
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 19 March 2019
Sec. Immunological Tolerance and Regulation
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00521
 Mélanie Lalfer1,2
Mélanie Lalfer1,2 Pascal Chappert1,2Maxime Carpentier1,2Dominique Urbain1,2
Pascal Chappert1,2Maxime Carpentier1,2Dominique Urbain1,2 Jean M. Davoust1,2*
Jean M. Davoust1,2* David-Alexandre Gross1,2*
David-Alexandre Gross1,2*Foxp3+ regulatory T cells (Tregs) play a major role in acquired immune tolerance to allogenic transplants. Their suppressive activity is thought to require T cell receptor (TCR)-driven antigen recognition; little, however, is known about the fraction of Tregs able to recognize alloantigens within this T cell subset primarily educated against self-antigens. Performing transfer experiments of Tregs or conventional T cells (Tconv) into both lymphoreplete and lymphopenic mice, we observed a similarly high proportion of cells signaling through their TCR and proliferating in allogenic hosts. Furthermore, using an in vivo proliferation assay with limited T cell numbers infused into lymphopenic mice, we found that the overall frequency of alloreactive Tregs was similar if not higher to that of alloreactive Tconv. Overall our study highlights a noticeably high level of alloreactive Foxp3+ regulatory T cells accounting for their predominant role in transplantation tolerance.
Foxp3+ regulatory T cells (Tregs) play a crucial role in the control of autoreactive T cells and in the maintenance of the immune system homeostasis (1–3). Beyond this critical role as regulators of self-tolerance, Tregs were also shown in mice to prevent or delay allograft rejection (4, 5) and suppress graft vs. host disease (GvHD) after allogenic bone marrow transplantation (6–9). The importance of regulatory T cells, in particular CD4+CD25+ T cells, exerting dominant tolerance against allograft was discovered more than three decades ago (10–12) and it was proposed early on to use these cells as a means to establish tissue-specific tolerance obviating or minimizing the need for immunosuppressive regimens (13). Adoptive immunotherapy with Tregs is now amenable in human for allogeneic hematopoietic stem cell transplantation, with recent clinical results reporting the prevention of acute and chronic GvHD (14) without loss of graft-vs. leukemia effect (15).
Optimal Tregs suppressive function relies on antigen (Ag) recognition as shown by their continuous T cell receptor (TCR) signaling requirement (16, 17) as well as by multiple evidences for their tissue specificity in models of autoimmunity (18), pregnancy (19), or cancer (20). The importance of Tregs specificity was also demonstrated in transplantation, as Tregs expanded with allogenic presenting cells proved to be more efficient than Tregs expanded with autologous cells or anti-CD3 beads to control GvHD (21) and prevent allograft rejection (5, 9).
The large fraction of alloreactive cells present within conventional CD4+ T cells (Tconv) has been appreciated for a long time as being responsible for organ rejection (22, 23) and is estimated to range between 1 and 10% of the entire T cell repertoire (24, 25). Little is known, however, about the proportion of alloreactive T cells within the Tregs compartment, which has a highly diversified TCR repertoire (26). Given the importance of their TCR in maintaining tolerance to self-antigens at the steady state and given the major role of Tregs in transplantation tolerance, we hypothesized that the repertoire of Tregs is also endowed with a substantial level of alloreactivity. In this study, we compared Tregs and Tconv side by side, and found that a similar proportion of both populations are able to signal through their TCR and expand in allogenic recipients.
All mice were used between 8 and 20 weeks of age. MHC haplotype antigens of C57Bl/6 (B6) mice are H-2Kb, H-2Db, I-Ab (H-2b). MHC haplotype of NOD and NSG mice are H-2Kd, H-2Db, I-Ag7 (H-2g7). B6-Foxp3EGFP (Foxp3tm1Mal) and NOD-Foxp3EGFP (Foxp3tm1Kuch) are normal mice with IRES-eGFP knock-in into the Foxp3 locus on the B6 and NOD backgrounds, respectively. B6-RAG (rag2−/−) and NOD-RAG (rag2−/−) mice are defective in T and B cells. B6-RAGγc (rag2−/− IL2Rg−/−) and NSG (prkdcscid IL2Rg−/−) are defective in T, B and NK cells. B6xNOD F1 mice were generated by crossing B6 mice with NOD mice. All mice were housed under specific pathogen-free conditions and handled in accordance with French and European directives. Animal experiments were approved by the Ethical Committee of Paris Descartes University (CEEA 34).
CD4+ T cells were enriched from pooled splenocytes and inguinal, brachial, and cervical lymph node cells using Dynal CD4 Negative Isolation Kit (Dynal Biotech). Viable CD4+ Foxp3-EGFP+ (Tregs) and CD4+ Foxp3-EGFP− (Tconv) cells were further sorted on a FACSAria (Becton Dickinson) or Sony SH800 (Sony). A purity of >99.9% Tregs or Tconv was regularly achieved. Of note, TCR Vβ3+ cells were excluded from the cell-sorted B6 CD4+ population to avoid potentially confounding superantigen-related stimulation systematically encountered in NSG hosts (data not shown). Cells were injected into the retro-orbital venous sinus in 0.2 ml PBS 1X.
