/nature12799
58. Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. (2003) 100:12989–94. doi: 10.1073/pnas.2134250100
59. Aboubaker Osman D, Bouzid F, Canaan S, Drancourt M. Smooth Tubercle Bacilli: Neglected opportunistic tropical pathogens. Front Public Health. (2015) 3:283. doi: 10.3389/fpubh.2015.00283
60. Gutierrez MC, Brisse S, Brosch R, Fabre M, Omais B, Marmiesse M, et al. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. (2005) 1:e5. doi: 10.1371/journal.ppat.0010005
61. Mortimer TD, Pepperell CS. Genomic signatures of distributive conjugal transfer among mycobacteria. Genome Biol Evol. (2014) 6:2489–500. doi: 10.1093/gbe/evu175
62. Boritsch EC, Khanna V, Pawlik A, Honore N, Navas VH, Ma L, et al. Key experimental evidence of chromosomal DNA transfer among selected tuberculosis-causing mycobacteria. Proc Natl Acad Sci USA. (2016) 113:9876–81. doi: 10.1073/pnas.1604921113
63. Firdessa R, Berg S, Hailu E, Schelling E, Gumi B, Erenso G, et al. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg Infect Dis. (2013) 19:460–3. doi: 10.3201/eid1903.120256
64. Vasconcellos SE, Huard RC, Niemann S, Kremer K, Santos AR, Suffys PN, et al. Distinct genotypic profiles of the two major clades of Mycobacterium africanum. BMC Infect Dis. (2010) 10:80. doi: 10.1186/1471-2334-10-80
65. Baker L, Brown T, Maiden MC, Drobniewski F. Silent nucleotide polymorphisms and a phylogeny for Mycobacterium tuberculosis. Emerg Infect Dis. (2004) 10:1568–77. doi: 10.3201/eid1009.040046
66. Filliol I, Motiwala AS, Cavatore M, Qi W, Hazbon MH, Bobadilla del Valle M, et al. (2006). Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J Bacteriol. 188, 759–772. doi: 10.1128/JB.188.2.759-772.2006
67. Gagneux S, DeRiemer K, Van T, Kato-Maeda M, de Jong BC, Narayanan S, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci USA. (2006) 103:2869–73. doi: 10.1073/pnas.0511240103
68. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. (1998) 393:537–44. doi: 10.1038/31159
69. Nusrath Unissa A, Hanna LE. Molecular mechanisms of action, resistance, detection to the first-line anti tuberculosis drugs: rifampicin and pyrazinamide in the post whole genome sequencing era. Tuberculosis. (2017) 105:96–107. doi: 10.1016/j.tube.2017.04.008
70. Reed MB, Pichler VK, McIntosh F, Mattia A, Fallow A, Masala S, et al. Major Mycobacterium tuberculosis lineages associate with patient country of origin. J Clin Microbiol. (2009) 47:1119–28. doi: 10.1128/JCM.02142-08
71. Luo T, Comas I, Luo D, Lu B, Wu J, Wei L, et al. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. Proc Natl Acad Sci USA. (2015) 112:8136–41. doi: 10.1073/pnas.1424063112
72. Merker M, Blin C, Mona S, Duforet-Frebourg N, Lecher S, Willery E, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. (2015) 47:242–9. doi: 10.1038/ng.3195
73. Coscolla M, Gagneux S. Consequences of genomic diversity in Mycobacterium tuberculosis. Seminars Immunol. (2014) 26:431–44. doi: 10.1016/j.smim.2014.09.012
74. Stucki D, Brites D, Jeljeli L, Coscolla M, Liu Q, Trauner A, et al. Mycobacterium tuberculosis lineage 4 comprises globally distributed and geographically restricted sublineages. Nat. Genet. (2016) 48:1535–43. doi: 10.1038/ng.3704
75. Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, et al. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nat Genet. (2010) 42:498–503. doi: 10.1038/ng.590
76. Saelens JW, Lau-Bonilla D, Moller A, Medina N, Guzman B, Calderon M, et al. Whole genome sequencing identifies circulating Beijing-lineage Mycobacterium tuberculosis strains in Guatemala and an associated urban outbreak. Tuberculosis. (2015) 95:810–6. doi: 10.1016/j.tube.2015.09.001
77. Saelens JW, Lau-Bonilla D, Moller A, Xet-Mull AM, Medina N, Guzman B, et al. Annotated genome sequences of 16 lineage 4 Mycobacterium tuberculosis strains from guatemala. Genome Announc. (2018) 6:e00024–18. doi: 10.1128/genomeA.00024-18
78. Krishnan N, Malaga W, Constant P, Caws M, Tran TH, Salmons J, et al. Mycobacterium tuberculosis lineage influences innate immune response and virulence and is associated with distinct cell envelope lipid profiles. PLoS ONE. (2011) 6:e23870. doi: 10.1371/journal.pone.0023870
79. Portevin D, Gagneux S, Comas I, Young D. Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. (2011) 7:e1001307. doi: 10.1371/journal.ppat.1001307
80. Wiens KE, Ernst JD. The mechanism for type I interferon induction by Mycobacterium tuberculosis is bacterial strain-dependent. PLoS Pathog. (2016) 12:e1005809. doi: 10.1371/journal.ppat.1005809
81. Collins FM, Smith MM. A comparative study of the virulence of Mycobacterium tuberculosis measured in mice and guinea pigs. Am Rev Respir Dis. (1969) 100:631–9.
