
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 March 2019
Sec. T Cell Biology
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00336
This article is part of the Research Topic Secondary Respiratory Infections in the Context of Acute and Chronic Pulmonary Diseases View all 13 articles
 Christophe Paget1,2
Christophe Paget1,2 François Trottein3,4,5,6,7*
François Trottein3,4,5,6,7*Despite the widespread application of vaccination programs and antiviral drug treatments, influenza viruses are still among the most harmful human pathogens. Indeed, influenza results in significant seasonal and pandemic morbidity and mortality. Furthermore, severe bacterial infections can occur in the aftermath of influenza virus infection, and contribute substantially to the excess morbidity and mortality associated with influenza. Here, we review the main features of influenza viruses and current knowledge about the mechanical and immune mechanisms that underlie post-influenza secondary bacterial infections. We present the emerging literature describing the role of “innate-like” unconventional T cells in post-influenza bacterial superinfection. Unconventional T cell populations span the border between the innate and adaptive arms of the immune system, and are prevalent in mucosal tissues (including the airways). They mainly comprise Natural Killer T cells, mucosal-associated invariant T cells and γδ T cells. We provide an overview of the principal functions that these cells play in pulmonary barrier functions and immunity, highlighting their unique ability to sense environmental factors and promote protection against respiratory bacterial infections. We focus on two major opportunistic pathogens involved in superinfections, namely Streptococcus pneumoniae and Staphylococcus aureus. We discuss mechanisms through which influenza viruses alter the antibacterial activity of unconventional T cells. Lastly, we discuss recent fundamental advances and possible therapeutic approaches in which unconventional T cells would be targeted to prevent post-influenza bacterial superinfections.
Respiratory infections are one the biggest health concerns worldwide. They account for a substantial rate of morbidity and mortality in Western and developing countries (1). Amongst respiratory pathogens, influenza viruses, commonly known as “the flu,” represent one the most important concern despite ongoing vaccine campaigns and anti-viral drugs. Each year, seasonal influenza infection affects 5 to 15% of the population and is a major contributor of pneumonia-related death worldwide (500,000 deaths per year) (2). Seasonal influenza is due to two main subtypes in humans, H1N1, and H3N2. Antigenic variations due to mutation in the hemagglutinin (HA) and neuraminidase (NA) genes, a phenomenon known as the antigenic drift, occur every year, and result in the circulation of new strains with sometime enhanced virulence and lethality potential. In parallel, in general every 10 to 20 years, new influenza subtypes distinct from circulating seasonal strains can emerge (due to antigenic shift) and provoke pandemic waves with sometime devastating consequences (3). Relative to seasonal influenza, pandemics exhibit a higher transmissibility and a higher rate of mortality, particularly among younger people who lack specific immunity against these new strains. Mortality attributed to influenza infection can be of high incidence during pandemics. During the Spanish flu (1918–1919), more than 40 million people died from influenza infection (4, 5). During the 2009 pandemic, influenza infection had a substantial impact on human mortality (3, 6). As discussed below, viral-bacteria pneumonia contribute significantly to morbidity and mortality during influenza epidemics and pandemics (5). Due to medical and economical burdens, and considering the threat of new pandemics and the emergence of antibiotic resistance, it is urgent to find novel options to fight against influenza infections and their complications, including secondary bacterial infections.
The influenza viruses (types A, B, and C) are negative sense single-stranded RNA viruses (7). These enveloped viruses belong to the family of Orthomyxoviridae. In humans, the most common cause of respiratory illness is influenza A virus (IAV) (8). To reach successful replication, sense messenger RNAs must be generated from the viral genome, a process due to the viral RNA polymerase. Sense messengers comprise eight RNA segments encoding eleven proteins (9, 10). The mature virion contains eight of these proteins surrounded by a protein envelope, which mainly includes two viral antigenic determinants: HA, which binds to terminal sialic acids expressed by airway and alveolar epithelial cells and NA, a critical enzyme necessary for releasing the viral progeny from infected cells. Eighteen HA subtypes and 11 NA subtypes have been identified to date. Two IAV subtypes (H1N1 and H3N2) along with one or two influenza B viruses co-circulate annually causing influenza epidemics. The primary targets (and site of replication) of IAV are airway and alveolar epithelial cells. Shortly after infection, the viral machinery, with the unintentionally help of host factors, is at work and generate the release of virions (11). Productive replication in epithelial cells results in cell death and to epithelial/endothelial damages leading to barrier rupture and exudation of fluids and proteins into the airways and alveolar spaces, greatly impairing gas exchanges (12). Meanwhile, an intense infiltration of immune cells (neutrophils, monocytes) occurs. Clinically, severe IAV infection can lead to acute respiratory distress syndrome, a severe form of respiratory failure associated with 40 % of mortality (5).
Innate immune response is rapidly triggered after IAV infection (13–18). This relies on the presence of viral RNA in the cytosol of infected cells and on different and complementary innate sensors including Toll-like receptors (TLRs; primarily TLR3 and TLR7), retinoic acid inducible gene-1 and inflammasomes. Activation of these innate sensors results on the production of massive amounts of type I and type III interferons (IFNs) as well as interleukin (IL)-1β and IL-18. Type I and type III IFNs, though autocrine and paracrine (myeloid cells) effects, elicit the production of a myriad of IFN-stimulated genes that strongly participate in virus clearance. Meanwhile, activation of inflammasomes and NF- κB promotes the release of pro-inflammatory cytokines and chemokines and the subsequent recruitment and activation of numerous immune cells such as monocytes/macrophages and neutrophils. These events limit and/or prevent viral entry and replication and attenuate the severity of the disease. All of these responses however contribute to tissue injury. For instance, inflammatory monocytes greatly participate in epithelial cell damage and death (19, 20). On the other hand, although they participate in virus clearance, neutrophils are also strong contributors of lung damage and lethality (21). While epithelial cells are critical to initiate innate immunity, other resident sentinel cells also play a role in virus clearance. Alveolar macrophages promote the elimination of viruses through the phagocytosis of collectin-opsonised viral particles or infected apoptotic cells (efferocytosis) and production of inflammatory cytokines and chemokines (22, 23). In parallel, other resident and/or recruited innate immune cells, including natural killer cells, unconventional T cells, and innate lymphoid cells play a part in disease outcomes (mouse model of influenza), an effect associated—or not—with effector functions (24–33). On the other hand, depending on the infectious dose, some of them (e.g., NK cells) may also participate in immunopathology (34, 35). Several days after IAV entry and elicitation of innate immunity, a strong antigen (Ag)-specific CD8+ T cell response develops in the lungs. In this phenomenon, the migration of antigen-loaded CD103+ dendritic cells to the draining lymph nodes is critical. Even though Ag-specific CD8+ T cells are sufficient to contain viruses (e.g., through lysis of infected cells), they also contribute to alveolar epithelium and endothelium damage (36, 37).
After the inflammatory burst and pulmonary tissue damage, a resolving/repair phase takes place, in general 7 to 14 days after the primary IAV infection. It leads to resolution of infiltrates and regeneration of damaged lung tissue thus restoring gas exchange. In this setting, murine studies of influenza infection suggested that CD8+ T cells, by producing the anti-inflammatory cytokine IL-10, are important to resolve inflammation (38). Activated macrophages can also promote the expansion of Foxp3-expressing regulatory T cells to suppress the deleterious production of inflammatory cytokines by neutrophils (39). By suppressing IL-17, a cytokine involved in neutrophil recruitment, type I IFNs also contribute to resolution of inflammation post-influenza. Recent data also indicate a role for M2 macrophages in this process (40). Unconventional T cells may also play a critical role in recovery from influenza infection (27, 33, 41, 42). Finally, through amphiregulin production, innate lymphoid cells restore airway epithelial integrity and tissue homeostasis during IAV infection (25). Resolution of inflammation during influenza infection is critical for lung resiliency and restoration of physiological functions (which can take several weeks). This regenerating response corresponds to a period of enhanced susceptibility to respiratory bacterial (particularly Gram-positive) infections. Indeed, this process creates a favorable environment to the emergence of opportunistic pathogens that can eventually result in bacterial superinfection, bacterial pneumonia and bacterial dissemination from the lungs. The two later are major contributors to lethality. Post-mortem examination of autopsy specimens collected during the last pandemic (as well as the 1918 pandemic) suggests that a substantial proportion of patients died from bacterial infections once the virus was cleared (5, 43). The most common bacteria found in autopsied individuals were Streptococcus pneumoniae (the pneumococcus) and Staphylococcus aureus, two major ubiquitous upper respiratory opportunistic pathogens. Two main mechanisms (mechanical and immunological) explain bacterial superinfection post-influenza: loss of the epithelial barrier function and altered innate immune defense. Before reviewing the role of “innate-like” unconventional T cells in this setting, we summarize the main mechanisms by which IAV favors secondary bacterial infection.
