 Ainara Castellanos-Rubio
Ainara Castellanos-Rubio Sankar Ghosh
Sankar Ghosh- 1Immunogenetics Research Laboratory, Department of Genetics, Physical Anthropology and Animal Physiology, University of the Basque Country, UPV/EHU, Leioa, Spain
- 2Functional Studies in Celiac Disease Group, BioCruces Health Research Institute, Barakaldo, Spain
- 3Department of Microbiology and Immunology, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, United States
Immune-mediated diseases, such as celiac disease, type 1 diabetes or multiple sclerosis, are a clinically heterogeneous group of diseases that share many key genetic triggers. Although the pathogenic mechanisms responsible for the development of immune mediated disorders is not totally understood, high-throughput genomic studies, such as GWAS and Immunochip, performed in the past few years have provided intriguing hints about underlying mechanisms and pathways that lead to disease. More than a hundred gene variants associated with disease susceptibility have been identified through such studies, but the progress toward understanding the underlying mechanisms has been slow. The majority of the identified risk variants are located in non-coding regions of the genome making it difficult to assign a molecular function to the SNPs. However, recent studies have revealed that many of the non-coding regions bearing disease-associated SNPs generate long non-coding RNAs (lncRNAs). LncRNAs have been implicated in several inflammatory diseases, and many of them have been shown to function as regulators of gene expression. Many of the disease associated SNPs located in lncRNAs modify their secondary structure, or influence expression levels, thereby affecting their regulatory function, hence contributing to the development of disease.
Introduction
Immune mediated disorders, such as celiac disease (CeD), inflammatory bowel diseases (IBD), atherosclerosis, rheumatoid arthritis (RA), type 1 diabetes (T1D) or multiple sclerosis (MS) among others, are a group of clinically heterogeneous diseases caused by dysfunction of the immune system. These disorders, share underlying pathogenic mechanisms that are not totally understood, although the general belief is that they develop due to an imbalance in the interaction between genetic and environmental factors (1, 2).
Immune mediated disorders share dysregulation of many key regulatory pathways and techniques, such as genome wide association studies (GWAS) coupled with next generation sequencing (NGS), have significantly increased our knowledge of genetic factors underlying such diseases (3). In the past decade or so, hundreds of risk alleles, both common and disease specific, have been identified by GWAS. Moreover, using the Immunochip platform in which 200,000 polymorphisms in 186 immune disease related regions were analyzed, additional immune disease associated variants were identified that revealed common susceptibility loci for several of these diseases (4–7). While these studies have helped identify immune disease conferring gene variants, the progress toward the understanding of the underlying mechanisms has remained limited. This difficulty is particularly exacerbated by the fact that around 90% of the SNPs associated with these diseases are located in non-coding regions, making it difficult to link them to specific biological pathways (8–10).
Advances in the sequencing and annotation of the human genome have revealed that many non-coding regions of the genome encode long non-coding RNAs (lncRNAs). The importance of lncRNA molecules in different biological processes is beginning to be appreciated although there is much that remains to be understood. LncRNAs are RNA molecules longer than 200 bp in length with no protein-coding potential. LncRNA expression is generally cell-lineage specific and they have diverse and still not very well-characterized mechanisms of action. The emerging view is that lncRNAs are fundamental regulators of transcription as they have been shown to control every level of the gene expression program. LncRNAs have been shown to control processes like protein synthesis, RNA maturation, and transport to regulate genes post-transcriptionally and they are also involved in transcriptional gene silencing through regulating the chromatin structure (11–13).
Many lncRNAs are enriched for disease-associated SNPs, suggesting that these SNPs might alter the function of lncRNAs e.g., by altering their secondary structures (14). Moreover, expression profile analyses of lncRNAs located in autoimmune disease-associated regions showed that lncRNAs are enriched in these loci, suggesting that lncRNAs may be crucial for interpreting GWAS findings (15). Disease associated SNPs can modify the lncRNA sequence or alter their gene expression levels, affecting their regulatory capacity, and alterations in the structure and function of lncRNAs have been associated with several immune-mediated diseases. However, the precise mechanism by which lncRNA variants contribute to the pathogenesis of disease remains unknown in the majority of cases (16).
As previously done with protein coding genes, intergenic SNPs have been analyzed in the context of lncRNA expression quantitative trait loci (eQTLs), namely, genetic variants that would explain variation in the lncRNA expression levels (17). More than 100 cis-eQTLs have been found in different tissues that appear to regulate the expression of nearby lncRNAs. In general, these eQTLs are lncRNA specific and do not regulate the expression of neighboring protein coding genes, but since lncRNAs can regulate the expression of protein-coding genes, both, located close by or farther away in the genome, it is possible that these SNPs also influence the function of protein-coding genes. Moreover, a considerable number of the lncRNA cis-eQTLs belong to disease-associated SNPs, making lncRNAs a potential link between non-coding SNPs and the expression of protein-coding genes (18–21) (Figure 1A).
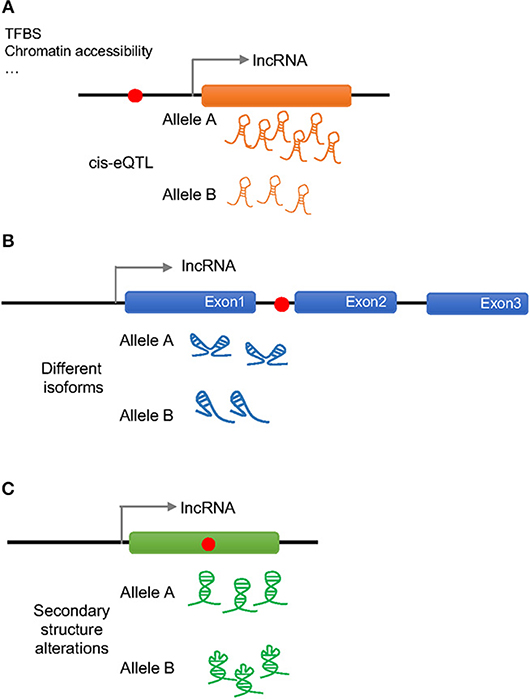
Figure 1. Possible effects of a disease associated SNP on lncRNA regulation and function. Red dots represent disease associated SNPs. (A) A SNP in the promoter region of a lncRNA can change the amount of transcribed lncRNA (cis-eQTL) by altering the binding of transcription factor binding sites (TFBS) or the chromatin accessibility altering downstream effects. (B) An intronic SNP in a lncRNA can influence the splicing of the lncRNA generating different isoforms that will act differently. (C) A SNP in the exonic sequence of a lncRNA can change its secondary structure altering the binding to the molecular partners.