Surface staining were performed in PBS 1X, with 2 mM EDTA and 0.1% BSA. Cell suspensions were first incubated with anti-FcγRIII/II (2.4G2, BioXcell) mAb for 15 min at 4°C and then stained for 30 min at 4°C with saturating amounts of indicated combinations of PE or AF700 anti-CD45.2 (104), e450, APC or PE-Cy7 anti-CD45.1 (A20), FITC, APC or PE-Cy7 anti-TCRβ (H57-597), e450, V500, or PE anti-CD4 (RM4-5). Dead cells were excluded using 7-actinomycin D (Sigma-Aldrich). For Nur77 and Foxp3 co-staining, cells were fixed 30 min at 4°C using Foxp3 Fixation/Permeabilization buffer (eBioscience) and labeled overnight with PE-conjugated anti-Nur77 (12–14) and APC-conjugated anti-Foxp3 mAb (FJK-16s) in Permeabilization Buffer (eBioscience). Antibodies were purchased from Becton Dickinson, eBioscience, or Sony Biotechnology. Dead cells were excluded using LIVE/DEAD® Fixable Near-IR staining (Life Technologies). VPD450-labeling (1 μM) was performed according to manufacturer instructions (Becton Dickinson).
For all statistical analyses, an unpaired t-test was performed within Excel software. In Figures 3A,C, 4C, mice were considered as negative (and plotted on the x axis) when the percentage of positive events found by FACS analysis in experimental group was inferior to three times the percentage of positive events detected in non-transferred NSG mice. This led to a detection limit of ~100 cells per spleen. Limiting dilution frequencies were calculated using L-Calc software (Stem-Cell Technologies).
Alloreactivity of Tconv and Tregs cells was assessed using adoptive transfer of T cells from C57Bl/6 (B6) mice (H-2b) into MHC-mismatched NOD mice (H-2g7). We first choose to measure both the upregulation of Nur77, an immediate early gene up-regulated by TCR stimulation in thymocytes and T cells shown to correlate nicely with TCR signal strength (27), and subsequent cell proliferation events. Splenocytes from B6 CD45.2+ mice were transferred into congenic B6 CD45.1+ mice or allogenic B6xNOD CD45.1+/− (B6xNOD) mice. These latter F1 mice were used as recipients to prevent NK cell-mediated rejection of donor T cells. As expected, Nur77 was induced in a higher proportion of Tconv transferred into allogenic hosts (10.6 ± 0.7%) compared to syngenic hosts (1.2 ± 0.4%) (Figures 1A,B). Importantly, a similar proportion of Tregs also upregulated Nur77 upon transfer into allogenic recipients (9.5 ± 0.8%), while a basal level of Nur77 upregulation was detected following transfer into syngenic mice (3.4 ± 1%), probably due to self-reactivity (27). In line with the comparable rates of early TCR signaling events detected in both populations, similar proliferation profiles were detected in both Tconv and Tregs 3 days after transfer into allogenic hosts (Figures 1C,D). Altogether, these results indicate that a similar, and sizeable, proportion of Tregs and Tconv, experience TCR-driven alloantigen recognition upon transfer in lymphoreplete hosts.
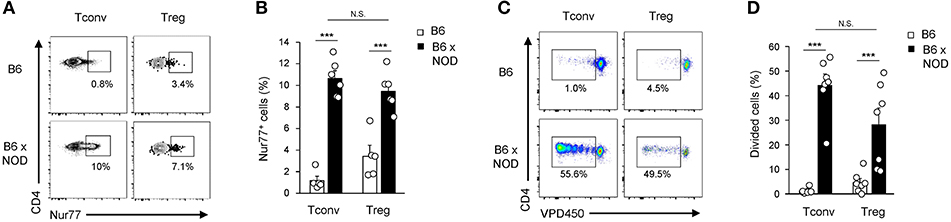
Figure 1. Activation and expansion of Tconv and Tregs in allogenic lymphoreplete hosts. (A,B) 20–40 × 106 B6 splenocytes (CD45.2) were transferred into syngenic B6 CD45.1 vs. allogenic B6xNOD (CD45.2/CD45.1) mice. Five hours post-transfer, mice were sacrificed and their splenocytes analyzed by FACS using AF700 anti-CD45.2, PE-Cy7 anti-CD45.1, FITC anti-TCRβ, e450 anti-CD4, PE anti-Nur77, and APC anti-Foxp3 mAb. (A) Representative dot plots gated on either Foxp3− (Tconv) or Foxp3+ (Tregs) live donor CD45.1−CD45.2+TCRβ+CD4+ cells and (B) percentages of Nur77+ cells in donor T cells recovered from indicated host are shown. (C,D) 20–30 × 106 VPD450-labeled Foxp3-EGFP splenocytes (CD45.2) were transferred into B6 CD45.1 vs. B6xNOD mice. Three days post-transfer, mice were sacrificed and their splenocytes analyzed by FACS using PE anti-CD45.2, APC anti-CD45.1, PE-Cy7 anti-TCRβ and V500 anti-CD4 mAb. (C) Representative dot plots gated on either Foxp3EGFP− or Foxp3EGFP+ live donor CD45.1−CD45.2+TCRβ+CD4+ cells and (D) percentages of divided donor T cells in individual mice are shown. Data are pooled from 3 to 4 independent experiments and represented as mean ± standard error of the mean (SEM). NS, non-significant; ***p < 0.001.