Several excellent reviews have described the current mechanistic understanding of how IAV enhances susceptibility to secondary bacterial infections (44–49). Current data, mostly derived from experimental (mouse) models, point toward a multifactorial mechanism. Briefly, IAV disrupts the functions of the respiratory barrier by inducing, in a direct or indirect (through inflammatory monocytes) fashion, epithelial cell death, and by degrading mucins (20, 50). This alteration leads to exposure of new attachment sites for bacteria and allows bacterial translocation (51–53). Influenza A virus can also alter respiratory ciliary function, thereby impairing the clearance of aspirated bacteria from the lungs (54). As stated above, alteration of the innate immune response is critical in post-influenza bacterial superinfections. In particular, poor bacterial control in the context of prior IAV infection is due to the loss and/or dysfunction of macrophages and neutrophils (55–59). For these later, their ability to sense and clear (phagocytosis and killing activity) bacteria is profoundly altered (60–62). Along with these effector cells, dysfunction of natural killer cells also depresses host's antibacterial capabilities (63). Some cytokines are critical in bacterial superinfection. The immune-suppressive cytokine IL-10 inhibits the functions of macrophages and neutrophils (64, 65). IL-27, another immunosuppressive cytokine downstream of type I interferon receptor (IFNAR) signaling pathway also impairs innate immune response against secondary bacterial challenge (66, 67). Type I interferons, which are massively produced during IAV infection to limit viral replication, are also detrimental in bacterial superinfection. Mechanistically, they inhibit the production of chemokines (CXCL1 and CXCL2) important for the recruitment of macrophages and neutrophils to the lung and impair their phagocytic responses (57, 68, 69). Of note, type III IFNs (which share similarities with type I IFNs) favor bacterial superinfection post-influenza by disrupting the nasal microbiome, which often includes potential pathogens (70). The underlying mechanisms are still elusive but may depend on altered barrier functions of the nasal epithelium and dysfunctional innate defense. Although IFN-γ is critical in host defense against respiratory bacterial infections, it might favor secondary bacterial infection, for instance by decreasing the expression of the scavenger receptor MARCO on macrophages (56). In fact, the role of IFN-γ in bacterial superinfection is controversial since a protective role has also been suggested (71). Finally, IAV infection reduces, through signal transducer of activation and transcription-1 (STAT-1), the production of Th17-related cytokines, a critical family of cytokines involved in the control of respiratory bacterial infections (66, 72–75). Hence, the accumulating literature (experimental models) provides a clearer understanding of mechanisms leading to bacterial superinfection and suggests several targets to prevent it. In humans, impairment of innate immunity by pre-existing viral (IAV) infections has also been shown to hamper the control of carriage load and clearance of upper respiratory bacteria such as S. pneumoniae (76). This, along with mechanical defects (respiratory ciliary and barrier functions), may favor bacterial superinfection and secondary bacterial pneumonia. While some progresses have been made recently, much remains to be learned about the way that the virus alters pulmonary barrier functions and undermines protective antibacterial immunity during IAV-bacterial (co)infection. As outlined below, recent evidences suggest that unconventional T cell functions are targeted during IAV infection, a process that may be important in secondary bacterial infections.
Natural killer T (NKT) cells represent a subset of lipid-reactive αβ T cells. In response to lipid Ags presented by the monomorphic Ag presenting molecule CD1d, NKT cells swiftly produce a large amount of cytokines, thus promoting and orientating immune responses (77). Lipid recognition by NKT cells is mediated by a conserved T cell receptor (TCR) repertoire. Natural killer T cells can be divided into two major populations: type I NKT cells and type II NKT cells. Type I NKT cells express a semi-invariant TCR α-chain (Vα14-Jα18 in mice and Vα24-Jα18 in humans) paired with a limited set of TCR β-chains (77, 78). These cells respond strongly to alpha-galactosylceramide (α-GalCer), a glycolipid under clinical development, particularly in cancer settings (79). Type I NKT cells also recognize endogenous lipids which are necessary for their selection in the thymus and for their activation at peripheral sites. Type I NKT cells can also react to microbial-derived lipids (80). Of importance, type I NKT cells also activate in response to a wide array of cytokines, including IL-12 and IL-23. Despite a relatively conserved TCR, type I NKT cells are heterogeneous and can be further divided into distinct subsets (81, 82). NKT cells produce a wide range of cytokines, with sometime opposite functions, a property that depends on the cell subset activated and on the nature of the stimulation (e.g., lipids and/or activating cytokines). Through this unique property, type I NKT cells can influence different types of immune responses ranging from T helper (Th)1-like, Th2-like, Th17-like, or T regulatory-like responses (83). This property is critical in pathological situations during which type I NKT cells can either exert positive or negative functions. Of note, type I NKT cells not only produce cytokines and display cytotoxic functions toward transformed cells and virally-infected cells (84). Type II NKT cells represent a much broader family of CD1d-restricted αβ T cells that react to lipids, but not to α-GalCer. They express a more diverse TCR repertoire that recognizes lipid Ags of various nature and origin (mammalian and microbial) (85). Due to the lack of specific tools, the functions of type II NKT cells have mainly been proposed indirectly by comparing the phenotypes observed in Jα18-deficient (which lack type I NKT cells) vs. CD1d-deficient (which lack both type I and type II NKT cells) mice in various settings. Type II NKT cells appear to share conserved phenotypic and functional features with type I NKT cells including an effector memory phenotype, cytotoxic potential and secretion of numerous cytokines/chemokines (85). Akin to type I NKT cells, type II NKT cells play important functions during (bacterial) infections. NKT cells, which are more abundant in mice relative to humans, populate both lymphoid tissues and mucosal sites, including the lungs (86, 87).
Mucosal-associated invariant T (MAIT) cells present many common features with NKT cells and γδ T cells including the capacity to rapidly react to non-peptide Ags. MAIT cells are defined by their restriction to the major histocompatibility complex class I-related molecule 1 (MR1) (88, 89). The majority of MAIT cells (referred to as classical MAIT cells) (90) express a semi-invariant TCR composed of a canonical TCRα-chain (Vα19-Jα33 in mice and Vα7.2-Jα33 in humans) associated with a restricted set of Vβ segments (88, 89, 91, 92). Through their TCR, MAIT cells recognize small intermediate metabolites from the riboflavin (vitamin B2) pathway of bacteria, mycobacteria and yeast (93–95). They can react to products derived from the non-enzymatic reaction between a riboflavin precursor and small aldehydes of both microbial and host origin. The high instability of these ligands has so far limited their use in the clinics. Reminiscent with NKT cells, MAIT cells can respond to TCR signals and/or to various activating cytokines, including IL-12 and IL-18 (96–98). Upon activation, MAIT cells produce large amounts of Th1- and Th17-related cytokines (99). Additionally, MAIT cells can kill bacteria-infected cells (100). Unlike NKT cells, MAIT cells are abundant in the blood (up to 10% of the T cell compartment) in humans. They are also present at mucosal sites, including the lungs (10% of respiratory mucosal T cells) (101), where they sense the environment and exert a role of sentinels of the immune system. Due to their scarce representation in common laboratory mouse strains (unlike NKT cells), understanding MAIT cell biology is challenging even using Mr1−/− mice. To better assess their role in preclinical models, transgenic mice (Vα19iTg x Cα−/−) displaying high content of MAIT cells have been developed (102). Given their cytokine profile and cytotoxic potential, MAIT cells intuitively emerged as a specialized cell population in host defense against bacteria.
γδ T cells represent approximately 1–10% of peripheral blood T cells in humans and are important components of both innate and adaptive immunity. They display vast effector and immune regulatory functions (103). Akin to other members of the unconventional T cell family, γδ T cells display a pre-activated status that allows rapid induction of effector functions following the detection of tissue stress (104–106). Another important feature of γδ T cells is their tropism for epithelial surfaces including lungs, to where they migrate shortly after development and persist as resident cells. They frequently express invariant or closely related γδ TCRs in a given tissue site (e.g., Vγ1, Vγ4, and Vγ6 in the mouse lung tissue), which confer them specific Ag recognition capabilities from one tissue to another (103). Reminiscent to other unconventional T cells, γδ T cells can kill infected cells and initiate adaptive immune responses through the release of substantial amounts of Th1- and Th17-related cytokines (103). Thus, γδ T cells have emerged as essential constituents of the antimicrobial immunity in both preclinical and clinical settings (107). While the Ags for mouse γδ TCRs have not been reported yet, human γδ T cell subsets (e.g., Vγ9Vδ2+ cells) can recognize both natural (of microbial and mammalian origins) and synthetic phosphoantigens (108). However, it is now clear that the phosphoantigens are not directly sensed by the γδ TCRs but rather require the involvement of butyrophilin BTN3A as an intermediate. The precise molecular mechanisms involved in this TCR-dependent γδ T cell activation are still a matter of debate (109). Whatever the mechanisms involved, phosphoantigens have been shown to strongly activate (in vivo and ex vivo) human Vγ9Vδ2 γδT cells to induce their proliferation and to increase their cytotoxic capacities as well as their cytokine secretion including IFN-γ and TNF-α. Given this, harnessing γδ T cell functions in therapeutic protocols is currently highly considered by clinicians especially in the context of cancer (110).
Evidence in both preclinical and clinical settings have suggested a key role for unconventional T cells in host response against lung bacterial pathogens. Here, we compared their mode of activation and functions during respiratory bacterial infections with a focus on the two major opportunistic pathogen bacteria implicated in bacterial superinfection post-influenza, namely S. pneumoniae, and S. aureus.
Streptococcus pneumoniae (also referred to as the pneumococcus) is the leading cause of community-acquired bacterial pneumonia worldwide (2 million deaths per year), with infants and the elderly exhibiting higher susceptibility. This Gram-positive bacterium, which comprises a group of more than 90 serotypes, colonizes asymptomatically nasopharynx of healthy individuals. However, when the immune equilibrium is broken, pneumococcus carriage can lead to mild disease such as otitis media or sinusitis and more occasionally turns into severe complications such as pneumonia, sepsis, and meningitis (111). Streptococcus pneumoniae is often found in biological fluids of hospitalized patients diagnosed for influenza infection as well as patients with exacerbated chronic lung inflammation (112, 113). Despite vaccination prevents pneumococcus spread and controls infections, the available vaccines have however some issues (114). In addition, the emergence of antibiotic-resistant strains represents an important threat for the management of pneumococcal infections in clinics (115).
