 Naeun Lee
Naeun Lee Donghyun Kim
Donghyun Kim Wan-Uk Kim
Wan-Uk Kim- 1Center for Integrative Rheumatoid Transcriptomics and Dynamics, The Catholic University of Korea, Seoul, South Korea
- 2Department of Microbiology and Immunology, Seoul National University College of Medicine, Seoul, South Korea
- 3Department of Biomedical Sciences, Seoul National University College of Medicine, Seoul, South Korea
- 4Institute of Infectious Diseases, Seoul National University College of Medicine, Seoul, South Korea
- 5Division of Rheumatology, Department of Internal Medicine, The Catholic University of Korea, Seoul, South Korea
The nuclear factor of activated T cells (NFAT5), also known as a tonicity-responsive enhancer-binding protein, was originally identified as a key transcription factor involved in maintaining cellular homeostasis against hypertonic and hyperosmotic environments. Although NFAT5 has been expressed and studied in various types of hyperosmolar tissues, evidence has emerged that NFAT5 plays a role in the development and activation of immune cells, especially T cells and macrophages. The immune-regulatory function of NFAT5 is achieved by inducing different target genes and different signaling pathways in both tonicity-dependent and -independent manners. Particularly in response to hyperosmotic stress, NFAT5 induces the generation of pathogenic TH17 cells and pro-inflammatory macrophages, contributing to autoimmune and inflammatory diseases. Meanwhile, with tonicity-independent stimuli, including activation of the Toll-like receptors and inflammatory cytokines, NFAT5 also can be activated and promotes immune cell survival, proliferation, migration, and angiogenesis. Moreover, under isotonic conditions, NFAT5 has been implicated in the pathogenesis of a variety of inflammatory and autoimmune diseases including rheumatoid arthritis. This review describes the current knowledge of NFAT5, focusing on its immune-regulatory functions, and it highlights the importance of NFAT5 as a novel therapeutic target for chronic inflammatory diseases.
Introduction
The nuclear factor of activated T cells-5 (NFAT5), also known as tonicity-responsive enhancer binding protein (TonEBP), was initially identified from the kidney medulla as a central regulator of cellular response to ambient hypertonicity (1). Under physiological conditions, the operation of urinary concentrating mechanisms in the kidney medulla often causes high osmolality—about four times that of the blood or higher—thereby maintaining constant body fluid volume and blood pressure (2). However, excess extracellular osmolality to renal cells drives double-strand DNA breaks and cell death (3). Fortunately, the high osmotic stress increases the amount and the nuclear translocation of NFAT5 at once that is directly responsible for accumulating organic osmolytes to restore homeostasis in the kidney medulla (1). Transcription factor NFAT5 induces the expression of a variety of osmoprotective genes, including ion transporters, aldose reductase, and heat shock protein 70, for intracellular distribution of compatible osmolytes and urea (4). The reduced intracellular ionic strength and urea concentration contribute to protecting the cells within the kidney medulla from the deadly effects of hypertonicity (4).
Gene, Protein, and Molecular Characteristics of NFAT5
The human Nfat5 gene exists in chromosome 16q22.1, and the mouse homolog is found in chromosome 8D (5). Various transcripts are made from the gene by alternative promoters and alternative splicing; thus far, 16 different variants have been reported. Among these, 12 transcripts have protein-coding potential, and the remaining variants appear not to encode proteins (6). The NFAT5 transcript is expressed in various human tissues such as the kidney, brain, heart, thymus, lung, and skeletal muscle (1, 7). In contrast to the ubiquitous mRNA expression, the abundant expression of NFAT5 protein is detected only in extract from the thymus, while there are much lower amounts in the testes, lung, liver, and brain and no expression in other tissues including lymph nodes (8).
NFAT5 belongs to the Rel family, in which members share the Rel-homology domain (RHD) responsible for DNA binding (Figure 1A) (9). The RHD of NFAT5 is highly similar to those of other NFAT members (NFAT1 to 4) but has minimal amino acid identity with that of NFκB (9). In addition to the RHD, the protein structure consists of a leucine-rich canonical nuclear export sequence (NES) located at the first 19 amino acids, an N-terminal compositionally serine/threonine and proline-rich region (transactivation domain 1; TAD1), an auxiliary export domain (AED), a consensus bipartite nuclear localization signal (NLS), a dimerization domain (DD) within the RHD, and a C-terminal low-complexity region (glutamine and serine/threonine-rich region, a TAD2) (10–13) (Figure 1A). However, NFAT5 protein lacks docking sites for calcineurin that is necessary for the nuclear translocation of the other NFAT proteins (Figure 1B), and thus the calcium/calcineurin signaling cascade is dispensable for activating the transcription factor.
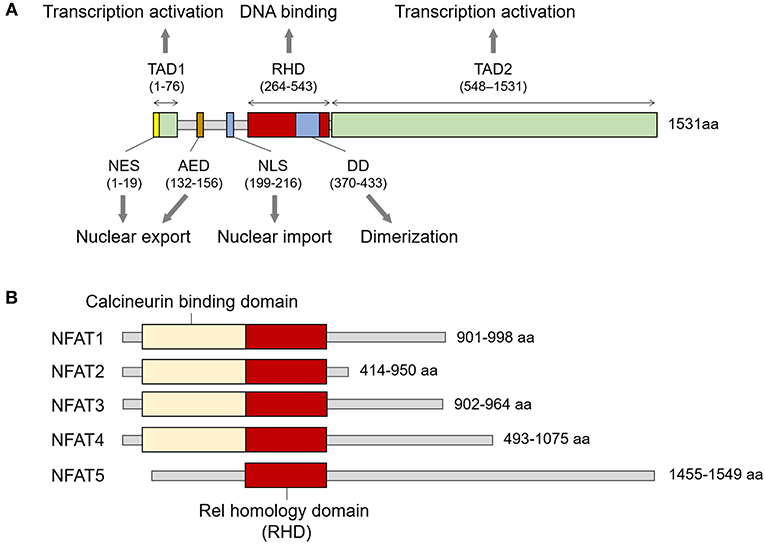
Figure 1. Structure of NFAT family members. (A) Signature domains in human NFAT5 protein and their function. NFAT5 contains several functional domains including NES, TAD1, AED, NLS, RHD, DD, and TAD2. The number of amino acids in parenthesis is based on KIAA0827 (1531 amino acid; GenBank accession no. AB020634). (B) Comparison of representative structures between NFAT members. All NFAT members share the Rel homology domain, but only NFAT5 does not have a calcineurin-binding domain. The minimum and maximum numbers of amino acids among representative transcript variants are presented.
NFAT5 protein exists in both cytoplasm and nucleus under isotonic conditions, but it is not in a static state; rather, it is in an active equilibrium state between cytoplasmic and nucleic protein (1, 11). The active nucleocytoplasmic shuttling under isotonic conditions is mediated by NLS and NES that are recognized by specific import and export receptors, respectively (11). Changes in extracellular tonicity rapidly alter the amount and ratio of NFAT5 between the cytosol and nucleus. Hypertonicity induces the transcription and translation of NFAT5 as well as leads to the translocation and accumulation of NFAT5 into nucleus (1, 4). As in isotonic condition, the NLS is indispensable for the nuclear import process induced by hyperosmotic stress (10). In contrast, hypotonic condition predominantly leads to the nuclear export of the transcription factor, which depends on the presence of AED but not of NES (11).
