 Sarah A. Robertson1*
Sarah A. Robertson1* Ella S. Green1
Ella S. Green1 Alison S. Care1
Alison S. Care1 Lachlan M. Moldenhauer1
Lachlan M. Moldenhauer1 Jelmer R. Prins2M. Louise Hull1,3
Jelmer R. Prins2M. Louise Hull1,3 Simon C. Barry1
Simon C. Barry1 Gustaaf Dekker1
Gustaaf Dekker1- 1Robinson Research Institute and Adelaide Medical School, University of Adelaide, Adelaide, SA, Australia
- 2University Medical Center Groningen, Groningen, Netherlands
- 3Women's and Children's Hospital, Adelaide, SA, Australia
Inflammation is a central feature and is implicated as a causal factor in preeclampsia and other hypertensive disorders of pregnancy. Inflammatory mediators and leukocytes, which are elevated in peripheral blood and gestational tissues, contribute to the uterine vascular anomalies and compromised placental function that characterize particularly the severe, early onset form of disease. Regulatory T (Treg) cells are central mediators of pregnancy tolerance and direct other immune cells to counteract inflammation and promote robust placentation. Treg cells are commonly perturbed in preeclampsia, and there is evidence Treg cell insufficiency predates onset of symptoms. A causal role is implied by mouse studies showing sufficient numbers of functionally competent Treg cells must be present in the uterus from conception, to support maternal vascular adaptation and prevent later placental inflammatory pathology. Treg cells may therefore provide a tractable target for both preventative strategies and treatment interventions in preeclampsia. Steps to boost Treg cell activity require investigation and could be incorporated into pregnancy planning and preconception care. Pharmacological interventions developed to target Treg cells in autoimmune conditions warrant consideration for evaluation, utilizing rigorous clinical trial methodology, and ensuring safety is paramount. Emerging cell therapy tools involving in vitro Treg cell generation and/or expansion may in time become relevant. The success of preventative and therapeutic approaches will depend on resolving several challenges including developing informative diagnostic tests for Treg cell activity applicable before conception or during early pregnancy, selection of relevant patient subgroups, and identification of appropriate windows of gestation for intervention.
Introduction
Preeclampsia and related hypertensive disorders complicate 3–5% of pregnancies. They are a leading cause of maternal deaths and perinatal morbidity and mortality (1) and are enormously expensive to health care systems, with an estimated cost in the US of $2.18 billion for the first 12 months of life alone (2). Preterm birth and fetal intrauterine growth restriction (IUGR) are common sequalae, causing developmental challenges for the neonate that adversely impact cardiovascular, metabolic, and neurodevelopmental health (3). Preeclampsia also has long-term consequences for maternal cardiovascular health (4). Despite extensive research, the pathophysiological origins of preeclampsia remain unclear and effective preventative interventions are lacking. Current clinical management is aimed at alleviating symptoms and delaying delivery, rather than preventing occurrence by modifying the underlying cause (5, 6).
An emerging view is that critical initiating events before pregnancy and in the conception and implantation phase determine preeclampsia susceptibility, eliciting changes in placental development much earlier in gestation than when symptoms appear (7–9). This is particularly the case for the severe, early onset form of preeclampsia where failed maternal vascular adaptation to pregnancy is implicated—but also likely contributes to later onset disease (10, 11). There is strong evidence that failure of the maternal immune response to adapt correctly in early pregnancy underpins the placental and cardiovascular anomalies that become evident in later gestation. Disturbance in the immune response appears to be central and causal of later placental and hypertensive symptoms (8, 12, 13).
The adaptive immune response, with its typical features of immunological priming and memory, appears integral to the pathophysiological origin of the condition. Preeclampsia is more common in first pregnancies, particularly after limited sexual contact with the conceiving partner due to short sexual cohabitation, use of barrier contraceptive methods or assisted reproduction (14–16). Prior pregnancy with the same partner offers protection, but this is partner-specific and is lost with a new partner, implying alloantigen specificity (17). Assisted reproduction with donor oocytes, where there is no prior contact with the donor's alloantigens, is associated with a 4.3-fold increase in preeclampsia compared to natural conception (18). The risk is also increased with donor sperm but this is reduced with multiple exposures to the same donor (19). Pregnancy-induced memory in T cells (20) and in uterine NK cells (21) likely contributes to the protective benefit of prior pregnancy, and mechanisms by which seminal fluid may also induce memory are emerging (22). Recognizing this protective role for the adaptive immune response offers scope for new approaches to tackle this prevalent condition.
All women show evidence of altered immunity and elevated inflammatory activation in pregnancy. Immune adaptation for pregnancy commences during the pre-implantation phase when conception and implantation evoke a controlled inflammatory response in the female reproductive tract, which must be rapidly resolved by specific cytokines and pro-tolerogenic mechanisms into an anti-inflammatory milieu, in order for pregnancy to progress (23). In preeclampsia there are excessive pro-inflammatory mediators and inappropriate activation of effector immune cells, detectable in peripheral blood and gestational tissues from the first trimester (24, 25), implying incomplete or insufficient establishment of anti-inflammatory mechanisms (Figure 1).
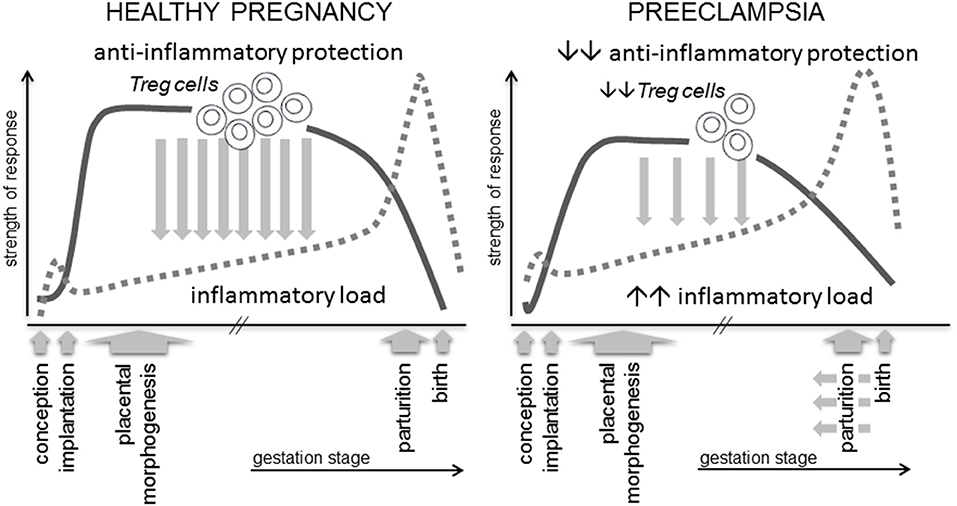
Figure 1. A robust immune response is essential for healthy pregnancy. Regulatory T cells (Treg cells) are a critical and rate-limiting element of the anti-inflammatory protection required to suppress inflammation and prevent adverse effects of anti-fetal effector immune responses. Treg cells arise as a consequence of an inflammation-like response during the peri-conception phase, and their abundance, suppressive function and stability are influenced by events prior to and during early pregnancy. Insufficient numbers, reduced suppressive function or instability are linked with preeclampsia and IUGR, potentially mediated by insufficient Treg cell capacity to support normal placental development and to suppress the elevated inflammatory load typical of this condition.
In healthy pregnancy, inflammation associated with conception and implantation is rapidly resolved, and then remains suppressed by anti-inflammatory protective mechanisms, amongst which the specialized subset of CD4+ T lymphocytes called regulatory T cells (Treg cells), are pivotal (26–28). Through their critical roles in constraining inflammation, suppressing effector immunity, and modulating vascular function, Treg cells are emerging along with uNK cells and macrophages as key coordinators of implantation and early placental development (23, 29–31).
In preeclampsia, Tregs in the maternal peripheral blood and decidua are fewer in number (26, 28, 32) and their suppressive function is impaired (33, 34), while pro-inflammatory Th17 cells (27), CD8+ effector T cells and trophoblast apoptosis (28) are increased. The underlying reasons are unclear, but number and functional capacity of Treg cells are known to vary between individuals and are influenced by agents and exposures identified as pre-pregnancy antecedents of preeclampsia and other adverse pregnancy outcomes. Risk factors for impaired Treg activity include elevated inflammatory load associated with obesity and metabolic dysfunction (35), autoimmune conditions and systemic inflammatory exposures (36, 37), nutritional deficiencies (38, 39), and age (40). Their abundance and phenotype in the uterus are furthermore regulated by relevant clinical factors including prior pregnancy, disparity between male and female partner alloantigens, and seminal fluid contact (23). Other obstetric disorders including fetal growth restriction, gestational diabetes, and spontaneous preterm birth also have an inflammatory etiology, but amongst these conditions, the causal link between Treg cell dysregulation and preeclampsia is most clear.
In this review, we make the case that interventions to boost the number, functional competence and stability of Treg cells may offer realistic preventative and therapeutic strategies to protect against preeclampsia in at-risk women. Several pharmacological agents and cell therapy approaches to target Treg cells are in clinical trials or under development for auto-immune disorders and organ transplantation (37, 41, 42). We argue that as Treg therapies move closer to reality in other clinical settings, these interventions warrant evaluation for their potential utility in preeclampsia and related obstetric disorders with an immune etiology.
Treg Cells—Essential for Maternal Adaptation to Pregnancy
Several mechanisms of active immune tolerance arise in early pregnancy to dampen inflammation and suppress allo-reactive immune responses that otherwise threaten conceptus survival. These include attenuated expression of polymorphic MHC molecules on placental tissues, trophoblast production of anti-inflammatory and pro-tolerogenic cytokines and hormones, and epigenetic modulation of decidual cell chemokine expression to prevent effector T cells (Teff) cells accumulating at the maternal-placental interface (43–45).
Amongst the various mechanisms of maternal tolerance, CD4+ Treg cells are essential for embryo implantation and early placental development (46, 47). Their capacity to constrain and resolve the inflammation elicited during embryo implantation, and suppress generation of immune effector cells in local lymph nodes, is pivotal to controlling inflammation and promoting immune tolerance over the course of gestation, until an inflammatory shift emerges again at parturition (Figure 1). This function is consistent with essential roles for Treg cells in immune homeostasis throughout the body, where they prevent autoimmunity to self-antigens, suppress Teff cells reacting to non-dangerous foreign antigens, regulate and limit excessive inflammation (48–50), and have important roles in tissue repair and homeostasis (51).
Different subsets of T cells with regulatory functions exist. CD4+ Treg cells, CD8+ Treg cells, gamma/delta T cells, Tr1 cells, and NKT cells can all exert suppressive functions and appear to operate collaboratively to control immune responses. CD4+ Treg cells are of particular interest because of their strong association with preeclampsia, and their potential for therapeutic manipulation (41). CD4+ Treg cells comprise about 1–3% of total T helper cells in humans and 3–10% in mice, and are defined by their expression of the master transcription factor Forkhead Box P3 (FOXP3). As well as FOXP3, CD4+ Treg cells constitutively express surface molecules including the IL2 receptor α-chain (CD25), the immune checkpoint receptor cytotoxic T-lymphocyte protein 4 (CTLA4), and glucocorticoid-induced tumor necrosis factor receptor (GITR), and in humans are CD127− or CD127low (49, 52).
There are two types of CD4+ Treg cells (referred to hereon as “Treg cells”). Thymus-derived Treg cells (tTreg cells) emerge from the thymus after self antigen-driven selection as functional suppressor T cells. Peripheral Treg cells (pTreg cells) differentiate from naïve CD4+ precursors after contact with antigens in peripheral lymph nodes or tissues (52). Differentiation of naïve CD4+ T cells into pTreg cells requires cognate antigen to be presented by antigen-presenting cells (APCs) such as pro-tolerogenic dendritic cells (tDCs) in the presence of IL2 and TGFB. The CD4 cells are thereby induced to express FOXP3 and become committed to suppressive function (53). These cells then promote a cycle of de novo Treg cell generation and drive the development of long-lasting immunologic memory, which is reinforced by persistent antigen exposure (54). Like pTreg, tTregs can also be induced to proliferate and acquire greater suppressive function by antigen contact in the periphery (51, 55, 56). In humans, tTregs and pTregs are not readily distinguishable but in mice, tTregs express neuropilin 1 (Nrp1) while pTregs are generally Nrp1 low or negative (52).
pTreg cells and tTreg cells exert anti-inflammatory and immune suppressive activity by secreting a range of soluble factors including IL10 and TGFB, as well as through cell contact-dependent mechanisms. Importantly, Treg cell suppressive function inhibits proliferation and cytokine release from pro-inflammatory CD4+ Teff cells, T helper 1 (Th1) and T helper 17 (Th17) cells, which typically produce pro-inflammatory IFNG and IL17, respectively. Activated Treg cells interact with DCs through CTLA4, to cause down-regulation of DC co-stimulatory molecules CD80 and CD86, which drive Teff cell activation (49).
Altered Treg Cells Accompany and may Precede Preeclampsia Onset in Women
In women, T cells comprise 10–20% of decidual immune cells in the first trimester (57). Many decidual T cells are CD8+, including regulatory subsets (58, 59). Amongst the CD4+ T cells, around 10–30% express FOXP3, which is a substantial enrichment compared to peripheral blood (60–62). The Tregs comprise of both tTregs and pTregs and exhibit heterogeneous phenotypes that vary across the menstrual cycle and phase of pregnancy (32, 63, 64).