In the mouse system, both type I NKT cells and γδT cells activate in response to S. pneumoniae (Figure 1). While type I NKT cells produce IFN-γ early after pneumococcal challenge, γδT cells produce IL-17A (66, 74, 116–118). Activation of type I NKT cells during S. pneumoniae infection depends on pneumococcal-derived lipid(s) (α-glucosyldiacylglycerol), cytokines (IL-12) or both according to the strain studied (86, 116, 119). Of note, we and others have highlighted the role of CD103+ dendritic cells in the activation of type I NKT cells during pneumococcal infection (119, 120). The mechanisms through which murine γδT cells activate (IL-17) mainly depends on IL-1β and IL-23 (117, 121). The lack of type I NKT cells (Jα18−/− mice) (119, 122, 123) or γδT cells (Tcrd−/− mice) (66, 117) results in higher bacterial loads and mortality. The underlying mechanisms of this protective activity rely on IFN-γ and IL-17 secretion and on the early recruitment of neutrophils. Hence, both type I NKT cells and γδT cells play a natural positive role in host defense against experimental pneumococcal infection (Figure 1). The potential role of type II NKT cells and MAIT cells during pneumococcal infection is still elusive. Of interest, S. pneumoniae expresses enzymes involved in the synthesis of riboflavin metabolites (124, 125) and human MAIT cells produce, in an MR1-dependent manner, IFN-γ in response to dendritic cells and airway epithelial cells exposed to S. pneumoniae (126, 127). Using Vα19iTg x Cα−/− mice, a small proportion of lung MAIT cells were shown to produce IFN-γ and IL-17A during pneumococcal infection (127). Although a more detailed kinetic analysis is required, these levels were relatively low compared to those produced by NKT cells and γδT cells. The use of Mr1−/− or Vα19iTg x Cα−/−Mr1−/− mice will be instrumental to address the role of MAIT cells during experimental pneumococcal infection.
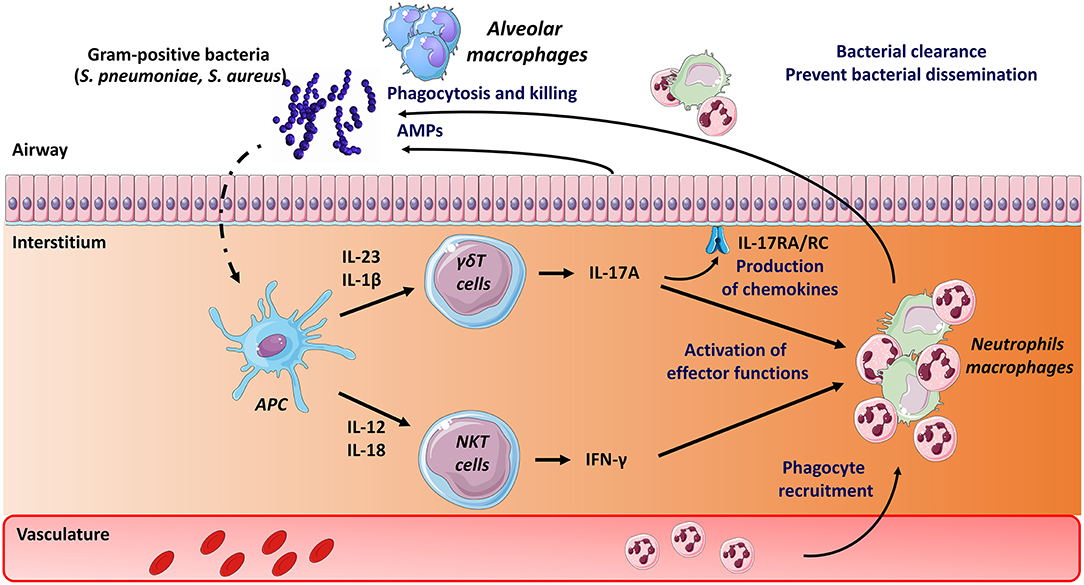
Figure 1. Mode of activation and role of unconventional T cells during pneumococcal and staphylococcal infections. Early after S. pneumoniae and S. Aureus infection, Ag presenting cells, including dendritic cells, produce a wide array of cytokines that activate pulmonary γδ T cells (IL-17A) and type I NKT cells (IFN-γ). Lipids from S. pneumoniae can also directly activate type I NKT cells through their TCR. The protective role mediated by γδ T cells and type I NKT cells comprises activation of innate effector cells, such as macrophages and neutrophils (IFN-γ, IL-17). The latter are rapidly recruited in response to chemokines produced, amongst other cell types, by epithelial cells (i.e., IL-17 receptor signaling pathways). Mechanisms leading to bacterial clearance include killing (bactericidal) activity of macrophages and neutrophils and release of antimicrobial peptides (AMPs). The potential role of MAIT cells in pneumococcal and staphylococcal infections is still unknown.
The potential effects of exogenous activation of unconventional T lymphocytes on pneumococcal infection have been examined. Inoculation of the type I NKT cell superagonist α-GalCer protects against lethal pneumococcal infection in the mouse system (123, 128). Mechanistically, this protective activity relies on respiratory CD103+ dendritic cells and on both IFN-γ and IL-17A production and neutrophils (128). The potential effect of exogenous γδ T cell and MAIT cell activation on host defense against pneumococcal infection is presently unknown. Despite emerging evidence for a critical role in host response to pneumococcus in experimental models, information regarding the phenotype and dynamics of unconventional T cells in patients with severe S. pneumoniae-driven pneumonia are rather limited. Of note, the level of circulating MAIT cells in critically ill patients with severe bacterial infection is markedly decreased compared to age-matched healthy controls (129). Although this decrease is less striking in patients with streptococcal infections, these data suggest that MAIT cells may migrate into the lungs, and thus may exert a potential role during pneumococcal infection.
Staphylococcus aureus is a Gram-positive bacterium with a potent pathogenic potential to cause a variety of community and hospital-acquired infections. In normal conditions, it commonly colonizes the upper airways. Under certain circumstances, including influenza infection, it can cause localized and serious invasive infections, as well as a severe septic shock syndrome (130). The frequency of these infections is increasing. The ability of S. aureus to form biofilms and the emergence of multidrug-resistant strains (e.g., methicillin-resistant Staphylococcus aureus) are the main reasons why their treatment is becoming more difficult. The capacity of S. aureus to become pathogenic is related to the expression of virulence factors, among which the production of a wide variety of toxins. Staphylococcal superantigens (SAgs) constitute a family of potent exotoxins secreted by S. aureus (131). They can cross-link MHC class II molecules with TCRs to stimulate an uncontrolled polyclonal activation of T lymphocytes (cytokine storm), potentially leading to severe illnesses including toxic shock syndrome.
Despite a relatively poor literature in the field, unconventional T cells might play role during S. aureus infection. They also recently emerged as potential targets of Staphylococcal SAgs. Indirect evidence suggest that IL-17 production by γδ T cells might be important in the control of S. aureus lung infection (72, 73) (Figure 1). Mice lacking γδ T cells have a reduced ability to clear bacteria and to control pulmonary inflammation (132). The role of NKT cells and MAIT cells in the control of S. aureus is still unknown. On the other hand, emerging evidence suggest that unconventional T cells (at least NKT cells and MAIT cells) are involved in toxic shock syndrome induced by Staphylococcal SAgs. Intranasal inoculation of Staphylococcal enterotoxin B promotes the activation of type I NKT cells and lung injury (133). Staphylococcal enterotoxin B activates mouse and human type I NKT cells via a MHC class II (but not CD1d) Vβ8-dependent pathway (134). More recently, Szabo et al., using SAg-sensitive HLA-DR4-transgenic mouse demonstrated that type I NKT cells are pathogenic (toxic shock syndrome) in response to Staphylococcal enterotoxin B (135). Of interest, administration of a Th2-polarizing glycolipid agonist for type I NKT cells reduced morbidity and mortality. Type I NKT cells may therefore constitute an attractive therapeutic target in SAg-mediated illnesses. Mouse and human MAIT cells can also activate in response to Sags in a largely TCR-independent, cytokine-driven manner (136). They produce a huge amount of pro-inflammatory cytokines and thereafter become unresponsive to stimulation with bacterial Ags. Through this mechanism, they might participate in cytokine storm and subsequent immunosuppression. Akin to type I NKT cells, MAIT cells may therefore provide an attractive therapeutic target for the management of both early and late phases of severe SAg-mediated illnesses.
As outlined below, mouse models of viral-bacterial infection have been used to assess the role of unconventional T cells in bacterial superinfection post-influenza. These cells are activated during influenza infection (24, 27–33, 137) and, through their ability to control barrier function, they may limit bacterial superinfection. On the other hand, although activation during influenza infection may preset their antibacterial effector functions, immune suppression arising from influenza counteracts their antibacterial potentials.
Disruption of the pulmonary barrier functions strongly contributes to enhanced bacterial colonization, bacterial superinfection and bacterial pneumonia in the context of prior influenza. Emerging evidences suggest that unconventional T cells play a natural role in the maintenance of tissue integrity and/or in tissue repair processes (138). Recent studies have addressed the role of unconventional T cells in tissue homeostasis and barrier functions during experimental influenza. Type I NKT cells and γδ T cells produce the tissue protective cytokine IL-22 (through IL-1β- and IL-23) during the early course of IAV infection (24, 139). Although IL-22 does not affect viral loads during influenza, several independent groups have demonstrated the protective effect of IL-22 against epithelial damages caused by viral replication (24, 139–143). The mechanisms through which IL-22 prevents epithelial barrier dysfunction during influenza infection might include an inhibitory effect on the recruitment of inflammatory monocytes and a direct effect on the expression of genes involved in barrier functions (143). Interleukin-22 might also participate in airway epithelial regeneration and barrier repair (141, 142). Interestingly, through its protective effect on barrier functions, IL-22 reduces secondary bacterial infection (139, 143). Of note, MAIT cells have recently been reported to accumulate in the lungs and to activate (through IL-18) during experimental IAV infection, a process associated with protection against a lethal viral challenge (33). Although not firmly established, MAIT cell activation during IAV infection may reduce pulmonary epithelial damage and reinforce barrier functions. Hence, by rapidly producing tissue protective factors, unconventional T cells, including NKT cells, γδ T cells, and MAIT cells, may limit the extent of secondary bacterial infection post-influenza. It is noteworthy that NKT cells can also indirectly activate the synthesis of protective barrier factors by other cells (e.g., amphiregulin by group 2 innate lymphoid cells) (25, 41). These functions might be exploited fortherapeutic purposes.