NFAT5 recognizes and binds to TGGAAANNYNY (N, any nucleotide; Y, any pyrimidine) sequences on DNA promoters similar to those recognized by NFAT1 (7, 9). NFAT5 acts as a dimeric form, as does NF-κB, and does not cooperate with FOS or JUN, whereas other NFAT members exist as a preformed monomer and often bind DNA together with Fos or Jun (9, 14).
Role of NFAT5 in the Immune System
Tonicity-Dependent Immune Regulation
High Salt and Th17 Polarity
The intake of too much dietary salt (sodium chloride, NaCl) has been implicated in various diseases including hypertension, stroke, coronary heart disease, heart failure, and renal disease (15, 16). In numerous instances, the disorders related to high salt intake are accompanied by the induction of TH17 cells that have a pathogenic role in the progression of chronic inflammatory and autoimmune diseases (17–19). When TH17 polarization was induced in vitro from human naïve CD4+ T cells by cytokines, a modest increase in NaCl concentration drove further augmentation in IL-17A and GM-CSF secretion (18). Even in healthy adults, dietary salt consumption gradually increases TH17 cells and reciprocally decreases Treg cells, although the TH17/Treg ratio is somewhat low on the first day of high salt loading (20).
Salt is a typical dietary nutrient that increases tonicity (15). Thus, simultaneously with the increase in TH17 cell population, the high salt consumption leads to the expression and transactivation of NFAT5. Moreover, several groups have shown that NFAT5 activation is pivotal in high salt-mediated TH17 polarization (18, 21). Gene silencing of Nfat5 in CD4+ T cells abrogates the expression of TH17-associated genes including Il17 and Rorγt (18, 21). Altogether, the high salt-NFAT5-TH17 axis may explain the pathogenesis of several inflammatory and autoimmune diseases (Figure 2).
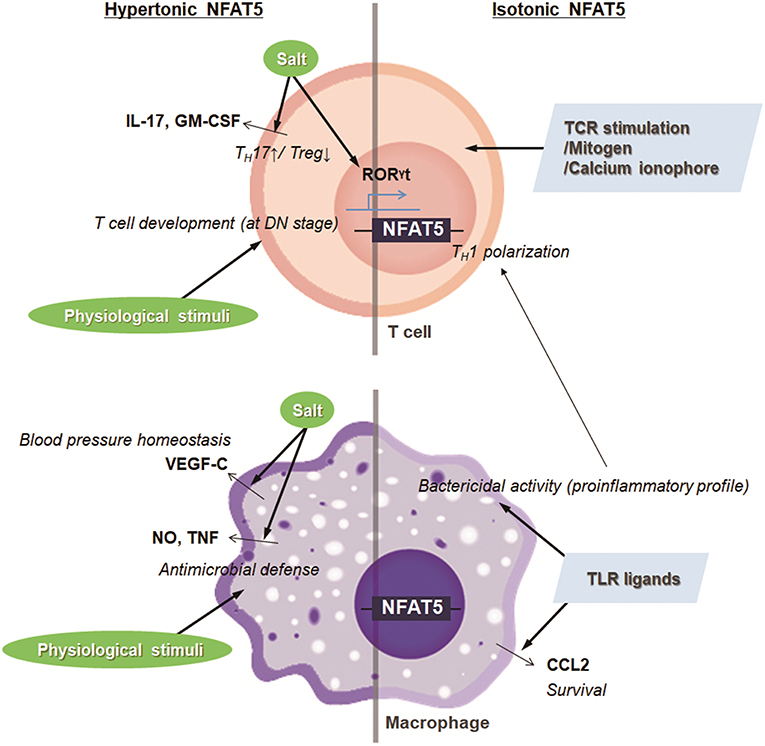
Figure 2. Targets and immunologic functions of hypertonic and isotonic NFAT5. NFAT5 induced by hypertonic or isotonic stimuli maintains blood pressure homeostasis, induces T cell development and TH17 cell differentiation, and enhances antimicrobial activity and cell survival of macrophages. In addition, NFAT5-mediated proinflammatory alteration in macrophages skews CD4+ T cells to TH1 polarization.
High Salt and Innate Immunity
The hyperosmolar state induced by salt intake also stimulates the expression and translocation of NFAT5 in innate cells, affecting physiologic and pathologic immune responses. In the macrophages, NaCl leads to the NFAT5-mediated induction of nitric oxide and TNF, which promotes cutaneous antimicrobial defense (Figure 2) (22). Also, under conditions of high salt intake, osmotic stress-induced NFAT5 contributes to the expression of vascular endothelial growth factor (VEGF)-C in macrophages (23). The VEGF-C restores blood pressure homeostasis by increasing the density and hyperplasia of the lymph capillary network, which protects against hypertension (Figure 2) (23). In invariant nature killer T cells, high salt condition inhibits TCR-dependent and TCR-independent IFN-γ production, which is dependent on NFAT5 (24). Although earlier researchers emphasize the protective and physiologic roles of NFAT5 in maintaining immune defense and body homeostasis, NFAT5 sometimes can critically mediate pathologic immune responses and tissue damage as well. For example, high extracellular NaCl concentration induces NLRP3 gene expression in retinal pigment epithelial cells that is partially dependent on the activities of NFAT5 (25); the retinal inflammation is thought to be associated with the development of age-related macular degeneration (25).
Physiological and Pathophysiological Osmotic Stress and Immune Regulation
Osmolality within lymphoid microenvironment, like the thymus and spleen, is significantly higher than that of serum, indicating that lymphocytes are exposed to physiologic osmotic stress (26). Moreover, sera obtained from NFAT5-deficient mice are markedly hypertonic and contain abnormally high concentrations of sodium, suggesting that NFAT5 might function as part of an osmotic stress response (27, 28). Simultaneously, NFAT5-haplo-deficient mice exhibited a 30–50% reduction in thymic and splenic cellularity evenly distributed among T cell subsets (26, 28). A similar phenotype was observed in transgenic mice which express a dominant inhibitory form of NFAT5 only in T lymphocytes by using the CD2 promoter (29). The decrease in thymic cellularity is associated with the reduced number of peripheral T cells (29). Consistently, conditional deletion of NFAT5 at the early double-negative thymocyte stage causes smaller thymi, fewer double positive and single positive thymocytes, and decreased cellularity in the lymphoid organ, although mice deficient in NFAT5 after double-positive thymocyte stage (CD4-Cre;Nfat5fl/fl mice) exhibit normal plasma tonicity and have only a mild naïve/memory imbalance in T cells (28, 30). Taken together, NFAT5 induced by physiological osmotic stress serves a survival signal during T cell development (Figure 2). As another example, renal sodium gradient induced by the physiological and pathophysiological situation causes chemokine secretion by the renal tubular epithelium, which recruits circulating monocyte-derived mononuclear phagocytes into the hypertonic region and stimulates antimicrobial activity of renal macrophages (31). Interestingly, NFAT5 is involved not only in the migration activity toward hypertonic spot but also in the antimicrobial function of renal macrophages (Figure 2) (31, 32). These findings indicate that NFAT5 has a pivotal role in regulating immune system in response to physiological and pathologic osmotic stress.