There is substantial evidence that many pregnant women with preeclampsia have fewer and less functionally competent Treg cells, accompanied by increased Teff cell activity, particularly Th1 and Th17 cells in decidual tissue and peripheral blood (26–28, 34, 65, 66). In a recent meta-analysis, a total of 17 independent primary studies were evaluated, and all but 2 showed consistent evidence of association between both severe, early-onset and late onset preeclampsia with fewer Treg cells in the third trimester (67). As well as reduced numbers, the suppressive function of Treg cells is often compromised in preeclampsia (33, 34, 68). The decrease in Treg cells may be proportional to the severity of disease (26), although relationship with time of disease onset and co-incidence of fetal growth restriction have not been consistently documented. There is evidence of an altered balance in Treg cell subsets in preeclampsia, with reports of fewer peripheral blood naïve HLADRneg CD45RA+ Treg cells (68, 69) and fewer CD45RA+CD31+ recent thymic emigrant Tregs (64) in peripheral blood. Decidual Treg populations may be differentially affected, given decidual tDCs exhibit a reduced capacity to induce pTreg in preeclampsia (32).
Treg cell changes become evident in peripheral blood and gestational tissues shortly after conception and accumulate in decidua reaching their highest levels in early to mid-gestation, before decreasing as term approaches (28, 61, 70). A recent study utilizing chorionic villous sampling (CVS) at week 10–12 of gestation, showed that women who progress to preeclampsia demonstrate dysregulated expression of decidual and immune cell genes from this early time (71). In another study, elevated expression of IL6 which counteracts Treg stability and promotes Th17 generation (72), as well as reduced numbers of alternatively activated M2 macrophage and T cell markers, were detected in CVS tissues of women who later develop preeclampsia associated with fetal growth restriction (IUGR) (73). Although longitudinal studies to track Treg cells over the course of gestation are not yet reported in women with preeclampsia, there is good evidence that low abundance of circulating Treg cells in the first trimester is predictive of miscarriage before 12 weeks (74). Collectively, these observations underpin a working hypothesis that disturbed immune adaptation in early pregnancy precedes impaired placental development, setting the scene for later emergence of preeclampsia and related complications of pregnancy (8, 10, 29, 75).
This fits an emerging paradigm which positions early pregnancy as the origin of disorders of deep placentation that underpin early onset, severe preeclampsia, and also contribute to IUGR, preterm labor, premature rupture of membranes, and late spontaneous abortion (11, 76, 77). So-called shallow placentation arises from insufficient trophoblast invasion and failure to adequately remodel spiral arteries and to achieve high capacity maternal blood flow, which further compromises placental development and function, and leads to IUGR (1, 7, 8).
Treg cells are emerging as key regulators in the decidual leukocyte network which controls implantation and placental development. Through interactive cross-regulation, growth factor secretion and extracellular matrix remodeling, this network controls the decidual immune environment which facilitates trophoblast invasion and cytotrophoblast shell development, and enables remodeling of the decidual vasculature to support placental development (10, 78).
Inappropriate function or insufficient numbers of Treg cells in the decidua are linked with inadequate extravillous trophoblast invasion, and poor spiral artery remodeling, in turn destabilizing placental development and resulting in “shallow” placentation (12, 79). There is also a clear link between Treg deficiency and both recurrent implantation failure and recurrent pregnancy loss, where more severe forms of impaired uterine receptivity arrest trophoblast invasion and early placental development (80, 81). Thus, it is not difficult to envisage how insufficient Treg cells in the preconception and peri-conception phase could be a key upstream trigger for the sequence of events leading to impaired vessel remodeling and shallow placentation, which ultimately cause the overt symptoms of preeclampsia in later gestation (Figure 2).
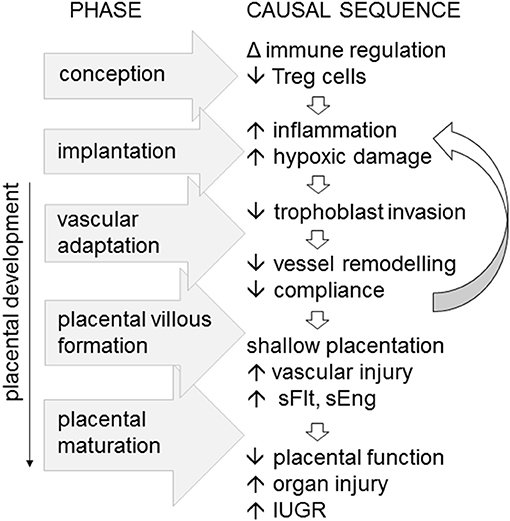
Figure 2. Immune imbalance associated with insufficient or incompetent Treg cells are implicated as an upstream cause of preeclampsia, particularly in severe, early onset disease. In a working model of the sequence of pathophysiological events, fewer Treg cells generated at conception impact the decidual environment at implantation, to limit trophoblast invasion, constrain maternal vascular adaptation, and vessel compliance. The resulting “shallow” placentation causes vascular inflammatory injury accompanied by elevated soluble Fms-like tyrosine kinase (sFlt) and soluble endoglin (eEng), reducing placental function, and causing maternal organ injury and in utero growth restriction (IUGR) of the fetus.
Treg cell Regulation of the Decidual Immune Environment
Mouse models have been instrumental for defining mechanisms through which Treg cells exert anti-inflammatory activity to influence the decidual environment and early placental development. Kinetic studies show that Treg cells accumulate in the uterine decidua from very early in pregnancy, and that these originate after naïve T cell activation and proliferation in local lymph nodes, causing numbers to expand through the first half of gestation (20, 46). After recruitment into the implantation site, Treg cells comprise around 30% of decidual T cells in the mouse (46).
Extensive experiments wherein Treg cells are selectively depleted, or overwhelmed by exacerbated Teff cell responses, show an essential role for Treg cells in preventing generation of destructive immunity to fetal alloantigens (82–85). Without sufficient Treg cells, an aggressive Th1 and Th17 mediated-response causes fetal loss in allogeneic but not syngeneic pregnancy (46).
Depending on the severity and timing of manipulation, Treg depletion can manifest as implantation failure, miscarriage or fetal growth restriction. Several studies show the pre- and peri-implantation phase is highly vulnerable. Administration of anti-CD25 Ab before or shortly after mating causes complete implantation failure (86–88). Depleting FOXP3+ cells from FOXP3-Dtr mice during early placentation increases later fetal resorption (85, 89), but depletion in mid-gestation only moderately reduces fetal viability (20), unless mice receive a second hit inflammatory challenge such that Treg depletion exacerbates the adverse impact (90–92). Mice deficient in T cells due to Rag1-null mutation are highly vulnerable to inflammation-induced fetal loss, but this is reversed by administering CD4+ T cells that differentiate to Tregs after transfer (90). Midgestation depletion of CD25+ cells using anti-CD25 mAb has a less severe impact than in early pregnancy, but this may be because Teff cells are also removed (86, 93). Additionally, other tolerogenic mechanisms including IL10 secretion by uNK cells (94) may compensate for Treg deficiency once placental development is complete.
Mouse models with a high rate of spontaneous fetal loss also demonstrate a critical role for Tregs in embryo implantation. CBA/J females mated with DBA/2J males have fewer decidual Tregs and elevated Th1 cells (87, 95). Adoptive transfer of Tregs from donor CBA/J females mated with Balb/c males boosts decidual Tregs and corrects fetal loss (87), but only if Treg transfer occurs before embryo implantation (87). These findings confirm that Tregs are most essential in the uterus during the peri-implantation period, consistent with a central role in orchestrating the transition to an anti-inflammatory mileu required for placental development (Figure 1).
Treg cells co-localize in clusters with uNK cells and other leukocytes in the human decidua basalis (78), where they exhibit activity expected to potently influence the local immune environment by enforcing an anti-inflammatory phenotype in other leukocyte lineages. In particular Tregs regulate uNK phenotype, through releasing TGFB and IL10 to control DC release of uNK viability factor IL15 (96), and suppress uNK cytolytic activity (91, 97). This may be particularly important in first pregnancy, given that uNK cells acquire memory and assume a more differentiated “trained” phenotype in subsequent pregnancies (21). Whether there is an interaction between antigen-experienced Tregs and trained uNK cells, remains to be investigated.
Treg cells also regulate M2 macrophages (98), mast cells (99), and tDCs, releasing heme oxygenase-1 which maintains immature DCs (100) and promotes indoleamine 2,3-dioxygenase (IDO) production to impair Th1 cell survival (101, 102). M2 macrophages and tDCs promote further Treg generation (98, 100) and produce an array of cytokines that reinforce a pro-tolerogenic decidual environment, including TGFB, CSF2 (GMCSF), IL4, IL10, CSF3 (GCSF), and prostaglandin E (103). Decidual Tregs also express other hallmark mediators of Teff suppression CD25, CTLA4, and PD-L1 (61, 91, 104–106).
Uterine NK cells and DCs are implicated as key regulators of decidual transformation (107–109) so through regulating uterine DC and uNK phenotype, Tregs would indirectly influence the extent and quality of the decidual response. Furthermore, trophoblasts engage with Tregs in a reciprocal interaction to modulate the secretory profile of both lineages (110). Together, these coordinated interactions allow Tregs to constrain inflammation and limit oxidative stress caused by trophoblast invasion during early placental development (13, 25, 111).
Treg cells Influence Maternal Vascular Remodeling and Early Placental Development
Treg cells are emerging as critical participants in the process of maternal vascular remodeling, through their modulating effects on the decidual leukocyte network (Figure 3). There is extensive evidence to demonstrate key roles for uNK cells (30, 112), macrophages (31), and mast cells (99) in decidual vessel transformation, and in collaborating with invading trophoblasts to restructure the endothelial surface and smooth muscle wall (7, 13, 104). As detailed above, Treg cells exert potent anti-inflammatory actions on uNK cells (91, 97), M2 macrophages (98), mast cells (99), and tDCs (100), thereby influencing the vascular remodeling process.
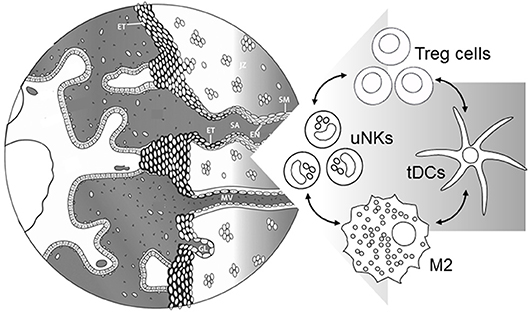
Figure 3. Events during early placental development require immune cells in the decidua to support maternal blood vessel (MV) adaptation and spiral artery (SA) transformation. Treg cells interact with uNK cells, M2 macrophages, and tDCs to suppress inflammation and provide secretory factors regulating extravillous trophoblast (ET) invasion, and functional changes in endothelial cells (EN) and surrounding smooth muscle (SM). Severe early-onset preeclampsia is accompanied by insufficient Treg cells, which in turn would be expected to contribute directly and indirectly through immune cell networks to impair vascular adaptations required for robust placental development. Left hand part of Figure is adapted from Steegers et al. (1).
This is unsurprising given growing evidence that Treg cells play important roles in modulating cardiovascular function, and vascular homeostasis throughout the body (113). In hypertensive mouse models, Treg cell infusion reduces blood pressure and vascular damage, and reverses hypertensive sequelae (114, 115). Treg cell-derived cytokines, particularly IL10 and TGFB, suppress inflammatory endothelial cell activation and inhibit development of atherosclerosis (113).
Rodent models of preeclampsia support a critical function for Treg cells in the pathophysiological events underlying abnormal placental development, through coordinated interactions with uNK cells, DCs, and macrophages (Figure 3). Experiments in mice deficient in T cells and/or NK cells show that T cells interact with uNK cells to influence the maternal hemodynamic response to pregnancy (116, 117). When cause-and-effect relationships are explored by antibody-mediated or genetic modulation of T cell subsets, Treg cells are implicated as having causal roles in the maternal and fetal symptoms of preeclampsia models.
Treg-deficient mice consistently show impaired uterine spiral arterial modification, reduced placental blood flow, and fetal growth restriction (85, 89, 118). Depletion of FOXP3+ Tregs in early pregnancy causes later dysfunction in uterine arteries accompanied by increased endothelin-1 production (89). Peripheral Treg cells are particularly implicated, as indicated by experiments in mice with a null mutation in the CNS1 gene which is a FOXP3 enhancer element essential for pTreg cell but dispensable for tTreg generation. CNS1 is only present in eutherian mammals, suggesting its introduction into the FOXP3 locus to enable pTreg generation, in turn facilitated evolution of placentation (85). In CNS1-null mice, impaired remodeling of material spiral arteries underpins defective placental development (85). Compromised trophoblast invasion and failed transformation of spiral arteries is also seen in mice where neutrophil depletion causes insufficiency of pro-angiogenic, neutrophil-induced Treg cells (118).
In the reduced uterine perfusion pressure (RUPP) model of preeclampsia in rats, reduced uterine artery flow is induced by clip placement on the abdominal aorta and right and left uterine artery arcades at day 14 of gestation, resulting in placental ischemia and oxidative stress. The model replicates human preeclampsia symptoms with hypertension accompanied by increased circulating VEGF, sEng, Flt1, and placental growth factor (PlGF), plus elevated inflammatory cytokines and IUGR. A substantial (~50%) reduction in decidual and placental Treg cells, and elevation in total CD4+ T cells and Th17 cells, is a consequence of the RUPP intervention. Remarkably, the preeclampsia symptoms induced in this model are T cell dependent since the RUPP intervention does not cause hypertension and IUGR in T cell deficient athymic rats, and disease can be induced by passive transfer of Th17 effector CD4+ T cells (119). Treg cell deficiency is a key driver of hypertension and IUGR and these symptoms are mitigated when Treg cells from pregnant control donor rats are administered shortly after the RUPP procedure (120). Treatment strategies applied to boost endogenous Treg cells, including IL10 administration (121) or low dose CD28 superagonist (120), also reduce hypertension and IUGR.