Alteration of innate immune defense also strongly contributes to bacterial superinfection post-influenza. As stated above, unconventional T cells play a part in host defense against S. pneumoniae and S. aureus. Here, we summarize host factors that (may) compromise their protective functions in the context of double viral and bacterial infection (mouse system). In this setting, IL-10 and type I IFNs appear to play a relevant role. During IAV infection, IL-10 is massively produced by innate and adaptive immune cells. This includes CD4+ (including regulatory T cells) and CD8+ T cells as well as NK cells and myeloid cells, mostly inflammatory monocytes (38, 119). Our data indicate that in the context of prior influenza, type I NKT cells fail to produce the protective cytokine IFN-γ (Figure 2), an effect associated with worse secondary pneumococcal infection (119). Blockade of IL-10 rescues activation of type I NKT cells (through restoration of IL-12 production by Ag-presenting cells), reduces bacterial outgrowth and dissemination and improves disease outcomes. Hence, the lack of type I NKT cell activation participates, at least in part, to bacterial (pneumococcal) superinfection post-influenza. Along with IL-10, type I IFNs favor bacterial superinfection post-influenza (57, 66, 68, 72–74, 144). γδ T cells appear to be the main target of type I IFNs. In the context of double IAV-bacterial (both pneumococcal and staphylococcal) infection, γδ T cells fail to secrete IL-17 in a type I IFN-dependent manner (66, 72, 74) (Figure 2). This ultimately leads to altered neutrophil recruitment and activity and to inhibition of the IL-17 antimicrobial pathway, including production of antimicrobial peptides. In this setting, the mode of action of type I IFNs is multiple. Type I IFNs can block the secretion of Th17-promoting cytokines IL-1β and IL-23 by Ag-presenting cells (72, 73). On the other hand, type I IFNs can directly target γδ T cells, via IFNAR, to inhibit IL-17 production (74) (Figure 2). Finally, type I IFNs indirectly inhibit IL-17 release by γδ T cells by promoting IL-27 production (66, 67). IL-27 targets γδ T cells to decrease expression of the IL-17-promoting factors RORγt and IL-23 receptor (66). The later mechanism is probably dominant as exogenous administration of IL-27 reverses the resistance phenotype of IFNAR-deficient mice upon post-influenza bacterial infection via down-regulating IL-17 production by γδ T cells and neutrophil response. Whether MAIT cell functions are affected by influenza, for instance through IL-10 or type I IFNs, is still ignored. Type III IFNs have also been shown to favor bacterial superinfection post-influenza (70). Regarding the role of unconventional T cells in barrier functions and innate antibacterial immunity, one can speculate that type III IFNs (like type I IFNs) also alter the functions of these cells to favor bacterial superinfection.
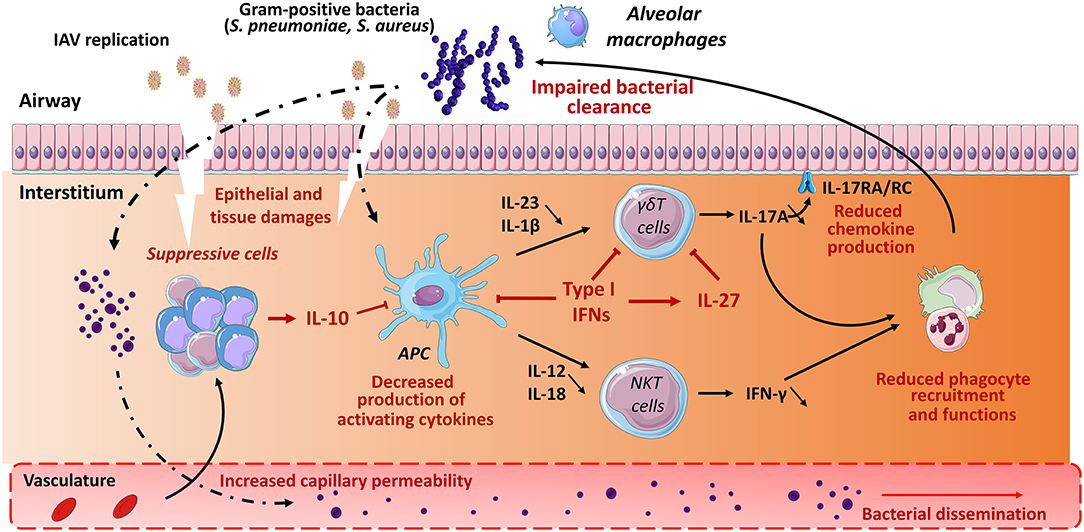
Figure 2. Role of unconventional T cells in bacterial superinfection post-influenza. Influenza A virus replicates in epithelial cells, thus leading to cellular damage and pulmonary inflammation. Along with typical inflammatory cells (neutrophils, inflammatory monocytes), the lungs is infiltrated with various populations of immune suppressive cells expressing for instance IL-10. The later inhibits the production of IFN-γ by type I NKT cells. Meanwhile, due to reduced IL-1β and IL-23 production, γδ T cells display a defective ability to release IL-17A. Multiple mechanisms are involved including a direct role for type I IFNs on Ag presenting cells (reduced IL-1β and IL-23) and γδ T cells (reduced IL-17A) and a promoting effect of type I IFNs on IL-27 synthesis. Interleukin-27 in turn targets γδ T cells to reduce IL-17A production. During IAV infection, there is also a numeric and/or functional defect of alveolar macrophages and neutrophils. As a result, the development of respiratory bacteria in the lung compartment is not controlled, leading to severe bacterial pneumonia and bacterial dissemination from the lungs. The potential positive role of γδ T cells and type I NKT cells (IL-22) in the maintenance of the epithelial barrier is not mentioned. The potential role of MAIT cells in bacterial superinfection is still unknown.
Whilst the use of experimental models suggests a role for NKT cells and γδ T cells in bacterial superinfection post-influenza (the potential role of MAIT cells has not yet been appreciated), few studies have so far investigated unconventional T cells during human influenza and secondary infections. MAIT cells are more abundant in the blood relative to NKT cells and, to a lesser extent, γδ T cells. Compared to healthy donors, the frequency of circulating MAIT cells decreased in patients hospitalized for severe pneumonia due to infection with the Asian lineage avian IAV (H7N9) (96). Of interest, individuals who recovered from pneumonia had a higher level of circulating MAIT cells compared with patients who succumbed (96). This study suggested a protective role of MAIT cells in human influenza. Another clinical study confirmed the reduced peripheral blood MAIT cell frequencies (and enhanced granzyme B expression) in patients with acute IAV infection (2009 H1N1 pandemic) (145). This decrease was even more pronounced in critically ill patients admitted in intensive care unit compared to patients with mild symptoms. Reduction of MAIT cell numbers during acute human influenza infection (critically ill patients) could impair protective anti-bacterial immunity increasing the risk of secondary bacterial infections, which would enhance disease severity and mortality. The frequency, number and functional state of NKT cells and γδ T cells during human influenza have not yet been examined. In influenza vaccinated individuals, γδ T cells proliferate and activate although this intensity weakens with age (146).
The unique biologic features of unconventional T cells are now being harnessed in the fight against cancer (79, 110, 147). Although this field is still in its infancy, exploitation of these cells in the management of lung infections appears to have therapeutic promise (148). Targeting unconventional T cells has several advantages. Firstly, the cells' restriction to non-polymorphic Ag-presenting molecules renders most patients eligible for unconventional T cell-based therapy using universal ligands. The question of whether unconventional T cells are potential immune targets in post-influenza bacterial superinfections has recently been addressed in preclinical models. The results suggest that these cells can indeed be exploited therapeutically. One major obstacle is the difficulty in balancing the induction of effective bacterial clearance and the avoidance of excessive inflammation. Cytokine-based strategies, neutralizing antibodies, and treatment with agonists that are specific for unconventional T cells have generated promising results over the last few years (mouse system).
The overexpression and/or inoculation of IL-23 or IL-1β restores the defective production of IL-17 by γδ T cells and T helper cells, and improves the clearance of pneumococci and staphylococci (72, 73). Furthermore, neutralization of IL-10 and IL-27 by blocking antibodies during the course of influenza restores the respective abilities of NKT cells and γδ T cells to combat secondary bacterial infections (66, 119). It is important to note that this approach is associated with better disease outcomes, including higher survival rates. In the mouse system, it has been suggested that treatment with the superagonist α-GalCer can enhance the beneficial activity of type I NKT cells. Inoculation of α-GalCer during IAV infection markedly reduces the bacterial (pneumococcal) burden in the lungs and bacterial dissemination from the lungs (149). However, the efficacy of this type of treatment is limited by its narrow therapeutic window; on day 7 (when susceptibility to superinfection peaks), α-GalCer has no effect. This is due to the disappearance of CD103+ dendritic cells (150)–a critical population involved in activation of type I NKT cells in the lungs—at this time point (128). In contrast, α-GalCer treatment early in the IAV infection (on day 4) or during the resolution phase (day 14) is associated with lower pneumococcal outgrowth and dissemination. Less intense viral-bacterial pneumonia and a lower morbidity rate were observed in superinfected mice treated with both α-GalCer and the anti-inflammatory corticosteroid dexamethasone (149). However, this combination therapy was not associated with a lower mortality rate during secondary bacterial superinfection. In contrast to type I NKT cells, the potential effects of agonists on γδ T cells and MAIT cells in the context of post-influenza secondary bacterial infections have yet to be investigated.