Interestingly, transfer into lymphopenic hosts avoids potential competition with other resident T cells (28) and mimics the lymphopenia conditions associated with transplant regimen. We therefore explored the expansion of Foxp3-EGFPhi-sorted B6 Tregs transferred into either allogenic NOD Prkdcscid IL2R (NSG) or syngenic C57Bl/6 Rag2−/− (B6-RAG) mice. IL2R mutation present in NSG mice was instrumental to prevent NK cell development and NK cell-mediated rejection of allogenic donor T cells. One week after transfer, twenty fold more Tregs were recovered in NSG mice as compared to B6-RAG mice (Figures 2A,B). As previously observed in lymphoreplete hosts, this was correlated with higher Nur77 expression and extensive proliferation up to day 7 for both Tregs and Tconv (data not shown). Reciprocal transfer experiments (H-2g7 -> H-2b) were performed to ensure that this alloreactive potential of Tregs is not specific to B6 Tregs and does not depend on particular hosts combinations. Confirming our previous findings, higher expansion of Tregs sorted from NOD-Foxp3EGFP mice was observed after transfer into allogenic B6-RAGγc recipients compared to syngenic NSG (Figure 2C) or NOD-RAG recipients (data not shown). Of importance, Foxp3-EGFP expression was maintained in more than 80% of Tregs in both syngenic and allogenic recipients, attesting their in vivo stability (Figures 2D–F). As expected, Tconv cells underwent strong expansion in NSG mice (Figures 2A,B), and almost no conversion of Tconv cells into Treg cells was observed in this setting (Figures 2D–E).
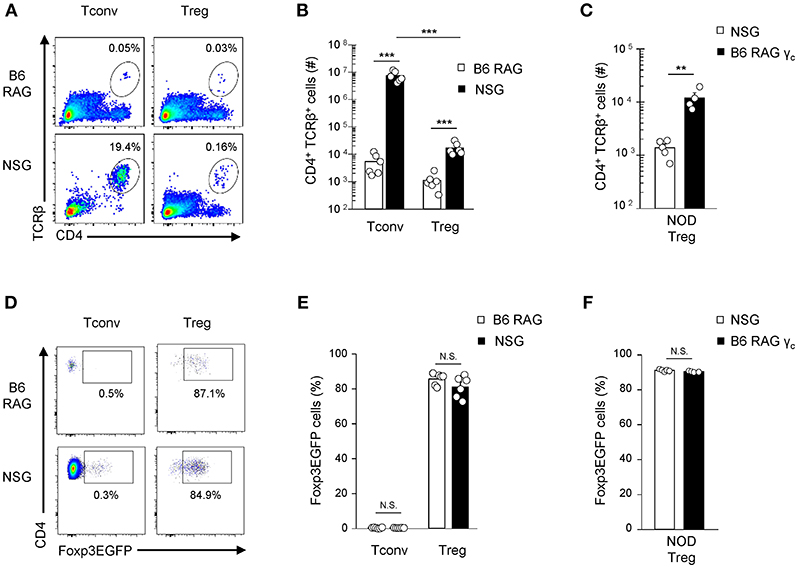
Figure 2. Expansion of both Tconv and Tregs in allogenic lymphopenic hosts. 1 × 105 B6-Tconv or Tregs were cell-sorted from B6-Foxp3EGFP mice and transferred into B6-RAG vs. NSG mice (A,B,D,E). 1 × 105 NOD-Tregs were cell-sorted from NOD-Foxp3EGFP mice and transferred into NSG vs. B6-RAGγc mice (C,F). Mice were sacrificed at day 7 and their splenocytes analyzed by FACS using PE anti-CD45.2, APC anti-CD45.1, PE-Cy7 anti-TCRβ and V500 anti-CD4 mAb. (A) Representative dot plots gated on live cells and (B,C) absolute numbers of recovered CD4+TCRβ+Foxp3EGFP− Treg and/or Foxp3EGFP+ Tconv in individual mice are shown. (D) Representative dot plots and (E,F) percentages of Foxp3EGFP expressing cells gated on CD4+TCRβ+ live cells. Data are pooled from 3 to 4 (A,B,D,E) or 1 (C,F) independent experiments and represented as mean ± standard error of the mean (SEM). NS, non-significant; **p < 0.01, ***p < 0.001.
To gain access to the frequency of alloreactive Tregs, we next performed an in vivo limiting dilution assay (LDA) in lymphopenic hosts, using low numbers of B6 T cells transferred into NSG or control B6-RAG mice. Seven days post-transfer, Tregs were found in a fraction of NSG mice receiving as little as 100 Tregs (4 out of 16 mice), while none were found in B6-RAG hosts receiving 10-times more Tregs, confirming at low cell numbers the predominant role of allogenic stimulation for Tregs expansion (Figure 3A). Indeed, the proportion of NSG mice in which Tregs were detected increased when more cells were injected. Linear regression of the frequency of negative mice vs. the numbers of injected T cells gave a proportion of 0.32 ± 0.08% of injected Tregs (f = 1/315) expanding in allogenic recipients (Figure 3D). Interestingly, in line with the high percentage of Foxp3+ cells observed upon transfer of high cell numbers (Figures 2D–F), Foxp3 expression was equally maintained at low numbers of injected Tregs (Figure 3B). This experiment was repeated with Tconv and a closely related percentage of 0.56 ± 0.12% alloreactive Tconv (f = 1/177) was found (Figures 3C,D).
