 Minghui He
Minghui He Lisa S. Westerberg
Lisa S. Westerberg- Department of Microbiology Tumor and Cell Biology, Karolinska Institutet, Stockholm, Sweden
The germinal center (GC) is a transient anatomical structure formed during the adaptive immune response that leads to antibody affinity maturation and serological memory. Recent works using two-photon microscopy reveals that the GC is a highly dynamic structure and GC B cells are highly motile. An efficient selection of high affinity B cells clones within the GC crucially relies on the interplay of proliferation, genome editing, cell-cell interaction, and migration. All these processes require actin cytoskeleton rearrangement to be well-coordinated. Dysregulated actin dynamics may impede on multiple stages during B cell affinity maturation, which could lead to aberrant GC response and result in autoimmunity and B cell malignancy. This review mainly focuses on the recent works that investigate the role of actin regulators during the GC response.
The germinal center (GC) is the site where B cells can modify their B cell receptor (BCR) affinity for antigen by expression of activation induced deaminase (AID), proliferation, and selection. The outcome will be plasma cells and memory B cells that have acquired B cell receptors (BCR) with higher affinity for antigen. During the last 10 years, the dynamics of GC B cells have been investigated by usage of intravital two photon microscopy and revealed an enormous dynamics of GC B cells in migration pattern and interactions with follicular dendritic cells (FDCs) and T follicular helper (Tfh) cells (1–3). A long-standing question about how the antigen is delivered to the FDC network has also been revealed. Small antigens can diffuse into the FDC network by the conduit system (4). Migratory B cells in the marginal zone (MZ) of the spleen and B cells close to the sinusoid macrophages in lymph nodes (LN) can capture antigen by the B cell complement receptors such as CD21 and deliver the antigen into the FDC network (5).
The GC reaction relies on the interplay between cell migration, cell-cell interaction, and cell proliferation. The GC is anatomically divided into the dark zone (DZ) and light zone (LZ). The DZ is the site where B cells have high expression of AID that induces somatic hypermutation (SHM) and Ig class switch recombination (Ig CSR) in the genes encoding the Ig heavy and light chains. The LZ is the site for B cell competition and selection to obtain B cells with highest affinity for antigen. Recent migratory B cell from the DZ compete for retrieval of native antigen on follicular dendritic cells (FDCs). BCR binding of antigen leads to endocytosis and processing of antigen for loading on MHC class II molecules (6–9). This process relies on that B cells form two types of immunological synapses, the first synapse will polarize the machinery for BCR endocytosis for antigen retrieval from FDCs and the second synapse is formed by MHC class II—peptide interaction with T cell receptors (TCR)s on Tfh cells (8). During extraction of antigen from the immune synapse by B cells, the strength and timing of mechanical forces in immune synapses can promote affinity discrimination (10, 11). The antigen presenting B cells interact with Tfh cells that provide co-stimulation and cytokines such as IL-21 and IL-4. The B cell expressing a BCR that have acquired highest affinity for the antigen will acquire more antigen for MHC class II presentation and outcompete B cells expressing a BCR with lower affinity for antigen (3). An estimated 10% of the B cells migrate back to the DZ (3, 12) to undergo more SHM to increase the BCR affinity for antigen. The B cells that have acquired higher affinity for antigen can undergo differentiation to plasma cells and memory cells (13). Whereas the differentiation program to become a plasma cell is defined in quite detail, the memory B cell differentiation program has only recently started to be identified. It is clear that the cell fate decisions that B cells make in the GC are well characterized and coordinated by expression of transcription factors. Pax5 is critical to maintain the GC B cell phenotype. Increased expression of IRF4 and downregulation of Pax5 is the first differentiation step toward plasmablasts and followed by upregulated expression of Blimp1 and Xbp1 in fully differentiated plasma cells. This induces a loss of B cell identity and plasma blasts leave the GC to migrate to the B-T cell bridging areas. The GC response is orchestrated by coordinated changes in cell shape to migrate between the DZ and LZ and to communicate with FDCs and Tfh cells in the LZ.
During the process of finding interaction partners, GC B cells rapidly change cell shape and polarization by forming leading edge protrusions and trailing uropods (14). It is therefore not surprising that inborn errors in genes that regulate the actin cytoskeleton lead to aberrant GC formation. What is perhaps more surprising is that specific mutations lead to development of autoreactive GCs, suggesting that the effects on discriminating the self and non-self B cell clones during the GC reaction is skewed. The importance of actin dynamics and generation of force in the B cell immune synapse has recently been described (11, 15). Investigation of patients with primary immunodeficiency diseases due to inborn errors in B cell responses provides important information about B cell dysfunction in severe disease (16). To understand aberrations in the GC reaction, animal models provide in depth analysis of the anatomical structure in secondary lymphoid organs and the outcome measured as plasma cell generation and antibody production (Figure 1). Here we review recent progress in understanding how cytoskeletal regulators leading to Arp2/3 mediated actin polymerization regulate the B cell fate during the GC response (Table 1). This axis of regulation to actin dynamics involves B cell receptor (BCR) signaling to guanine exchange factors (GEFs) that activate the small GTPases of the Ras homology (Rho) family. Rho GTPases binds to and activates the Wiskott-Aldrich syndrome (WASp) family proteins for actin polymerization by the Arp2/3 complex.
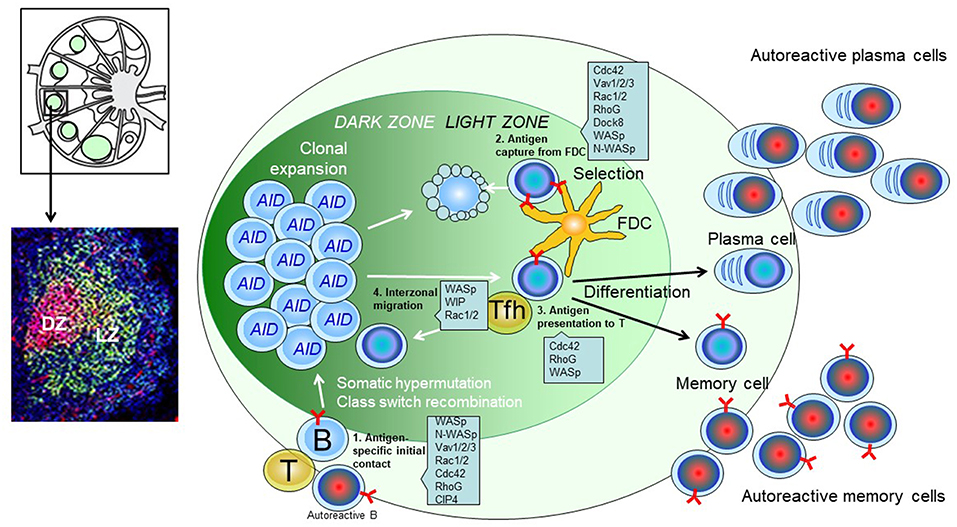
Figure 1. Predicted role of actin regulators during the GC response of B cells. (Upper left): Schematics of the lymph node structure. (Lower left): Immunohistochemistry of a single GC. Red: GL7; Green: CD21/35; Blue: B220. (Right): Schematic of a GC. Antigen-engaged B cells meet antigen experienced Tfh cells and enter the B cell follicle to initiate a GC response. B cells in dark zone express AID (activation-induced cytidine deaminase) and undergo somatic hypermutation. B cell clones that successfully mutate the BCR migrate to light zone, capture antigens deposit on the surface of FDC (follicular dendric cells), and present the antigen to Tfh (T follicular helper) cells. The positive selected B cell clones can differentiate into plasma cells, memory cells or migrate back to dark zone for further mutations and selection.