Tonicity-Independent Immune Regulation
In addition to the enhanced extracellular tonicities, isotonic factors other than tonicity are involved in NFAT5 abundance and activity in some cells and tissues that are presumably well-conserved by isotonic body fluids (27). Isotonic stimuli-induced NFAT5 has been implicated in various physiological and pathological responses that have no relevance to osmotic stress. For example, several researchers have confirmed that NFAT5 plays a pivotal role in the pathogenesis of RA, a tonicity-independent disorder, using various experimental arthritis models (33, 34). Upon toll-like receptor (TLR) stimulation in the macrophages, NFAT5 is activated through xanthine oxidase-reactive oxygen species (ROS)-p38MAPK cascade (33). The protein provides apoptotic resistance to RA macrophages by inducing CCL2 secretion (Figure 2) (33). Like that in macrophages, inflammatory stimuli-induced NFAT5 activation promotes the expression of tissue factor and CCL2 in the FLSs of RA patients (RA-FLSs), causing the hypermotility and invasiveness that are pathological features of RA (35). Next, during vascular injury, platelets, and other cells of the vasculature secrete platelet-derived growth factor-BB (PDGF-BB), which drives the proliferation and migration of vascular smooth muscle cells (SMCs) (36). PDGF-BB increases NFAT5 protein expression and its reporter activity in SMCs, which mediates SMC migration (36). Angiotensin II, another factor involved in vascular injury, promotes SMC contraction and hypertrophy (36). The contractile SMC phenotype is dependent on the angiotensin II-mediated NFAT5 nuclear import and its transcriptional activity (36). Some inflammatory molecules, such as TNF-α, have been known to cause intervertebral disc degeneration, triggering chronic back pain (37). In the process, the inflammatory responses to TNF-α in NP cells are mediated by induced NFAT5 transcription (37). Two growth factors, bone morphogenetic protein 2 (BMP-2) and transforming growth factor-β (TGF-β), were found to significantly increase both NFAT5 protein expression and transactivation in nucleus pulposus cells located at the center of the intervertebral disc, which influences disk cell function through chondroitin sulfate and aggrecan synthesis (38). Inflammatory stimuli such as lipopolysaccharide (LPS), IFN-γ, and IL-4 also induce the expression and activation of the transcription factor in microglia, resident immune cells of the central nervous system (39). In the same context, ischemic brain injury induces NFAT5 expression, which is required for achieving an osmotic equilibrium (39, 40). Besides, clustering of α6β4 integrin in carcinomas enhances NFAT5 protein and hence NFAT5-dependent transcription, which promotes carcinoma invasion into surrounding tissues (41). NFAT5 is also required for systemic inflammation and septic shock associated with NF-κB enhanceosome in myeloid cells (42). LPS stimulation after classical or alternative polarization in macrophages enhances NFAT5 expression (43). The increased transcription factor in macrophages enhances the proinflammatory profile such as bactericidal capability and promotes more dominant Th1 polarization compared with Th2 responses (Figure 2) (43). In T lymphocytes, isotonic stimuli, such as cross-linking with T cell receptors, mitogen, and calcium ionophore, are also able to induce NFAT5 expression (Figure 2) (8). Taken together, regardless of osmotic stress, various physiological and pathophysiological stimuli trigger the NFAT5 expression and activity implicated in various immune responses and disease pathogenesis (Figure 2).
Context-Dependent Regulation and Functions of NFAT5
Interestingly, NFAT5 induced by isotonic stimuli (isotonic NFAT5) and osmotic NFAT5 shows distinct differences in the lymphocytes. For instance, in hypertonic culture media, the proliferation of NFAT5-deficient CD4+ T cells was markedly reduced as compared to that of normal CD4+ T cells, resulting from osmoadaptive functions of NFAT5 (44). In contrast, there were no differences in T cell proliferation regardless of NFAT5 expression under isotonic culture condition (44). In addition, NFAT5 induced by high salt media skews CD4+ T cells toward the TH17 phenotype (18, 21), whereas under isotonic normal media, the depletion of NFAT5 in CD4+ T cells enhances interferon γ (IFNγ) and IL-17 expression and reduces Treg responses (21). Like T cells, NFAT5 has a critical role in the B-cell proliferation induced by LPS treatment under hypertonic conditions but not isotonic media (45). In macrophages, both LPS and NaCl reciprocally inhibit the expression of downstream target genes (46). In this process, NFAT5 contributes to context-dependent inhibition through the activation of different ROS sources (46).
It is unclear how exactly isotonic NFAT5 differs from osmotic NFAT5 with regard to its functional and molecular outcomes. As mentioned above, we suggested that ROS function as molecular sensors to discriminate between isotonic and osmotic stimuli, directing NFAT5 activity toward differential responses in a context-dependent manner (46). Alternatively, cell proliferation-inducing stimuli, including PDGF, TGF-β, and TNF-α, may increase in active macromolecular biosynthesis, leading to a hyperosmolar state, albeit very transiently, in the cells stimulated with such stimuli. Although hypertonic stress may not approach that within the kidney, another possibility is that environmental osmotic stress might be exacerbated by the depletion of intracellular osmolytes and reduction in available cell volume caused by the massive induction of intracellular protein biosynthesis during cellular hyperproliferation. An earlier finding that the liver, a non-proliferative but metabolically active tissue, shows hyperosmolarity supports this notion (27). Despite these possibilities, the exact difference between isotonic and osmotic NFAT5 in the regulation and function still remains to be unveiled.
Role of NFAT5 in the Pathogenesis of Autoimmune and Inflammatory Diseases
NFAT5 and Chronic Arthritis
RA is a chronic inflammatory disease characterized by the expansion of inflammatory immune cells as well as synoviocytes, leading to the destruction of bone and cartilage (47, 48). RA can present with numerous autoimmune features, including producing autoantibodies such as rheumatoid factors and anti-citrullinated protein antibodies (ACPA) (47, 48). Although the etiology of RA remains elusive, interplay between genetic susceptibility and environmental factors seems to trigger disease onset. Environmental risk factors such as smoking, air pollutants, and the microbiome can drive the epigenetic modification, increasing the risk for developing RA in individuals with genetic susceptibility, including certain MHC (e.g., HLA-DR1 and DR4) or non-MHC molecules associated with autoimmune responses (47). In particular, cigarette smoking, one of the most prominent environmental risk factors for RA, increases citrullination to self-proteins and the formation of ACPA (48). As a result of autoimmune responses to citrullinated proteins, a variety of immune cells including macrophages, B cells, and T cells heavily infiltrate the synovial tissues and are involved in the initiation and perpetuation of RA. These cells can activate each other, constructing a very complex inter- and intracellular network of pro-inflammatory cytokines and chemokines including IL-1β, TNF-α, IL-6, and CCL2 (47). Chronic exposure to these pro-inflammatory cytokines, growth factors, and hypoxia may lead to abnormal proliferation of synovial fibroblasts, a pathologic hallmark of RA, and confer on these cells apoptotic resistance (35).