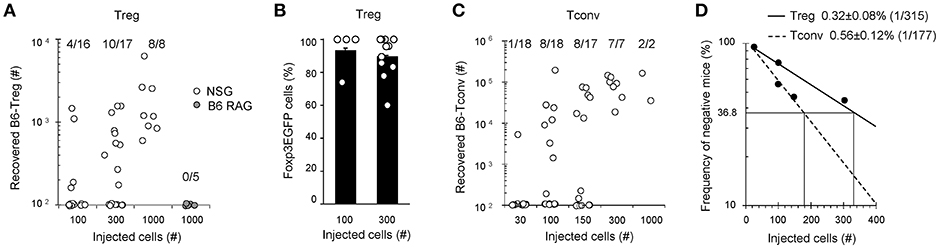
Figure 3. Frequencies of alloreactive Tconv and Tregs. Indicated numbers (30–1000) of cell-sorted Tregs or Tconv were transferred into NSG mice. At day 7, mice were sacrificed and their splenocytes analyzed by FACS using APC anti-TCRβ and PE anti-CD4 mAb. (A,C) Absolute numbers of recovered live CD4+TCRβ+Foxp3+ Tregs and CD4+TCRβ+Foxp3− Tconv in individual mice are shown. (B) Percentage of Foxp3EGFP+ cells into individual mice injected with 100 or 300 Tregs that exhibit CD4+TCRβ+ events. (D) Numbers of injected T cells are plotted against the log frequency of negative mice. Mean ± standard error of the mean (SEM) is shown. Data are pooled from 4 independent experiments.
Next, to compare Tregs and Tconv survival upon transfer into lymphopenic mice, 5 × 104 Tconv and 5 × 104 Tregs were co-injected into NSG or B6-RAG mice and their relative proportions determined 16 h later, prior to any cell expansion. At this early time point, Tregs recovery in secondary lymphoid organs amounted to 12.4 ± 0.8% and 18.4 ± 1.9% relative to that of Tconv in NSG and B6-RAG mice, respectively (Figures 4A,B). This lower survival was not due to cell-sorting procedure since direct injection of B6 splenocytes into NSG or B6-RAG mice yielded a comparable Tregs over Tconv survival ratio of 18.8 ± 2.3% and 19.5 ± 1.8%, respectively (data not shown). Of note, it has been reported that lower Tregs survival could result from ARTC2-mediated ADP-ribosylation of P2X7 during murine primary lymphocytes preparation (29). Finally, to estimate the absolute level of Tconv survival after transfer into lymphopenic hosts, we transferred limited amounts of Rag−/− TCR transgenic Marilyn T cells, all reactive to the HY male antigen, and found that a proportion of 12.2 ± 2.5% (f = 1/8) of injected Marilyn Tconv survived and underwent expansion in male B6-RAG mice (Figures 4C,D).
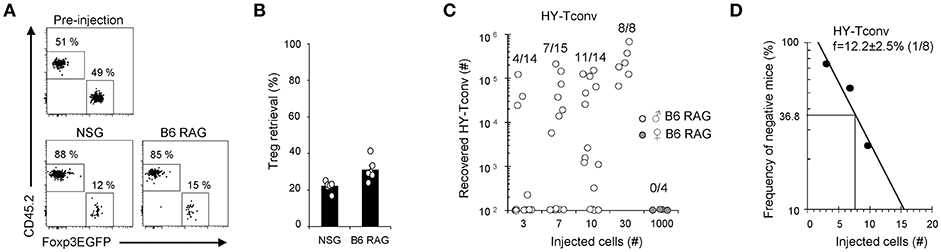
Figure 4. Adoptive transfer survival of Tconv and Tregs in lymphopenic hosts. (A,B) 5 × 104 Tregs (CD45.1) were mixed with 5 × 104 Tconv (CD45.2) and transferred into male NSG or B6-RAG mice. One day later, mice were sacrificed and their splenocytes analyzed by FACS using PE anti-CD4, APC anti-TCRβ, e450 anti-CD45.1 and AF700 anti-CD45.2 mAb. (A) Representative dot plots gated on live donor CD4+ TCRβ+ cells. (B) Histogram shows Tregs retrieval as a percentage of Tconv. Mean ± standard error of the mean (SEM) is shown. (C,D) Indicated numbers (3–1000) of CD4+ RAG2−/− Marilyn T cells were transferred into male or female B6-RAG mice. At day 7, mice were sacrificed and their splenocytes analyzed by FACS using APC anti-TCRβ and PE anti-CD4 mAb. (C) Absolute numbers of recovered live CD4+TCRβ+ Marylin T cells in individual mice are shown. (D) Numbers of injected Marilyn T cells are plotted against the log frequency of negative mice. Data are pooled from 2 (A,B) or 4 (C,D) independent experiments.
