 Xin Chen
Xin Chen Xiaomin Song
Xiaomin Song Kang Li*
Kang Li* Tong Zhang
Tong Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 February 2019
Sec. Molecular Innate Immunity
Volume 10 - 2019 | https://doi.org/10.3389/fimmu.2019.00292
This article is part of the Research Topic Roles of Fc Receptors in Disease and Therapy View all 25 articles
T cells play critical roles in anti-tumor immunity. Up-regulation of immune checkpoint molecules (PD-1, PD-L1, CTLA-4, TIM-3, Lag-3, TIGIT, CD73, VISTA, B7-H3) in the tumor microenvironment is an important mechanism that restrains effector T cells from the anti-tumor activity. To date, immune checkpoint antibodies have demonstrated significant clinical benefits for cancer patients treated with mono- or combination immunotherapies. However, many tumors do not respond to the treatment well, and merely blocking the immune suppression pathways by checkpoint-regulatory antibodies may not render optimal tumor growth inhibition. Binding of the antibody Fc-hinge region to Fc gamma receptors (FcγRs) has been shown to exert a profound impact on antibody function and in vivo efficacy. Investigation of immune checkpoint antibodies regarding their effector functions and impact on therapeutic efficacy has gained more attention in recent years. In this review, we discuss Fc variants of antibodies against immune checkpoint targets and the potential mechanisms of how FcγR-binding could influence the anti-tumor activity of these antibodies.
Immune checkpoints refer to multiple inhibitory pathways that control the immune system to maintain self-tolerance and modulate the intensity of physiological immune responses in order to minimize pathological damage (1–3). Antagonizing antibodies against immune checkpoint inhibitory molecules has achieved great success in cancer treatment (1, 2). However, many tumors do not respond to the treatment, and antibody optimization (especially in the isotype selection) is essential for improving outcomes (4, 5). target-binding specificity, imparted by the antibody's variable region, is well-known to be critical for the primary functional activities of the antibody. However, mounting evidence has shown that the antibody's constant region also plays a crucial role, much of which is mediated through interaction of the crystallizable fragment (Fc) with Fcγ receptors (FcγRs) (6). Fc endows IgG antibodies with effector functions, which include antibody dependent-cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), antibody-dependent cellular phagocytosis (ADCP), Induction of cytokines/chemokines and endocytosis of opsonized targets (7).
To date, therapeutic IgG antibodies (either approved or in clinical development) belong to the IgG1, IgG2 or IgG4 subclasses. Each IgG isotype has a distinct binding affinity to the various FcγRs, which are expressed differently on immune cells. A combination of these features leads to diverse and highly regulated antibody responses.
Antagonizing antibodies against major T-cell inhibitory pathways, such as PD-1/PD-L1 and CTLA-4, have become important parts of cancer therapeutics (1). Consequently, the next wave of therapeutic antibodies targeting alternative immunosuppression pathways (e.g., LAG-3, TIM-3, B7-H3, VISTA, CD73) are rapidly emerging (8). The majority of the immune checkpoint antibodies have low or significantly reduced binding to FcγRs to avoid potential ADCC and CDC, especially when the target molecule is expressed on effector T cells (9). However, for targets such as CTLA-4, TIGIT, and VISTA, competent Fc is required for optimal anti-tumor immune responses in various mouse models (10–12). The mechanisms of action (MOA) may involve the killing of regulatory T cells (Tregs), promoting immune synapse formation and production of pro-inflammatory cytokines due to cross-linking of FcγRs with the competent Fc.
In this article, we summarize the major properties of different IgG isotypes and FcγRs, describe the MOA of different immune checkpoint targets in inhibiting anti-tumor immunity and review the recent studies on the important roles of either binding or not binding to FcγRs in immune checkpoint antibody therapy. It should be noted that many of the findings come from mouse models; the clinical significance of these findings has yet to be determined.
In humans, there are four isotypes of IgG (IgG1-4), differing from the other in their binding profiles to various FcγRs and to complement subunits, such as C1q. IgG1 has the highest affinity to all FcγRs and C1q, leading to significant effector functions, such as ADCC, ADCP, and CDC (5, 13). Although human IgG3 can also mediate competent effector functions, it has a very long hinge region and complex disulfide bonds, resulting in significantly greater polymorphism, which may increase the risk of immunogenicity. Therefore, the IgG3 isotype is rarely chosen in antibody therapeutics (14) and is not further discussed in this review. In comparison, IgG2 and IgG4 induce significantly weaker or no ADCC and CDC (13). The binding features of different IgG isotypes to various FcγRs are summarized in Table 1 and discussed below.
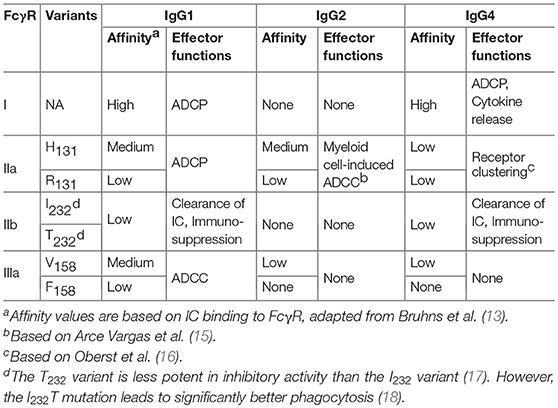
Table 1. Binding activities of human FcγR to IgG isotypes and resulting effector functions.
The overall structures of IgG1, IgG2, and IgG4 are very similar with more than 90% sequence homology. The major differences reside in the hinge region and CH2 domain, which form primary binding sites to FcγRs (19–21). The hinge region also functions as a flexible linker between the Fab and Fc portion.
In addition to differential binding affinity to FcγRs, IgG4, and IgG2 demonstrate other unique features. IgG4 has a unique S228 in the hinge region, which allows for interchangeable disulfide bond configurations and formation of “half-antibodies” (22). In vivo, IgG4 with different specificity may shuffle, resulting in monovalent-bispecific antibodies (a process called “Fab-arm exchange”) (23). S228P mutation of IgG4 can efficiently eliminate fab-arm change. Therefore, the majority of recently approved therapeutic IgG4 antibodies adopt an S228P mutation (24). In IgG2, several disulfide bond isomers (IgG2A, IgG2B, and IgG2A/B) can be formed (25, 26). Many factors such as cell culture conditions or thermal stress contribute to the formation and equilibrium of different isomers (27). In vivo, IgG2A isomer can convert to the form of IgG2B (28). Among the three isomers, IgG2B has the most compact structure (26). In addition, as compared to the form of IgG2A, the IgG2B conformation imparts super-agonistic properties to immunostimulatory antibodies, such as anti-CD40 antibodies (29). The feature of IgG2 isomer transformation is FcγR-independent and its activity has been demonstrated for IgG2 CD40 mAb in the clinical trial CP870-893 (29).
In mice, IgG2A functionally resembles human IgG1, whereas mouse IgG1 is considered the closest functional equivalent of human IgG4. The D265A mutation can further reduce the affinity of mouse IgG1 for the Fc receptor, leading to a “silent Fc” and antibodies harboring this mutation have been widely used in mouse models to evaluate the effects of FcγR-binding on in vivo therapeutic efficacy (30–32).
Based on the differences in structure, function, and affinity for IgG binding, FcγRs are classified into three major groups: FcγRI, FcγRII (FcγRIIa and FcγRIIb) and FcγRIII (FcγRIIIa and FcγRIIIb) (13). Among them, FcγRI, FcγRIIa, and FcγRIIIa are activating receptors containing the signal transduction motif, immunoreceptor tyrosine-based activation motif (ITAM), in the γ subunit of FcγRI and FcγRIIIa, or in the cytoplasmic tail of FcγRIIa (14). In contrast, FcγRIIb is an inhibitory receptor. Cross-linking of FcγRIIb leads to the phosphorylation of the immunoreceptor tyrosine-based inhibitory motif (ITIM) and inhibitory signaling transduction (33).
FcγRI is a high-affinity Fc receptor for both the monomeric IgG and immune complex (IC) (13). The affinities of FcγRI to IgG1 or IgG4 are similar (KD of 1–10 nM). In contrast, FcγRI has no binding to IgG2. FcγRI is mainly expressed on monocytes/macrophages, dendritic cells (DCs), and activated neutrophils. One of the major functions of FcγRI is to activate myeloid cells to phagocytose IgG1 and IgG-bound target cells via ADCP (34). Due to high-affinity binding of FcγRI to monomeric IgG and high serum concentrations of IgG (~15 mg/mL), it is believed that most FcγRI is occupied by endogenous IgG (35). However, a recent study has shown that stimulation of myeloid cells with cytokines, such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ), could induce the clustering of FcγRI and increase the binding of FcγRI to ICs (36). Multiple studies have also shown that FcγRI plays an important role in modulating immune responses in autoimmune diseases, inflammation, and antibody therapy (37–39).