Recent researchers have demonstrated that NFAT5 activation by tonicity-independent, isotonic stimuli is a key regulator of RA pathology. Masuda et al. first identified that NFAT5 mRNA is expressed in the RA synovia, particularly at sites of bone destruction (49). Along with this, our group has shown that NFAT5 is highly expressed in the RA-FLSs and that its expression in RA-FLSs is upregulated by the stimulation of pro-inflammatory cytokines such as IL-1β, TNF-α, and TGF-β (33, 35). Through the global gene expression profiling of RA-FLSs, we also found that NFAT5 is a major transcription factor for regulating the migration and invasion, an aggressive phenotype of RA-FLSs, which are mediated by the upregulation of CCL2- and tissue factor expressions as its downstream target genes (35). Thus, we presume that NFAT5 plays an essential role in promoting RA-FLS migration and invasion.
Transcriptomic analysis of RA macrophages revealed that NFAT5 is also a master regulator for the pathology of RA macrophages (34). In that analysis, differentially expressed genes (DEGs) in RA macrophages overlapped significantly with the DEGs governed by NFAT5 (34). The overlapping DEGs represented a variety of biologic processes such as cell proliferation, apoptosis, and death (34). Moreover, NFAT5-deficient macrophages showed reduced survival and proliferation, suggesting that NFAT5 promotes the survival of macrophages (34). Additionally, as mentioned earlier, NFAT5 in macrophages can be activated by isotonic TLR ligands (e.g., LPS) as well as high salt (33, 50). TLR-stimulated NFAT5 activates a sets of downstream target genes including Nos2, Il6, and Tnf that differ from osmolarity-associated target molecules, leading to macrophage activation and TLR-induced arthritis (33). Along with this, NFAT5-deficiency reduces disease severity in mice with chronic inflammatory arthritis dependent on TLR-2 and 4 (51). Taken together, we consider that NFAT5 is an important regulator of macrophage activation and survival and that it thereby contributes to RA pathology.
Of interest, recent studies have shown that smoking together with high sodium intake is associated with the development of ACPA-positive RA (52, 53). As described in the discussion of High salt and TH17 polarity, sodium chloride drives the development of pathogenic TH17 cells through the serum glucocorticoid kinase 1 (SGK1)-mediated pathway; SGK1 is an inducible salt-sensing kinase (18, 54). Conversely, high salt prevents the suppressive function of Foxp3+ Treg cells (55). These reports provide important evidence that high salt itself favors the polarization of immune cells, including T helper cells, toward pro-inflammatory phenotypes while suppressing the activation of Treg, promoting immune-mediated diseases. Although the role of NFAT5 in high salt-induced development of TH17 cells in autoimmune diseases remains elusive, it is conceivable that tonicity-dependent NFAT5 activation has a pivotal role in initiating RA in smokers, who often enjoy eating salty food because it is crucial to both high salt-induced activation of TH17 cells (56) and osmo-protection (57).
Taken together, given the effects of NFAT5 on immunity under isotonic and hypertonic conditions, we believe that the pathologic role of NFAT5 in RA is not limited to a single type of immune cell. Rather, overall RA pathology can be demonstrated to result from the combinatory action of NFAT5 on multiple types of cells including macrophages, TH17 cells, Treg cells, and synoviocytes.
NFAT5 and Diabetes Mellitus
Type 1 diabetes (T1D) is well-known as a T-cell mediated autoimmune disorder characterized by destroying insulin-producing β cells in the pancreas, which causes multiple complications such as retinopathy and nephropathy (58). T1D as a metabolic disorder interrupts blood glucose homeostasis, resulting in hyperglycemia owing to a lack of insulin. Hyperglycemia induces metabolic influx associated with increased osmotic stress, which leads to the development of diabetes-related microvascular complications (59). As discussed above, hyperosmotic stress itself functions as a potent inflammatory stimulus to release pro-inflammatory cytokines (60). In terms of tonicity-dependent mechanisms, intracellular osmolarity tightly regulates the expression of aldose reductase (AR), which is a key inducer of hyperglycemia-induced oxidative stress as well as a target gene of NFAT5 under hypertonic conditions in both peripheral blood mononuclear cells and human mesangial cells (59). Consistently, in streptozotocin-induced diabetic retinopathy, NFAT5 deficiency decreases AR expression and alleviates the retinopathy (61). Furthermore, NFAT5 deficiency attenuates insulin resistance and reduces macrophage infiltration to adipose tissues in mice with high-fat diet- or streptozotocin-induced diabetes (62).
In terms of tonicity-independent mechanisms, the progression of T1D accompanies impaired immune tolerance and subsequent aberrant immune activation. Several studies in animal models showed that T1D could be induced by the interaction among several types of immune cells, including CD4+, CD8+ T cells, and macrophages. In particular, CD8+ T cells are mainly involved in destroying pancreatic β cells through diverse pathways, including IFN-γ production and expression of death receptor FAS (58). IFN-γ produced by CD8+ T cells can drive IL-1β and TNF-α production in macrophages, leading to β cell apoptosis (58). In contrast, Treg cells counterbalance the activation of effector T cells to prevent the onset of T1D (63, 64). For example, studies in a Treg cell-deficient animal model showed broken immune tolerance and exacerbated disease development under diabetes-prone conditions (65). Thus, enhancing the function of Treg cells may be suggested as a potential approach to treating T1D (65). Recently, a new mechanism by which microRNA181a activity reduced the induction of Treg cells to break peripheral tolerance was revealed in a T1D animal model (66). Of interest, miRNA181a promotes the expression of NFAT5 in a tonicity-independent manner, which substantially reduced the frequency of Treg cells. The authors proposed that microRNA181a-mediated NFAT5 interferes with inducing Treg cells and thereby contributes to T1D (66).
Collectively, through tonicity-dependent and -independent mechanisms, NFAT5 regulates the development of T1D, another model of autoimmune disease, which seems to be mediated by the induction of AR (under hypertonic conditions) and expansion of Treg cells (under isotonic conditions). In this respect, targeting NFAT5 signaling using siRNA or small molecules may limit islet autoimmunity.