Although the above-mentioned findings (the restoration of NKT cell and/or γδ T cell functions) have revealed a novel aspect of immunotherapy against superinfection in animal models, their clinical relevance remains to be proven. In the search for an effective balance between effective bacterial clearance and the avoidance of excessive inflammation, it is likely that additional therapeutic approaches (e.g., anti-inflammatory drugs) will have to be implemented. One can also speculate that combination treatment with antibiotics might enhance the efficacy of immunotherapy. It was recently shown that the application of a combination of antibiotics and immune stimulators (e.g., Toll-like receptor agonists) improved the outcome of post-influenza bacterial superinfection in a murine system (151). On the same lines, it would be useful to study the effects of a combination of an agonist (e.g., α-GalCer) and an antibiotic. Another key challenge relates to cell targeting. As discussed above, unconventional T cells are heterogeneous, and comprise subpopulations with sometime opposite functions. It will be necessary to target subpopulations of interest (e.g., IL-17 producers) more accurately by engineering T-helper-polarizing agonists (which have only been developed for NKT cells so far) and/or co-factors polarizing their functions. This would open the way to immunotherapies tailored to match a patient's immune profile. Importantly, it seems that this type of treatment must also take account of the nature of secondary bacterial infection. As discussed above, Staphylococcal SAgs have a critical role in the pathogenesis of S. aureus infections. In this setting, antagonists or T-helper-polarizing agonists could be used to manipulate type I NKT cells and MAIT cells—both of which are hyper-responsive to SAgs.
Unconventional T cells have attracted growing interest from researchers and clinicians. The literature on the cells' roles in immune and inflammatory responses has grown tremendously over the last 10 years. In view of their immunoregulatory potential, unconventional T cells are well poised to help fight lung infections and the latter's complications. However, there is a paucity of preclinical and clinical research on the cells' potential roles in the context of influenza and secondary bacterial infections. Further research into (i) the role of unconventional T cells in bacterial (super) infections of the respiratory tract and (ii) how influenza modulates the cells' functions is now needed. Furthermore, the use of novel mouse models will be essential for defining the respective roles of unconventional T cells and their subsets in influenza and secondary bacterial infections. Given that the mechanisms of post-influenza bacterial superinfections are multifactorial (with the exploitation of mechanical and/or immune alterations in the host), future therapeutics will probably have to include several components that target several host factors in addition to the viruses and bacteria themselves. Although this approach is in its infancy, the manipulation of unconventional T cells during influenza (cytokines, α-GalCer) has shown its potential in the fight against secondary infections. As mentioned above, this strategy is not problem-free, and must be considered with caution. Research on the effects of combining immunostimulatory factors with antimicrobial drugs (e.g., antibiotics) should be encouraged, and might help to lessen the development of drug resistance. Given the physiological role of unconventional T cells in tissue repair and barrier functions, strategies for promoting these functions might also be of value. Lastly, given the role of type I NKT cells and MAIT cells in the cytokine storm that follows exposure to Staphylococcal SAgs, the manipulation of these cells might help to control the outcomes of secondary staphylococcal infection—including necrotizing pneumonia. As discussed in this review, there is also a critical knowledge gap between preclinical and clinical studies; hence, analyses of the frequency/number and functional states of patients' unconventional T cells should be encouraged. Counts of circulating unconventional T cells are not negligible; considering the critical role they exert in many diseases, one can expect to see some major breakthroughs in the near future. Promising research initiatives might include a complete analysis of the whole family of unconventional T cells, i.e., NKT cells, group 1 CD1-restricted T cells, MAIT cells and γδ T cells. High-throughput RNA sequencing (at the bulk population and single-cell levels) and the computer modeling of cytokine signatures in patients should also be encouraged. Although the work will be time-consuming and arduous, it might translate into improved clinical outcomes.
In conclusion, we critically analyzed the available evidence on the potential role of unconventional T cells in post-influenza bacterial superinfections. In view of these cells' extraordinary immunostimulatory and immunoregulatory properties and the proven safety of unconventional T cell agonists, further research in this field should be encouraged.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was supported by the Inserm, the CNRS, the University of Lille Nord de France, the Pasteur Institute of Lille, and the Agence Nationale de la Recherche (ANR-17-CE15-0020-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to colleagues whose works could not be cited due to space constraints. FT is supported by the CNRS and CP by the INSERM. The figure was created using the vector image bank of Servier Medical Art (http://smart.servier.com/). Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
1. Quinton LJ, Walkey AJ, Mizgerd JP. Integrative physiology of pneumonia. Physiol Rev. (2018) 98:1417–64. doi: 10.1152/physrev.00032.2017
2. Fiers W, De Filette M, Birkett A, Neirynck S, Min Jou W. A “universal” human influenza A vaccine. Virus Res. (2004) 103:173–6. doi: 10.1016/j.virusres.2004.02.030
3. Zimmer SM, Burke DS. Historical perspective–Emergence of influenza A (H1N1) viruses. N Engl J Med. (2009) 361:279–85. doi: 10.1056/NEJMra0904322
4. Johnson NPAS, Mueller J. Updating the accounts: global mortality of the 1918-1920 “Spanish” influenza pandemic. Bull Hist Med. (2002) 76:105–15. doi: 10.1353/bhm.2002.0022
5. Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. (2008) 198:962–70. doi: 10.1086/591708
6. Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng P-Y, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect Dis. (2012) 12:687–95. doi: 10.1016/S1473-3099(12)70121-4
7. Bouvier NM, Palese P. The biology of influenza viruses. Vaccine. (2008) 26 (Suppl. 4):D49–53. doi: 10.1016/j.vaccine.2008.07.039
8. Paules C, Subbarao K. Influenza. Lancet Lond Engl. (2017) 390:697–708. doi: 10.1016/S0140-6736(17)30129-0
9. Hutchinson EC, Fodor E. Transport of the influenza virus genome from nucleus to nucleus. Viruses. (2013) 5:2424–46. doi: 10.3390/v5102424
10. Dou D, Revol R, Östbye H, Wang H, Daniels R. Influenza a virus cell entry, replication, virion assembly and movement. Front Immunol. (2018) 9:1581. doi: 10.3389/fimmu.2018.01581
11. Edinger TO, Pohl MO, Stertz S. Entry of influenza A virus: host factors and antiviral targets. J Gen Virol. (2014) 95:263–77. doi: 10.1099/vir.0.059477-0
12. Short KR, Kroeze EJBV, Fouchier RAM, Kuiken T. Pathogenesis of influenza-induced acute respiratory distress syndrome. Lancet Infect Dis. (2014) 14:57–69. doi: 10.1016/S1473-3099(13)70286-X
13. Damjanovic D, Small C-L, Jeyanathan M, Jeyananthan M, McCormick S, Xing Z. Immunopathology in influenza virus infection: uncoupling the friend from foe. Clin Immunol Orlando Fla. (2012) 144:57–69. doi: 10.1016/j.clim.2012.05.005
14. Yoo J-K, Kim TS, Hufford MM, Braciale TJ. Viral infection of the lung: host response and sequelae. J Allergy Clin Immunol. (2013) 132:1263–76; quiz 1277. doi: 10.1016/j.jaci.2013.06.006
16. Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. (2014) 14:315–28. doi: 10.1038/nri3665
17. Herold S, Becker C, Ridge KM, Budinger GRS. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J. (2015) 45:1463–78. doi: 10.1183/09031936.00186214
18. Tripathi S, White MR, Hartshorn KL. The amazing innate immune response to influenza A virus infection. Innate Immun. (2015) 21:73–98. doi: 10.1177/1753425913508992
19. Herold S, Steinmueller M, von Wulffen W, Cakarova L, Pinto R, Pleschka S, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med. (2008) 205:3065–77. doi: 10.1084/jem.20080201
20. Ellis GT, Davidson S, Crotta S, Branzk N, Papayannopoulos V, Wack A. TRAIL+ monocytes and monocyte-related cells cause lung damage and thereby increase susceptibility to influenza-Streptococcus pneumoniae coinfection. EMBO Rep. (2015) 16:1203–18. doi: 10.15252/embr.201540473
21. Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. (2013) 154:197–212. doi: 10.1016/j.cell.2013.06.013
22. Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol. (2007) 178:2448–57. doi: 10.4049/jimmunol.178.4.2448
23. Hillaire MLB, Haagsman HP, Osterhaus ADME, Rimmelzwaan GF, van Eijk M. Pulmonary surfactant protein D in first-line innate defence against influenza A virus infections. J Innate Immun. (2013) 5:197–208. doi: 10.1159/000346374
24. Paget C, Ivanov S, Fontaine J, Renneson J, Blanc F, Pichavant M, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem. (2012) 287:8816–29. doi: 10.1074/jbc.M111.304758
25. Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. (2011) 12:1045–54. doi: 10.1038/ni.2131
26. Stein-Streilein J, Guffee J. In vivo treatment of mice and hamsters with antibodies to asialo GM1 increases morbidity and mortality to pulmonary influenza infection. J Immunol. (1986) 136:1435–41.