82. Mitchison DA, Wallace JG, Bhatia AL, Selkon JB, Subbaiah TV, Lancaster MC. A comparison of the virulence in guinea-pigs of South Indian and British tubercle bacilli. Tubercle. (1960) 41:1–22. doi: 10.1016/S0041-3879(60)80019-0
83. Grange JM, Aber VR, Allen BW, Mitchison DA, Goren MB. The correlation of bacteriophage types of Mycobacterium tuberculosis with guinea-pig virulence and in vitro-indicators of virulence. J General Microbiol. (1978) 108:1–7. doi: 10.1099/00221287-108-1-1
84. Valway SE, Sanchez MP, Shinnick TF, Orme I, Agerton T, Hoy D, et al. An outbreak involving extensive transmission of a virulent strain of Mycobacterium tuberculosis. N Engl J Med. (1998) 338:633–9. doi: 10.1056/NEJM199803053381001
85. van Soolingen D, Hermans PW, de Haas PE, Soll DR, van Embden JD. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J Clin Microbiol. (1991) 29:2578–86.
86. Li Q, Whalen CC, Albert JM, Larkin R, Zukowski L, Cave MD, et al. Differences in rate and variability of intracellular growth of a panel of Mycobacterium tuberculosis clinical isolates within a human monocyte model. Infect Immunity. (2002) 70:6489–93. doi: 10.1128/IAI.70.11.6489-6493.2002
87. Lopez B, Aguilar D, Orozco H, Burger M, Espitia C, Ritacco V, et al. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol. (2003) 133:30–7. doi: 10.1046/j.1365-2249.2003.02171.x
88. Tsenova L, Ellison E, Harbacheuski R, Moreira AL, Kurepina N, Reed MB, et al. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis. (2005) 192:98–106. doi: 10.1086/430614
89. Reiling N, Homolka S, Walter K, Brandenburg J, Niwinski L, Ernst M, et al. Clade-specific virulence patterns of Mycobacterium tuberculosis complex strains in human primary macrophages and aerogenically infected mice. MBio. (2013) 4:e00250–13. doi: 10.1128/mBio.00250-13
90. Ribeiro SC, Gomes LL, Amaral EP, Andrade MR, Almeida FM, Rezende AL, et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol. (2014) 52:2615–24. doi: 10.1128/JCM.00498-14
91. Manca C, Tsenova L, Barry CE 3rd, Bergtold A, Freeman S, Haslett PA, et al. Mycobacterium tuberculosis CDC1551 induces a more vigorous host response in vivo and in vitro, but is not more virulent than other clinical isolates. J Immunol. (1999) 162:6740–6.
92. Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, et al. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha /beta. Proc Natl Acad Sci USA. (2001) 98:5752–7. doi: 10.1073/pnas.091096998
93. Via LE, Weiner DM, Schimel D, Lin PL, Dayao E, Tankersley SL, et al. Differential virulence and disease progression following Mycobacterium tuberculosis complex infection of the common marmoset (Callithrix jacchus). Infect Immunity. (2013) 81:2909–19. doi: 10.1128/IAI.00632-13
94. Banuls AL, Sanou A, Anh NT, Godreuil S. Mycobacterium tuberculosis: ecology and evolution of a human bacterium. J Med Microbiol. (2015) 64:1261–9. doi: 10.1099/jmm.0.000171
95. Forrellad MA, Klepp LI, Gioffre A, Sabio y Garcia J, Morbidoni HR, de la Paz Santangelo M, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. (2013) 4:3–66. doi: 10.4161/viru.22329
96. Minnikin DE, Kremer L, Dover LG, Besra GS. The methyl-branched fortifications of Mycobacterium tuberculosis. Chem Biol. (2002) 9:545–53. doi: 10.1016/S1074-5521(02)00142-4
97. Sani M, Houben EN, Geurtsen J, Pierson J, de Punder K, van Zon M, et al. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog. (2010) 6:e1000794. doi: 10.1371/journal.ppat.1000794
98. Jackson M. The mycobacterial cell envelope-lipids. Cold Spring Harb Perspect Med.(2014) 4:a021105. doi: 10.1101/cshperspect.a021105
99. Azad AK, Sirakova TD, Fernandes ND, Kolattukudy PE. Gene knockout reveals a novel gene cluster for the synthesis of a class of cell wall lipids unique to pathogenic mycobacteria. J Biol Chem. (1997) 272:16741–5. doi: 10.1074/jbc.272.27.16741
100. Ferreras JA, Stirrett KL, Lu X, Ryu J-S, Soll CE, Tan DS, et al. Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chem Biol. (2008) 15:51–61. doi: 10.1016/j.chembiol.2007.11.010
101. Turner J, Torrelles JB. Mannose-capped lipoarabinomannan in Mycobacterium tuberculosis pathogenesis. Pathog Dis. (2018) 76. doi: 10.1093/femspd/fty026
102. Khoo KH, Tang JB, Chatterjee D. Variation in mannose-capped terminal arabinan motifs of lipoarabinomannans from clinical isolates of Mycobacterium tuberculosis and Mycobacterium avium complex. J Biol Chem. (2001) 276:3863–71. doi: 10.1074/jbc.M004010200
103. Torrelles JB, Knaup R, Kolareth A, Slepushkina T, Kaufman TM, Kang P, et al. Identification of Mycobacterium tuberculosis clinical isolates with altered phagocytosis by human macrophages due to a truncated lipoarabinomannan. J Biol Chem. (2008) 283:31417–28. doi: 10.1074/jbc.M806350200
104. Cox JS, Chen B, McNeil M, Jacobs WRJr. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. (1999) 402:79–83. doi: 10.1038/47042
105. Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Ave P, et al. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol. (2004) 6:277–87. doi: 10.1046/j.1462-5822.2004.00368.x
106. Barczak AK, Avraham R, Singh S, Luo SS, Zhang WR, Bray MA, et al. Systematic, multiparametric analysis of Mycobacterium tuberculosis intracellular infection offers insight into coordinated virulence. PLoS Pathog. (2017) 13:e1006363. doi: 10.1371/journal.ppat.1006363
107. Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. (2004) 431:84–7. doi: 10.1038/nature02837
108. Cambier CJ, O'Leary SM, O'Sullivan MP, Keane J, Ramakrishnan L. Phenolic glycolipid facilitates mycobacterial escape from microbicidal tissue-resident macrophages. Immunity. (2017) 47:552–565.e554. doi: 10.1016/j.immuni.2017.08.003
109. Huet G, Constant P, Malaga W, Laneelle MA, Kremer K, van Soolingen D, et al. A lipid profile typifies the Beijing strains of Mycobacterium tuberculosis: identification of a mutation responsible for a modification of the structures of phthiocerol dimycocerosates and phenolic glycolipids. J Biol Chem. (2009) 284:27101–13. doi: 10.1074/jbc.M109.041939