NFAT5 and Multiple Sclerosis
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) that is also triggered by several environmental factors in genetically susceptible individuals. Similar to RA, autoreactive T cells play a central role in MS pathogenesis; in particular, pathogenic TH17 cells have been implicated in the disease progression (67). Recent studies have suggested that high salt induces the generation of IL-17-producing CD4+ TH17 cells in the CNS and exacerbates the development of experimental autoimmune encephalomyelitis (EAE), a mouse model of MS dependent on TH17 cells (18, 54). Similar to the pathogenic pathway of RA, high salt results in increased p38-MAPK and NFAT5 expression in this model, promoting the expression of SGK1, a downstream target of NFAT5 (54, 57), indicating that EAE is dependent on a high salt-NFAT5-SGK1- TH17 axis. In parallel with the findings in animal models, sodium concentrations in urine from MS patients shows a positive correlation with disease progression, although there was no association between sodium intake and onset risk in pediatric MS patients (57, 68). Taken together, although further investigation is required, aforementioned studies highlight the importance of osmotic NFAT5 in the pathogenesis of MS.
NFAT5 and Tumor Immunity
In addition to autoimmune diseases, accumulating evidence demonstrates that NFAT5 is associated with the progression of various types of tumors, including breast cancer, hepatocellular carcinoma, and lung cancer (69–71). As mentioned above, it has been revealed that high salt promotes inflammatory responses by inducing a variety of pro-inflammatory mediators, including inducible nitric oxide synthases, IL-6, and TNF-α in innate immune cells. Additionally, high salt directly induces the expression of VEGF in breast tumor cells, resulting in angiogenesis and immune dysfunction (70). Interestingly, sodium concentrations in breast tumors were shown to be higher than those in normal cells (70, 72). Accordingly, NFAT5, a representative osmo-sensitive transcription factor, has been proposed to mediate tumor progression. Indeed, a positive correlation was found between the expression of NFAT5 and inflammatory breast cancer (73). NFAT5 expression is dramatically elevated in hepatocellular carcinoma and lung adenocarcinoma cells compared with normal tissues (69, 71). Moreover, inhibiting NFAT5 expression reduces the proliferation and migration of lung adenocarcinoma cells, which is accompanied by suppressing the expression of aquaporin-5 (AQP5), a water transporter depending on the osmotic gradient (69). It also attenuates hepatocellular carcinogenesis, recurrence, and metastasis (71). It is implicated in the relationship of NFAT5 expression in tumor cells and tumorigenesis in terms of tonicity-dependent manners.
López-Rodríguez et al. recently published a tonicity-independent mechanism in which NFAT5-sufficient macrophages drive a pro-inflammatory function to promote IFNγ-expressing TH1 polarization via up-regulating IL-12 production (43). In contrast, NFAT5-deficient macrophages exhibit reduced pro-inflammatory function, resulting in enhanced tumor growth in Lewis lung carcinoma and ID8 ovarian carcinoma models (43). Intriguingly, NFAT5-deficient macrophages show the reduced infiltration of effector CD8+ T cells into the tumor site (43), which is accordant with our findings of NFAT5 regulation of FLS migration (35). Together, in a sharp contrast to the tumorigenic role of NFAT5 in the cancer cells, NFAT5 in immune cells seems to escalate antitumor immunity by polarizing pro-inflammatory macrophages as well as promoting accumulation of CD8+ T cells adjacent to tumor masses. Therefore, in light of the dynamic interaction of cancer cells with microenvironments, it should be judiciously determined what the net in vivo effect of NFAT5 inhibition for cancer progression is.
Perspectives on NFAT5 as a Therapeutic Target of Autoimmune and Inflammatory Diseases
Here, we review the essential role of NFAT5 in immunity and autoimmune diseases. Given the pivotal role of NFAT5 in macrophage activation and survival, Treg cell generation, and TH17 differentiation, it is an appropriate therapeutic target for autoimmune diseases. Indeed, we have demonstrated that transfection of NFAT5 siRNAs into RA synoviocytes and endothelial cells inhibits their proliferation and survival and impedes angiogenic processes (35, 51). Again, mice injected with NFAT5 siRNAs showed reduced streptozotocin-induced diabetic retinopathy (61), suggesting that transcriptional knockdown of NFAT5 using siRNAs can be a useful strategy to treat NFAT5-dependent autoimmune inflammatory diseases including RA and diabetes mellitus.
Some drugs or drug candidates also have been investigated to test their capacity to inhibit NFAT5 expression and/or activation. For example, metformin, a well-known anti-diabetic drug, inhibits the expression of hypertonicity-induced NFAT5 and its downstream target genes, and it enhances the apoptosis of renal medullary interstitial cells in T2D mice (74). Recently, our research group identified two small molecules, KRN2 and KRN5, to specifically suppress NFAT5 expression via a high-throughput screening system; KRN2 is 13-(2-fluorobenzyl)-berberine, a derivative of berberine, and KRN5 is an oral derivative of KRN2 (50). KRN2 selectively inhibits the transcriptional activation of NFAT5 by blocking NF-κB binding to the NFAT5 promoter region, downregulating the expression of pro-inflammatory NFAT5-target genes in macrophages including Nos2 and Il6 (50). Importantly, KRN2 selectively inhibited the activation of inflammatory NFAT5 induced by TLR ligands without hampering high-salt-induced NFAT5 and its target gene expressions (50). Intra-peritoneal or oral administration of KRN2 and KRN5 markedly suppresses the progression of chronic inflammatory arthritis (50). Moreover, KRN2 effectively inhibits the pro-migratory capacity of synoviocytes induced by TGF-β as well as suppressing NFAT5 expression in FLSs (35), suggesting that KRN2 can be a potential therapeutic agent to block FLS invasiveness. Subsequently, Serr at al. demonstrated that injecting KRN2 into diabetes-prone mice dramatically promoted peripheral Treg cells and alleviated the infiltration of auto-immune inflammatory cells in the pancreas (66). Interestingly, KRN2 decreases NFAT5 expression in an infiltrated T cell-specific manner and has no adverse effects on metabolic parameters.
Taken together, we anticipate that anti-NFAT5 blockades (e.g., small molecules including KRN2 or KRN5 and small hairpin RNAs) will be novel candidates in the treatment of autoimmune diseases such as RA, T1D, and MS in which NFAT5 and its targets play a key role. The immunological roles of NFAT5 and the action of NFAT5 siRNA or KRN2 under pathological conditions is summarized in Figure 3.