Combining the survival ratio of Tregs over Tconv (Figures 4A,B) with the survival of Marilyn Tconv (Figures 4C,D), our results indicate that only 1.5 ± 0.4% of Tregs (i.e., 12.4 ± 0.8% × 12.2 ± 2.5%) were able to survive after adoptive transfer into NSG mice. This survival efficiency is markedly different from that of Tconv, leading to corrected alloreactive frequencies for Tregs and Tconv of 21.0 ± 10.7% (0.32 ± 0.08% over 1.5 ± 0.4% surviving cells) and 4.6 ± 1.9% (0.56 ± 0.12% over 12.2 ± 2.5% surviving cells), respectively.
To compare the frequency of alloreactive Tregs and Tconv in vivo, we used two different methods: an indirect measure of TCR activation (Nur77) and a limiting dilution assay. While powerful, LDA has rarely been used in vivo, because of two experimental constraints. First, detection of limited number of T cells is laborious in normal hosts. Second, classical analysis at 4 weeks takes into account proliferation events resulting from both cognate MHC/antigen-TCR interactions and the so-called homeostatic proliferation, which is mainly driven by available environmental resources (30).
Using lymphopenic hosts and focusing on early time points, we estimated that 4.6 ± 1.9% of Tconv that survived the initial transfer underwent alloantigen-driven expansion. This result is in the same range, and somewhat lower, than the frequency found using Nur77 measurement in lymphoreplete hosts (10.6 ± 0.7%), and is in agreement with other studies using different approaches such as measurement of prolonged interactions using time-lapse microscopy (5–15%) (31), CFSE dye dilution after in vivo transfer (0.7–21% depending on the calculation method) or in vitro mixed lymphocyte reactions (4.0–4.6%) (24, 25).
Regarding Treg cells, a similarly high frequency of alloreactive cells was detected in vivo by Nur77 measurement (9.5 ± 0.8%), in agreement with a previous study using an in vitro analysis of the reactivity of hybridoma cells derived from a limited set of Tconv and Tregs (32). Taking into account the lesser survival efficiency of Tregs, the percentage of alloreactive Tregs (21.0 ± 10.7%) appears higher than that of Tconv (4.6 ± 1.9%). This difference largely depends on the survival measurement that could be affected by different parameters such as differential migration patterns into non-lymphoid tissues between Tregs and Tconv (33). Nevertheless, a higher alloreactivity was also suggested using an in vivo MLR, which does not take into account inhibitory effects due to T cell competition (34). Interestingly, no major difference were observed when other T cell subsets were compared: frequencies of alloreactive CD4+ and CD8+ were in the same range (3.9% for the CD4+, 2.5% for the CD8+), and naïve and memory cells contribute equally to the allo-response (35). Overall, assays based on early TCR signaling measurement (Nur77, hybridoma) give rise to underestimated fractions of alloreactive Tregs as compared to assays based on T cell expansion (LDA, MLR), a fact that could be explained by the particular TCR machinery of Tregs (36).
Our findings are mainly based on transfer of B6 Tregs into MHC-mismatched NOD mice (H-2b -> H-2g7). Nevertheless, alloreactivity of Tregs has been noticed in other MHC combinations, with B6 Tregs alloreactivity being observed against H-2d and H-2k haplotypes (32, 34), and in our reciprocal transfer experiments (H-2g7 -> H-2b), which evidenced equally high alloreactive Tregs expansion (Figure 2C). Finally, it should be noted that to prevent NK cell-mediated rejection of allogenic T cells, we used IL2R lymphopenic hosts, devoid of NK cells. Resulting from the absence of functional receptors, these mice could have increased levels of cytokines such as IL-7, which is important for Tregs maintenance (37). Here, significant differences were still observed for the expansion of NOD-Foxp3EGFP Tregs adoptively transferred into allogenic B6-RAGγc (IL2R) mice compared to syngenic mice being either IL2R (NSG) (Figure 2C) or IL2R (NOD RAG) (data not shown), ruling out this potential non-MHC-dependent strain-specific effect.
The TCR repertoire of Tregs and Tconv are vast and diverse but poorly overlapping (26). Whether cross-reactivity that applies to the Tconv subset and explain their alloreactivity (38) equally applies to the Tregs compartment is unknown. The high frequency of allogenic Tregs found here can have different explanations. First, it has been shown that dual TCR T cells are predisposed to alloreactivity (39), and a vast majority of Tregs harbor two different TCRα chain (40). Second, Tregs are more resistant to deletional process in the thymus (41) and most of the TCRα chains that normally promote negative selection confer a high level of alloreactivity (42).