110. Constant P, Perez E, Malaga W, Laneelle MA, Saurel O, Daffe M, et al. Role of the pks15/1 gene in the biosynthesis of phenolglycolipids in the Mycobacterium tuberculosis complex. Evidence that all strains synthesize glycosylated p-hydroxybenzoic methyl esters and that strains devoid of phenolglycolipids harbor a frameshift mutation in the pks15/1 gene. J Biol Chem. (2002) 277:38148–58. doi: 10.1074/jbc.M206538200
111. Dao DN, Sweeney K, Hsu T, Gurcha SS, Nascimento IP, Roshevsky D, et al. Mycolic acid modification by the mmaA4 gene of M. tuberculosis modulates IL-12 production. PLoS Pathog. (2008) 4:e1000081. doi: 10.1371/journal.ppat.1000081
112. Nigou J, Zelle-Rieser C, Gilleron M, Thurnher M, Puzo G. Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J Immunol. (2001) 166:7477–85. doi: 10.4049/jimmunol.166.12.7477
113. Axelrod S, Oschkinat H, Enders J, Schlegel B, Brinkmann V, Kaufmann SH, et al. Delay of phagosome maturation by a mycobacterial lipid is reversed by nitric oxide. Cell Microbiol. (2008) 10:1530–45. doi: 10.1111/j.1462-5822.2008.01147.x
114. Hunter RL, Olsen M, Jagannath C, Actor JK. Trehalose 6,6'-dimycolate and lipid in the pathogenesis of caseating granulomas of tuberculosis in mice. Am J Pathol. (2006) 168:1249–61. doi: 10.2353/ajpath.2006.050848
115. Indrigo J, Hunter RLJr, Actor JK. Cord factor trehalose 6,6'-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages. Microbiology. (2003) 149:2049–59. doi: 10.1099/mic.0.26226-0
116. Middlebrook G, Dubos RJ, Pierce C. Virulence and morphological characteristics of Mammalian Tubercle Bacilli. J Exp Med. (1947) 86:175–84. doi: 10.1084/jem.86.2.175
117. Patin EC, Geffken AC, Willcocks S, Leschczyk C, Haas A, Nimmerjahn F, et al. Trehalose dimycolate interferes with FcgammaR-mediated phagosome maturation through Mincle, SHP-1 and FcgammaRIIB signalling. PLoS ONE. (2017) 12:e0174973. doi: 10.1371/journal.pone.0174973
118. Sakamoto K, Kim MJ, Rhoades ER, Allavena RE, Ehrt S, Wainwright HC, et al. Mycobacterial trehalose dimycolate reprograms macrophage global gene expression and activates matrix metalloproteinases. Infect Immunity. (2013) 81:764–76. doi: 10.1128/IAI.00906-12
119. Glickman MS, Cox JS, Jacobs WRJr. A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. (2000) 5:717–27. doi: 10.1016/S1097-2765(00)80250-6
120. Rao V, Fujiwara N, Porcelli SA, Glickman MS. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. (2005) 201:535–43. doi: 10.1084/jem.20041668
121. Oehlers SH, Cronan MR, Beerman RW, Johnson MG, Huang J, Kontos CD, et al. Infection-induced vascular permeability aids mycobacterial growth. J Infect Dis. (2017) 215:813–7. doi: 10.1093/infdis/jiw355
122. Oehlers SH, Cronan MR, Scott NR, Thomas MI, Okuda KS, Walton EM, et al. Interception of host angiogenic signalling limits mycobacterial growth. Nature. (2015) 517:612–5. doi: 10.1038/nature13967
123. Walton EM, Cronan MR, Cambier CJ, Rossi A, Marass M, Foglia MD, et al. Cyclopropane modification of trehalose dimycolate drives granuloma angiogenesis and mycobacterial growth through vegf signaling. Cell Host Microbe. 24:514–25 e516. doi: 10.1016/j.chom.2018.09.004
124. Groschel MI, Sayes F, Simeone R, Majlessi L, Brosch R. ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol. (2016) 14:677–91. doi: 10.1038/nrmicro.2016.131
125. Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis J Bacteriol. (1996) 178:1274–82. doi: 10.1128/jb.178.5.1274-1282.1996
126. Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA. (2003) 100:12420–5. doi: 10.1073/pnas.1635213100
127. Lewis KN, Liao R, Guinn KM, Hickey MJ, Smith S, Behr MA, et al. Deletion of RD1 from Mycobacterium tuberculosis mimics bacille Calmette-Guerin attenuation. J Infect Dis. (2003) 187:117–23. doi: 10.1086/345862
128. Gey Van Pittius NC, Gamieldien J, Hide W, Brown GD, Siezen RJ, Beyers AD. The ESAT-6 gene cluster of Mycobacterium tuberculosis and other high G+C Gram-positive bacteria. Genome Biol. (2001) 2:RESEARCH0044.