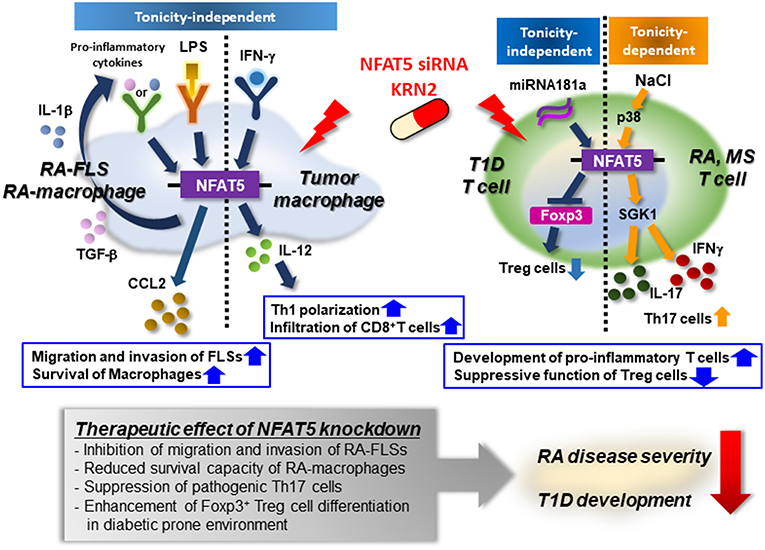
Figure 3. Mode of action of NFAT5 as a therapeutic target for autoimmune diseases. Pro-inflammatory cytokines (e.g., IL-1β and TGF-β) or TLR ligand (e.g., LPS) enhances the expression of NFAT5 in rheumatoid arthritis (RA)-FLSs and RA macrophages. Up-regulated NFAT5, in turn, produces CCL2, promoting RA-FLS migration and invasion and conferring on RA-macrophages apoptotic resistance. IFN-γ promotes NFAT5-dependent IL-12 production, which mediates TH1 polarization, and the infiltration of CD8+ T cells into tumor microenvironments. High salt also increases the expression of NFAT5 via the p38-mediated pathway, resulting in the development of pro-inflammatory T cells that produce IL-17 and/or IFNγ. Furthermore, in diabetes-prone mice, NFAT5 suppresses the differentiation of Foxp3+ Treg cells, breaking peripheral tolerance. As a therapeutic target, NFAT5 siRNA or KRN2 prevents the pathologic action of NFAT5 and alleviates the progression of RA and T1D. Therefore, NFAT5 blockades may be considered a promising strategy to treat autoimmune diseases, including RA and T1D.
Author Contributions
NL, DK, and W-UK designed the concept of the review. NL and DK wrote the manuscript and depicted the figures. W-UK provided critical revisions to the final manuscript. All authors approved the final version of the manuscript.
Funding
This work was supported by grants from the Research Resettlement Fund for the new faculty of Seoul National University, the Liu Inkyung Memorial Endowment Fund through Seoul National University in 2018, and the National Research Foundation of Korea funded by the Ministry of Science and ICT (2015R1A3A2032927, 2015R1C1A2A01055547, 2017R1D1A1B04033009, and 2018R1A1A3A04078559).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank all members of the Center for Integrative Rheumatoid Transcriptomics and Dynamics for their help and advice.
References
1. Miyakawa H, Woo SK, Dahl SC, Handler JS, Kwon HM. Tonicity-responsive enhancer binding protein, a Rel-like protein that stimulates transcription in response to hypertonicity. Proc Natl Acad Sci USA. (1999) 96:2538–42. doi: 10.1073/pnas.96.5.2538
2. Sands JM, Layton HE. The physiology of urinary concentration: an update. Semin Nephrol. (2009) 29:178–95. doi: 10.1016/j.semnephrol.2009.03.008
3. Michea L, Ferguson DR, Peters EM, Andrews PM, Kirby MR, Burg MB. Cell cycle delay and apoptosis are induced by high salt and urea in renal medullary cells. Am J Physiol Renal Physiol. (2000) 278:F209–18 doi: 10.1152/ajprenal.2000.278.2.F209
4. Woo S, Lee S, Kwon MH. TonEBP transcriptional activator in the cellular response to increased osmolality. Pflügers Archiv. (2002) 444:579–85. doi: 10.1007/s00424-002-0849-2
5. Hebinck A, Dalski A, Engel H, Mattei MG, Hawken R, Schwinger E, et al. Assignment of transcription factor NFAT5 to human chromosome 16q22.1, murine chromosome 8D and porcine chromosome 6p1.4 and comparison of the polyglutamine domains. Cytogenet Genome Res. (2000) 90:68–70. doi: 10.1159/000015665
6. Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. (2006) 7:S12. doi: 10.1186/gb-2006-7-s1-s12
7. López-Rodríguez C, Aramburu J, Rakeman AS, Rao A. NFAT5, a constitutively nuclear NFAT protein that does not cooperate with Fos and Jun. Proc Natl Acad Sci USA. (1999) 96:7214–19.
8. Trama J, Lu Q, Hawley RG, Ho SN. The NFAT-Related protein NFATL1 (TonEBP/NFAT5) is induced upon T Cell activation in a calcineurin-dependent manner. J Immunol. (2000) 165:4884–94. doi: 10.4049/jimmunol.165.9.4884
9. Stroud JC, Lopez-Rodriguez C, Rao A, Chen L. Structure of a TonEBP–DNA complex reveals DNA encircled by a transcription factor. Nat Struct Biol. (2002) 9:90. doi: 10.1038/nsb749
10. Eisenhaber B, Sammer M, Lua WH, Benetka W, Liew LL, Yu W, et al. Nuclear import of a lipid-modified transcription factor. Cell Cycle (2011) 10:3897–911. doi: 10.4161/cc.10.22.18043
11. Tong EHY, Guo J-J, Huang A-L, Liu H, Hu C-D, Chung SSM, et al. Regulation of nucleocytoplasmic trafficking of transcription factor OREBP/TonEBP/NFAT5. J Biol Chem. (2006) 281:23870–79. doi: 10.1074/jbc.M602556200
12. Cheung CYK, Ko BCB. NFAT5 in cellular adaptation to hypertonic stress – regulations and functional significance. J Mol Signal. (2013) 8:5. doi: 10.1186/1750-2187-8-5
13. Ferraris JD, Williams CK, Persaud P, Zhang Z, Chen Y, Burg MB. Activity of the TonEBP/OREBP transactivation domain varies directly with extracellular NaCl concentration. Proc Natl Acad Sci USA. (2002) 99:739–44. doi: 10.1073/pnas.241637298
14. López-Rodriguez C, Aramburu J, Jin L, Rakeman AS, Michino M, Rao A. Bridging the NFAT and NF-κB Families: NFAT5 dimerization regulates cytokine gene transcription in response to osmotic stress. Immunity (2001) 15:47–58. doi: 10.1016/S1074-7613(01)00165-0
15. Farquhar WB, Edwards DG, Jurkovitz CT, Weintraub WS. Dietary sodium and health: more than just blood pressure. J Am Coll Cardiol. (2015) 65:1042–50. doi: 10.1016/j.jacc.2014.12.039
16. Ritz E, Koleganova N, Piecha G. Role of sodium intake in the progression of chronic kidney disease. J Ren Nutr. (2009) 19:61–2. doi: 10.1053/j.jrn.2008.10.007
17. Dar HY, Singh A, Shukla P, Anupam R, Mondal RK, Mishra PK, et al. High dietary salt intake correlates with modulated Th17-Treg cell balance resulting in enhanced bone loss and impaired bone-microarchitecture in male mice. Sci Rep. (2018) 8:2503. doi: 10.1038/s41598-018-20896-y
18. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature (2013) 496:518–22. doi: 10.1038/nature11868
19. Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. (2015) 88:776–84. doi: 10.1038/ki.2015.200