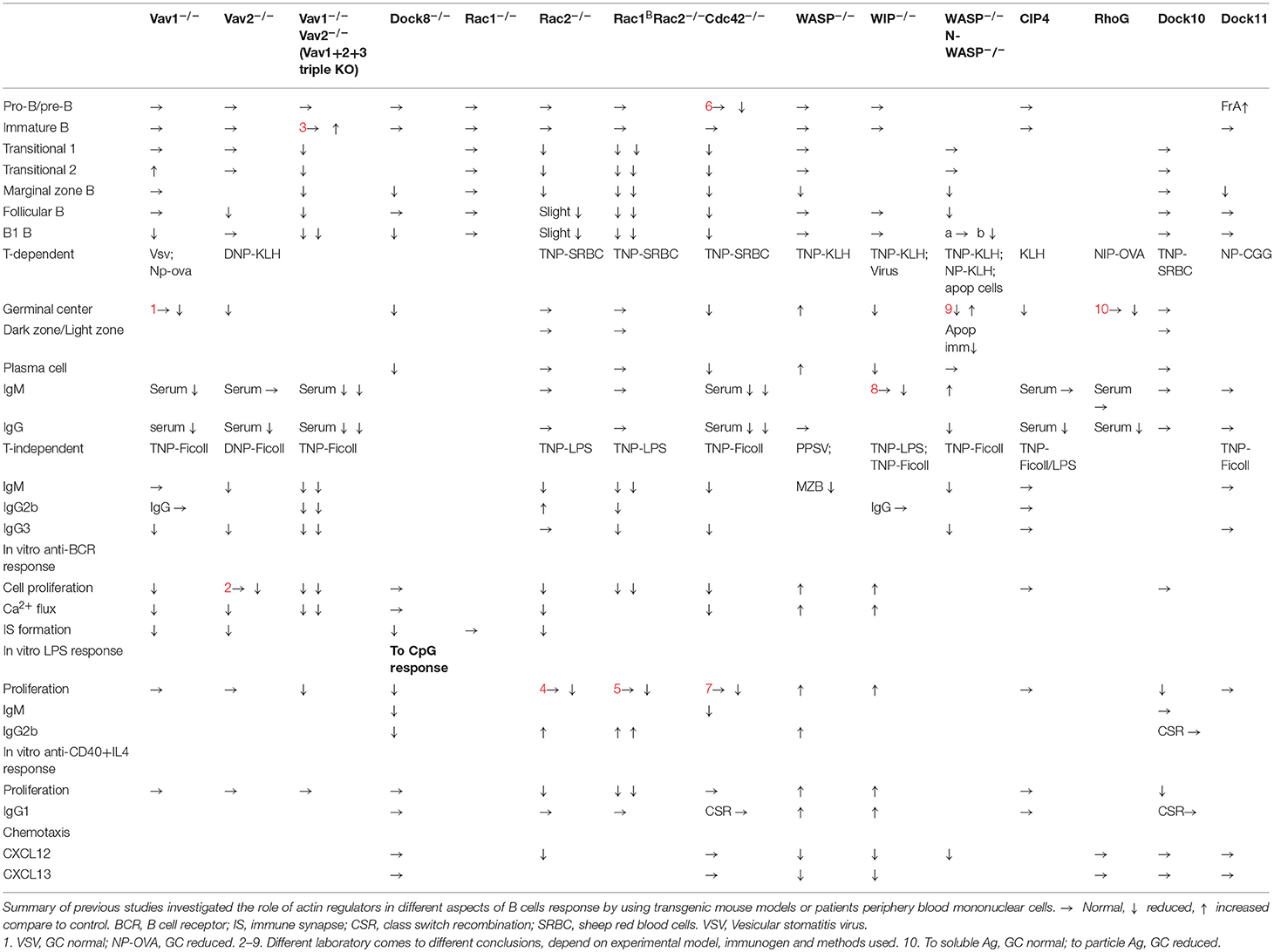
Table 1. B cell development and periphery B cell response in actin regulator deficiency.
GEFs: Dock Family and Vav1-3
GEFs activate small GTPases by stimulating the exchange of guanosine diphosphate (GDP) to guanosine triphosphate (GTP). GTPase activating proteins (GAPs) stimulate GTP hydrolysis thereby reinstating the GDP-bound form of the GTPases to terminate their signaling. Regulated by GEFs and GAPs, the Rho family GTPases cycle between a GDP-bound inactive form and a GTP-bound active form (17). The GEFs that regulate Rho GTPases called RhoGEFs fall into two different classes: the dedicator of cytokinesis (Dock) family of proteins including Dock 1–11 and the diffuse B-cell lymphoma (Dbl) family including Vav1-3.
Dock Family Proteins
Dock8, a GEF for Rho GTPases, was first identified in a yeast two hybrid screening for Cdc42 binding partners (18). Dock8 deficiency in patients leads to multiple immune disorders including T and B cell deficiency, increased cutaneous viral infection, severe atopy with elevated serum IgE level, and compromised antibody response (19, 20). Detailed analysis of patient PBMCs reveal a reduced proportion of T cells and slightly elevated CD19+ B cells. However, among the periphery blood B cells in Dock8 deficient patients, there is almost a complete lack of CD27+ B cells including switched memory (IgD−CD27+) B cells and non-switched memory (MZ like, IgD+CD27+) B cells. This is associated with reduced serum IgG and IgM response to vaccination and lack of serological memory in the patients (21). These data suggests that Dock8 deficiency leads to a compromised GC response. In depth studies of a Dock8−/− mice shows reduced naïve T cells, MZ B, and B1 B cell subsets. Upon antigen challenge, the Dock8−/− B cell response in the extrafollicular pathway is comparable to that of wildtype B cells. However, the GC response and antibody affinity maturation of Dock8−/− B cells is greatly compromised although the rate of SHM is comparable to wildtype cells. The reduced GC response is probably not due to compromised entry of Dock8−/− B cells into the GCs. Because during the early GC response (day 2–5), Dock8−/− and wildtype B cells occupy the GC area equally well. However, at the later time points, Dock8−/− GC B cells gradually lose the competition, suggesting a critical role of Dock8 for GC B cell persistence or survival. This defect may be caused by the compromised immune synapse formed during the selection stage of GC B cells in the LZ, which may provide crucial survival signal to the GC B cells (22).
Dock2
Dock2 is predominantly expressed in hematopoietic cells and human loss-of-function mutations result in early onset of invasive bacterial and viral infection, T cell lymphopenia, and decreased antibody responses (23). Detailed analysis of Dock2-deficient patient cells reveal defective T cell and B cell responses upon antigen stimulation as a result of impaired Rac activation and actin polymerization. Analysis of B cell specific Dock2−/− (CD19-Cre x Dock2fl/fl) mice and cell lines have identified a critical role of Dock2 in B cells during the antigen induced immune synapse formation, cell proliferation, and plasma cell differentiation (24, 25). CD19-Cre x Dock2fl/fl mice have normal B cell development in bone marrow from the pro-/pre-B cell stage to the immature B cell stage. However, there is a dramatic decrease in the mature B cell subsets including transitional B cell, marginal zone B, and follicular B cells (25, 26). This could at least partly result from compromised cell migration to chemokines of Dock2−/− B cells (26, 27). CD19-Cre x Dock2fl/fl mice have decreased IgG1 and IgG2b antibody response to T cell dependent (TD) antigen. Examination of the GC response show that Dock2 deficiency does not affect GC B cell formation and Ig class switching, whereas the GC B cell proliferation and differentiation into plasma cells are greatly compromised (25). This could be caused by a defective immune synapse formation at the selection stage in the LZ and therefore lack of survival and differentiation signal from the Tfh cells.
Dock10
Other proteins in the Dock family have been associated with B cell biology and the GC response. In a screen for genes upregulated by IL-4 activation of B cells, Dock10 was one of the highest expressed genes (28, 29). Dock10−/− mice have reduced numbers of B cells in secondary lymphoid organs, and FO B cells display elevated expression of membrane CD23 (30). These results suggest that Dock10 plays a role in B-cell lymphopoiesis in secondary lymphoid tissue. However, specific deletion of Dock10 in B cells was associated with a mild phenotype with normal B cell development and normal B cell spreading, polarization, motility, chemotaxis, aggregation, and Ig class switching. Dock10B B cells showed lower proliferation in response to anti-CD40 and IL-4 stimulation in vitro and Dock10B mice had reduced IgG response to NP-KLH in vivo (28). This suggest that IL-4 induced activation of B cells was decreased both in vitro and in vivo but that most B cell responses were functional in the absence of Dock10, rising the interesting question if the closest homologs to Dock10, Dock 9, and Dock11 may have redundant activity in B cells.
Dock11 is highly expressed in lymphocytes and Dock11-deficient mice have reduced development of splenic MZ B cells (31). Dock11−/− mice show a normal antibody response to T cell independent (TI) antigens and TD antigens, TNP-LPS, TNP-Ficoll, and NP-CGG (32). This indicates that Dock11−/− mice have a normal GC response although generation of high affinity antibodies was not examined in detail.