Both FcγRIIa and FcγRIIIa are low-affinity FcγRs, which bind weakly to monomeric IgG, but strongly to IC. FcγRIIa and FcγRIIIa receptors are primarily expressed on monocytes/macrophages, dendritic cells, natural killer cells and platelets. FcγR polymorphisms exist in FcγRIIa and FcγRIIIa receptors, resulting in two isoforms of each receptor: H131 and R131 of FcγRIIa(40), V158 and F158 of FcγRIIIa (41), respectively. FcγRIIa-H131 variant is considered a high responder as compared to R131 variant (low responder) due to a higher affinity for IgG1 and increased effector functions (such as phagocytosis) (13, 22). Similar to FcγRI, FcγRIIa is one of the major phagocytic FcγRs that mediates ADCP. In human, FcγRIIIa is the primary receptor for NK- and macrophage-mediated ADCC. FcγRIIIa-V158 variant (high responder) has a higher affinity for IgG1 and can also interact with IgG4 (13). Functionally, IgG-induced NK cell activity is increased in FcγRIIIA-V/V158 homozygotes compared with FcγRIIIA-F/F158 individuals (42).
FcγRIIb is expressed on many types of immune cells including B cells, DCs, monocytes/macrophages, mast cells and basophils (33). In addition, FcγRIIb was found to be expressed on liver sinusoidal endothelial cells (LSEC) and plays an important role in IC clearance (43). On B cells, FcγRIIb functions as a primary inhibitory FcγR to suppress B cell activation and antigen internalization after binding to the immune complex (33). FcγRIIb also inhibits the type I interferon production by DCs. The binding affinities of monomeric IgG to FcγRIIb are extremely low (KA ≈ 2 x 105M−1), whereas the affinities of IC to FcγRIIb are significantly higher (13). Despite the critical roles of FcγRIIb in the negative regulation of immune responses, several studies have shown that FcγRIIb is required for the induction of efficient anti-tumor activity by agonistic anti-TNF receptor superfamily-antibody therapeutics such as anti-CD40 antibodies (44, 45). The overall binding features of human FcγR to IgG isotypes are summarized in Table 1.
In addition to the FcγRs described above, in mice, there is a unique FcγR (i.e., FcγRIV), whose expression is restricted to myeloid lineage cells (46). FcγRIV bind to mouse IgG2a and IgG2b with intermediate affinity and plays critical roles in IgG2a- and IgG2b-mediated in vivo efficacy (46, 47). Mouse FcγRIV is functionally similar to human FcγRIIIa, but not expressed on natural killer cells (47). In a mouse model, anti-CTLA-4 antibody-mediated depletion of Tregs is largely dependent on FcγRIV (10).
Several modifications to IgG can directly affect their binding to FcγRs. The N297A mutation was the first mutation to be described with significantly reduced FcγR-binding (48). It was later demonstrated that mutations of residues 234 and 235 in the lower hinge region (EU numbering system) to alanine could also lead to significantly reduced FcγR-binding; the L234A/L235A double mutation on the human IgG1 backbone is also known as the “LALA” mutation (49). In addition, hybrid antibody isotype IgG2m4, which is based on the IgG2 with four key amino acid residue changes derived from IgG4 (H268Q, V309L, A330S, and P331S), has been shown to have significantly reduced FcγR binding (50).
CTLA-4 (cytotoxic T-lymphocyte-4, or CD152) is a member of the Ig superfamily, which plays a critical role in inhibiting T-cell immunity (51). The ligands are the B7 family members, CD80 (B7-1) and CD86 (B7-2). As a CTLA-4-related protein, CD28 is constitutively expressed on naïve T cells and enhances T-cell activation when engaged by B7-1/2 on antigen-presenting cells (APC) (52, 53). In contrast, CTLA-4 surface expression increases in a day or two after T cell activation (51, 52). CTLA-4 is also highly expressed on Tregs and plays an important role in the homeostasis and suppressive functions of Tregs (54). There is no known canonical immunoreceptor tyrosine-based inhibitory (ITIM) motif in the cytoplasmic tail of CTLA-4 (55). The exact signaling pathway of CTLA-4 upon engagement with its ligands still remains largely unknown. Accumulating evidence suggested that CTLA-4 primarily exerted its inhibitory functions by competing off CD28 binding to CD80 and/or CD86, due to the higher affinity of CTLA4 to CD80 or CD86 (55). In addition, CTLA-4 has been shown to down-regulate CD80 and CD86 on APC, thus inhibiting CD28-mediated co-stimulation (54).
In mouse tumor models (melanoma and colorectal cancer), several groups have clearly shown that surrogate anti-CTLA-4 antibody-mediated anti-tumor efficacy is dependent on Fc effector functions and correlate with depletion of tumor-infiltrating Tregs (10, 30, 56) (Figure 1A). In 2011, the FDA approved the first anti-CTLA-4 antibody, ipilimumab (IgG1 wild-type), for the treatment of melanoma. Furthermore, the combination of PD-1 blockade with ipilimumab demonstrated increased, durable anti-tumor activity in renal cell carcinoma and non-small cell lung cancer (NSCLC) (57, 58). Interestingly, anti-CTLA-4 clones, which lose the ability to block the B7-CTLA-4 interaction, remain fully active in inducing tumor rejection, suggesting that other mechanisms are involved in anti-CTLA-4 antibody-mediated anti-tumor efficacy besides the blocking of B7-CTLA-4 (59). In an ex-vivo assay, melanoma patient-derived non-classical monocytes could kill Tregs via ADCC (60). In addition, patients who responded to ipilimumab tended to have a higher percentage of CD14+CD16+ monocytes in the periphery. Using human FcγR-transgenic mice, Arce Vargas et al. clearly demonstrated that antibodies with isotypes equivalent to ipilimumab increased the CD8+ to Treg ratio by depleting intra-tumoral Tregs to promote tumor rejection (15). Furthermore, a response to ipilimumab in melanoma patients is associated with a high-affinity FcγRIIIa (CD16-V158) polymorphism. A second anti-CTLA-4 mAb, tremelimumab, is a human IgG2 isotype with minimal FcγRIIIa-mediated ADCC effects (61). However, anti-mouse CTLA-4 antibody with human IgG2 isotype could also deplete Tregs in human FcγR-transgenic mice in a FcγRIIa-dependent manner (15). Despite the convincing data from mouse models, there has not been direct evidence indicating that anti-CTLA-4 immunotherapy could efficiently deplete Tregs in human cancers (62, 63).
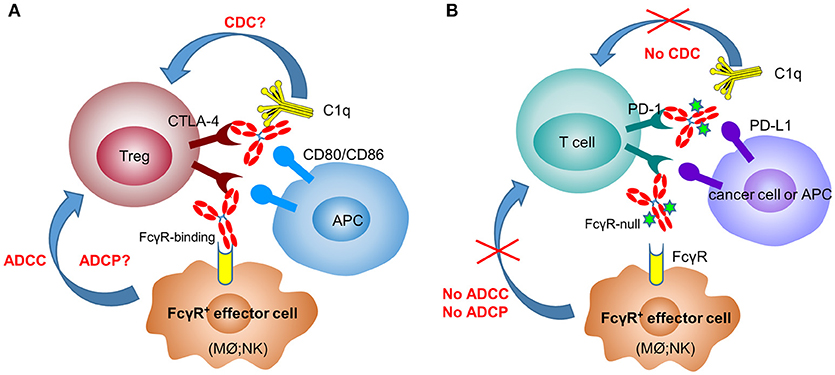
Figure 1. Anti-CTLA-4 and anti-PD-1 therapeutic antibodies have differential FcγR-binding requirement for optimal activity. In the mechanisms of action of anti-CTLA-4 antibodies (A), depletion of Tregs after engaging FcγR+ effector cells [macrophages (Mϕ) and NK cells] plays a critical role in their efficacy. In contrast, anti-PD-1 antibodies need to have the Fc-mediated effector functions (ADCC, ADCP, and CDC) removed to avoid the killing of PD-1+ T cells by FcγR+ effector cells (B).
In recent years, immune therapy targeting the PD-1/PD-L1 pathway has become a backbone clinical strategy for cancer treatment. Programmed cell death 1 (PD-1) is an inhibitory immune modulatory receptor (64–66). It is inducibly expressed on activated T, NK, and B lymphocytes (67), macrophages, DCs (68), and monocytes (69) as an immune suppressor for both adaptive and innate immune responses. PD-1 is highly expressed on tumor-specific T cells. Engagement of PD-1 by its ligands, PD-L1 (70) or PD-L2 (71, 72) leads to the exhaustion of T cell function and immune tolerance in the tumor microenvironment. Blockade of PD-1 pathway has been shown to restore the function of “exhausted” T cells, resulting in significant anti-tumor activity (70, 73). To date, five PD-1 antibodies have been approved and many others are under development for the treatment of a broad spectrum of cancers (Table 2). Most of these anti-PD-1 antibodies are of IgG4 isotype with the S228P mutation (IgG4 S228P), which has similar effector-binding properties as the natural IgG4 with reduced ADCC and “null” CDC, but still retaining high affinity to FcγRI and binding to FcγRIIb. In the MC38 mouse model, Dahan et al. reported that engagement of FcγRs reduced the anti-tumor activity of an anti-PD-1 antibody by eliminating CD8+ tumor-infiltrating lymphocytes (TILs) via ADCC in a FcγRI-dependent manner (9). In addition, engagement of FcγRIIb by an anti-PD-1 antibody could also decrease its anti-tumor activities. Arlauckas et al. demonstrated that anti-PD-1 antibodies can be captured from the T-cell surface by FcγR-bearing macrophages. The blockade of FcγRs could thus prolong the binding of the anti-PD-1 antibody to CD8+ TILs and enhance the anti-tumor activity in vivo (74). A preclinical study by our group also suggested that FcγRI binding had a negative impact on the anti-tumor activity of anti-PD-1 antibodies in a humanized xenograft model. The binding could induce FcγRI+ macrophages to phagocytose PD-1+ T cells via ADCP and reverse the function of an anti-PD-1 antibody from blocking to activating (37). Recently, several published research papers documented the phenomenon that the hyperprogression frequencies of certain cancer types treated with FDA-approved anti-PD-1 antibodies were substantially higher than the control chemotherapy group (75–77). Lo Russo et al. linked the interaction between the anti-PD-1 antibody and FcγR+ macrophages to the hyperprogression in NSCLC during PD-1 blockade therapy (78). Based on these observations, an anti-PD-1 antibody with pure blocking activity would be more desirable, since an anti-PD-1 antibody with FcγR-binding activity can mediate cross-linking between PD-1+ T-cells and FcγR+ macrophages, induce the depletion of PD-1+ T effector cells, and thus compromise the T-cell activity of tumor growth inhibition (9, 37, 74) (Figure 1B).