Disease associated SNPs have been also suggested to be involved in “splicing models,” in which the presence of different alleles would influence the splicing of the lncRNA by regulating exon skipping (22) (Figure 1B). In this way, different isoforms of the lncRNA would present different affinity for their binding partners affecting the regulation of downstream events. It has been observed, that when human endothelial cells are stimulated with lipopolysaccharide (LPS) several lncRNAs show splice variant-specific expression at different stimulation time points (23), underlining the importance of SNP regulated splicing in lncRNA function.
As it is generally believed that lncRNA molecules adopt specific secondary and tertiary structures to execute their functions, it is likely that disease associated SNPs have an impact on lncRNA structure (Figure 1C). The analysis of secondary structures has largely been performed using computational tools, and several tools can predict changes in lncRNA structures caused by the presence of different alleles of a certain SNP (24–26). For example, GWAS SNPs associated with IBD and T1D have been shown to disrupt the structure of an lncRNA implicated in the pathogenesis of both diseases, which associates with the BACH2 gene (27). However, this field is still in its infancy and the principles that underlie the impact of SNPs on lncRNA structure and function remains to be fully established.
In this article, we have reviewed the link between four intergenic GWAS variants that are located within lncRNA sequences, which have been associated with inflammatory diseases, and we discuss the studies that have been carried out to characterize their contribution to the development of disease pathogenesis. As of now, these four inflammatory-disease associated SNPs are the best mechanistically characterized in the context of lncRNA function.
Lnc13 and Celiac Disease Susceptibility
Celiac disease is a complex, chronic, immune-mediated disease that affects ~1% of the population and develops in genetically susceptible individuals in response to ingested gluten proteins from wheat, barley, and rye (28). The strongest genetic association, around 40% of the genetic risk (29), maps to the human leukocyte antigen (HLA) region in chromosome 6p21, and virtually all CeD patients carry HLA-DQ2 or HLA-DQ8 heterodimers (30, 31).
Two GWA studies, together with the Immunochip project, have identified a total of 39 non-HLA loci associated with the genetic risk of CeD (32–34). Only 3 of the CeD associated SNPs are linked to protein-altering variants located in exonic regions, although some potentially causative coding genes have been proposed, mainly related to the immune response, due to the existence of signals near their 5′ or 3′ regulatory regions. Although some lncRNAs have been related to celiac disease pathogenesis due to the location of an associated SNP within their transcriptional region, and differential expression found in samples from CeD patients (35, 36), the exact mechanism by which they contribute to disease development is not understood.
The only functionally characterized lncRNA harboring a CeD associated intergenic SNP so far has been found linked to the NF-κB pathway (37), which is known to be constitutively active in the CeD mucosa (38, 39). This lncRNA, named lnc13, harbors the SNP rs917997 and it is located in the associated region 2q12, with the sense sequence overlapping the coding gene IL18RAP that had been proposed, but never firmly confirmed, as the functional candidate gene in the region (40–42).
This lncRNA is expressed in different human cells and tissues, including mononuclear cells in the lamina propria, where it was observed to be localized in the nucleus. Lnc13 quantification in small intestinal biopsy samples from celiac patients and controls showed markedly lower levels of this lncRNA in CeD samples, contrary to the expression of the coding mRNA, IL18RAP (42). In fact, it is known that IL18RAP expression is induced in response to inflammation via NF-κB in certain immune cells (43). The characterization of the regulation, function and mechanisms of action of lnc13 revealed that under basal conditions lnc13 represses the expression of certain CeD related genes (STAT1, MYD88, IL1RA, and TRAF2) via its interaction with hnRNPD (Heterogeneous Nuclear Ribonucleoprotein D), a nuclear AU1 rich RNA binding protein, and HDAC1 (Histone Deacetylase 1), a histone deacetylase which negatively regulates transcription, proteins on the chromatin (Figure 2A). In response to inflammatory stimuli, lnc13 is degraded by Decapping enzyme 2 (DCP2), releasing the protein complex from chromatin and allowing the expression of the proinflammatory genes (37).