Vav1, Vav2, and Vav3
Vav proteins were first described as proto-oncogenes acting as substrates for tyrosine protein kinase activity (33). Recent studies examining the role of Vav family proteins, including Vav1, Vav2, and Vav3, in lymphocytes have revealed their critical function to link lymphocyte antigen receptor activation to actin cytoskeleton dynamics. Vav1, Vav2, and Vav3 share more than 50% homology in the protein sequences, all of which are composed of a Dbl-homologous (DH) domain, pleckstrin homology (PH) domain, SH2/SH3 domain, proline rich area, and a calponin homology (CH) domain (34). Reduced Vav1 expression has been detected in common variable immunodeficiency (CVID) patients with defective TCR mediated signaling (35). Vav1 expression is mainly restricted to the haematopoietic lineage cells (36). Although Vav1 has been shown to play a critical role in T cell development and activation by regulation of TCR signaling, B cell development of Vav1−/− mice seems largely unaltered, except a profound reduction of B1 B cells in the peritoneal cavity (37–39). The in vivo response of Vav1−/− B cells to T-independent antigens (both TI-1 and TI-2) is comparable to wildtype cells as measured by production of antigen specific IgM. However, despite normal formation of GCs in response to vesicular stomatitis virus (VSV), antigen specific IgG responses are reduced. In response to NIP-OVA, Vav1−/− mice completely lack GCs, which probably leads to reduced antigen specific IgG1 and IgG2b. Vav1 is highly expressed in all haematopoietic cells, whereas Vav2 shows the highest expression in splenic mature B cells when compared to other B cell subsets, suggesting an important role of Vav2 in mature B cell homeostasis. Consistently, Vav2−/− mice seem to have a development block from the immature/transitional B cell stage to the mature B cell stage. There is also reduced response to both TI and TD antigens of Vav2−/− B cells when compared to wildtype cells. In response to TNP-KLH, Vav2−/− mice show an 80% reduction in the GC B cells. Because T cell subsets and function are suggested to be unaltered in Vav2−/− mice, it is likely that the compromised GC response in Vav2−/− mice results from a B cell intrinsic defect (40, 41).
All three proteins of Vav1, Vav2, and Vav3 are quickly phosphorylated after the antigen receptor engagement. Since previous data demonstrates relative mild defect in Vav1−/− and Vav2−/− single knockout mice, Vav1, Vav2, and Vav3 may have functional redundancy downstream of BCR activation. The collected experimental data so far supports this hypothesis. Vav1−/− Vav2−/− double knockout mice and Vav1−/− Vav2−/− Vav3−/− triple knockout mice have a more severe B cell deficiency, including a developmental block at the immature/transitional B cell stage in bone marrow and spleen, reduced serum level of IgM and IgG, defective response to TI and TD antigens and greatly compromised cell proliferation and calcium flux upon BCR stimulation (42).
Small Rho GTPases
The Rho family belongs to the Ras super family of small GTPases and like other Ras-related proteins, most of the Rho GTPases adopt either active GTP-bound or inactive GDP-bound conformational states. The important role of the small Rho GTPases in regulation of actin dynamics was first characterized by Alan Hall and coworkers that showed induction of specific actin structures when microinjected into fibroblasts (43–46). Cell division control protein 42 homolog (Cdc42), Ras-related C3 botulinum toxin substrate 1 (Rac1), and Ras homolog gene family, member A (RhoA) has been the prototypic members of the family of small Rho GTPases. Cdc42 microinjection into fibroblasts induces membrane filopodia and Cdc42 regulates cell polarity and cell division (44). Rac1 induces membrane ruffles and lamellipodia and RhoA regulates stress fiber formation (46, 47). It was later shown that such actin dependent structures is induced by Cdc42, Rac1, and RhoA in other cell types including B cells (48, 49). Studies from many laboratories have revealed extensive cross-talk among the Rho GTPases, not the least in hematopoietic cells that express many variants of the Rho GTPases (50).
Cdc42
The small GTPase Cdc42 can mediate the interaction between actin and microtubules and regulate cell shape and polarity. Cdc42 coordinates actin polymerization by direct binding to WASp and N-WASp (51–53) and coordinates the microtubule cytoskeleton by binding to the Cdc42 interacting protein (CIP4) that directly regulates microtubule assembly (54, 55). In vitro, dominant negative mutants of Cdc42 interfere with B cell formation of cytoskeletal responses such as formation of filopodia, and cell polarization and migration (48, 49). Two patients with unrelated Cdc42 mutations have been reported recently (56, 57). The patients are characterized with developmental delay, macro thrombocytopenia, and lymphedema. Repeated upper respiratory infection and chronic leukocytopenia has been observed in one of the patients, indicating a mild form of immunodeficiency. Using animal models, Cdc42 has a non-redundant role during B cell development since deletion in early B cell progenitors results in a severe reduction in the numbers of mature B cells (58, 59). Using CD19-Cre for deletion of a floxed Cdc42 allele, Cdc42-deficient B cells have decreased phosphorylation of Akt upon BCR activation and reduced BAFFR signaling leading to reduced proliferation and increased apoptosis (58). Mice with B cell-specific deletion of Cdc42 induced a reduced antibody response to TNP-Ficoll and NP-KLH. Early deletion of Cdc42 during B cell development using mb1-Cre x Cdc42flox/flox mice, led to reduced B cell number in spleen and LN and antibody titers reaching the detection limit (59). This led to abolished capacity to generate a high affinity antibody response to NP-KLH and reduced GC response to Influenza A virus. Together this suggests that Cdc42 serves an important role during B cell development in the bone marrow. Using the super resolution microscopy technique dSTORM, Cdc42 KO B cells showed increased dispersion of IgM nanoclusters and decreased BCR induced signaling leading to reduced internalization of antigen (59). Using two-photon microscopy, Cdc42 KO B cells formed fewer contacts with antigen-specific T cells (59).
Cdc42−/− B cells migrate normally to chemokines in vitro (58, 60), but have reduced capacity to home to the B cell follicles in the spleen (60). To exclude the effect of Cdc42 deletion on B cell development and the effect of Cdc42 deficiency on positioning in LNs and splenic white pulp, inducible deletion of Cdc42 by crossing Cdc42flox/flox mice with mb1-Cre-ERT2 mice was employed (60). This approach allowed for specific deletion of Cdc42 in B cells that had already entered the B cell follicles. Inducible deletion of Cdc42 in B cells led to reduced number of splenic MZ B cells and follicular B cells. Upon antigen challenge with the particulate antigen sheep red blood cells (SRBC), Cdc42B−ERT2 had reduced formation of GCs. In response to NP-KLH, Cdc42B−ERT2 B cells showed reduced capacity to induce NP-specific antibodies. This was associated with reduced capacity to present antigenic peptides to T cells in vitro (60). Moreover, Cdc42B−ERT2 B cells failed to form membrane extensions rich in tubulin and formed only short membrane protrusions that do not contain tubulin.
Together, these studies suggest that Cdc42 plays a role both during B cell development and in GC response and Cdc42 deficient B cells fail to regulate formation of membrane extensions and to interact with T follicular helper cells.