20. Luo T, Ji W-j, Yuan F, Guo Z-z, Li Y-x, Dong Y, et al. Th17/Treg imbalance induced by dietary salt variation indicates inflammation of target organs in humans. Sci Rep. (2016) 6:26767. doi: 10.1038/srep26767
21. Alberdi M, Iglesias M, Tejedor S, Merino R, Lopez-Rodriguez C, Aramburu J. Context-dependent regulation of Th17-associated genes and IFNgamma expression by the transcription factor NFAT5. Immunol Cell Biol. (2017) 95:56–67. doi: 10.1038/icb.2016.69
22. Jantsch J, Schatz V, Friedrich D, Schroder A, Kopp C, Siegert I, et al. Cutaneous Na+ storage strengthens the antimicrobial barrier function of the skin and boosts macrophage-driven host defense. Cell Metab. (2015) 21:493–501. doi: 10.1016/j.cmet.2015.02.003
23. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. (2009) 15:545–52. doi: 10.1038/nm.1960
24. Jeong D, Kim HY, Chung DH. Sodium chloride inhibits IFN-gamma, but not IL-4, production by invariant NKT cells. J Leukoc Biol. (2018) 103:99–106. doi: 10.1002/JLB.3A0217-076R
25. Prager P, Hollborn M, Steffen A, Wiedemann P, Kohen L, Bringmann A. P2Y1 receptor signaling contributes to high salt-induced priming of the NLRP3 inflammasome in retinal pigment epithelial cells. PLoS ONE (2016) 11:e0165653. doi: 10.1371/journal.pone.0165653
26. Go WY, Liu X, Roti MA, Liu F, Ho SN. NFAT5/TonEBP mutant mice define osmotic stress as a critical feature of the lymphoid microenvironment. Proc Natl Acad Sci USA. (2004) 101:10673–8. doi: 10.1073/pnas.0403139101
27. Zhang Z, Ferraris JD, Brooks HL, Brisc I, Burg MB. Expression of osmotic stress-related genes in tissues of normal and hyposmotic rats. Am J Physiol Renal Physiol. (2003) 285:F688–93. doi: 10.1152/ajprenal.00028.2003
28. Berga-Bolaños R, Drews-Elger K, Aramburu J, López-Rodríguez C. NFAT5 regulates T lymphocyte homeostasis and CD24-dependent T cell expansion under pathologic hypernatremia. J Immunol. (2010) 185:6624–35. doi: 10.4049/jimmunol.1001232
29. Trama J, Go WY, Ho SN. The osmoprotective function of the NFAT5 transcription factor in T cell development and activation. J Immunol. (2002) 169:5477–88. doi: 10.4049/jimmunol.169.10.5477
30. Berga-Bolaños R, Alberdi M, Buxadé M, Aramburu J, López-Rodríguez C. NFAT5 induction by the pre–T-cell receptor serves as a selective survival signal in T-lymphocyte development. Proc Natl Acad Sci USA. (2013) 110:16091–6.
31. Berry MR, Mathews RJ, Ferdinand JR, Jing C, Loudon KW, Wlodek E, et al. Renal sodium gradient orchestrates a dynamic antibacterial defense zone. Cell (2017) 170:860–74.e19. doi: 10.1016/j.cell.2017.07.022
32. Muller S, Quast T, Schroder A, Hucke S, Klotz L, Jantsch J, et al. Salt-dependent chemotaxis of macrophages. PLoS ONE (2013) 8:e73439. doi: 10.1371/journal.pone.0073439
33. Kim N-H, Choi S, Han E-J, Hong B-K, Choi SY, Kwon HM, et al. The xanthine oxidase–NFAT5 pathway regulates macrophage activation and TLR-induced inflammatory arthritis. Eur J Immunol. (2014) 44:2721–36. doi: 10.1002/eji.201343669
34. Choi S, You S, Kim D, Choi SY, Kwon HM, Kim HS, et al. Transcription factor NFAT5 promotes macrophage survival in rheumatoid arthritis. J Clin Invest. (2017) 127:954–69. doi: 10.1172/JCI87880
35. Lee S, Kong JS, You S, Kwon HM, Yoo SA, Cho CS, et al. Transcription factor NFAT5 promotes migration and invasion of rheumatoid synoviocytes via coagulation factor III and CCL2. J Immunol. (2018) 201:359–70. doi: 10.4049/jimmunol.1701097
36. Halterman JA, Kwon HM, Zargham R, Bortz PD, Wamhoff BR. Nuclear factor of activated T cells 5 regulates vascular smooth muscle cell phenotypic modulation. Arterioscler Thromb Vasc Biol. (2011) 31:2287–96. doi: 10.1161/ATVBAHA.111.232165
37. Johnson ZI, Doolittle AC, Snuggs JW, Shapiro IM, Le Maitre CL, Risbud MV. TNF-alpha promotes nuclear enrichment of the transcription factor TonEBP/NFAT5 to selectively control inflammatory but not osmoregulatory responses in nucleus pulposus cells. J Biol Chem. (2017) 292:17561–75. doi: 10.1074/jbc.M117.790378
38. Hiyama A, Gogate SS, Gajghate S, Mochida J, Shapiro IM, Risbud MV. BMP-2 and TGF-β stimulate expression of β1,3-glucuronosyl transferase 1 (GlcAT-1) in nucleus pulposus cells through AP1, TonEBP, and Sp1: Role of MAPKs. J Bone Miner Res. (2010) 25:1179–90. doi: 10.1359/jbmr.091202
39. Jeong GR, Im SK, Bae YH, Park ES, Jin BK, Kwon HM, et al. Inflammatory signals induce the expression of tonicity-responsive enhancer binding protein (TonEBP) in microglia. J Neuroimmunol. (2016) 295–296:21–9. doi: 10.1016/j.jneuroim.2016.04.009
40. Neuhofer W. Role of NFAT5 in inflammatory disorders associated with osmotic stress. Curr Genomics (2010) 11:584–90. doi: 10.2174/138920210793360961
41. Jauliac S, López-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. (2002) 4:540. doi: 10.1038/ncb816
42. Lee HH, Sanada S, An SM, Ye BJ, Lee JH, Seo YK, et al. LPS-induced NFkappaB enhanceosome requires TonEBP/NFAT5 without DNA binding. Sci Rep. (2016) 6:24921. doi: 10.1038/srep24921
43. Tellechea M, Buxadé M, Tejedor S, Aramburu J, López-Rodríguez C. NFAT5-regulated macrophage polarization supports the proinflammatory function of macrophages and T lymphocytes. J Immunol. (2018) 200:305–15. doi: 10.4049/jimmunol.1601942
44. Drews-Elger K, Ortells MC, Rao A, López-Rodriguez C, Aramburu J. The transcription factor NFAT5 is required for cyclin expression and cell cycle progression in cells exposed to hypertonic stress. PLoS ONE (2009) 4:e5245. doi: 10.1371/journal.pone.0005245
45. Rokvic L, Titze J, Schuh W, Jäck H-M. The importance of the NFAT5/TonEBP-mediated osmotic stress response in B cells. J Immunol. (2017) 198(1 Suppl.):152.5.