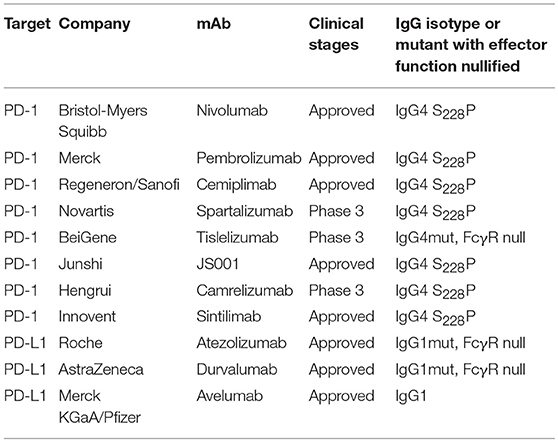
Table 2. Select PD-1 and PD-L1 antibodies under development for cancer treatment.
Programmed death ligand 1 (PD-L1) is constitutively expressed by immune cells of myeloid lineages (79) and the cells at immune-privileged sites (80, 81). It is also inducibly expressed on T, NK and B lymphocytes, epithelial and endothelial cells upon stimulation by pro-inflammatory factors, such as IFN-γ and TNF-α (82). PD-L1 is the main ligand of PD-1, and the PD-L1/PD-1 axis is the major controller of the peripheral immune tolerance (65). In tumors, PD-L1 is expressed on both tumor cells (83) and tumor-infiltrating immune cells and can suppress anti-tumor immunity independently (84). Unlike anti-PD-1 antibodies, the three approved PD-L1 antibodies have differentiated FcγR-binding properties (Table 2). Atezolizumab and durvalumab are designed to eliminate FcγR-binding and effector functions (85, 86), while avelumab retains intact Fc functions (87). Recent preclinical data suggested that the engagement of FcγRs could augment the anti-tumor activity of anti-PD-L1 antibodies via the ADCC effect against the PD-L1+ immune suppressive myeloid cells (88) or tumor cells (89). However, it is also speculated that the effector function could be detrimental to the anti-tumor immunity due to the depletion of PD-L1+ APC cells and T effector cells. To understand the role of FcγR-binding on anti-PD-L1 anti-tumor efficacy, future studies are needed to elucidate the expression of PD-L1 in the tumor microenvironment and the effect of anti-PD-L1 antibody treatment.
TIM-3 (T cell immunoglobulin and mucin-domain containing-3, also known as HAVCR2) is a member of the T-cell immunoglobulin- and mucin-domain-containing family that plays an important role in promoting T-cell exhaustion in both chronic viral infections and tumor escape from immune surveillance (90, 91). It is primarily expressed on immune cells, such as T cells, NK cells, DCs, and monocytes/macrophages (92). When expressed on effector T cells, activation of TIM-3 has been shown to reduce cytokine production, T-cell proliferation, and cytotoxicity, all of which could be rescued by TIM-3 blocking antibodies (93, 94). TIM-3 is also expressed on FoxP3+ Treg cells, especially in human tumor tissues, and is correlated with poor clinical parameters (95, 96).
Four TIM-3 ligands have been identified, which include PtdSer, Gal-9, carcinoembryonic antigen-related cell adhesion molecule 1, and high mobility group box 1 (97). To date, the detailed mechanisms of TIM-3 signaling remain unclear. Upregulation of TIM-3 expression in TILs, macrophages, and tumor cells has been reported in many types of cancers (98–101). Increased expression of TIM-3 in those cancers is associated with a poor prognosis and/or patient survival.
Following PD-1 antibody blockade, TIM-3 expression has been shown to be upregulated on TILs from both patient samples and animal models, resulting in “adaptive resistance” to anti-PD-1 treatment (102–104). Blockade of the TIM-3 receptor alone or in combination with PD-1/PD-L1 blockade has been shown both in vitro and in vivo to rescue functionally “exhausted” T cells (3, 93, 105).
In pre-clinical mouse models of colorectal cancer (MC38 and CT26), the effects of “silent” Fc vs. “competent” Fc on TIM-3 antibody-mediated anti-tumor activity with or without anti-PD-1 antibody treatment were evaluated by several groups (106, 107). The results showed that the combination of “Fc-silent” TIM-3 Ab with PD-1 Ab led to significantly more synergistic tumor-inhibitory effects than the one with “competent” Fc, while TIM-3 blocking Ab monotherapy demonstrated marginal anti-tumor efficacy. The exact mechanisms of Fc effector functions (ADCC and/or ADCP) in the negative regulation of anti-TIM-3 antibody-mediated anti-tumor efficacy remain unknown.
To date, the first-in-human phase 1/2 clinical trials have been initiated for four anti-TIM-3 antibodies: TSR-022 (NCT02817633), MBG543 (NCT02608268), BMS-986258 (NCT03446040), and LY3321367 (NCT03099109). TESARO recently released the clinical data of TSR-022, in monotherapy or in combination with an anti-PD-1 antibody (TSR-042) in patients who progressed following anti-PD-1 treatment (108). The results showed that the combination of TSR-022 and TSR-042 (500 mg) was generally well-tolerated in both NSCLC and melanoma patients, and clinical activities have been observed in the combination therapy, especially at a high dose of TSR-022 (300 mg) with an objective response rate (ORR) of 15% (3/20) and 40% stable disease (8/20) (108).
LAG-3 (Lymphocyte activation gene-3, or CD223) is a member of the immunoglobulin superfamily (IgSF) (109). The immune-regulatory roles of LAG-3 were demonstrated in LAG-3 knockout mice, in which increased susceptibility to autoimmune diseases was observed (110, 111). LAG-3 is primarily expressed on activated T, natural killer (NK), and plasmacytoid dendritic cells (pDC), but not on resting T cells (109, 112). In addition, LAG-3 expression on Tregs is positively correlated with their immune-suppressive activity (113). Sequence homology analysis revealed that LAG-3 is structurally related to CD4, but with higher affinity (60 nM) to MHC class II (MHC-II) molecules, thus inhibiting CD4-MHC-II interaction and negatively regulating T-cell receptor (TCR) signaling (109, 114). In addition, LAG-3 can exert negative regulation of CD8+ T cells via CD4+ T cell-dependent and/or independent manners (115, 116). Similar to PD-1, LAG-3 is expressed on tumor-infiltrating lymphocytes (TILs), but to a less extent. Besides MHC-II molecules, LAG-3 has been shown to bind to galectin-3 (Gal-3) and LSECtin (115, 117). The exact biological function of these two ligands binding to LAG-3 remains unknown. Recently, fibrinogen-like protein 1 (FGL1) has been identified as a novel high-affinity ligand for LAG-3 (118). In vitro, FGL1 could induce T-cell inhibition in a LAG-3-dependent manner. In the MC38 colorectal cancer model, ablation of FGL1-LAG-3 interaction with either anti-FGL1 or anti-LAG-3 blocking antibodies inhibits tumor growth.
In mouse tumor models (Sa1N fibrosarcoma, MC38 colorectal cancer, and MBT-2 bladder cancer), dual blockade of LAG-3 and PD-1 receptors with blocking antibodies has shown to significantly improve the anti-tumor activity than either antibody alone (111, 119). In a study by Jun et al., a pair of anti-mouse LAG-3 surrogate antibodies with IgG1 (D265A) [anti-mLAG-3 IgG1(D265A)] or IgG2a (anti-mLAG-3 IgG2a) isotypes were generated based on a commercial clone (C9B7W). Comparative study of these two antibodies either alone or in combination with anti-mouse PD-1 antibody in the CT26 mouse colorectal cancer model showed that anti-mouse LAG-3 antibody with minimal Fc effector functions [IgG1 (D265A)] had anti-tumor efficacy, and the one with effector function (IgG2a) had no apparent tumor inhibitory effect (120). In addition, when combined with PD-1 blocking antibody, anti-mLAG-3 IgG1 (D265A) showed significantly synergistic anti-tumor effects, whereas anti-mLAG-3 IgG2a with intact effector function in combination with an anti-mouse PD-1 antibody was less efficacious than anti-mouse PD-1 alone, suggesting that the effector function of LAG-3 antibody might interfere with anti-mouse PD-1 mediated efficacy. The anti-tumor efficacy of anti-mouse LAG-3 antibodies without effector functions was also observed by other groups (119, 121, 122).