Rac1 and Rac2
The Rac proteins were first identified in Snyderman's laboratory in 1989 (61). Sequence analysis reveals more than 90% homologous region between Rac1 and Rac2 proteins. A point mutation that leads to a dominant negative form of Rac2 (D57N) has been identified in infant patients characterized with recurrent bacterial infection and failure of wound healing resulting from defective neutrophil function (62–64). Although there is reduced T and B cell count in the patient, serum Ig level is normal except for one patient that had hypogammaglobulinemia (64). One of the patients harboring a homozygous mutation in Rac2 (W56X) that leads to a complete loss of the protein developed progressive B cell lymphopenia and hypogammaglobinemia (64). Based on studies of mice that lack Rac1 and Rac2, their function in multiple cellular processes, including proliferation, survival, adhesion, and migration have been implicated. In contrast to the B cell specific Rac1 knockout mice that do not present an obvious alteration of B cell functionality, Rac2 deficiency or combined deficiency of Rac1 and Rac2 (Rac1BRac2−/−) leads to developmental block of B cells at the immature/transitional B cell stage. A study by Tybulewicz et al. shows that this is probably not due to a differentiation arrest of the transitional B cells, since ectopic expression of the anti-apoptotic gene Bcl-xl can partly rescue the differentiation defect of the Rac1BRac2−/− immature/transitional B cells. Instead, the defective migration toward chemokines is likely to be the reason why Rac1BRac2−/− B cells are unable to enter the white pulp where crucial survival signals to the mature B cells are available. This leads to a large reduction of the mature B cell population in the spleen including marginal zone B cells and follicular B cells (65). Defective entry of mature B cells into the white pulp makes it difficult to study the role of Rac1 and Rac2 in antigen-activated B cells. To circumvent this issue, Rac proteins were inducibly deleted by Tamoxifen in the mature B cell population (Rac1B−ERT2Rac2−/−) (66). The TI response to TNP-LPS of Rac1B−ERT2Rac2−/− B cells is greatly compromised, with reduced level of antigen specific IgM and IgG3, whereas the TD response to TNP-SRBC in these mice seems comparable to wildtype mice, with a normal GC response and plasma cell output. Notably, Rac1B−ERT2Rac2−/− mice have increased serum titer of antigen specific IgG2b. In vitro analysis of Ig class switching reveals that the Rac1B−ERT2Rac2−/− B cells have increased capacity to switch to IgG2b, possibly attributed to increased gamma2b germline transcript. In addition, B cell activation induced by BCR cross-linking is compromised in Rac1B−ERT2Rac2−/− B cells and associated with reduced cell proliferation and survival. This could be caused by compromised BCR signaling and upregulation of BAFF-R.
CIP4
CIP4 (Cdc42 interacting protein 4) belongs to the Fes–CIP4 homology-Bin/Amphyphysin/Rvsp (F-BAR) family of proteins, which includes FBP17 (formin binding protein 17), and Toca-1 (transducer of Cdc42-dependent actin assembly 1). CIP4 interacts with Cdc42 and is a downstream target of activated GTP-bound Cdc42 (54). Similar to mice with Cdc42-deficient B cells, mice completely devoid of CIP4 have normal B and T cell development but reduced germinal center formation and decreased production of high affinity IgG in response to NP-KLH (67). Since CIP4 was deleted in all cells, the specific role of CIP4 in GC B cells and T cells was not examined. CIP4-deficient T cells had decreased migration and integrin-mediated adhesion under sheer forces, suggesting a defect in entry of Tfh cells into the GC.
TC10/RhoG
TC10/RhoG is an atypical Rho GTPase identified as a member of the ras homolog gene family (68). TC10/RhoG is a member of the Rho family of GTPases that shares 72–62% sequence identity with Rac1 and Cdc42, respectively (69). In contrast to the marked defect of Cdc42-deficient B cells, specific deletion of TC10 had little effect on B cell development or differentiation into GC B cells, indicating that Cdc42 may compensate for loss of TC10 (70). Indeed, deletion of both Cdc42 and TC10 in B cells led to much reduced B cell proliferation in response to LPS and CpG stimulation.
WASp Family of Actin Regulators
The Rho GTPases activate the Wiskott-Aldrich syndrome protein (WASp) family of actin regulators. The WASp family of proteins includes WASp, neuronal (N)-WASp, and WASp-family verprolin-homologous protein (WAVE)/suppressor of the cyclic AMP receptor (SCAR) 1–3, WASp and SCAR homolog (WASH), and junction-mediating and regulatory protein (JMY) (71–73). WASp family proteins are characterized by high homology in the C-terminal domain consisting of the verprolin cofilin acidic (VCA) domain though which they can bind to globular actin and the Arp2/3 complex. The N-terminus of the protein show higher variability likely linked to cell-specific functions. At rest, WASp and N-WASp resides in an auto-inhibited conformation due to an intramolecular interaction between the VCA domain and the GTPase-binding domain (74–76). Upon binding of Cdc42, the auto-inhibited conformation is released and exposes the VCA domain that allows for recruitment of the Arp2/3 complex and actin polymerization. Rac1 and Rac2 regulate activation of the multimeric WAVE/Scar regulatory complex to stimulate actin polymerization by the VCA domain (77–79). WASp was the first identified member due to that its loss-of-function leads to the severe immunodeficiency disease Wiskott-Aldrich syndrome (WAS), initially described by Alfred Wiskott in 1937 and Robert Aldrich in 1954 (Wiskott A, Familiärer, angeborener Morbus Werlhofii? Monatsschr Kinderheilkd 1937; 68:212-216; Aldrich RA, Pediatrics 1954; 13:133–139).
WASp and N-WASp
WASp is uniquely expressed in hematopoietic lineage cells whereas N-WASp that shares 50% homology with WASp in the amino acid sequence is ubiquitously expressed. Humoral immunodeficiency caused by mutations in the WAS gene encoding WASp is associated with failure to respond to common pathogens and up to 40–70% of patients developing autoimmune disease with high titers of autoantibodies (80–85).
WAS patients have normal to slightly reduced absolute numbers of circulating B cells, however, have reduced MZ B cells and dysmorphic GC in spleen (80, 86). Although the proportion of memory B cells remains intact, WAS patient memory B cells have reduced responsiveness to BCR activation probably due to impaired BCR signaling (87). WASp−/− mice have normal B cell development and FO B cells, but reduced number of MZ B cells and MZ precursor T2-MZP cells (88–90). This leads to reduced capacity to respond to TI antigens TNP-Ficoll and TNP-dextran, likely due to a combined effect of reduced number of MZ B cells and decreased antigen delivery by the MZ B cells to the B cell follicle (88, 90, 91). WASp−/− mice have slightly reduced capacity to form high affinity IgG antibodies to TD antigen NP-KLH and particulate antigen SRBCs (88, 90–92). WASp−/− B cells have decreased formation of the immune synapse upon BCR activation in vitro (89, 93) and reduced capacity to from long membrane extensions (49). Despite this defects in the BCR response, WASp−/− B cells can present antigen and induce T cell activation similar to wildtype B cells, at least in vitro (94, 95). WASp acts as a negative regulator for autoreactive B cells since both WAS patients and WASp−/− mice develop broad range IgM and IgG autoantibodies associated in mice with spontaneous generation of GCs (81, 85, 95). Moreover, WASp−/− B cells are hyper responsive to B cell receptor and Toll-like receptor (TLR) signals in vitro, thereby promoting a B cell–intrinsic break in tolerance. To understand the B cell intrinsic defects, WASpflox/flox mice were bred mb1-Cre mice to delete WASp specifically in B cells. These WASpB mice have high titers of autoreactive IgM and IgG and form large GCs in the absence of antigen challenge (91, 96). To reveal the unique and redundant role of WASp and N-WASp in the GC response, WASp−/− mice or WASpflox/flox mice were bred to N-WASpflox/flox mice and CD19-Cre or mb1-Cre to delete WASp and N-WASp specifically in B cells. Analysis of WASp−/−N-WASpB and WASpBN-WASpB mice revealed a reduced response to NP-KLH with small GCs that lost LZ and DZ integrity and failure to generated high affinity NP-specific IgG antibodies (95, 97). Strikingly, N-WASp deletion in WASp−/− B cells lowered the autoreactive antibodies and GCs, suggesting that N-WASp deletion protects mice from developing autoimmune disease (95, 97). Interestingly, N-WASp-deleted B cells (that express normal WASp) have increased BCR synapse response associated with development of autoantibodies in N-WASpB mice (93). This indicates that WASp and N-WASp serve both unique and redundant roles in BCR signaling to B cell activation. WASp-deficient follicular T (Tfh) cells show defective activation and proliferation and is likely to contribute to altered antibody production in WAS patients and WASp−/− mice (98). Moreover, WASp deficiency in regulatory B cells leads to exacerbated experimental autoimmune arthritis (99).
The WAS gene is localized on the X chromosome and only boys are affected by WAS mutations. Studies of asymptomatic female WAS carriers has revealed that while haematopoietic stem cells have largely random X chromosome inactivation, there is a strong selective advantage for B and T cells that express WASp during development and differentiation (88, 89). By analysis of WASp+/− heterozygous mice and WT:WASp−/− bone marrow chimeric mice, a strong advantage was detected for WASp-expressing FO B cells and MZ B cells in the spleen, as well as GC B cells in Peyer's patches (88, 89). It was later shown that WASp−/− B cells competed equally well with wildtype B cells among GC B cells, both DZ and LZ cells, whereas WASp−/−N-WASpB had selective disadvantage in contribution to the GC B cells (95). This suggests that WASp together with N-WASp are needed for a normal GC response to prevent selection of autoreactive B cells. Gene therapy for WAS patients is currently evaluated in several centers and has shown success and ameliorate the autoreactive B cells. Gene therapy may provide a future curative option alongside haematopoietic stem cell transplantation (100).