129. Newton-Foot M, Warren RM, Sampson SL, van Helden PD, Gey van Pittius NC. The plasmid-mediated evolution of the mycobacterial ESX (Type VII) secretion systems. BMC Evol Biol. (2016) 16:62. doi: 10.1186/s12862-016-0631-2
130. Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jimenez C, et al. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol. (2009) 73:329–40. doi: 10.1111/j.1365-2958.2009.06783.x
131. Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, van der Wel N, et l. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J Biol Chem. (2011) 286:19024–34. doi: 10.1074/jbc.M110.204966
132. Korotkova N, Freire D, Phan TH, Ummels R, Creekmore CC, Evans TJ, et al. Structure of the Mycobacterium tuberculosis type VII secretion system chaperone EspG5 in complex with PE25-PPE41 dimer. Mol Microbiol. (2014) 94:367–82. doi: 10.1111/mmi.12770
133. Abdallah AM, Verboom T, Hannes F, Safi M, Strong M, Eisenberg D, et al. A specific secretion system mediates PPE41 transport in pathogenic mycobacteria. Mol Microbiol. (2006) 62:667–79. doi: 10.1111/j.1365-2958.2006.05409.x
134. Bottai D, Brosch R. Mycobacterial PE, PPE and ESX clusters: novel insights into the secretion of these most unusual protein families. Mol Microbiol. (2009) 73:325–8. doi: 10.1111/j.1365-2958.2009.06784.x
135. Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol. (2004) 53:1677–93. doi: 10.1111/j.1365-2958.2004.04261.x
136. Wagner JM, Evans TJ, Korotkov KV. Crystal structure of the N-terminal domain of EccA(1) ATPase from the ESX-1 secretion system of Mycobacterium tuberculosis. Proteins. (2014) 82:159–63. doi: 10.1002/prot.24351
137. Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol. (2012) 83:1195–209. doi: 10.1111/j.1365-2958.2012.08001.x
138. Brodin P, Majlessi L, Marsollier L, de Jonge MI, Bottai D, Demangel C, et al. Dissection of ESAT-6 system 1 of Mycobacterium tuberculosis and impact on immunogenicity and virulence. Infect Immunity. (2006) 74:88–98. doi: 10.1128/IAI.74.1.88-98.2006
139. Houben EN, Bestebroer J, Ummels R, Wilson L, Piersma SR, Jimenez CR, et al. Composition of the type VII secretion system membrane complex. Mol Microbiol. (2012) 86:472–84. doi: 10.1111/j.1365-2958.2012.08206.x
140. Siegrist MS, Steigedal M, Ahmad R, Mehra A, Dragset MS, Schuster BM, et al. Mycobacterial Esx-3 requires multiple components for iron acquisition. mBio. (2014) 5:e01073–14. doi: 10.1128/mBio.01073-14
141. Stanley SA, Raghavan S, Hwang WW, Cox JS. Acute infection and macrophage subversion by Mycobacterium tuberculosis require a specialized secretion system. Proc Natl Acad Sci USA. (2003) 100:13001–6. doi: 10.1073/pnas.2235593100
142. Brown GD, Dave JA, Gey van Pittius NC, Stevens L, Ehlers MR, Beyers AD. The mycosins of Mycobacterium tuberculosis H37Rv: a family of subtilisin-like serine proteases. Gene. (2000) 254:147–55. doi: 10.1016/S0378-1119(00)00277-8
143. Dave JA, Gey van Pittius NC, Beyers AD, Ehlers MR, Brown GD. Mycosin-1, a subtilisin-like serine protease of Mycobacterium tuberculosis, is cell wall-associated and expressed during infection of macrophages. BMC Microbiol. (2002) 2:30. doi: 10.1186/1471-2180-2-30
144. Daffe M. The cell envelope of tubercle bacilli. Tuberculosis. (2015) 95(Suppl. 1):S155–8. doi: 10.1016/j.tube.2015.02.024
145. Lou Y, Rybniker J, Sala C, Cole ST. EspC forms a filamentous structure in the cell envelope of Mycobacterium tuberculosis and impacts ESX-1 secretion. Mol Microbiol. (2017) 103:26–38. doi: 10.1111/mmi.13575
146. Pallen MJ. The ESAT-6/WXG100 superfamily – and a new Gram-positive secretion system? Trends Microbiol. (2002) 10:209–12. doi: 10.1016/S0966-842X(02)02345-4
147. Fortune SM, Jaeger A, Sarracino DA, Chase MR, Sassetti CM, Sherman DR, et al. Mutually dependent secretion of proteins required for mycobacterial virulence. Proc Natl Acad Sci USA. (2005) 102:10676–81. doi: 10.1073/pnas.0504922102