Rodent models show that the protective effects of Treg cells are crucial from the early implantation phase, when vascular adaptation and early placental development begin. Several studies using different approaches to deplete Treg cells at various time points show the peri-implantation phase is most severely affected, with extensive Treg cell depletion at embryo implantation causing complete implantation failure (46, 86, 87). Experiments in the abortion-prone CBA/J x DBA/2 mouse model indicate transferred Treg cells can rescue the underlying placental defect, but only if Treg cells are transferred from healthy pregnant mice at or before the time of embryo implantation (87). Treg cell replacement influences other immune cells in the decidua, including mast cells, to repair placental and vascular defects and prevent sFlt elevation and fetal loss (99). Consistent with a critical role for peri-implantation Treg cells, a genetic model of preeclampsia involving overexpression of human angiotensinogen and renin in rats showed greater responsiveness to Treg cell therapy when it was applied in early gestation (122).
Even subtle disturbances to the T cell response in early pregnancy may impact later pregnancy progression. This may be the consequence of an altered environment during T cell activation, as we have recently demonstrated for CD8+ T cells in pregnancy (123). Other studies in mice show that reduced numbers or altered function of Treg cells at conception can disrupt fetal-placental development without immediate adverse effects, but with a legacy that becomes apparent in mid- or late gestation, particularly when a “second-hit” inflammatory challenge is applied (106, 124).
The Treg Cell Response is Determined in the Pre- and Peri-conception Phase
The conditions under which the Treg cell populations of pregnancy originate are likely to be critical to the reduced quantity and impaired quality of the Treg response in preeclampsia. Uterine recruitment of Tregs in readiness for possible embryo implantation commences in the proliferative phase of each cycle, with an estrogen-driven increase peaking around ovulation (125). CD4+ FOXP3+ cells, thought to be pTregs based on expression of the Helios marker, are a major subset amongst the expanding Treg populations in blood and decidua in early human pregnancy (32). Helios+ Treg cells appear to be preferentially recruited into the decidua in the first trimester (63). Amongst peripheral blood tTregs the population of CD45RA+CD31+ cells, which have recently emigrated from the thymus, expand prominently in the first trimester and differentiate into CD45RA−CD31− memory Tregs (64).
The majority of decidual T cells in women have a memory phenotype (CD45RA− or CD45RO+) (59, 126) and show evidence of fetal antigen specificity (62), which indicates antigen exposure must occur to elicit the full Treg cell response. HLA-C is the only polymorphic HLA expressed by human placental trophoblasts, and fetal-maternal HLA-C mismatch is associated with a greater expansion in decidual Tregs (127). Many decidual Tregs show fetal HLA-C antigen specificity (62, 128), but whether other reproductive or tissue antigens are involved has not been investigated.
In preeclampsia, Treg cell deficiency is most pronounced in pTreg cells (32), as well as CD45RA+CD31+ recent thymic emigrant tTregs less able to acquire a memory phenotype (64). This implies there may be an underlying problem with antigen priming. Consistent with this, dysfunctional DCs with reduced HLA-G and ILT4 (32), and/or insufficent PD-L1 (129), have been reported in preeclampsia.
Contact with fetal alloantigens must occur under conditions that favor antigen presentation and stable Treg cell (not Teff cell) development. These conditions occur in two waves in the reproductive process. Paternally-derived transplantation antigens shared by the fetus are first and most frequently contacted during transmission of seminal fluid at coitus, at conception and in pre-conception cycles (22). Seminal fluid primes the activation of pTregs that are specific for paternal transplantation antigens which will later be expressed by fetal and placental cells. Additionally, once pregnancy is established and maternal blood comes into contact with the syncytiotrophoblast surface, placental exosomes are released into maternal blood, providing a second wave of alloantigen exposure (130, 131).
Again, mouse models have been informative in tracing the origins and regulation of Treg cells and point to specific events as critical for generation of the Treg cell pool in early pregnancy (132). The two stages of T cell activation can be tracked through the first half of gestation using T cell transgenic mice (93). The strength of seminal fluid as the initial priming event is first seen as a burst of T cell proliferation in the peri-conception phase, evident in cells recovered from the uterus-draining para-aortic lymph nodes (dLN) on day 3.5 post-coitus (pc), followed by a steady progressive increase during the post-implantation phase once placental morphogenesis is complete (93).
The first wave of proliferation of Treg cells can be detected within days of insemination in the lymph nodes draining the reproductive tract, in the peripheral blood, and spleen (46, 133). Seminal fluid contains paternal alloantigens and high levels of TGFB, and elicits an inflammation-like response in female reproductive tract tissues. DCs and macrophages recruited into female tissues take up seminal fluid alloantigens, traffic to the dLN and present antigen to naive T cells (93). Treg expansion is maximized in allogeneic compared with syngeneic matings, demonstrating a contribution of male alloantigens (134), but endogenous antigens might also contribute to the activation and expansion of Tregs in early pregnancy (135). Amongst the responding pTregs, paternal antigen-reactive pTreg cells are selectively enriched (133, 136). Data from mice with a mutation in the CNS1 gene show elevated fetal loss when pTreg alone are deficient, suggesting that pTreg cells have non-redundant functions important for viviparous pregnancy (85).
A population of tTreg cells of thymic origin also expand systemically prior to conception. These cells are recruited into the uterus after proliferation in the dLN during the estrous stage of the reproductive cycle in mice, in response to rising estrogen at ovulation (125, 137). After mating, factors in seminal fluid induce tTreg to proliferate and express elevated FOXP3 and CTLA4, both markers of suppressive competence, accompanied by demethylation of the Treg-specific demethylation region (TSDR) in the FOXP3 locus (138). This expansion of tTreg cells occurs in parallel with the seminal fluid antigen-driven expansion of pTreg cells. This population may well have different functional qualities to pTreg, although these are still to be defined.
After recirculation via peripheral blood, Treg cells are recruited into the fetal-maternal interface in response to chemokines secreted by uterine epithelial cells including CCL19 (133), and may be stimulated to undergo further rounds of proliferation locally in the uterine tissue (46). The resulting expansion of the Treg cell pool induces a state of hypo-responsiveness to paternal alloantigens, concurrent with embryo implantation when the conceptus first contacts maternal tissues (136, 139). The kinetics of Treg cell induction in the peri-conception phase ensures sufficient abundance of Treg cells in the endometrium at embryo implantation, when their function is most critical (86, 87, 140). Continued release of paternally-inherited alloantigen from trophoblasts over the course of pregnancy sustains the T cell response until post-partum (20, 93). After birth, a population of paternal alloantigen-reactive Tregs are sustained, and in the event of a subsequent mating with a male expressing the same alloantigens, there is accelerated expansion of Treg cells driven by proliferation of fetal-specific Treg cells retained from the prior pregnancy (20).
Mouse studies imply that the immune response initiated at seminal fluid priming is a crucial initiating step and highly vulnerable phase for Treg cell tolerance to be established. In particular, responding pTreg cells require appropriate environmental signals including the cytokines IL2 and TGFB, to ensure naïve T cells differentiate into Treg cells and not Th1 or Th17 effector T cells (93, 133). Both the size of the Treg cell pool and the suppressive competence of pTreg cells will be determined by the strength of the antigenic challenge, and the nature of the cytokine context in which antigen contact occurs—parameters which are determined by seminal fluid composition as well as female tract factors. Since newly generated pTreg cells that have only recently commenced FOXP3 expression appear more vulnerable to phenotype switching and lineage instability (141), the extent to which pTreg cells primed at coitus will commit to a secure Treg fate will be substantially influenced by the conception environment. Relevant factors impacting this environment would include MHC disparity between male and female partners, the abundance and phenotype of DCs involved in antigen presentation, and bioavailability of local cytokines, hormones and other positive and negative regulators including microRNAs and the local microbiome (23).
Similar events occur in women, where the cervical immune response to seminal fluid mirrors the mouse response, causing elevated cytokine production, recruitment of leukocytes and T cell activation (22, 142), consistent with prior seminal fluid contact contributing to priming the paternal antigen-specific Treg cell response of pregnancy (62). It is yet to be proven that seminal fluid induces pTreg cells in women, and other factors must contribute to uterine Treg accumulation since IVF pregnancy can be established without seminal plasma contact. Expansion of uterine Tregs after conception may be further facilitated by human chorionic gonadotropin (hCG) secreted by invading placental trophoblasts (143). This builds on the hormone-driven expansion of Treg cells in the follicular phase of the menstrual cycle, correlating with progressively elevating serum E2 levels (125). However, the in vivo cervical response and an array of in vitro studies demonstrating that seminal fluid skews DC cells to an tDC phenotype and induces Treg cells in vitro, is consistent with a key role for seminal fluid in women (22, 144, 145). A priming effect of seminal fluid contact in women also explains the benefit of cumulative seminal fluid contact with the conceiving partner in protecting from preeclampsia (17, 146).
Is Insufficient Priming a Cause of Treg Cell Deficiency in Preeclampsia?
An important question is why some women have fewer Treg cells and/or impaired Treg function at the outset of pregnancy. The nature and significance of factors that cause variation in the uterine Treg cell response are unclear and require investigation. As detailed above, antigen priming in the appropriate environmental context is a critical factor in the strength and quality of any peripheral tissue Treg cell response. In the reproductive tissues, the strength and quality of antigen and immune-regulatory signals in the female reproductive mucosa during priming would be paramount, as well as the number and timing of prior exposures to the conceiving partner's seminal fluid and any previous pregnancies with that partner (25).
This raises the possibility that some women develop preeclampsia after conceiving without adequate prior priming to male partner alloantigens. It seems likely that pTregs reacting with paternal alloantigens would be more vulnerable than tTregs to variations in population size, antigen experience and memory, functional competence, and stability. Because newly generated pTregs are particularly susceptible to phenotype-switching and lineage instability (141), the priming environment would be a key determinant of a secure fate amongst pTreg with male partner alloantigen specificity. Recent evidence in mice that seminal fluid contact regulates tTreg cells, inducing proliferation and reinforcing a suppressive phenotype through epigenetic modulation, suggests tTreg as well as pTreg are impacted (138).
Priming may be dysregulated due to seminal fluid composition or female responsiveness to seminal fluid signals (22, 25). It has been shown that recurrent miscarriage patients produce more CD4+IL17+ and CD4+IFNG+ cells and fewer CD4+CD25+FOXP3+ Tregs, compared to fertile controls, when CD4+ T cells are cultured with DCs and partner's seminal fluid antigens (147). The balance of immune-regulatory agents in seminal fluid, particularly pro-tolerogenic TGFB, varies between men, and within men over time (148). The anti-tolerogenic cytokine IFNG, which drives generation of Th1 immunity, fluctuates substantially and can become elevated in seminal fluid in the event of infection or other inflammatory conditions (149). IFNG interferes with synthesis of CSF2 required to drive the T cell proliferative response at conception (150, 151), skews Th0 differentiation toward Th17 cells (48, 152), and increases Treg susceptibility to transdifferentiate into Th17 cells (153).
Clinical and Lifestyle Factors Impacting the Treg Cell Response
A suboptimal Treg cell adaptation for pregnancy could also occur in women due to intrinsic Treg deficiency. The specific factors determining between individual variation in Treg numbers and functional capacity are yet to be fully defined. An interaction between genetic, epigenetic, and environmental factors seems likely, based on data from animal models and limited studies in population cohorts (154). The thymic output of tTreg, and peripheral tissue induction of pTreg cells, are independently regulated and can be affected by a range of metabolic and nutritional parameters, inflammatory exposures, autoimmune conditions, and age (35–40). Common health conditions that affect the immune system including intestinal microbial dysbiosis and dietary deficiencies, particularly vitamin A and vitamin D, have been associated with poor adaptive immunity and may be a common cause of compromised Treg activity (38). Exposure to sunlight (55) and exercise (56) are also recently identified to support Treg homeostasis, but how these are linked to variation in human Treg parameters are yet to be defined.
In hyper-inflammatory conditions caused by autoimmune, infectious or metabolic disorders some pTreg cells exhibit phenotypic plasticity and instability, with increased disposition to shift phenotype, or lose FOXP3 expression and become reprogrammed into a Teff fate (154). Studies in mice and humans demonstrate that FOXP3+ T cells can be induced by inflammatory stimuli to express IL17 and IFNG characteristic of Teff cells (155, 156), and may then transdifferentiate into effector Th17-like cells, known as “exTregs,” which can amplify inflammatory pathology (157).
Epigenetic regulation of FOXP3 through demethylation of the TSDR region is a key factor in the resilience of Tregs to inflammatory stress, that controls whether T cells can express sufficient FOXP3 to overrule Teff functions and maintain a Treg suppressive phenotype (158). Increased TSDR methylation and associated underexpression of FOXP3 is a feature of some autoimmune conditions (159).
There is little information on whether Treg cells exhibit signaling defects, lineage instability or methylation changes in preeclampsia. Given the evidence of elevated Th1 and Th17 cells counteracting the decrease in Treg number and function in preeclampsia, defects in both Treg cell induction and/or stability seem plausible, and would explain the concurrent reduction in Treg cell suppressive function (34, 68). Although large populations have not been examined, exploratory studies in preeclampsia suggest an elevated incidence of gene variants within the promoter region of FOXP3 that may affect expression levels and hence Treg stability (160, 161). Elevated IL6 trans-signaling, which is known to promote Treg instability and transdifferentiation, has been described in women with recurrent miscarriage (162). IL6 is associated with reduced TGFB output and IL2-mediated STAT5 signaling (163), and is a possible candidate contributing to impaired Treg capacity in preeclampsia, given that elevated expression of IL6 is seen in gestational tissues of women who later develop the condition (73).