Our results also showed that frequency of Tregs reactive to self-Ags (B6 syngenic hosts) was lower than that reactive to alloantigens (NOD allogenic hosts), as measured by Nur77 expression, VPD450 dye dilution and early cell expansion capacity. Of note, Tregs emerge in the thymus upon agonist self-antigen recognition (43), require continuous TCR signaling to exert their function in the periphery (16, 17) and demonstrate tissue specificity in models of autoimmunity (18), pregnancy (19), or cancer (20), but these experimental observations provided only indirect proofs of recognition of self-Ags. Intriguingly, hybridoma cells derived from either Tregs (32) or “would be” Tregs (44) were found poorly reactive against syngenic splenocytes, and in contrast to Tconv, “would be” Tregs, which express a non-functional Foxp3 protein, were not pathogenic after transfer into lymphopenic mice (45, 46). However, two naturally-expressed Ags have been recently identified as being recognized by Tregs: TCAF3, a prostate-specific protein, which is expressed in the thymus in an Aire-dependant manner (47) and MOG, which is a minor component of the myelin expressed in the central nervous system (48). The fraction of Tregs recognizing such tissue-specific Ags is not known and it is possible that only a minor fraction of the Tregs pool possess this kind of antigenic specificity. Indeed, exogenous Ags-specific Tregs were also implicated in limiting pathological immunity toward environmental antigens including commensal bacteria (49) and allergens (50). Overall, two non-mutually exclusive parameters could explain the lower reactivity of Tregs toward self-Ags found here, even when assessed in recipient lymph nodes (data not shown): (1) lower and limiting abundance of self-peptides-MHC complexes as compared to allogenic MHC, (2) lower TCR avidity for self-Ags than for alloantigens.
Foxp3+CD4+ regulatory T cells, formerly known as CD4+CD25+, have been identified as a major component required for transplantation tolerance (51–53). These cells are mobilized by short-term immunosuppressive drugs and promote long-term tolerance through “linked suppression” and “infectious tolerance” mechanisms relying on their specific immunosuppressive properties (13). This advantageous Ag-specific activity of Tregs was also observed in other models, such as type 1 diabetes (54) or gene transfer (55), but depend on the expression of transgenic TCRs. In the transplantation field, protocols aiming at recruiting alloantigen-specific Tregs were particularly efficient to prevent both allograft rejection and GvHD in mice (5–9) and to promote tolerance induction in human (56, 57). Altogether, our results reveal an important level of alloreactivity within the vast self-specific Tregs repertoire, in line with and supporting their predominant role in controlling adverse immune responses in organ transplantation.
All datasets generated for this study are included in the manuscript.
ML, PC, MC, and DU performed experiments. ML analyzed and prepared figures. PC, JD, and D-AG wrote the manuscript.
This work was supported by the Association Française contre les Myopathies (AFM) and Agence Nationale de la Recherche (ANR-11-JSV3, ANR-15-CE15-0005-02). ML and MC were supported by the French Ministry of Research and AFM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Corinne Cordier, Olivier Pellé and Jérôme Mégret for cell sorting, and Lucienne Chatenoud, Hanem Sadek, Isabelle André-Schmutz, and Marie Cherrier for providing us with NSG and B6-RAGγc mice.
1. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. (2003) 299:1057–61. doi: 10.1126/science.1079490
2. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+regulatory T cells. Nat Immunol. (2003) 4:330–6. doi: 10.1038/ni904
3. Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+T regulatory cells. Nat Immunol. (2003) 4:337–42. doi: 10.1038/ni909
4. Graca L, Thompson S, Lin CY, Adams E, Cobbold SP, Waldmann H. Both CD4+CD25+and CD4+CD25− regulatory cells mediate dominant transplantation tolerance. J Immunol. (2002) 168:5558–65. doi: 10.4049/jimmunol.168.11.5558
5. Joffre O, Gorsse N, Romagnoli P, Hudrisier D, van Meerwijk JP. Induction of antigen-specific tolerance to bone marrow allografts with CD4+CD25+ T lymphocytes. Blood. (2004) 103:4216–21. doi: 10.1182/blood-2004-01-0005
6. Hoffmann P, Ermann J, Edinger M, Fathman CG, Strober S. Donor-type CD4+CD25+ regulatory T cells suppress lethal acute graft- versus-host disease after allogeneic bone marrow transplantation. J Exp Med. (2002) 196:389–99. doi: 10.1084/jem.20020399
7. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4+CD25+ immunoregulatory T Cells: new therapeutics for graft- versus-host disease. J Exp Med. (2002) 196:401–6. doi: 10.1084/jem.20020090
8. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat Med. (2003) 9:1144–50. doi: 10.1038/nm915