148. Renshaw PS, Lightbody KL, Veverka V, Muskett FW, Kelly G, Frenkiel TA, et al. Structure and function of the complex formed by the tuberculosis virulence factors CFP-10 and ESAT-6. EMBO J. 2005) 24:2491–8. doi: 10.1038/sj.emboj.7600732
149. Renshaw PS, Panagiotidou P, Whelan A, Gordon SV, Hewinson RG, Williamson RA, et al. Conclusive evidence that the major T-cell antigens of the Mycobacterium tuberculosis complex ESAT-6 and CFP-10 form a tight, 1:1 complex and characterization of the structural properties of ESAT-6, CFP-10, and the ESAT-6*CFP-10 complex. Implications for pathogenesis and virulence. J Biol Chem. (2002) 277:21598–603. doi: 10.1074/jbc.M201625200
150. Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbroucke-Grauls CM, Luirink J, et al. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci USA. (2012) 109:11342–7. doi: 10.1073/pnas.1119453109
151. Derrick SC, Morris SL. The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. (2007) 9:1547–55. doi: 10.1111/j.1462-5822.2007.00892.x
152. Smith J, Manoranjan J, Pan M, Bohsali A, Xu J, Liu J, et al. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immunity. (2008) 76:5478–87. doi: 10.1128/IAI.00614-08
153. de Jonge MI, Pehau-Arnaudet G, Fretz MM, Romain F, Bottai D, Brodin P, et al. ESAT-6 from Mycobacterium tuberculosis dissociates from its putative chaperone CFP-10 under acidic conditions and exhibits membrane-lysing activity. J Bacteriol. (2007) 189:6028–34. doi: 10.1128/JB.00469-07
154. Conrad WH, Osman MM, Shanahan JK, Chu F, Takaki KK, Cameron J, et al. Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc Natl Acad Sci USA. (2017) 114:1371–6. doi: 10.1073/pnas.1620133114
155. Houben D, Demangel C, van Ingen J, Perez J, Baldeon L, Abdallah AM, et al. ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. (2012) 14:1287–98. doi: 10.1111/j.1462-5822.2012.01799.x
156. Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, et al. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. (2012) 8:e1002507. doi: 10.1371/journal.ppat.1002507
157. van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, et al. M. tuberculosis and M leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. (2007) 129:1287–98. doi: 10.1016/j.cell.2007.05.059
158. Solomonson M, Setiaputra D, Makepeace KA, Lameignere E, Petrotchenko EV, Conrady DG, et al. Structure of EspB from the ESX-1 type VII secretion system and insights into its export mechanism. Structure. (2015) 23:571–83. doi: 10.1016/j.str.2015.01.002
159. Volkman HE, Pozos TC, Zheng J, Davis JM, Rawls JF, Ramakrishnan L. Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science. (2010) 327:466–9. doi: 10.1126/science.1179663
160. Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, Brodin P, et al. Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS Pathog. (2008) 4:e33. doi: 10.1371/journal.ppat.0040033
161. Perez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martin C. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol. (2001) 41:179–87. doi: 10.1046/j.1365-2958.2001.02500.x
162. Gonzalo-Asensio J, Malaga W, Pawlik A, Astarie-Dequeker C, Passemar C, Moreau F, et al. Evolutionary history of tuberculosis shaped by conserved mutations in the PhoPR virulence regulator. Proc Natl Acad Sci USA. (2014) 111:11491–6. doi: 10.1073/pnas.1406693111
163. Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol. (2006) 6:95. doi: 10.1186/1471-2148-6-95
164. Brennan MJ. The enigmatic PE/PPE multigene family of Mycobacteria and tuberculosis vaccination. Infect Immunity. (2017) 85:e00969–16. doi: 10.1128/IAI.00969-16
165. Fishbein S, van Wyk N, Warren RM, Sampson SL. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol. (2015) 96:901–16. doi: 10.1111/mmi.12981