Potential Interventions to Target Treg Cells in Pregnancy
Recognition that excessive inflammation secondary to insufficient anti-inflammatory protection is a key driver of preeclampsia gives rise to the prospect of targeting the immune response to prevent or suppress progression of the disease. In particular, Treg cells provide an attractive target, because of (1) the clear link between compromised Treg cells and preeclampsia; (2) a logical mechanistic pathway placing insufficient Treg cells as an upstream event in the placental and systemic pathophysiological sequalae; (3) compelling evidence from preclinical rodent models showing that insufficient Treg cells can elicit preeclampsia-like symptoms, while boosting Treg cells mitigates symptoms, and (4) encouraging progress in development of Treg cell therapies for other autoimmune and inflammatory conditions.
Interventions to boost Treg cell populations and their suppressive competence are under development and show promise in autoimmunity and tissue transplantation (41, 42), and more recently have been considered for cardiovascular disease (113). Treg cell therapies relevant to preeclampsia could take one of three alternative approaches: (1) lifestyle and health advice during preconception planning to assist immune adaptation to pregnancy; (2) neutraceutical, pharmacological, or other strategies to increase Treg cell numbers and/or function in an antigen non-specific, systemic manner, or (3) cell therapy treatments that involve ex vivo generation and/or expanding Treg cells in a highly-individualized process. These clearly represent different degrees of technical challenge, invasiveness, cost and risk. While lifestyle adjustments or dietary supplements are generally safe and tractable, cell therapies are labor-intensive, expensive, and higher risk.
The evidence base for approaches to target Tregs in other clinical settings is building (41, 42), but to date little consideration has been given to applications in reproductive conditions. To advance new treatments targeting Treg cells for preeclampsia prevention and mitigation, research on several fronts is required. Most immediate goals should be to develop appropriate diagnostics, and to investigate and validate pre-pregnancy planning interventions to boost Treg cells. There should also be careful consideration of the rationale for initiating clinical studies, using robust clinical trial methodology, to evaluate pharmacological and/or cell therapy treatments for application when Treg deficiencies are not responsive to lower intervention approaches.
Diagnosis of Treg Cell Deficiency
To progress understanding of Treg cell insufficiency in preeclampsia, and to develop therapeutic options targeting these, it is essential that effective diagnostic tools are developed and validated. These should detect common and informative defects in Treg cell parameters that define competency for healthy pregnancy, and ideally be applied to peripheral blood if preference to endometrial biopsies. Treg tests should be appropriate for routine use during pregnancy planning or early after conception, to provide a therapeutic window for early treatment interventions to prevent progression to miscarriage or later obstetric conditions. To date little work has been done to investigate Treg cell deficiency before conception, or in early pregnancy, in the blood or endometrium of women who go on to develop preeclampsia. Ongoing studies to address the pre-pregnancy origins of preeclampsia may begin to address this (9).
A recent meta-analysis of Treg cell parameters quantified in preeclampsia highlights the considerable variability in markers that different groups have measured to date (67). A useful step will be to develop a consensus definition of minimum essential Treg markers to facilitate harmonization across future studies, and to determine the best stage in preconception cycles or early pregnancy for analysis (164). Given the significance of tTreg cells vs. pTreg cells, and of naïve vs. memory cells in preeclampsia (68, 69), extensive marker panels to discriminate these subsets in flow cytometry-based tests will be most informative. Along with standard markers CD4, CD25, CD127, and FOXP3, markers that reflect memory, suppressive capacity, and activation status amongst Treg cells should be measured. These may include GITR which is emerging as a superior marker of active, functional Tregs (165), plus CTLA4, CD45RO, HLADR, and potentially intracellular cytokines or transcription factors which appear particularly informative in the preeclampsia setting (27, 34).
Ideally, Treg cell assays should also inform on suppressive competence in a paternal antigen-specific manner. Assessment of suppressive function by in vitro-based assays and/or analysis of FOXP3 methylation status has been the gold standard for assessment of suppressive potential, but new markers such GITR, CD154, and PI16 may supersede these tests and be more amenable to a clinical diagnostic setting (165–167). With increasing availability of tetramer-based diagnostic tools for identifying TCR specificity, identification of partner alloantigen-reactive Treg cells may in time become feasible.
Optimal Timing for Interventions
A major challenge for developing strategies to target Treg cells in pregnancy is timing—how and when would Treg cell deficiency be diagnosed, and how would this relate to the window of opportunity for intervention? With evidence that Treg cells are most critical during the implantation and early placentation phase of pregnancy, interventions in cycles prior to conception, or as soon as possible after conception would likely be most advantageous. The timing would need to align with hormone-driven regulation across the menstrual cycle, and might leverage events controlling estrogen and progesterone-regulated expansion of the Treg pool (125). Interventions would need to be coupled with Treg screening of high risk women during pre-pregnancy planning or early after conception to allow the best chance to identify patient subgroups that could be amenable to therapy.
Treg Cells and Preconception Care
A tractable approach worthy of further investigation is advice on immune system health and boosting immune priming during preconception planning. In nulliparous women, the available evidence suggests on average, 3–6 months of sexual cohabitation without using barrier contraceptives is required for sufficient seminal fluid priming to minimize the chance of preeclampsia (15). Consistent with this, in a recent study of 340 women, women in the highest 10th percentile of exposure to partner's seminal fluid had a 70% reduced odds of preeclampsia relative to women in the lowest 25th percentile (168). Thus, advising nulliparous women to avoid use of barrier contraceptive methods and to consider increasing vaginal coitus prior to conceiving may reduce preeclampsia risk. A key question is the impact of different non-barrier approaches to facilitating immune priming, such as oral contraceptive pill or intrauterine device, which both deliver immune-modulating hormones. Further studies are required to evaluate the impact on preeclampsia rates of preconception advice on seminal fluid contact and contraceptive choice, and to investigate whether a partner-specific Treg cell response is involved.
Detecting and correcting any immune imbalance due to clinical, nutritional and/or, lifestyle factors is also likely to be effective for pregnancy planning and reducing susceptibility to preeclampsia. Elevated inflammatory load due to chronic infection, smoking, diabetic and pre-diabetic conditions, obesity and/or microbiome dysbiosis in women would be expected to adversely affect intrinsic Treg cell parameters and responsiveness to priming (35, 169), while in men these conditions may increase seminal fluid IFNG and reduce capacity to elicit a healthy female response (149). A range of autoimmune conditions known to impact reproductive function likely have a shared underlying etiology and correcting the immune disorder with validated approaches would reasonably yield dividends for pregnancy health (170). Microbiome disorders, and vitamin and micronutrient deficiencies also affect Treg cells, and treating these might have utility in boosting Treg cell activity in the reproductive setting, as has been shown for some other immune disorders (38). It will be important for future studies to trial the efficacy of alternative approaches to pre-pregnancy care, in order to determine the most effective interventions.
Pharmaceutical Interventions to Expand Treg Cells for Pregnancy
High-risk women with a previous pre-eclamptic pregnancy are an obvious target for preconception care to boost immune tolerance. However, duration of sexual cohabitation is unlikely to be limiting in this patient group, and couple-intrinsic issues such as insufficient HLA disparity between partners, or HLA incompatibility resulting in low immunogenicity of male alloantigens, could theoretically interfere with priming and expansion of the Treg cell pool. In selected women with a demonstrated intrinsic Treg deficiency, approaches that target Treg cells might warrant consideration.
Agents of potential utility to induce Treg cell-mediated tolerance in women include cytokines and other biological agents. Two cytokines that been used clinically to attempt to enhance embryo implantation and placentation, CSF3 (171) and CSF2 (172), act on myeloid immune cells and promote recruitment and function of tDCs in the reproductive tract mucosa. Mouse studies are consistent with their fertility-promoting effects being mediated via tDC-mediated induction of Treg cells (151, 173), but their effect on T cells has not been studied in women. IL10 and several microRNAs that act to expand the Treg cell pool and increase functional competence, and are known to be induced naturally in the female tract response to seminal fluid, are also worthy of investigation (106, 174).
Several existing drugs deployed in pregnancy may act at least partly through Treg cells. Studies in mice suggest that progesterone mediates suppression of the Teff cell response, affecting CD4+ T cell and Treg cell phenotype (175, 176). Progesterone effectively suppresses the generation of Th1 cells and Th17 cells and induces Treg cell differentiation (177–179). Treg cells induced by progesterone have increased capacity to suppress the activation and expansion of Teff cells (177, 178). This fits with evidence of progesterone-regulated increases in uterine Treg cell populations in mice and in women (125, 137). Physiological levels of progesterone increase the functional population of CD4+FOXP3+ cells in pseudopregnant mice and increase the splenic CD4+FOXP3+ cell proportions in mid gestation (180). Progesterone also acts to selectively repress IFNG gene expression in CD4+ T cells (181), allowing enhanced induction of Treg cells and suppression of Th1 and Th17 differentiation (84, 182). A Cochrane meta-analysis demonstrated a benefit of progesterone for reducing recurrent miscarriage in women (183), but whether this impacts Treg cells is unknown. Furthermore, the outcomes in this setting are confounded by the large proportion of losses related to embryo chromosomal abnormalities, rather than immune dysregulation in the endometrium (184). There is no proven clinical benefit of progesterone in preeclampsia, and a Cochrane review did not find sufficient evidence to support its clinical use to prevent preeclampsia when administration was commenced between 16 and 28 weeks gestation in 4 clinical trials (185). Administration in early pregnancy would likely be required to improve Treg cells at the relevant developmental phase, but the effect of early administration of progesterone on susceptibility to preeclampsia has not been assessed.
Although immune suppressive glucocorticoid drugs conventionally used in autoimmune conditions are sometimes administered in assisted reproduction settings, these suppress both Treg cells and Teff cells, and carry risks when used in pregnancy (186). Intravenous immunoglobulins (IVIg) and Intralipid have also been empirically used in artificial reproductive technology settings to enhance implantation and in recurrent miscarriage clinics to reduce miscarriage rates (187). IVIg did not demonstrate an improvement in livebirth outcomes in 8 small studies in 303 women who suffered recurrent miscarriage (188). Although there is some evidence to suggest that intralipid infusions are associated with immune suppression and alter NK cell activity (189), their effects on Treg cell parameters has not been measured and their clinical benefit for implantation disorders or miscarriage is unproven in clinical trials. The impact of administering intralipid and IVIg infusion in early pregnancy on the development of early or late onset preeclampsia has not been assessed.
There are several drugs under development for autoimmune diseases, including immune checkpoint regulators and other immune-active biologics, that may afford greater selectivity for Treg cells than the immune modulating treatments described above (42). These approaches may be worthy of cautious evaluation in reproductive conditions. Drugs targeting immune checkpoint regulators CTLA4 and PD-1 offer enormous potential, and studies in preclinical models offer encouragement. A recent study in rats where preeclampsia-like symptoms are induced by L-NAME administration showed treatment with PD-L1-Fc protein was effective in reversing Treg/Th17 imbalance and mitigating placental damage (129). Substantial promise for a CD28 superagonist treatment was demonstrated in a rat model of preeclampsia induced by overexpression of human angiotensinogen. Administration of CD28 superagonist was highly effective in increasing Treg cells and alleviating maternal hypertension, proteinuria and IUGR, particularly when treatment was applied from the pre-implantation phase (122). Low dose IL2 has been used to expand Tregs in several conditions, including in abortion-prone mice where protection against fetal loss was achieved (135).
Other relevant agents include humanized antibodies against T cell markers such as anti-CD3, anti-CD52, and anti-CD45 RO/RA which reestablish immune tolerance by selectively depleting Teff cells and retaining Treg cells (37). Other approaches utilize cytokine specific monoclonal antibodies to promote Treg cells—these include anti-TNFA which is approved for use in rheumatoid arthritis and Crohn's Disease, or protolerogenic cytokines such as TGFB and IL10 (37).
Epigenetic regulation of FOXP3 to impart elevated suppressive function and stability in Tregs is another candidate approach. Inhibitors of DNA methyltransferases such as 5-aza-2′-deoxycytidine (Aza), or factors involved in DNA methylation such as Ten-eleven translocation (TET) protein, have been utilized in vitro to drive hypomethylation of the FOXP3 locus, resulting in strong, stable FOXP3 expression in Treg cells (190–192). In mouse models, administration of DNMT inhibitors enhances Treg number, FOXP3 expression and suppressive capacity which assists in reducing inflammation associated with LPS-induced lung injury (193), and prolongs cardiac allograft survival (194). Histone deacetylase (HDACs) inhibitors have also been shown to boost Treg cells and improve suppressive function, resulting in decreased inflammatory bowel disease and increase tissue graft survival in mice (195). These agents carry risks as well as potential, so any application in a reproductive setting would need to be carefully evaluated, initially in preclinical studies.
Cell Therapy Interventions to Boost Treg Cells for Pregnancy
Cell therapy provides a challenging but highly personalized and thus potentially more effective approach to tackling Treg-mediated conditions (37). Cell therapy involves either (i) isolating in vivo differentiated Treg cells and expanding them ex vivo or (ii) generating and expanding pTreg cells in vitro, before subsequent reinfusion. These approaches are in development for transplantation and severe autoimmune disease, but would currently be difficult to justify for a non-life-threatening pregnancy condition. However, given that in preeclampsia the Teff response is not overwhelming, once Treg cell therapy becomes a reality it may prove to be more amenable than other conditions where an extreme immune deviation is beyond rescue (37, 41).
A substantial benefit of cell therapy is that antigen-specific Treg cells can be manipulated without systemic effects on the immune response, with lower risk of off target effects in the mother and fetus than with pharmacological approaches. Studies in type 1 diabetes and other diseases show that T cell receptor (TCR) reactivity with relevant antigens in the target tissue improve Treg cell recruitment and capacity to persist and execute effective suppression, with a low chance of non-specific immune suppression (41, 196). This is a challenge for many disease conditions that might be considered for Treg cell therapy, when Treg cells reactive to tissue-specific antigens are rare. However, the relevant antigens in pregnancy are paternal alloantigens where the starting frequency is much higher. A large proportion of naturally-occurring naïve CD4+ T cells, pTreg cells and tTreg cells react with allo-antigens and could readily be expanded amongst polyclonal populations. Furthermore, because of their capacity to suppress immune responses in an antigen non-specific manner (bystander suppression) and their capacity to skew T cell responses to other tissue antigens toward tolerance (infectious tolerance), it is possible to regulate the immune response in a whole organ using Treg cells reactive with a single antigen (37, 41). In pregnancy, this means that Treg cells reactive with just one or a subset of paternally-inherited fetal alloantigens, or perhaps even a male minor histocompatibility antigen such as H-Y, could reasonably be effective in suppressing immune responses to a wide range of placental and fetal antigens.