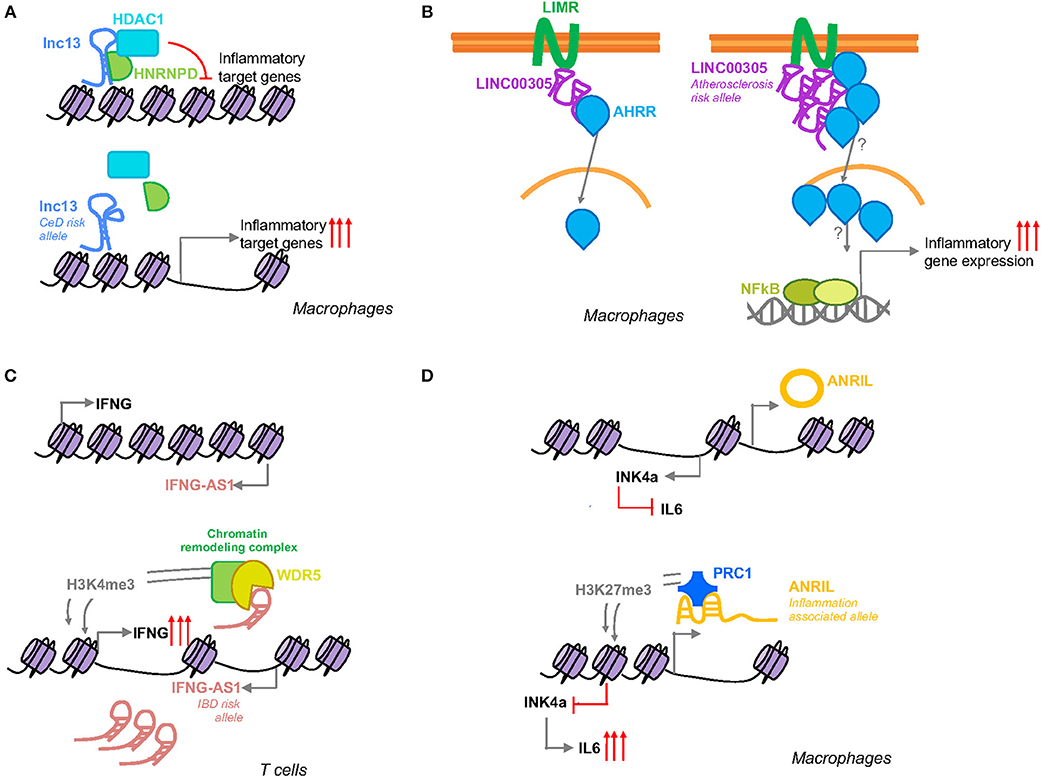
Figure 2. Schematic representation of the function of inflammation associated SNP harboring lncRNAs. (A) Lnc13 harbors a CeD associated SNP that changes the secondary structure of the lncRNA modifying its binding with the proteins hnRNPD and HDAC1 and regulating the expression of disease related inflammatory genes. (B) LINC00305 interacts with the transmembrane protein LIMR facilitating the binding of this protein to AHRR that in turn induces the translocation of the latter to the nucleus inducing NFκB and subsequent inflammatory gene expression. Atherosclerosis patients present higher levels of this lincRNA which could be influenced by a SNP located in the promoter region of LINC00305. (C) IFNG-AS1 is closely located to IFNG gene. Activation of its transcription leads to induction of IFNG by WDR5 mediated H3K4me3 methylation. IBD patients present higher levels of IFNG-AS1 that could be related to a disease associated SNP located in the enhancer region of the lncRNA. (D) Suggested SNP related splicing model for ANRIL mediated inflammation regulation. The inflammation associated allele will affect ANRIL splicing generating a linear ANRIL that will interact with a member of the PRC1 complex mediating an epigenetic transcriptional repression of the INK4a gene via H3K27me3.
Although the GWAS disease association has been generally attributed to the SNP rs917997 (33), located 1.5 kb away from the coding gene, linkage analysis of the lnc13 region revealed that there are a total of six SNPs in total linkage disequilibrium within the lncRNA sequence. The nucleotide changes in lnc13 cause a disruption of the secondary structure of this lncRNA decreasing its affinity to bind hnRNPD and chromatin, resulting in higher expression of the proinflammatory genes (37). Thus, patients with the risk haplotype have a higher basal expression of CeD related inflammatory genes, thereby increasing their predisposition to develop the disease (Figure 2A).
The SNPs in lnc13 have also been associated with other inflammatory diseases, such as T1D, Crohn's disease or rheumatoid arthritis (44–46). Interestingly, while the risk signal in CeD corresponds to the T allele, the C allele is the risk allele in T1D, suggesting that the function of the lncRNA may be cell specific, but equally affected by the SNPs in different inflammatory diseases.
In summary, it is known that lnc13 and the CeD associated SNP rs917997 contribute to the pathology of celiac disease by regulating expression of certain immune related genes that play a role in the development of inflammation in the intestinal epithelia. However, other cell-type and allele specific functions cannot be excluded, due to the association of this SNP with other inflammatory conditions.
LINC00305 and Inflammatory Response in Atherosclerosis
Atherosclerosis is a complex, chronic disease of the arterial wall triggered by multiple factors including amongst others, inflammation and lipid metabolism (47). Monocyte-mediated inflammation plays a critical role in atherosclerosis due to the secretion of proinflammatory cytokines and amplification of local inflammation (48).
Although a locus in the 9p21.3 region is the strongest genetic factor for atherosclerosis described so far, GWAS have led to the identification of a substantial number of additional genetic loci associated with atherosclerosis and atherosclerosis-related complications (49). Analysis of atherosclerosis GWAS SNPs, revealed that the SNP rs2850711 is located within an intronic sequence of a long intergenic non-coding RNA named LINC00305. This lincRNA was found to be overexpressed in atherosclerotic plaques and in peripheral blood mononuclear cells (PBMCs) from patients, supporting its role in the disease. Analysis of LINC00305 in the cell types that composed the plaques revealed that monocytes are the primary cell type expressing this lncRNA and that its expression was induced in response to stimulation with lipopolysaccharide (LPS). Further functional studies, demonstrated that LINC00305 interacts with the transmembrane protein LIMR (Lipocalin-1 Interacting Membrane Receptor) enhancing its interaction with AHRR (Aryl-Hydrocarbon Receptor Repressor) which at the same time promotes NF-κB activation and subsequent inflammatory gene expression (Figure 2B) (50).
The development of atherosclerotic plaques is induced by the change in phenotype of the vascular smooth muscle cells in response to the cytokines secreted by inflammatory cells (51). To investigate the functional significance of LINC00305 in the pathogenesis of atherosclerosis, the lincRNA was stably overexpressed in human monocytes and these were co-cultured with human aortic smooth muscle cells. It was observed that those muscle cells that were cultured in the presence of the monocytes overexpressing LINC00305 showed less expression of their basal markers, thus suggesting that they were switching to the pathogenic phenotype (50). Independent studies, have shown that overexpression of this lincRNA induces apoptosis in hypoxia induced endothelial cells (52). Further analysis of the role of this lincRNA in the regulation of apoptosis, revealed that it acts as an endogenous sponge for miR-136 which had been previously related to apoptosis in the context of atherosclerosis (52, 53).