27. Carding SR, Allan W, Kyes S, Hayday A, Bottomly K, Doherty PC. Late dominance of the inflammatory process in murine influenza by gamma/delta + T cells. J Exp Med. (1990) 172:1225–31. doi: 10.1084/jem.172.4.1225
28. Eichelberger M, Doherty PC. Gamma delta T cells from influenza-infected mice develop a natural killer cell phenotype following culture. Cell Immunol. (1994) 159:94–102. doi: 10.1006/cimm.1994.1298
29. Tu W, Zheng J, Liu Y, Sia SF, Liu M, Qin G, et al. The aminobisphosphonate pamidronate controls influenza pathogenesis by expanding a gammadelta T cell population in humanized mice. J Exp Med. (2011) 208:1511–22. doi: 10.1084/jem.20110226
30. De Santo C, Salio M, Masri SH, Lee LY-H, Dong T, Speak AO, et al. Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J Clin Invest. (2008) 118:4036–48. doi: 10.1172/JCI36264
31. Paget C, Ivanov S, Fontaine J, Blanc F, Pichavant M, Renneson J, et al. Potential role of invariant NKT cells in the control of pulmonary inflammation and CD8+ T cell response during acute influenza A virus H3N2 pneumonia. J Immunol. (2011) 186:5590–602. doi: 10.4049/jimmunol.1002348
32. Kok WL, Denney L, Benam K, Cole S, Clelland C, McMichael AJ, et al. Pivotal advance: invariant NKT cells reduce accumulation of inflammatory monocytes in the lungs and decrease immune-pathology during severe influenza A virus infection. J Leukoc Biol. (2012) 91:357–68. doi: 10.1189/jlb.0411184
33. van Wilgenburg B, Loh L, Chen Z, Pediongco TJ, Wang H, Shi M, et al. MAIT cells contribute to protection against lethal influenza infection in vivo. Nat Commun. (2018) 9:4706. doi: 10.1038/s41467-018-07207-9
34. Abdul-Careem MF, Mian MF, Yue G, Gillgrass A, Chenoweth MJ, Barra NG, et al. Critical role of natural killer cells in lung immunopathology during influenza infection in mice. J Infect Dis. (2012) 206:167–77. doi: 10.1093/infdis/jis340
35. Zhou G, Juang SWW, Kane KP. NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol. (2013) 43:929–38. doi: 10.1002/eji.201242620
36. Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol. (1997) 159:5197–200.
37. Brincks EL, Katewa A, Kucaba TA, Griffith TS, Legge KL. CD8 T cells utilize TRAIL to control influenza virus infection. J Immunol. (2008) 181:4918–25. doi: 10.4049/jimmunol.181.7.4918
38. Sun J, Madan R, Karp CL, Braciale TJ. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat Med. (2009) 15:277–84. doi: 10.1038/nm.1929
39. Moser EK, Hufford MM, Braciale TJ. Late engagement of CD86 after influenza virus clearance promotes recovery in a FoxP3+ regulatory T cell dependent manner. PLoS Pathog. (2014) 10:e1004315. doi: 10.1371/journal.ppat.1004315
40. Chen WH, Toapanta FR, Shirey KA, Zhang L, Giannelou A, Page C, et al. Potential role for alternatively activated macrophages in the secondary bacterial infection during recovery from influenza. Immunol Lett. (2012) 141:227–34. doi: 10.1016/j.imlet.2011.10.009
41. Gorski SA, Hahn YS, Braciale TJ. Group 2 innate lymphoid cell production of IL-5 is regulated by NKT cells during influenza virus infection. PLoS Pathog. (2013) 9:e1003615. doi: 10.1371/journal.ppat.1003615
42. Nüssing S, Sant S, Koutsakos M, Subbarao K, Nguyen THO, Kedzierska K. Innate and adaptive T cells in influenza disease. Front Med. (2018) 12:34–47. doi: 10.1007/s11684-017-0606-8
43. Taubenberger JK, Morens DM. Influenza revisited. Emerg Infect Dis. (2006) 12:1–2. doi: 10.3201/eid1201.051442
44. Snelgrove RJ, Godlee A, Hussell T. Airway immune homeostasis and implications for influenza-induced inflammation. Trends Immunol. (2011) 32:328–34. doi: 10.1016/j.it.2011.04.006
45. Metzger DW, Sun K. Immune dysfunction and bacterial coinfections following influenza. J Immunol. (2013) 191:2047–52. doi: 10.4049/jimmunol.1301152
46. Habibzay M, Weiss G, Hussell T. Bacterial superinfection following lung inflammatory disorders. Future Microbiol. (2013) 8:247–56. doi: 10.2217/fmb.12.143
47. McCullers JA. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat Rev Microbiol. (2014) 12:252–62. doi: 10.1038/nrmicro3231
48. Rynda-Apple A, Robinson KM, Alcorn JF. Influenza and bacterial superinfection: illuminating the immunologic mechanisms of disease. Infect Immun. (2015) 83:3764–70. doi: 10.1128/IAI.00298-15
49. Robinson KM, Kolls JK, Alcorn JF. The immunology of influenza virus-associated bacterial pneumonia. Curr Opin Immunol. (2015) 34:59–67. doi: 10.1016/j.coi.2015.02.002
50. Wheeler AH, Nungester WJ. Effect of mucin on influenza virus infection in hamsters. Science. (1942) 96:92–3. doi: 10.1126/science.96.2482.92
51. Cundell DR, Gerard NP, Gerard C, Idanpaan-Heikkila I, Tuomanen EI. Streptococcus pneumoniae anchor to activated human cells by the receptor for platelet-activating factor. Nature. (1995) 377:435–8. doi: 10.1038/377435a0
52. McCullers JA, Rehg JE. Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J Infect Dis. (2002) 186:341–50. doi: 10.1086/341462
53. Rijneveld AW, Weijer S, Florquin S, Speelman P, Shimizu T, Ishii S, et al. Improved host defense against pneumococcal pneumonia in platelet-activating factor receptor-deficient mice. J Infect Dis. (2004) 189:711–6. doi: 10.1086/381392
54. Pittet LA, Hall-Stoodley L, Rutkowski MR, Harmsen AG. Influenza virus infection decreases tracheal mucociliary velocity and clearance of Streptococcus pneumoniae. Am J Respir Cell Mol Biol. (2010) 42:450–60. doi: 10.1165/rcmb.2007-0417OC
55. McNamee LA, Harmsen AG. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect Immun. (2006) 74:6707–21. doi: 10.1128/IAI.00789-06
56. Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. (2008) 14:558–64. doi: 10.1038/nm1765
57. Shahangian A, Chow EK, Tian X, Kang JR, Ghaffari A, Liu SY, et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest. (2009) 119:1910–20. doi: 10.1172/JCI35412
58. Ghoneim HE, Thomas PG, McCullers JA. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J Immunol. (2013) 191:1250–59. doi: 10.4049/jimmunol.1300014
59. Bansal S, Yajjala VK, Bauer C, Sun K. IL-1 signaling prevents alveolar macrophage depletion during influenza and Streptococcus pneumoniae coinfection. J Immunol. (2018) 200:1425–33. doi: 10.4049/jimmunol.1700210
60. Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. (2008) 205:323–9. doi: 10.1084/jem.20070891
61. Subramaniam R, Barnes PF, Fletcher K, Boggaram V, Hillberry Z, Neuenschwander P, et al. Protecting against post-influenza bacterial pneumonia by increasing phagocyte recruitment and ROS production. J Infect Dis. (2014) 209:1827–36. doi: 10.1093/infdis/jit830
62. Sun K, Metzger DW. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J Immunol. (2014) 192:3301–7. doi: 10.4049/jimmunol.1303049
63. Small C-L, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG, et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. (2010) 184:2048–56. doi: 10.4049/jimmunol.0902772
64. van der Sluijs KF, van Elden LJR, Nijhuis M, Schuurman R, Pater JM, Florquin S, et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J Immunol. (2004) 172:7603–9. doi: 10.4049/jimmunol.172.12.7603
65. van der Sluijs KF, Nijhuis M, Levels JHM, Florquin S, Mellor AL, Jansen HM, et al. Influenza-induced expression of indoleamine 2,3-dioxygenase enhances interleukin-10 production and bacterial outgrowth during secondary pneumococcal pneumonia. J Infect Dis. (2006) 193:214–22. doi: 10.1086/498911
66. Cao J, Wang D, Xu F, Gong Y, Wang H, Song Z, et al. Activation of IL-27 signalling promotes development of postinfluenza pneumococcal pneumonia. EMBO Mol Med. (2014) 6:120–40. doi: 10.1002/emmm.201302890
67. Robinson KM, Lee B, Scheller EV, Mandalapu S, Enelow RI, Kolls JK, et al. The role of IL-27 in susceptibility to post-influenza Staphylococcus aureus pneumonia. Respir Res. (2015) 16:10. doi: 10.1186/s12931-015-0168-8
68. Nakamura S, Davis KM, Weiser JN. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J Clin Invest. (2011) 121:3657–65. doi: 10.1172/JCI57762
69. Schliehe C, Flynn EK, Vilagos B, Richson U, Swaminanthan S, Bosnjak B, et al. The methyltransferase Setdb2 mediates virus-induced susceptibility to bacterial superinfection. Nat Immunol. (2015) 16:67–74. doi: 10.1038/ni.3046
70. Planet PJ, Parker D, Cohen TS, Smith H, Leon JD, Ryan C, et al. Lambda interferon restructures the nasal microbiome and increases susceptibility to Staphylococcus aureus superinfection. mBio. (2016) 7:e01939–15. doi: 10.1128/mBio.01939-15
71. Blevins LK, Wren JT, Holbrook BC, Hayward SL, Swords WE, Parks GD, et al. Coinfection with Streptococcus pneumoniae negatively modulates the size and composition of the ongoing influenza-specific CD8+ T cell response. J Immunol. (2014) 193:5076–87. doi: 10.4049/jimmunol.1400529
72. Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. (2011) 186:1666–74. doi: 10.4049/jimmunol.1002194
73. Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, et al. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1β production in mice. J Immunol. (2013) 191:5153–9. doi: 10.4049/jimmunol.1301237
74. Li W, Moltedo B, Moran TM. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of γδ T cells. J Virol. (2012) 86:12304–12. doi: 10.1128/JVI.01269-12
75. Lee B, Gopal R, Manni ML, McHugh KJ, Mandalapu S, Robinson KM, et al. STAT1 is required for suppression of type 17 immunity during influenza and bacterial superinfection. Immuno Horizons. (2017) 1:81–91. doi: 10.4049/immunohorizons.1700030
76. Jochems SP, Marcon F, Carniel BF, Holloway M, Mitsi E, Smith E, et al. Inflammation induced by influenza virus impairs human innate immune control of pneumococcus. Nat Immunol. (2018) 19:1299–308. doi: 10.1038/s41590-018-0231-y
77. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
78. Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol. (2010) 11:197–206. doi: 10.1038/ni.1841
79. Bedard M, Salio M, Cerundolo V. Harnessing the power of Iinvariant Natural Killer T cells in cancer immunotherapy. Front Immunol. (2017) 8:1829. doi: 10.3389/fimmu.2017.01829
80. Van Kaer L, Parekh VV, Wu L. The response of CD1d-restricted invariant NKT cells to microbial pathogens and their products. Front Immunol. (2015) 6:226. doi: 10.3389/fimmu.2015.00226
81. Lee YJ, Holzapfel KL, Zhu J, Jameson SC, Hogquist KA. Steady-state production of IL-4 modulates immunity in mouse strains and is determined by lineage diversity of iNKT cells. Nat Immunol. (2013) 14:1146–54. doi: 10.1038/ni.2731
82. Engel I, Seumois G, Chavez L, Samaniego-Castruita D, White B, Chawla A, et al. Innate-like functions of natural killer T cell subsets result from highly divergent gene programs. Nat Immunol. (2016) 17:728–39. doi: 10.1038/ni.3437
83. Matsuda JL, Mallevaey T, Scott-Browne J, Gapin L. CD1d-restricted iNKT cells, the “Swiss-Army knife” of the immune system. Curr Opin Immunol. (2008) 20:358–68. doi: 10.1016/j.coi.2008.03.018
84. Metelitsa LS, Naidenko OV, Kant A, Wu HW, Loza MJ, Perussia B, et al. Human NKT cells mediate antitumor cytotoxicity directly by recognizing target cell CD1d with bound ligand or indirectly by producing IL-2 to activate NK cells. J Immunol. (2001) 167:3114–22. doi: 10.4049/jimmunol.167.6.3114
85. Macho-Fernandez E, Brigl M. The extended family of CD1d-restricted NKT cells: sifting through a mixed bag of TCRs, antigens, and functions. Front Immunol. (2015) 6:362. doi: 10.3389/fimmu.2015.00362
86. Thanabalasuriar A, Neupane AS, Wang J, Krummel MF, Kubes P. iNKT cell emigration out of the lung vasculature requires neutrophils and monocyte-derived dendritic cells in inflammation. Cell Rep. (2016) 16:3260–72. doi: 10.1016/j.celrep.2016.07.052
87. Scanlon ST, Thomas SY, Ferreira CM, Bai L, Krausz T, Savage PB, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med. (2011) 208:2113–24. doi: 10.1084/jem.20110522
88. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature. (2003) 422:164–9. doi: 10.1038/nature01433
89. Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med. (1999) 189:1907–21. doi: 10.1084/jem.189.12.1907
90. Gherardin NA, McCluskey J, Rossjohn J, Godfrey DI. The diverse family of MR1-restricted T cells. J Immunol. (2018) 201:2862–71. doi: 10.4049/jimmunol.1801091
91. Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, et al. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med. (2013) 210:2305–20. doi: 10.1084/jem.20130958
92. Eckle SBG, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HEG, Reantragoon R, et al. A molecular basis underpinning the T cell receptor heterogeneity of mucosal-associated invariant T cells. J Exp Med. (2014) 211:1585–1600. doi: 10.1084/jem.20140484
93. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. (2012) 491:717–23. doi: 10.1038/nature11605
94. Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V, et al. MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog. (2013) 9:e1003681. doi: 10.1371/journal.ppat.1003681
95. Gold MC, McLaren JE, Reistetter JA, Smyk-Pearson S, Ladell K, Swarbrick GM, et al. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J Exp Med. (2014) 211:1601–10. doi: 10.1084/jem.20140507
96. Loh L, Wang Z, Sant S, Koutsakos M, Jegaskanda S, Corbett AJ, et al. Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc Natl Acad Sci USA. (2016) 113:10133–8. doi: 10.1073/pnas.1610750113
97. Tang X-Z, Jo J, Tan AT, Sandalova E, Chia A, Tan KC, et al. IL-7 licenses activation of human liver intrasinusoidal mucosal-associated invariant T cells. J Immunol. (2013) 190:3142–52. doi: 10.4049/jimmunol.1203218
98. Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol. (2010) 11:701–8. doi: 10.1038/ni.1890
99. Salio M, Silk JD, Jones EY, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol. (2014) 32:323–66. doi: 10.1146/annurev-immunol-032713-120243
100. Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. (2010) 8:e1000407. doi: 10.1371/journal.pbio.1000407
101. Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SBG, Meehan B, et al. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med. (2015) 212:1095–108. doi: 10.1084/jem.20142110
102. Kawachi I, Maldonado J, Strader C, Gilfillan S. MR1-restricted V alpha 19i mucosal-associated invariant T cells are innate T cells in the gut lamina propria that provide a rapid and diverse cytokine response. J Immunol. (2006) 176:1618–27. doi: 10.4049/jimmunol.176.3.1618
103. Bonneville M, O'Brien RL, Born WK. Gammadelta T cell effector functions: a blend of innate programming and acquired plasticity. Nat Rev Immunol. (2010) 10:467–478. doi: 10.1038/nri2781
104. Nielsen MM, Witherden DA, Havran WL. γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat Rev Immunol. (2017) 17:733–45. doi: 10.1038/nri.2017.101
105. Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity. (2009) 31:184–96. doi: 10.1016/j.immuni.2009.08.006
106. Hayday A, Tigelaar R. Immunoregulation in the tissues by gammadelta T cells. Nat Rev Immunol. (2003) 3:233–42. doi: 10.1038/nri1030
107. Chien Y, Meyer C, Bonneville M. γδ T cells: first line of defense and beyond. Annu Rev Immunol. (2014) 32:121–55. doi: 10.1146/annurev-immunol-032713-120216
108. Gu S, Nawrocka W, Adams EJ. Sensing of pyrophosphate metabolites by Vγ9Vδ2 T cells. Front Immunol. (2014) 5:688. doi: 10.3389/fimmu.2014.00688
109. Adams EJ, Gu S, Luoma AM. Human gamma delta T cells: evolution and ligand recognition. Cell Immunol. (2015) 296:31–40. doi: 10.1016/j.cellimm.2015.04.008
110. Silva-Santos B, Serre K, Norell H. γδ T cells in cancer. Nat Rev Immunol. (2015) 15:683–91. doi: 10.1038/nri3904
111. Weiser JN, Ferreira DM, Paton JC. Streptococcus pneumoniae: transmission, colonization and invasion. Nat Rev Microbiol. (2018) 16:355–67. doi: 10.1038/s41579-018-0001-8
112. Morris DE, Cleary DW, Clarke SC. Secondary bacterial infections associated with Influenza pandemics. Front Microbiol. (2017) 8:1041. doi: 10.3389/fmicb.2017.01041
113. Domenech A, Ardanuy C, Tercero A, García-Somoza D, Santos S, Liñares J. Dynamics of the pneumococcal population causing acute exacerbations in COPD patients in a Barcelona hospital (2009-12): comparison with 2001-04 and 2005-08 periods. J Antimicrob Chemother. (2014) 69:932–9. doi: 10.1093/jac/dkt476
114. Moffitt K, Malley R. Rationale and prospects for novel pneumococcal vaccines. Hum Vaccines Immunother. (2016) 12:383–92. doi: 10.1080/21645515.2015.1087625
115. Cherazard R, Epstein M, Doan T-L, Salim T, Bharti S, Smith MA. Antimicrobial resistant streptococcus pneumoniae: prevalence, mechanisms, and clinical implications. Am J Ther. (2017) 24:e361–9. doi: 10.1097/MJT.0000000000000551
116. Kinjo Y, Illarionov P, Vela JL, Pei B, Girardi E, Li X, et al. Invariant natural killer T cells recognize glycolipids from pathogenic Gram-positive bacteria. Nat Immunol. (2011) 12:966–74. doi: 10.1038/ni.2096
117. Hassane M, Demon D, Soulard D, Fontaine J, Keller LE, Patin EC, et al. Neutrophilic NLRP3 inflammasome-dependent IL-1β secretion regulates the γδT17 cell response in respiratory bacterial infections. Mucosal Immunol. (2017) 10:1056–68. doi: 10.1038/mi.2016.113
118. Trottein F, Paget C. Natural Killer T Cells and mucosal-associated invariant T cells in lung infections. Front Immunol. (2018) 9:1750. doi: 10.3389/fimmu.2018.01750