Enormous potential is offered by new gene modification developments in generating alloantigen-specific Treg cells using chimeric antigen receptor technology. This approach overcomes the challenge of the low frequency of antigen-reactive T cells occurring naturally in peripheral blood, by genetically manipulating Treg cells with self-specificity to express either a TCR complex, or a chimeric antigen receptor (CAR) reactive to specific antigens. Use of CAR technology can reliably generate potent, functionally competent, and stable alloantigen-specific human Treg cells that have utility in a wide range of human autoimmune diseases (197). Ongoing clinical trials are showing exciting results in Crohn's disease and are likely to soon be applied in the tissue transplant setting (198). There is a prospect that in time, obstetric disorders may be amongst the range of diseases to benefit from CAR T cell therapy—but again, this must wait until relevant reproductive antigens are identified, and can be targeted in a patient-specific manner.
Conclusions
Immune imbalance or “maladaptation” has been implicated as central and causal in disease development in preeclampsia, and Treg cells are identified as a pivotal immune cell lineage. Their unique combination of anti-inflammatory, and immune modulatory properties affords Treg cells a potent capacity to support maternal vascular adaptation and placental development, suppress inflammation and sustain maternal tolerance of the fetus. The effects of Treg cells appear most critical at the time of pregnancy establishment and during early placental morphogenesis. Insufficient or dysfunctional Tregs provides a mechanism through which environmental, metabolic, and genetic factors can converge to increase disease risk (154), likely interacting with clinical factors such as prior pregnancy and immune compatibility between partners, which are known to be important pre-pregnancy antecedents of preeclampsia (10).
Given the rapid advances in Treg cell immunology including informative diagnostics based on flow cytometry of peripheral blood, and development of a range of low and high intervention treatments, the prospect of targeting Treg cells in at-risk women to treat early placental disturbances and effectively mitigate preeclampsia onset, warrants evaluation. It will be important to focus on developing diagnostics and interventions for application before or during early pregnancy, to divert the course of disease development before placental or fetal injury occurs. Proof-of-concept experiments in rodent models of preeclampsia already demonstrate the utility of biological agents PF-L1 Fc (129), CD28 superligand (122), and low dose IL2 (135) to boost Treg cell numbers and stability.
Experimental evaluation of any strategy to increase Tregs in a human reproductive setting must take a highly cautious approach and be founded in robust clinical trial design principles. It is critical that safety for mothers and infants is paramount, and the different risk-benefit ratio of reproductive and obstetric conditions, compared to life-threatening immune diseases, is recognized. Possible adverse consequences of artificially boosting maternal Treg cells, including reduced pathogen defense (199) or even reduced immune surveillance against malignancy (200) would need to be considered. Notwithstanding the substantial work to be done to evaluate alternative approaches and identify responsive patient groups, there is an imperative to invest in developing immune therapy options with the goal to reduce the morbidity and mortality associated with preeclampsia.
Author Contributions
SR, EG, AC, and LM assembled and interpreted the relevant literature and prepared manuscript drafts and Figures. JP, MH, SB, and GD provided expert knowledge on content and revised manuscript drafts.
Funding
This research is funded by NHMRC Project Grant APP1099461 to SR and SB.
Conflict of Interest Statement
SR receives income from Origio A/S.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Steegers EA, von Dadelszen P, Duvekot JJ, Pijnenborg R. Pre-eclampsia. Lancet. (2010) 376:631–44. doi: 10.1016/S0140-6736(10)60279-6
2. Stevens W, Shih T, Incerti D, Ton TGN, Lee HC, Peneva D, et al. Short-term costs of preeclampsia to the United States health care system. Am J Obstet Gynecol. (2017) 2017:32. doi: 10.1016/j.ajog.2017.04.032
3. Saigal S, Doyle LW. An overview of mortality and sequelae of preterm birth from infancy to adulthood. Lancet. (2008) 371:261–9. doi: 10.1016/S0140-6736(08)60136-1
4. Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. (2009) 33:130–7. doi: 10.1053/j.semperi.2009.02.010
5. Turanov AA, Lo A, Hassler MR, Makris A, Ashar-Patel A, Alterman JF, et al. RNAi modulation of placental sFLT1 for the treatment of preeclampsia. Nat Biotechnol. (2018) 2018:4297. doi: 10.1038/nbt.4297
6. Robertson SA. Preventing preeclampsia by silencing sFLT1? N Engl J Med. (2019) 36:12. doi: 10.1056/NEJMcibr1817501
7. Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension. (2008) 51:970–5. doi: 10.1161/HYPERTENSIONAHA.107.107607
8. Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. (2010) 63:534–43. doi: 10.1111/j.1600-0897.2010.00831.x
9. Wen SW, Xie RH, Tan H, Walker MC, Smith GN, Retnakaran R. Preeclampsia and gestational diabetes mellitus: pre-conception origins? Med Hypotheses. (2012) 79:120–5. doi: 10.1016/j.mehy.2012.04.019
10. Roberts JM, Redman CW. Global Pregnancy Collaboration symposium: prepregnancy and very early pregnancy antecedents of adverse pregnancy outcomes: overview and recommendations. Placenta. (2017) 60:1039. doi: 10.1016/j.placenta.2017.07.012
11. Brosens I, Pijnenborg R, Vercruysse L, Romero R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol. (2011) 204:193–201. doi: 10.1016/j.ajog.2010.08.009
12. Harmon AC, Cornelius DC, Amaral LM, Faulkner JL, Cunningham MW Jr, Wallace K, et al. The role of inflammation in the pathology of preeclampsia. Clin Sci. (2016) 130:409–19. doi: 10.1042/CS20150702
13. Saito S, Shiozaki A, Nakashima A, Sakai M, Sasaki Y. The role of the immune system in preeclampsia. Mol Aspects Med. (2007) 28:192–209. doi: 10.1016/j.mam.2007.02.006
14. Wang JX, Knottnerus AM, Schuit G, Norman RJ, Chan A, Dekker GA. Surgically obtained sperm, and risk of gestational hypertension and pre-eclampsia. Lancet. (2002) 359:673–4. doi: 10.1016/S0140-6736(02)07804-2
15. Kho EM, McCowan LM, North RA, Roberts CT, Chan E, Black MA, et al. Duration of sexual relationship and its effect on preeclampsia and small for gestational age perinatal outcome. J Reprod Immunol. (2009) 82:66–73. doi: 10.1016/j.jri.2009.04.011
16. Klonoff-Cohen HS, Savitz DA, Celafo RC, McCann MF. An epidemiologic study of contraception and preeclampsia. JAMA. (1989) 262:3143–7. doi: 10.1001/jama.1989.03430220066032
17. Dekker G. The partner's role in the etiology of preeclampsia. J Reprod Immunol. (2002) 57:203–15. doi: 10.1016/S0165-0378(02)00039-6
18. Masoudian P, Nasr A, de Nanassy J, Fung-Kee-Fung K, Bainbridge SA, Demellawy DE. Oocyte donation pregnancies and the risk of preeclampsia or gestational hypertension: a systematic review and metaanalysis. Am J Obstet Gynecol. (2016) 214:328–39. doi: 10.1016/j.ajog.2015.11.020
19. Kyrou D, Kolibianakis EM, Devroey P, Fatemi HM. Is the use of donor sperm associated with a higher incidence of preeclampsia in women who achieve pregnancy after intrauterine insemination? Fertil Steril. (2010) 93:1124–7. doi: 10.1016/j.fertnstert.2008.12.021
20. Rowe JH, Ertelt JM, Xin L, Way SS. Pregnancy imprints regulatory memory that sustains anergy to fetal antigen. Nature. (2012) 490:102–6. doi: 10.1038/nature11462
21. Gamliel M, Goldman-Wohl D, Isaacson B, Gur C, Stein N, Yamin R, et al. Trained memory of human uterine NK cells enhances their function in subsequent pregnancies. Immunity. (2018) 48:951–962 e5. doi: 10.1016/j.immuni.2018.03.030
22. Robertson SA, Sharkey DJ. Seminal fluid and fertility in women. Fertil Steril. (2016) 106:511–9. doi: 10.1016/j.fertnstert.2016.07.1101
23. Robertson SA, Care AS, Moldenhauer LM. Regulatory T cells in embryo implantation and the immune response to pregnancy. J Clin Invest. (2018) 128:4224–35. doi: 10.1172/JCI122182
24. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an excessive maternal inflammatory response to pregnancy. Am J Obstet Gynecol. (1999) 180:499–506. doi: 10.1016/S0002-9378(99)70239-5
25. Saito S, Sakai M, Sasaki Y, Nakashima A, Shiozaki A. Inadequate tolerance induction may induce pre-eclampsia. J Reprod Immunol. (2007) 76:30–9. doi: 10.1016/j.jri.2007.08.002
26. Sasaki Y, Darmochwal-Kolarz D, Suzuki D, Sakai M, Ito M, Shima T, et al. Proportion of peripheral blood and decidual CD4(+) CD25(bright) regulatory T cells in pre-eclampsia. Clin Exp Immunol. (2007) 149:139–45. doi: 10.1111/j.1365-2249.2007.03397.x
27. Santner-Nanan B, Peek MJ, Khanam R, Richarts L, Zhu E, Fazekas de St Groth B, Nanan R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J Immunol. (2009) 2009:1154. doi: 10.4049/jimmunol.0901154
28. Quinn KH, Lacoursiere DY, Cui L, Bui J, Parast MM. The unique pathophysiology of early-onset severe preeclampsia: role of decidual T regulatory cells. J Reprod Immunol. (2011) 91:76–82. doi: 10.1016/j.jri.2011.05.006
29. LaMarca B, Cornelius DC, Harmon AC, Amaral LM, Cunningham MW, Faulkner JL, et al. Identifying immune mechanisms mediating the hypertension during preeclampsia. Am J Physiol Regul Integr Comp Physiol. (2016) 311:R1–9. doi: 10.1152/ajpregu.00052.2016
30. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. (2006) 12:1065–74. doi: 10.1038/nm1452
31. Lash GE, Pitman H, Morgan HL, Innes BA, Agwu CN, Bulmer JN. Decidual macrophages: key regulators of vascular remodeling in human pregnancy. J Leukoc Biol. (2016) 100:315–25. doi: 10.1189/jlb.1A0815-351R
32. Hsu P, Santner-Nanan B, Dahlstrom JE, Fadia M, Chandra A, Peek M, et al. Altered decidual DC-SIGN+ antigen-presenting cells and impaired regulatory T-cell induction in preeclampsia. Am J Pathol. (2012) 181:2149–60. doi: 10.1016/j.ajpath.2012.08.032
33. Darmochwal-Kolarz D, Kludka-Sternik M, Tabarkiewicz J, Kolarz B, Rolinski J, Leszczynska-Gorzelak B, et al. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol. (2012) 93:75–81. doi: 10.1016/j.jri.2012.01.006
34. Steinborn A, Schmitt E, Kisielewicz A, Rechenberg S, Seissler N, Mahnke K, et al. Pregnancy-associated diseases are characterized by the composition of the systemic regulatory T cell (Treg) pool with distinct subsets of Tregs. Clin Exp Immunol. (2012) 167:84–98. doi: 10.1111/j.1365-2249.2011.04493.x
35. Carbone F, La Rocca C, De Candia P, Procaccini C, Colamatteo A, Micillo T, et al. Metabolic control of immune tolerance in health and autoimmunity. Semin Immunol. (2016) 28:491–504. doi: 10.1016/j.smim.2016.09.006
36. Murai M, Krause P, Cheroutre H, Kronenberg M. Regulatory T-cell stability and plasticity in mucosal and systemic immune systems. Mucosal Immunol. (2010) 3:443–9. doi: 10.1038/mi.2010.27
37. Bluestone JA, Trotta E, Xu D. The therapeutic potential of regulatory T cells for the treatment of autoimmune disease. Expert Opin Ther Targets. (2015) 19:1091–103. doi: 10.1517/14728222.2015.1037282
38. Issazadeh-Navikas S, Teimer R, Bockermann R. Influence of dietary components on regulatory T cells. Mol Med. (2012) 18:95–110. doi: 10.2119/molmed.2011.00311
39. Kim W, Lee H. Advances in nutritional research on regulatory T-cells. Nutrients. (2013) 5:4305–15. doi: 10.3390/nu5114305
40. Jagger A, Shimojima Y, Goronzy JJ, Weyand CM. Regulatory T cells and the immune aging process: a mini-review. Gerontology. (2014) 60:130–7. doi: 10.1159/000355303