Although it is quite clear that LINC00305 plays a functional role in development of atherosclerosis by inducing production of inflammatory cytokines in monocytes, and by regulating apoptosis via miR-136, the role of the associated SNP in the function of the lincRNA remains to be elucidated. The GWAS SNP rs2850711 is transmitted in a linkage disequilibrium (LD) block of a total of 16 SNPs, all of which are located within introns, and hence probably do not influence the secondary structure of the lincRNA. Although there are no in vitro molecular studies evaluating this possibility, it is noteworthy that one of the associated SNPs lies within an experimentally confirmed USF2 (Upstream Transcription Factor 2) binding region (54). As USF2 is a protein that has been associated with cholesterol metabolism and atherosclerosis development (55), further mechanistic studies assessing the contribution of the SNP alleles in the function and regulation of the lincRNA are necessary to understand how the SNPs in non-coding regions identified by GWAS influence the inflammatory environment in atherosclerosis.
IFNG-AS1 (NeST or Tmevpg1) and Ulcerative Colitis
Inflammatory bowel diseases (IBD) are chronic common inflammatory gastrointestinal disorders clinically comprised of Crohn's disease (CD) and ulcerative colitis (UC) (56). These diseases are believed to develop due to inappropriate inflammatory responses to intestinal microbes and foreign antigens in genetically susceptible individuals (57, 58).
Meta-analyses of multiple GWAS have implicated 163 genetic loci in IBD susceptibility. Although functional analysis of the associated SNPs have revealed multiple pathophysiological mechanisms, the function for many of the genes in close association with these loci are yet to be determined (59, 60). It has been observed that several lncRNAs are differentially expressed in inflammatory bowel disease, and that the expression profiling of lncRNAs can be useful to stratify IBD patients from healthy controls (27, 61, 62).
When comparing the genomic location of differentially expressed lncRNAs with those of IBD susceptibility loci, IFNG-AS1 (also called NeST or Tmevpg1) was found to fulfill both criteria (61). The IBD associated SNP rs7134599 is located in the region 12q15 in close proximity to the inflammatory cytokine IFNG. This SNP is in total LD with 10 other SNPs within the lncRNA gene. Additionally, IFNG-AS1 is significantly overexpressed in intestinal samples of ulcerative colitis patients and its expression appears to correlate with the elevated levels of IFNG, IL1, IL6, and TNF-α observed in patients (61). Increased expression level of this lncRNA has also been related to other inflammatory diseases, such as Hashimoto's thyroiditis or Sjögren syndrome (63, 64) although the mechanisms by which it contributes to development of these diseases remain unclear.
IFNG-AS1 gene was first related with the immune response in the context of susceptibility to persistent Theiler's virus infection. It was observed that IFNG-AS1 is expressed in immune cells of mouse and human origin and it was speculated that this lncRNA may regulate the expression of IFNG (65). Further studies, demonstrated that IFNG-AS1 contributes to IFNG expression regulation as part of the Th1 differentiation program and that T-bet guides epigenetic remodeling of the lncRNA enhancers, leading to recruitment of stimulus-inducible transcription factors, such as NF-κB (66, 67). More recently, it was observed that different mouse strains with different genotype composition of IFNG-AS1 were sufficient to confer disparate immune-related phenotypes. Specifically, certain alleles, derived from SJL/J mouse strain, were responsible for the failure to clear Theiler's virus, but at the same time they conferred resistance to lethal infection with Salmonella enterica Typhimurium and induced synthesis of Ifng in CD8+ T cells. Functional analysis performed in this same study, showed that IFNG-AS1 is a nuclear lncRNA that can act in trans. IFNG-AS1 binds WDR5, a component of active chromatin remodeling complexes increasing H3K4me3 methylation which in turn programs an active chromatin state that induces Ifng gene transcription (68) (Figure 2C).
Although IFNG-AS1 is differentially expressed in IBD samples and harbors a disease associated SNP (61) the exact impact of the different alleles in the development of the disease has not been assessed so far. In silico interaction evaluation of human IFNG-AS1 lncRNA and WDR5 protein using CatRapid (69), an algorithm that estimates the binding propensity of protein-RNA pairs, states that these two molecules are also able to interact, suggesting that it may act in a similar way as described in mice. Analysis of the location of the SNPs that are in LD with the associated SNP rs7134599 reveals that all of them are located in intronic regions, suggesting that they will most likely not affect the secondary structure of the RNA molecule. However, analysis of the region using HaploReg v4.1 (70) shows that 4 of the SNPs are located within enhancer histone marks and all of them are predicted to disturb a protein binding motif that could change the regulation of the lncRNA expression, thereby influencing the levels of IFNG.
Thus, INFG-AS1 is clearly involved in the immune response and inflammatory processes involved in disease, and although in silico data point to a disturbance of lncRNA expression regulation mediated by the IBD associated SNPs uncovered by GWAS, the true relevance of these SNPs have still to be experimentally confirmed.
ANRIL and Inflammation
The antisense non-coding RNA in the INK4 locus or ANRIL was first described in melanoma patients (71) and since its discovery it has been shown to be involved in several types of cancers (72). This lncRNA is located in the 9p21 region, that has been associated by GWAS not only to cancer but also to other diseases that are related with inflammation, such as coronary artery disease (73) or type 2 diabetes (T2D) (74). ANRIL is expressed as either linear or circular forms, that have been observed to have opposing effects in disease development (75), making the deciphering of the functionality of this lncRNA and the involvement of its related SNPs highly complicated.
ANRIL has been described to interact with CBX7 (Chromobox 7), one of the members of the polycomb repressive complex 1 (PRC1). CBX7 binds both, ANRIL and H3K27me3 to mediate an epigenetic transcriptional repression of the INK4a (Inhibitor of CDK4) gene, which is located adjacent to the ANRIL gene (Figure 2D) (76). INK4a is a cell cycle inhibitor that is lost in a wide spectrum of cancers (77), but it has been also been reported to act as an anti-inflammatory molecule that is able to suppress the production of IL-6 in macrophages (78). ANRIL itself has been also shown to regulate the inflammatory response by its interaction with the YY1 (Yin Yang 1) protein (79), a transcription repressor involved in cancer development and immune processes (80).