9. Taylor PA, Panoskaltsis-Mortari A, Swedin JM, Lucas PJ, Gress RE, Levine BL, et al. L-Selectin(hi) but not the L-selectin(lo) CD4+25+ T-regulatory cells are potent inhibitors of GVHD and BM graft rejection. Blood. (2004) 104:3804–12. doi: 10.1182/blood-2004-05-1850
10. Kilshaw PJ, Brent L, Pinto M. Suppressor T cells in mice made unresponsive to skin allografts. Nature. (1975) 255:489–91. doi: 10.1038/255489a0
11. Hall BM, Jelbart ME, Gurley KE, Dorsch SE. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. Mediation of specific suppression by T helper/inducer cells. J Exp Med. (1985) 162:1683–94. doi: 10.1084/jem.162.5.1683
12. Hall BM, Pearce NW, Gurley KE, Dorsch SE. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. III. Further characterization of the CD4+ suppressor cell and its mechanisms of action. J Exp Med. (1990) 171:141–57. doi: 10.1084/jem.171.1.141
13. Qin S, Cobbold SP, Pope H, Elliott J, Kioussis D, Davies J, et al. “Infectious” transplantation tolerance. Science. (1993) 259:974–7. doi: 10.1126/science.8094901
14. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood. (2016) 127:1044–51. doi: 10.1182/blood-2015-06-653667
15. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. (2014) 124:638–44. doi: 10.1182/blood-2014-03-564401
16. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity. (2014) 41:722–36. doi: 10.1016/j.immuni.2014.10.012
17. Levine AG, Arvey A, Jin W, Rudensky AY. Continuous requirement for the TCR in regulatory T cell function. Nat Immunol. (2014) 15:1070–8. doi: 10.1038/ni.3004
18. Samy ET, Parker LA, Sharp CP, Tung KS. Continuous control of autoimmune disease by antigen-dependent polyclonal CD4+CD25+ regulatory T cells in the regional lymph node. J Exp Med. (2005) 202:771–81. doi: 10.1084/jem.20041033
19. Rowe JH, Ertelt JM, Xin L, Way SS. Pregnancy imprints regulatory memory that sustains anergy to fetal antigen. Nature. (2012) 490:102–6. doi: 10.1038/nature11462
20. Malchow S, Leventhal DS, Nishi S, Fischer BI, Shen L, Paner GP, et al. Aire-dependent thymic development of tumor-associated regulatory T cells. Science. (2013) 339:1219–24. doi: 10.1126/science.1233913
21. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-type specific CD4+CD25+ regulatory T cells favor immune reconstitution and control graft-versus-host disease while maintaining graft-versus-leukemia. J Clin Invest. (2003) 112:1688–96. doi: 10.1172/JCI17702
22. Simonsen M. The clonal selection hypothesis evaluated by grafted cells reacting against their hosts. Cold Spring Harb Symp Quant Biol. (1967) 32:517–23. doi: 10.1101/SQB.1967.032.01.063
23. Wilson DB, Blyth JNP. Quantitative studies on the mixed lymphocyte interaction in rats. 3. Kinetics of the response. J Exp Med. (1968) 128:1157–81.
24. Noorchashm H, Lieu YK, Rostami SY, Song HK, Greeley SA, Bazel S, et al. A direct method for the calculation of alloreactive CD4+ T cell precursor frequency. Transplantation. (1999) 67:1281–4. doi: 10.1097/00007890-199905150-00015
25. Suchin EJ, Langmuir PB, Palmer E, Sayegh MH, Wells AD, Turka LA. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. J Immunol. (2001) 166:973–81. doi: 10.4049/jimmunol.166.2.973
26. Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. (2004) 21:267–77. doi: 10.1016/j.immuni.2004.07.009
27. Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, et al. T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med. (2011) 208:1279–89. doi: 10.1084/jem.20110308
28. Singh NJ, Bando JK, Schwartz RH. Subsets of nonclonal neighboring CD4+ T cells specifically regulate the frequency of individual antigen-reactive T cells. Immunity. (2012) 37:735–46. doi: 10.1016/j.immuni.2012.08.008
29. Rissiek B, Danquah W, Haag F, Koch-Nolte F. Technical Advance: a new cell preparation strategy that greatly improves the yield of vital and functional Tregs and NKT cells. J Leukoc Biol. (2014) 95:543–9. doi: 10.1189/jlb.0713407
30. Min B, Yamane H, Hu-Li J, Paul WE. Spontaneous and homeostatic proliferation of CD4 T cells are regulated by different mechanisms. J Immunol. (2005) 174:6039–44. doi: 10.4049/jimmunol.174.10.6039
31. Juvet SC, Sanderson S, Hester J, Wood KJ, Bushell A. Quantification of CD4+T cell alloreactivity and its control by regulatory t cells using time-lapse microscopy and immune synapse detection. Am J Transplant. (2016) 16:1394–407. doi: 10.1111/ajt.13607
32. Pacholczyk R, Kern J, Singh N, Iwashima M, Kraj P, Ignatowicz L. Nonself-antigens are the cognate specificities of Foxp3(+) regulatory T cells. Immunity. (2007) 27:493–504 doi: 10.1016/j.immuni.2007.07.019
33. Campbell DJ. Control of regulatory T cell migration, function, and homeostasis. J Immunol. (2015) 195:2507–13. doi: 10.4049/jimmunol.1500801
34. Lee K, Nguyen V, Lee KM, Kang SM, Tang Q. Attenuation of donor-reactive T cells allows effective control of allograft rejection using regulatory T cell therapy. Am J Transplant. (2014) 14:27–38. doi: 10.1111/ajt.12509