41. Allan SE, Broady R, Gregori S, Himmel ME, Locke N, Roncarolo MG, et al. CD4+ T-regulatory cells: toward therapy for human diseases. Immunol Rev. (2008) 223:391–421. doi: 10.1111/j.1600-065X.2008.00634.x
42. Furukawa A, Wisel SA, Tang Q. Impact of immune-modulatory drugs on regulatory T cell. Transplantation. (2016) 100:2288–300. doi: 10.1097/TP.0000000000001379
43. Trowsdale J, Betz AG. Mother's little helpers: mechanisms of maternal-fetal tolerance. Nat Immunol. (2006) 7:241–6. doi: 10.1038/ni1317
44. Guleria I, Sayegh MH. Maternal acceptance of the fetus: true human tolerance. J Immunol. (2007) 178:3345–51. doi: 10.4049/jimmunol.178.6.3345
45. Nancy P, Tagliani E, Tay CS, Asp P, Levy DE, Erlebacher A. Chemokine gene silencing in decidual stromal cells limits T cell access to the maternal-fetal interface. Science. (2012) 336:1317–21. doi: 10.1126/science.1220030
46. Aluvihare VR, Kallikourdis M, Betz AG. Regulatory T cells mediate maternal tolerance to the fetus. Nat Immunol. (2004) 5:266–71. doi: 10.1038/ni1037
47. Guerin LR, Prins JR, Robertson SA. Regulatory T-cells and immune tolerance in pregnancy: a new target for infertility treatment? Hum Reprod Update. (2009) 15:517–35. doi: 10.1093/humupd/dmp004
48. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
49. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
50. Rudensky AY. Regulatory T cells and Foxp3. Immunol Rev. (2011) 241:260–8. doi: 10.1111/j.1600-065X.2011.01018.x
51. Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, et al. A distinct function of regulatory T cells in tissue protection. Cell. (2015) 162:1078–89. doi: 10.1016/j.cell.2015.08.021
52. Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. (2012) 30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623
53. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
54. Waldmann H, Adams E, Fairchild P, Cobbold S. Infectious tolerance and the long-term acceptance of transplanted tissue. Immunol Rev. (2006) 212:301–13. doi: 10.1111/j.0105-2896.2006.00406.x
55. Yamazaki S, Nishioka A, Kasuya S, Ohkura N, Hemmi H, Kaisho T, et al. Homeostasis of thymus-derived Foxp3+ regulatory T cells is controlled by ultraviolet B exposure in the skin. J Immunol. (2014) 193:5488–97. doi: 10.4049/jimmunol.1400985
56. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A special population of regulatory T cells potentiates muscle repair. Cell. (2013) 155:1282–95. doi: 10.1016/j.cell.2013.10.054
57. Williams PJ, Searle RF, Robson SC, Innes BA, Bulmer JN. Decidual leucocyte populations in early to late gestation normal human pregnancy. J Reprod Immunol. (2009) 82:24–31. doi: 10.1016/j.jri.2009.08.001
58. Shao L, Jacobs AR, Johnson VV, Mayer L. Activation of CD8+ regulatory T cells by human placental trophoblasts. J Immunol. (2005) 174:7539–47. doi: 10.4049/jimmunol.174.12.7539
59. Tilburgs T, Schonkeren D, Eikmans M, Nagtzaam NM, Datema G, Swings GM, et al. Human decidual tissue contains differentiated CD8+ effector-memory T cells with unique properties. J Immunol. (2010) 185:4470–7. doi: 10.4049/jimmunol.0903597
60. Dimova T, Nagaeva O, Stenqvist AC, Hedlund M, Kjellberg L, Strand M, et al. Maternal Foxp3 expressing CD4+ CD25+ and CD4+ CD25- regulatory T-cell populations are enriched in human early normal pregnancy decidua: a phenotypic study of paired decidual and peripheral blood samples. Am J Reprod Immunol. (2011) 66(Suppl 1):44–56. doi: 10.1111/j.1600-0897.2011.01046.x
61. Mjosberg J, Berg G, Jenmalm MC, Ernerudh J. FOXP3+ regulatory T cells and T helper 1, T helper 2, and T helper 17 cells in human early pregnancy decidua. Biol Reprod. (2010) 82:698–705. doi: 10.1095/biolreprod.109.081208
62. Tilburgs T, Roelen DL, van der Mast BJ, de Groot-Swings GM, Kleijburg C, Scherjon S, et al. Evidence for a selective migration of fetus-specific CD4+CD25bright regulatory T cells from the peripheral blood to the decidua in human pregnancy. J Immunol. (2008) 180:5737–45. doi: 10.4049/jimmunol.180.8.5737
63. Inada K, Shima T, Ito M, Ushijima A, Saito S. Helios-positive functional regulatory T cells are decreased in decidua of miscarriage cases with normal fetal chromosomal content. J Reprod Immunol. (2015) 107:10–9. doi: 10.1016/j.jri.2014.09.053
64. Wagner MI, Jost M, Spratte J, Schaier M, Mahnke K, Meuer S, et al. Differentiation of ICOS+ and ICOS- recent thymic emigrant regulatory T cells (RTE T regs) during normal pregnancy, pre-eclampsia and HELLP syndrome. Clin Exp Immunol. (2016) 183:129–42. doi: 10.1111/cei.12693
65. Sargent IL, Borzychowski AM, Redman CWG. Immunoregulation in normal pregnancy and pre-eclampsia: an overview. Reprod BioMed Online. (2006) 13:680–6. doi: 10.1016/S1472-6483(10)60659-1
66. Hudic I, Fatusic Z. Progesterone - induced blocking factor (PIBF) and Th(1)/Th(2) cytokine in women with threatened spontaneous abortion. J Perinat Med. (2009) 37:338–42. doi: 10.1515/jpm.2009.061
67. Rahimzadeh M, Norouzian M, Arabpour F, Naderi N. Regulatory T-cells and preeclampsia: an overview of literature. Expert Rev Clin Immunol. (2016) 12:209–27. doi: 10.1586/1744666X.2016.1105740
68. Steinborn A, Haensch GM, Mahnke K, Schmitt E, Toermer A, Meuer S, et al. Distinct subsets of regulatory T cells during pregnancy: is the imbalance of these subsets involved in the pathogenesis of preeclampsia? Clin Immunol. (2008) 129:401–12. doi: 10.1016/j.clim.2008.07.032
69. Toldi G, Vasarhelyi ZE, Rigo J Jr, Orban C, Tamassy Z, Bajnok A, et al. Prevalence of regulatory T-cell subtypes in preeclampsia. Am J Reprod Immunol. (2015) 74:110–5. doi: 10.1111/aji.12380
70. Mjosberg J, Svensson J, Johansson E, Hellstrom L, Casas R, Jenmalm MC, et al. Systemic reduction of functionally suppressive CD4dimCD25highFoxp3+ Tregs in human second trimester pregnancy is induced by progesterone and 17beta-estradiol. J Immunol. (2009) 183:759–69. doi: 10.4049/jimmunol.0803654
71. Rabaglino MB, Post Uiterweer ED, Jeyabalan A, Hogge WA, Conrad KP. Bioinformatics approach reveals evidence for impaired endometrial maturation before and during early pregnancy in women who developed preeclampsia. Hypertension. (2015) 65:421–9. doi: 10.1161/HYPERTENSIONAHA.114.04481
72. Prins JR, Gomez-Lopez N, Robertson SA. Interleukin-6 in pregnancy and gestational disorders. J Reprod Immunol. (2012) 95:1–14. doi: 10.1016/j.jri.2012.05.004
73. Prins JR, Faas MM, Melgert BN, Huitema S, Timmer A, Hylkema MN, et al. Altered expression of immune-associated genes in first-trimester human decidua of pregnancies later complicated with hypertension or foetal growth restriction. Placenta. (2012) 33:453–5. doi: 10.1016/j.placenta.2012.02.010
74. Winger EE, Reed JL. Low circulating CD4(+) CD25(+) Foxp3(+) T regulatory cell levels predict miscarriage risk in newly pregnant women with a history of failure. Am J Reprod Immunol. (2011) 66:320–8. doi: 10.1111/j.1600-0897.2011.00992.x
75. Ahn H, Park J, Gilman-Sachs A, Kwak-Kim J. Immunologic characteristics of preeclampsia, a comprehensive review. Am J Reprod Immunol. (2011) 65:377–94. doi: 10.1111/j.1600-0897.2010.00913.x
76. Romero R, Kusanovic JP, Chaiworapongsa T, Hassan SS. Placental bed disorders in preterm labor, preterm PROM, spontaneous abortion and abruptio placentae. Best Pract Res Clin Obstet Gynaecol. (2011) 25:313–27. doi: 10.1016/j.bpobgyn.2011.02.006
77. Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. (2014) 345:760–5. doi: 10.1126/science.1251816
78. Zhang J, Dunk C, Croy AB, Lye SJ. To serve and to protect: the role of decidual innate immune cells on human pregnancy. Cell Tissue Res. (2016) 363:249–65. doi: 10.1007/s00441-015-2315-4
79. Nevers T, Kalkunte S, Sharma S. Uterine regulatory T cells, IL-10 and hypertension. Am J Reprod Immunol. (2011) 66(Suppl 1):88–92. doi: 10.1111/j.1600-0897.2011.01040.x
80. Jasper MJ, Tremellen KP, Robertson SA. Primary unexplained infertility is associated with reduced expression of the T-regulatory cell transcription factor Foxp3 in endometrial tissue. Mol Hum Reprod. (2006) 12:301–8. doi: 10.1093/molehr/gal032
81. Kwak-Kim J, Bao S, Lee SK, Kim JW, Gilman-Sachs A. Immunological modes of pregnancy loss: inflammation, immune effectors, and stress. Am J Reprod Immunol. (2014) 72:129–40. doi: 10.1111/aji.12234
82. Zenclussen AC, Fest S, Joachim R, Klapp BF, Arck PC. Introducing a mouse model for pre-eclampsia: adoptive transfer of activated Th1 cells leads to pre-eclampsia-like symptoms exclusively in pregnant mice. Eur J Immunol. (2004) 34:377–87. doi: 10.1002/eji.200324469
83. Wegorzewska M, Nijagal A, Wong CM, Le T, Lescano N, Tang Q, et al. Fetal intervention increases maternal T cell awareness of the foreign conceptus and can lead to immune-mediated fetal demise. J Immunol. (2014) 192:1938–45. doi: 10.4049/jimmunol.1302403
84. Xin L, Ertelt JM, Rowe JH, Jiang TT, Kinder JM, Chaturvedi V, et al. Cutting edge: committed Th1 CD4+ T cell differentiation blocks pregnancy-induced Foxp3 expression with antigen-specific fetal loss. J Immunol. (2014) 192:2970–4. doi: 10.4049/jimmunol.1302678
85. Samstein RM, Josefowicz SZ, Arvey A, Treuting PM, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal-fetal conflict. Cell. (2012) 150:29–38. doi: 10.1016/j.cell.2012.05.031
86. Shima T, Sasaki Y, Itoh M, Nakashima A, Ishii N, Sugamura K, et al. Regulatory T cells are necessary for implantation and maintenance of early pregnancy but not late pregnancy in allogeneic mice. J Reprod Immunol. (2010) 85:121–9. doi: 10.1016/j.jri.2010.02.006
87. Zenclussen AC, Gerlof K, Zenclussen ML, Sollwedel A, Bertoja AZ, Ritter T, et al. Abnormal T-cell reactivity against paternal antigens in spontaneous abortion: adoptive transfer of pregnancy-induced CD4+CD25+ T regulatory cells prevents fetal rejection in a murine abortion model. Am J Pathol. (2005) 166:811–22. doi: 10.1016/S0002-9440(10)62302-4
88. Darrasse-Jeze G, Klatzmann D, Charlotte F, Salomon BL, Cohen JL. CD4+CD25+ regulatory/suppressor T cells prevent allogeneic fetus rejection in mice. Immunol Lett. (2006) 102:106–9. doi: 10.1016/j.imlet.2005.07.002
89. Care AS, Bourque SL, Morton JS, Hjartarson EP, Robertson SA, Davidge ST. Reduction in regulatory T cells in early pregnancy causes uterine artery dysfunction in mice. Hypertension. (2018) 72:177–87. doi: 10.1161/HYPERTENSIONAHA.118.10858
90. Bizargity P, Del Rio R, Phillippe M, Teuscher C, Bonney EA. Resistance to lipopolysaccharide-induced preterm delivery mediated by regulatory T cell function in mice. Biol Reprod. (2009) 80:874–81. doi: 10.1095/biolreprod.108.074294
91. Li L, Tu J, Jiang Y, Zhou J, Schust DJ. Regulatory T cells decrease invariant natural killer T cell-mediated pregnancy loss in mice. Mucosal Immunol. (2017) 10:613–23. doi: 10.1038/mi.2016.84
92. Lin Y, Liu X, Shan B, Wu J, Sharma S, Sun Y. Prevention of CpG-induced pregnancy disruption by adoptive transfer of in vitro-induced regulatory T cells. PLoS ONE. (2014) 9:e94702. doi: 10.1371/journal.pone.0094702
93. Moldenhauer LM, Diener KR, Thring DM, Brown MP, Hayball JD, Robertson SA. Cross-presentation of male seminal fluid antigens elicits T cell activation to initiate the female immune response to pregnancy. J Immunol. (2009) 182:8080–93. doi: 10.4049/jimmunol.0804018
94. Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol. (2005) 175:4084–90. doi: 10.4049/jimmunol.175.6.4084
95. Thuere C, Zenclussen ML, Schumacher A, Langwisch S, Schulte-Wrede U, Teles A, et al. Kinetics of regulatory T cells during murine pregnancy. Am J Reprod Immunol. (2007) 58:514–23. doi: 10.1111/j.1600-0897.2007.00538.x
96. Terme M, Chaput N, Combadiere B, Ma A, Ohteki T, Zitvogel L. Regulatory T cells control dendritic cell/NK cell cross-talk in lymph nodes at the steady state by inhibiting CD4+ self-reactive T cells. J Immunol. (2008) 180:4679–86. doi: 10.4049/jimmunol.180.7.4679
97. Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. (2005) 202:1075–85. doi: 10.1084/jem.20051511
98. Vacca P, Cantoni C, Vitale M, Prato C, Canegallo F, Fenoglio D, et al. Crosstalk between decidual NK and CD14+ myelomonocytic cells results in induction of Tregs and immunosuppression. Proc Natl Acad Sci USA. (2010) 107:11918–23. doi: 10.1073/pnas.1001749107
99. Woidacki K, Meyer N, Schumacher A, Goldschmidt A, Maurer M, Zenclussen AC. Transfer of regulatory T cells into abortion-prone mice promotes the expansion of uterine mast cells and normalizes early pregnancy angiogenesis. Sci Rep. (2015) 5:13938. doi: 10.1038/srep13938
100. Schumacher A, Wafula PO, Teles A, El-Mousleh T, Linzke N, Zenclussen ML, et al. Blockage of heme oxygenase-1 abrogates the protective effect of regulatory T cells on murine pregnancy and promotes the maturation of dendritic cells. PLoS ONE. (2012) 7:e42301. doi: 10.1371/journal.pone.0042301
101. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. (2003) 4:1206–12. doi: 10.1038/ni1003
102. Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. (1998) 281:1191–3. doi: 10.1126/science.281.5380.1191
103. Blois SM, Kammerer U, Alba Soto C, Tometten MC, Shaikly V, Barrientos G, et al. Dendritic cells: key to fetal tolerance? Biol Reprod. (2007) 77:590–8. doi: 10.1095/biolreprod.107.060632
104. Zhang Y, Liu Z, Tian M, Hu X, Wang L, Ji J, et al. The altered PD-1/PD-L1 pathway delivers the ‘one-two punch’ effects to promote the Treg/Th17 imbalance in pre-eclampsia. Cell Mol Immunol. (2017) 2017:70. doi: 10.1038/cmi.2017.70
105. Blois SM, Ilarregui JM, Tometten M, Garcia M, Orsal AS, Cordo-Russo R, et al. A pivotal role for galectin-1 in fetomaternal tolerance. Nat Med. (2007) 13:1450–7. doi: 10.1038/nm1680
106. Prins JR, Zhang B, Schjenken JE, Guerin LR, Barry SC, Robertson SA. Unstable Foxp3+ regulatory T cells and altered dendritic cells are associated with lipopolysaccharide-induced fetal loss in pregnant interleukin 10-deficient mice. Biol Reprod. (2015) 93:95. doi: 10.1095/biolreprod.115.128694
107. Plaks V, Birnberg T, Berkutzki T, Sela S, BenYashar A, Kalchenko V, et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J Clin Invest. (2008) 118:3954–65. doi: 10.1172/JCI36682
108. Tirado-Gonzalez I, Barrientos G, Freitag N, Otto T, Thijssen VL, Moschansky P, et al. Uterine NK cells are critical in shaping DC immunogenic functions compatible with pregnancy progression. PLoS ONE. (2012) 7:e46755. doi: 10.1371/journal.pone.0046755
109. Blois SM, Klapp BF, Barrientos G. Decidualization and angiogenesis in early pregnancy: unravelling the functions of DC and NK cells. J Reprod Immunol. (2011) 88:86–92. doi: 10.1016/j.jri.2010.11.002
110. Du MR, Guo PF, Piao HL, Wang SC, Sun C, Jin LP, et al. Embryonic trophoblasts induce decidual regulatory T cell differentiation and maternal-fetal tolerance through thymic stromal lymphopoietin instructing dendritic cells. J Immunol. (2014) 192:1502–11. doi: 10.4049/jimmunol.1203425
111. Redman CW, Sargent IL. Placental stress and pre-eclampsia: a revised view. Placenta. (2009) 30(Suppl A):S38–42. doi: 10.1016/j.placenta.2008.11.021
112. Ashkar AA, Di Santo JP, Croy BA. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J Exp Med. (2000) 192:259–70. doi: 10.1084/jem.192.2.259
113. Yamashita T, Sasaki N, Kasahara K, Hirata K. Anti-inflammatory and immune-modulatory therapies for preventing atherosclerotic cardiovascular disease. J Cardiol. (2015) 66:1–8. doi: 10.1016/j.jjcc.2015.02.002
114. Matrougui K, Abd Elmageed Z, Kassan M, Choi S, Nair D, Gonzalez-Villalobos RA, et al. Natural regulatory T cells control coronary arteriolar endothelial dysfunction in hypertensive mice. Am J Pathol. (2011) 178:434–41. doi: 10.1016/j.ajpath.2010.11.034
115. Maganto-Garcia E, Bu DX, Tarrio ML, Alcaide P, Newton G, Griffin GK, et al. Foxp3+-inducible regulatory T cells suppress endothelial activation and leukocyte recruitment. J Immunol. (2011) 187:3521–9. doi: 10.4049/jimmunol.1003947
116. Croy BA, Burke SD, Barrette VF, Zhang J, Hatta K, Smith GN, et al. Identification of the primary outcomes that result from deficient spiral arterial modification in pregnant mice. Pregnancy Hypertens. (2011) 1:87–94. doi: 10.1016/j.preghy.2010.10.002
117. Burke SD, Barrette VF, Carter AL, Gravel J, Adams MA, Croy BA. Cardiovascular adaptations of pregnancy in T and B cell-deficient mice. Biol Reprod. (2011) 85:605–14. doi: 10.1095/biolreprod.111.092668
118. Nadkarni S, Smith J, Sferruzzi-Perri AN, Ledwozyw A, Kishore M, Haas R, et al. Neutrophils induce proangiogenic T cells with a regulatory phenotype in pregnancy. Proc Natl Acad Sci USA. (2016) 113:E8415–24. doi: 10.1073/pnas.1611944114
119. Cornelius DC, Amaral LM, Wallace K, Campbell N, Thomas AJ, Scott J, et al. Reduced uterine perfusion pressure T-helper 17 cells cause pathophysiology associated with preeclampsia during pregnancy. Am J Physiol Regul Integr Comp Physiol. (2016) 311:R1192–9. doi: 10.1152/ajpregu.00117.2016
120. Cornelius DC, Amaral LM, Harmon A, Wallace K, Thomas AJ, Campbell N, et al. An increased population of regulatory T cells improves the pathophysiology of placental ischemia in a rat model of preeclampsia. Am J Physiol Regul Integr Comp Physiol. (2015) 309:R884–91. doi: 10.1152/ajpregu.00154.2015
121. Harmon A, Cornelius D, Amaral L, Paige A, Herse F, Ibrahim T, et al. IL-10 supplementation increases Tregs and decreases hypertension in the RUPP rat model of preeclampsia. Hypertens Pregnancy. (2015) 34:291–306. doi: 10.3109/10641955.2015.1032054
122. Przybyl L, Ibrahim T, Haase N, Golic M, Rugor J, Luft FC, et al. Regulatory T cells ameliorate intrauterine growth retardation in a transgenic rat model for preeclampsia. Hypertension. (2015) 65:1298–306. doi: 10.1161/HYPERTENSIONAHA.114.04892
123. Moldenhauer LM, Diener KR, Hayball JD, Robertson SA. An immunogenic phenotype in paternal antigen-specific CD8+ T cells at embryo implantation elicits later fetal loss in mice. Immunol Cell Biol. (2017) 2017:41. doi: 10.1038/icb.2017.41
124. Robertson SA, Moldenhauer LM. Immunological determinants of implantation success. Int J Dev Biol. (2014) 58:205–17. doi: 10.1387/ijdb.140096sr
125. Arruvito L, Sanz M, Banham AH, Fainboim L. Expansion of CD4+CD25+and FOXP3+ regulatory T cells during the follicular phase of the menstrual cycle: implications for human reproduction. J Immunol. (2007) 178:2572–8. doi: 10.4049/jimmunol.178.4.2572
126. Saito S, Nishikawa K, Morii T, Narita N, Enomoto M, Ito A, et al. A study of CD45RO, CD45RA and CD29 antigen expression on human decidual T cells in an early stage of pregnancy. Immunol Lett. (1994) 40:193–7. doi: 10.1016/0165-2478(93)00019-A
127. Tilburgs T, Scherjon SA, van der Mast BJ, Haasnoot GW, Versteeg VDV-M, Roelen D, et al. Fetal-maternal HLA-C mismatch is associated with decidual T cell activation and induction of functional T regulatory cells. J Reprod Immunol. (2009) 82:148–57. doi: 10.1016/j.jri.2009.05.003
128. Mjosberg J, Berg G, Ernerudh J, Ekerfelt C. CD4+ CD25+ regulatory T cells in human pregnancy: development of a Treg-MLC-ELISPOT suppression assay and indications of paternal specific Tregs. Immunology. (2007) 120:456–66. doi: 10.1111/j.1365-2567.2006.02529.x
129. Tian M, Zhang Y, Liu Z, Sun G, Mor G, Liao A. The PD-1/PD-L1 inhibitory pathway is altered in pre-eclampsia and regulates T cell responses in pre-eclamptic rats. Sci Rep. (2016) 6:27683. doi: 10.1038/srep27683
130. Petroff MG. Review: fetal antigens–identity, origins, and influences on the maternal immune system. Placenta. (2011) 32(Suppl 2):S176–81. doi: 10.1016/j.placenta.2010.12.014
131. Chamley LW, Chen Q, Ding J, Stone PR, Abumaree M. Trophoblast deportation: just a waste disposal system or antigen sharing? J Reprod Immunol. (2011) 88:99–105. doi: 10.1016/j.jri.2011.01.002
132. Moldenhauer LM, Hayball JD, Robertson SA. Utilising T cell receptor transgenic mice to define mechanisms of maternal T cell tolerance in pregnancy. J Reprod Immunol. (2010) 87:1–13. doi: 10.1016/j.jri.2010.05.007
133. Guerin LR, Moldenhauer LM, Prins JR, Bromfield JJ, Hayball JD, Robertson SA. Seminal fluid regulates accumulation of FOXP3+ regulatory T cells in the preimplantation mouse uterus through expanding the FOXP3+ cell pool and CCL19-mediated recruitment. Biol Reprod. (2011) 85:397–408. doi: 10.1095/biolreprod.110.088591
134. Zhao JX, Zeng YY, Liu Y. Fetal alloantigen is responsible for the expansion of the CD4(+)CD25(+) regulatory T cell pool during pregnancy. J Reprod Immunol. (2007) 75:71–81. doi: 10.1016/j.jri.2007.06.052
135. Chen T, Darrasse-Jeze G, Bergot AS, Courau T, Churlaud G, Valdivia K, et al. Self-specific memory regulatory T cells protect embryos at implantation in mice. J Immunol. (2013) 191:2273–81. doi: 10.4049/jimmunol.1202413
136. Shima T, Inada K, Nakashima A, Ushijima A, Ito M, Yoshino O, et al. Paternal antigen-specific proliferating regulatory T cells are increased in uterine-draining lymph nodes just before implantation and in pregnant uterus just after implantation by seminal plasma-priming in allogeneic mouse pregnancy. J Reprod Immunol. (2015) 108:72–82. doi: 10.1016/j.jri.2015.02.005
137. Kallikourdis M, Betz AG. Periodic accumulation of regulatory T cells in the uterus: preparation for the implantation of a semi-allogeneic fetus? PLoS ONE. (2007) 2:e382. doi: 10.1371/journal.pone.0000382
138. Moldenhauer LM, Schjenken JE, Hope CM, Green ES, Zhang B, Eldi P, et al. Thymus-derived regulatory T cells exhibit Foxp3 epigenetic modification and phenotype attenuation after mating in mice. J Immunol. (2019) 4:253. doi: 10.3389/fimmu.2013.00253
139. Robertson SA, Guerin LR, Bromfield JJ, Branson KM, Ahlström AC, AS Care. Seminal fluid drives expansion of the CD4+CD25+ T regulatory cell pool and induces tolerance to paternal alloantigens in mice. Biol Reprod. (2009) 80:1036–45. doi: 10.1095/biolreprod.108.074658
140. Johansson M, Bromfield JJ, Jasper MJ, Robertson SA. Semen activates the female immune response during early pregnancy in mice. Immunology. (2004) 112:290–300. doi: 10.1111/j.1365-2567.2004.01876.x
141. Hori S. Lineage stability and phenotypic plasticity of Foxp3(+) regulatory T cells. Immunol Rev. (2014) 259:159–72. doi: 10.1111/imr.12175
142. Sharkey DJ, Tremellen KP, Jasper MJ, Gemzell-Danielsson K, Robertson SA. Seminal fluid induces leukocyte recruitment and cytokine and chemokine mRNA expression in the human cervix after coitus. J Immunol. (2012) 188:2445–54. doi: 10.4049/jimmunol.1102736
143. Schumacher A, Heinze K, Witte J, Poloski E, Linzke N, Woidacki K, et al. Human chorionic gonadotropin as a central regulator of pregnancy immune tolerance. J Immunol. (2013) 190:2650–8. doi: 10.4049/jimmunol.1202698
144. Balandya E, Wieland-Alter W, Sanders K, Lahey T. Human seminal plasma fosters CD4(+) regulatory T-cell phenotype and transforming growth factor-beta1 expression. Am J Reprod Immunol. (2012) 68:322–30. doi: 10.1111/j.1600-0897.2012.01176.x
145. Meuleman T, Snaterse G, van Beelen E, Anholts JD, Pilgram GS, van der Westerlaken LA, et al. The immunomodulating effect of seminal plasma on T cells. J Reprod Immunol. (2015) 110:109–16. doi: 10.1016/j.jri.2015.01.012
146. Triche EW, Harland KK, Field EH, Rubenstein LM, Saftlas AF. Maternal-fetal HLA sharing and preeclampsia: variation in effects by seminal fluid exposure in a case-control study of nulliparous women in Iowa. J Reprod Immunol. (2014) 101–102:111–9. doi: 10.1016/j.jri.2013.06.004
147. Liu C, Wang XZ, Sun XB. Assessment of sperm antigen specific T regulatory cells in women with recurrent miscarriage. Early Hum Dev. (2012) 2012:003. doi: 10.1016/j.earlhumdev.2012.08.003