166. Tiwari BM, Kannan N, Vemu L, Raghunand TR. The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages. PLoS ONE. (2012) 7:e51686. doi: 10.1371/journal.pone.0051686
167. Tiwari B, Ramakrishnan UM, Raghunand TR. The Mycobacterium tuberculosis protein pair PE9 (Rv1088)-PE10 (Rv1089) forms heterodimers and induces macrophage apoptosis through Toll-like receptor 4. Cell Microbiol. (2015) 17:1653–69. doi: 10.1111/cmi.12462
168. Tiwari B, Soory A, Raghunand TR. An immunomodulatory role for the Mycobacterium tuberculosis region of difference 1 locus proteins PE35 (Rv3872) and PPE68 (Rv3873). Febs J. (2014) 281:1556–70. doi: 10.1111/febs.12723
169. Ramakrishnan L, Federspiel NA, Falkow S. Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science. (2000) 288:1436–9. doi: 10.1126/science.288.5470.1436
170. Ates LS, Dippenaar A, Ummels R, Piersma SR, van der Woude AD, van der Kuij K, et al. Mutations in ppe38 block PE_PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat Microbiol. (2018) 3:181–8. doi: 10.1038/s41564-017-0090-6
171. Romero MM, Balboa L, Basile JI, López B, Ritacco V, de la Barrera SS, et al. Clinical isolates of Mycobacterium tuberculosis differ in their ability to induce respiratory burst and apoptosis in neutrophils as a possible mechanism of immune escape. Clin Dev Immunol. (2012) 2012:11. doi: 10.1155/2012/152546
172. Cooper AM, Segal BH, Frank AA, Holland SM, Orme IM. Transient loss of resistance to pulmonary tuberculosis in p47(phox-/-) mice. Infect Immunity. (2000) 68:1231–4. doi: 10.1128/IAI.68.3.1231-1234.2000
173. Olive AJ, Sassetti CM. Tolerating the Unwelcome Guest; How the Host Withstands Persistent Mycobacterium tuberculosis. Front Immunol. (2018) 9:2094. doi: 10.3389/fimmu.2018.02094
174. Olive AJ, Smith CM, Kiritsy MC, Sassetti CM. The phagocyte oxidase controls tolerance to Mycobacterium tuberculosis infection. J Immunol. (2018) 201:1705–16. doi: 10.4049/jimmunol.1800202
175. Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. (2012) 148:434–46. doi: 10.1016/j.cell.2011.12.023
176. Tobin DM, Vary JCJr, Ray JP, Walsh GS, Dunstan SJ, Bang ND, et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. (2010) 140:717–30. doi: 10.1016/j.cell.2010.02.013
177. Divangahi M, Khan N, Kaufmann E. Beyond Killing Mycobacterium tuberculosis: Disease Tolerance. Front Immunol. (2018) 9:2976. doi: 10.3389/fimmu.2018.02976
178. van Crevel R, Parwati I, Sahiratmadja E, Marzuki S, Ottenhoff TH, Netea MG, et al. Infection with Mycobacterium tuberculosis Beijing genotype strains is associated with polymorphisms in SLC11A1/NRAMP1 in Indonesian patients with tuberculosis. J Infect Dis. (2009) 200:1671–4. doi: 10.1086/648477
179. Intemann CD, Thye T, Niemann S, Browne EN, Amanua Chinbuah M, Enimil A, et al. Autophagy gene variant IRGM−261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains PLoS Pathog. (2009) 5:e1000577. doi: 10.1371/journal.ppat.1000577
180. Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet. (2014) 46:279–86. doi: 10.1038/ng.2878
181. Guerra-Assuncao JA, Crampin AC, Houben RM, Mzembe T, Mallard K, Coll F, et al. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. eLife. (2015) 4:e05166. doi: 10.7554/eLife.05166
Keywords: Mycobacterium tuberculosis, evolution, host tolerance, clinical phenotypes, mycobacteria, mycobacterial genomes
Citation: Saelens JW, Viswanathan G and Tobin DM (2019) Mycobacterial Evolution Intersects With Host Tolerance. Front. Immunol. 10:528. doi: 10.3389/fimmu.2019.00528
Received: 20 November 2018; Accepted: 27 February 2019;
Published: 22 March 2019.
Edited by:
Maziar Divangahi, McGill University, CanadaReviewed by:
Catherine Astarie-Dequeker, Centre National de la Recherche Scientifique (CNRS), FranceAndrew J. Olive, Michigan State University, United States
Copyright © 2019 Saelens, Viswanathan and Tobin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David M. Tobin, ZGF2aWQudG9iaW5AZHVrZS5lZHU=