As of now, there are six LAG-3 antibodies being evaluated in clinical trials. All these LAG-3 antibodies have Fc with either reduced or “null” effector functions. Preliminary data showed that combining anti-LAG-3 therapy (BMS-986016) with nivolumab in melanoma patients refractory to PD-1/PD-L1 treatment could help patients overcome resistance and restore T-cell function with an ORR up to 18%, especially in patients with high LAG-3 expression (≥1%)(123).
TIGIT (T cell immunoglobulin and ITIM domain, also known as WUCAM or Vstm3) is a member of the CD28 family of proteins that play an important role in inhibiting T- and NK cell-mediated functional activities in anti-tumor immunity (124–126). TIGIT is mainly expressed on T and NK cells. T cells in the tumor microenvironment (3) often co-express TIGIT with other “checkpoint” inhibitory immune receptors, such as PD-1, LAG-3, and TIM-3 (93, 127).
Two TIGIT ligands, CD155 (PVR) and CD112 (PVRL2, nectin-2), have been identified; they are primarily expressed on APCs (such as dendritic cells and macrophages) and tumor cells (125, 126, 128, 129). The binding affinity of TIGIT to CD155 (Kd: ~1nM) is much higher than to CD112. Whether the TIGIT: CD112 interaction is functionally relevant in mediating inhibitory signals is yet to be determined. High-affinity binding of TIGIT to CD155 could compete with another co-stimulatory receptor, CD226 (DNAM-1), which binds to the same ligands with lower affinity (Kd: ~100nM) and delivers a positive signal (130), therefore reducing T- or NK-activation. In addition, the interaction between TIGIT and PVR on dendritic cells (DCs) could deliver a “reverse signaling” in DCs, leading to reduced DC activity and T-cell activation (126). TIGIT expression on Tregs has been associated with a highly immune-suppressive phenotype in tumor tissue and TIGIT signaling in Tregs may favor Treg stability (131, 132).
Blockade of the TIGIT receptor alone or in combination with PD-1/PD-L1 blockade could rescue functionally “exhausted” T cells both in vitro and in vivo (133, 134). In the CT26 cancer model, Fc with effector functions is critical for TIGIT antibody-mediated anti-tumor activity (11, 135). The TIGIT antibody with wild-type (WT) human IgG1 Fc (EOS884448) has been shown to be capable of preferentially depleting Treg cells in vitro (11). The authors demonstrated that the surrogate mouse TIGIT antibody of the mIgG2a isotype has potent anti-tumor activity either as monotherapy or in combination with a PD-1 antibody. In contrast, the mouse anti-TIGIT antibody with Fc devoid of effector functions did not show any of the anti-tumor efficacies, indicating that Fc-mediated effector functions are required for TIGIT antibody-mediated anti-tumor effects. In addition, the observed efficacy was associated with increased activity of effector T cells (CD8+ and CD4+) and also with Treg depletion within the TME. Argast et al. also observed that effector functions were critical for TIGIT antibody-induced in vivo efficacy (135).
To date, there are six TIGIT antibodies (see Table 3) in clinical trials, with different IgG isotypes or mutant forms. The most advanced, MTIG7192 (NCT03563716), is in a phase 2 trial in combination with the anti-PD-L1 antibody atezolizumab for treatment of advanced NSCLC. How the effector functions affect clinical activities remains to be seen.
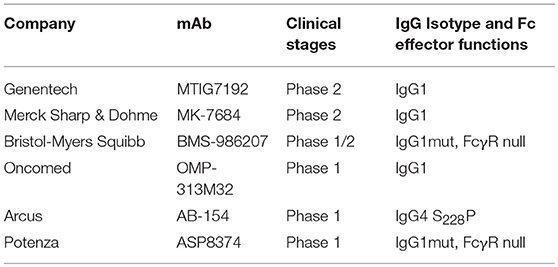
Table 3. Anti-TIGIT in clinical trials.
CD73 (also known as 5'-ecto-nucleotidase, or NT5E) is a glycosylphosphatidylinositol (136) anchored cell surface protein, which has both enzymatic and non-enzymatic functions (137). As a nucleotidase, it catalyzes the extracellular dephosphorylation of adenosine monophosphate (AMP) to adenosine. Adenosine is believed to be an immunosuppressive molecule inhibiting CD8+ T cells, NK cells, and dendritic cells, while promoting the proliferation of immunosuppressive cells (138, 139). In some cases, CD73 can be shed from the cell surface with retained enzymatic activity (140). Expression of CD73 varies on normal tissues but remains at constitutively high levels on many types of cancer cells. High CD73 expression has been shown to be correlated with unfavorable clinical outcomes (141–147), which is consistent with the immunosuppressive role of adenosine.
Three CD73 blocking antibodies have been entered into clinical trials (i.e., BMS-986179, CPI-006, and MEDI9447). Compared with small-molecule inhibitors, anti-CD73 mAbs offer the possibility of directly targeting both enzymatic and non-enzymatic CD73 pathways (148). In vitro data showed that MEDI9447 (human IgG1 variant) could inhibit the enzymatic activity of both soluble- and membrane-bound CD73 through prevention of the conformational transition of CD73 to an active state, and could induce internalization of membrane-bound CD73, and restore T-cell proliferation from the inhibition by AMP (149, 150). In a mouse model, MEDI9447 monotherapy showed significant anti-tumor efficacy, which was further increased when combined with a PD-1 antibody (150). In the Fc region of MEDI9447, triple mutations (L234F/L235E/P331S) were introduced to eliminate its binding to FcγRs (Including FcγRI, FcγRIIa, and FcγRIIIa) and C1q (150, 151). Similarly, CPI-006 from Corvus is also an IgG1 isotype with a “silent” Fc. It could fully block the production of adenosine by inhibiting the enzymatic activity of CD73 (IC50, 17nM) without internalization, while also activate B cells independent of adenosine reduction (152).
Another anti-CD73 antibody, BMS-986179, is an IgG2/IgG1 hybrid with a “null” effector function. BMS-986179 could not only inhibit CD73 enzymatic function but also induce rapid, near-complete internalization (153). The disulfide bond isomerization of IgG2 is thought to be the major mechanism for BMS-986179-induced CD73 efficient clustering and internalization. Results from mouse models indicated that the combination of PD-1 blockade and a surrogate anti-mouse-CD73 antibody treatment resulted in more enhanced anti-tumor efficacy than either treatment alone (153). In a phase 1/2a study (NCT02754141), 59 patients with advanced solid tumors were treated either alone with BMS-986179 or in combination with nivolumab. Preliminary results showed that both the monotherapy of BMS-986179 and the combination were well-tolerated and clinical activities were observed with 7 partial responses and 10 stable diseases (154).
VISTA (V-domain Ig-containing Suppressor of T cell Activation, also known as B7-H5, B7H5, C10orf54, DD1alpha, GI24, PD-1H, PP2135, SISP1) is a type I transmembrane protein with a single extracellular IgV domain, functioning as a negative regulator of T-cell immunity. It is predominantly expressed on hematopoietic cells, at the highest level on myeloid cells and at lower levels on T cells (155). In vitro studies indicated that not only could VISTA-Ig inhibit T-cell activation and proliferation, but it could also induce Treg differentiation (155). The receptor for VISTA remains unknown. Results from murine models suggested that VISTA and PD-1 suppressed T-cell function in a synergistic manner, providing the possibility of combined therapy targeting both VISTA and PD-1 to enhance anti-tumor immunity (156).
To date, JNJ-61610588, a fully human IgG1 antibody (with wild-type Fc) is the only anti-VISTA monoclonal antibody in a clinical trial (NCT02671955). A preliminary study showed that JNJ-61610588 could induce monocytes and T-cell activation, as well as T-cell proliferation in vitro (12). Interestingly, active Fc and Fc receptor crosslinking is required for the efficacy, since neither the silent Fc version of VSTB140, with an IgG2 sigma constant region, nor the Fc blocking of JNJ-61610588 exhibited activity. Consistent with in vitro findings, the anti-tumor activity of JNJ-61610588 in mouse tumor models was observed. The exact mechanisms and clinical evidence remain to be seen.
B7-H3 (Human B7 homolog 3, also known as CD276) is a member of the B7 family of immune proteins. The majority of studies suggest that B7-H3 is an immune checkpoint molecule (157–159), although it was initially characterized as a co-stimulatory molecule for T-cell activation and IFNγ production (160). The B7-H3 receptor expressed on T cells remains to be identified (161). B7-H3 has limited expression on normal tissues but is preferentially expressed on a wide spectrum of cancer cells and tumor vasculature, which is associated with poor outcomes in multiple cancers (162–168).