WASp-interacting Protein (WIP)
WIP was originally cloned as a WASp interacting protein using a yeast two-hybrid system. WIP interacts with the N-terminal WASp homology domain (WH) 1 domain of both WASp and N-WASp and is essential for their stability (101–103). Three pedigrees of WIP deficient patients have been reported so far (104–106). Their symptoms highly resemble those of WAS patients, however, with milder thrombocytopenia and earlier onset of severe infections and T cell deficiency (107). Similar to WAS patients, WIP deficient patients have elevated serum IgE titer and normal to elevated IgG and IgM antibody titer, suggesting abnormal B cell responses (105, 107). WIP−/− B cells show reduced B cell homing, chemotaxis, survival, and differentiation due to an altered CD19 cell surface dynamics (108). Upon NP-KLH immunization, WIP−/− mice failed to form GCs and have reduced NP-specific antibody responses. This was caused by reduced activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) in WIP−/− B cells. WIP has important function in B cells, independent of its binding to WASp, by direct binding to actin (109). B cells expressing WIP lacking the actin binding domain (ABD) of WIP (WIPΔABD) have reduced BCR induced actin foci and reduced signaling with PI3K to p-Akt. Using NP-KLH immunization of WT:WIP−/− mixed bone marrow chimeras, WIP−/− B cells are less efficient at differentiating into GC B cells in a competitive environment. However, in a non-competitive setting, GC responses are comparable to WT mice but WIP−/− mice are impaired in producing high-affinity antibodies (109). It was recently shown in T cells that WIP bridges Dock8 to WASp and actin and that Dock8 GEF activity is essential for TCR-driven WASp activation and F-actin assembly (110). It is plausible that WIP serves a similar function in BCR signaling.
Conclusions and Future Perspectives
Positive selection of B cells in GCs depends on the BCR affinity and requires help from Tfh cells. Selected B cells have three possible fates: to become a plasma cell, a memory cell, or to re-enter the DZ for more rounds of mutation and selection. Absolute high affinity is suggested to drive GC B cells to differentiate into plasma cells, whereas relatively lower affinity lead to differentiation into memory B cells. However, several questions remain elusive about how variable BCR affinity is discriminated and how cell fate decisions within the GCs are regulated. Some recent studies suggests that the actin regulators are involved in the antigen retrieval of GC B cells from FDC by polarization of the lysosomes to the BCR-antigen immune complex and by generating mechanic forces. This raises the interesting question of whether dysregulated actin dynamics can directly influence the fate decision of GC B cells and eventually impact on the quality and efficacy of humoral immune responses.
Deficiency in cytoskeletal regulation often influences the cell fate decision to become a FO B cell or MZ B cells. Mice devoid of Dock8, Cdc42, Rac2, WASp, WASp plus N-WASp, and WIP have reduced number of MZ B cells. Reduced MZ B cells may lead to decreased delivery of antigens to the FDC network, as is the case for mice lacking WASp and WASp plus N-WASp in B cells (88, 92). The reduction in MZ B cells may be related to changes in BCR signaling strength. Data support that the strength of BCR signaling in the transitional B cells that enter secondary lymphoid organs is important. MZ B cells are favored by low BCR signaling whereas FO B cells depend on high BCR signaling (111). Within the GC, BCR signaling may be of less importance and BCR as an endocytic receptor for antigen capture, processing, and presentation may be more important during affinity maturation (6). In contrast with naive and memory B cells, which extract antigen in the synapse center, GC B cells extract antigen using several small peripheral clusters. Both naive and GC B cell synapses require proximal BCR signaling, but GC B cells signal less through the protein kinase Cβ-NFκB allowing them to more stringently regulate antigen binding (10). A unifying conclusion from the studies discussed here is that there is enormous redundancy in signaling pathways leading to Arp2/3 mediated actin polymerization (Figure 2). However, approaches to use double-deficiency of two potential redundant factor such as WASp and N-WASp have led to surprising results. This is likely due to that the signaling threshold for BCR activation is fine-tuned to achieve a balance between antigen affinity and antigen extraction to avoid differentiation of autoreactive B cells and malignant B cell clones.
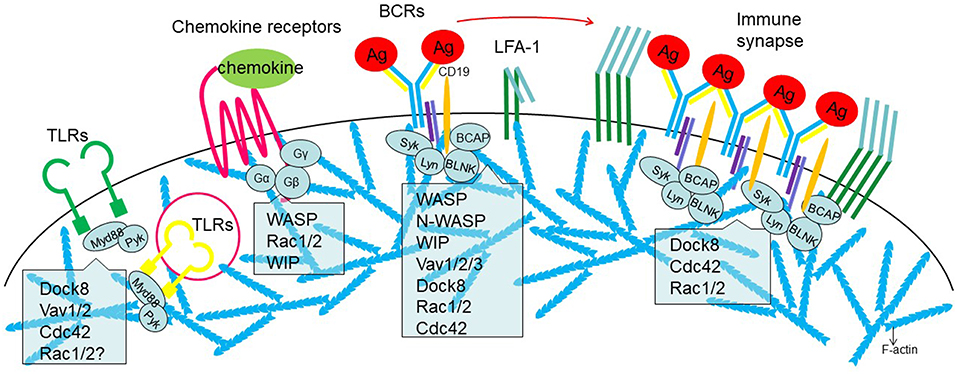
Figure 2. Involvement of actin regulators in the receptor signaling during B cell activation. From Left to right, surface or cytosolic Toll-like receptors (TLRs), chemokine receptors, B cell receptors (BCRs), and immune synapse. Role of actin regulators discussed is indicated.
Author Contributions
Both authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a postdoctoral fellowship from Olle Engqvist Byggmästare foundation to MH, the O. E. and Edla Johanssons foundation and Lars Hierta Memorial Foundation to MH, the Swedish Medical Society, Groschinsky Foundation and Åke Wiberg Foundation, the Swedish Cancer Society, the Swedish Research Council, the Swedish Childhood Cancer Fund, Karolinska Institutet, the European Commission 7th framework program, Åke Olsson foundation, Jeansson foundation, and Bergvall Foundation to LW. LW is a Ragnar Söderberg fellow in Medicine.