The influence on gene expression of the variants within ANRIL region have been analyzed in a variety of tissues and cells, but the results have been inconsistent (75). Several SNPs have been described to be involved in alternative splicing events, and modifying ANRIL structure has been suggested to lead to changes in its function and consequent regulation of downstream inflammatory genes (Figure 2D) (22). Although the exact mechanism by which the SNPs within ANRIL confer susceptibility to disease has not been firmly established, it seems clear that the disease associated SNPs are related with ANRIL expression, suggesting that modulation of its expression mediates disease susceptibility.
One example of SNP-dependent ANRIL related inflammation is the correlation between the lead periodontitis associated SNP, rs1333048, and the levels of the C-reactive protein. Periodontitis is a complex, chronic inflammatory disease associated with increased concentration of high-sensitive C-reactive protein (hsCRP), a marker for systemic inflammation. It was found that AA-genotype of ANRIL rs1333048 is associated with significantly elevated hsCRP plasma levels in patients with periodontitis (81). However, the functional relationship between the SNP, ANRIL and the hsCRP molecule has not been clearly established.
Another disease in which ANRIL has been functionally implicated is Type 2 diabetes (T2D). Although the major causes of T2D are insulin resistance and beta cell dysfunction, recent evidence implicates the immune system in the pathogenesis of this disease that can be considered as an autoinflammatory disease (82). T2D associated SNPs in the ANRIL locus were evaluated, and it was observed that the risk genotype was correlated with increased levels of ANRIL expression. Moreover, although the associated SNPs did not seem to influence insulin secretion, it was observed that they affect human beta cell proliferation index, with homozygous risk alleles showing approximately half of the proliferation capacity observed in the presence of the protective alleles (83). Although this study suggests that ANRIL lncRNA may play a role in human islets and uncovers a link between T2D associated SNPs and beta cell proliferation, once again, the functional relationship between the SNPs and the biological process is still not understood.
Additionally, ANRIL is significantly downregulated in the inflamed intestinal mucosa of Crohn's and inflammatory bowel disease patients (84). At the same time its reduced levels in rats have been related to prevention of coronary atherosclerosis due to lower expression of inflammatory factors (85) which are upregulated in patients with coronary artery disease (86).
It therefore seems clear that disease associated SNPs in ANRIL lncRNA influence its function in the context of inflammatory diseases. However, the involvement of ANRIL in inflammation and the influence of the GWAS SNPs in the function of the different isoforms of ANRIL needs further investigation.
Concluding Remarks
Although our knowledge about the genetic variants contributing to immune mediated diseases has increased considerably in the last decade, the intergenic location of the great majority of the associated SNPs has made it difficult to decipher their functional roles in disease development. As disease associated SNPs are enriched within lncRNAs, and as many of these RNA molecules have been implicated in the regulation of inflammatory processes, a new field of study focused on the influence of disease-associated SNPs in the function of inflammation-related lncRNAs has been opened. Interestingly, such lncRNAs have been linked to major immune-mediated diseases as celiac disease, type 1 diabetes or rheumatoid arthritis. The experimental approaches utilized so far have been mainly focused on the expression analysis of the SNP harboring lncRNA in diseased tissues, but functional studies evaluating the contribution of each allele to lncRNA function, and thus to disease development, is mostly missing. In general, the function of the lncRNA itself, and the mechanisms by which they contribute to inflammatory disease development, are mostly uncharacterized. Analyzing the position and the linkage disequilibrium block of the associated SNP within the lncRNA sequence can help predict the functional impact of the allelic variant. Associated SNPs may not only affect the expression of the lncRNA itself, but also their splicing, their secondary structure or their ability to regulate expression of downstream genes. Thus, approaches that evaluate the functional differences of the lncRNA alleles are necessary in order to understand how the disease-associated SNPs affect the function of such inflammation related lncRNAs.
As our knowledge about the molecular mechanisms by which the inflammation related lncRNAs exert their biological functions increases, so will our understanding of how the disease associated SNPs influence lncRNA function thereby opening up the possibility for targeting such lncRNAs for diagnostic and therapeutic purposes.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
AC-R is funded by an Ikerbasque Research Fellowship, SG is funded by grant R01 DK102180 (NIH).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Cotsapas C, Hafler DA. Immune-mediated disease genetics: the shared basis of pathogenesis. Trends Immunol. (2013) 34:22–6. doi: 10.1016/J.IT.2012.09.001