148. Sharkey DJ, Tremellen KP, Briggs NE, Dekker GA, Robertson SA. Seminal plasma transforming growth factor-beta, activin A and follistatin fluctuate within men over time. Hum Reprod. (2016) 31:2183–91. doi: 10.1093/humrep/dew185
149. Sharkey DJ, Tremellen KP, Briggs NE, Dekker GA, Robertson SA. Seminal plasma pro-inflammatory cytokines interferon-gamma (IFNG) and C-X-C motif chemokine ligand 8 (CXCL8) fluctuate over time within men. Hum Reprod. (2017) 2017:1–9. doi: 10.1093/humrep/dex106
150. Sharkey DJ, Glynn DJ, Schjenken JE, Tremellen KP, Robertson SA. Interferon-gamma inhibits seminal plasma induction of colony-stimulating factor 2 in mouse and human reproductive tract epithelial cells. Biol Reprod. (2018) 99:514–26. doi: 10.1093/biolre/ioy071
151. Moldenhauer LM, Keenihan SN, Hayball JD, Robertson SA. GM-CSF is an essential regulator of T cell activation competence in uterine dendritic cells during early pregnancy in mice. J Immunol. (2010) 185:7085–96. doi: 10.4049/jimmunol.1001374
152. Kryczek I, Bruce AT, Gudjonsson JE, Johnston A, Aphale A, Vatan L, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol. (2008) 181:4733–41. doi: 10.4049/jimmunol.181.7.4733
153. Zhao J, Zhao J, Perlman S. Differential effects of IL-12 on Tregs and non-Treg T cells: roles of IFN-γ, IL-2 and IL-2R. PLoS ONE. (2012) 7:e46241. doi: 10.1371/journal.pone.0046241
154. Sadlon T, Brown CY, Bandara V, Hope CM, Schjenken JE, Pederson SM, et al. Unravelling the molecular basis for regulatory T-cell plasticity and loss of function in disease. Clin Trans Immunol. (2018) 7:e1011. doi: 10.1002/cti2.1011
155. Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA. (2009) 106:4793–8. doi: 10.1073/pnas.0900408106
156. Feng T, Cao AT, Weaver CT, Elson CO, Cong Y. Interleukin-12 converts Foxp3+ regulatory T cells to interferon-gamma-producing Foxp3+ T cells that inhibit colitis. Gastroenterology. (2011) 140:2031–43. doi: 10.1053/j.gastro.2011.03.009
157. Komatsu N, Okamoto K, Sawa S, Nakashima T, Oh-hora M, Kodama T, et al. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med. (2014) 20:62–8. doi: 10.1038/nm.3432
158. Beyer M, Thabet Y, Muller RU, Sadlon T, Classen S, Lahl K, et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat Immunol. (2011) 12:898–907. doi: 10.1038/ni.2084
159. Ngalamika O, Liang G, Zhao M, Yu X, Yang Y, Yin H, et al. Peripheral whole blood FOXP3 TSDR methylation: a potential marker in severity assessment of autoimmune diseases and chronic infections. Immunol Invest. (2015) 44:126–36. doi: 10.3109/08820139.2014.938165
160. Chen X, Gan T, Liao Z, Chen S, Xiao J. Foxp3 (-/ATT) polymorphism contributes to the susceptibility of preeclampsia. PLoS ONE. (2013) 8:e59696. doi: 10.1371/journal.pone.0059696
161. Norouzian M, Rahimzadeh M, Rajaee M, Arabpour F, Naderi N. FoxP3 gene promoter polymorphism affects susceptibility to preeclampsia. Hum Immunol. (2016) 77:1232–8. doi: 10.1016/j.humimm.2016.09.001
162. Arruvito L, Billordo A, Capucchio M, Prada ME, Fainboim L. IL-6 trans-signaling and the frequency of CD4+FOXP3+ cells in women with reproductive failure. J Reprod Immunol. (2009) 82:158–65. doi: 10.1016/j.jri.2009.04.010
163. Arruvito L, Sotelo AI, Billordo A, Fainboim L. A physiological role for inducible FOXP3(+) Treg cells. Lessons from women with reproductive failure. Clin Immunol. (2010) 136:432–41. doi: 10.1016/j.clim.2010.05.002
164. Prins JR, Holvast F, van 't Hooft J, Bos AF, Ganzevoort JW, Scherjon SA, et al. Development of a core outcome set for immunomodulation in pregnancy (COSIMPREG): a protocol for a systematic review and Delphi study. BMJ Open. (2018) 8:e021619. doi: 10.1136/bmjopen-2018-021619
165. Petrillo MG, Ronchetti S, Ricci E, Alunno A, Gerli R, Nocentini G, et al. GITR+ regulatory T cells in the treatment of autoimmune diseases. Autoimmun Rev. (2015) 14:117–26. doi: 10.1016/j.autrev.2014.10.011
166. Hill D, Eastaff-Leung N, Bresatz-Atkins S, Warner N, Ruitenberg J, Krumbiegel D, et al. Inhibition of activation induced CD154 on CD4+ CD25- cells: a valid surrogate for human Treg suppressor function. Immunol Cell Biol. (2012) 90:812–21. doi: 10.1038/icb.2012.18
167. Nicholson IC, Mavrangelos C, Bird DR, Bresatz-Atkins S, Eastaff-Leung NG, Grose R, et al. PI16 is expressed by a subset of human memory Treg with enhanced migration to CCL17 and CCL20. Cell Immunol. (2012) 275:12–8. doi: 10.1016/j.cellimm.2012.04.002
168. Saftlas AF, Rubenstein L, Prater K, Harland KK, Field E, Triche EW. Cumulative exposure to paternal seminal fluid prior to conception and subsequent risk of preeclampsia. J Reprod Immunol. (2014) 101–102:104–10. doi: 10.1016/j.jri.2013.07.006
169. Brown HM, Green ES, Tan TCY, Gonzalez MB, Rumbold AR, Hull ML, Norman RJ, et al. Periconception onset diabetes is associated with embryopathy and fetal growth retardation, reproductive tract hyperglycosylation and impaired immune adaptation to pregnancy. Sci Rep. (2018) 8:2114. doi: 10.1038/s41598-018-19263-8
170. Gleicher N, el-Roeiy A. The reproductive autoimmune failure syndrome. Am J Obstet Gynecol. (1988) 159:223–7. doi: 10.1016/0002-9378(88)90525-X
171. Scarpellini F, Sbracia M. Use of granulocyte colony-stimulating factor for the treatment of unexplained recurrent miscarriage: a randomised controlled trial. Hum Reprod. (2009) 24:2703–8. doi: 10.1093/humrep/dep240
172. Ziebe S, Loft A, Povlsen BB, Erb K, Agerholm I, Aasted M, et al. A randomized clinical trial to evaluate the effect of granulocyte-macrophage colony-stimulating factor (GM-CSF) in embryo culture medium for in vitro fertilization. Fertil Steril. (2013) 99:1600–9. doi: 10.1016/j.fertnstert.2012.12.043
173. Rahmati M, Petitbarat M, Dubanchet S, Bensussan A, Chaouat G, Ledee N. Colony Stimulating Factors 1:2, 3 and early pregnancy steps: from bench to bedside. J Reprod Immunol. (2015) 109:1–6. doi: 10.1016/j.jri.2015.01.005
174. Schjenken JE, Zhang B, Chan HY, Sharkey DJ, Fullston T, Robertson SA. miRNA regulation of immune tolerance in early pregnancy. Am J Reprod Immunol. (2016) 75:272–80. doi: 10.1111/aji.12490
175. Szekeres-Bartho J. Progesterone-mediated immunomodulation in pregnancy: its relevance to leukocyte immunotherapy of recurrent miscarriage. Immunotherapy. (2009) 1:873–82. doi: 10.2217/imt.09.54
176. Druckmann R, Druckmann M-A. Progesterone and the immunology of pregnancy. J Steroid Biochem Mol Biol. (2005) 97:389–96. doi: 10.1016/j.jsbmb.2005.08.010
177. Lee JH, Ulrich B, Cho J, Park J, Kim CH. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J Immunol. (2011) 187:1778–87. doi: 10.4049/jimmunol.1003919
178. Lee JH, Lydon JP, Kim CH. Progesterone suppresses the mTOR pathway and promotes generation of induced regulatory T cells with increased stability. Eur J Immunol. (2012) 42:2683–96. doi: 10.1002/eji.201142317
179. Piccinni M-P, Scaletti C, Maggi E, Romagnani S. Role of hormone-controlled Th1- and Th2-type cytokines in successful pregnancy. J Neuroimmunol. (2000) 109:30–3. doi: 10.1016/S0165-5728(00)00299-X
180. Mao G, Wang J, Kang Y, Tai P, Wen J, Zou Q, et al. Progesterone increases systemic and local uterine proportions of CD4+CD25+ Treg cells during midterm pregnancy in mice. Endocrinology. (2010) 151:5477–88. doi: 10.1210/en.2010-0426
181. Hughes GC, Clark EA, Wong AH. The intracellular progesterone receptor regulates CD4+ T cells and T cell-dependent antibody responses. J Leukoc Biol. (2013) 93:369–75. doi: 10.1189/jlb.1012491
182. Thangamani S, Kim M, Son Y, Huang X, Kim H, Lee JH, et al. Cutting edge: progesterone directly upregulates vitamin D receptor gene expression for efficient regulation of T cells by calcitriol. J Immunol. (2015) 194:883–6. doi: 10.4049/jimmunol.1401923
183. Haas DM, Hathaway TJ, Ramsey PS. Progestogen for preventing miscarriage in women with recurrent miscarriage of unclear etiology. Cochrane Database Syst Rev. (2018) 10:CD003511. doi: 10.1002/14651858.CD003511.pub4
184. Ogasawara M, Aoki K, Okada S, Suzumori K. Embryonic karyotype of abortuses in relation to the number of previous miscarriages. Fertil Steril. (2000) 73:300–4. doi: 10.1016/S0015-0282(99)00495-1
185. Meher S, Duley L. Progesterone for preventing pre-eclampsia and its complications. Cochrane Database Syst Rev. (2006) 4:CD006175. doi: 10.1002/14651858.CD006175
186. Robertson SA, Jin M, Yu D, Moldenhauer LM, Davies MJ, Hull ML, et al. Corticosteroid therapy in assisted reproduction - immune suppression is a faulty premise. Hum Reprod. (2016) 31:2164–73. doi: 10.1093/humrep/dew186
187. Coulam CB, Acacio B. Does immunotherapy for treatment of reproductive failure enhance live births? Am J Reprod Immunol. (2012) 67:296–304. doi: 10.1111/j.1600-0897.2012.01111.x
188. Wong LF, Porter TF, Scott JR. Immunotherapy for recurrent miscarriage. Cochrane Database Syst Rev. (2014) 10:CD000112. doi: 10.1002/14651858.CD000112.pub3
189. Roussev RG, Acacio B, Ng SC, Coulam CB. Duration of intralipid's suppressive effect on NK cell's functional activity. Am J Reprod Immunol. (2008) 60:258–63. doi: 10.1111/j.1600-0897.2008.00621.x
190. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol. (2009) 182:259–73. doi: 10.4049/jimmunol.182.1.259
191. Lu CH, Wu CJ, Chan CC, Nguyen DT, Lin KR, Lin SJ, et al. DNA methyltransferase inhibitor promotes human CD4(+)CD25(h)FOXP3(+) regulatory T lymphocyte induction under suboptimal TCR stimulation. Front Immunol. (2016) 7:488. doi: 10.3389/fimmu.2016.00488
192. Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda KIAkanuma T, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol. (2017) 29:365–75. doi: 10.1093/intimm/dxx049
193. Singer BD, Mock JR, Aggarwal NRGaribaldi BT, Sidhaye VK, Florez MA, et al. Regulatory T cell DNA methyltransferase inhibition accelerates resolution of lung inflammation. Am J Respir Cell Mol Biol. (2015) 52:641–52. doi: 10.1165/rcmb.2014-0327OC
194. Cheng C, Wang S, Ye P, Huang X, Liu Z, Wu J, et al. 'Default' generated neonatal regulatory T cells are hypomethylated at conserved non-coding sequence 2 and promote long-term cardiac allograft survival. Immunology. (2014) 143:618–30. doi: 10.1111/imm.12343
195. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. (2007) 13:1299–307. doi: 10.1038/nm1652
196. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. (2004) 199:1455–65. doi: 10.1084/jem.20040139
197. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. (2016) 126:1413–24. doi: 10.1172/JCI82771
198. Boardman D, Maher J, Lechler R, Smyth L, Lombardi G. Antigen-specificity using chimeric antigen receptors: the future of regulatory T-cell therapy? Biochem Soc Trans. (2016) 44:342–8. doi: 10.1042/BST20150247
199. Rowe JH, Ertelt JM, Xin L, Way S. Regulatory T cells and the immune pathogenesis of prenatal infection. Reproduction. (2013) 2013:262. doi: 10.1530/REP-13-0262
Keywords: pregnancy, preeclampsia, placenta, embryo implantation, maternal vascular adaptation, inflammation, Treg cells, immune tolerance
Citation: Robertson SA, Green ES, Care AS, Moldenhauer LM, Prins JR, Hull ML, Barry SC and Dekker G (2019) Therapeutic Potential of Regulatory T Cells in Preeclampsia—Opportunities and Challenges. Front. Immunol. 10:478. doi: 10.3389/fimmu.2019.00478
Received: 06 November 2018; Accepted: 21 February 2019;
Published: 21 March 2019.
Edited by:
Julia Szekeres-Bartho, University of Pécs, HungaryReviewed by:
Gerard Chaouat, INSERM U976 Immunologie, Dermatologie, Oncologie, FranceSurendra Sharma, Women & Infants Hospital of Rhode Island, United States
Copyright © 2019 Robertson, Green, Care, Moldenhauer, Prins, Hull, Barry and Dekker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah A. Robertson, c2FyYWgucm9iZXJ0c29uQGFkZWxhaWRlLmVkdS5hdQ==