References
1. Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, et al. T follicular helper cell dynamics in germinal centers. Science. (2013) 341:673–7. doi: 10.1126/science.1241680
2. Shulman Z, Gitlin AD, Weinstein JS, Lainez B, Esplugues E, Flavell RA, et al. Dynamic signaling by T follicular helper cells during germinal center B cell selection. Science. (2014) 345:1058–62. doi: 10.1126/science.1257861
3. Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, et al. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. (2010) 143:592–605. doi: 10.1016/j.cell.2010.10.032
4. Roozendaal R, Mempel TR, Pitcher LA, Gonzalez SF, Verschoor A, Mebius RE, et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity. (2009) 30:264–76. doi: 10.1016/j.immuni.2008.12.014
5. Cinamon G, Zachariah MA, Lam OM, Foss FW Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. (2008) 9:54–62. doi: 10.1038/ni1542
6. Khalil AM, Cambier JC, Shlomchik MJ. B cell receptor signal transduction in the GC is short-circuited by high phosphatase activity. Science. (2012) 336:1178–81. doi: 10.1126/science.1213368
7. Chaturvedi A, Martz R, Dorward D, Waisberg M, Pierce SK. Endocytosed BCRs sequentially regulate MAPK and Akt signaling pathways from intracellular compartments. Nat Immunol. (2011) 12:1119–26. doi: 10.1038/ni.2116
8. Yuseff MI, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity. (2011) 35:361–74. doi: 10.1016/j.immuni.2011.07.008
9. Krautler NJ, Suan D, Butt D, Bourne K, Hermes JR, Chan TD, et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J Exp Med. (2017) 214:1259–67. doi: 10.1084/jem.20161533
10. Nowosad CR, Spillane KM, Tolar P. Germinal center B cells recognize antigen through a specialized immune synapse architecture. Nat Immunol. (2016) 17:870–7. doi: 10.1038/ni.3458
11. Natkanski E, Lee WY, Mistry B, Casal A, Molloy JE, Tolar P. B cells use mechanical energy to discriminate antigen affinities. Science. (2013) 340:1587–90. doi: 10.1126/science.1237572
12. Hauser AE, Junt T, Mempel TR, Sneddon MW, Kleinstein SH, Henrickson SE, et al. Definition of germinal-center B cell migration in vivo reveals predominant intrazonal circulation patterns. Immunity. (2007) 26:655–67. doi: 10.1016/j.immuni.2007.04.008
13. Mesin L, Ersching J, Victora GD. Germinal center B cell dynamics. Immunity. (2016) 45:471–82. doi: 10.1016/j.immuni.2016.09.001
14. Schwickert TA, Victora GD, Fooksman DR, Kamphorst AO, Mugnier MR, Gitlin AD, et al. A dynamic T cell-limited checkpoint regulates affinity-dependent B cell entry into the germinal center. J Exp Med. (2011) 208:1243–52. doi: 10.1084/jem.20102477
15. Tolar P. Cytoskeletal control of B cell responses to antigens. Nat Rev Immunol. (2017) 17:621–34. doi: 10.1038/nri.2017.67
16. Smith T, Cunningham-Rundles C. Primary B-cell immunodeficiencies. Hum Immunol. (2009) 27:199–227. doi: 10.1146/annurev.immunol.021908.132649
17. Sadok A, Marshall CJ. Rho GTPases: masters of cell migration. Small GTPases. (2014) 5:e29710. doi: 10.4161/sgtp.29710
18. Ruusala A, Aspenstrom P. Isolation and characterisation of DOCK8, a member of the DOCK180-related regulators of cell morphology. FEBS Lett. (2004) 572:159–66. doi: 10.1016/j.febslet.2004.06.095
19. Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, et al. Combined immunodeficiency associated with DOCK8 mutations. N Engl J Med. (2009) 361:2046–55. doi: 10.1056/NEJMoa0905506
20. Su HC, Jing H, Zhang Q. DOCK8 deficiency. Ann N Y Acad Sci. (2011) 1246:26–33. doi: 10.1111/j.1749-6632.2011.06295.x
21. Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat Immunol. (2012) 13:612–20. doi: 10.1038/ni.2305
22. Randall KL, Lambe T, Johnson AL, Treanor B, Kucharska E, Domaschenz H, et al. Dock8 mutations cripple B cell immunological synapses, germinal centers and long-lived antibody production. Nat Immunol. (2009) 10:1283–91. doi: 10.1038/ni.1820
23. Dobbs K, Dominguez Conde C, Zhang SY, Parolini S, Audry M, Chou J, et al. Inherited DOCK2 Deficiency in Patients with Early-Onset Invasive Infections. N Engl J Med. (2015) 372:2409–22. doi: 10.1056/NEJMoa1413462
24. Wang J, Xu L, Shaheen S, Liu S, Zheng W, Sun X, et al. Growth of B cell receptor microclusters is regulated by PIP2 and PIP3 equilibrium and Dock2 recruitment and activation. Cell Rep. (2017) 21:2541–57. doi: 10.1016/j.celrep.2017.10.117
25. Ushijima M, Uruno T, Nishikimi A, Sanematsu F, Kamikaseda Y, Kunimura K, et al. The Rac activator DOCK2 mediates plasma cell differentiation and IgG antibody production. Front Immunol. (2018) 9:243. doi: 10.3389/fimmu.2018.00243
26. Fukui Y, Hashimoto O, Sanui T, Oono T, Koga H, Abe M, et al. Haematopoietic cell-specific CDM family protein DOCK2 is essential for lymphocyte migration. Nature. (2001) 412:826–31. doi: 10.1038/35090591
27. Nombela-Arrieta C, Lacalle RA, Montoya MC, Kunisaki Y, Megias D, Marques M, et al. Differential requirements for DOCK2 and phosphoinositide-3-kinase gamma during T and B lymphocyte homing. Immunity. (2004) 21:429–41. doi: 10.1016/j.immuni.2004.07.012
28. Gerasimcik N, He M, Baptista MAP, Severinson E, Westerberg LS. Deletion of Dock10 in B cells results in normal development but a mild deficiency upon in vivo and in vitro stimulations. Front Immunol. (2017) 8:491. doi: 10.3389/fimmu.2017.00491
29. Yelo E, Bernardo MV, Gimeno L, Alcaraz-Garcia MJ, Majado MJ, Parrado A. Dock10, a novel CZH protein selectively induced by interleukin-4 in human B lymphocytes. Mol Immunol. (2008) 45:3411–8. doi: 10.1016/j.molimm.2008.04.003
30. Garcia-Serna AM, Alcaraz-Garcia MJ, Ruiz-Lafuente N, Sebastian-Ruiz S, Martinez CM, Moya-Quiles MR, et al. Dock10 regulates CD23 expression and sustains B-cell lymphopoiesis in secondary lymphoid tissue. Immunobiology. (2016) 221:1343–50. doi: 10.1016/j.imbio.2016.07.015
31. Nishikimi A, Meller N, Uekawa N, Isobe K, Schwartz MA, Maruyama M. Zizimin2: a novel, DOCK180-related Cdc42 guanine nucleotide exchange factor expressed predominantly in lymphocytes. FEBS Lett. (2005) 579:1039–46. doi: 10.1016/j.febslet.2005.01.006
32. Matsuda T, Yanase S, Takaoka A, Maruyama M. The immunosenescence-related gene Zizimin2 is associated with early bone marrow B cell development and marginal zone B cell formation. Immun Ageing I A. (2015) 12:1. doi: 10.1186/s12979-015-0028-x
33. Katzav S, Martin-Zanca D, Barbacid M. vav, a novel human oncogene derived from a locus ubiquitously expressed in hematopoietic cells. EMBO J. (1989) 8:2283–90. doi: 10.1002/j.1460-2075.1989.tb08354.x
34. Bustelo XR. Vav family exchange factors: an integrated regulatory and functional view. Small GTPases. (2014) 5:9. doi: 10.4161/21541248.2014.973757
35. Paccani SR, Boncristiano M, Patrussi L, Ulivieri C, Wack A, Valensin S, et al. Defective Vav expression and impaired F-actin reorganization in a subset of patients with common variable immunodeficiency characterized by T-cell defects. Blood. (2005) 106:626–34. doi: 10.1182/blood-2004-05-2051
36. Bustelo XR. The VAV family of signal transduction molecules. Crit Rev Oncogene. (1996) 7:65–88. doi: 10.1615/CritRevOncog.v7.i1-2.50
37. Tarakhovsky A, Turner M, Schaal S, Mee PJ, Duddy LP, Rajewsky K, et al. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature. (1995) 374:467–70. doi: 10.1038/374467a0
38. Zhang R, Alt FW, Davidson L, Orkin SH, Swat W. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the vav proto-oncogene. Nature. (1995) 374:470–3. doi: 10.1038/374470a0
39. Bachmann MF, Nitschke L, Krawczyk C, Tedford K, Ohashi PS, Fischer KD, et al. The guanine-nucleotide exchange factor Vav is a crucial regulator of B cell receptor activation and B cell responses to nonrepetitive antigens. J Immunol. (1999) 163:137–42.
40. Tedford K, Nitschke L, Girkontaite I, Charlesworth A, Chan G, Sakk V, et al. Compensation between Vav-1 and Vav-2 in B cell development and antigen receptor signaling. Nat Immunol. (2001) 2:548–55. doi: 10.1038/88756
41. Doody GM, Bell SE, Vigorito E, Clayton E, McAdam S, Tooze R, et al. Signal transduction through Vav-2 participates in humoral immune responses and B cell maturation. Nat Immunol. (2001) 2:542–7. doi: 10.1038/88748
42. Fujikawa K, Miletic AV, Alt FW, Faccio R, Brown T, Hoog J, et al. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J Exp Med. (2003) 198:1595–608. doi: 10.1084/jem.20030874
43. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. (2005) 21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721
44. Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. (1995) 81:53–62. doi: 10.1016/0092-8674(95)90370-4
45. Ridley AJ, Hall A. Distinct patterns of actin organization regulated by the small GTP-binding proteins Rac and Rho. Cold Spring Harbor Symposia Quant Biol. (1992) 57:661–71. doi: 10.1101/SQB.1992.057.01.072
46. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. (1992) 70:389–99. doi: 10.1016/0092-8674(92)90163-7
47. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. (1992) 70:401–10. doi: 10.1016/0092-8674(92)90164-8
48. Yoshida H, Tomiyama Y, Ishikawa J, Oritani K, Matsumura I, Shiraga M, et al. Integrin-associated protein/CD47 regulates motile activity in human B-cell lines through CDC42. Blood. (2000) 96:234–41.