2. Wu X, Chen H, Xu H. The genomic landscape of human immune-mediated diseases. J Hum Genet. (2015) 60:675–81. doi: 10.1038/jhg.2015.99
3. Diaz-Gallo L-M, Martin J. Common genes in autoimmune diseases: a link between immune-mediated diseases. Expert Rev Clin Immunol. (2012) 8:107–9. doi: 10.1586/eci.11.90
4. Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. (2013) 14:661–73. doi: 10.1038/nrg3502
5. Ricaño-Ponce I, Wijmenga C. Mapping of immune-mediated disease genes. Annu Rev Genomics Hum Genet. (2013) 14:325–53. doi: 10.1146/annurev-genom-091212-153450
6. Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. (2008) 322:881–8. doi: 10.1126/science.1156409
7. Burton PR, Clayton DG, Cardon LR, Craddock N, Deloukas P, Duncanson A, et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. doi: 10.1038/nature05911
8. Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. (2015) 518:337–43. doi: 10.1038/nature13835
9. Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin. (2015) 8:57. doi: 10.1186/s13072-015-0050-4
10. Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci USA. (2009) 106:9362–7. doi: 10.1073/pnas.0903103106
11. Chen YG, Satpathy AT, Chang HY. Gene regulation in the immune system by long noncoding RNAs. Nat Immunol. (2017) 18:962–72. doi: 10.1038/ni.3771
12. Mowel WK, Kotzin JJ, McCright SJ, Neal VD, Henao-Mejia J. Control of immune cell homeostasis and function by lncRNAs. Trends Immunol. (2018) 39:55–69. doi: 10.1016/j.it.2017.08.009
13. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. (2011) 43:904–14. doi: 10.1016/j.molcel.2011.08.018
14. Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. (2013) 9:e1003569. doi: 10.1371/journal.pgen.1003569
15. Hrdlickova B, Kumar V, Kanduri K, Zhernakova DV, Tripathi S, Karjalainen J, et al. Expression profiles of long non-coding RNAs located in autoimmune disease-associated regions reveal immune cell-type specificity. Genome Med. (2014) 6:88. doi: 10.1186/s13073-014-0088-0
16. Li X, Wu Z, Fu X, Han W. lncRNAs: Insights into their function and mechanics in underlying disorders. Mutat Res Mutat Res. (2014) 762:1–21. doi: 10.1016/j.mrrev.2014.04.002
17. Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans R Soc Lond B Biol Sci. (2013) 368:20120362. doi: 10.1098/rstb.2012.0362
18. Popadin K, Gutierrez-Arcelus M, Dermitzakis ET, Antonarakis SE. Genetic and epigenetic regulation of human lincRNA gene expression. Am J Hum Genet. (2013) 93:1015–26. doi: 10.1016/j.ajhg.2013.10.022
19. McDowell I, Pai A, Guo C, Vockley CM, Brown CD, Reddy TE, et al. Many long intergenic non-coding RNAs distally regulate mRNA gene expression levels. bioRxiv. (2016) 044719. doi: 10.1101/044719
20. Tan JY, Smith AAT, Ferreira da Silva M, Matthey-Doret C, Rueedi R, Sönmez R, et al. cis-Acting complex-trait-associated lincrna expression correlates with modulation of chromosomal architecture. Cell Rep. (2017) 18:2280–8. doi: 10.1016/J.CELREP.2017.02.009
21. Kumar V, Westra H-J, Karjalainen J, Zhernakova DV, Esko T, Hrdlickova B, et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. (2013) 9:e1003201. doi: 10.1371/journal.pgen.1003201
22. Aguilo F, Di Cecilia S, Walsh MJ. Long non-coding RNA ANRIL and polycomb in human cancers and cardiovascular disease. Curr Top Microbiol Immunol. (2016) 394:29–39. doi: 10.1007/82_2015_455
23. Chowdhury IH, Narra HP, Sahni A, Khanipov K, Schroeder CLC, Patel J, Fofanov et al. Expression profiling of long noncoding RNA splice variants in human microvascular endothelial cells: lipopolysaccharide effects in vitro. Mediators Inflamm. (2017) 2017:1–18. doi: 10.1155/2017/3427461
24. Gong J, Liu W, Zhang J, Miao X, Guo A-Y. lncRNASNP: a database of SNPs in lncRNAs and their potential functions in human and mouse. Nucleic Acids Res. (2015) 43:D181–6. doi: 10.1093/nar/gku1000
25. Ren C, An G, Zhao C, Ouyang Z, Bo X, Shu W. Lnc2Catlas: an atlas of long noncoding RNAs associated with risk of cancers. Sci Rep. (2018) 8:1909. doi: 10.1038/s41598-018-20232-4
26. Halvorsen M, Martin JS, Broadaway S, Laederach A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. (2010) 6:e1001074. doi: 10.1371/journal.pgen.1001074
27. Mirza AH, Kaur S, Brorsson CA, Pociot F. Effects of GWAS-associated genetic variants on lncRNAs within IBD and T1D candidate loci. PLoS ONE. (2014) 9:e0105723. doi: 10.1371/journal.pone.0105723
28. Husby S, Koletzko S, Korponay-szabó IR, Mearin Ml, Phillips A, Shamir R, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr. (2012) 54:136–60. doi: 10.1097/mpg.0b013e31821a23d0
29. Gutierrez-Achury J, Zhernakova A, Pulit SL, Trynka G, Hunt KA, Romanos J, et al. Fine mapping in the MHC region accounts for 18% additional genetic risk for celiac disease. Nat Genet. (2015) 47:577–8. doi: 10.1038/ng.3268
30. Spurkland A, Sollid LM, Polanco I, Vartdal F, Thorsby E. HLA-DR and -DQ genotypes of celiac disease patients serologically typed to be non-DR3 or non-DR5/7. Hum Immunol. (1992) 35:188–92. doi: 10.1016/0198-8859(92)90104-U
31. Sollid LM, Markussen G, Ek J, Gjerde H, Vartdal F, Thorsby E. Evidence for a primary association of celiac disease to a particular HLA-DQ alpha/beta heterodimer. J Exp Med. (1989) 169:345–50. doi: 10.1084/JEM.169.1.345
32. van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, et al. A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet. (2007) 39:827–9. doi: 10.1038/ng2058
33. Hunt KA, Zhernakova A, Turner G, Heap GAR, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. (2008) 40:395–402. doi: 10.1038/ng.102