MGA271 (or enoblituzumab), is an Fc-enhanced humanized IgG1 anti-B7-H3 antibody developed by MacroGenics. Mutations were introduced in the IgG1 Fc domain to increase its affinity to FcγRIIIa but decrease the affinity to FcγRIIb (169). Enhanced ADCC against a wide arrange of B7-H3 positive tumor cell lines (including prostate, lung, breast, colon, bladder, renal cancers and melanoma) was observed across all the donors with different FcγRIIIa polymorphisms (low-affinity 158F homozygous, high-affinity 158V homozygous, and 158F/V heterozygous). Consistent with in vitro data, greater anti-tumor efficacy was observed in the group with MGA271 than the one with wildtype IgG1 Fc in human FcγRIIIa-158F-transgenic mice (170). Initial evidence of anti-tumor activity was observed in a clinical trial with MGA271, with no dose-limiting toxicities or severe immune-related side effects (171).
In this review, we have summarized recent advances in the study of FcγR-binding on checkpoint antibody therapy. For targets such as CTLA-4, multiple studies indicated the critical role of competent IgG1-Fc for anti-CTLA-4 antibody-mediated intratumoral depletion of Tregs via ADCC (10, 15). This MOA may largely be attributed to the preferential surface expression of CTLA-4 on Tregs and the presence of significant numbers of CD16+ macrophages inside tumors (15). In mouse models, anti-CTLA-4 mAbs do not block CTLA-4-B7 interaction, yet they remain active in anti-tumor efficacy, suggesting that intratumoral depletion of Tregs by anti-CTLA-4 antibodies might be the primary MOA (172). A similar phenomenon was observed for TIGIT or VISTA in mouse models, in which their antibody-elicited anti-tumor efficacy is mainly dependent on Fc-mediated effector functions (11).
So far, five approved anti-PD-1 mAbs (nivolumab, pembrolizumab, and cemiplimab) are of human IgG4 isotype. The choice was made primarily based on the fact that the affinity of IgG4 to FcγRIIIa is very low, inducing little ADCC (13). However, IgG4 binds to FcγRI with high affinity, which can negatively impact the efficacy of PD-1 therapy (9, 37). Moreover, IgG4 can also bind to FcγRIIb, leading to reduced anti-tumor efficacy, likely through the induction of a more immunosuppressive environment (9, 78). Therefore, an IgG variant of the anti-PD-1 antibody with null FcγR-binding is expected to be the optimal candidate for therapeutic blocking of PD-1 without the unwanted engagement of FcγR pathways. A similar rationale applies to co-inhibitory receptors TIM-3 and LAG-3, in which blocking antibody-mediated anti-tumor efficacy might be compromised when the Fc maintains intact effector functions.
Three PD-L1-targeting mAbs have been approved: atezolizumab, durvalumab (IgG1 variant with null or reduced Fc-FcγR binding), and avelumab (wild-type IgG1, ADCC-enabling) (173). Comparison of clinical activities of these mAbs may provide important insight into the contribution of FcγRs for the anti-PD-L1 treatment of human cancers.
It should be noted that most of the findings in this review about the role of IgG antibody and FcγR binding on immune-oncology therapy were obtained from mouse models (some even in human FcγR-transgenic mice). There are several factors that need to be taken into consideration, including, how well the mouse FcγR expression pattern (including transgenic human FcγRs) mimics the human counterpart, especially in cancer patients, and how different the abundance and distribution of FcγR+ effector cells (e.g., NK cells and macrophages) are in mice vs. in humans in the TME. Studies on the impact of human FcγR polymorphisms (FcγRIIIa-V158 vs. F158; FcγRIIa-H131 vs. R131) on clinical activity may also shed light on the MOA of immune checkpoint-targeted antibodies (15). In addition, ex vivo assays using human tumor samples and targeted antibodies in various settings may provide useful insight into this matter.
In summary, the triggering of effector functions on IgG and FcγR interactions is a complex process; the overall outcome may be dependent on the target expression level, distribution, and abundance of T cells, and the FcγR+ effector cells (NK cells and macrophages) inside tumors. Further investigation through clinical pathology and pharmacology studies is needed to assess the translational applicability of these findings in mouse models to human cancer treatment.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This review is supported by BeiGene internal funds.
This work was funded by BeiGene. All authors have an ownership interest in BeiGene.
1. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
2. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell (2015) 27:450–61. doi: 10.1016/j.ccell.2015.03.001
3. Dietze KK, Zelinskyy G, Liu J, Kretzmer F, Schimmer S, Dittmer U. Combining regulatory T cell depletion and inhibitory receptor blockade improves reactivation of exhausted virus-specific CD8+ T cells and efficiently reduces chronic retroviral loads. PLoS Pathog. (2013) 9:e1003798. doi: 10.1371/journal.ppat.1003798
4. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
5. Beers SA, Glennie MJ, White AL. Influence of immunoglobulin isotype on therapeutic antibody function. Blood (2016) 127:1097–101. doi: 10.1182/blood-2015-09-625343
6. Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol. (2016) 40:62–9. doi: 10.1016/j.coi.2016.03.002
7. Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol. (2010) 10:301–16. doi: 10.1038/nri2761
8. Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J Hematol Oncol. (2018) 11:39. doi: 10.1186/s13045-018-0582-8
9. Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcgammaRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell (2015) 28:543. doi: 10.1016/j.ccell.2015.09.011
10. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. (2013) 210:1695–710. doi: 10.1084/jem.20130579
11. Leroy X, Hoofd C, Cuende J, Denies S, Rabolli V, Preillon J, et al. Abstract LB-114: a-TIGIT antagonist antibody EOS884448 shows dual mechanism of action by restoration of T cell effector functions and preferential depletion of Treg. Cancer Res. (2018) 78(13 Supplement):LB-114-LB-14. doi: 10.1158/1538-7445.Am2018-lb-114
12. Snyder LA. JNJ-61610588: A human anti-VISTA antibody induces antitumor responses via a unique mechanism of action. In: AACR Annual Meeting 2016. New Orleans, LA (2016).
13. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113:3716–25. doi: 10.1182/blood-2008-09-179754
14. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
15. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc Effector function contributes to the activity of human anti–CTLA-4 antibodies. Cancer Cell (2018) 33:649–63.e4. doi: 10.1016/j.ccell.2018.02.010
16. Oberst MD, Auge C, Morris C, Kentner S, Mulgrew K, McGlinchey K, et al. Potent Immune Modulation by MEDI6383, an Engineered Human OX40 Ligand IgG4P Fc Fusion Protein. Mol Cancer Ther. (2018) 17:1024–38. doi: 10.1158/1535-7163.MCT-17-0200
17. Kono H, Kyogoku C, Suzuki T, Tsuchiya N, Honda H, Yamamoto K, et al. FcgammaRIIB Ile232Thr transmembrane polymorphism associated with human systemic lupus erythematosus decreases affinity to lipid rafts and attenuates inhibitory effects on B cell receptor signaling. Hum Mol Genet. (2005) 14:2881–92. doi: 10.1093/hmg/ddi320
18. Clatworthy MR, Willcocks L, Urban B, Langhorne J, Williams TN, Peshu N, et al. Systemic lupus erythematosus-associated defects in the inhibitory receptor FcgammaRIIb reduce susceptibility to malaria. Proc Natl Acad Sci USA. (2007) 104:7169–74. doi: 10.1073/pnas.0608889104
19. Lu J, Chu J, Zou Z, Hamacher NB, Rixon MW, Sun PD. Structure of FcgammaRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc Natl Acad Sci USA. (2015) 112:833–8. doi: 10.1073/pnas.1418812112
20. Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. (2001) 276:6591–604. doi: 10.1074/jbc.M009483200
21. Wines BD, Powell MS, Parren PW, Barnes N, Hogarth PM. The IgG Fc contains distinct Fc receptor (FcR) binding sites: the leukocyte receptors Fc gamma RI and Fc gamma RIIa bind to a region in the Fc distinct from that recognized by neonatal FcR and protein A. J Immunol. (2000) 164:5313–8. doi: 10.4049/jimmunol.164.10.5313
22. Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology (2002) 105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x
23. Labrijn AF, Buijsse AO, van den Bremer ET, Verwilligen AY, Bleeker WK, Thorpe SJ, et al. Therapeutic IgG4 antibodies engage in Fab-arm exchange with endogenous human IgG4 in vivo. Nat Biotechnol. (2009) 27:767–71. doi: 10.1038/nbt.1553
24. Yang X, Wang F, Zhang Y, Wang L, Antonenko S, Zhang S, et al. Comprehensive analysis of the therapeutic igg4 antibody pembrolizumab: hinge modification blocks half molecule exchange in vitro and in vivo. J Pharm Sci. (2015) 104:4002–14. doi: 10.1002/jps.24620
25. Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, et al. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem. (2008) 283:16206–15. doi: 10.1074/jbc.M709988200
26. Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, et al. Human IgG2 antibodies display disulfide-mediated structural isoforms. J Biol Chem. (2008) 283:16194–205. doi: 10.1074/jbc.M709987200
27. Liu H, May K. Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs (2012) 4:17–23. doi: 10.4161/mabs.4.1.18347
28. Liu YD, Chen X, Enk JZ, Plant M, Dillon TM, Flynn GC. Human IgG2 antibody disulfide rearrangement in vivo. J Biol Chem. (2008) 283:29266–72. doi: 10.1074/jbc.M804787200
29. White AL, Chan HT, French RR, Willoughby J, Mockridge CI, Roghanian A, et al. Conformation of the human immunoglobulin G2 hinge imparts superagonistic properties to immunostimulatory anticancer antibodies. Cancer Cell (2015) 27:138–48. doi: 10.1016/j.ccell.2014.11.001
30. Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. (2013) 1:32–42. doi: 10.1158/2326-6066.CIR-13-0013
31. Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, et al. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol. (2008) 181:6664–9. doi: 10.4049/jimmunol.181.9.6664
32. Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. (2000) 6:443–6. doi: 10.1038/74704
33. Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. (2010) 10:328–43. doi: 10.1038/nri2762
34. Swisher JF, Feldman GM. The many faces of FcgammaRI: implications for therapeutic antibody function. Immunol Rev. (2015) 268:160–74. doi: 10.1111/imr.12334
35. Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. (2008) 8:34–47. doi: 10.1038/nri2206
36. Brandsma AM, Schwartz SL, Wester MJ, Valley CC, Blezer GLA, Vidarsson G, et al. Mechanisms of inside-out signaling of the high-affinity IgG receptor FcgammaRI. Sci Signal. (2018) 11:eaaq0891. doi: 10.1126/scisignal.aaq0891
37. Zhang T, Song X, Xu L, Ma J, Zhang Y, Gong W, et al. The binding of an anti-PD-1 antibody to FcgammaRIota has a profound impact on its biological functions. Cancer Immunol Immunother. (2018) 67:1079–90. doi: 10.1007/s00262-018-2160-x
38. Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, et al. FcγRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity (2002) 16:391–402. doi: 10.1016/S1074-7613(02)00294-7
39. Mancardi DA, Albanesi M, Jonsson F, Iannascoli B, Van Rooijen N, Kang X, et al. The high-affinity human IgG receptor FcgammaRI (CD64) promotes IgG-mediated inflammation, anaphylaxis, and antitumor immunotherapy. Blood (2013) 121:1563–73. doi: 10.1182/blood-2012-07-442541
40. Clark MR, Stuart SG, Kimberly RP, Ory PA, Goldstein IM. A single amino acid distinguishes the high-responder from the low-responder form of Fc receptor II on human monocytes. Eur J Immunol. (1991) 21:1911–6. doi: 10.1002/eji.1830210820
41. Koene HR, Kleijer M, Algra J, Roos D, von dem Borne AE, de Haas M. Fc gammaRIIIa-158V/F polymorphism influences the binding of IgG by natural killer cell Fc gammaRIIIa, independently of the Fc gammaRIIIa-48L/R/H phenotype. Blood (1997) 90:1109–14.
42. Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, et al. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. (1997) 100:1059–70. doi: 10.1172/JCI119616
43. Ganesan LP, Kim J, Wu Y, Mohanty S, Phillips GS, Birmingham DJ, et al. FcgammaRIIb on liver sinusoidal endothelium clears small immune complexes. J Immunol. (2012) 189:4981–8. doi: 10.4049/jimmunol.1202017
44. Li F, Ravetch JV. Antitumor activities of agonistic anti-TNFR antibodies require differential FcgammaRIIB coengagement in vivo. Proc Natl Acad Sci USA. (2013) 110:19501–6. doi: 10.1073/pnas.1319502110
45. Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proc Natl Acad Sci USA. (2012) 109:10966–71. doi: 10.1073/pnas.1208698109
46. Nimmerjahn F, Bruhns P, Horiuchi K, Ravetch JV. FcgammaRIV: a novel FcR with distinct IgG subclass specificity. Immunity (2005) 23:41–51. doi: 10.1016/j.immuni.2005.05.010
47. Nimmerjahn F, Lux A, Albert H, Woigk M, Lehmann C, Dudziak D, et al. FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci USA. (2010) 107:19396–401. doi: 10.1073/pnas.1014515107
48. Bolt S, Routledge E, Lloyd I, Chatenoud L, Pope H, Gorman SD, et al. The generation of a humanized, non-mitogenic CD3 monoclonal antibody which retains in vitro immunosuppressive properties. Eur J Immunol. (1993) 23:403–11. doi: 10.1002/eji.1830230216
49. Xu D, Alegre ML, Varga SS, Rothermel AL, Collins AM, Pulito VL, et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol. (2000) 200:16–26. doi: 10.1006/cimm.2000.1617
50. An Z, Forrest G, Moore R, Cukan M, Haytko P, Huang L, et al. IgG2m4, an engineered antibody isotype with reduced Fc function. MAbs (2009) 1:572–9. doi: 10.4161/mabs.1.6.10185
51. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol. (2006) 24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535
52. Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7-1 and B7-2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity (2004) 21:401–13. doi: 10.1016/j.immuni.2004.06.017
53. Jansson A, Barnes E, Klenerman P, Harlen M, Sorensen P, Davis SJ, et al. A theoretical framework for quantitative analysis of the molecular basis of costimulation. J Immunol. (2005) 175:1575–85. doi: 10.4049/jimmunol.175.3.1575
54. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science (2008) 322:271–5. doi: 10.1126/science.1160062
55. Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. (2015) 36:63–70. doi: 10.1016/j.it.2014.12.001
56. Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, et al. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. (2013) 210:1685–93. doi: 10.1084/jem.20130573
57. Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med. (2018) 378:1277–90. doi: 10.1056/NEJMoa1712126
58. Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. (2018) 378:2093–104. doi: 10.1056/NEJMoa1801946
59. Du X, Liu M, Su J, Zhang P, Tang F, Ye P, et al. Uncoupling therapeutic from immunotherapy-related adverse effects for safer and effective anti-CTLA-4 antibodies in CTLA4 humanized mice. Cell Res. (2018) 28:433–47. doi: 10.1038/s41422-018-0012-z
60. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA. (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
61. Hanson DC, Canniff PC, Primiano MJ, Donovan CB, Gardner JP, Natoli EJ, et al. Preclinical in vitro characterization of anti-CTLA4 therapeutic antibody CP-675,206. In: AACR Annual Meeting 2004. Orlando, FL (2004). p. 877–77.
62. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 Immunotherapy does not deplete FOXP3(+) regulatory T Cells (Tregs) in human cancers. Clin Cancer Res. (2019) 25:1233–8. doi: 10.1158/1078-0432.CCR-18-0762
63. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA. (2008) 105:14987–92. doi: 10.1073/pnas.0806075105
64. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. (1992) 11:3887–95.
65. Yao S, Chen L. PD-1 as an immune modulatory receptor. Cancer J. (2014) 20:262–4. doi: 10.1097/PPO.0000000000000060
66. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51.
67. Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. (1996) 8:765–72.
68. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood (2009) 113:5811–8. doi: 10.1182/blood-2009-02-203141
69. Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. (2010) 16:452–9. doi: 10.1038/nm.2106
70. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
71. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood (2009) 114:1537–44. doi: 10.1182/blood-2008-12-195792
72. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. (2001) 2:261–8. doi: 10.1038/85330
73. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. (2012) 366:2443–54. doi: 10.1056/NEJMoa1200690
74. Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, et al. In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy. Sci Transl Med. (2017) 9:eaal3604. doi: 10.1126/scitranslmed.aal3604
75. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive Disease is a new pattern of progression in cancer patients treated by anti-PD-1/PD-L1. Clin Cancer Res. (2017) 23:1920–28. doi: 10.1158/1078-0432.CCR-16-1741
76. Saada-Bouzid E, Defaucheux C, Karabajakian A, Coloma VP, Servois V, Paoletti X, et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann Oncol. (2017) 28:1605–11. doi: 10.1093/annonc/mdx178
77. Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. Hyperprogressive disease in patients with advanced non-small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol. (2018) 4:1543–52. doi: 10.1001/jamaoncol.2018.3676
78. Lo Russo G, Moro M, Sommariva M, Cancila V, Boeri M, Centonze G, et al. Antibody-Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non-small cell lung cancer subsequent to PD-1/PD-L1 blockade. Clin Cancer Res. (2018) 25:989–99. doi: 10.1158/1078-0432.CCR-18-1390
79. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. (1999) 5:1365–9. doi: 10.1038/70932
80. Hori J, Wang M, Miyashita M, Tanemoto K, Takahashi H, Takemori T, et al. B7-H1-induced apoptosis as a mechanism of immune privilege of corneal allografts. J Immunol. (2006) 177:5928–35. doi: 10.4049/jimmunol.177.9.5928
81. Petroff MG, Chen L, Phillips TA, Hunt JS. B7 family molecules: novel immunomodulators at the maternal-fetal interface. Placenta (2002) 23(Suppl. A):S95–101). doi: 10.1053/plac.2002.0813
82. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
83. Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. (2015) 14:847–56. doi: 10.1158/1535-7163.MCT-14-0983
84. Kowanetz M, Zou W, Gettinger SN, Koeppen H, Kockx M, Schmid P, et al. Differential regulation of PD-L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti-PD-L1). Proc Natl Acad Sci USA. (2018) 115:E10119–26. doi: 10.1073/pnas.1802166115
85. Inman BA, Longo TA, Ramalingam S, Harrison MR. Atezolizumab: a PD-L1-blocking antibody for bladder cancer. Clin Cancer Res. (2017) 23:1886–90. doi: 10.1158/1078-0432.CCR-16-1417
86. Ibrahim R, Stewart R, Shalabi A. PD-L1 blockade for cancer treatment: MEDI4736. Semin Oncol. (2015) 42:474–83. doi: 10.1053/j.seminoncol.2015.02.007
87. Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. (2015) 3:1148–57. doi: 10.1158/2326-6066.CIR-15-0059
88. Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcgammaRs modulate the anti-tumor activity of antibodies targeting the PD-1/PD-L1 axis. Cancer Cell (2015) 28:285–95. doi: 10.1016/j.ccell.2015.08.004
89. Goletz C, Lischke T, Harnack U, Schiele P, Danielczyk A, Ruhmann J, et al. Glyco-Engineered anti-human programmed death-ligand 1 antibody mediates stronger CD8 T cell activation than its normal glycosylated and non-glycosylated counterparts. Front Immunol. (2018) 9:1614. doi: 10.3389/fimmu.2018.01614
90. Sabatos CA, Chakravarti S, Cha E, Schubart A, Sanchez-Fueyo A, Zheng XX, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. (2003) 4:1102–10. doi: 10.1038/ni988
91. Anderson AC, Anderson DE. TIM-3 in autoimmunity. Curr Opin Immunol. (2006) 18:665–9. doi: 10.1016/j.coi.2006.09.009
92. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
93. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
94. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
95. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE (2012) 7:e30676. doi: 10.1371/journal.pone.0030676
96. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PLoS ONE (2013) 8:e58006. doi: 10.1371/journal.pone.0058006
97. Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. (2017) 276:97–111. doi: 10.1111/imr.12520
98. Zhuang X, Zhang X, Xia X, Zhang C, Liang X, Gao L, et al. Ectopic expression of TIM-3 in lung cancers: a potential independent prognostic factor for patients with NSCLC. Am J Clin Pathol. (2012) 137:978–85. doi: 10.1309/AJCP9Q6OVLVSHTMY
99. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology (2012) 56:1342–51. doi: 10.1002/hep.25777
100. Jiang J, Jin MS, Kong F, Cao D, Ma HX, Jia Z, et al. Decreased galectin-9 and increased Tim-3 expression are related to poor prognosis in gastric cancer. PLoS ONE (2013) 8:e81799. doi: 10.1371/journal.pone.0081799
101. Komohara Y, Morita T, Annan DA, Horlad H, Ohnishi K, Yamada S, et al. The coordinated actions of TIM-3 on cancer and myeloid cells in the regulation of tumorigenicity and clinical prognosis in clear cell renal cell carcinomas. Cancer Immunol Res. (2015) 3:999–1007. doi: 10.1158/2326-6066.CIR-14-0156
102. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti-PD1 therapy by Tim-3 upregulation is mediated by the PI3K-Akt pathway in head and neck cancer. Oncoimmunology (2017) 6:e1261779. doi: 10.1080/2162402X.2016.1261779
103. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. (2016) 7:10501. doi: 10.1038/ncomms10501
104. Datar I, Mani N, Henick BS, Wurtz A, Kaftan E, Herbst RS, et al. Measurement of PD-1, TIM-3 and LAG-3 protein in non-small cell lung carcinomas (NSCLCs) with acquired resistance to PD-1 axis blockers. J Clin Oncol. (2017) 35(15_suppl.):e14611–11. doi: 10.1200/JCO.2017.35.15_suppl.e14611
105. Golden-Mason L, Palmer BE, Kassam N, Townshend-Bulson L, Livingston S, McMahon BJ, et al. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J Virol. (2009) 83:9122–30. doi: 10.1128/JVI.00639-09
106. Waight J, Iyer P, Breous-Nystrom E, Riordan C, Findeis M, Underwood D, et al. Abstract 3825: INCAGN02390, a novel antagonist antibody that targets the co-inhibitory receptor TIM-3. Cancer Res. (2018) 78(13 Supplement):3825–25. doi: 10.1158/1538-7445.Am2018-3825
107. Kehry M, Horlick R, Bowers P, Jun T, da Silva Correia J, Graves J, et al. Abstract 271: Targeting PD-1, TIM-3 and LAG-3 in combination for improved immunotherapy combinations. Cancer Res. (2015) 75(15 Supplement):271–71. doi: 10.1158/1538-7445.Am2015-271
108. Davar D, Boasberg P, Eroglu Z, Falchook G, Gainor J, Hamilton E, et al. A phase 1 study of TSR-022 (Anti-TIM-3) in combination with TSR-042 (Anti-PD-1). In 2018 Annual Meeting of the Society for Immunotherapy of Cancer (SITC) Conference. Washington, DC (2018).
109. Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. (1990) 171:1393–405. doi: 10.1084/jem.171.5.1393
110. Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. (2011) 208:395–407. doi: 10.1084/jem.20100466
111. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. (2012) 72:917–27. doi: 10.1158/0008-5472.CAN-11-1620
112. Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. (2004) 172:5450–5. doi: 10.4049/jimmunol.172.9.5450
113. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in regulatory T cells. Immunity (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
114. Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol. (1995) 25:2718–21. doi: 10.1002/eji.1830250949
115. Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci USA. (2010) 107:7875–80. doi: 10.1073/pnas.1003345107
116. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest. (2007) 117:3383–92. doi: 10.1172/JCI31184
117. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. (2015) 3:412–23. doi: 10.1158/2326-6066.CIR-14-0150
118. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al. Fibrinogen-like Protein 1 is a major immune inhibitory ligand of LAG-3. Cell (2019) 176:334–47.e12. doi: 10.1016/j.cell.2018.11.010
119. Haines BB, Javaid S, Cui L, Hirsch H, Cemerski S, McClanahan T, et al. Abstract 4714: blockade of LAG-3 amplifies immune activation signatures and augments curative antitumor responses to anti-PD-1 therapy in immune competent mouse models of cancer. Cancer Res. (2017) 77(13 Supplement):4714–14. doi: 10.1158/1538-7445.Am2017-4714
120. Jun HT, Kehry M, Bowers P, King DJ. Antibodies Directed Against Lymphocyte Activation Gene 3 (lag-3). San Diego, CA: US20180127496A (2018).
121. Zettl M, Wurm M, Schaaf O, Tirapu I, Mostböck S, Reschke M, et al. Abstract 4547: characterization of the LAG-3 targeting antibody BI 754111 in monotherapy and in combination with the anti-PD-1 antibody BI 754091. Cancer Res. (2018) 78(13 Supplement):4547–47. doi: 10.1158/1538-7445.Am2018-4547
122. Savitsky D, Ward R, Riordan C, Mundt C, Jennings S, Connolly J, et al. Abstract 3819: INCAGN02385 is an antagonist antibody targeting the co-inhibitory receptor LAG-3 for the treatment of human malignancies. Cancer Res. (2018) 78(13 Supplement):3819–19. doi: 10.1158/1538-7445.Am2018-3819
123. Ascierto PA, Bono P, Bhatia S, Melero I, Nyakas MS, Svane I, et al. Efficacy of BMS-986016, a monoclonal antibody that targets lymphocyte activation gene-3 (LAG-3), in combination with nivolumab in pts with melanoma who progressed during prior anti-PD-1/PD-L1 therapy (mel prior IO) in all-comer and biomarker-enriched populations. Ann Oncol. (2017) 28(suppl_5):v605–49. doi: 10.1093/annonc/mdx440
124. Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur J Immunol. (2009) 39:695–703. doi: 10.1002/eji.200839116
125. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci USA. (2009) 106:17858–63. doi: 10.1073/pnas.0903474106
126. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. (2009) 10:48–57. doi: 10.1038/ni.1674
127. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest. (2014) 124:2246–59. doi: 10.1172/JCI73639
128. Casado JG, Pawelec G, Morgado S, Sanchez-Correa B, Delgado E, Gayoso I, et al. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell-mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol Immunother. (2009) 58:1517–26. doi: 10.1007/s00262-009-0682-y
129. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol. (2011) 41:902–15. doi: 10.1002/eji.201041136
130. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J Exp Med. (2003) 198:557–67. doi: 10.1084/jem.20030788
131. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/JCI81187
132. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
133. Johnston RJ, Comps-Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell (2014) 26:923–37. doi: 10.1016/j.ccell.2014.10.018
134. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Invest. (2015) 125:2046–58. doi: 10.1172/JCI80445
135. Argast GM, Cancilla B, Cattaruzza F, Yeung P, Scolan El, Harris R, et al. Abstract 5627: anti-TIGIT biomarker study: inhibition of TIGIT induces loss of Tregs from tumors and requires effector function for tumor growth inhibition. Cancer Res. (2018) 78(13 Supplement):5627–27. doi: 10.1158/1538-7445.Am2018-5627
136. Barnhart BC, Sega E, Yamniuk A, Hatcher S, Lei M, Ghermazien H, et al. Abstract 1476: a therapeutic antibody that inhibits CD73 activity by dual mechanisms. Cancer Res. (2016) 76(14 Supplement):1476–76. doi: 10.1158/1538-7445.am2016-1476
137. Gao ZW, Wang HP, Lin F, Wang X, Long M, Zhang HZ, et al. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer (2017) 17:135. doi: 10.1186/s12885-017-3128-5
138. Antonioli L, Yegutkin GG, Pacher P, Blandizzi C, Hasko G. Anti-CD73 in cancer immunotherapy: awakening new opportunities. Trends Cancer (2016) 2:95–109. doi: 10.1016/j.trecan.2016.01.003
139. Gao ZW, Dong K, Zhang HZ. The roles of CD73 in cancer. Biomed Res Int. (2014) 2014:460654. doi: 10.1155/2014/460654
140. Klemens MR, Sherman WR, Holmberg NJ, Ruedi JM, Low MG, Thompson LF. Characterization of soluble vs membrane-bound human placental 5'-nucleotidase. Biochem Biophys Res Commun. (1990) 172:1371–7.