49. Westerberg L, Greicius G, Snapper SB, Aspenstrom P, Severinson E. Cdc42, Rac1, and the Wiskott-Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood. (2001) 98:1086–94. doi: 10.1182/blood.V98.4.1086
50. Baptista MA, Westerberg LS. Activation of compensatory pathways via Rac2 in the absence of the Cdc42 effector Wiskott-Aldrich syndrome protein in Dendritic cells. Small GTPases. (2017) 2017:1–8. doi: 10.1080/21541248.2016.1275363
51. Aspenstrom P, Lindberg U, Hall A. Two GTPases, Cdc42 and Rac, bind directly to a protein implicated in the immunodeficiency disorder Wiskott-Aldrich syndrome. Curr Biol. (1996) 6:70–5. doi: 10.1016/S0960-9822(02)00423-2
52. Symons M, Derry JM, Karlak B, Jiang S, Lemahieu V, McCormick F, et al. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. (1996) 84:723–34. doi: 10.1016/S0092-8674(00)81050-8
53. Kolluri R, Tolias KF, Carpenter CL, Rosen FS, Kirchhausen T. Direct interaction of the Wiskott-Aldrich syndrome protein with the GTPase Cdc42. Proc Natl Acad Sci USA. (1996) 93:5615–8. doi: 10.1073/pnas.93.11.5615
54. Aspenstrom P. A Cdc42 target protein with homology to the non-kinase domain of FER has a potential role in regulating the actin cytoskeleton. Curr Biol. (1997) 7:479–87. doi: 10.1016/S0960-9822(06)00219-3
55. Tian L, Nelson DL, Stewart DM. Cdc42-interacting protein 4 mediates binding of the Wiskott-Aldrich syndrome protein to microtubules. J Biol Chem. (2000) 275:7854–61. doi: 10.1074/jbc.275.11.7854
56. Takenouchi T, Kosaki R, Niizuma T, Hata K, Kosaki K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: yet another locus for thrombocytopenia and developmental delay. Am J Med Genet A. (2015) 167A:2822–5. doi: 10.1002/ajmg.a.37275
57. Takenouchi T, Okamoto N, Ida S, Uehara T, Kosaki K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am J Med Genet A. (2016) 170A:852–5. doi: 10.1002/ajmg.a.37526
58. Guo FK, Velu CS, Grimes HL, Zheng Y. Rho GTPase Cdc42 is essential for B-lymphocyte development and activation. Blood. (2009) 114:2909–16. doi: 10.1182/blood-2009-04-214676
59. Burbage M, Keppler SJ, Gasparrini F, Martinez-Martin N, Gaya M, Feest C, et al. Cdc42 is a key regulator of B cell differentiation and is required for antiviral humoral immunity. J Exp Med. (2015) 212:53–72. doi: 10.1084/jem.20141143
60. Gerasimcik N, Dahlberg CI, Baptista MA, Massaad MJ, Geha RS, Westerberg LS, et al. The Rho GTPase Cdc42 is essential for the activation and function of mature B cells. J Immunol. (2015) 194:4750–8. doi: 10.4049/jimmunol.1401634
61. Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. (1989) 264:16378–82.
62. Ambruso DR, Knall C, Abell AN, Panepinto J, Kurkchubasche A, Thurman G, et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc Natl Acad Sci USA. (2000) 97:4654–9. doi: 10.1073/pnas.080074897
63. Accetta D, Syverson G, Bonacci B, Reddy S, Bengtson C, Surfus J, et al. Human phagocyte defect caused by a Rac2 mutation detected by means of neonatal screening for T-cell lymphopenia. J Allergy Clin Immunol. (2011) 127:535–8 e531-532. doi: 10.1016/j.jaci.2010.10.013
64. Alkhairy OK, Rezaei N, Graham RR, Abolhassani H, Borte S, Hultenby K, et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J Allergy Clin Immunol. (2015) 135:1380–4 e1381-1385. doi: 10.1016/j.jaci.2014.10.039
65. Henderson RB, Grys K, Vehlow A, de Bettignies C, Zachacz A, Henley T, et al. A novel Rac-dependent checkpoint in B cell development controls entry into the splenic white pulp and cell survival. J Exp Med. (2010) 207:837–53. doi: 10.1084/jem.20091489
66. Gerasimcik N, He M, Dahlberg CIM, Kuznetsov NV, Severinson E, Westerberg LS. The small Rho GTPases Rac1 and Rac2 are important for T-cell independent antigen responses and for suppressing switching to IgG2b in mice. Front Immunol. (2017) 8:1264. doi: 10.3389/fimmu.2017.01264
67. Koduru S, Kumar L, Massaad MJ, Ramesh N, Le Bras S, Ozcan E, et al. Cdc42 interacting protein 4 (CIP4) is essential for integrin-dependent T-cell trafficking. Proc Natl Acad Sci USA. (2010) 107:16252–6. doi: 10.1073/pnas.1002747107
68. Vincent S, Jeanteur P, Fort P. Growth-regulated expression of rhoG, a new member of the ras homolog gene family. Mol Cell Biol. (1992) 12:3138–48. doi: 10.1128/MCB.12.7.3138
69. Gauthier-Rouviere C, Vignal E, Meriane M, Roux P, Montcourier P, Fort P. RhoG GTPase controls a pathway that independently activates Rac1 and Cdc42Hs. Mol Biol Cell. (1998) 9:1379–94. doi: 10.1091/mbc.9.6.1379
70. Burbage M, Keppler SJ, Montaner B, Mattila PK, Batista FD. The small Rho GTPase TC10 modulates B cell immune responses. J Immunol. (2017) 199:1682–95. doi: 10.4049/jimmunol.1602167
71. Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat Rev Immunol. (2010) 10:182–92. doi: 10.1038/nri2724
72. Moulding DA, Record J, Malinova D, Thrasher AJ. Actin cytoskeletal defects in immunodeficiency. Immunol Rev. (2013) 256:282–99. doi: 10.1111/imr.12114
73. Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. (2007) 8:37–48. doi: 10.1038/nrm2069
74. Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. (2000) 404:151–8. doi: 10.1038/35004513
75. Torres E, Rosen MK. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol Cell. (2003) 11:1215–27. doi: 10.1016/S1097-2765(03)00139-4
76. Rohatgi R, Ma L, Miki H, Lopez M, Kirchhausen T, Takenawa T, et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell. (1999) 97:221–31. doi: 10.1016/S0092-8674(00)80732-1
77. Machesky LM, Insall RH. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. (1998) 8:1347–56. doi: 10.1016/S0960-9822(98)00015-3
78. Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. (1998) 17:6932–41. doi: 10.1093/emboj/17.23.6932
79. Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. (2002) 418:790–3. doi: 10.1038/nature00859
80. Castiello MC, Bosticardo M, Pala F, Catucci M, Chamberlain N, van Zelm MC, et al. Wiskott-Aldrich Syndrome protein deficiency perturbs the homeostasis of B-cell compartment in humans. J Autoimmun. (2014) 50:42–50. doi: 10.1016/j.jaut.2013.10.006
81. Castiello MC, Pala F, Sereni L, Draghici E, Inverso D, Sauer AV, et al. In vivo chronic stimulation unveils autoreactive potential of wiskott-aldrich syndrome protein-deficient B cells. Front Immunol. 8:490. doi: 10.3389/fimmu.2017.00490
82. Liu DW, Zhang ZY, Zhao Q, Jiang LP, Liu W, Tu WW, et al. Wiskott-Aldrich syndrome/X-linked thrombocytopenia in China: clinical characteristic and genotype-phenotype correlation. Pediatr Blood Cancer. (2015) 62:1601–8. doi: 10.1002/pbc.25559
83. Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, et al. Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. (2003) 111:e622–7. doi: 10.1542/peds.111.5.e622
84. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr. (1994) 125:876–85. doi: 10.1016/S0022-3476(05)82002-5