34. Dubois PCA, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. (2010) 42:295–302. doi: 10.1038/ng.543
35. Plaza-Izurieta L, Fernandez-Jimenez N, Irastorza I, Jauregi-Miguel A, Romero-Garmendia I, Vitoria JC, et al. Expression analysis in intestinal mucosa reveals complex relations among genes under the association peaks in celiac disease. Eur J Hum Genet. (2015) 23:1100–5. doi: 10.1038/ejhg.2014.244
36. Santin I, Jauregi-miguel A, Velayos T, Castellanos-rubio A, Garcia-etxebarria K, Romero-garmendia I, et al. Celiac disease-associated lncRNA named HCG14 regulates NOD1 expression in intestinal cells. J Pediatr Gastroenterol Nutr. (2018) 67:225–31. doi: 10.1097/mpg.0000000000001970
37. Castellanos-Rubio A, Fernandez-Jimenez N, Kratchmarov R, Luo X, Bhagat G, Green PHR, et al. A long noncoding RNA associated with susceptibility to celiac disease. Science. (2016) 352:91–5. doi: 10.1126/science.aad0467
38. Maiuri MC, De Stefano D, Mele G, Fecarotta S, Greco L, Troncone R, et al. Nuclear factor κB is activated in small intestinal mucosa of celiac patients. J Mol Med. (2003) 81:373–9. doi: 10.1007/s00109-003-0440-0
39. Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L, Irastorza I, Elcoroaristizabal X, Jauregi-Miguel A, et al. Coregulation and modulation of NF B-related genes in celiac disease: uncovered aspects of gut mucosal inflammation. Hum Mol Genet. (2014) 23:1298–310. doi: 10.1093/hmg/ddt520
40. Heap GA, Trynka G, Jansen RC, Bruinenberg M, Swertz MA, Dinesen LC, et al. Complex nature of SNP genotype effects on gene expression in primary human leucocytes. BMC Med Genomics. (2009) 2:1. doi: 10.1186/1755-8794-2-1
41. Myhr CB, Hulme MA, Wasserfall CH, Hong PJ, Lakshmi PS, Schatz DA, et al. The autoimmune disease-associated SNP rs917997 of IL18RAP controls IFNγ production by PBMC. J Autoimmun. (2013) 44:8–12. doi: 10.1016/J.JAUT.2013.06.001
42. Plaza-Izurieta L, Castellanos-Rubio A, Irastorza I, Fernández-Jimenez N, Gutierrez G, CEGEC Bilbao JR. Revisiting genome wide association studies (GWAS) in coeliac disease: replication study in Spanish population and expression analysis of candidate genes. J Med Genet. (2011) 48:493–6. doi: 10.1136/jmg.2011.089714
43. Sareneva T, Julkunen I, Matikainen S. IFN-alpha and IL-12 induce IL-18 receptor gene expression in human NK and T cells. J Immunol. (2000) 165:1933–8. Available online at: http://www.ncbi.nlm.nih.gov/pubmed/10925275 (accessed January 17, 2019).
44. Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JHM, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. (2008) 359:2767–77. doi: 10.1056/NEJMoa0807917
45. Zhernakova A, Festen EM, Franke L, Trynka G, van Diemen CC, Monsuur AJ, et al. Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet. (2008) 82:1202–10. doi: 10.1016/J.AJHG.2008.03.016
46. Coenen MJH, Trynka G, Heskamp S, Franke B, van Diemen CC, Smolonska J, et al. Common and different genetic background for rheumatoid arthritis and coeliac disease. Hum Mol Genet. (2009) 18:4195–203. doi: 10.1093/hmg/ddp365
47. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. (2011) 473:317–25. doi: 10.1038/nature10146
48. Ghattas A, Griffiths HR, Devitt A, Lip GYH, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol. (2013) 62:1541–51. doi: 10.1016/J.JACC.2013.07.043
49. Holdt LM, Teupser D. From genotype to phenotype in human atherosclerosis–recent findings. Curr Opin Lipidol. (2013) 24:410–8. doi: 10.1097/MOL.0b013e3283654e7c
50. Zhang D-D, Wang W-T, Xiong J, Xie X-M, Cui S-S, Zhao Z-G, et al. Long noncoding RNA LINC00305 promotes inflammation by activating the AHRR-NF-κB pathway in human monocytes. Sci Rep. (2017) 7:46204. doi: 10.1038/srep46204
51. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
52. Zhang B-Y, Jin Z, Zhao Z. Long intergenic noncoding RNA 00305 sponges miR-136 to regulate the hypoxia induced apoptosis of vascular endothelial cells. Biomed Pharmacother. (2017) 94:238–43. doi: 10.1016/J.BIOPHA.2017.07.099
53. Zhang C, Kang K, Li X, Xie B. MicroRNA-136 promotes vascular muscle cell proliferation through the ERK1/2 pathway by targeting PPP2R2A in atherosclerosis. Curr Vasc Pharmacol. (2015) 13:405–12. Available onlinev at: http://www.ncbi.nlm.nih.gov/pubmed/25409743 (accessed May 18, 2018).
54. Consortium TEP. An integrated encyclopedia of DNA elements in the human genome. Nature. (2012) 489:57–74. doi: 10.1038/nature11247
55. Pan X, Jiang X-C, Hussain MM. Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation. (2013) 128:1758–69. doi: 10.1161/CIRCULATIONAHA.113.002885
56. Fusunyan RD, Sanderson IR. Inflammatory bowel disease. In: Peter J, editor. Encyclopedia of Immunology. London: Elsevier (1998). p. 1375–81. doi: 10.1006/rwei.1999.0351
57. Eckburg PB, Relman DA. The role of microbes in Crohn's disease. Clin Infect Dis. (2007) 44:256–62. doi: 10.1086/510385
58. Weinstock JV. Helminths and mucosal immune modulation. Ann N Y Acad Sci. (2006) 1072:356–64. doi: 10.1196/annals.1326.033
59. Ye BD, McGovern DPB. Genetic variation in IBD: progress, clues to pathogenesis and possible clinical utility. Expert Rev Clin Immunol. (2016) 12:1091–107. doi: 10.1080/1744666X.2016.1184972
60. Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. (2012) 491:119–24. doi: 10.1038/nature11582