46. Kim NH, Hong BK, Choi SY, Moo Kwon H, Cho CS, Yi EC, et al. Reactive oxygen species regulate context-dependent inhibition of NFAT5 target genes. Exp Mol Med. (2013) 45:e32 doi: 10.1038/emm.2013.61
47. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
48. Arend WP, Firestein GS. Pre-rheumatoid arthritis: predisposition and transition to clinical synovitis. Nat Rev Rheumatol. (2012) 8:573–86. doi: 10.1038/nrrheum.2012.134
49. Masuda K, Masuda R, Neidhart M, Simmen BR, Michel BA, Muller-Ladner U, et al. Molecular profile of synovial fibroblasts in rheumatoid arthritis depends on the stage of proliferation. Arthritis Res. (2002) 4:R8. doi: 10.1186/ar427
50. Han EJ, Kim HY, Lee N, Kim NH, Yoo SA, Kwon HM, et al. Suppression of NFAT5-mediated inflammation and chronic arthritis by novel kappaB-binding inhibitors. EBioMedicine (2017) 18:261–73. doi: 10.1016/j.ebiom.2017.03.039
51. Yoon HJ, You S, Yoo SA, Kim NH, Kwon HM, Yoon CH, et al. NF-AT5 is a critical regulator of inflammatory arthritis. Arthritis Rheum. (2011) 63:1843–52. doi: 10.1002/art.30229
52. Sundström B, Johansson I, Rantapää-Dahlqvist S. Interaction between dietary sodium and smoking increases the risk for rheumatoid arthritis: results from a nested case-control study. Rheumatology (Oxford) (2015) 54:487–93. doi: 10.1093/rheumatology/keu330
53. Salgado E, Bes-Rastrollo M, de Irala J, Carmona L, Gómez-Reino JJ. High sodium intake is associated with self-reported rheumatoid arthritis: a cross sectional and case control analysis within the SUN cohort. Medicine (Baltimore) (2015) 94:e924. doi: 10.1097/MD.0000000000000924
54. Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature (2013) 496:513–7. doi: 10.1038/nature11984
55. Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. (2015) 125:4212–22. doi: 10.1172/JCI81151
56. Sharif K, Amital H, Shoenfeld Y. The role of dietary sodium in autoimmune diseases: the salty truth. Autoimmun Rev. (2018) 17:1069–1073. doi: 10.1016/j.autrev.2018.05.007
57. Toussirot E, Béreau M, Vauchy C, Saas P. Could sodium chloride be an environmental trigger for immune-mediated diseases? An overview of the experimental and clinical evidence. Front Physiol. (2018) 9:440. doi: 10.3389/fphys.2018.00440
58. Lehuen A, Diana J, Zaccone P, Cooke A. Immune cell crosstalk in type 1 diabetes. Nat Rev Immunol. (2010) 10:501–13. doi: 10.1038/nri2787
59. Yang B, Hodgkinson AD, Oates PJ, Kwon HM, Millward BA, Demaine AG. Elevated activity of transcription factor nuclear factor of activated T-cells 5 (NFAT5) and diabetic nephropathy. Diabetes (2006) 55:1450–5. doi: 10.2337/db05-1260
60. Brocker C, Thompson DC, Vasiliou V. The role of hyperosmotic stress in inflammation and disease. Biomol Concepts (2012) 3:345–64. doi: 10.1515/bmc-2012-0001
61. Park J, Kim H, Park SY, Lim SW, Kim YS, Lee DH, et al. Tonicity-responsive enhancer binding protein regulates the expression of aldose reductase and protein kinase C delta in a mouse model of diabetic retinopathy. Exp Eye Res. (2014) 122:13–9. doi: 10.1016/j.exer.2014.03.001
62. Lee JY, Jeong EA, Kim KE, Yi CO, Jin Z, Lee JE, et al. TonEBP/NFAT5 haploinsufficiency attenuates hippocampal inflammation in high-fat diet/streptozotocin-induced diabetic mice. Sci Rep. (2017) 7:7837. doi: 10.1038/s41598-017-08319-w
63. Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity (2000) 12:431–40. doi: 10.1016/S1074-7613(00)80195-8
64. Cabrera SM, Rigby MR, Mirmira RG. Targeting regulatory T cells in the treatment of type 1 diabetes mellitus. Curr Mol Med. (2012) 12:1261–72. doi: 10.2174/156652412803833634
65. Tang Q, Bluestone JA. The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. (2008) 9:239–44. doi: 10.1038/ni1572
66. Serr I, Scherm MG, Zahm AM, Schug J, Flynn VK, Hippich M, et al. A miRNA181a/NFAT5 axis links impaired T cell tolerance induction with autoimmune type 1 diabetes. Sci Transl Med. (2018) 10:eaag1782. doi: 10.1126/scitranslmed.aag1782
67. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron (2018) 97:742–68. doi: 10.1016/j.neuron.2018.01.021
68. Farez MF, Fiol MP, Gaitán MI, Quintana FJ, Correale J. Sodium intake is associated with increased disease activity in multiple sclerosis. J Neurol Neurosurg Psychiatry (2015) 86:26–31. doi: 10.1136/jnnp-2014-307928
69. Guo K, Jin F. NFAT5 promotes proliferation and migration of lung adenocarcinoma cells in part through regulating AQP5 expression. Biochem Biophys Res Commun. (2015) 465:644–9. doi: 10.1016/j.bbrc.2015.08.078
70. Amara S, Alotaibi D, Tiriveedhi V. NFAT5/STAT3 interaction mediates synergism of high salt with IL-17 towards induction of VEGF-A expression in breast cancer cells. Oncol Lett. (2016) 12:933–43. doi: 10.3892/ol.2016.4713
71. Lee JH, Suh JH, Choi SY, Kang HJ, Lee HH, Ye BJ, et al. Tonicity-responsive enhancer-binding protein promotes hepatocellular carcinogenesis, recurrence and metastasis. Gut (2018) 68:347–58. doi: 10.1136/gutjnl-2017-315348
72. Sparks RL, Pool TB, Smith NK, Cameron IL. Effects of amiloride on tumor growth and intracellular element content of tumor cells in vivo. Cancer Res. (1983) 43:73–7.
73. Remo A, Simeone I, Pancione M, Parcesepe P, Finetti P, Cerulo L, et al. Systems biology analysis reveals NFAT5 as a novel biomarker and master regulator of inflammatory breast cancer. J Transl Med. (2015) 13:138. doi: 10.1186/s12967-015-0492-2
Keywords: NFAT5, hyperosmolarity, immune regulation, autoimmune diseases, therapeutic target
Citation: Lee N, Kim D and Kim W-U (2019) Role of NFAT5 in the Immune System and Pathogenesis of Autoimmune Diseases. Front. Immunol. 10:270. doi: 10.3389/fimmu.2019.00270
Received: 20 November 2018; Accepted: 31 January 2019;
Published: 19 February 2019.
Edited by:
Takayuki Yoshimoto, Tokyo Medical University, JapanCopyright © 2019 Lee, Kim and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wan-Uk Kim, d2FuNzI1QGNhdGhvbGljLmFjLmty
†These authors have contributed equally to this work