85. Crestani E, Volpi S, Candotti F, Giliani S, Notarangelo LD, Chu J, et al. Broad spectrum of autoantibodies in patients with Wiskott-Aldrich syndrome and X-linked thrombocytopenia. J Allergy Clin Immunol. (2015) 136:1401–4 e1401-1403. doi: 10.1016/j.jaci.2015.08.010
86. Vermi W, Blanzuoli L, Kraus MD, Grigolato P, Donato F, Loffredo G, et al. The spleen in the Wiskott-Aldrich syndrome: histopathologic abnormalities of the white pulp correlate with the clinical phenotype of the disease. Am J Surg Pathol. (1999) 23:182–91. doi: 10.1097/00000478-199902000-00007
87. Bai X, Zhang Y, Huang L, Wang J, Li W, Niu L, et al. The early activation of memory B cells from Wiskott-Aldrich syndrome patients is suppressed by CD19 downregulation. Blood. (2016) 128:1723–34. doi: 10.1182/blood-2016-03-703579
88. Westerberg LS, de. la Fuente MA, Wermeling F, Ochs HD, Karlsson MC, Snapper SB, et al. WASP confers selective advantage for specific hematopoietic cell populations and serves a unique role in marginal zone B-cell homeostasis and function. Blood. (2008) 112:4139–47. doi: 10.1182/blood-2008-02-140715
89. Meyer-Bahlburg A, Becker-Herman S, Humblet-Baron S, Khim S, Weber M, Bouma G, et al. Wiskott-Aldrich syndrome protein deficiency in B cells results in impaired peripheral homeostasis. Blood. (2008) 112:4158–69. doi: 10.1182/blood-2008-02-140814
90. Westerberg L, Larsson M, Hardy SJ, Fernandez C, Thrasher AJ, Severinson E. Wiskott-Aldrich syndrome protein deficiency leads to reduced B-cell adhesion, migration, and homing, and a delayed humoral immune response. Blood. (2005) 105:1144–52. doi: 10.1182/blood-2004-03-1003
91. Recher M, Burns SO, de. la Fuente MA, Volpi S, Dahlberg C, Walter JE, et al. B cell-intrinsic deficiency of the Wiskott-Aldrich syndrome protein (WASp) causes severe abnormalities of the peripheral B-cell compartment in mice. Blood. (2012) 119:2819–28. doi: 10.1182/blood-2011-09-379412
92. Westerberg LS, Dahlberg C, Baptista M, Moran CJ, Detre C, Keszei M, et al. Wiskott-Aldrich syndrome protein (WASP) and N-WASP are critical for peripheral B-cell development and function. Blood. (2012) 119:3966–74. doi: 10.1182/blood-2010-09-308197
93. Liu C, Bai X, Wu J, Sharma S, Upadhyaya A, Dahlberg CI, et al. N-wasp is essential for the negative regulation of B cell receptor signaling. PLoS Biol. (2013) 11:e1001704. doi: 10.1371/journal.pbio.1001704
94. Westerberg L, Wallin RP, Greicius G, Ljunggren HG, Severinson E. Efficient antigen presentation of soluble, but not particulate, antigen in the absence of Wiskott-Aldrich syndrome protein. Immunology. (2003) 109:384–91. doi: 10.1046/j.1365-2567.2003.01668.x
95. Dahlberg CI, Torres ML, Petersen SH, Baptista MA, Keszei M, Volpi S, et al. Deletion of WASp and N-WASp in B cells cripples the germinal center response and results in production of IgM autoantibodies. J Autoimmun. (2015) 62:81–92. doi: 10.1016/j.jaut.2015.06.003
96. Becker-Herman S, Meyer-Bahlburg A, Schwartz MA, Jackson SW, Hudkins KL, Liu C, et al. WASp-deficient B cells play a critical, cell-intrinsic role in triggering autoimmunity. J Exp Med. (2011) 208:2033–42. doi: 10.1084/jem.20110200
97. Volpi S, Santori E, Abernethy K, Mizui M, Dahlberg CI, Recher M, et al. N-WASP is required for B-cell-mediated autoimmunity in Wiskott-Aldrich syndrome. Blood. (2016) 127:216–20. doi: 10.1182/blood-2015-05-643817
98. Zhang X, Dai R, Li W, Zhao H, Zhang Y, Zhou L, et al. Abnormalities of follicular helper T-cell number and function in Wiskott-Aldrich syndrome. Blood. (2016) 127:3180–91. doi: 10.1182/blood-2015-06-652636
99. Bouma G, Carter NA, Recher M, Malinova D, Adriani M, Notarangelo LD, et al. Exacerbated experimental arthritis in Wiskott-Aldrich syndrome protein deficiency: modulatory role of regulatory B cells. Eur J Immunol. (2014) 44:2692–702. doi: 10.1002/eji.201344245
100. Worth AJ, Thrasher AJ. Current and emerging treatment options for Wiskott-Aldrich syndrome. Expert Rev Clin Immunol. (2015) 11:1015–32. doi: 10.1586/1744666X.2015.1062366
101. Ramesh N, Anton IM, Hartwig JH, Geha RS. WIP, a protein associated with wiskott-aldrich syndrome protein, induces actin polymerization and redistribution in lymphoid cells. Proc Natl Acad Sci USA. (1997) 94:14671–6. doi: 10.1073/pnas.94.26.14671
102. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. (2013) 1285:26–43. doi: 10.1111/nyas.12049
103. Martinez-Quiles N, Rohatgi R, Anton IM, Medina M, Saville SP, Miki H, et al. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nat Cell Biol. (2001) 3:484–91. doi: 10.1038/35074551
104. Al-Mousa H, Hawwari A, Al-Ghonaium A, Al-Saud B, Al-Dhekri H, Al-Muhsen S, et al. Hematopoietic stem cell transplantation corrects WIP deficiency. J Allergy Clin Immunol. (2017) 139:1039–40 e1034. doi: 10.1016/j.jaci.2016.08.036
105. Lanzi G, Moratto D, Vairo D, Masneri S, Delmonte O, Paganini T, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. (2012) 209:29–34. doi: 10.1084/jem.20110896
106. Pfajfer L, Seidel MG, Houmadi R, Rey-Barroso J, Hirschmugl T, Salzer E, et al. WIP deficiency severely affects human lymphocyte architecture during migration and synapse assembly. Blood. (2017) 130:1949–53. doi: 10.1182/blood-2017-04-777383
107. Schwinger W, Urban C, Ulreich R, Sperl D, Karastaneva A, Strenger V, et al. The Phenotype and treatment of WIP deficiency: literature synopsis and review of a patient with pre-transplant serial donor lymphocyte infusions to eliminate CMV. Front Immunol. (2018) 9:2554. doi: 10.3389/fimmu.2018.02554
108. Keppler SJ, Gasparrini F, Burbage M, Aggarwal S, Frederico B, Geha RS, et al. Wiskott-Aldrich syndrome interacting protein deficiency uncovers the role of the Co-receptor CD19 as a generic hub for PI3 kinase signaling in B cells. Immunity. (2015) 43:660–73. doi: 10.1016/j.immuni.2015.09.004
109. Keppler SJ, Burbage M, Gasparrini F, Hartjes L, Aggarwal S, Massaad MJ, et al. The lack of WIP binding to actin results in impaired B cell migration and altered humoral immune responses. Cell Rep. (2018) 24:619–29. doi: 10.1016/j.celrep.2018.06.051
110. Janssen E, Tohme M, Hedayat M, Leick M, Kumari S, Ramesh N, et al. A DOCK8-WIP-WASp complex links T cell receptors to the actin cytoskeleton. J Clin Invest. (2016) 126:3837–51. doi: 10.1172/JCI85774
Keywords: germinal center, B cell receptor, immune synapse, actin cytoskeleton, antibodies
Citation: He M and Westerberg LS (2019) Congenital Defects in Actin Dynamics of Germinal Center B Cells. Front. Immunol. 10:296. doi: 10.3389/fimmu.2019.00296
Received: 14 November 2018; Accepted: 05 February 2019;
Published: 06 March 2019.
Edited by:
Tae Jin Kim, Sungkyunkwan University, South KoreaReviewed by:
Masaki Hikida, Akita University, JapanStuart G. Tangye, Garvan Institute of Medical Research, Australia
Copyright © 2019 He and Westerberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minghui He, TWluZ2h1aS5IZUBraS5zZQ==
Lisa S. Westerberg, TGlzYS5XZXN0ZXJiZXJnQGtpLnNl