61. Padua D, Mahurkar-Joshi S, Law IKM, Polytarchou C, Vu JP, Pisegna JR, et al. A long noncoding RNA signature for ulcerative colitis identifies IFNG-AS1 as an enhancer of inflammation. Am J Physiol Gastrointest Liver Physiol. (2016) 311:G446–57. doi: 10.1152/ajpgi.00212.2016
62. Wu F, Huang Y, Dong F, Kwon JH. Ulcerative colitis-associated long noncoding RNA, BC012900, regulates intestinal epithelial cell apoptosis. Inflamm Bowel Dis. (2016) 22:782–95. doi: 10.1097/MIB.0000000000000691
63. Peng H, Liu Y, Tian J, Ma J, Tang X, Rui K, et al. The long noncoding RNA IFNG-AS1 promotes T helper type 1 cells response in patients with Hashimoto's thyroiditis. Sci Rep. (2015) 5:17702. doi: 10.1038/srep17702
64. Wang J, Peng H, Tian J, Ma J, Tang X, Rui K, et al. Upregulation of long noncoding RNA TMEVPG1 enhances T helper type 1 cell response in patients with Sjögren syndrome. Immunol Res. (2016) 64:489–96. doi: 10.1007/s12026-015-8715-4
65. Vigneau S, Rohrlich P-S, Brahic M, Bureau J-F. Tmevpg1, a candidate gene for the control of Theiler's virus persistence, could be implicated in the regulation of gamma interferon. J Virol. (2003) 77:5632–8. doi: 10.1128/JVI.77.10.5632-5638.2003
66. Collier SP, Collins PL, Williams CL, Boothby MR, Aune TM. Cutting edge: influence of Tmevpg1, a long intergenic noncoding RNA, on the expression of Ifng by Th1 cells. J Immunol. (2012) 189:2084–8. doi: 10.4049/jimmunol.1200774
67. Collier SP, Henderson MA, Tossberg JT, Aune TM. Regulation of the Th1 genomic locus from Ifng through Tmevpg1 by T-bet. J Immunol. (2014) 193:3959–65. doi: 10.4049/jimmunol.1401099
68. Gomez JA, Wapinski OL, Yang YW, Bureau J-FO, Gopinath S, Monack DM, et al. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-γ locus. Cell. (2013) 152:743–54. doi: 10.1016/j.cell.2013.01.015
69. Agostini F, Zanzoni A, Klus P, Marchese D, Cirillo D, Tartaglia GG. catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics. (2013) 29:2928–30. doi: 10.1093/bioinformatics/btt495
70. Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. (2012) 40:D930–4. doi: 10.1093/nar/gkr917
71. Pasmant E, Laurendeau I, Heron D, Vidaud M, Vidaud D, Bieche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. (2007) 67:3963–9. doi: 10.1158/0008-5472.CAN-06-2004
72. Li Z, Yu X, Shen J. ANRIL: a pivotal tumor suppressor long non-coding RNA in human cancers. Tumor Biol. (2016) 37:5657–61. doi: 10.1007/s13277-016-4808-5
73. McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, et al. A common allele on chromosome 9 associated with coronary heart disease. Science. (2007) 316:1488–91. doi: 10.1126/science.1142447
74. Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. (2007) 316:1336–41. doi: 10.1126/science.1142364
75. Aarabi G, Zeller T, Heydecke G, Munz M, Schäfer A, Seedorf U. Roles of the Chr.9p21.3 ANRIL locus in regulating inflammation and implications for anti-inflammatory drug target identification. Front Cardiovasc Med. (2018) 5:47. doi: 10.3389/fcvm.2018.00047
76. Yap KL, Li S, Muñoz-Cabello AM, Raguz S, Zeng L, Mujtaba S, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. (2010) 38:662–74. doi: 10.1016/j.molcel.2010.03.021
77. Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. (2006) 127:265–75. doi: 10.1016/j.cell.2006.10.003
78. Murakami Y, Mizoguchi F, Saito T, Miyasaka N, Kohsaka H. p16INK4a exerts an anti-inflammatory effect through accelerated IRAK1 degradation in macrophages. J Immunol. (2012) 189:5066–72. doi: 10.4049/jimmunol.1103156
79. Zhou X, Han X, Wittfeldt A, Sun J, Liu C, Wang X, et al. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-κB pathway. RNA Biol. (2016) 13:98–108. doi: 10.1080/15476286.2015.1122164
80. Lin J, He Y, Chen J, Zeng Z, Yang B, Ou Q. A critical role of transcription factor YY1 in rheumatoid arthritis by regulation of interleukin-6. J Autoimmun. (2017) 77:67–75. doi: 10.1016/J.JAUT.2016.10.008
81. Teeuw WJ, Laine ML, Bizzarro S, Loos BG. A lead ANRIL polymorphism is associated with elevated CRP levels in periodontitis: a pilot case-control study. PLoS ONE. (2015) 10:e0137335. doi: 10.1371/journal.pone.0137335
82. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. (2011) 11:98–107. doi: 10.1038/nri2925
83. Kong Y, Sharma RB, Ly S, Stamateris RE, Jesdale WM, Alonso LC. CDKN2A/B T2D genome-wide association study risk SNPs impact locus gene expression and proliferation in human islets. Diabetes. (2018) 67:872–84. doi: 10.2337/db17-1055
84. Mirza AH, Berthelsen CH, Seemann SE, Pan X, Frederiksen KS, Vilien M, et al. Transcriptomic landscape of lncRNAs in inflammatory bowel disease. Genome Med. (2015) 7:39. doi: 10.1186/s13073-015-0162-2
85. Song C-L, Wang J-P, Xue X, Liu N, Zhang X-H, Zhao Z, et al. Effect of circular ANRIL on the inflammatory response of vascular endothelial cells in a rat model of coronary atherosclerosis. Cell Physiol Biochem. (2017) 42:1202–12. doi: 10.1159/000478918
Keywords: lncRNA, linc RNA, inflammation, inflammatory disease, GWAS, SNP
Citation: Castellanos-Rubio A and Ghosh S (2019) Disease-Associated SNPs in Inflammation-Related lncRNAs. Front. Immunol. 10:420. doi: 10.3389/fimmu.2019.00420
Received: 27 September 2018; Accepted: 18 February 2019;
Published: 08 March 2019.
Edited by:
Jorge Henao-Mejia, University of Pennsylvania, United StatesReviewed by:
Trinath Jamma, Birla Institute of Technology and Science, IndiaRenata Meirelles Pereira, Federal University of Rio de Janeiro, Brazil
Copyright © 2019 Castellanos-Rubio and Ghosh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sankar Ghosh, c2cyNzE1QGNvbHVtYmlhLmVkdQ==