35. Macedo C, Orkis EA, Popescu I, Elinoff BD, Zeevi A, Shapiro R, et al. Contribution of naive and memory T-cell populations to the human alloimmune response. Am J Transplant. (2009) 9:2057–66. doi: 10.1111/j.1600-6143.2009.02742.x
36. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol. (2016) 16:220–33. doi: 10.1038/nri.2016.26
37. Simonetta F, Gestermann N, Martinet KZ, Boniotto M, Tissieres P, Seddon B, et al. Interleukin-7 influences FOXP3+CD4+ regulatory T cells peripheral homeostasis. PLoS ONE. (2012) 7:e36596. doi: 10.1371/journal.pone.0036596
38. Degauque N, Brouard S, Soulillou JP. Cross-reactivity of TCR repertoire: current concepts, challenges, and implication for allotransplantation. Front Immunol. (2016) 7:89. doi: 10.3389/fimmu.2016.00089
39. Balakrishnan A, Morris GP. The highly alloreactive nature of dual TCR T cells. Curr Opin Organ Transplant. (2016) 21:22–8. doi: 10.1097/MOT.0000000000000261
40. Tuovinen H, Salminen JT, Arstila TP. Most human thymic and peripheral-blood CD4+ CD25+ regulatory T cells express 2 T-cell receptors. Blood. (2006) 108:4063–70. doi: 10.1182/blood-2006-04-016105
41. van Santen HM, Benoist C, Mathis D. Number of T reg cells that differentiate does not increase upon encounter of agonist ligand on thymic epithelial cells. J Exp Med. (2004) 200:1221–30. doi: 10.1084/jem.20041022
42. McDonald BD, Bunker JJ, Erickson SA, Oh-Hora M, Bendelac A. Crossreactive alphabeta T cell receptors are the predominant targets of thymocyte negative selection. Immunity. (2015) 43:859–69. doi: 10.1016/j.immuni.2015.09.009
43. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. (2001) 2:301–6. doi: 10.1038/86302
44. Kuczma M, Podolsky R, Garge N, Daniely D, Pacholczyk R, Ignatowicz L, et al. Foxp3-deficient regulatory T cells do not revert into conventional effector CD4+ T cells but constitute a unique cell subset. J Immunol. (2009) 183:3731–41. doi: 10.4049/jimmunol.0800601
45. Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, et al. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. (2007) 8:359–68. doi: 10.1038/ni1445
46. Lahl K, Mayer CT, Bopp T, Huehn J, Loddenkemper C, Eberl G, et al. Nonfunctional regulatory T cells and defective control of Th2 cytokine production in natural scurfy mutant mice. J Immunol. (2009) 183:5662–72. doi: 10.4049/jimmunol.0803762
47. Leonard JD, Gilmore DC, Dileepan T, Nawrocka WI, Chao JL, Schoenbach MH, et al. Identification of natural regulatory T cell epitopes reveals convergence on a dominant autoantigen. Immunity. (2017). 47:107–17. doi: 10.1016/j.immuni.2017.06.015
48. Kieback E, Hilgenberg E, Stervbo U, Lampropoulou V, Shen P, Bunse M, et al. Thymus-derived regulatory T cells are positively selected on natural self-antigen through cognate interactions of high functional avidity. Immunity. (2016) 44:1114–26. doi: 10.1016/j.immuni.2016.04.018
49. Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, et al. Peripheral education of the immune system by colonic commensal microbiota. Nature. (2011) 478:250–4. doi: 10.1038/nature10434
50. Bacher P, Heinrich F, Stervbo U, Nienen M, Vahldieck M, Iwert C, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell. (2016) 167:1067–1078.e16 doi: 10.1016/j.cell.2016.09.050
51. Wood KJ, Sakaguchi S. Regulatory T cells in transplantation tolerance. Nat Rev Immunol. (2003) 3:199–210. doi: 10.1038/nri1027
52. Waldmann H, Graca L, Cobbold S, Adams E, Tone M, Tone Y. Regulatory T cells and organ transplantation. Semin Immunol. (2004) 16:119–26. doi: 10.1016/j.smim.2003.12.007
53. Hall BM. CD4+CD25+ T Regulatory cells in transplantation tolerance: 25 years on. Transplantation. (2016) 100:2533–47. doi: 10.1097/TP.0000000000001436
54. Tarbell KV, Yamazaki S, Olson K, Toy P, Steinman RM. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med. (2004) 199:1467–77. doi: 10.1084/jem.20040180
55. Gross DA, Leboeuf M, Gjata B, Danos O, Davoust J. CD4+CD25+ regulatory T cells inhibit immune-mediated transgene rejection. Blood. (2003) 102:4326–8. doi: 10.1182/blood-2003-05-1454
56. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology. (2016) 64:632–43. doi: 10.1002/hep.28459
Keywords: T cells, Foxp3+ regulatory T cells, TCR, alloreactivity, transplantation
Citation: Lalfer M, Chappert P, Carpentier M, Urbain D, Davoust JM and Gross D-A (2019) Foxp3+ Regulatory and Conventional CD4+ T Cells Display Similarly High Frequencies of Alloantigen-Reactive Cells. Front. Immunol. 10:521. doi: 10.3389/fimmu.2019.00521
Received: 10 December 2018; Accepted: 26 February 2019;
Published: 19 March 2019.
Edited by:
Shohei Hori, The University of Tokyo, JapanReviewed by:
Nick David Jones, University of Birmingham, United KingdomCopyright © 2019 Lalfer, Chappert, Carpentier, Urbain, Davoust and Gross. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean M. Davoust, amVhbi5kYXZvdXN0QGluc2VybS5mcg==
David-Alexandre Gross, ZGF2aWQuZ3Jvc3NAaW5zZXJtLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.