119. Barthelemy A, Ivanov S, Fontaine J, Soulard D, Bouabe H, Paget C, et al. Influenza A virus-induced release of interleukin-10 inhibits the anti-microbial activities of invariant natural killer T cells during invasive pneumococcal superinfection. Mucosal Immunol. (2017) 10:460–9. doi: 10.1038/mi.2016.49
120. King IL, Amiel E, Tighe M, Mohrs K, Veerapen N, Besra G, et al. The mechanism of splenic invariant NKT cell activation dictates localization in vivo. J Immunol Baltim. (2013) 191:572–82. doi: 10.4049/jimmunol.1300299
121. Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KHG. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. (2009) 31:331–41. doi: 10.1016/j.immuni.2009.08.001
122. Kawakami K, Kinjo Y, Uezu K, Miyagi K, Kinjo T, Yara S, et al. Interferon-gamma production and host protective response against Mycobacterium tuberculosis in mice lacking both IL-12p40 and IL-18. Microbes Infect. (2004) 6:339–49. doi: 10.1016/j.micinf.2004.01.003
123. Nakamatsu M, Yamamoto N, Hatta M, Nakasone C, Kinjo T, Miyagi K, et al. Role of interferon-gamma in Valpha14+ natural killer T cell-mediated host defense against Streptococcus pneumoniae infection in murine lungs. Microbes Infect. (2007) 9:364–74. doi: 10.1016/j.micinf.2006.12.003
124. Tettelin H, Nelson KE, Paulsen IT, Eisen JA, Read TD, Peterson S, et al. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science. (2001) 293:498–506. doi: 10.1126/science.1061217
125. Lanie JA, Ng W-L, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, et al. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol. (2007) 189:38–51. doi: 10.1128/JB.01148-06
126. Kurioka A, van Wilgenburg B, Javan RR, Hoyle R, van Tonder AJ, Harrold CL, et al. Diverse streptococcus pneumoniae strains drive a mucosal-associated invariant T-cell response through major histocompatibility complex class I-related molecule-dependent and cytokine-driven pathways. J Infect Dis. (2018) 217:988–99. doi: 10.1093/infdis/jix647
127. Hartmann N, McMurtrey C, Sorensen ML, Huber ME, Kurapova R, Coleman FT, et al. Riboflavin metabolism variation among clinical isolates of Streptococcus pneumoniae results in differential activation of MAIT cells. Am J Respir Cell Mol Biol. (2018) 58:767–76. doi: 10.1165/rcmb.2017-0290OC
128. Ivanov S, Fontaine J, Paget C, Macho Fernandez E, Van Maele L, Renneson J, et al. Key role for respiratory CD103(+) dendritic cells, IFN-γ, and IL-17 in protection against Streptococcus pneumoniae infection in response to α-galactosylceramide. J Infect Dis. (2012) 206:723–34. doi: 10.1093/infdis/jis413
129. Grimaldi D, Le Bourhis L, Sauneuf B, Dechartres A, Rousseau C, Ouaaz F, et al. Specific MAIT cell behaviour among innate-like T lymphocytes in critically ill patients with severe infections. Intensive Care Med. (2014) 40:192–201. doi: 10.1007/s00134-013-3163-x
130. Krismer B, Weidenmaier C, Zipperer A, Peschel A. The commensal lifestyle of Staphylococcus aureus and its interactions with the nasal microbiota. Nat Rev Microbiol. (2017) 15:675–87. doi: 10.1038/nrmicro.2017.104
131. Tuffs SW, Haeryfar SMM, McCormick JK. Manipulation of innate and adaptive immunity by Staphylococcal superantigens. Pathog Basel Switz. (2018) 7:E53. doi: 10.3390/pathogens7020053
132. Cheng P, Liu T, Zhou W-Y, Zhuang Y, Peng L, Zhang J-Y, et al. Role of gamma-delta T cells in host response against Staphylococcus aureus-induced pneumonia. BMC Immunol. (2012) 13:38. doi: 10.1186/1471-2172-13-38
133. Rieder SA, Nagarkatti P, Nagarkatti M. CD1d-independent activation of invariant natural killer T cells by staphylococcal enterotoxin B through major histocompatibility complex class II/T cell receptor interaction results in acute lung injury. Infect Immun. (2011) 79:3141–8. doi: 10.1128/IAI.00177-11
134. Hayworth JL, Mazzuca DM, Maleki Vareki S, Welch I, McCormick JK, Haeryfar SMM. CD1d-independent activation of mouse and human iNKT cells by bacterial superantigens. Immunol Cell Biol. (2012) 90:699–709. doi: 10.1038/icb.2011.90
135. Szabo PA, Rudak PT, Choi J, Xu SX, Schaub R, Singh B, et al. Invariant Natural Killer T Cells are pathogenic in the HLA-DR4-transgenic humanized mouse model of toxic shock syndrome and can be targeted to reduce morbidity. J Infect Dis. (2017) 215:824–9. doi: 10.1093/infdis/jiw646
136. Shaler CR, Choi J, Rudak PT, Memarnejadian A, Szabo PA, Tun-Abraham ME, et al. MAIT cells launch a rapid, robust and distinct hyperinflammatory response to bacterial superantigens and quickly acquire an anergic phenotype that impedes their cognate antimicrobial function: Defining a novel mechanism of superantigen-induced immunopathology and immunosuppression. PLoS Biol. (2017) 15:e2001930. doi: 10.1371/journal.pbio.2001930
137. Costanzo AE, Taylor KR, Dutt S, Han PP, Fujioka K, Jameson JM. Obesity impairs γδ T cell homeostasis and antiviral function in humans. PLoS ONE. (2015) 10:e0120918. doi: 10.1371/journal.pone.0120918
138. Fan X, Rudensky AY. Hallmarks of tissue-resident lymphocytes. Cell. (2016) 164:1198–211. doi: 10.1016/j.cell.2016.02.048
139. Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, et al. Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol. (2013) 87:6911–24. doi: 10.1128/JVI.02943-12
140. Guo H, Topham DJ. Interleukin-22 (IL-22) production by pulmonary Natural Killer cells and the potential role of IL-22 during primary influenza virus infection. J Virol. (2010) 84:7750–9. doi: 10.1128/JVI.00187-10
141. Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, et al. IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol. (2013) 182:1286–96. doi: 10.1016/j.ajpath.2012.12.007
142. Kumar P, Thakar MS, Ouyang W, Malarkannan S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. (2013) 6:69–82. doi: 10.1038/mi.2012.49
143. Barthelemy A, Sencio V, Soulard D, Deruyter L, Faveeuw C, Le Goffic R, et al. Interleukin-22 immunotherapy during severe influenza enhances lung tissue integrity and reduces secondary bacterial systemic invasion. Infect Immun. (2018) 86:e00706–17. doi: 10.1128/IAI.00706-17
144. Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, et al. Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am J Physiol Lung Cell Mol Physiol. (2015) 309:L158–167. doi: 10.1152/ajplung.00338.2014
145. van Wilgenburg B, Scherwitzl I, Hutchinson EC, Leng T, Kurioka A, Kulicke C, et al. MAIT cells are activated during human viral infections. Nat Commun. (2016) 7:11653. doi: 10.1038/ncomms11653
146. Stervbo U, Pohlmann D, Baron U, Bozzetti C, Jürchott K, Mälzer JN, et al. Age dependent differences in the kinetics of γδ T cells after influenza vaccination. PLoS ONE. (2017) 12:e0181161. doi: 10.1371/journal.pone.0181161
147. Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity. (2018) 48:453–73. doi: 10.1016/j.immuni.2018.03.009
148. Paget C, Chow MT, Gherardin NA, Beavis PA, Uldrich AP, Duret H, et al. CD3bright signals on γδ T cells identify IL-17A-producing Vγ6Vδ1+ T cells. Immunol Cell Biol. (2015) 93:198–212. doi: 10.1038/icb.2014.94
149. Barthelemy A, Ivanov S, Hassane M, Fontaine J, Heurtault B, Frisch B, et al. Exogenous activation of invariant Natural Killer T Cells by α-galactosylceramide reduces pneumococcal outgrowth and issemination postinfluenza. mBio. (2016) 7:e01440–16. doi: 10.1128/mBio.01440-16
150. Beshara R, Sencio V, Soulard D, Barthélémy A, Fontaine J, Pinteau T, et al. Alteration of Flt3-Ligand-dependent de novo generation of conventional dendritic cells during influenza infection contributes to respiratory bacterial superinfection. PLoS Pathog. (2018) 14:e1007360. doi: 10.1371/journal.ppat.1007360
151. Porte R, Fougeron D, Muñoz-Wolf N, Tabareau J, Georgel A-F, Wallet F, et al. A Toll-Like Receptor 5 agonist improves the efficacy of antibiotics in treatment of primary and influenza virus-associated pneumococcal mouse infections. Antimicrob Agents Chemother. (2015) 59:6064–72. doi: 10.1128/AAC.01210-15
Keywords: unconventional T cells, influenza A virus, secondary bacterial infection, Streptococcus pneumoniae, Staphylococcus aureus, immune suppression, barrier function, immunotherapy
Citation: Paget C and Trottein F (2019) Mechanisms of Bacterial Superinfection Post-influenza: A Role for Unconventional T Cells. Front. Immunol. 10:336. doi: 10.3389/fimmu.2019.00336
Received: 05 October 2018; Accepted: 08 February 2019;
Published: 01 March 2019.
Edited by:
David Lewinsohn, Oregon Health & Science University, United StatesReviewed by:
Siobhan Cowley, State Food and Drug Administration, ChinaCopyright © 2019 Paget and Trottein. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: François Trottein, ZnJhbmNvaXMudHJvdHRlaW5AcGFzdGV1ci1saWxsZS5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.