141. Inoue Y, Yoshimura K, Kurabe N, Kahyo T, Kawase A, Tanahashi M, et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget (2017) 8:8738–51. doi: 10.18632/oncotarget.14434
142. Leclerc BG, Charlebois R, Chouinard G, Allard B, Pommey S, Saad F, et al. CD73 Expression is an independent prognostic factor in prostate cancer. Clin Cancer Res. (2016) 22:158–66. doi: 10.1158/1078-0432.CCR-15-1181
143. Loi S, Pommey S, Haibe-Kains B, Beavis PA, Darcy PK, Smyth MJ, et al. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc Natl Acad Sci USA. (2013) 110:11091–6. doi: 10.1073/pnas.1222251110
144. Gaudreau PO, Allard B, Turcotte M, Stagg J. CD73-adenosine reduces immune responses and survival in ovarian cancer patients. Oncoimmunology (2016) 5:e1127496. doi: 10.1080/2162402X.2015.1127496
145. Wu XR, He XS, Chen YF, Yuan RX, Zeng Y, Lian L, et al. High expression of CD73 as a poor prognostic biomarker in human colorectal cancer. J Surg Oncol. (2012) 106:130–7. doi: 10.1002/jso.23056
146. Morello S, Capone M, Sorrentino C, Giannarelli D, Madonna G, Mallardo D, et al. Soluble CD73 as biomarker in patients with metastatic melanoma patients treated with nivolumab. J Transl Med. (2017) 15:244. doi: 10.1186/s12967-017-1348-8
147. Ren ZH, Lin CZ, Cao W, Yang R, Lu W, Liu ZQ, et al. CD73 is associated with poor prognosis in HNSCC. Oncotarget (2016) 7:61690–702. doi: 10.18632/oncotarget.11435
148. Vijayan D, Barkauskas DS, Stannard K, Sult E, Buonpane R, Takeda K, et al. Selective activation of anti-CD73 mechanisms in control of primary tumors and metastases. Oncoimmunology (2017) 6:e1312044. doi: 10.1080/2162402X.2017.1312044
149. Geoghegan JC, Diedrich G, Lu X, Rosenthal K, Sachsenmeier KF, Wu H, et al. Inhibition of CD73 AMP hydrolysis by a therapeutic antibody with a dual, non-competitive mechanism of action. MAbs (2016) 8:454–67. doi: 10.1080/19420862.2016.1143182
150. Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology (2016) 5:e1208875. doi: 10.1080/2162402X.2016.1208875
151. Oganesyan V, Gao C, Shirinian L, Wu H, Dall'Acqua WF. Structural characterization of a human Fc fragment engineered for lack of effector functions. Acta Crystallogr D Biol Crystallogr. (2008) 64(Pt 6):700–4. doi: 10.1107/S0907444908007877
152. Piccione EC, Hotson A, Mikesell G, Daine-Matsuoka B, Gu C, Dao-Pick T, et al. Preclinical and initial phase I clinical characterization of CPI-006: an anti-CD73 monoclonal antibody with unique immunostimulatory activity. In 2018 Annual Meeting of the Society for Immunotherapy of Cancer (SITC) Conference. Washington, DC (2018).
153. Barnhart BC, Sega E, Yamniuk A, Hatcher S, Lei M, Ghermazien H, et al. Abstract 1476: a therapeutic antibody that inhibits CD73 activity by dual mechanisms. Cancer Res. (2016) 76:1476. doi: 10.1158/1538-7445.AM2016-1476
154. Siu LL, Burris H, Le DT, Hollebecque A, Steeghs N, Delord JP, et al. Abstract CT180: preliminary phase 1 profile of BMS-986179, an anti-CD73 antibody, in combination with nivolumab in patients with advanced solid tumors. Cancer Res. (2018) 78:CT180. doi: 10.1158/1538-7445.AM2018-CT180
155. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. (2014) 74:1924–32. doi: 10.1158/0008-5472.CAN-13-1504
156. Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci USA. (2015) 112:6682–7. doi: 10.1073/pnas.1420370112
157. Vigdorovich V, Ramagopal UA, Lazar-Molnar E, Sylvestre E, Lee JS, Hofmeyer KA, et al. Structure and T cell inhibition properties of B7 family member, B7-H3. Structure (2013) 21:707–17. doi: 10.1016/j.str.2013.03.003
158. Lee YH, Martin-Orozco N, Zheng P, Li J, Zhang P, Tan H, et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. (2017) 27:1034–45. doi: 10.1038/cr.2017.90
159. Prasad DV, Nguyen T, Li Z, Yang Y, Duong J, Wang Y, et al. Murine B7-H3 is a negative regulator of T cells. J Immunol. (2004) 173:2500–6. doi: 10.4049/jimmunol.173.4.2500
160. Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB, Liu D, et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. (2001) 2:269–74. doi: 10.1038/85339
161. Sun M, Richards S, Prasad DV, Mai XM, Rudensky A, Dong C. Characterization of mouse and human B7-H3 genes. J Immunol. (2002) 168:6294–7. doi: 10.4049/jimmunol.168.12.6294
162. Zhang G, Xu Y, Lu X, Huang H, Zhou Y, Lu B, et al. Diagnosis value of serum B7-H3 expression in non-small cell lung cancer. Lung Cancer (2009) 66:245–9. doi: 10.1016/j.lungcan.2009.01.017
163. Zang X, Thompson RH, Al-Ahmadie HA, Serio AM, Reuter VE, Eastham JA, et al. B7-H3 and B7x are highly expressed in human prostate cancer and associated with disease spread and poor outcome. Proc Natl Acad Sci USA. (2007) 104:19458–63. doi: 10.1073/pnas.0709802104
164. Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM, Frigola X, et al. Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res. (2008) 14:5150–7. doi: 10.1158/1078-0432.CCR-08-0536
165. Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X, Inman BA, et al. B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res. (2007) 67:7893–900. doi: 10.1158/0008-5472.CAN-07-1068
166. Sun Y, Wang Y, Zhao J, Gu M, Giscombe R, Lefvert AK, et al. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer (2006) 53:143–51. doi: 10.1016/j.lungcan.2006.05.012
167. Zang X, Sullivan PS, Soslow RA, Waitz R, Reuter VE, Wilton A, et al. Tumor associated endothelial expression of B7-H3 predicts survival in ovarian carcinomas. Mod Pathol. (2010) 23:1104–12. doi: 10.1038/modpathol.2010.95
168. Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell (2017) 31:501–15 e8. doi: 10.1016/j.ccell.2017.03.005
169. Rizvi NA, Loo D, Baughman JE, Yun S, Chen F, Moore PA, et al. A phase 1 study of enoblituzumab in combination with pembrolizumab in patients with advanced B7-H3-expressing cancers. J Clin Oncol. (2016) 34(15_suppl):TPS3104–TPS04. doi: 10.1200/JCO.2016.34.15_suppl.TPS3104
170. Loo D, Alderson RF, Chen FZ, Huang L, Zhang W, Gorlatov S, et al. Development of an Fc-enhanced anti-B7-H3 monoclonal antibody with potent antitumor activity. Clin Cancer Res. (2012) 18:3834–45. doi: 10.1158/1078-0432.CCR-12-0715
171. Powderly J, Cote G, Flaherty K, Szmulewitz RZ, Ribas A, Weber J, et al. Interim results of an ongoing Phase I, dose escalation study of MGA271 (Fc-optimized humanized anti-B7-H3 monoclonal antibody) in patients with refractory B7-H3-expressing neoplasms or neoplasms whose vasculature expresses B7-H3. J ImmunoTher Cancer (2015) 3(Suppl 2): O8. doi: 10.1186/2051-1426-3-S2-O8
172. Du X, Tang F, Liu M, Su J, Zhang Y, Wu W, et al. A reappraisal of CTLA-4 checkpoint blockade in cancer immunotherapy. Cell Res. (2018) 28:416–32. doi: 10.1038/s41422-018-0011-0
Keywords: FcγR, checkpoint blockade, antibody therapy, cancer immunotherapy, IgG isotype
Citation: Chen X, Song X, Li K and Zhang T (2019) FcγR-Binding Is an Important Functional Attribute for Immune Checkpoint Antibodies in Cancer Immunotherapy. Front. Immunol. 10:292. doi: 10.3389/fimmu.2019.00292
Received: 30 November 2018; Accepted: 05 February 2019;
Published: 26 February 2019.
Edited by:
Mark S. Cragg, University of Southampton, United KingdomReviewed by:
Aida Karachi, University of Florida, United StatesCopyright © 2019 Chen, Song, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kang Li, a2FuZy5saUBiZWlnZW5lLmNvbQ==
Tong Zhang, dG9uZy56aGFuZ0BiZWlnZW5lLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.